Embed Size (px)
Citation preview

Comparative Modelling Between Hc-Stp1 And Crystal
Structure Protein Phosphatase 1(PP1) Using 3e7a As a
Template.
BIOINFORMATICS
7307 BPS
BALVINDER KAUR MIHIDA SINGH
2819317
2012

ABSTRACT.
This study was mainly done to structurally analyse the parasite protein and to construct a
design for the protein. In order to choose the right template, the Hc-STP-1 gene product was
Blast against the non redundant nucleotide to get the protein sequence. (ADJ96628).
Trichostrongylus vitrinus had the highest sequence homology agains Haemonchus contortus.
Hc-STP-1 is devided into 4 categories PP1, PP2, PP2B, and PP2C. Studies have shown high
similarities in its active site. Therefore 3e7a was taken as the template for Hc-STP1 due to
Protein protease 1 (PP1) which is bound to Nodularin-R. Multiple sequence alignment was
generated using Hc-Stp1 and 3e7a in CLUSTAL W. Then, the secondary structure was
predicted using PSIPRED for both Hc-Stp1 and 3e7a. The secondary prediction was used to
highlight the Alpha-helixes and Beta-strands which iis used to predict the catalytic domain.
For Hc-Stp1 the sequence starts at 1 and ends at 294. For the template, the sequence starts
from 7 and ends at 299. An input file is then crated which consist of pir file, inp file and a
pdb file which will be used in MODELLER 9.10 to compute 20 models and the lowest
energy was selected which was 1555.4291. The geometry of the final volume was predicted
using Ramachandran plot which showed 3 outliers. Overall, further investigation needs to be
done, to evaluate the outliers because it can’t be used as a target for antihelmint therapy.

CHAPTER 1
INTRODUCTION
1.1 Introduction to Haemonchus contortus.
Haemonchus contortus is a nematode parasite of the small ruminant from the order
Strongylida and the family Trichostrongylidae. It is also known as wire or barber’s pole
worm. Haemonchus contortus infacts goats and sheeps. Its larvae has four stages L1, L2, L3
and L4. The first two stages L1 and L2 of the larvae is known as rhabditiform and once it
transforms into the third stage L3, it becomes an infective stage known as filiariform. At this
stage it usually can be found on grass which the goats ingest. In the goats abomasums the
third stage larvae will transform to forth stage L4 which is the adult stage. In its adult form
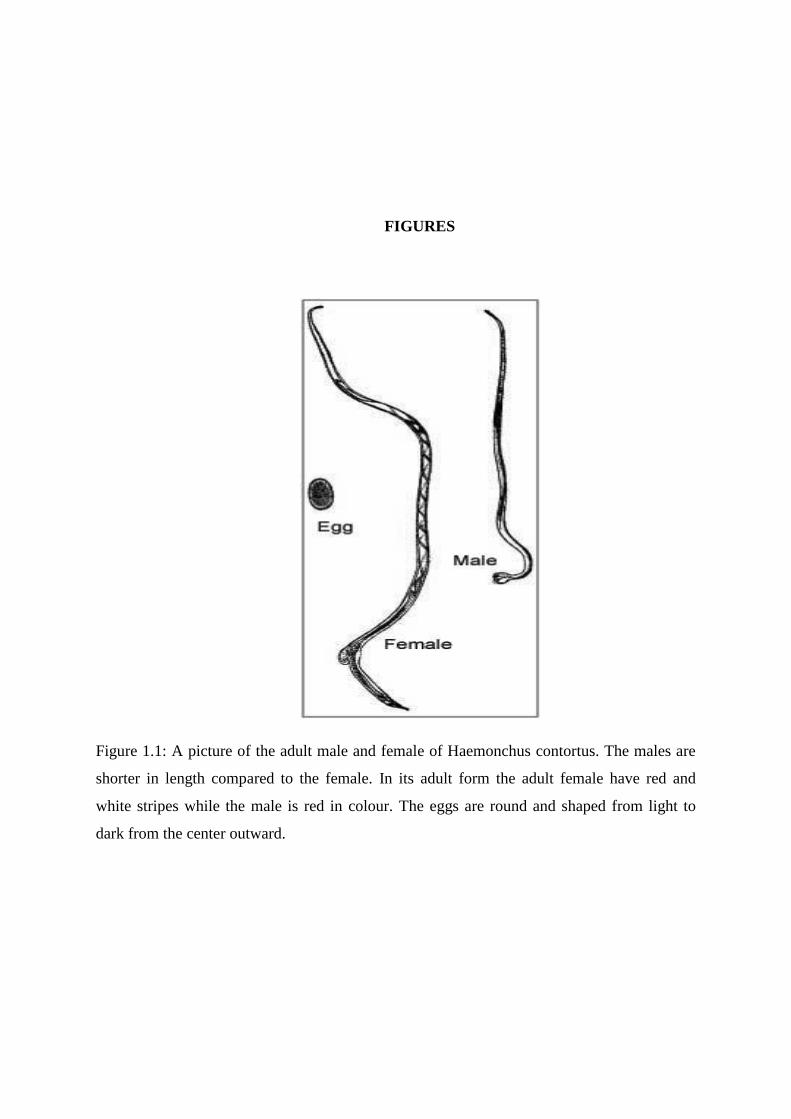
the adult female have red and white stripes while the male is red in colour. (Figure 1.1)
A recent study done by Bronwyn on a full lenght complementary DNA encodes for a
serine/threonine phosphatase (Hc-STP-1) was shown in adult male and its fourth stage larvae
and not in the female. In this case bioinformatics is used to further understand the molecular
biology of Haemonchus contortus(Campbell et al., 2010)
According to Peter, Haemonchus contortus in recent years has shown resistance to
antihelmint drugs. One of the main reasons to why this particular nematode needs to be
focused on is because it is the most pathogenic parasite of the small ruminant which has
become more common in northern Europe. Its free living stage does not suit to cold and dry
climate. When an individual is infected, mostly results in mixed infection with other
nematode parasites(Waller and P., 2005)

