Embed Size (px)
Citation preview

1
Cos’è un database Una banca dati è una collezione di informazioni, organizzate in maniera da facilitare l’accesso casuale, la ricerca ed eventualmente l’utilizzo a fini statistici.
Nelle banche dati più semplici, spesso definite sequenziali, i dati sono
organizzati in una tabella, all’interno della quale, in ciascuna riga è conservato un elemento della banca (entry): nome, cognome, numero di telefono,
indirizzo, città. A ciascun elemento, quindi, sono associate caratteristiche
differenti conservate in colonne successive diverse. Questa impostazione e’ sufficiente nei casi più semplici e crea un tabulato, ma porta alla frequente
ripetizione dello stesse informazioni in righe diverse (es. alcuni cognomi ed alcuni indirizzi simili e la città che è sempre la stessa e si ripete, come si vede
in figura 1.
Il modello relazionale, riportato in figura 2,
supera questo limite separando le informazioni in tabelle diverse. Gli stessi dati
di figura 1 sono organizzati in due tabelle corrispondenti a “persone” e “indirizzi”. Le entries presenti nelle due tabelle sono collegate da link tra

2
identificativi numerici (id) assegnati a ciascuna entry, che vengono definiti
relazioni. In figura 3
il processo viene ripetuto introducendo una nuova tabella, quella della città. Questo modello evita la ripetizione dei dati e permette una migliore
organizzazione dei dati stessi, perchè spinge ad identificare meglio le diverse entità presenti nella banca dati e permette di porre al database delle queries
con vincoli, per es. evidenziare tutti gli abitanti della stessa città che si chiamano Mario o che abitano nella stessa strada. Permettono, cioè, di ricavare
subset di informazioni, già presenti nel database, ma non facilmente
evidenziabili, creando in tal modo nuova informazione.
Cosa sono le Banche Dati Biologiche
In informatica, il termine database, tradotto in italiano con banca dati, base
di dati o anche base dati, indica un insieme di dati riguardanti uno stesso argomento, o più argomenti correlati tra loro, strutturata in modo tale da
consentire l'uso dei dati stessi (e il loro aggiornamento) da parte di applicazioni software.
La base di dati, oltre ai dati veri e propri, deve contenere anche le informazioni
sulle loro rappresentazioni e sulle relazioni che li legano.
In un sistema informatico, una base di dati può essere manipolata
direttamente dai programmi applicativi, interfacciandosi con il sistema operativo. Tale strategia era quella adottata universalmente fino agli anni
sessanta, ed è tuttora impiegata quando i dati hanno una struttura molto
semplice, o quando sono elaborati da un solo programma applicativo.
Tuttavia, a partire dalla fine degli anni sessanta, per gestire basi di dati
complesse condivise da più applicazioni, si sono utilizzati appositi sistemi

3
software, detti sistemi per la gestione di basi di dati (in inglese "Database
Management System" o "DBMS").
Una delle attività principali dei bioinformatici consiste nella progettazione,
costruzione e uso di banche dati di interesse biologico. Gli avanzamenti della biologia molecolare e dell'ingegneria genetica degli ultimi anni sono stati
accompagnati da tecnologie strumentali sempre più sofisticate. Tutto questo sta portando ad una enorme produzione di dati, inimmaginabile fino a poco
tempo fa.
Questa figura è stata tratta da un lavoro di Mark Boguski (www.sciencemag.org); si può osservare che stiamo vivendo un momento
molto particolare in cui la quantità di dati prodotti (sequenze di DNA nell'esempio) aumenta in modo molto più veloce rispetto al numero di
pubblicazioni scientifiche.
Il grafico si riferisce ai dati di sequenze di DNA che a loro volta corrispondono a geni e a proteine. Oltre ai dati di sequenze si stanno producendo molti altri dati
in modo sistematico, ad esempio sull'espressione genica mediante la tecnica dei chip di DNA (microarray), oppure di espressione e interazione proteica con
le tecniche della proteomica.
Non avrebbe senso aumentare il numero di pubblicazioni scientifiche perchè
non si può pensare che si possano leggere milioni di articoli diversi. E' quindi necessario disporre di nuovi sistemi di immagazzinamento e accesso
all'informazione. Questa esigenza trova una risposta nelle banche dati che nel settore biologico si stanno diffondendo moltissimo e sono diventate uno
strumento indispensabile per la ricerca e per la diffusione dei risultati.
Una banca dati biologica raccoglie informazioni e dati che possono essere derivati dalla letteratura o da analisi effettuate in laboratorio (analisi in vitro o
in vivo) oppure attraverso applicazioni di analisi bioinformatiche, dette analisi

4
in silico (si dice "in silico", in quanto i processori dei calcolatori sono costituiti
da silicio) e dalla letteratura scientifica. Le banche dati sono progettate come contenitori costruiti per immagazzinare dati in modo efficiente e razionale al
fine di renderli facilmente accessibili a tutti gli utenti: ricercatori, medici,
studenti, etc.
Entry
Ogni banca dati biologica ha un elemento principale attorno a cui viene costruita la entry, contenente informazioni sull’oggetto caratteristico della
banca dati (ad esempio: sequenze nucleotidiche o referenze bibliografiche) insieme a tutte le altre informazioni che si riferiscono a quella entry in
particolare). Una entry di una banca dati di sequenze nucleotidiche potrebbe contenere, oltre alla sequenza di una molecola di DNA, il nome dell’organismo
cui la sequenza appartiene, la lista degli articoli che riportano dati su quella sequenza, le caratteristiche funzionali (cioè si tratta di un gene o di una
sequenza non codificante) e ogni altra informazione ritenuta di interesse.
Esempio di entry:
In una banca dati di sequenze di acidi nucleici l’elemento centrale è la
sequenza nucleotidica di DNA o RNA a cui si associano annotazioni con le quali si classifica l’elemento come ad esempio il nome della specie, la
funzione, le referenze bibliografiche, ecc. In una banca dati dei promotori eucaristici l’elemento centrale è il
promotore. Ogni entry racchiude quindi le informazioni che caratterizzano l’elemento, cioè gli attributi dell’elemento centrale.
Per definire la struttura di una banca dati si definiscono gli attributi e il formato con cui queste informazioni verranno organizzate. La maggior parte della
banche dati biologiche possono essere usate dalla comunità scientifica in formato flat-file, cioè un file sequenziale in cui ogni classe di formazione è
riportata su una o più linee consecutive identificate da un codice a sinistra che caratterizza gli attributi annotati sulla linea.
Questo formato è molto utilizzato perché è molto leggibile e analizzabile con programmi che estraggono dalla banca dati informazioni specifiche. Prima tutte
le banche dati biologiche erano in formato flat-file, oggi invece si usano i DBMS ovvero i Database Management System per disegnare banche dati sempre più
complesse.
Cross-referencing
Uno dei problemi più grandi è il bisogno di avere accesso in modo immediato a informazioni distribuite fra varie banche dati.

5
Una soluzione è offerta dal cross-referencing (riferimento crociato) ovvero trovare
collegamento tra i dati attraverso delle righe che relazionano i dati annotati da una entry di una specifica banca dati con altri dati presenti in altre entries in altre banche
dati. Su internet questo meccanismo viene implementato con l’hypertext link, ovvero i link.
Esistono anche altre soluzioni più complesse come il rilascio dei dati in formato XML (eXtensible Markup Language) che è un linguaggio simile all HTML ma che si può
facilmente riportare a qualsiasi sistema
Nascita delle banche dati biologiche
1965: Margareth Dayhoff compila un atlante di proteine omologhe studiando le relazioni tra le sequenze primarie
Inizio anni 70: L’atlante viene reso pubblico in versione elettronica nella banca dati
NBRF.
E' questa la nascita della prima banca dati proteica. Ancora non ci sono dati di sequenziamento nucleotidico nella banca, sono tutti dati di natura biochimica classica, ma l’idea di rendere disponibili in modo libero dei dati accumulati e organizzati è alla
base del concetto che muove gli organizzatori e i curatori delle banche dati, e che muove anche i fondi per la loro gestione
Nasce inoltre la tecnologia del DNA ricombinante, che permette di manipolare le
sequenze nucleotidiche e di capire la struttura, la funzione e l’organizzazione del DNA.
Fine anni 70: pubblicazione dei primi dati genomici, con le prime sequenze nucleotidiche codificanti liberamente accessibili attraverso i rudimenti della rete
disponibili a quel tempo tra le varie università.
2001: il Consorzio Pubblico Internazionale e la Celera Genomics forniscono dati del genoma umano completo, aprendo la strada ai progetti di sequenziamento a tappeto.
Successivamente, l’approccio biotecnologico ha fornito una serie imponente di dati di natura proteomica grazie all’analisi spettrometrica e all’elettroforesi 2-D, ed una serie
altrettanto vasta di dati di trascrittomica grazie alla tecnologia dei microarrays.
Insieme ai dati nasce l’esigenza di sistemi di archiviazione e di ritrovamento facili e esaustivi, in modo da averli a disposizione in ogni istante, dato che sebbene ci siano tantissime informazioni, ognuna deve essere validata e confermata, essendo per la
maggior parte dati grezzi non rielaborati.
Conoscere il dato non significa capire il dato, serve sempre un approccio sperimentale classico perchè questo sia veramente verificato
Interrogazione delle banche dati

