Embed Size (px)
Citation preview

Comparison of Hybrid DensityFunctional, Hartree–Fock, andGW Calculations on NiO
C. H. PATTERSONUniversity of Florida, Quantum Theory Project, P. O. Box 118435, Gainesville, Florida 32611-8435
Received 1 May 2006; accepted 15 May 2006Published online 26 July 2006 in Wiley InterScience (www.interscience.wiley.com).DOI 10.1002/qua.21136
ABSTRACT: The electronic band structure of NiO in the ferromagnetic state iscalculated using the B3LYP hybrid density functional, the Hartree–Fock (HF) method,and the GW approximation (GWA) with dielectric functions constructed using eitherB3LYP or HF wave functions and energy eigenvalues. The band structure from theGWA calculation based on B3LYP wave functions is quite similar to the parent B3LYPband structure; the main differences are in valence bandwidth and in the upward shiftof the empty minority spin Ni 3d bands relative to the same bands in the B3LYPcalculation. The band structure from the GWA calculation based on HF wave functionsdiffers in that there are large upward shifts in valence band positions in the GWAcalculation relative to the HF calculation, which result from screening of the bareexchange term in the Fock operator. Matrix elements of the HF exchange operator areobtained using wave functions from self-consistent HF or B3LYP calculations.Magnitudes of these matrix elements for several states at the � point of the Brillouinzone are compared, and it is found that the only major difference occurs in the emptyminority spin bands derived from Ni 3d states of e symmetry. © 2006 Wiley Periodicals,Inc. Int J Quantum Chem 106: 3383–3386, 2006
Key words: B3LYP functional; GW approximation; nickel oxide; perturbation theory
Introduction
N ickel oxide (NiO) is an anti-ferromagnetictransition metal oxide whose electronic struc-
ture has been studied using a wide range of theo-
retical techniques [1–9]. There have been a numberof ab initio Hartree–Fock (HF) and density func-tional theory (DFT) studies of NiO. It is well knownthat these approaches over- or underestimate thebandgap to a large extent. In fact, the local spindensity approximation (LSDA) to DFT predicts thatNiO in a ferromagnetic (FM) state is metallic [3],while in the anti-ferromagnetic-II (AF-II) state it ispredicted to have a small bandgap of 0.3 eV [8]; thebandgap for the AF-II state is observed to be �4.3
Correspondence to: C. H. Patterson; e-mail: [email protected]
Contract grant sponsor: Irish Higher Education Authority.Contract grant number: PRTLI-HTAC2.
International Journal of Quantum Chemistry, Vol 106, 3383–3386 (2006)© 2006 Wiley Periodicals, Inc.

eV [10]. Gross errors like this are a common prob-lem in mean field ab initio predictions of propertiesof magnetic transition metal oxides. A reliable ap-proach is required that can be used to predict elec-tronic structures of transition metal oxides cor-rectly.
The GW approximation (GWA) can be viewed asa generalization of HF theory in which the HFexchange is screened; practical experience with theGWA would suggest that it is screened to a largeextent (by more than 50%) in materials withbandgaps of �10 eV. The B3LYP functional con-tains an HF exchange with a weight of 0.2, whichcan be regarded as a uniform screening of exchangeby 80%. In contrast, the extent to which the ex-change potential felt by an electron in the GWA isscreened depends on the energy and wave vector ofthe electron. Wave functions and energy eigenval-ues from self-consistent B3LYP calculations maytherefore present good starting points for perturba-tive GWA calculations on transition metal oxides.The GWA has previously been applied to NiOwithin perturbative [1, 2, 5] and self-consistent field(SCF) schemes [9].
The GWA self-energy operator can be repre-sented as a sum of Feynman ring diagramssummed to infinite order. When it is applied as aperturbation theory beginning from a DFT meanfield Hamiltonian, the zeroth-order Hamiltoniancontains the direct mean field Coulomb interaction,denoted �(0) in Figure 1, plus the self-consistentKohn–Sham potential and the bandgap will be un-derestimated. The perturbative correction to theDFT energy eigenvalues, ��nk, consists of the dif-ference in diagonal matrix elements of the GWA
self-energy operator, �, and the Kohn–Sham poten-tial, vxc,
��nk � ��nk�� � vxc��nk�, (1)
�1, 2 � �iGo1, 2W1, 2, (2)
W1, 2 � � d3��11, 3v3, 2, (3)
where �nk is a mean field wave function.In contrast, when a GWA calculation is per-
formed beginning with a HF mean field Hamilto-nian, both the direct Coulomb interaction and ex-change are part of the HF mean field Hamiltonian(i.e., the two diagrams on the left of Fig. 1); this canresult in a gross overestimation of the bandgap. Thefirst terms that enter the self-energy in perturbationtheory, beginning from a HF mean field Hamilto-nian, are the two diagrams on the right in Figure 1,although only ring diagrams are retained in theGWA. In practice, summation of ring diagrams toinfinite order is achieved by inverting a dielectricmatrix [�(1, 2) in Eq. (3)], and the end result is thatthe bare exchange is “screened” by the inverse di-electric matrix.