1.2 Background of STP-1.
Serine/threonine phosphatase 1 (STP-1) can be classed into 4 categories of proteins which
are PP1, PP2, PP2B and PP2C. PP1 and PP2 are holoenzymes where these proteins require
catalytic protein and regulating protein to be linked together for the targeting and regulation
of their activity. Looking at the catalytic site, the structural difference is only identified in the
ligand –binding interface during the 3 dimensional structure modelling.
On the other hand, Protein Phosphatase (PP), one of its important fundamental is
Phosphorylation/ dephosphorylation of protein. Protein phosphatase is usually involved in
cell diviation, ion channel electrophysiology, neuronal activity, apotosis, and exocytosis.
Protein phosphatase then can be further categorised into two types, tyrosine phosphatase and
serine/threonine phosphatase which is located in the cytoplasm of the cell. Its main function
is in signalling transduction/ transcriptional activation. It works when protein kinase transfers
ATP to phosphate and then further into protein. So, it is important to develop a technique for
the functional analysis of STPs and PP which will enable the insights of the biological
target(Campbell et al., 2011)
1.3 Gene and protein.
The gene of Haemonchus contortus was taken from genbank its accession number is
GQ 280009. It’s a messenger RNA (mRNA) of 951bp with an e-value of 0.0 and an identity
of 100%. (Figure 1.3.1).
This gene is specifically transcribed in males of adult and larvae stage 4 but not in the
adult female and larvae stage. It has identity of 50-90% to a wide range of taxonomic groups
such as amoebae, amphibians, arthropods, choanoflagellate, chordates, echinoderms, fish,
fungi, mammals, nematodes, plants, plathyhelminths, protozoa, and yeast. Its gene is also
transcribed in the same manner as Trichostrongylus vitrinus Tv-Stp-1 and also
Oesophagostomum dentatum Od-mpp-1

Protein found in genbank is 316 a.a long and its accession number is ADJ96628. Protein
location is fron 1..316 and its product is serine/threonine phosphatase 1from the family of
metallophosphatase superfamily. (Figure 1.3.2)
Hc-STP-1 is usually involved in metal ion binding and protein donation for catalytic
activity. In addition to this, it also has high sequence identity to Caenorhabditis elegans which
reveals a presence of conserved motifs.
1.4 Objective
Objective of this assignment is to structurally analyse the parasite protein and to construct
a design for the protein.

CHAPTER 2
MATERIALS AND METHODS.
2.1 Materials.
Table 2.1: List of materials used to analyse the protein.
Link.
BLAST (p and n) http://blast.ncbi.nlm.nih.gov/
Protein Data Bank (PDB) http://www.rcsb.org/pdb/home/home.do
Pfam http://pfam.sanger.ac.uk/
SMART http://smart.embl-heidelberg.de/
Sequence alignment:
CLUSTAL W
http://www.ebi.ac.uk/Tools/msa/clustalw2/
PSIPRED http://bioinf.cs.ucl.ac.uk/psipred/
MODELLER. To generate a homology model
http://salilab.org/modeller/
Pymol and chimera Download to visualize the pdb files