6
Lo scopo di interrogare una banca dati è quello di ottenere informazioni da
esse, attraverso sistemi informatici, e da altre banche dati cui è correlata.
Uno dei principali problemi legati alle banche dati biologiche è quello
dellanomenclatura. Non esiste uno standard nell’assegnazione di nomi ai geni; uno stesso gene può avere diversi nomi (Es. TRF2 è anche noto come
TLP o TLF), o uno stesso nome può individuare diversi geni (Es. TRF sta per TBP Related Factor ma anche per Transferrina o ancora per Telomeric Repeat
Binding Factor). Occorre quindi un modo per individuare univocamente i geni e le proteine, e per gestire la grande quantità di informazioni ad essi legate:
nelle banche dati primarie ogni elemento (gene, sequenza, etc) è individuato univocamente da un accession number.
Per realizzare l'estrazione di dati esistono vari sistemi fra cui i più efficienti
sono SRS ed ENTREZ. Altri sistemi altrettanto validi sono ACNUC eAceDB.
L’interrogazione di una banca dati può avvenire in maniera banale, inserendo il nome ricercato in una finestra di tipo text-search oppure tramite la
sottomissione di forms in cui inserire varie informazioni sulla nostra ricerca. La logica di criterio è quella booleana che effettua intersezioni (operatore AND),
somme (operatore OR), ed esclusioni (operatore BUT NOT), di insiemi di dati.
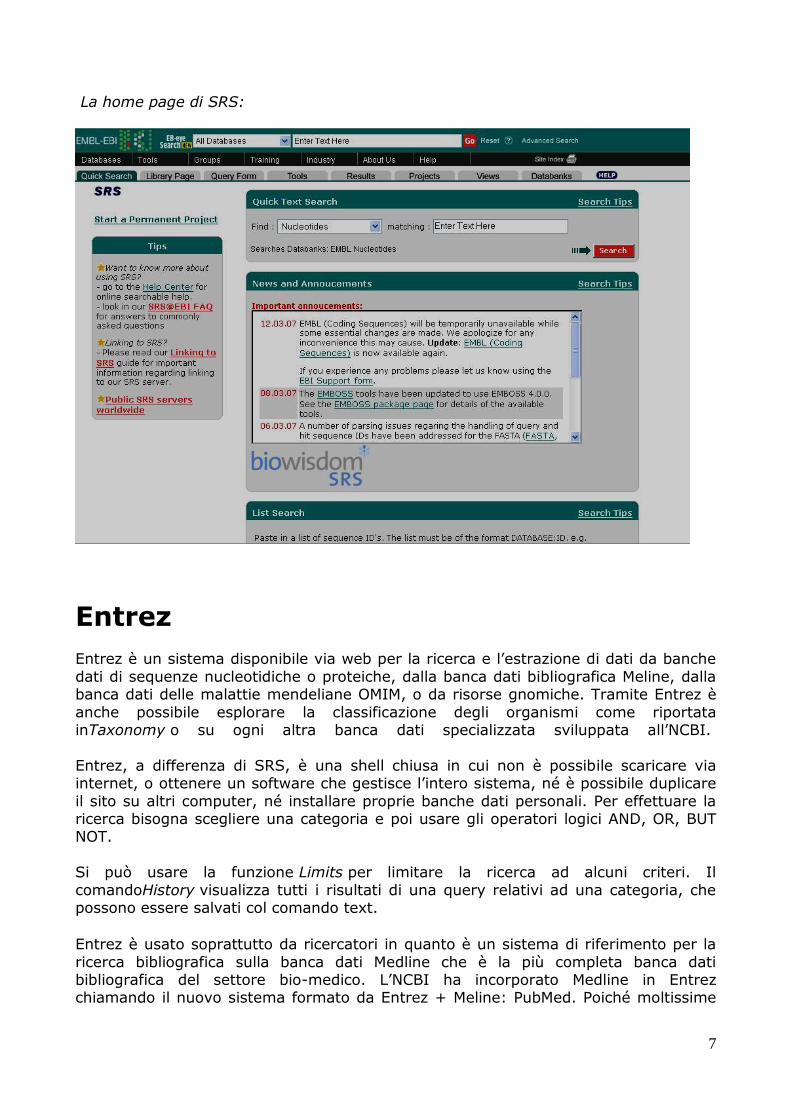
SRS
SRS (Sequence Retrieval System) è un sistema per la ricerca e l’estrazione di dati biologici via web. Esso consente di interrogare più banche dati differenti purché
abbiano almeno un informazione comune. SRS inoltre consente la navigazione attraverso varie banche dati sfruttando il cross-referencing. Può essere installato su
diversi server e interagire con altri server SRS o altre banche dati, con pochi accorgimenti.
Si può attivare una sessione cliccando su start, visualizzando così la top page in cui è possibile scegliere tra le varie banche dati. Una volta scelte le banche dati è possibile
effettuare una query riempiendo l’apposito form.
Completato il form e cliccando su submit query verranno visualizzati tutti i risultati con le varie possibilità di visualizzazione, inoltre sarà possibile salvare i dati con il
comando save.
Altri comandi utili sono link con cui si ottengono vari link ad altre banche dati per ottenere ulteriori informazioni, launch con cui è possibile lanciare un programma di analisi e projects con cui vengono salvati tutti i dati relativi alla sessione nel caso in
cui la stessa ricerca debba essere ripetuta più volte.
7
La home page di SRS:
Entrez
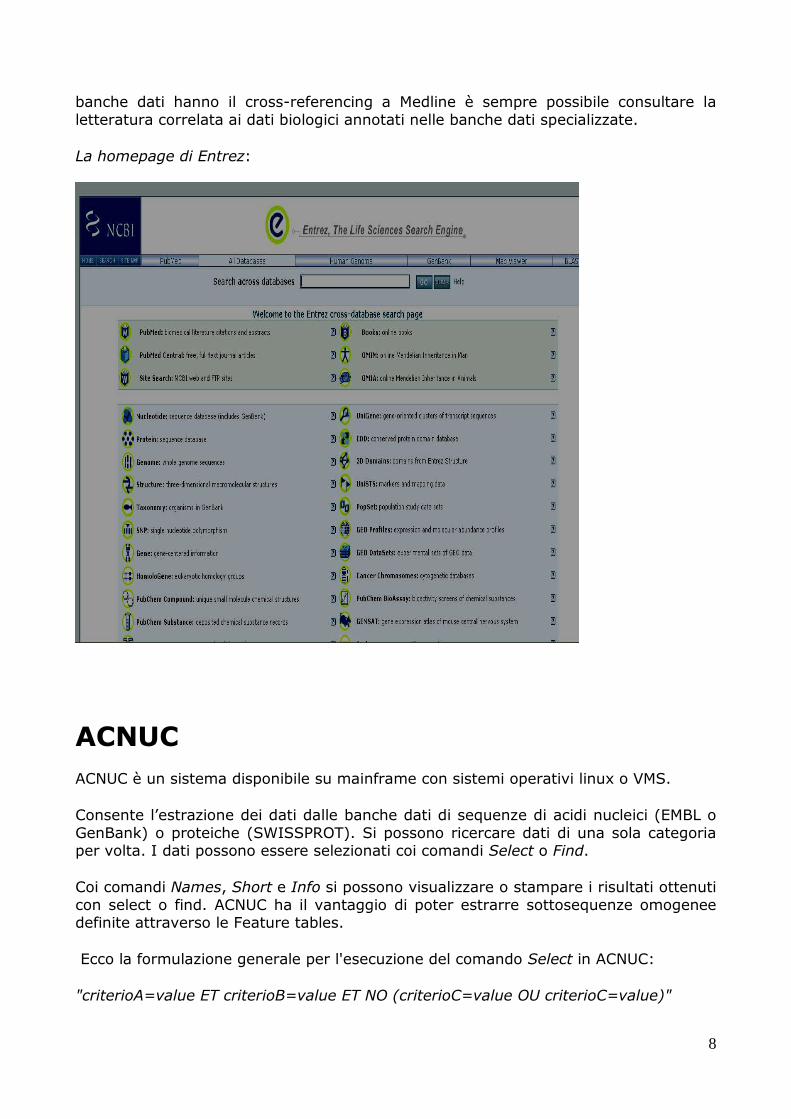
Entrez è un sistema disponibile via web per la ricerca e l’estrazione di dati da banche
dati di sequenze nucleotidiche o proteiche, dalla banca dati bibliografica Meline, dalla banca dati delle malattie mendeliane OMIM, o da risorse gnomiche. Tramite Entrez è
anche possibile esplorare la classificazione degli organismi come riportata inTaxonomy o su ogni altra banca dati specializzata sviluppata all’NCBI.
Entrez, a differenza di SRS, è una shell chiusa in cui non è possibile scaricare via internet, o ottenere un software che gestisce l’intero sistema, né è possibile duplicare
il sito su altri computer, né installare proprie banche dati personali. Per effettuare la ricerca bisogna scegliere una categoria e poi usare gli operatori logici AND, OR, BUT NOT.
Si può usare la funzione Limits per limitare la ricerca ad alcuni criteri. Il
comandoHistory visualizza tutti i risultati di una query relativi ad una categoria, che possono essere salvati col comando text.
Entrez è usato soprattutto da ricercatori in quanto è un sistema di riferimento per la
ricerca bibliografica sulla banca dati Medline che è la più completa banca dati bibliografica del settore bio-medico. L’NCBI ha incorporato Medline in Entrez chiamando il nuovo sistema formato da Entrez + Meline: PubMed. Poiché moltissime
8
banche dati hanno il cross-referencing a Medline è sempre possibile consultare la
letteratura correlata ai dati biologici annotati nelle banche dati specializzate.
La homepage di Entrez:
ACNUC
ACNUC è un sistema disponibile su mainframe con sistemi operativi linux o VMS.
Consente l’estrazione dei dati dalle banche dati di sequenze di acidi nucleici (EMBL o
GenBank) o proteiche (SWISSPROT). Si possono ricercare dati di una sola categoria per volta. I dati possono essere selezionati coi comandi Select o Find.
Coi comandi Names, Short e Info si possono visualizzare o stampare i risultati ottenuti
con select o find. ACNUC ha il vantaggio di poter estrarre sottosequenze omogenee definite attraverso le Feature tables.
Ecco la formulazione generale per l'esecuzione del comando Select in ACNUC:
"criterioA=value ET criterioB=value ET NO (criterioC=value OU criterioC=value)"

9
Qui invece la sintassi descrittiva del comando select in ACNUC per la ricerca di
sequenze umane codificanti globine con esclusione delle alpha e beta globine
"sp=homo sapiens" ET k=globin ET NO (k=alpha globin OU k=beta globin)
eDB
AceDB era stato sviluppato inizialmente per la gestione dei dati di mappaggio e sequenziamento del genoma Caenorhabditis elegans.
Oggi è adottato per altri progetti genomici. AceDB comprende programmi per la
strutturazione in formato AceDb di nuove banche dati per l’interrogazione e l’analisi dei dati in AceDB. Si può scaricare il pacchetto con questi programmi per
ricercare dei dati o anche per aggiornare il database via web.
Banche dati primarie e banche dati specializzate
Le banche dati possono essere di due tipi: primarie o specializzate.
Le banche dati primarie contengono informazioni e annotazioni molto generiche delle sequenze di acidi nucleici (DNA e RNA). Le principali banche dati primarie più importanti sono la EMBL datalibrary, la GenBank e la DDBJ.
Nel 1981 nasce nel Laboratorio Europeo di Biologia Molecolare ad Heidelberg
(Germania) l’EMBL-datalibrary, 519 entries con sequenze di DNA e RNA, l'autore è Kurt Stueber.
La EMBL datalibrary è la banca dati europea costituita nel 1980 nel laboratorio Europeo di Biologia Molecolare di Heidelberg (Germania) e comprende numerose fonti,
le seguenti:

10
La GenBank è la corrispondente banca americana costituita nel 1982 da Walter Goad
La DDBJ infine è la corrispondente giapponese della GenBank, nata nel 1986 dal National Institute of Genetics in Mishima (Giappone).
Fra le tre banche dati è stato stipulato un accordo internazionale per cui il contenuto dei dati di sequenza presenti nelle tre banche dati è quasi del tutto coincidente in
quanto gli aggiornamenti quotidiani apportati in ciascuna banca dati vengono automaticamente trasmessi alle altre due.
Le banche dati specializzate si sono sviluppate successivamente e raccolgono insiemi di dati omogenei dal punto di vista tassonomico e/o funzionale disponibili nelle
Banche dati Primarie e/o in Letteratura, o derivanti da vari approcci sperimentali,

11
rivisti e annotati con informazioni di valore aggiunto.
Un elenco dettagliato e aggiornato di tutte le banche dati biologiche disponibili e
operative si può ottenere consultando la compilazione di banche dati sviluppata da Baxevanis in concomitanza con la pubblicazione annuale del volume speciale pubblicato annualmente da Nucleic Acids Research.
Esistono anche banche dati a supporto di analisi sperimentali di routine. Ad esempio la
REBASE è una banca dati che elenca tutti i nomi degli enzimi di restrizione isolati.
Un esempio di entry proteica EMBL (flat-file)
ACCESSION: AAC74054
indica il numero di accesso
ORGANISM: Escherichia coli K12 Bacteria; Proteobacteria; gamma subdivision; Enterobacteriaceae; Escherichia indica l’organismo a cui appartiene e la sua tassonomia.

