Embed Size (px)
Citation preview

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 1 of 96
DELIVERABLE
D1.2 – Regulatory constraints
Project Acronym: UNCAP
Grant Agreement number: 643555
Project Title: Ubiquitous iNteroperable Care for Ageing People
Revision:
Authors: Elisa Morganti, Claudio Eccher, Simona Anzivino, Francesco Tessarolo (FBK); Maša Isakovič (UL), Polona Lah (VOG); George Spyroglou (Bioassist); Evangelia Romanopoulou, Panagiotis Bamidis (AUTH); Saso Koceski, Natasa Koceska (GDU); Valentina Conotter (SocialIT), Giuseppe Conti (TRILOGIS)[…]
Project co-funded by the Horizon 2020 Framework Programme of the European Union
Dissemination Level P Public X C Confidential, only for members of the consortium and the Commission Services
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 2 of 96
1. Revision history and statement of originality
Revision history
Rev Date Author Organization Description
V0.1 16/03/2015 Simona Anzivino FBK First Draft
V0.2 23/03/2015 Maša Isakovič UL Slovenia regulatory constraints
V0.3 31/03/2015 Saso Koceski GDU Macedonia regulatory constraints
V0.4 01/04/2015 George Spyroglou BioAssist Greece regulatory constraints
V0.5 02/04/205 Evangelia Romanopoulou
AUTH Thessaloniki specific constraints
V0.6 08/04/2015 Simona Anzivino FBK Medical devices
V0.7 09/04/2015 Polona Lah VOG Slovenia ethical laws
V1.0 09/04/2015 Claudio Eccher FBK European Ethical regulations
v1.1 17/04/2015 Giuseppe Conti FBK Review
V1.2 22/04/2015 Saso Koceski UGD Macedonia ethical regulations
V1.3 22/04/2015 Francesco Tessarolo FBK Regulations concerning medical devices
Conducting clinical trials in Italy
V2.0 22/04/2015 Elisa Morganti FBK Review
V2.1 23/04/2015 Elisa Morganti, Claudio Eccher
FBK Conclusions
V2.2 28/04/2015 Elisa Morganti FBK Final version
V2.3 29/04/2015 Giuseppe Conti Trilogis Final review
V2.4 30/10/2015 Elisa Morganti, Jovan Stevovic
FBK, CHINO Added contribution to reviewer’s comments
Statement of originality This deliverable contains original unpublished work except where clearly indicated otherwise. Acknowledgement of previously published material and of the work of others has been made through appropriate citation, quotation or both.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 3 of 96
2. List of references
Number Full Reference
1 World Medical Association Declaration of Helsinki. Ethical Principles for Medical Research Involving Human Subjects.
Available online at: www.wma.net/en/30publications/10policies/b3/17c.pdf
2 Directive 2001/20/EC of the European Parliament and of the Council of 4 April 200. Available online at: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2001:121:0034:0044:en:PDF
3 COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 and amendments http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CONSLEG:1993L0042:20071011:en:PDF
4 ISO14155 Available online at: http://www.iso.org/iso/catalogue_detail?csnumber=45557
5 Clinical Evidence – Key Definitions and Concepts Study Group 5 Final Document, Global Health Task Force SG5/N1R8. May 2007
6 MEDDEV 2.7/4 December 2010 GUIDELINES ON MEDICAL DEVICES GUIDELINES ON CLINICAL INVESTIGATION: A GUIDE FOR MANUFACTURERS AND NOTIFIED BODIES
7 CLINICAL INVESTIGATION OF MEDICAL DEVICES IN EUROPE An overview of requirements, study structure, existing data and common pitfalls. Author: Jaap Laufer, MD, PharmD Vice President of Regulatory and Clinical Affairs at Emergo
8 ICH Good clinical practice. Available online at: http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/good-clinical-practice.html.
9 Decreto Legislativo 24 giugno 2003. Available online at: http://www.camera.it/parlam/leggi/deleghe/testi/03211dl.htm
10 Decreto lgs. 24 febbraio 1997. Available online at: http://www.camera.it/parlam/leggi/deleghe/97046dl.htm
11 Decreto Legislativo 25 gennaio 2010, n. 37. Available online at: http://www.camera.it/parlam/leggi/deleghe/10037dl.htm
12 Code of Medical Ethics. Greek Government Gazette. 28 Nov 2005;287(1):5391-5402.
13 Law no. 3304/2005 (Greece). Available online at: http://www.ilo.org/dyn/natlex/natlex4.detail?p_lang=en&p_isn=83985&p_country=GRC&p_count=610&p_classification=05&p_classcount=9
14 Medical products act (Germany). Available online at: http://www.gesetze-im-internet.de/englisch_amg/englisch_amg.html#p0917