2.2 Method.
Protein structures can be categorized into 4 stages which are primary structure,
secondary structure, tertiary structure and quaternary structure.
2.2.1 Primary structure.
Primary structure is the simplest level with amino acid residues linked together by
peptide bond. The gene product that was given was H.contortus Stp-1, the nucleotide
sequence was Blast using Blast n and gene prediction seen in genbank , accession number
GQ 280009. The sequence belonged to the nematode parasite Haemonchus contortus which
is 951bp long and its product is serine/threonine phosphatase 1 (STP-1). Once the organism
was identified the protein sequence was taken in fasta format, accession number ADJ96628
which is 316 a.a long. When the protein sequence is blast using blast p, Trichostrongylus
vitrinus (accession number CAM84509) has the closest identity to Haemonchus contortus,
with the maximum identity of 91% and e-value of 0.0.
Since there was a study done by Campbell et al., 2010, which indicates that
Trichostrongylus vitrinus and Haemonchus contortus have maximum homology since the
product is Tv-Stp-1 and it’s from the same family as Hc-Stp-1 which is MPP_Superfamily,
Metallophosphatase superfamily. Sequence is then, analysed using pfam to see the conserved
domain and SMART to see the trasmembrane. .
When the structures were analysed by Campbell et al., 2010, an appropriate structural
template is selected. This is the first step of protein structural modelling. A pdb template of
3e7a was used. This code is taken from the protein data bank (PDB). 3e7a template is said to
have a homology model for Hc-STP-1 and Tv-STP-1. The active site and the catalytic
residues were conserved which infers an enzymatic activity consistent with serine/threonine
phosphatase. (Campbell et al., 2010)

Then, a Position Specific Iterated Blast (PSI-BLAST), was done to see the
difference between 1s70 and 3e7a. 1s70 is a Chain A, complex between protein
Serine/threonine phosphatase (Delta) and The myosin phosphatase targeting subunit 1
(Mypt1), whereas, 3e7a is Chain A, crystal structure of protein phosphatase-1 bound to the
natural toxin Nodularin-R. The templates were analysed using Pymol and Chimera.
Using the Hc-STP-1 sequence from genbank accession number ADJ96628 and the
sequence of 3e7a from the protein data bank (PDB) accession code 3e7a a multiple sequence
alignment was done using CLUSTAL W. Once the alignment is collected, the individual
sequence is then run using PSIPRED to get its secondary structure.
2.2.2 Secondary structure.
Secondary structure is used to do local conformation of a peptide chain. It is a
highly regular and repeated arrangement of amino acid residues stabilized by hydrogen bond
between carbonyl oxygen and amino hydrogen which will be stabilized by noncovalent
forces. Its main element is the α-helices, β-sheets and coils. PSIPRED is a web based
program that predicts protein secondary structure using evolutionary information and neutral
networks. The alignment is derived fron PSI-Blast database search(Xiong, 2006).
2.2.3 Tertiary structure and Quaternary structure.
Once the secondary structure has been predicted, pir file, Inp file and a pdb file
containing atoms are made which will be used in MODELLER and compute 20 models to
generate a tertiary structure. A tertiary structure is a three dimensional arrangement of various
secondary structural elements and connecting region which assembly the amino acid of a
single polypeptide chain. Homology modelling which predicts the protein structure based on
sequence homology with known structures(Xiong, 2006)
Generation of a homology model is done using MODELLER the three main fails
are needed which are a pir file, inp file and a pdb file with the atoms of the known protein.
Then, the lowest energy is selected. A Quarternaty structure will be generated. Quaternary
structure refers to the association of several polypeptide chains into a polypeptide chains
called monomers or subunits. Finally the geometry of the final model is checked using
Ramachandran plot.

CHAPTER 3
RESULTS
3.1 Gene and protein.
Hc-STP-1 have a high sequence homolygy to Tv-STP-1( Figure3.1.1) with an e-
value of 0.0 and a maximum identity of 91%. Hc-STP-1 has a function of dissecting
phosphatase based cell functions and signalling pathways. In addition to this, it is also used as
a treatment for cancer due to the lead compound in the protein (Kelker et al., 2009)
There was 1 significant domain when the protein sequence is run in Pfam (Figure
3.1.2) the significant domain found was metallophos which is a calcineurin- like
phosphoesterase. Its alignment start from 52 to 246 with a bit score of 145.6 and an e-value
of 1.1e-42. The domain has a predicted active site of 119 with coordinates from 51 to 247.
The most active site for this conserved region is the metal chelating residue. One of the
drawback od Pfam is that it misses out on the transmembrane domain.
SMART showed a domain with the query sequence of 316 residues known as
PP2Ac domaim (Figure 3.1.3) from position 24 to 295 with an e-value of 3.20e – 150. Its a
protein phosphatase 2A homologues catalytic domain from the large family of
serine/threonine phosphatase that includes PP1, PP2A, and PP2B (calcineurin).PP2A is a
trimeric enzyme that consist of a core catalytic subunit. Protein phosphorylation has a major
role in regulationg the cell function. Kinase and phosphatase are the major enzymes that are
involved (Stone et al., 1987)