One of the difficulties in performing ab initiomany-body calculations on transition metal oxidesis that the dielectric matrix contains a denominatorthat depends on the energy differences of occupiedand unoccupied states of the mean field Hamilto-nian. A dielectric matrix constructed using DFTsingle-particle wave functions and energy eigenval-ues will predict too much screening of the exchangeterm in the Hamiltonian, while one derived from aHF Hamiltonian will predict too little. The B3LYPhybrid density functional [11] contains a combina-tion of HF exchange plus Slater and Becke exchangefunctionals, as well as a correlation potential. Theweight of the HF exchange in the B3LYP functionalis 0.2. This functional has been shown to result inpredictions of bandgaps of oxides that comparefavorably with experiment [3]. It has also beenshown that the bandgap of NiO varies nearly lin-early with the proportion of HF exchange, x, in aB3LYP-like family of hybrid functionals [3], whereonly the proportion of HF exchange is modified.Since the bandgap is predicted fairly accurately,wave functions and energy eigenvalues derivedfrom B3LYP calculations are likely to be a betterstarting point for a perturbative GWA calculationthan those from either DFT or HF. In this work, we
FIGURE 1. Contributions to the self-energy up to sec-ond order.
PATTERSON
3384 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 106, NO. 15

present GWA calculations for NiO in the FM statebeginning from B3LYP and HF self-consistentHamiltonians. We also present matrix elements ofthe exchange operator using B3LYP and HF self-consistent wave functions as a measure of the ex-tent to which the corresponding wave functionsdiffer.
HF, B3LYP, and GWA Calculations onNiO
Perturbative GWA calculations were performedfor NiO in the rocksalt structure with a lattice con-stant of 4.1767 Å, using the Crystal [12] code togenerate mean field wave functions and the Excitoncode [13] for the GWA calculations. All electronbasis sets were used for Ni [14] and O ions [14], andthe O basis was supplemented with a d polarizationfunction with exponent 0.5. Wave functions andenergy eigenvalues used to calculate the polariz-ability and dielectric function were derived fromeither unrestricted HF (x � 1.0) or B3LYP (x � 0.2)self-consistent calculations or a version of B3LYP inwhich x � 0.07. GWA band structures and theparent mean field HF or B3LYP band structures areshown in Figures 2 and 3. The dominant electronicconfiguration in NiO is 1 Ni 3d e2t3 O 2p3 2 Ni t3
O 2p3, where arrows indicate relative spin orienta-tion. The degeneracies of occupied valence states atthe � point of the Brillouin zone are therefore 2-, 3-,and 3-fold, respectively, for majority spin states and
3- and 3-fold, respectively, for minority spin states,in order of increasing energy. The atom-projecteddensity of states for NiO shows that most occupiedO 2p states lie at �5 eV below the valence bandmaximum (VBM), which is of d character [4]. At the� point of the Brillouin zone, the highest occupiedstate of FM NiO has O 2p character and lies at �5eV below the VBM. In the B3LYP calculation, thedirect and indirect bandgaps for majority spins are6.8 and 4.4 eV, and for minority spins the direct andindirect gaps are 4.0 and 3.7 eV. There is a singlelow-lying conduction band for the majority spins,while there is a similar state for the minority spinsplus the empty 3d states of e character at the � point.The band structure in the GWA calculation derivedfrom B3LYP wave functions, denoted GWA(B3LYP), and the parent B3LYP band structure arevery similar (Fig. 2). The main difference in theB3LYP and GWA (B3LYP) band structures is thatthere is a significant upward shift of the empty Ni3d bands of e character in the GWA (B3LYP) calcu-lation, compared with the B3LYP calculation; a sim-ilar upward shift was reported recently for a GWAcalculation on NiO which used a GGA-DFT meanfield Hamiltonian as the starting point for the GWAcalculation [5].