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 4 of 96
15 Regulation for the Application of Good Clinical Practice of Clinical Medications for Human Use (2012) (Germany). Available online at:
http://www.gesetze-im-internet.de/bundesrecht/gcp-v/gesamt.pdf
16 Principles and Responsibilities When Carrying Out Clinical Studies (2013) (Germany). Available online at:
http://www.gesundheitsforschung-bmbf.de/_media/Grundsaetze_und_Verantwortlichkeiten_20130424.pdf
17 Act on Medical Devices (2014) (German). Available online at: http://bundesrecht.juris.de/mpg/index.html
18 Romanian legislation. Available online at: http://www.anm.ro/en/html/legislation_minister_orders.html
19 The Constitution of The Republic of Slovenia. Available online at: http://unpan1.un.org/intradoc/groups/public/documents/UNTC/UNPAN014895.pdf
20 Slovenia Research and Development act Official Journal L 121, 1/5/2001 p.34–44
21 PERSONAL DATA PROTECTION ACT OF THE REPUBLIC OF SLOVENIA, Ministry of Justice of the Republic of Slovenia, 2013, https://www.ip-rs.si/fileadmin/user_upload/doc/ZVOP-1_in_ZVOP-1a__English_/Personal_Data_Protection_Act_of_Slovenia_status_2013_final_eng.doc
22 Charter of Fundamental Rights of the European Union Available online at: http://www.europarl.europa.eu/charter/pdf/text_en.pdf
23 Convention on Human Rights and Biomedicine. Available online at: http://conventions.coe.int/Treaty/en/Treaties/Html/164.htm.
24 European Social Charter. Available online at: https://www.coe.int/t/dghl/monitoring/socialcharter/Presentation/ESCRBooklet/English.pdf
25 Additional Protocol to the Convention on Human Rights Biomedicine, concerning Biomedical Research. Available online at: http://conventions.coe.int/Treaty/en/Treaties/Html/195.htm
26 Slovenian Code of Medical Deontology (Kodeks medicinske deontologije Slovenije). Zdravni√ka zbornica Slovenije, 1992.
27 Slovenia Code of ethical principles in social care Official Gazette RS, No. 59/2002
28 Закон за лекови и медицински помагала (Службен весник на РМ број). Available online at: https://lekovi.zdravstvo.gov.mk/documents/2
29 Правилник за налинот и постапката за клиничките испитувања на лековите и содината на докуметацијата Available online at: https://lekovi.zdravstvo.gov.mk/documents/1/1
30 Упатство за начелата на добрата клиничка пракса (Службен весник на РМ бој 62/2009) Available online at:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 5 of 96
https://lekovi.zdravstvo.gov.mk/documents.documentcomponent:downloadfile/817325622?t:ac=1/1
31 Convention for the Protection of Individuals with regard to Automatic Processing of Personal Data of the Council of Europe of 1 January 1981, Strasbourg.
conventions.coe.int/Treaty/EN/Treaties/Html/108.htm
32 Directive 95/46/EC. Available online at:
eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:31995L0046:en:HTML
33 Directive 2002/58/EC. Available online at:
eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=CELEX:32002L0058:en:HTML
34 Data Protection, Information Privacy, and Security Measures: an essay on the European and the Italian Legal Frameworks, Paolo Guarda. Available online at: eprints.biblio.unitn.it/1524/1/DataProtection_SecurityMeasures_Guarda.pdf
35 Available online at: europa.eu/rapid/press-release_IP-12-46_en.htm
36 Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the regions safeguarding privacy in a connected world a European data protection framework for the 21st century (COM/2012/09 final). Available online at: http://eur-lex.europa.eu/legal-content/en/ALL/?uri=CELEX:52012DC0009
37 Available online at: ec.europa.eu/justice/policies/privacy/docs/wpdocs/2011/wp187_en.pdf
38 Italian Personal Data Protection Code. Legislative Decree no. 196 of 30 June 2003. Available online at: www.privacy.it/privacycode-en.html
39 Law 2472/97. Available online at: www.dpa.gr/pls/portal/docs/PAGE/APDPX/ENGLISH_INDEX/LEGAL%20FRAMEWORK/LAW%202472-97-APRIL010-EN%20_2_.PDF
40 P. Kalampouka-Giannopoulou, Protection of the patient as a consumer, Nomiki Bibliothiki, Athens, 2011, p. 149, 166
41 G.Vasilakopoulos, Security of electronic medical records: International trends and Greek reality in medical confidentiality, Sakkoulas Athens – Thessaloniki, 2006, p.306.
42 Annual Report of DPA, 2011, p.68. Available online at: www.dpa.gr/pls/portal/docs/PAGE/APDPX/ANNUALREPORTS/AR2011/ARXH_PROSTASIAS_2011.PDF
43 P. Tsantila, Ch. Latsiou, Medical confidentiality in light of the personal data protection, Review of the Social Security Law, Vol.3-4, 2011, pp 161-167.
44 Law 2071/1992. Available online at: www.elinyae.gr/el/item_details.jsp?item_id=2736&cat_id=686
45 Available online at:
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 6 of 96
www.dgipi.ro/administrare/_uploads/_documente/26_20101019161007094188400_5.pdf
46 Available online at: www.spitalalba.ro/wp/wp-content/uploads/2013/07/Legea-nr.46-din-21-ianuarie-2003-Legea-drepturilor-pacientului.pdf
47 Available online at: dataprotection.ro/servlet/ViewDocument?id=35
48 Available online at: dataprotection.ro/servlet/ViewDocument?id=451
49 Available online at: dataprotection.ro/servlet/ViewDocument?id=861
50 Available online at: dataprotection.ro/servlet/ViewDocument?id=859
51 Available online at: www.legi-internet.ro/legislatie-itc/criminalitate-informatica/prevederi-legislative-privind-prevenirea-si-combaterea-criminalitatii-informatice/legea-1612003-pentru-prevenirea-si-sanctionarea-coruptiei.html
52 Available online at: www.legi-internet.ro/legislatie-itc/date-cu-caracter-personal/codul-civil.html
53 Overview of the national laws on electronic health records in the EU Member States, National Report for Slovenia, 2014, http://ec.europa.eu/health/ehealth/docs/laws_slovenia_en.pdf
54 Available online at: eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2007:247:0021:0055:en:PDF
55 Available online at: eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:1993:169:0001:0043:EN:PDF
56 Law on Personal Data Protection in the Republic of Macedonia, available online at:
http://www.dzlp.mk/sites/default/files/Law_on_Personal_Data_Protection_Cleared_version_0.pdf
57 Charter of Fundamental Rights of the European Union.
Available online at: www.europarl.europa.eu/charter/pdf/text_en.pdf
58 European Convention for the Protection of Human Rights and Fundamental Freedoms.
Available online at: www.echr.coe.int/Documents/Convention_ENG.pdf
59 ITU-R Radio Regulations. Available online at: www.itu.int/pub/R-REG-RR/en
60 WAI (Web Accessibility Initiative). Available online at: www.w3.org/WAI/
61 WCAG (Web Content Accessibility Guidelines). Available online at: www.w3.org/WAI/intro/wcag
62 UAAG (User Agent Accessibility Guidelines). Available online at: www.w3.org/WAI/intro/uaag

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 7 of 96
63 ATAG (Authoring Tool Accessibility Guidelines). Available online at: www.w3.org/WAI/intro/atag.php
64 WAI-ARIA (Accessible Rich Internet Applications Suite). Available online at: www.w3.org/WAI/intro/aria
65 Available online at: eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv:OJ.L_.2014.158.01.0001.01.ENG
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 8 of 96
3. Table of Acronyms
Acronym Description
ATAG Authoring Tool Accessibility Guidelines
BDSG Bundesdatenschutzgesetz (Federal Data Protection Act)
CEN European Committee for Standardization
CENELEC European Committee for Electrotechnical Standardization
CEPT European Conference of Postal and Telecommunication Administration
ECC Electronic Communications Committee
EEA European Economic Area
EMC Electromagnetic compatibility
HIPAA Health Insurance Portability and Accountability Act
ICH The International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use
ICT Information Communication Technologies
IDCP Italian Data Protection Code
ISM Industrial, Scientific and Medical
ISO International Standard Organization
JCI Joined Commission International
MDD Medical Devices Directive
RF Radiofrequency
SAR Specific Absorption Rate
SRD Short-Range Devices
UAAG User Agent Accessibility Guidelines
W3C World Wide Web Consortium
WAI Web accessibility initiative
WAI-ARIA Accessible Rich Internet Applications Suite
WCAG Web Content Accessibility Guidelines

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 9 of 96
WMA World Medical Association

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 10 of 96
4. Executive Abstract
This document reports on the regulatory elements that pilots will have to comply with in terms of 1) hardware characteristics (e.g. to ensure suitability with home or residential care environments); 2) usability; 3) compliancy with clinical/care rules; 4) data accessibility; 5) privacy & safety.
FBK with the collaboration of all partners collected all the national and local constraints to ensure that the pilots will be compliant with the norms and laws in place at the various sites.
The deliverable is divided in three main parts: Chapter 8 deals with ethical constraints regulations at both European and national level starting from Helsinki Declaration and the Directive 2001/20 /EC concerning the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use. Since this last directive is focused on pharmaceutical trials, an in-depth analysis about regulations concerning the use of medical devices is reported in section 8.3. This is followed by an overview on how the European directive has been adopted in the several participating countries.
Issues related to privacy and security are described in Chapter 9, including data management, data protection and informed consent regulations both at European and national level.
Chapter 10 is dedicated to the definition of Medical devices and the steps required to obtain European Certification. Finally, on Chapter 11 we reported further regulations concerning electromagnetic compatibility, usability and inclusiveness.
It should be noticed that most of the reported text is either an excerpt of national legal texts or a translation of them.
Chapters 9 and 11 have been adapted from i-locate (FP7 project) deliverable on legal requirements with the permission of the coordinator (http://www.i-locate.eu/data/uploads/2015/01/D.1.2-Regulatory-constraints-rev-2015-01-201.pdf).