3.2 Structure predictions.
Since there was significance between Hc-STP-1 and Tv-STP-1, the pdb code for
this protein is 1s70 (Figure 3.2.1) was taken from protein data bank (PDB). Its structure has 2
chains A and B from Homo sapiens. The A chain is a serine/threonine phosphatase PP1-beta
catalytic subunit and 130 kDa myosin-binding subunit of smooth muscle myosine
phosphatase for chain B. Compared to the pdb code 3e7a (Figure 3.2.2) which has 4 chains
A, B, C, and D. Chain A and B is a serine/threonine phosphatase PP1-alpha catalytic subunit
and its chain C and D is a Nodularin-R from homo sapiens presenting an anti parallel β-sheet
when visualized using Pymol.
Hc-STP-1 protein sequence is blast again using blast p but using position specific
Iterated Blast (PSI-BLAST) to see the comparison between this two pdb codes. (Table 3.2.1)
3e7a has shown a better homology of 57%. Further comparison was done by calculating the
mach using Chimera (Figure 3.2.3). Both the pdb code had a match.
Table 3.2.1: Difference between PDB accession code 1s70 and 3e7a using PSI-BLAST .
PDB accession code. E-value Maximum identity
(%)
1s70 1e-121 56%
3e7a 2e-120 57%
The template 3e7a encodes for Protein Phosphatase 1(PP1) which functions in tissues
and regulates pathway ranging from cell cycle progression to carbohydrate metabolism.
Previous studies have shown that PP1 has advantages to be used as a therapeutic agent for
cancer. Most widely studies classes of PP1 first is the cyclic hepta-peptide microsystic sp and
Nodularia sp. Second is the Ocadaic acid COA , polyether fatty acids from the marine dino-
flagellates prorocentrum sp and dinophysis. Third is calyculin A octamethyl
polyhydroxylated fatty acids from marine sponges. Catalytic subunit of PP1 consist of 10α-
helices and 3 β-sheets which consist of 14 β-strands. PP1 has three major active sites which
are the hydrophobic groove, C-terminal and acidic groove (Kelker et al., 2009)

Multiple sequence alignment using CLUSTAL W was used to predict the sequence
alignment for Hc-STP-1 and 3e7a (Figure 3.2.4). The alignment can be said thet it was well
conserved. A secondary prediction was made using PSIPRED for Hc-STP-1 and 3e7a
(Figure 3.2.5) and (Figure 3.2.6) and the α-helices and the β-strands was highlighted on the
sequence alignment (Figure 3.2.7). From this alignment a pir file (Figure 3.2.8), an inp file
(Figure 3.2.9) and an atom file from PDB are made. Independent homology models were then
computed with Modeller 9.10. Twenty models were predicted and the lowest energy was
taken to obtain a structure (Figure 3.2.10). In this case the lowest energy was produced at
B99990020 of 1555.4291. This lowest energy gave a quaternary structure which was named
Hc-Stp1_3e7a.pdb (Figure 3.2.11). Ramachandran plot was done (Figure 3.2.12) to complete
this modelling and evaluate the overall geometry of the structure. It’s a two dimentional
scatter plot showing torsion angles of each amino acid. Number of residues in the favoured
region was 279 (95.5%), number of residues in the allowed region was 10 (3.4%) and number
of residues in outlier region was 3 (1%).

CHAPTER 4
DISCUSSION.
Studies have shown that PP1, PP2A, and PP2B have highly similar active site. Due to
this highly similar active site, There are three factors to this, first the binding of the molecular
toxin to the PP active site, second is the interaction of the molecular toxin with β-12 to β-13
loop which is situated at residue 268 to 281 of PP1 in the template 3e7a, and third is the
molecular toxin with the hydrophobic groove. In addition to this the template 3e7a was used
for this analysis because PP1 provides multiple significance for serine/threonine protein
phoshatase- specific inhibitors to be generated. These inhibitors are highly selective for PP1
holoenzymes.
Since they require PP1 and PP2 to bind to the template, 3e7a consist of molecular
toxin that modulates PP1 activity. Hence, a structure based alignment was generated using
the human PP1 alpha catalytic subunit. Till date, all reported PP1 structures are homologus
despite it has been crystallized in disparate crystallization conditions or by forming crystals in
different space groups or crystallized with different ligands. Due to the lack of changes, 3e7a
was chosen to be the right template (Kelker et al., 2009)
Ramachandran plot was made and resulted in Number of residues in the favoured
region was 279 (95.5%), number of residues in the allowed region was 10 (3.4%) and number
of residues in outlier region was 3 (1%). The 3 outliers were Leucine, Asparagine and
Threonine. As we know only glycine in the outliers are acceptable, but not the others. If there
is presence of outliers, the structure needs to be corrected. In this case, leucine is a