The situation is somewhat different in the HFand GWA (HF) band structures: the HF direct andindirect bandgaps are 15.0 and 13.5 eV for majorityspins, and the direct and indirect gaps are both 14.6eV for minority spins. When dielectric screening isadded in the GWA (HF) calculation, there is a large
FIGURE 2. Majority (left) and minority spin (right)band structures for FM NiO. GWA (B3LYP) calculation(solid lines) B3LYP calculation (dashed lines). The Fermilevel for the GWA (B3LYP) calculation is at �3.7 eVand for the B3LYP calculation it is at �3.4 eV.
FIGURE 3. Majority (left) and minority spin (right)band structures for FM NiO. GWA (HF) calculation(solid lines) Hartree–Fock calculation (dashed lines). TheFermi level for the GWA (HF) calculation is at �4.8 eVand for the HF calculation it is at �8.8 eV.
COMPARISON OF HYBRID DFT, HF, AND GW ON NiO
VOL. 106, NO. 15 DOI 10.1002/qua INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 3385
upward shift of the valence bands by �4 eV andsome band narrowing.
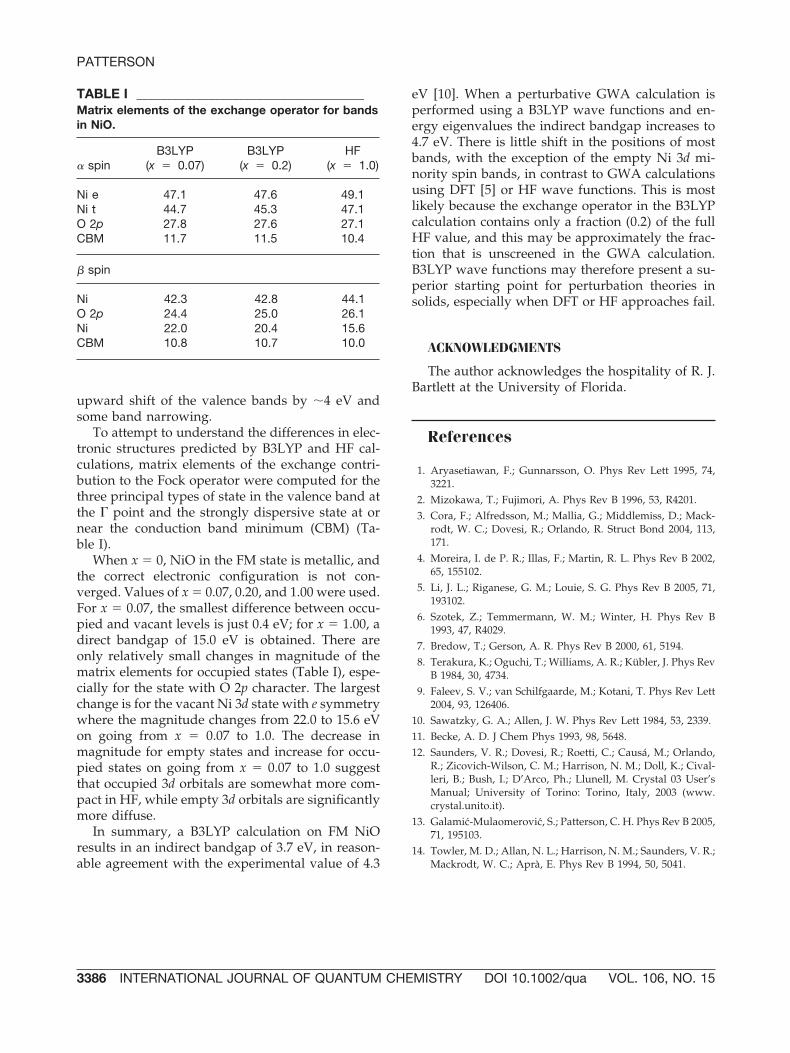
To attempt to understand the differences in elec-tronic structures predicted by B3LYP and HF cal-culations, matrix elements of the exchange contri-bution to the Fock operator were computed for thethree principal types of state in the valence band atthe � point and the strongly dispersive state at ornear the conduction band minimum (CBM) (Ta-ble I).