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 11 of 96
5. Table of Content
1. Revision history and statement of originality ............................................. 2
Revision history ............................................................................................. 2Statement of originality .................................................................................. 2
2. List of references........................................................................................ 3
3. Table of Acronyms ...................................................................................... 8
4. Executive Abstract .................................................................................... 10
5. Table of Content ....................................................................................... 11
6. Table of Figures ........................................................................................ 12
7. Ethical constraints and regulations .......................................................... 13
The Declaration of Helsinki ............................................................................ 13European regulation ..................................................................................... 16Regulations concerning Medical Devices .......................................................... 21
Clinical Investigation ............................................................................................. 22National regulations ..................................................................................... 27
Italy ................................................................................................................... 27Greece ................................................................................................................ 27
Thessaloniki ...................................................................................................... 28Germany ............................................................................................................. 29Romania ............................................................................................................. 29Slovenia .............................................................................................................. 29Macedonia ........................................................................................................... 30
8. Security and privacy regulations .............................................................. 30
Personal data management: privacy and security ............................................. 30Europe ................................................................................................................ 30Italy ................................................................................................................... 33Greece ................................................................................................................ 36Germany ............................................................................................................. 38Romania ............................................................................................................. 46Slovenia .............................................................................................................. 54Macedonia ........................................................................................................... 61
9. Medical devices ........................................................................................ 67
Certification ................................................................................................ 67
10. Other requirements ................................................................................ 69
Electromagnetic compatibility requirements ..................................................... 69Usability and Inclusiveness ........................................................................... 70
Web accessibility initiative (WAI) ............................................................................ 70

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 12 of 96
Web pages or web-based interfaces ............................................................... 70
11. Conclusions ............................................................................................ 74
12. Ethical regulations: Implications for the pilots ....................................... 77
13. Privacy and security regulations: Implications for the architecture and the pilots ...................................................................................................... 79
16. Analysis of the very local regulatory constraints and requirements ........ 85
Annex I Conducting Clinical investigations in Italy ....................................... 91
6. Table of Figures
Figure1:Clinicalinvestigationprocess...........................................................................................................25

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 13 of 96
7. Ethical constraints and regulations
We have identified four levels of ethics constraint: declarations of ethical principles, European directives, National laws, and Local norms.
Within this document we report only the parts of the declarations, European directives, National laws and, in general any other regulation, significant to the UNCAP project. For the complete documents, one can refer to the original documents, either reported as attachments to this document or available, where possible, online.
The Declaration of Helsinki The set of ethical principles regulating human experimentation are established in the Declaration of Helsinki developed for the medical community by the World Medical Association (WMA) [1]. The Declaration is widely regarded as the cornerstone document on human research ethics.
Since its general adoption in June 1964, the Declaration has undergone seven revisions (the most recent being adopted at the General Assembly in October 2013). Although the Declaration, and successive revisions, is morally binding for physician, it does not constitute a legally binding instrument under the international law.
These concepts are stated in the Preamble, as follows.
• Art. 1. The World Medical Association (WMA) has developed the Declaration of Helsinki as a statement of ethical principles for medical research involving human subjects, including research on identifiable human material and data.
• Art. 2. Consistent with the mandate of the WMA, the Declaration is addressed primarily to physicians. The WMA encourages others who are involved in medical research involving human subjects to adopt these principles.
On the other hand, the Declaration draws its authority from the degree to which it has been coded or influenced, namely national or regional legislation and regulations. In this regard, the Declaration recommends that ethical considerations must always take precedence over laws and regulations.
• Art. 9. It is the duty of physicians who are involved in medical research to protect life, health, dignity, integrity, right to self-determination, privacy, and confidentiality of personal information of research subjects […].
The fundamental principle to consider in doing research involving human subjects is the respect for the individual, as stated in Section “General principles”, in particular Articles 6, 7 and 8, as follows.
• Art. 6. The primary purpose of medical research involving human subjects is to understand the causes, development and effects of diseases and improve preventive, diagnostic and therapeutic interventions (methods, procedures and treatments). Even the best-proven interventions must be evaluated continually through research for their safety, effectiveness, efficiency, accessibility and quality.
• Art. 7. Medical research is subject to ethical standards that promote and ensure respect for all human subjects and protect their health and rights.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 14 of 96
• Art. 8. While the primary purpose of medical research is to generate new knowledge, this goal can never take precedence over the rights and interests of individual research subjects.
The second principle of the Declaration is the right of the subject of research to self-determination and the right to make informed decisions (Articles 25, 26 and 31). This applies to participation in research, both initially and during the course of the research. In particular, according to Article 26, the subject must express explicit consent in the appropriate form.
• Art. 25. Participation by individuals capable of giving informed consent as subjects in medical research must be voluntary. Although it may be appropriate to consult family members or community leaders, no individual person capable of giving informed consent may be enrolled in a research study unless he or she freely agrees.
• Art. 26. In medical research involving human subjects capable of giving informed consent, each potential subject must be adequately informed of the aims, methods, sources of funding, any possible conflicts of interest, institutional affiliations of the researcher, the anticipated benefits and potential risks of the study and the discomfort it may entail, post-study provisions and any other relevant aspects of the study. The potential subject must be informed of the right to refuse to participate in the study or to withdraw consent to participate at any time without reprisal. Special attention should be paid to the specific information needs of individual potential subjects as well as to the methods used to deliver the information. After ensuring that the potential subject has understood the information, the physician, or another appropriately qualified individual, must then seek the potential subject’s freely-given informed consent, preferably in writing. If the consent cannot be expressed in writing, the non-written consent must be formally documented and witnessed. All medical research subjects should be given the option of being informed about the general outcome and results of the study.
• Art. 31. The physician must fully inform the patient which aspects of their care are related to the research. The refusal of a patient to participate in a study or the patient’s decision to withdraw from the study must never adversely affect the patient-physician relationship.
The Declaration recommends special vigilance on the recognition of the vulnerable individuals and groups (Articles 19 and 20) and, consequently, their special protection, which must be carefully considered, given that the UNCAP project deals with cognitively impaired patients:
• Art. 19. Some groups and individuals are particularly vulnerable and may have an increased likelihood of being hurt or of incurring additional harm. For this reason all vulnerable groups and individuals should receive specifically considered protection.
• Art. 20. Medical research with a vulnerable group is only justified if the research is responsive to the health needs or priorities of this group and the research cannot be carried out in a non-vulnerable group. In addition, this group should stand to benefit from the knowledge, practices or interventions that result from the research.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 15 of 96
Accordingly, special operational procedures must be adopted if research requires involvement of participants who are incompetent, physically or mentally incapable of giving consent, or if the participants are younger than 18 (Articles 27, 28). In this case, an allowance should be considered for surrogate consent by an individual acting in the subject’s best interest. In this case, the consent of the subject should still be obtained, whenever possible (Article 29, 30).
• Art. 27. When seeking informed consent for participation in a research study, the physician must be particularly cautious if the potential subject is in a dependent relationship with the physician or may consent under duress. In such situations, the informed consent must be sought by an appropriately qualified individual who is completely independent of this relationship.
• Art. 28. For a potential research subject who is incapable of giving informed consent, the physician must seek informed consent from the legally authorised representative. These individuals must not be included in a research study that has no likelihood of benefit for them unless it is intended to promote the health of the group represented by the potential subject. The research cannot instead be performed with persons not capable of providing informed consent and the research does not only entail minimal risk and minimal burden.
• Art. 29. When a potential research subject who is deemed incapable of giving informed consent is able to give assent to decisions about participation in research, the physician must seek that assent in addition to the consent of the legally authorised representative. The potential subject’s dissent should be respected.
• Art. 30. Research involving subjects who are physically or mentally incapable of giving consent (for example unconscious patients) may be carried on only if the physical or mental condition that prevents giving informed consent is a necessary characteristic of the research group. In such circumstances, the physician must seek informed consent from the legally authorised representative. If no such representative is available and if the research cannot be delayed, the study may proceed without informed consent provided that the specific reasons for involving subjects includes a condition that renders them unable to give informed consent have been stated in the research protocol and the study has been approved by a research ethics committee. Consent to remain within the research must be obtained as soon as possible from the subject or a legally authorized representative.
As regards the design and approval of the research study, the principle is that of minimization of the risks and careful evaluation of the benefits (Articles 17, 18), appropriateness and necessity of the study (Articles 21, 22), and the requirement that the study protocol must be discussed and approved by an “independent” and “duly qualified” Ethics committee (Article 23).
• Art. 17. All medical research involving human subjects must be preceded by careful assessment of predictable risks and burdens to the individuals and groups involved in the research in comparison with foreseeable benefits to them and to other individuals or groups affected by the condition under investigation. Measures to minimize the risks must be implemented. The risks must be continuously monitored, assessed and documented by the researcher.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 16 of 96
• Art. 18. Physicians may not be involved in a research study involving human subjects unless they are confident that the risks have been adequately assessed and can be satisfactorily managed. When the risks are found to outweigh the potential benefits or when there is a conclusive proof of definitive outcomes, physicians must assess whether to continue, modify or immediately stop the study.
• Art. 21. Medical research involving human subjects must conform to generally accepted scientific principles, be based on a thorough knowledge of the scientific literature, other relevant sources of information, and adequate laboratory and, as appropriate, animal experimentation. The welfare of animals used for research must be respected.
• Art. 22. The design and performance of each research study involving human subjects must be clearly described and justified in a research protocol. The protocol should contain a statement of the ethical considerations involved and should indicate how the principles in this Declaration have been addressed. The protocol should include information regarding funding, sponsors, institutional affiliations, potential conflicts of interest, incentives for subjects and information regarding provisions for treating and/or compensating subjects who are harmed as a consequence of participation in the research study. In clinical trials, the protocol must also describe appropriate arrangements for post-trial provisions.
• Art. 23. The research protocol must be submitted for consideration, comment, guidance and approval to the concerned research ethics committee before the study begins. This committee must be transparent in its functioning, must be independent of the researcher, the sponsor and any other undue influence and must be duly qualified. The committee must take into consideration the laws and regulations of the country or countries where the research is to be performed as well as applicable international norms and standards. The latter must not be allowed to reduce or eliminate any of the protections for research subjects set forth in the Declaration. The committee must have the right to monitor on-going studies. The researcher must provide monitoring information to the committee, especially information about any serious adverse events. No amendment to the protocol may be made without consideration and approval by the committee. After the end of the study, the researchers must submit a final report to the committee containing a summary of the study’s findings and conclusions.
Finally, the Declaration provide recommendations on the protection of the privacy regarding the subjects of the research, any issue strictly related to the protection of privacy in the case of data treatment with ICT systems (see Chapter 9):
• Art. 24. Every precaution must be taken to protect the privacy of research subjects and the confidentiality of their personal information.
European regulation The principles stated in the Declaration of Helsinki are acknowledged in the preamble (point 2) of the Directive 2001/20/EC of the European Parliament and of the Council of 4 April 2001 “on the approximation of the laws, regulations and administrative