hydrophobic amino acid, threonine helps maintain the protein balanceand it plays a major
role in the human system by helping the production of antibodies.
This PDB model is not a good model to be used as a target for drug usage due to the
outliers. Despite the fact that the catalytic residues between Hc-Stp1 and the template 3e7a
were highly conserved they are still not a good target for drug usage. One factor that may be
the reason to this is that the template 3e7a is shorter than the target sequence Hc-Stp1.
Another factor can be due to the N-terminal and the C-terminal which were suppose to bind
to the protein. It could be that these terminals can’t regulate their activity when constructing
a therapeutic drug. Another reason could be the polar or the hydrophobic residues in the core
of the protein will minimize the contact with the hydrophobic residues.
Previous study shows that the PP1 gene in Hc-Stp1 encodes for approximately 50%
phosphatase and 30% kinase which is linked to the sperm production in the nematode parasite
Haemonchus contortus. There is currently no effective approach for investigating the gene of
this particular nematode. The reason to this is still not clear. Perhaps by reflecting the
pathways for growth, development and survival of the nematode could be further investigated
as there is still a wide area of problem with the antihelmint resistance in Haemonchus
contortus (Campbell et al., 2010)
There is no quaternary structure for this protein because only one chain was analysed.
One major disadvantage of this protein is that it’s a large protein, thats the reason to why only
part of the sequence is used which interacts with the active site. The crystal structure at
resolution 1.63 is said to have properties that will increase the production of PP1(Kelker et
al., 2009). So in order to target an appropriate antihelmint drug another template will have to
be used or the alignment of the target and template will be modified. A higher resolution may
have a positive effect to the protein.
Nowadays there are automated modelling which is used to predict a model. In this
case, it can be applied, but there are advantages as well as disadvantages. Advantage is that it
is fast and chances of error during creating input files can be avoided. Its disadvantage is that,
won’t be able to master the technique in making and correcting the input files. Further
analysis should be done to predict a suitable antihelmint therapy. Overall the objective of this
analysis was achieved.
FIGURES
Figure 1.1: A picture of the adult male and female of Haemonchus contortus. The males are
shorter in length compared to the female. In its adult form the adult female have red and
white stripes while the male is red in colour. The eggs are round and shaped from light to
dark from the center outward.

Figure 1.3.1: shows the gene in Genbank, its accession number is GQ 280009, from the
organism Haemonchus contortus, it’s an mRNA of 951bp. The gene product is
Serine/threonine phosphatase 1.

Figure 1.3.2: Haemonchus contortus protein sequence with 316 a.a long and its accession
number is ADJ96628. Its product is Hc-STP-1 and at region 6..294. Its amino acid sequence
will be obtained in FASTA format.

Figure 3.1.1: Results from BLAST indicates that there is high sequence similarities between
Hc-STP-1 accession number ADJ96628 and Tv-STP-1 accession number CAM84509. There
is a maximum identity of 91%. A PSI-BLAST later was done to see if it’s suitable to be used
as a template.
Figure 3.1.2: Results obtained from Pfam to evaluate the presence of significant domains.
Significant domain found was metallophos which is a calcineurin- like phosphoesterase. Its
alignment start from 52 to 246 with a bit score of 145.6 and an e-value of 1.1e-42. The
domain has a predicted active site of 119 with coordinates from 51 to 247. The most active
site for this conserved region is the metal chelating residue.
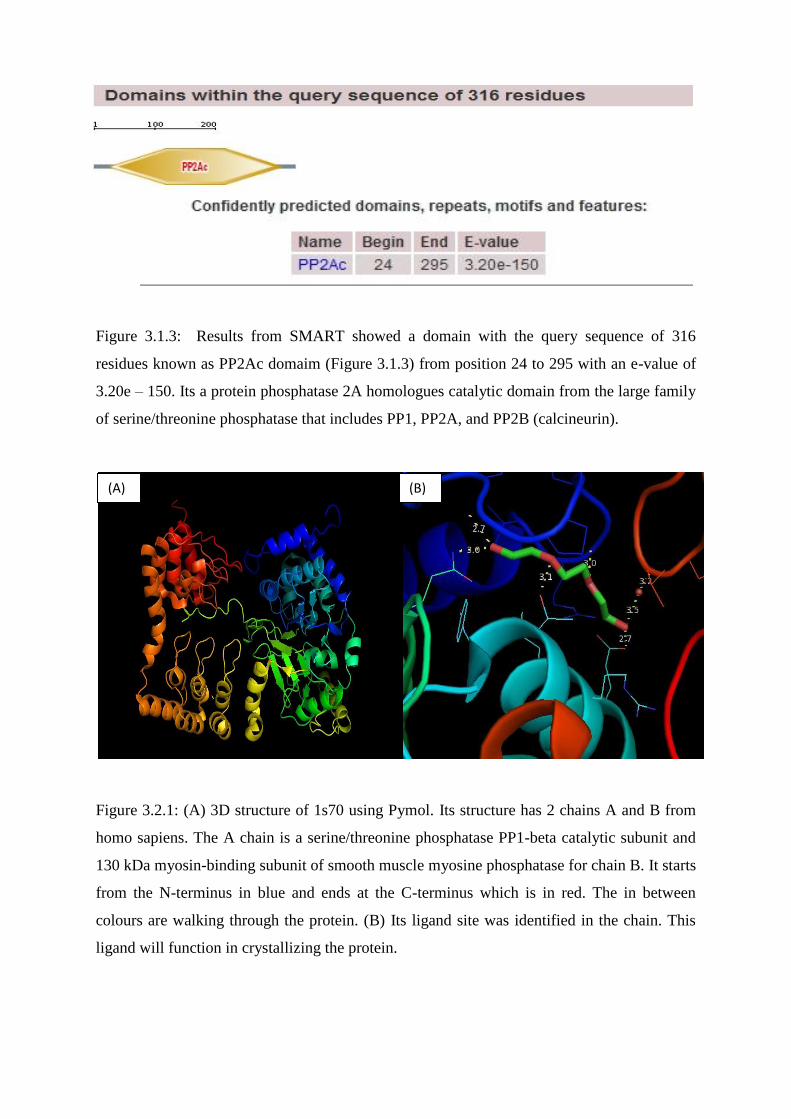
Figure 3.1.3: Results from SMART showed a domain with the query sequence of 316
residues known as PP2Ac domaim (Figure 3.1.3) from position 24 to 295 with an e-value of
3.20e – 150. Its a protein phosphatase 2A homologues catalytic domain from the large family
of serine/threonine phosphatase that includes PP1, PP2A, and PP2B (calcineurin).
Figure 3.2.1: (A) 3D structure of 1s70 using Pymol. Its structure has 2 chains A and B from
homo sapiens. The A chain is a serine/threonine phosphatase PP1-beta catalytic subunit and
130 kDa myosin-binding subunit of smooth muscle myosine phosphatase for chain B. It starts
from the N-terminus in blue and ends at the C-terminus which is in red. The in between
colours are walking through the protein. (B) Its ligand site was identified in the chain. This
ligand will function in crystallizing the protein.
(A) (B)