12

13

14
Qualsiasi cosa è standardizzata, dai tags agli spazi ed ai segni di punteggiatura.
Questo permette ai programmi di RETRIEVAL, cioè di ricerca, di trovare rapidamente ciò che si cerca.
Banche dati di sequenze nucleotidiche
Le banche dati di sequenze nucleotidiche sono la EMBL, la GenBank e la DDBJ.
Poiché sono coincidenti (vedi banche dati primarie e specializzate) ne descriveremo solo una: la EMBL.
Le entries nella banca dati EMBL sono classificate in divisioni identificate da un codice
a 3 lettere annotato nella riga ID. Il raggruppamento nelle varie divisioni è basato prevalentemente sulla tassonomia tranne in alcuni casi come il gruppo delle EST (frammenti di sequenze espresse), delle HTG (sequenze derivate da progetti
genomici), e altre ancora.
Le banche dati di sequenze nucleotidiche (o primarie) sono aggiornate quasi via internet interamente dai ricercatori produttori di nuove sequenze.

15
Sono state organizzate nei primi anni in modo non molto accurato in termini di
annotazioni e ridondanza di informazioni. Per questo motivo le analisi statistiche sui campioni estratti in modo automatico sono poco attendibili.
In ogni caso il numero totale di specie differenti rappresentate nella banca dati EMBL
ammonta a circa 87000.
Banche dati di sequenze proteiche
Le banche dati proteiche sono il secondo grande aggregato di dati biologici. Esse raccolgono sia sequenze proteiche ottenute dalla sperimentazione della sequenza
amminoacidica, sia dalla traduzione di nucleotidiche. Qui si trovano i dati estratti dalle banche dati di acidi nucleici relativi a proteine che vengono poi accuratamente validati e arricchiti di informazioni specifiche.
Le banche dati di sequenze proteiche più importanti sono la SWISSPORT,
laTREMBL e la PIR
La PIR, Protein Information Resource è sviluppata in collaborazione fra due grossi
centri: la Georgetown University negli USA e il MIPS a Monaco di Baviera.
Questa è una banca dati valida per il livello di annotazioni e il livello di aggiornamento ma è poco integrata con altre banche dati.
La SWISSPORT è la banca dati di proteine di riferimento per tutti gli studi correlati in
silicio di proteine e patterns proteici.E' sviluppata in Svizzera a Ginevra dal gruppo di Amos Bairoch che afferisce all’istituto nazionale SIB.
La entry in SWISSPORT differisce per quella in EMBL soprattutto per le features che in questo caso descrivono la presenza nella proteina di amminoacidi modificati, regioni
peptidiche, domini strutturali, siti di splicing proteici, polimorfismi e altri segnali e dati rilevanti per la struttura della proteina. C’è grande cura per l’annotazione del nome
della proteina ma un problema è che spesso allo stesso gene vengono dati nomi differenti, rendendo difficile la ricerca per nome del gene. Per risolvere il problema è stato costituito un consorzio: Gene Ontology (GO). Sul sito del consorzio è disponibile
un dizionario con tutti i nomi dei geni.
In SWISSPROT sono riportate anche le informazioni relative ad alterazioni della proteina.; tali informazioni derivano da OMIM (On-line Mendelian Inheritance in Man).
L’aggiornamento di SWISSPROT avviene tramite l’EBI dove viene sviluppata un’altra banca dati di proteine, TREMBL, che altro non è che il risultato della traduzione
automatica in amminoacidi di tutte le sequenze annotate nella banca dati EMBL come sequenze codificanti di proteine. Una parte di queste sequenze costituisce la

16
SPTREMBL che si occupa delle proteine immunologiche,la REMTREMBL invece si
occupa delle proteine brevettate e a frammenti non caratterizzanti.
Se si vuole consultare contemporaneamente SWISSPROT e SPTREMBL si fa riferimento a SWALL che è una raccolta di sequenze proteiche ridondante e non accurata in tutti i suoi elementi
Banche dati di motivi e domini proteici
La comparazione per individuare caratteristiche strutturali e funzionali già riscontrate
in altre sequenze ed annotate in specifiche banche dati si può effettuare attraverso l’applicazione di tecniche di ricerca di similarità, oppure, nel caso in cui tale ricerca
non evidenzia sequenze simili a quelle in oggetto, attraverso l’applicazione di tecniche di ricerca di segnali (pattern recognition) basate su algoritmi più o meno complessi.
L’approccio pattern recognition consente di ritrovare segnali, motivi o domini
strutturali e funzionali che si conservano nel tempo anche quando le sequenze hanno subito una divergenza tale da aver perso in buona parte le caratteristiche di similarità globale.
I motivi sono combinazioni regolari di strutture secondarie. Possiedono determinate
funzioni biologiche. Esempi di motivi sono: elica-loop-elica, o zinc finger, o i motivi BH della famiglia BCL2.
I domini sono invece regioni ampie e discrete di una proteina in grado di assumere
una struttura terziaria (quindi di effettuare folding) fisicamente separata e distinguibile da altre parti della stessa molecola. A volte è possibile definire
funzionalmente un dominio quando questo definisce una particolare funzione della proteina
A questo proposito esistono diverse banche dati specializzate che annotano
informazioni relative a motivi e domini funzionali. Tra queste un gruppo cospicuo è stato integrato in InterPRO, una risorsa bioinformatica, sviluppata dall’EBI, che
consente di ricercare contemporaneamente su più banche dati, distribuite su calcolatori diversi e strutturate in modo differente, informazioni funzionali e strutturali relative ad una proteina o ad una famiglia di proteine.
La ricerca dei dati in InterPRO si può effettuare attraverso un sistema di semplice ricerca basato su componenti del DBMS Oracle o attraverso il sito SRS dell’EBI. Inoltre attraverso il software InterPROscan è possibile ricercare motivi strutturali e funzionali
annotati nelle banche dati integrate in InterPRO al fine di caratterizzare dal punto di vista funzionale nuove proteine derivate da progetti di sequenziamento genomico.
Le banche dati integrate in InterPRO sono :
PROSITE, Pfam, PRINTS, ProDom,SMART e TIGRFAMs.
Ognuna di queste banche dati è stata prodotta a partire da dati di sequenze proteiche annotati in SWISSPROT e TREMBL, che sono la fonte primaria per i dati disponibili
attraverso InterPRO. A causa della mancanza di una concordanza nella definizione dei
17
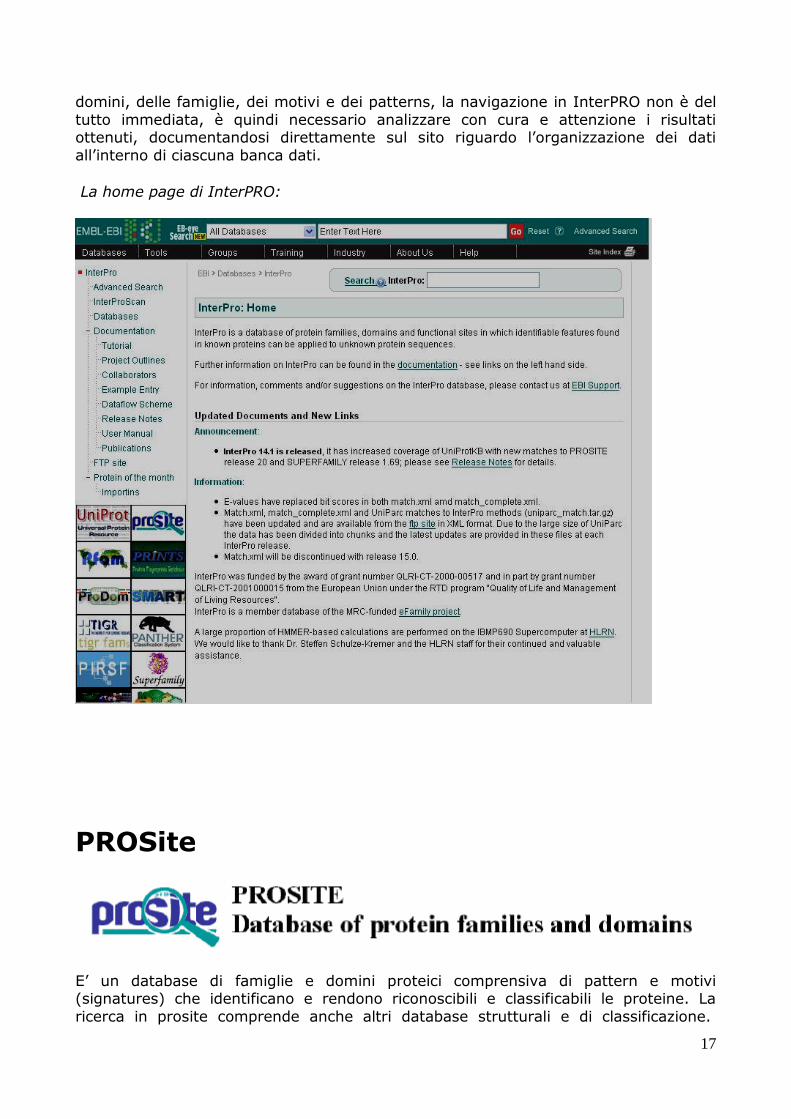
domini, delle famiglie, dei motivi e dei patterns, la navigazione in InterPRO non è del
tutto immediata, è quindi necessario analizzare con cura e attenzione i risultati ottenuti, documentandosi direttamente sul sito riguardo l’organizzazione dei dati
all’interno di ciascuna banca dati.
La home page di InterPRO:
PROSite
E’ un database di famiglie e domini proteici comprensiva di pattern e motivi (signatures) che identificano e rendono riconoscibili e classificabili le proteine. La
ricerca in prosite comprende anche altri database strutturali e di classificazione.