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 17 of 96
provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use” [2]:
• Art. 2. The accepted basis for the conduct of clinical trials in humans is founded in the protection of human rights and the dignity of the human being with regard to the application of biology and medicine, as for instance reflected in the 1996 version of the Helsinki Declaration. The clinical trial subject's protection is safeguarded through risk assessment based on the results of toxicological experiments prior to any clinical trial, screening by ethics committees and Member States' competent authorities, and rules on the protection of personal data.
The Directive explicitly specifies the applicability in the member States (Article 3, section 1):
• Art. 3.1. This Directive shall apply without prejudice to the national provisions on the protection of clinical trial subjects if they are more comprehensive than the provisions of this Directive and consistent with the procedures and time-scales specified therein. Member States shall, insofar as they have not already done so, adopt detailed rules to protect from abuse individuals who are incapable of giving their informed consent.
The scope of the Directive is given in Article 1, Section 1:
• Art. 1.1. This Directive establishes specific provisions regarding the conduct of clinical trials, including multi-centre trials, on human subjects involving medicinal products as defined in Article 1 of Directive 65/65/EEC, in particular relating to the implementation of good clinical practice. This Directive does not apply to non-interventional trials.
Article 2 provides some definition of the concepts referred to in the Directive. Of particular importance is the definition of what it is meant by “clinical trial” and “intervention trial” (letter a, b, and c):
• (a) ‘Clinical trial’: any investigation in human subjects intended to discover or verify the clinical, pharmacological and/or other pharmacodynamics effects of one or more investigational medicinal product(s), and/or to identify any adverse reactions to one or more investigational medicinal product(s) and/or to study absorption, distribution, metabolism and excretion of one or more investigational medicinal product(s) with the object of ascertaining its (their) safety and/or efficacy. This includes clinical trials carried out in either one site or multiple sites, whether in one or more than one Member State.
• (b) ‘Multi-centre clinical trial’: a clinical trial conducted according to a single protocol but at more than one site, and therefore by more than one investigator, in which the trial sites may be located in a single Member State, in a number of Member States and/or in Member States and third countries.
• (c) ‘Non-interventional trial’: a study where the medicinal product(s) is (are) prescribed in the usual manner in accordance with the terms of the marketing authorisation. The assignment of the patient to a particular therapeutic strategy is not decided in advance by a trial protocol but falls within current practice and the prescription of the medicine is clearly separated from the decision to include the patient in the study. No additional diagnostic or

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 18 of 96
monitoring procedures shall be applied to the patients and epidemiological methods shall be used for the analysis of collected data;
Of particular interest for the project are also the definitions of informed consent and ethics committee (letter j and k):
• (j) ‘Informed consent’: decision, which must be written, dated and signed, to take part in a clinical trial, taken freely after being duly informed of its nature, significance, implications and risks and appropriately documented, by any person capable of giving consent or, where the person is not capable of giving consent, by his or her legal representative. If the person concerned is unable to write, oral consent in the presence of at least one witness may be given in exceptional cases, as provided for in national legislation.
• (k) ‘Ethics committee’: an independent body in a Member State, consisting of healthcare professionals and nonmedical members, whose responsibility it is to protect the rights, safety and wellbeing of human subjects involved in a trial and to provide public assurance of that protection, by, among other things, expressing an opinion on the trial protocol, the suitability of the investigators and the adequacy of facilities, and on the methods and documents to be used to inform trial subjects and obtain their informed consent.
The principle of the protection of the rights of the subject, the grounds of the Helsinki Declaration, is stated in Article 3 (Protection of clinical trial subjects). Aside the already cited subsection 1, the successive subsections follow the Directive. Of particular interest are the following subsections:
• Art 3.2. A clinical trial may be undertaken only if, in particular:
a) the foreseeable risks and inconveniences have been weighed against the anticipated benefit for the individual trial subject and other present and future patients. A clinical trial may be initiated only if the Ethics Committee and/or the competent authority comes to the conclusion that the anticipated therapeutic and public health benefits justify the risks and may be continued only if compliance with this requirement is permanently monitored;
b) the trial subject or, when the person is not able to give informed consent, his legal representative has had the opportunity, in a prior interview with the investigator or a member of the investigating team, to understand the objectives, risks and inconveniences of the trial, and the conditions under which it is to be conducted and has also been informed of his right to withdraw from the trial at any time;
c) the rights of the subject to physical and mental integrity, to privacy and to the protection of the data concerning him in accordance with Directive 95/46/EC are safeguarded;
d) the trial subject or, when the person is not able to give informed consent, his legal representative has given his written consent after being informed of the nature, significance, implications and risks of the clinical trial; if the individual is unable to write, oral consent in the presence of at least one witness may be given in exceptional cases, as provided for in national legislation;