Figure 3.2.2: 3e7a has 4 chains A, B, C, and D. Chain A and B is a serine/threonine
phosphatase PP1-alpha catalytic subunit and its chain C and D is a Nodularin-R from Homo
sapiens. For all its chain it has an anti parallel β-sheet. The loop region helps to bind the
protein. It has several left handed helixes. It starts from the N-terminus in blue and ends at the
C-terminus which is in red. The in between colours are walking through the protein.
Figure 3.2.3: Comparison was done between 1s70 (grey) and 3e7a (blue). Both the pdb code
was analysed using Chimera program. Using the match maker, a match was made and the
results were that both the pdb code looked homologus, so it can be said that it was from the
same protein.

Figure 3.2.4: once the template was selected, multiple sequence alignment was done using
Clustal W, it can be said that the alignment is well conserved. The top alignment of
ADJ96628 represents Hc-Stp1 and the lower alignment represents the template used which is
3e7a. The results obtained are said to be well conserved because (*) represents a match
between the amino acids, (:) means that there is no match, but it shares the amino acid
properties, it’s properties matches to a very high extent, and (.) means it’s not a match but
there are very slight similarities in the amino acid properties.(-) represents the gaps or
mismatches. Phylogenetic tree, there were no didtance between the two sequence, both
showed a result of 0.22241.

Figure 3.2.5: Secondary structure prediction for 3e7a using PSIPRED. ( H ) represents
the α-helix in the amino acid sequence, as shown above, the the highlighted region is above
the amino acid ( E) represents the β-strands which will show the pattern of hydrophobic
and hydrophilic regions. (C) represents the Coiled coils region. This secondary structure is
then taken and highlighted on the amino sequence aligned using ClustalW based on the
colour codes.

Figure 3.2.6: Secondary structure prediction for Hc-Stp1 using PSIPRED. ( H)
represents the α-helix in the amino acid sequence, as shown above, the the highlighted region
is above the amino acid. ( E) represents the β-strands which will show the pattern of
hydrophobic and hydrophilic regions. (C) represents the Coiled coils region. This secondary
structure is then taken and highlighted on the amino sequence aligned using ClustalW based
on the colour codes.