18
ProDom
ProDom è un database che raccoglie i dati relativi a famiglie di proteine generate automaticamente dall' applicazione di PSI-Blast, una versione di gapped-Blast che,
confrontando una sequenza proteica detta sequenza sonda, in un database di proteine, raccoglie un multi-allineamento di tutte le sequenze proteiche per le quali Blast ha determinato uno score più alto di una certa soglia detta threshold.
Il risultato è un profilo generato a partire dal multi-allineamento trovato, il quale viene
poi utilizzato per rilanciare Blast su tutto il database di proteine per individuare nuove sequenze correlate a quelle già allineate. Le nuove sequenze trovate vengono
aggiunte al multi-allineamento al fine di ottimizzare ulteriormente il profilo. Questa procedura si ripetute finché non si raggiunge una certa convergenza. Ad ogni iterazione l’utente può decidere di eliminare sequenze incluse che ritiene non essere
adatte alla propria ricerca.
ProDOM è generato a partire da profili ottenuti dal multi-allineamento di famiglie di proteine annotate in PfamA. Infine ProDOM ha recentemente introdotto un nuovo
sottoinsieme definito ProDOMCG che annota domini proteici di organismi per i quali è stato sequenziato il genoma completo.
Pfam

19
Pfam è una banca dati di famiglie di proteine accomunate da elementi strutturali e
funzionali. Ogni entry in Pfam è formata da un tipo che può essere famiglia, dominio, repeat o motivo.
Il tipo famiglia raggruppa le sequenze proteiche che hanno in comune gli stessi
domini; Il tipo dominio definisce una unità strutturale che può essere presente in famiglie differenti; Il tipo repeats raggruppa elementi funzionali attivi e presenti in
copie multiple in proteine globulari; Il tipo motivi include pattern componenti blocchi strutturali non associati a proteine globulari.
Ogni dominio in Pfam ha dei limiti ottenuti dal database SCOP, un database gerarchico delle strutture di proteine nel quale si trovano le classificazioni dei domini strutturali.
Poiché una stessa funzione proteica può essere caratterizzata da più domini, i links fra Pfam e SCOP possono essere molti a molti. Le famiglie di proteine non classificabili
secondo i criteri su citati, ma che comunque sono state prodotte automaticamente attraverso l’applicazione di PSI-BLAST e quindi annotate in ProDOM, sono annotate in Pfam nel sottoinsieme Pfam-B, un database meno accurato ma comunque di supporto
all’analisi proteomica.
Pfam annota anche gruppi di proteine classificate per la presenza di regioni non definibili come domini, come per esempio gli elementi trans-membrana, i peptidi di
segnale, regioni a struttura random e regioni a bassa complessità: questi patterns sono determinati attraverso l’applicazione di softwares specifici quali TMHMM, SignalP,
ncoil e SEG e sono annotati nella sezione Non Pfam regions.
Prints
PRINTS è un database che raccoglie sequenze proteiche in clusters definiti da un
Fingerprint comune, cioè un insieme di più motivi conservati e dedotti dall’osservazione di un multi-allineamento ottenuto applicando algoritmi per la ricerca di similarità locali; Il multi-allineamento prodotto non include gaps.
I clusters sono classificati in una forma gerarchica di superfamiglie, famiglie
e sottofamiglie. Il numero di famiglie annotate in PRINTS è ridotto rispetto a Pfam e ProDOM in quanto i dati, prodotti in modo automatico, successivamente sono rivisti manualmente e annotati con dati biologici derivati dalla letteratura e da ulteriori
analisi.
PRINTS può essere interrogato utilizzando un semplice sistema di ricerca testuale secondo criteri diversificati oppure è possibile effettuare una ricerca di similarità tramite Blast o applicare software specifici quali Fpscan, MulScan, GraphScan e
FingerScan che ricercano, con modalità differenziate, in una nuova sequenza di cui di voglia caratterizzare la struttura e la funzione, fingerprints già annotati in PRINTS.

20
SMART
SMART (Simple Modular Architecture Research Tool) è una risorsa Web che raccoglie
dati relativi a domini proteici e consente la ricerca di domini in nuove sequenze proteiche.
SMART per ogni famiglia di proteine associate a un dominio, annota informazioni sulla funzione sulla localizzazione cellulare, sulla struttura terziaria in cui è coinvolto il
dominio e su relazioni filogenetiche fra le specie da cui sono derivati le proteine componenti la famiglia. Le informazioni vengono annotate a mano dopo accurate
valutazioni. SMART raccoglie anche informazioni correlate a OMIM per quei domini dove sono state
riscontrate mutazioni associate a fenotipi patologici. SMART cura particolarmente domini associati a elementi mobili presenti nei genomi eucarioti e annota anche gli
elementi intrinsechi quali i peptidi segnale, gli elementi transmembrana e le regioni a struttura random.
TIGRFAMs
TIGRFAMs è una collezione di famiglie di proteine prodotta mediante annotazione biologica di semplici multi-allineamenti proteici o di profili ottenuti
dalla ottimizzazione di multi-allineamenti attraverso Hidden Markov Models.
Un’ultima banca dati da citare non integrata in InterPRO ma con cross-referencing a InterPRO, è ClusTR , un database di cluster di proteine
multiallineate, prodotti automaticamente a partire dagli accoppiamenti fra tutte le proteine annotate in SWISSPROT + TREMBL. Il database è sviluppato
dall’EBI
Banche dati di strutture proteiche
La conoscenza di motivi strutturali delle proteine è di grande importanza per la comprensione funzionale delle biosequenze. Per dati strutturali di una proteina si intendono la distribuzione spaziale degli atomi componenti gli amminoacidi e quindi
degli amminoacidi stessi.
Tali dati corrispondono alle coordinate atomiche determinate attraverso analisi cristallografiche ai raggi X o mediante applicazione di tecniche di spettroscopia NMR su
proteine cristallizzate.

21
L’unica banca dati che raccoglie tutte tali informazioni è la banca dati PDB che a
gennaio 2002 riportava più di 16.000 strutture proteiche.
Tale banca dati è un riferimento unico per tutti gli studi strutturali. I produttori di nuovi dati possono sottomettere nuovi dati utilizzando il sistema web ADIT (AutoDep
Input Tool).
Presso il sito del PDB sono disponibili dati statistici relativi al numero di strutture e alla loro distribuzione nelle varie classi di macromolecole. Nell’ambito dei dati strutturali le banche dati di notevole importanza sono: MMDB, CATH, DSSP, SCOP, MSDB.
Banche dati biologiche per il sistema immunitario
L’immunologia è una branca della moderna ricerca biomedica che si basa, tra le altre cose, sullo studio funzionale e strutturale delle macromolecole biologiche e sull’analisi
di variabilità molecolare associata alle risposte immunitarie.
Alcuni dei più rilevanti database nel settore immunologico sono: IMGT, MHCpep, FIMM e MPID.
IMGT è il database internazionale di ImmunoGenetica e accoglie dati relativi alle
ImmunoGlobuline, ai recettori delle cellule T(TCR) e al maggiore complesso di istocompatibilità di classe I e II. Il database riporta dati relativi alle sequenze, ai
genomi, alle strutture e alla variabilità delle macromolecole immunologiche umane e di altri vertebrati. Il sito di IMGT consente di accedere al database per effettuare ricerca di dati, ricerca di similarità e altre specifiche analisi in silico. Il database
contiene anche un sottodatabase IMGT/HLA esclusivo per il complesso HLA. IMGT è sviluppato in Francia dal gruppo di M.P.Lefranc a Montpellier ma in stretta
collaborazione con l’EBI e con il gruppo SWISSPORT.
I peptidi generati dal processamento di proteine antigeniche legano le molecole costituenti il complesso di maggiore istocompatibilità (MHC) che li presenta sulla
superficie cellulare per il riconoscimento dei recettori delle cellule T. Tali peptidi sono definiti epitopi T-cell e sono caratterizzati da una estrema variabilità composizionale associata anche alla variabilità delle molecole MHC.
MHCpep è un database che annota i dati di sequenza dei peptidi che legano molecole
di MHC di uomo, topo, e in minima parte anche di ratto e di altri primati. Ogni entry è associata a uno specifico peptide che lega uno specifico allele MHC. Sono annotate
anche informazioni sull’attività di legame e sui metodi con cui i peptidi sono stati determinati. Infine ciascun peptide è correlato attraverso le linee di cross-referencig alla banca dati SWISSPROT in modo da mettere in relazione il peptide con la
corrispondente proteina sorgente. L’aggiornamento di MHCpep è bloccato a luglio

22
1998; dati più recenti possono essere ottenuti dal database FIMM.
FIMM è un database di antigeni, molecole MHC, peptidi associati alle molecole MHC e
dati correlati a patologie. A differenza di MHCpep che è un database disponibile in formato flat-file, scaricabile dalla rete e quindi consultabile a misura delle esigenze dell’utente, FIMM è strutturato in un pacchetto chiuso, secondo gli schemi delle
cosiddette data-warehouse che consentono la ricerca e l’analisi dei dati esclusivamente secondo percorsi pre-progettati dal produttore del pacchetto stesso.
Per esempio non è possibile estrarre l’intero database o un intero sottoinsieme del database come per esempio tutti i peptidi leganti MHC di classe I, ma è solo possibile consultare il database ed effettuare analisi molto mirate e specifiche.
MPID (MHC Peptide Interactions DB) annota informazioni relative alle correlazioni
sequenza-struttura-funzione per i peptidi che legano MHC. MPID riporta in particolare tutte le strutture delle proteine contenenti peptidi che legano i complessi MHC e
informazioni slla caratterizzazione strutturale delle interazioni complesso-peptidi. Le strutture sono dedotte dal database PDB
Banche Dati mitocondriali
Gli organismi eucariotici contengono nel citoplasma delle loro cellule organuli di
vario tipo fra cui i mitocondri, il cui ruolo è di assoluta importanza in moltissimi processi metabolici e di funzionalità della cellula. Le numerose e interessanti
proprietà del mitocondrio fra cui le piccole dimensioni del suo genoma hanno favorito numerosi studi e anche grandi e coordinati progetti di sequenziamento
dei genomi mitocondriali di vari organismi. Numerose informazioni sono disponibili tramite le banche dati specializzate come le seguenti:
GOBASE (Organelle Genome Database) è una risorsa genomica che
raccoglie dati sui genomi di cloroplasti e mitocondri. I nomi dei geni sono annotati secondo un vocabolario controllato definito da esperti.
MITOMAP (Human Mitochondrial Genome Database) è un report
aggiornato ai dati pubblicati di tutte le variazioni riscontrate sul DNA mitocondriale di soggetti affetti da patologie e su soggetti i cui campioni
sono stati prelevati per studi di genetica di popolazione. I dati sono annotati in forma tabellare e possono essere estratti attraverso l’utilizzo
di un sistema di interrogazione semplice. Non è presenta alcuna relazione tra i dati per cui non è possibile effettuare statistiche sulla
frequenza di variabilità di ciascun sito del genoma in cui siano state riscontrate e annotate mutazioni.
Human MitBASE è una banca dati nata per raccogliere in un'unica risorsa integrata i dati sul mitocondrio di tutti gli organismi eucariotici. I
dati sono organizzati in base a ogni individuo, alla sua origine geografica e alla sua descrizione dei dati clinici associati. Ogni entry raccoglie
moltissime informazioni associate all’individuo e ciò implica un notevole

23
dispendio di risorse umane e una difficoltà di mantenimento della banca
dati stessa, che risulta meno aggiornata rispetto a MITOMAP.
HrvBase è una banca dati che raccoglie i multi-allineamenti delle
sequenze relative alle regioni di controllo del genoma mitocondriale dei
primati.
MITOP raccoglie informazioni su geni correlati alla funzionalità del
mitocondrio di uomo, topo, lievito, Caenorhabditis elegans e Neurospora crassa. Ogni entry è associata a una proteina della quale sono annotate
la classe funzionale, il codice dell’enzima, il complesso proteico di appartenenza della proteina, il peso molecolare, il punto isolettrico, etc.
MitoNuc una banca dati di geni nucleari di metazoi per il mitocondrio. I dati sono estratti da SWISSPROT come sequenze mitocondriali di
metazoi e vengono quindi accuratamente controllati e annotati con informazioni specifiche. Per quanto riguarda le proteine umane è
riportata la localizzazione del gene sul genoma umano ottenuta attraverso analisi effettuate su Ensembl.
AMmtDB è la banca dati dei multi-allineamenti di geni codificati da genomi mitocondriali di Metazoi. Ogni entry è gene e classe-tassonomica
specifica.
MITOCHONDRIOME è un sito web che raccoglie banche dati mitocondriali e informazioni correlate. Attraverso tale sito si accede alle
banche dati Human_MitBase, MITONUC e AMmtDB oltre a dati ottenuti dall’analisi di variabilità e complessità di geni e egenomi mitocondriali di
metazoi.
PLMitRNA è una banca dati di molecole e geni di tRNA identificati nei
mitocondri di tutte le piante verdi. Informazioni caratterizzanti il gene o la molecola sono annotate e possono esssere utilizzate per la ricerca dei
dati. I tRNA possono essere selezionati per nome della specie o per raggruppamento tassonomico. Il multiallineamento di ciascun cluster di
tRNA omologhi è anche disponibile.
Risorse Genomiche
Il progresso dei risultati ottenuti dai progetti genomici ha dato un grande grande impulso alla bioinformatica.
Le risorse genomiche sono siti dove è possibile reperire dati relativi al
mappaggio e al sequenziamento genomico ed eventualmente altre informazioni
correlate. Le tipologie di tali risorse sono:
Risorse integrate dove sono disponibili dati relativi a tutti i genomi attualmente in fase di studio (Entrez_Genomes o EBI_Genome)
Risorse relative ai genomi di determinate categorie di organismi