When x � 0, NiO in the FM state is metallic, andthe correct electronic configuration is not con-verged. Values of x � 0.07, 0.20, and 1.00 were used.For x � 0.07, the smallest difference between occu-pied and vacant levels is just 0.4 eV; for x � 1.00, adirect bandgap of 15.0 eV is obtained. There areonly relatively small changes in magnitude of thematrix elements for occupied states (Table I), espe-cially for the state with O 2p character. The largestchange is for the vacant Ni 3d state with e symmetrywhere the magnitude changes from 22.0 to 15.6 eVon going from x � 0.07 to 1.0. The decrease inmagnitude for empty states and increase for occu-pied states on going from x � 0.07 to 1.0 suggestthat occupied 3d orbitals are somewhat more com-pact in HF, while empty 3d orbitals are significantlymore diffuse.
In summary, a B3LYP calculation on FM NiOresults in an indirect bandgap of 3.7 eV, in reason-able agreement with the experimental value of 4.3
eV [10]. When a perturbative GWA calculation isperformed using a B3LYP wave functions and en-ergy eigenvalues the indirect bandgap increases to4.7 eV. There is little shift in the positions of mostbands, with the exception of the empty Ni 3d mi-nority spin bands, in contrast to GWA calculationsusing DFT [5] or HF wave functions. This is mostlikely because the exchange operator in the B3LYPcalculation contains only a fraction (0.2) of the fullHF value, and this may be approximately the frac-tion that is unscreened in the GWA calculation.B3LYP wave functions may therefore present a su-perior starting point for perturbation theories insolids, especially when DFT or HF approaches fail.
ACKNOWLEDGMENTS
The author acknowledges the hospitality of R. J.Bartlett at the University of Florida.
References
1. Aryasetiawan, F.; Gunnarsson, O. Phys Rev Lett 1995, 74,3221.
2. Mizokawa, T.; Fujimori, A. Phys Rev B 1996, 53, R4201.3. Cora, F.; Alfredsson, M.; Mallia, G.; Middlemiss, D.; Mack-
rodt, W. C.; Dovesi, R.; Orlando, R. Struct Bond 2004, 113,171.
4. Moreira, I. de P. R.; Illas, F.; Martin, R. L. Phys Rev B 2002,65, 155102.
5. Li, J. L.; Riganese, G. M.; Louie, S. G. Phys Rev B 2005, 71,193102.
6. Szotek, Z.; Temmermann, W. M.; Winter, H. Phys Rev B1993, 47, R4029.
7. Bredow, T.; Gerson, A. R. Phys Rev B 2000, 61, 5194.8. Terakura, K.; Oguchi, T.; Williams, A. R.; Kubler, J. Phys Rev
B 1984, 30, 4734.9. Faleev, S. V.; van Schilfgaarde, M.; Kotani, T. Phys Rev Lett
2004, 93, 126406.10. Sawatzky, G. A.; Allen, J. W. Phys Rev Lett 1984, 53, 2339.11. Becke, A. D. J Chem Phys 1993, 98, 5648.12. Saunders, V. R.; Dovesi, R.; Roetti, C.; Causa, M.; Orlando,
R.; Zicovich-Wilson, C. M.; Harrison, N. M.; Doll, K.; Cival-leri, B.; Bush, I.; D’Arco, Ph.; Llunell, M. Crystal 03 User’sManual; University of Torino: Torino, Italy, 2003 (www.crystal.unito.it).
13. Galamic-Mulaomerovic, S.; Patterson, C. H. Phys Rev B 2005,71, 195103.
14. Towler, M. D.; Allan, N. L.; Harrison, N. M.; Saunders, V. R.;Mackrodt, W. C.; Apra, E. Phys Rev B 1994, 50, 5041.
TABLE I ______________________________________Matrix elements of the exchange operator for bandsin NiO.
� spinB3LYP
(x � 0.07)B3LYP
(x � 0.2)HF
(x � 1.0)
Ni e 47.1 47.6 49.1Ni t 44.7 45.3 47.1O 2p 27.8 27.6 27.1CBM 11.7 11.5 10.4
� spin
Ni 42.3 42.8 44.1O 2p 24.4 25.0 26.1Ni 22.0 20.4 15.6CBM 10.8 10.7 10.0
PATTERSON
3386 INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY DOI 10.1002/qua VOL. 106, NO. 15





