Hc-Stp1 -----MDPTQLITNLLNVGLPDKGLTKTVSENDIMEVLGKAREMFLSQPP
3E7A GHMGSLNLDSIIGRLLEVQGSRPGKNVQLTENEIRGLCLKSREIFLSQPI
Hc-Stp1 MVELDSPVKICGDTHGQYIDLLRLFNKGGFPPLSNYLFLGDYVDRGKQNL
3E7A LLELEAPLKICGDIHGQYYDLLRLFEYGGFPPESNYLFLGDYVDRGKQSL
Hc-Stp1 EVILLMIAYKLRFPKNFFLLRGNHECANVNRAYGFYEECNRRYQSQRMWQ
3E7A ETICLLLAYKIKYPENFFLLRGNHECASINRIYGFYDECKRRYN-IKLWK
Hc-Stp1 AFQDVLCVMPLTALVSDKILCMHGGLSPHLQSLDQLRNITRPTDALGATL
3E7A TFTDCFNCLPIAAIVDEKIFCCHGGLSPDLQSMEQIRRIMRPTDVPDQGL
Hc-Stp1 EMDLLWADPVIGLNGFQANIRGASYGFGPDILAKYCQLLNIDLVARAHQV
3E7A LCDLLWSDPDKDVQGWGENDRGVSFTFGAEVVAKFLHKHDLDLICRAHQV
Hc-Stp1 VQDGYEFFGGRKLVTIFSAPHYCGQFDNAAAMMTVDENLQCSFDAFRPSC
3E7A VEDGYEFFAKRQLVTLFSAPNYCGEFDNAGAMMSVDETLMCSFQILKPAD
Hc-Stp1 AKPQPKIVATSMGSPGAPPCQ
3E7A ---------------------
Alpha-helix
Beta-strand
Figure 3.2.7: This is a structured based sequence alignment in word format, as shown above
there are high similarities between target Hc-Stp1 and the template 3e7a. The green
highlighted regions are the alpha-helix and the red highlight regions are beta-strand. It has a
N-terminal and C-terminal. Hc-Stp1 is longer than 3e7a. The enzymatic domain is sitting
from the start till the end.
>P1; Hc-Stp1
sequence:Hc-Stp1:1:A: 294:A: Hc-Stp1:H.contortus:0:0
MDTPQLITNLLNVGLPDKGLTKTVSENDIMEVLGKAREMFLSQPP
MVELDSPVKICGDTHGQYIDLLRLFNKGGFPPLSNYLFLGDYVDRGKQNL
EVILLMIAYKLRFPKNFFLLRGNHECANVNRAYGFYEECNRRYQSQRMWQ
AFQDVLCVMPLTALVSDKILCMHGGLSPHLQSLDQLRNITRPTDALGATL
EMDLLWADPVIGLNGFQANIRGASYGFGPDILAKYCQLLNIDLVARAHQV
VQDGYEFFGGRKLVTIFSAPHYCGQFDNAAAMMTVDENLQCSFDAFRPS*
>P1;3E7A
structureX:3E7A:7:A:299:A:3E7A:H.sapiens:1.63:0
LNLDSIIGRLLEVQGSRPGKNVQLTENEIRGLCLKSREIFLSQPI
LLELEAPLKICGDIHGQYYDLLRLFEYGGFPPESNYLFLGDYVDRGKQSL
ETICLLLAYKIKYPENFFLLRGNHECASINRIYGFYDECKRRYN-IKLWK
TFTDCFNCLPIAAIVDEKIFCCHGGLSPDLQSMEQIRRIMRPTDVPDQGL
LCDLLWSDPDKDVQGWGENDRGVSFTFGAEVVAKFLHKHDLDLICRAHQV
VEDGYEFFAKRQLVTLFSAPNYCGEFDNAGAMMSVDETLMCSFQILKPA*
Figure 3.2.8: Pir file has two important parts, first is the known target and second is the
template. Title must be given forth Modeller program to know which is the target sequence
and which is the template, Command line for Hc-Stp1 consist of residue number which is 1
Catalytic
domain
Start
End
Title
Command line
Command line
Amino acid sequence
Amino acid sequence