24
Risorse organismo specifiche che hanno la caratteristica comune di
poter scaricare sul proprio computer la sequenza dell’intero genoma o di parti di esse individuate dalla localizzazione cromosomiale o da uno
specifico marker. E' poi sempre possibile effettuare ricerche di similarità
di sequenza contro l’intero genoma o parti di esso mediante l’applicazione dei metodi FASTA e/o Blast.
Banche Dati di Geni
Numerose Banche Dati di geni sono stati sviluppate a partire prevalentemente da dati genomici o comunque da dati annotati nelle banche dati primarie.
Ricordiamo tra gli altri: LocusLink, RefSeq,UniGENE, COGs,GENES ed
euGENES. LocusLink è uno dei database sviluppati all’NCBI nell’attività di
annotazione curata dei dati genomici. Vengono annotati, per ogni locus genetico (ogni elemento funzionale di un genoma), il nome ufficiale ed
eventuali sinonimi, il codice della classificazione internazionale degli enzimi, se trattiamo degli enzimi, il link a OMIM, gli Accession_numbers
delle sequenze nucleotidiche associate al locus e annotate nelle banche
dati primarie e il link alle banche dati RefSeq e UniGene. COGs riporta una compilazione di geni ortologhi codificanti proteine
relativi a organismi completamente sequenziati oppure clusters di geni paraloghi conservati in almeno 3 organismi differenti e significativamente
distanti fra loro; queste ultime condizioni assicurano l’appartenenza delle proteine paraloghe a un dominio comune ancestrale.
GENES annota le informazioni relative a tutti i geni identificati sui genomi completi sia di procarioti sia di eucarioti.
EuGENES è ancora una banca dati di geni e genomi relativi a 7 organismi eucariotici e descrive circa 150.000 geni noti, predetti o non
classificati.
Banche dati di patterns nucleotidici
Insieme alle banche dati dei geni abbiamo le banche dati di patterns nucleotidici o di regioni funzionali del gene associati a specifiche funzioni regolatorie e di controllo: EPD, TRANSFAC, UTRdb, TRANSTERM,
TRANSCOMPEL
EPD è una delle prime banche dati specializzate progettata, annota le
info bibliografiche e sperimentali sui promotori eucariotici

25
TRANSFAC è la banca dati dei fattori di trascrizione che annota dati sui
fattori proteici e sui corrispondenti siti di legame sul DNA coinvolto nell’attivazione e la regolazione della Trascrizione. Dal suo sito è possibile
ottenere una scheda in formato flat-file con le caratteristiche
dell'elemento.
UTRdb svolge un ruolo importante poiché annota tutte le sequenze non tradotte dei messaggeri eucariotici derivate dalla banca dati primaria
EMBL.
TRASTERM è la banca dati degli elementi che regolano la traduzione e le
modificazioni post-traduzionali. Gli elementi sono classificati dal punto di vista funzionale e strutturale, raggruppando gli elementi in categorie
TRANScompel è la banca dati degli elementi compositi coinvoli nella regolazione della trascrizione. Elementi regolatori compositi (CE)
annotano due siti di legame situati in posizioni vicine nella unità trascrizionale e che legano due distinti fattori di trascrizione ma
controllano in modo combinato la regolazione della trascrizione.
Banche Dati del trascrittoma
In questi ultimi anni si stanno realizzando le banche dati del trascrittoma,
ovvero dell’insieme di tutti i trascritti di un dato organismo ottenuti attraverso il sequenziamento delle EST (Expressed Sequenze Tags) o dei cDNA completi.
Alcune tra le più importanti sono dbEST e UniGENE.
dbEST raccoglie tutta la mole di dati relativi alle EST, ottenute tramite il sequenziamento parziale di cloni di cDNA
UniGENE raggruppa sequenze geniche trascritte dedotte da sequenziamento di cDNA o di EST di uomo, topo, ratto, Drosophila,
Anopheles, danio renio, Arabidopsis e altri organismi modello, in clusters teoricamente corrispondenti a un singoolo gene, attraverso criteri di
similarità o provenienza da uno stesso clone

26
Banche Dati di profili di espressione
La tecnologia dei microarrays permette in un solo esperimento di quantificare i trascritti di un intero genoma (il trascrittoma) e quindi di confrontare la
variabilità di espressione di ciascun gene in tessuti diversi, in individui diversi, in stati patologici diversi. In pratica consente di associare il livello di
espressione di un gene al corrispondente fenotipo. Molte delle altre risorse dei profili di espressione sono invece prodotte in modo non coordinato.
Si è dato quindi avvio a progetti coordinati per la raccolta di questi dati, progetti che si stanno concretizzando nella realizzazione di tre banche
dati:GEO, ArrayExpress e KEGG/Expression
GEO (Gene Expression Omnibus) è sviluppato all’NCBI come risorsa eterogenea per la sottomissione e il retrieval di dati correlati a
esperimenti basati sulla tecnologia dei microarrays e preposti allo studio di espressione di geni e di ibridizzazione fra genomi. I dati sono
classificati in 3 categorie: platform (dati su tutte le sonde molecolari identificative di ciascuno spot per l’allestimento di un microarray),
samples (dati sulle molecole che devono essere analizzate) e series (tutti
i dati relativi a un esperimento).
ArrayExpress è l’equivalente europeo di GEO e raccoglie dati eterogenei su profili di espressione. E’ strutturato utilizzando il DMBS Oracle
secondo una definizione a oggetti. Riporta tutti i dati su interi
esperimenti e anche le immagini grezze del profilo come viene prodotto dall’esperimento. Il database può essere interrogato attraverso un
sistema semplice di ricerca testuale ed è interfacciato al sistema Expression Profiler che consente di analizzare i profili di espressione e di
effettuare confronti tra differenti esperimenti. Le informazioni annotate in ArrayExpress sono raggruppabili in tre grandi categorie: Experiment,
Array e Protocol.
KEGG/Expression è un database che raccoglie dati sui profili di
espressione ottenuti con la tecnica dei microarrays in vari laboratori giapponesi.
Banche Dati di polimorfismi e mutazioni
L’annotazione nelle banche dati di eventi generativi di mutazioni e polimorfismo è di rilevante importanza sia per studi di genetica di popolazione
sia per studi di associazione fra mutazione e fenotipi con diversificate

27
manifestazioni cliniche.
Il termine mutazione indica la differenza puntuale riscontrata in un campione
rispetto al genoma di riferimento a causa di disfunzioni di un gene e quindi di
manifestazioni di fenotipi patologici. Il terminepolimorfismo invece indica l’evento che lascia inalterata la funzionalità del gene. Una variazione che in una
popolazione si riscontra con una frequenza superiore all’ 1% è considerata polimorfismo. Recentemente è stato introdotto un nuovo termine o meglio
acronimo: SNP (Single Nucleotdice Polymorphism) e che dovrebbe indicare tutti i polimorfismi associati al cambiamento di un solo nucleotide.
Per studiare la variabilità popolazionale in modo coordinato è stato creato il
database HGVbase, che annota tutti i dati derivati da studi di variabilità popolazionale. Parallelamente è nato il database dbSNPs che annota dati di
SNPs, ma anche polimorfismi di regioni e mutazioni associate all’insorgenza di una specifica patologia.
Altre banche dati disponibili nell’ambito della variabilità sono HGDM, OMIM, Pharmacogenetics e Genes and Diseases
HGMD ( Human Gene Mutation Database) raccoglie dati sulle mutazioni riportate come causa di alterazioni e disfuzioni di geni nucleari in malattie ereditarie. Non vengono annotate mutazioni somatiche o del
DNA mitocondriale, inoltre sono annotate solo mutazioni sperimentalmente determinate sul DNA e non sulla proteina. Ogni
mutazione è annotata una sola volta nella banca dati per evitare
confusioni tra mutazioni frequenti e ereditarie. Questo impedisce però di effettuare valutazioni statistiche di variabilità in base ai dati annotati in
HGMD.
OMIM (Online Mendeliam Inheritance in Man) raccoglie informazioni
correlate alle malattie genetiche di origine Mendeliana. Sono raccolti dati non solo sulle malattie genetiche di origine autosomica ma anche sulle
malattie associate ad alterazioni dei cromosomi X e Y del mitocondrio. Sono curate le annotazioni dei dati attraverso l’uso di un vocabolario
controllato relativo ai nomi dei geni. Genes and Disease è una risorsa di dati sviluppata in base alla
patologia, da cui si arriva al gene e a informazioni correlate annotate in altre banche dati fra cui OMIM.
Pharmacogenetics è una risorsa creata da una rete di laboratori di ricerca per la raccolta integrata di dati genomici, clinici e descrittivi del
fenotipo derivati da studi di farmacogenomica.

28
Banche Dati di pathways metabolici
Questi tipi di banche dati studiano i processi metabolici. L’idea è quella di realizzare network di dati biologici nei quali siano annotati i processi di
interazione fra le molecole, per favorire la comprensione dei processi di regolazione dell’espressione genica e i processi post-traduzione relativi al
trasporto e al metabolismo delle proteine. Esempi ne sono le banche datiKEGG, EcoCyc, ENZYME e Ligand.
ENZYME riporta in una struttura gerarchica la classificazione internazionale degli enzimi. Ogni entry rporta un id corrispondente all’EC
number, il nome dell’enzima e i suoi sinonimi, l’attività catalitica, gli eventuali cofattori, il cross-referencing alla banca dati delle proteine e
alla banca dati OMIM.
Ligand è la banca dati dei composti chimici e delle reazioni coinvolte nei
processi metabolici.
EcoCyc è un database di un organismo modello, l’Escherichia coli, che
annota dati non solo genomici e proteomici, ma anche quelli relativi ai processi metabolici, al trasporto e alla regolazione dell’espressione dei
geni di Escherichia coli. Vengono annotati una grande quantità di geni la cui funzione è stata determinata sperimentalmente, quindi è un’ottima
risorsa per predire nuovi geni in genomi di altri organismi microbici. Questa banca dati è un valido modello utilizzabile come strumento
didattico per lo studio e l’approfondimento delle scienze Biochimiche.
KEGG è l’enciclopedia di Kyoto di geni e genomi ed è una risorsa
integrata di banche dati correlate ai genomi completamente sequenziati o in fase di completamento. Lo scopo di tale banca dati è creare una rete
tra le varie classi di dati per la comprensione dei meccanismi preposti alla funzionalità delle cellule e degli organismi a partire dai dati genomici.
I database integrati in KEGG sono SSDB, Genes, Pathways, Kegg/Expression e Ligand.
Banche Dati mitocondriali
Gli organismi eucariotici contengono nel citoplasma delle loro cellule organuli di vario tipo fra cui i mitocondri, il cui ruolo è di assoluta importanza in moltissimi
processi metabolici e di funzionalità della cellula. Le numerose e interessanti proprietà del mitocondrio fra cui le piccole dimensioni del suo genoma hanno