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 19 of 96
e) the subject may without any resulting detriment withdraw from the clinical trial at any time by revoking his informed consent;
• Art 3.4. In the case of other persons incapable of giving their consent, such as persons with dementia, psychiatric patients, etc., inclusion in clinical trials in such cases should be on an even more restrictive basis. Medicinal products for trials may be administered to all such individuals only when there are grounds for assuming that the direct benefit to the patient outweighs the risks. Moreover, in such cases the written consent of the patient's legal representative, given in cooperation with the treating doctor, is necessary before participation in any such clinical trial.
• Art 3.11. As a rule, authorisation should be implicit, i.e. if there has been a vote in favour by the Ethics Committee and the competent authority has not objected within a given period, it should be possible to begin the clinical trials. In exceptional cases raising especially complex problems, explicit written authorisation should, however, be required.
Note that the Subsection 3.2.a mandates to submit the trial to an Ethics committee or, in particular cases to a “competent authority” of the Member State (see also Article 6), whose individuation is left, evidently, to each State.
For the purposes of this document, it is worth reporting the provisions of Article 5 (Clinical trials on incapacitated adults not able to give informed legal consent), since the subjects of research in UNCAP are elderly people with Mild Cognitive Impairment:
• Art. 5. In the case of other persons incapable of giving informed legal consent, all relevant requirements listed for persons capable of giving such consent shall apply. In addition to these requirements, inclusion in clinical trials of incapacitated adults who have not given or not refused informed consent before the onset of their incapacity shall be allowed only if:
a) the informed consent of the legal representative has been obtained; consent must represent the subject's presumed will and may be revoked at any time, without detriment to the subject;
b) the person not able to give informed legal consent has received information according to his/her capacity of understanding regarding the trial, the risks and the benefits;
c) the explicit wish of a subject who is capable of forming an opinion and assessing this information to refuse participation in, or to be withdrawn from, the clinical trial at any time is considered by the investigator or where appropriate the principal investigator;
d) no incentives or financial inducements are given except compensation;
e) such research is essential to validate data obtained in clinical trials on persons able to give informed consent or by other research methods and relates directly to a life-threatening or debilitating clinical condition from which the incapacitated adult concerned suffers;
f) clinical trials have been designed to minimise pain, discomfort, fear and any other foreseeable risk in relation to the disease and developmental stage; both the risk threshold and the degree of distress shall be specially defined and constantly monitored;

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 20 of 96
g) the Ethics Committee, with expertise in the relevant disease and the patient population concerned or after taking advice in clinical, ethical and psychosocial questions in the field of the relevant disease and patient population concerned, has endorsed the protocol;
h) the interests of the patient always prevail over those of science and society; and
i) there are grounds for expecting that administering the medicinal product to be tested will produce a benefit to the patient outweighing the risks or produce no risk at all.
The rules for the establishment, composition and activities of the Ethics Committee in the Member States are given in Article 6 (Ethics Committee):
• Art 6.1. For the purposes of implementation of the clinical trials, Member States shall take the measures necessary for establishment and operation of Ethics Committees.
• Art 6.2. The Ethics Committee shall give its opinion, before a clinical trial commences, on any issue requested.
• Art 6.3. In preparing its opinion, the Ethics Committee shall consider, in particular:
a) the relevance of the clinical trial and the trial design;
b) whether the evaluation of the anticipated benefits and risks as required under Article (3)(2)(a) is satisfactory and whether the conclusions are justified;
c) the protocol;
d) the suitability of the investigator and supporting staff;
e) the investigator's brochure;
f) the quality of the facilities;
g) the adequacy and completeness of the written information to be given and the procedure to be followed for the purpose of obtaining informed consent and the justification for the research on persons incapable of giving informed consent as regards the specific restrictions laid down in Article 3;
h) provision for indemnity or compensation in the event of injury or death attributable to a clinical trial;
i) any insurance or indemnity to cover the liability of the investigator and sponsor;
j) the amounts and, where appropriate, the arrangements for rewarding or compensating investigators and trial subjects and the relevant aspects of any agreement between the sponsor and the site;
k) the arrangements for the recruitment of subjects.
• Art 6.4. Notwithstanding the provisions of this Article, a Member State may decide that the competent authority it has designated for the purpose of Article 9 shall be responsible for the consideration of, and the giving of an opinion on, the matters referred to in paragraph 3(h), (i) and (j) of this Article. When a

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 21 of 96
Member State avails itself of this provision, it shall notify the Commission, the other Member States and the Agency.
• Art 6.5. The Ethics Committee shall have a maximum of 60 days from the date of receipt of a valid application to give its reasoned opinion to the applicant and the competent authority in the Member State concerned.
Article 7 (Single opinion) provides regulation of multi-centric studies1 (defined in Article 2, Subsection b):
• Art 7. For multi-centre clinical trials limited to the territory of a single Member State, Member States shall establish a procedure providing, notwithstanding the number of Ethics Committees, for the adoption of a single opinion for that Member State. In the case of multi-centre clinical trials carried out in more than one Member State simultaneously, a single opinion shall be given for each Member State concerned by the clinical trial.
Future legal framework
The EU clinical Trials Directive has been criticised by stakeholders because of its disharmonised interpretation, and delays especially for launching multi-national trials. In fact, the authorisation procedures are performed separately in each Member State. In order to overcome this problem, the European Commission published a proposal for an EU Clinical Trials Regulation on 17 July 2012, then adopted on 16 April 2014. Currently, it is expected that the new regulatory framework will apply by mid-2016.
The major novelties of the proposed regulation are the application procedure for initial authorisation via a single entry point, with one single decision issued by each concerned Member State, and within defined deadlines applicable for all Member States. In addition, the Regulation also establishes that Member States shall cooperate on safety information assessment [65].
Regulations concerning Medical Devices The regulation of the medical use and clinical investigations with medical devices is set up in the COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 concerning medical devices. Directive 98/79/EC of 27 October 1998, Directive 2000/70/EC 16 November 2000, Directive 2001/104/EC 7 December 2001, Regulation (EC) No 1882/2003 of 29 September 2003 and Directive 2007/47/EC of 5 September 2007 subsequently amended parts of this directive [3]. Here we report the parts of interest of 93/42/EEC in the context of UNCAP, which is updated with the sections amended by the more recent directives cited above.
First of all, the directive gives the definition of medical devices.
1 In the UNCAP project it is foreseen to carry out clinical studies in the healthcare organizations inside the territory of the Member states and clinical studies in the territory of the Member States participating to the project. Since these clinical studies, however, are in principle different as regards both the interventions and the measured outcomes, in our opinion they can hardly be defined multi-centric studies and the Article 7 should be not applicable.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 22 of 96
Art 1 (Definitions, scope). This Directive shall apply to medical devices and their accessories. For the purposes of this Directive, accessories shall be treated as medical devices in their own right. Both medical devices and accessories shall hereinafter be termed devices.
For the purposes of this Directive, the following definitions shall apply:
a) ‘medical device’ means any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
o diagnosis, prevention, monitoring, treatment or alleviation of disease,
o diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
o investigation, replacement or modification of the anatomy or of a physiological process,
o control of conception, and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means;
• Article 9 states that devices shall be divided into Classes I, IIa, IIb and III. Classification shall be carried out in accordance with Annex IX.
Clinical Investigation
Article 15 regulates the use of medical devices in clinical investigations.
Art 15 (Clinical investigation):
1) In the case of devices intended for clinical investigations, the manufacturer or the authorised representative, established in the Community, should follow the procedure referred to in Annex VIII and notify the competent authorities of the Member States in which the investigations are to be conducted by means of the statement mentioned in Section 2.2 of Annex VIII.
2) In the case of devices falling within Class III and implantable and long-term invasive devices falling within Class IIa or IIb, the manufacturer may commence the relevant clinical investigation at the end of a period of 60 days after notification, unless the competent authorities have notified him within that period of a decision to the contrary based on considerations of public health or public policy. Member States may however authorise manufacturers to commence the relevant clinical investigations before the expiry of the period of 60 days, insofar as the relevant ethics committee has issued a favourable opinion on the programme of investigation in question, including its review of the clinical investigation plan.
3) In the case of devices other than those referred to in paragraph 2, Member States may authorise manufacturers to commence clinical investigations