that indicates the first residue in the alignment. Followed by chain id which is ‘A’, because
we are using the ‘A’ chain not ‘B’, followed by the last residue number in the alignment
which is 294. PDB file has been used as the major reference to create this command line.
Sequence is from the organism H.contortus with resolution of structure 0.0. Amino acid
sequence is attached and at the end will have to add (*) which indicated the end of the
command. Command line for 3e7a consist of residue number which is 7 that indicates the
first residue in the alignment. Followed by chain id which is ‘A’, followed by the last residue
number in the alignment which is 299. Sequence is from the organism Homo sapiens with
resolution of structure 1.63.0. Amino acid sequence is attached and at the end will have to
add (*).
from modeller import *
from modeller.automodel import *
env = environ()
a = automodel(env, alnfile='Hc-Stp1_3E7A.pir',
knowns='3E7A', sequence='Hc-Stp1',
assess_methods=(assess.DOPE, assess.GA341))
a.starting_model = 1
a.ending_model = 20
a.make()
Figure 3.2.9: an input script will be needed for MODELLER. Have to add cooments such as
the name of the pir file and the pdb file containing atoms for 3e7a. In this case the pir file
was saved as Hc-Stp1_3E7A.pir and the pdb file was saved as 3E7A. The sequence used for
the study was Hc-Stp1, and 20 models were calculated to get the energy level using
MODELLER.
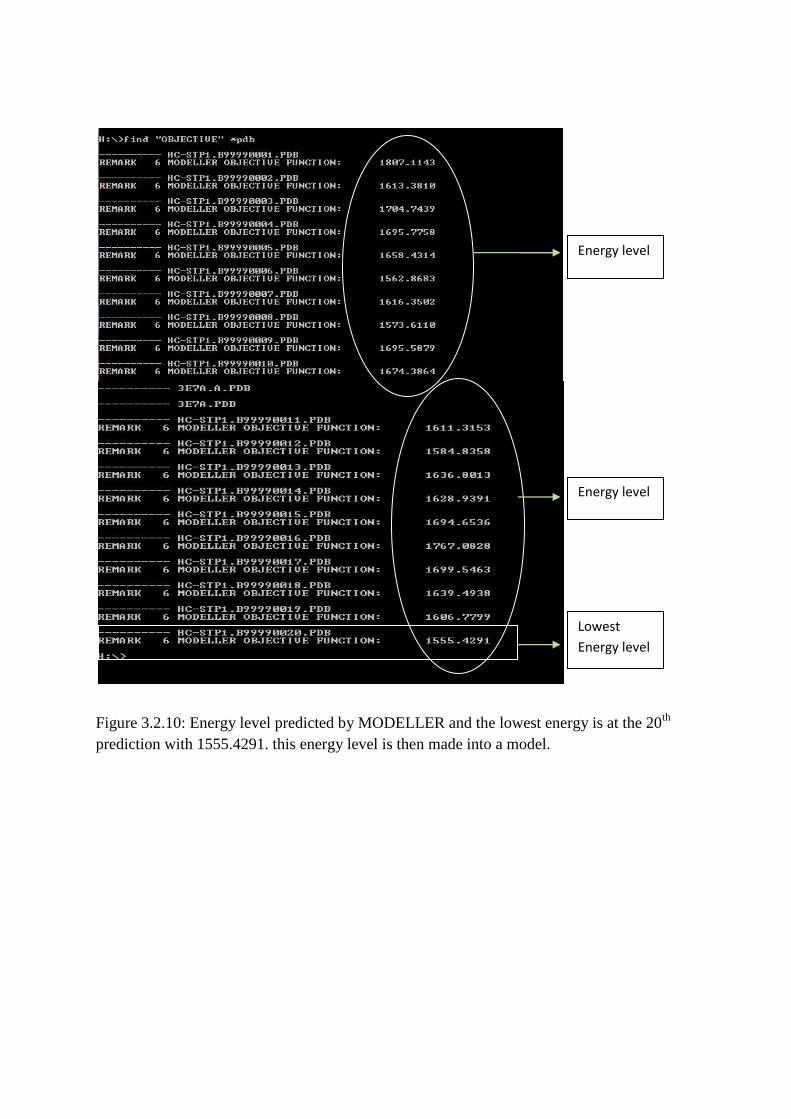
Figure 3.2.10: Energy level predicted by MODELLER and the lowest energy is at the 20th
prediction with 1555.4291. this energy level is then made into a model.
Energy level
Energy level
Lowest
Energy level

Figure 3.2.11: Hc-Stp1_3e7a.pdb (A) The model generated by Pymol shows a globular
protein of cone shaped cleft which highlights the loop region in green, alpha-helixes in red
and beta-sheet in yellow. (B) Cartoon structure for the same model shows an it is a 3 strand
anti-parallel beta-sheet. Starts from N-terminal (blue) and ends at C-terminal (red), the in
between colours are walking through the protein.
(A) (B)

Figure 3.2.12: Ramachandran plot evaluation to evaluate the overall geometry of the
structure. It’s a two dimentional scatter plot showing torsion angles of each amino acid.
Number of residues in the favoured region was 279 (95.5%), number of residues in the
allowed region was 10 (3.4%) and number of residues in outlier region was 3 (1%). The 3
outliers were Leucine, Asparagine, and Threonine.

REFERENCE.
CAMPBELL, B. E., HOFMANN, A., MCCLUSKEY, A. & GASSER, R. B. 2011. Serine/threonine phosphatases in socioeconomically important parasitic nematodes--prospects as novel drug targets? Biotechnol Adv, 29, 28-39.
CAMPBELL, B. E., RABELO, E. M., HOFMANN, A., HU, M. & GASSER, R. B. 2010. Characterization of a
Caenorhabditis elegans glc seven-like phosphatase (gsp) orthologue from Haemonchus contortus (Nematoda). Mol Cell Probes, 24, 178-89.
KELKER, M. S., PAGE, R. & PETI, W. 2009. Crystal structures of protein phosphatase-1 bound to
nodularin-R and tautomycin: a novel scaffold for structure-based drug design of serine/threonine phosphatase inhibitors. J Mol Biol, 385, 11-21.
STONE, S. R., HOFSTEENGE, J. & HEMMINGS, B. A. 1987. Molecular cloning of cDNAs encoding two
isoforms of the catalytic subunit of protein phosphatase 2A. Biochemistry, 26, 7215-20. WALLER, P. J. & P., C. 2005. Haemonchus contortus: Parasite problem No. 1 from Tropics - Polar
Circle. Problems and prospects for control based on epidemiology. Tropical Biomedicine, 22, 131-37.
XIONG, J. 2006. Essential Bioinformatics, New york, CAMBRIDGE UNIVERSITY PRESS.





