29
favorito numerosi studi e anche grandi e coordinati progetti di sequenziamento
dei genomi mitocondriali di vari organismi. Numerose informazioni sono disponibili tramite le banche dati specializzate come le seguenti:
GOBASE (Organelle Genome Database) è una risorsa genomica che raccoglie dati sui genomi di cloroplasti e mitocondri. I nomi dei geni sono
annotati secondo un vocabolario controllato definito da esperti.
MITOMAP (Human Mitochondrial Genome Database) è un report
aggiornato ai dati pubblicati di tutte le variazioni riscontrate sul DNA mitocondriale di soggetti affetti da patologie e su soggetti i cui campioni
sono stati prelevati per studi di genetica di popolazione. I dati sono annotati in forma tabellare e possono essere estratti attraverso l’utilizzo
di un sistema di interrogazione semplice. Non è presenta alcuna relazione tra i dati per cui non è possibile effettuare statistiche sulla
frequenza di variabilità di ciascun sito del genoma in cui siano state riscontrate e annotate mutazioni.
Human MitBASE è una banca dati nata per raccogliere in un'unica risorsa integrata i dati sul mitocondrio di tutti gli organismi eucariotici. I
dati sono organizzati in base a ogni individuo, alla sua origine geografica e alla sua descrizione dei dati clinici associati. Ogni entry raccoglie
moltissime informazioni associate all’individuo e ciò implica un notevole dispendio di risorse umane e una difficoltà di mantenimento della banca
dati stessa, che risulta meno aggiornata rispetto a MITOMAP.
HrvBase è una banca dati che raccoglie i multi-allineamenti delle sequenze relative alle regioni di controllo del genoma mitocondriale dei primati.
MITOP raccoglie informazioni su geni correlati alla funzionalità del mitocondrio di uomo, topo, lievito, Caenorhabditis elegans e Neurospora
crassa. Ogni entry è associata a una proteina della quale sono annotate la classe funzionale, il codice dell’enzima, il complesso proteico di
appartenenza della proteina, il peso molecolare, il punto isolettrico, etc.
MitoNuc una banca dati di geni nucleari di metazoi per il mitocondrio. I dati sono estratti da SWISSPROT come sequenze mitocondriali di

30
metazoi e vengono quindi accuratamente controllati e annotati con
informazioni specifiche. Per quanto riguarda le proteine umane è riportata la localizzazione del gene sul genoma umano ottenuta
attraverso analisi effettuate su Ensembl.
AMmtDB è la banca dati dei multi-allineamenti di geni codificati da genomi mitocondriali di Metazoi. Ogni entry è gene e classe-tassonomica specifica.
MITOCHONDRIOME è un sito web che raccoglie banche dati mitocondriali e informazioni correlate. Attraverso tale sito si accede alle
banche dati Human_MitBase, MITONUC e AMmtDB oltre a dati ottenuti dall’analisi di variabilità e complessità di geni e egenomi mitocondriali di
metazoi.
PLMitRNA è una banca dati di molecole e geni di tRNA identificati nei mitocondri di tutte le piante verdi. Informazioni caratterizzanti il gene o
la molecola sono annotate e possono esssere utilizzate per la ricerca dei dati. I tRNA possono essere selezionati per nome della specie o per
raggruppamento tassonomico. Il multiallineamento di ciascun cluster di tRNA omologhi è anche disponibile.
Link utili EBI (European Bioinformatics Institute):
http://www.ebi.ac.uk/
NCBI (National Center for Biotechnology Information): http://www.ncbi.nlm.nih.gov/
SRS (Sequence Retrieval System):
http://srs.ebi.ac.uk/
MitBASE (banca dati integrata di sequenze di DNA mitocondriale):
http://www3.ebi.ac.uk/Research/Mitbase/mitbase.pl
Human MitBASE (banca dati di varianti di DNA mitocondriale Umane associate a studi di genetica di popolazione e a studi sulle patologie mitocondriali):
http://srs.ebi.ac.uk/srs6bin/cgi-bin/wgetz?-page+LibInfo+-
lib+HUMAN_MITBASE
MITONUC (banca dati di geni nucleari coinvolti nella Biogenesi del
Mitocondrio):

31
http://bio-www.ba.cnr.it:8000/BioWWW/#MitoNuc
Wikipedia, l'enciclopedia libera:
http://it.wikipedia.org
BANCHE DATI BIBLIOGRAFICHE
MEDLINE (Medical Literature, Analysis, and Retrieval System Online)
MeSH (Medical Subject Headings )
Differenza tra PubMed e Medline
database di NLM delle citazioni su riviste e abstract che coprono 4500 riviste
pubblicate negli USA e in altri 70 paesi a partire dal 1966. Per poter accedere a Medline tramite web si utilizza PubMed.
Oltre a fornire accesso a Medline, Pubmed fornisce accesso ad altre info (citazioni fuori argomento)
Ricerche bibliografiche
PubMed (NCBI)
NLM GAteway Journals Databases (NCBI)
BOOKSHELF: collezione di testi biomedici; possibilità di ricerca online.
Gene Ontology
vocabolario descrittivo controllato delle funzioni molecolari, dei processi metabolici e delle localizzazioni cellulari di ciascun gene e del suo prodotto
indirizzo. http://www.geneontology.org/
BANCHE DATI ACIDI NUCLEICI
Banche dati primarie
EMBL:
http://www.ebi.ac.uk/cgi-bin/sva/sva.pl/
GenBank:
http://www.ncbi.nlm.nih.gov/Genbank/index.html
DDBJ:

32
http://www.ddbj.nig.ac.jp/
BANCHE DATI GENOMICHE
GDB http://www.gdb.org/
MGI http://www.informatics.jax.org/
SGD http://www.yeastgenome.org//
Banche dati derivate: malattie genetiche
OMIM Online Mendelian Inheritance in Man
indirizzo: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM
FAQ: http://www.ncbi.nlm.nih.gov/Omim/omimfaq.html
esempio: http://www.ncbi.nlm.nih.gov/Omim/omimhelp.html#QuickstartTutorial
BANCHE DATI DI GENI E TRASCRITTI
UniGene database di sequenze geniche trascritte dedotte da cDNA ed EST (ricavate dai databases primari) raggruppate in cluster che teoricamente
corrispondono ad un singolo gene.
http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unigene
LocusLink: database degli elementi funzionale del genoma (loci genetici).
RIMOSSO NEL 2005
http://www.ncbi.nlm.nih.gov/LocusLink/
RefSeq: Dati di sequenze nucleotidiche associati a: genoma, cromosoma,
regione genomica, mRNA, proteina Ad ogni entry è associata la sequenza più completa fra le ridondanti
http://www.ncbi.nlm.nih.gov/RefSeq/
dbEST: http://www.ncbi.nlm.nih.gov/dbEST/
BANCHE DATI DI MUTAZIONI E POLIMORFISMI
HGVbase: Annota i dati derivati da studi di variabilità della popolazione
ricavate dalla sottomissione degli autori o attraverso la consultazione della

33
letteratura
indirizzo: http://hgvbase.cgb.ki.se/
dbSNPs : annotazione di Single Nucleotide Polymorphisms; riporta anche
polimorfismi di regioni e mutazioni associate all'insorgenza di una specifica patologia, basandosi sul principio che la variazione della sequenza sia associata
a fenotipi ereditabili. Si vuole accelerare la scoperta di geni-malattia.
DIFFERENZA DA OMIM: non si basa sull'analisi di pedigree, ma sulla tipizzazione di DNA ottenuti da un campione casuale di individui (più facile).
http://www.ncbi.nlm.nih.gov/SNP/
HGMD (Human gene Mutation Databases)
indirizzo: http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html
BANCHE DATI DI REGIONI FUNZIONALI
UTRdb: database delle regioni non tradotte dei trascritti (regolazione dei processi post-trascrizionali, compartimentazione dei trascritti, stabilità)
Promemoria: diversi link utili in
http://www.ba.itb.cnr.it/BIGHome/ita/Sezione.htm
indirizzo: http://bighost.area.ba.cnr.it/BIG/UTRHome/
EPD (Eukaryotic Promotor Database) : collezione di promotori eucariotici per la
POL II annotati e non ridondanti, il cui codone d'inizio trascrizione è stato determinato sperimentalmente.
indirizzo: http://www.epd.isb-sib.ch/
TRANSFAC : Banca dati dei fattori di trascrizione. Annota i dati sui fattori proteici e i corrispondenti siti di legame sul DNA coinvolti nell'attivazione o
regolazione della trascrizione
indirizzo: http://www.gene-regulation.com/
TRANSTERM : Banca dati degli elementi che regolano la traduzione e le
modificazioni pos-trascrizionali. Ricavato dalla banca primaria GenBank
indirizzo: http://cbcb.umd.edu/software/transterm/
BANCHE DATI DI SEQUENZE PROTEICHE
SWISS-PROT: http://us.expasy.org/sprot/

34
banca dati di riferimento per analisi in silico. Le annotazioni sono: AC; nome;
dati generali (features: aminoacidi modificati, regioni peptidiche di isoforme, domini strutturali, siti di splicing proteici, polimorfismi ecc.; informazioni su
malattie ereditarie (OMIM). Cross-link con altre banche dati (Pfam ,PRINTS,
ProDom, SMART, PROSITE, ecc)
The UniProt Knowledgebase consists of:
UniProtKB/Swiss-Prot; a curated protein sequence database which strives to provide a high level of annotation (such as the description of the function of a
protein, its domains structure, post-translational modifications, variants, etc.),
a minimal level of redundancy and high level of integration with other databases
UniProtKB/TrEMBL; a computer-annotated supplement of Swiss-Prot that
contains all the translations of EMBL nucleotide sequence entries not yet integrated in Swiss-Prot.
TrEMBL contains the translations of all coding sequences (CDS) present in the EMBL Nucleotide Sequence Database, which are not yet integrated into
SwissProt.
PIR Protein Infromation Resource : http://pir.georgetown.edu/pirwww/
PIR offers a wide variety of resources mainly oriented to assist the propagation
and standarization of protein annotation. Among these are: PIRSF, which provides curated protein families with rules for functional site and protein
name; iProLink, that supports text mining in the area of literature-based database curation, named entity recognition, and protein ontology
development; and iProClass, which contains value-added annotation reports for UniProt proteins.
UNIPROT: http://www.pir.uniprot.org/
UniProt (Universal Protein Resource) is the world's most comprehensive catalog of information on proteins. It is a central repository of protein
sequence and function created by joining the information contained in Swiss-Prot, TrEMBL, and PIR.
INTERPRO
consorzio di database; integra numerose banche dati proteiche (PROSITE, Pfam,PRINTS,ProDom,SMART,TIGRFAMs).
Consente di ricercare contemporaneamente informazioni funzionali e strutture relative ad una proteina o a una famiglia di proteine su più banche dati
distribuite anche su calcolatori differenti e strutturate in modo differente. La ricerca dei dati può essere fatta attraverso il sito SRS dell'EBI.
Attraverso il software InterPROScan è possibile ricercare motivi strutturali e