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 23 of 96
immediately after the date of notification, provided that the ethics committee concerned has issued a favourable opinion on the programme of investigation in question including its review of the clinical investigation plan.
4) The authorization referred to in paragraph 2 and paragraph 3, may be made subject to authorization from the competent authority.
5) The clinical investigations must be conducted in accordance with the provisions of Annex X. The measures designed to amend nonessential elements of this Directive, inter alia by supplementing it, relating to the provisions on clinical investigation in Annex X shall be adopted in accordance with the regulatory procedure with scrutiny referred to in Article 7(3).
The Article refers to Annex VIII (section 1) and Annex X (Section 5). The Annex VIII concerns the statement that the manufacturer or his authorized must draw up for custom-made devices or for devices intended for clinical investigations.
A clinical investigation is defined as a “Systematic investigation in one or more human subjects, undertaken to assess the safety or performance of a medical devices” (ISO 14155 [4]). A clinical investigation is carried out either on a Non-CE-marked device or on a CE-marked device that is used out with indication of use.
The crucial point for the UNCAP project is stated in Article 2 (Clinical Investigation) of Annex X
Art 2.1. Objectives. The objectives of clinical investigation are:
o to verify that, under normal conditions of use, the performance of the devices conform to those referred to in Section 3 of Annex I, and
o to determine any undesirable side-effects, under normal conditions of use, and assess whether they constitute risks when weighed against the intended performance of the device.
Art 2.2. Ethical considerations. Clinical investigations must be carried out in accordance with the Helsinki Declaration adopted by the 18th World Medical Assembly in Helsinki, Finland, in 1964, as last amended by the World Medical Assembly. It is mandatory that all measures relating to the protection of human subjects are carried out in the spirit of the Helsinki Declaration. This includes every step in the clinical investigation from first consideration of the need and justification of the study to publication of the results.
Art 2.3. Methods
o 2.3.1. Clinical investigations must be performed on the basis of an appropriate plan of investigation reflecting the latest scientific and technical knowledge and defined in such a way as to confirm or refute the manufacturer's claims for the device; these investigations must include an adequate number of observations to guarantee the scientific validity of the conclusions.
o 2.3.2. The procedures used to perform the investigations must be appropriate to the device under examination.
o 2.3.3. Clinical investigations must be performed in circumstances similar to the normal conditions of use of the device.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 24 of 96
o 2.3.4. All the appropriate features, including those involving the safety and performances of the device, and its effect on patients must be examined.
o 2.3.5. All serious adverse events must be fully recorded and immediately notified to all competent authorities of the Member States in which the clinical investigation is being performed.
o 2.3.6. The investigations must be performed under the responsibility of a medical practitioner or another authorized qualified person in an appropriate environment. The medical practitioner or other authorized person must have access to the technical and clinical data regarding the device.
o 2.3.7. The written report, signed by the medical practitioner or other authorized person responsible, must contain a critical evaluation of all the data collected during the clinical investigation.
From what stated in Section 2.2 and, partly, in 2.3.1 it is clear that from the ethical perspective the clinical investigation with medical devices can be related to the human experimentation of drugs; hence, it is regulated by the European Directive 2001/20/EC [2] and by the National laws that have adopted it.
The diversity of medical devices and the technologies on which they are based pose special challenges for manufacturers, conformity assessment bodies and regulators alike when trying to identify what should constitute evidence sufficient to demonstrate compliance with the Essential Principles. Some technologies have been available for many years and are well characterised from a clinical safety and performance viewpoint. On the other hand, many devices utilise new, state-of-the-art technology that has had little prior application in the treatment of humans. Furthermore, their intended purpose and clinical application can vary widely with results influenced by a wide range of different and differently experienced end-users. Given the complexity of the medical devices milieu, the assessment of what is acceptable clinical evidence for demonstrating compliance with the Essential Principles must be undertaken on a case-by-case basis. [5]
According to Annex 1.I.5a of directive 90/385/EEC and Annex I.I.6a of directive 93/42/EEC demonstration of conformity with the essential requirements must include a clinical evaluation in accordance with Annex 7/Annex X of the respective directive. As a general rule, confirmation of conformity with the requirements concerning the characteristics and performances referred to in sections I.1 and I.3 of annex I of directive 90/385/EEC and in sections I.1 and I.2 of annex I of directive 93/42/EEC under the normal conditions of use of the device, and the evaluation of the side effects and of the acceptability of the benefit/risk ratio referred to in Section I.5/I.6 of Annex I of directives 90/385/EEC and 93/42/EEC respectively, must be based on clinical data. The kind and amount of clinical data needed will primarily depend on the specifics of the clinical claims with regard to clinical performance, considerations of clinical safety, including determination of undesirable side-effects and on risk management output, namely determination of residual risks and favourable benefit/risk ratio. [6]
Risk Management is the core analysis from which all the testing, including clinical evaluation, originates. Consequently, if one or more of the risks associated with the device can only be evaluated by a clinical investigation, then that investigation by

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 25 of 96
necessity will have to happen. Risk Management file should be peer reviewed within the context of the Technical File at a relatively early stage, in order to ensure that all testing is scheduled in a timely fashion and the Conformity Assessment is not unduly delayed by flawed planning [7].
When must/should a clinical investigation be undertaken?
The Conformity Assessment process for active implantable medical devices as well as for class III and implantable medical devices requires that a clinical investigation is undertaken unless it is duly justified to rely on existing data (section 1.2 of Annex 7 of directive 90/385/EEC and section I.1a of Annex X of directive 93/42/EEC). Any such justification will have to be based on a proper clinical evaluation. Depending on clinical claims, risk management outcome and on the results of the clinical evaluation, clinical investigations may also have to be performed for non-implantable medical devices of classes I, IIa and IIb. Additional clinical investigations may be feasible to corroborate the existing clinical evidence with regard to aspects of clinical performance, safety, benefit/risk-ratio or to determine relative effectiveness and safety with suitable comparators. [6]
At the onset of the clinical investigation process, it is necessary to define the overall regulatory strategy for the specific device. Additionally, the clinical strategy should be taken into account because the two strategies are interdependent and run concurrently. Once defined, the clinical study can be designed, set-up and implemented after all the required approval has been obtained. Throughout the whole life cycle, the clinical investigation needs to be closely managed to ensure the quality and integrity of the data. Routine monitoring and, if relevant, quality audit should be performed. Data collection and recording, with statistical analysis performed according to a predefined plan is essential. The results can then be included in the final study report. When the study report is generated, it forms part of the regulatory submission and when appropriate, the results can be published. The entire process is conducted under the umbrella of Regulatory and ethical compliance.
Figure 1: Clinical investigation process
Regulatory and ethical compliance
Clinical investigation compliance with the relevant directive, harmonized standards and local ethical considerations is essential. Without these, the clinical investigation will not obtain the necessary approvals and the returned data will not be acceptable (e.g. for obtaining CE-mark approval).

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 26 of 96
Clinical studies have to comply with the specific EU directive on medical devices:
• MDD – 93/385/EEC, 2007/47/EEC [3]
In addition, minimum requirements indicated in the harmonized standards should be satisfied:
• ISO 14155:2011 – clinical investigation of medical devices for human subjects – Good clinical practice [4]
Moreover, clinical studies should comply with the ethical considerations of the:
• Declaration of Helsinki [1]
• ICH, Good Clinical Practice [8]
A prevision for adequate compensation of study subjects in case of adverse events should be considered by stipulating a specific insurance.
Finally, the sponsor and the clinical investigator have to obtain all the relevant approvals (Ethical committee, Competent authority and local institution) and to comply with these conditions before starting the study.
Approval process for clinical investigations
Clinical investigations require approval from the competent authority, from Ethics and from the local institution (e.g. hospital) where the study is conducted.
In some EU countries, additional approvals are required.
Competent authority
Competent authorities are interested in public health and safety and, as such, they approve the study design and investigational devices.
Ethics committees
Ethics committees are interested mainly in the safety and wellbeing of the study subjects. They approve the study design and Patient Information sheet and Consent form.
Local institutions (Hospitals)
Local institutions approve the study, the resources at the facility, the investigator and the ability of the investigator of to perform the study specific evaluations that could be over and above the routine standard of care. Usually the local institution set up an agreement or contract with the clinical investigator and the sponsor.
Requirement before approval
• Regulatory and clinical strategy must be defined
• Clinical document finalized in file format (Clinical Investigation Protocol, Case Report Form, Clinical Investigator Brochure, Patient Informed Consent, etc.)
• All supporting pre-clinical data on device available (as required for CE mark technical dossier: e.g.: bench testing, electrical safety testing, biocompatibility testing, in-vitro and in-vivo testing and any previous clinical data according to device characteristics and classification)

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 27 of 96
• Investigators and investigation site selected
• Investigator CVs available
• Confidentiality agreements and financial disclosure in place
• Ethics committees selected, and submission deadlines and meeting date checked out
• Devices or well-advanced prototypes available
• Labelling requirements and shipping/customs documents in place
• Patients’ insurance/indemnity in place (country/site specific)
• Accredited translation services identified and documents needing translation available (Patient information and Patient informed consent, Instruction for use, Questionnaires etc. In general all document that should be provided to the patient must be in local language)
• Local country knowledge/assistance arranged
National regulations
Italy
In Italy, the European Directive has been adopted through the Decreto Legislativo (law decreet) of 24 June 2003, n. 211 "Attuazione della direttiva 2001/20/CE relativa all'applicazione della buona pratica clinica nell'esecuzione delle sperimentazioni cliniche di medicinali per uso clinico" [9] (Implementation of the directive 2001/20/EC related to application of best practice, and execution of clinical trials of medical treatments for clinical use). In substance, besides few subsections that refer to Member States, the legislative decree is the almost a verbatim translation of the European Directive.
The Directive 93/42/CEE was adopted with the Decreto legislativo 24 febbraio 1997, n. 46 [10] “ATTUAZIONE DELLA DIRETTIVA 93/42/CEE CONCERNENTE I DISPOSITIVI MEDICI” and with the Decreto Legislativo 25 gennaio 2010, n. 37[11] "Attuazione della direttiva 2007/47/CE che modifica le direttive 90/385/CEE per il ravvicinamento delle legislazioni degli stati membri relative ai dispositivi medici impiantabili attivi, 93/42/CE concernente i dispositivi medici e 98/8/CE relativa all'immissione sul mercato dei biocidi", which adopted the amendments set up in Directive 2007/47/CE.
Again, these legislative decrees are almost the verbatim translation of the corresponding European Directives.
Details about how to conduct a clinical investigation in Italy are reported in Annex I.
Greece
In Greece, according to the Medical Law code, and especially the Law number 3418 dated 28 November 2005 [12] (number of edition 287), the following articles give directions on ethics concerning the clinical trials and related issues.
Within article 25 of the Medical Law code of the Hellenic Republic there is a description of the Clinical Research with new medications or new technological medical monitoring

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 28 of 96
methods. Specifically the clinical research with new medications or application of new monitoring methods are allowed only if:
• a) The specifications of the clinical research abide with the specifications and processes that are defined by the respective authorities of European Union.
• b) There are strong indications that the use of the Application will increase the possibility of survival, or health recovery, or generally improve the health of population.
Moreover, if the patient does not want to participate in the related programme or use the device, the doctor should respect his/her opinion without influencing in any manner the trustful relationship between the doctor and the patient. The doctor should not also apply new methods or diagnostic devices to patients if she/he does not know for sure the consequences of these devices that use. He should use them only for the benefits of patients.
In the article 26 of the Medical Law code of the Hellenic Republic there is a description of the Biomedicine Research. According to that, we have the following directions.
It is allowed to make Biomedical research and clinical research on human beings with the prerequisite that the doctor follows, as guiding principle, the protection of human life and the dignity of human being. No other kind of interchange is allowed in order to conduct the research except for the purpose of the research. The medical research and clinical trial should be stopped if the extension of the research becomes dangerous for the health of the human being. The doctor should make it open and clear the results of their research concerning the use of biomedicine tools and methods to the medical community. Before making these results available to the public, he/she should discuss and listen to the opinion of his/her colleagues. The doctor should also mention the company or institute that has technically helped or has provided financial help to the research. The doctor may also mention people/colleagues who have helped within the research. The doctors, who are responsible for issuing of medical magazines, should investigate the correctness and the rules that should follow for the result of each medical practice and research. Activities should abide to articles from 24 to 26 of the Greek Medical Law code.
Law no. 3304 / 27th January 2005 [13] ratifies the protection of the elderly people with regard to equal treatment of people regardless of their racial or ethnic origin, religion or belief, disability, age or sexual orientation. Ageing is not necessarily connected to body injury or disability. Unfortunately, age discrimination (negative and positive social aspects of aging) is prevalent worldwide and it results in prejudice, discrimination and violation of the elderlies’ rights. The purpose of this law is to guarantee and protect the individual’s fundamental rights and freedoms, especially the right to personal health and care, independent living, community inclusion, as well as adequate living standards. The law deals with the respect of personal, family and private life, of death with dignity, according to a person’s beliefs, values and preferences.
Thessaloniki
In order to comply with local policies, an ethical approval by the Bioethical committee of the Aristotle University of Thessaloniki is required before conducting user studies regarding privacy & security, as well as informed consent for data treatment. This board considers all ethical considerations such as informed consent, data protection,