35
funzionali annotati nelle banche dati integrate in InterPRO al fine di
caratterizzare dal punto di vista funzionale nuove proteine derivate a progetti di sequenziamento genomico
indirizzo: http://www.ebi.ac.uk/interpro/
BANCHE DATI DI DOMINI PROTEICI
PROSITE Patterns amminoacidici. L'entry è divisa in due parti:
1) PDOCxx: documentazione: funzionalità del pattern ; bibliografia NiceSite View (PSxx): annotazione del pattern (secondo regole della sequenza
consenso); tutte le entry in SWISSPROT dove il pattern è localizzato (per ogni entry SWISSPROT riportato la funzionalità riscontrata in letteratura (T)=True
(F)=false (P)=potential ; cross-reference in SWISSPROT e PDB
indirizzo: http://us.expasy.org/prosite/
Pfam banca dati di famiglie di proteine accomunate da elementi strutturali e
funzionali . Si divide in due parti:
PfamA: Ogni entry è caratterizzata da:
- famiglia: sequenze proteiche accomunate dagli stessi domini
- dominio: unità strutturale che può essere presente in famiglie differenti - repeats raggruppa elementi funzionali attivi e presenti in multi copia in
proteine globulari - motivi pattern componenti blocchi strutturali non associati a proteine
globulari PfamB:
famigli proteiche ottenute automaticamente (PSI-BLAST e annotate in ProDOM)
Questa parte del database è memo accurata della PfamA
indirizzo: http://www.sanger.ac.uk/Software/Pfam/
ProDOM famiglie proteiche generate automaticamente da PSI_BLAST; il
database è generato a partire da multiallineamento di famiglie di proteine annotate in PfamA
indirizzo: http://prodes.toulouse.inra.fr/prodom/current/html/home.php
SMART Raccoglie dati relativi a domini proteici e consente la ricerca di domini in nuove sequenze proteiche
Sono annotate per ogni famiglia di proteine associate ad un dominio informazioni quali:
- funzione - localizzazione cellulare

36
- struttura terziaria (del dominio)
- relazioni filogenetiche tra le specie da cui sono derivate le proteine
indirizzo: http://smart.embl-heidelberg.de/
PRINTS Raccoglie sequenze proteiche in cluster definiti da un comune
Fingerprint (fingerprint è l'insieme di più motivi conservati e dedotti dal multiallineamento utilizzando similarità locale)
I cluster ottenuti definiscono: - superfamiglie
- famiglie
- sottofamiglie associa a queste famiglie la loro funzionalità.
E' un database ridotto rispetto a Pfam e proDOM, ma i dati sono vagliati in manuale e annotati con dati biologici derivati dalla letteratura e da ulteriore
analisi
indirizzo: http://bioinf.man.ac.uk/dbbrowser/PRINTS/
BANCHE DATI DI STRUTTURE PROTEICHE
PDB
È la principale banca dati delle strutture proteiche.
Struttura di una proteina: distribuzione spaziale degli atomi che compongono la proteina (coordinate atomiche determinate con analisi cristallografiche con
raggi X o NMR indirizzo: http://www.rcsb.org/pdb/
Altre banche dati di struttura ricavate da PDB: MMDB (Molecular Modeling DataBase) (NCBI)
Strutture ricavate da PDB escludendo i modelli teorici e validate da diverse procedure.
Contiene inoltre una definizione uniforme di strutture secondarie ecc.
indirizzo: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=Structure
DSSP (Dictionary of Protein Secondary structure)
Ad ogni entry di PDB sono associate le informazioni sulle relative strutture secondarie
indirizzo: http://www.cmbi.kun.nl/gv/dssp/
HSSP (Homology derived Secondary Structure of Proteins) Contiene informazioni per costruire modelli di proteina a struttura non nota ma
che abbiano una buona identità di sequenza.
indirizzo: http://www.sander.ebi.ac.uk/hssp/

37
FSSP (Fold classification based on Structure Structure alignment of Protein):
classificazione della struttura sulla base della loro similarità (ricavato in modo automatico dal programma DALI)
indirizzo: http://www.embl-ebi.ac.uk/dali/
SCOP (Structural Classification Of Protein): Organizza le strutture proteiche gerarchicamente seguendo criteri evolutivi e di
similarità strutturale. Si base sui singoli domini raggruppandoli in famigli di domini simili. Queste
famiglie sono organizzate in superfamiglie che sono raggruppate a loro volta in
fold. I fold simili sono organizzati in classi: Classi: alpha, beta, alpha/beta, alpha + beta (in base alla struttura secondaria
della proteina) Fold
Superfamiglie Famiglie
Domini
indirizzo: http://scop.mrc-lmb.cam.ac.uk/scop/
CATH (Class Architecture,Topology, Homologus superfamilies)
Simile a SCOP ma le strutture sono ricavate con il programma SSAP.
indirizzo: http://www.biochem.ucl.ac.uk/bsm/cath/
BANCHE DATI MITOCONDRIALI
MITOMAP
http://www.mitomap.org/
GOBASE
http://megasun.bch.umontreal.ca/gobase/
HUMAN MITOCHONDRIAL PROTEIN DATABASE
http://bioinfo.nist.gov:8080/examples/servlets/index.html
SISTEMI DI INTERROGAZIONE DELLE BANCHE DATI BIOLOGICHE
SRS (Sequence Retrievel System): ( http://srs.ebi.ac.uk o http://bighost.area.ba.cnr.it/srs6/)
Consente di interrogare contemporaneamente più banche biologiche Sfruttando i meccanismi di cross-referencing consente la navigazione tra

38
banche di dati differenti
Può essere installato su qualsiasi server e può essere integrato con altre banche dati
Una volta scelte la/le banche dati si può fare l'interrogazione con le
- Standard Query Form: la relativa Query Page consente di selezionare i dati impostando 4 differenti criteri usando AND, OR, BUT NOT. All'interno di ciascun
criterio è possibile selezionare più valori combinandoli con '&' (AND), '|' (OR), '!' (BUT NOT)
- Extended Query Form: si adatta agli attributi delle banche prescelte. Consente di utilizzare tutti i criteri possibili di selezione che accumunano tali
banche utilizzando gli stessi operatori della Standard Query Form. Dal menù View è possibile selezionare le banche e i campi desiderati da
visualizzare Si possono salvare i dati sul proprio PC con lo stesso criterio adottato per la
visualizzazione. Altre funzioni di SRS:
Result: si visualizzano i risultati delle varie selezioni fatte in una sezione. Link: si ottengono dati presenti in altre banche correlati ai dati alla query
prescelta
Launch: (non sempre presente) consente di applicare programmi di analisi ai dati di sequenza associati alla query (Blast, Fasta, Clustaw, ecc)
Projects: consente di salvare in un file tutte le fasi delle sessioni SRS per poi riutilizzarlo in sessioni successive consentendo di aggioranre i dati associati al
progetto previa selezione dalla top page delle banche dati coinvolte.
Entrez Ricerca ed estrazione dei dati da bache dati di sequenze nucleotidiche o
proteiche, dalla banca dati bibliografica Medline, dalla banca dati delle malattie mendeliane OMIM o da risorse genomiche. Possibilità di esplorare ogni altra
banca sviluppata all'NCBI (es. la classificazione degli organismi riportata in
Taxonomy ) A differenza di SRS, Entrez è una shell chiusa, non è possibile scaricare il
software che gestisce il sistema, quindi non è possibile duplicare Entrez in altri computer e non è possibile integrarlo con dati personali.
Ricerca dati previa scelta della categoria: nucleotide, protein, PubMed, genomes ecc.) usando gli operatori booleani.
La ricera può essere ottimizzata mediante la funzione Limits che consente di limitare la ricerca a solo alcuni criteri o a sottoinsiemi selezionati di dati.
Consente di effettuare una navigazione fra le diverse banche disponibili. Con il comando History si visualizzano tutte le query selezionate nell'ambito di
una categoria di dati (nucleotide, protein, ecc) I dati ottentuti e associati a ciascuna query possono essere visualizzati e
salvati sul proprio PC.
RISORSE GENOMICHE INTEGRATE
ENSEMBL

39
http://www.ensembl.org
Raccolta e annotazioni di dati genomici: - umano
- topo - pesce zebra (Danio retrio)
- Anopheles gambiae Riporta dati sul mappaggio dei geni e polimorfismi
È possibile visualizzare ed estrarre dati relativi ai geni come i suoi trascritti, le sue proteine, domini funzionali delle proteine, mutazioni o SNPs e correlazione
con le malattie genetiche. E' possibile effettuare una ricerca di similarità con una sequenza mediante
BLAST i cui risultati sono visualizzati sui cromosomi con differenti colori. I dati presenti in Ensembl possono essere completamente scaricati in locale
GENOME BROWSER
http://genome.ucsc.edu/
MIRROR ITALIANO: http://genome.cribi.unipd.it/
Link utili
SRS italiano: http://bighost.area.ba.cnr.it/srs6/
Come usare le banche dati
I seguenti files formato "pdf " sono tratti dal sito http://www.ceebt.embo.org"Continuing Education for European Biology Teachers" e mostrano come utilizzare le banche dati SwissProt, OMIM e PubMed nel dettaglio.
(vedere file Pdf a parte)
1. Swiss Prot
2. OMIM 3. PubMed
GLOSSARIO (voci tratte da Wikipedia)
Genoma: è l'insieme dei geni di un organismo vivente.
L'informazione genetica è portata dalla molecola di Acido desossiribonucleico (DNA) che,
associato aproteine, è il principale costituente dei cromosomi degli Eucarioti. L'insieme dei
cromosomi, o comunque delle molecole di DNA, determina il patrimonio genetico. Il DNA è
latore dell'informazione ereditaria il cui meccanismo d'azione è stato ampiamente decifrato
grazie allo studio degli Acidi nucleici.

40
Il patrimonio genetico strutturale è scritto nella catena del DNA con un codice detto Codice
genetico che mette in corrispondenza le quattro basi azotate che entrano nella composizione
del DNA stesso con gli amminoacidi. Ciascuna parola del codice è costituita da una serie di tre
basi detta tripletta. Ognuna di esse indica agli organi effettori (RNA e ribosomi) che deve
essere preso un determinato amminoacido e legato alla catena polipeptidica che si sta
costruendo. Da ciò si coglie che il fenomeno genetico fondamentale, a livello cellulare, è la sintesi delle proteine.
Proteoma: Il termine proteoma, coniato da Mark Wilkins nel 1995 (1), è usato per descrivere
l'insieme delle proteine di un organismo o di un sistema biologico, ovvero le proteine prodotte
dalgenoma. Il termine è stato applicato a diversi tipi di sistemi biologici. Esiste un proteoma
cellulare, che è un insieme di proteine trovate in un particolare tipo di cellule in particolari
condizioni ambientali, come ad esempio sotto esposizione ad una stimolazione ormonale. Può
anche essere utile considerare il proteoma completo di un organismo, che può essere
immaginato come l'insieme globale delle proteine di tutti i proteomi cellulari. Questo è, grosso
modo, l'equivalente proteico del genoma. Il termine "proteoma" è stato usato anche per
riferirsi all'insieme delle proteine di un sistema biologico sub-cellulare: ad esempio l'insieme
delle proteine di un virus può essere detto proteoma virale.
Il proteoma è più grande del genoma, specialmente negli eucarioti, perché ci sono
più proteine chegeni. Ciò è dovuto all'accoppiamento dei geni ed alle modificazioni post-traslazionali come laglicosilazione o la fosforilazione.
Il proteoma mostra almeno due livelli di complessità che mancano al genoma. Mentre il
genoma è definito da una sequenza di nucleotidi, il proteoma non si limita alla somma delle
sequenze di proteine presenti. La conoscenza del proteoma richiede di conoscere, oltre alle strutture delle proteine del proteoma, anche le interazioni funzionali tra le proteine stesse.
Lo studio del proteoma è detto Proteomica. Esso è stato a lungo praticato con la separazione
delleproteine per mezzo della elettroforesi bidimensionale su gel. Nella prima dimensione,
le proteine sono separate per punto isoelettrico, che distingue le proteine sulla base del loro
pH. Nella seconda dimensione le proteine sono separate per massa molecolare usando l'SDS-
PAGE. Il gel è colorato conCoomassie Blue o argento per visualizzare le proteine. Le macchie sul gel sono proteine che sono migrate in posizioni specifiche.
Lo spettrometro di massa ha migliorato la proteomica. La tecnica nota come Peptide mass
fingerprinting identifica una proteina scindendola in brevi segmenti peptidici e successivamente
deducendo l'identità della proteina confrontando le masse dei peptidi con quelle di un database
di riferimento. La spettrometria di massa, d'altra parte, può fornire informazioni sulle sequenze
dapeptidi singoli isolandoli, trattandoli con un gas inerte e quindi catalogando i frammenti ioniciprodotti.
In Silico: La locuzione latina in silico, tradotta letteralmente, significa nel silicio.
La locuzione, comparsa di recente in letteratura scientifica, è usata per indicare fenomeni
biologici riprodotti in una simulazione matematica al computer, invece che in provetta o in un
essere vivente. Infatti il silicio, è la sostanza di cui sono fatti i componenti elettronici all'interno
della quasi totalità dei computer, anche se il concetto di simulazione matematica non ha niente
a che fare con il silicio. Al contrario, se il fenomeno biologico si riproduce in provetta, si dice "in
vitro", mentre se si riproduce in un essere vivente si dice "In vivo".
La ricerca scientifica in silico è ovviamente il frutto della recente informatizzatione della ricerca.
Inbiologia hanno assunto un importanza fondamentale le basi di dati che contengono i dati
disequenziamento del DNA e i livelli di espressione genica di un gene. Inoltre sono stati creati
numerosiprogrammi informatici per l'analisi e l'interpretazione di dati sperimentali. La
ricerca in silico consiste dunque nell'uso di tali programmi e raccolte di dati allo scopo di

41
ricavare nuove informazioni dalla comparazione, interpretazione, analisi ed interconnessione
dei dati.
Nucleotidi: I nucleotidi sono i monomeri che formano i polimeri DNA e RNA a seconda che lo
zucchero sia desossiribosio o ribosio. Sono costituiti da un gruppo fosfato, da uno zucchero
pentosio (desossiribosio o ribosio appunto) e da una base azotata. I nucleotidi sono
degli esteri fosforici deinucleosidi, costituiti da tre subunità: una base azotata (purina o
pirimidina), uno zucchero a cinque atomi di carbonio (pentosio) e un gruppo fosfato. Il
pentosio può essere ribosio o desossiribosio. Il nucleotide è inoltre il monomero costitutivo degli acidi nucleici (DNA e RNA).
La presenza del residuo fosforico conferisce carattere fortemente acido ai nucleotidi (per
questo noti anche come acido adenilico o acido guanilico). L'aggiunta di uno o di due altri
residui fosforici nella catena (fosforilazione ossidativa) produce i nucleoside-di- e trifosfati (NDP e NTP), i quali svolgono un ruolo fondamentale nel metabolismo energetico della cellula.
Mirror: In Informatica il termine mirror viene utilizzato per indicare una copia esatta di un insieme di dati.
L'utilizzo più conosciuto di questo termine lo si trova nei mirror dei server internet: un intero
sito (pagine HTML ma anche semplici file binari) viene copiato su di un altro computer per
essere reso accessibile anche da altre fonti. Il motivo principale dell'utilizzo dei mirror sta nel
sovraccarico del sito originale che spesso non possiede una banda sufficiente per l'utenza a cui
è destinato. Attraverso questo meccanismo (il cui aggiornamento viene svolto
automaticamente a intervalli regolari) una stessa risorsa può essere disponibile a un maggior numero di utenti.
Amminoacidi: in chimica gli amminoacidi (o aminoacidi) sono molecole che nella loro
struttura recano sia il gruppo funzionale dell'ammina (-NH2) che quello dell'acido carbossilico (-COOH).
In biochimica il termine amminoacidi si riferisce più spesso agli L-α-amminoacidi, cioè quelli il
cui gruppo amminico ed il cui gruppo carbossilico sono legati allo stesso atomo di carbonio (chiamato appunto carbonio α) in configurazione L
Peptidi: sono molecole di peso ≤5000 dalton, costituiti da una catena di
pochi amminoacidi (fino a 100 circa), che si uniscono tra di loro attraverso un legame
peptidico. A loro volta queste catene peptidiche si uniscono formando le proteine.
Tra i peptidi troviamo l'ossitocina (ormone neuroipofisario), le bradichinine (antinfiammatorio
tissutale), gli ormoni (o fattori) ipotalmici (che favoriscono/inibiscono la sintesi di ormoni ipofisiari),ilglutatione, le encefaline (analgesici naturali prodotti nel Sistema nervoso centrale).
Splicing: Il termine splicing (saldatura) indica, nella lingua inglese, la maturazione del trascritto primario dei geni discontinui.
La maggior parte dei geni eucariotici conta regioni presenti nel mRNA maturo (esoni) e altre
non presenti (introni). Alcuni introni sono presenti anche nei geni degli archeobatteri, mentre
sono assenti in quelli degli eubatteri. Dopo la trascrizione da parte della RNA polimerasi il
trascritto primario va incontro a numerose modificazioni. Prima fra tutte l’eliminazione degli
introni, denominata splicing.
Fingerprint: La fingerprint (impronta digitale) in informatica è una
sequenza alfanumerica o stringadi bit di lunghezza prefissata che identifica in maniera univoca
un certo file. Viene utilizzata per garantire l'autenticità e la sicurezza dei file ma soprattutto per identificare rapidamente file distribuiti in rete tramite sistemi di file-sharing.

42
Eucariota: Il termine eukaryota (o eucariote, eucariota, o eukarya) deriva dalla fusione dei
due termini greci "Eu", bene e "Carion", nucleo.
Le cellule, in base alla loro organizzazione interna, possono essere distinte in due grandi
categorie: le cellule procariote, esclusive dei Procarioti (rappresentati dal regno Monera), e le
cellule eucariote, che sono invece caratteristiche di organismi viventi più evoluti, gli
Eucarioti.Tali organismi sono classificati nei rimanenti quattro regni dei viventi
(Protisti, Piante, Funghi e Animali).
Il dominio Eukaryota comprende organismi, mono- o pluricellulari, costituiti da cellule
eucariote che hanno come caratteristica principale la presenza di un nucleo, ben definito e
isolato dal resto della cellula tramite l'involucro nucleare, nel quale è racchiuso la maggior
parte del materiale genetico, ilDNA (una parte è contenuta nei mitocondri).
Queste cellule presentano una dimensione maggiore (solitamente il loro asse maggiore è
compreso fra i 10 e i 50 µm) e sono dotate di un citoplasma compartimentato da membrane
interne che delimitano degli organuli specializzati. Ognuno degli organuli immersi nel citoplasma è deputato a svolgere una particolare funzione.
Gli organuli presenti in tutte le cellule eucariote sono un nucleo, diversi mitocondri, le cisterne
delreticolo endoplasmatico e dell'apparato di Golgi, vacuoli (vescicole temporanee tra cui
i lisosomi). Sono presenti aggregati complessi come i ribosomi, e varie fibre proteiche che costituiscono il citoscheletro.
Gli Eucarioti si distinguono dai Procarioti anche per numerose caratteristiche a livello molecolare quali, ad esempio:
diverse proprietà delle sequenze genomiche regolatrici geni organizzati in "introni ed esoni" con conseguente processamento (splicing) del
trascritto primario trascrizione e traduzione di un trascritto sono eventi separati nello spazio e nel tempo i trascritti eucariotici non sono (quasi) mai policistronici, ossia portano una sola ORF percentuale di DNA non codificante molto più elevata DNA associato ad istoni diversa percentuale di G-C nel genoma presenza di colesterolo nella membrana cellulare.
Solo negli Eucarioti si ha riproduzione sessuale: le cellule eucariote presentano due modi di
divisione: la mitosi e la meiosi. Tutte le cellule possono dividersi attraverso il processo di mitosi, ma solo quelle diploidi possono subire la meiosi.
Cromosoma: in biologia, il cromosoma è un corpuscolo che appare nel nucleo di una cellula eucariota durante la mitosi o la meiosi.
In greco "chroma" significa "colore" e soma significa "corpo". Essi sono colorati da sostanze
apposite, perché siano visibili al microscopio. Sono costituiti da un filamento di DNA e da
proteine.
I cromosomi sono spesso presenti in coppie, 23 nella specie umana, di cui 22 coppie sono
cromosomi omologhi (cioè simili) detti autosomi ed una coppia di cromosomi diversi che sono i
cromosomi sessuali. Tutti i cromosomi sono portatori dei caratteri ereditari. Le cellule che
hanno coppie di cromosomi omologhi sono dette diploidi (2n), mentre sono definite aploidi (n)
quelle che possiedono solo un cromosoma per tipo.
I nuclei delle cellula eucariotica contengono un materiale che si colora intensamente con certi
coloranti istologici e viene quindi detto cromatina. Durante l'interfase la cromatina non rivela

43
alcuna struttura, se non la differenziazione tra una componente maggioritaria più lassa
(eucromatina) e una più condensata (eterocromatina).
Durante la divisione cellulare la cromatina si suddivide in un numero ben definito di corpiccioli
con la dimensione maggiore dell'ordine del micrometro: i cromosomi. Nella cellula appena
formata, i cromosomi hanno forma di bastoncelli: l'unica struttura evidente al microscopio è una strozzatura detta centromero.
Nella metafase i cromosomi hanno una forma a X, dovuta al fatto che si sono quasi
completamente duplicati e risultano formati da due cromatidi identici, uniti per il centromero che si divide per ultimo.
Al microscopio ottico, i cromosomi sono distinguibili tra loro per le dimensioni e per la "forma",
ossia per la posizione del centromero. Ulteriori distinzioni si possono effettuare con opportuni
trattamenti chimici, che evidenziano un bandeggio: l'alternanza di bande con diversa
pigmentazione.
Numero e struttura dei cromosomi costituiscono il cariotipo, ben evidenziabile (e fotografabile) durante la metafase, in cui i cromosomi si dispongono nella piastra metafasica.
La cromatina è costituita permanentemente da DNA e proteine. Il Dna è avvolto attorno a
cilindretti formati dai quattro tipi di istoni, proteine basiche: tale fibra fondamentale si chiama cromonema ed è ulteriormente avvolta in strutture di ordine superiore.