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 29 of 96
the type of participants used, and the content of the study (in particular potential deception or harm). Typical times to obtain approval may vary between one week and one month.
In addition, all trials are going to be recorded through a registry of publicly and privately supported clinical studies on human participants (called ClinicalTrials.gov) under the account of Aristotle University of Thessaloniki.
Germany
Clinical trials with human subjects in Germany are regulated by the Medicinal Products Act (2014) “PROTECTION OF HUMAN SUBJECTS IN CLINICAL TRIALS”: [14]
• Section 40: General conditions for the clinical trial
• Section 41: Special conditions for the clinical trial
• Section 42: Ethics committee procedure, procedure for authorisation by the higher federal authority
Other relevant regulations are:
• Regulation for the Application of Good Clinical Practice of Clinical Medications for Human Use (2012) [15]
• Principles and Responsibilities When Carrying Out Clinical Studies (2013) [16]
The act on Medical Devices, instead, regulates clinical investigation with medical devices. [16]
Romania
The EU directives 2001/20/EC and 2003/94/EC are transposed verbatim as reported in:
• Order 904/25Jul2006 on Approval of Rules Relating to the Implementation of Good Clinical Practice in the Conduct of Clinical Trials on Medicinal Products for Human Use
• Order 905/25Jul2006 on Approval of the Principles and Guidelines for Good Manufacturing Practice in Respect of Medicinal Products for Human Use and Investigational Medicinal Products for Human Use [18]
Slovenia
In the Republic of Slovenia, the field of ethics in research is dealt with by the following regulations and bylaws:
• The Constitution of The Republic of Slovenia [19].
• Research and Development Act [20].
• Personal data protection act of the Republic of Slovenia [21]
• Charter of Fundamental Rights of the European Union [22].
• Convention on the Protection of Human Rights and Dignity of the Human Being with regard to the Application of Biology and Medicine: Convention on Human Rights and Biomedicine [23].

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 30 of 96
• The European Social Charter [24].
• World Medical Association Declaration Of Helsinki, Ethical Principles for Medical Research Involving Human Subjects [1]
• Additional Protocol to the Convention on Human Rights Biomedicine, concerning Biomedical Research [25].
• Code of Medical Ethics Slovenia [26].
• Code of ethical principles in social care [27].
Macedonia DIRECTIVE 2001/20/EC OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL of 4 April 2001 on the approximation of the laws, regulations and administrative provisions of the Member States relating to the implementation of good clinical practice in the conduct of clinical trials on medicinal products for human use) is transposed into the national legislative. The legislative decree is almost verbatim translation of the European Directive in the following documents:
1. Law on drugs and medical devices, Official Gazette of Republic of Macedonia (29/2009 106/2007, 88/2010,36/2011, 53/2011, 136/2011, 11/2012 , 147/2013, 164/2013, 27/2014, 43/20140) [28]
2. Regulations on the procedure for clinical trials of drugs and content of documentation, Official Gazette 29/2009 [29]
3. Guidelines for the principles of good clinical practice, Official Gazette 62/2009 [30]
Moreover, Republic of Macedonia is respecting the RECOMMENDATION No. R (97) 5 OF THE COMMITTEE OF MINISTERS TO MEMBER STATES ON THE PROTECTION OF MEDICAL DATA (Adopted by the committee of Ministers on 13 February 1997 at the 584th meeting of the Ministers’ Deputies) which is applicable to the collection and automatic processing of medical data, in a specific context outside the health-care sector.
The RECOMMENDATION No. R (83) 10, OF THE COMMITTEE OF MINISTERS TO MEMBER STATES ON THE PROTECTION OF PERSONAL DATA USED FOR SCIENTIFIC RESEARCH AND STATISTICS (Adopted by the Committee of Ministers on 23 September 1983 at the 362nd meeting of the Ministers’ Deputies) is also respected in Macedonia.
8. Security and privacy regulations
Personal data management: privacy and security
Europe
Convention for the Protection of Individuals with regard to Automatic Processing of Personal Data of the Council of Europe of 1 January 1981
The Convention for the Protection of Individuals with regard to Automatic Processing of Personal Data [31] states “There is a need for such legal rules in view of the increasing use made of computers for administrative purposes. Compared with

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 31 of 96
manual files, automated files have a vastly superior storage capability and offer possibilities for a much wider variety of transactions, which they can perform at high speed. Further growth of automatic data processing in the administrative field is expected in the coming years inter alia as a result of the lowering of data processing costs, the availability of "intelligent" data processing devices and the establishment of new telecommunication facilities for data transmission”.
The convention addresses also trans-border flow of personal data undergoing automatic processing or collected with the goal of being processed in an automatic manner.
The legal framework on privacy and security issues related to data protection within the EU is essentially represented by the “Data Protection Directive” and the “ePrivacy Directive”, as detailed hereafter.
Data Protection Directive (95/46/EC) and ePrivacy Directive (2002/58/EC)
The Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 on the protection of individuals with regard to the processing of personal data and on the free movement of such data [32] and the Directive 2002/58/EC of the European Parliament and of the Council of 12 July 2002 concerning the processing of personal data and the protection of privacy in the electronic communications sector (Directive on privacy and Electronic Communications) [33], provide indications about data processing.
The privacy principles are summarized as follows:
• The collection and processing of personal data shall neither intrude on the data subjects’ privacy nor interfere with their autonomy and integrity.
• Personal data shall be collected and processed only after the person involved provides explicit consent.
• Personal data shall be collected for specified, lawful and legitimate purposes.
• The collection and processing of personal data shall be limited to the minimum necessary for achieving the specific purpose. This includes that personal data shall be retained only for the time necessary to achieve the specific purpose.
• The disclosure of personal data to third parties shall be restricted and only occur upon certain conditions.
• Personal data shall be accurate, relevant, and complete with respect to the purposes for which they are collected and processed.
• The data subject shall be able to check and influence the processing of his/her personal data.
• The processing of personal data, which are particularly sensitive for the data subject, shall be subject to more stringent protection measures than other personal data.
• Personal data shall be processed in a way that guarantees a level of security appropriate to the risks presented by the processing and the nature of the data [34].
In relation to security of personal data processing, the main reference at the EU level is Article 17 of Directive 95/46/EC according to which:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 32 of 96
• “Member States shall provide that the controller must implement appropriate technical and organizational measures to protect personal data against accidental or unlawful destruction or accidental loss, alteration, unauthorized disclosure or access, in particular where the processing involves the transmission of data over a network, and against all other unlawful forms of processing. Having regard to the state of the art and the cost of their implementation, such measures shall ensure a level of security appropriate to the risks represented by the processing and the nature of the data to be protected”.
• “The Member States shall provide that the controller must, where processing is carried out on his/her behalf, choose a processor providing sufficient guarantees in respect of the technical security measures and organizational measures governing the processing to be carried out, and must ensure compliance with those measures”.
Future legal framework
On 25 January 2012, the European Commission proposed a comprehensive reform of the EU's 1995 data protection rules to strengthen online privacy rights and boost Europe's digital economy [35]. The Commission opted for a Regulation instead of a Directive. In fact, Regulation will directly and equally apply in all Member States. This should overcome the current diverging national interpretations as well as different local enforcement of the provisions.
An overall agreement is expected by the end of this year (2015), and after formal approval/official publication it is expected a 2 year transitional period will apply.
The major novelty of the proposed regulation is reported in the Communication from the Commission to the European Parliament, the Council, the European Economic and Social Committee and the Committee of the regions on “safeguarding privacy in a connected world a European data protection framework for the 21st century” (COM/2012/09 final) [36]. This includes:
• A single set of rules on data protection, valid across the EU instead of the current obligation of all companies to notify all data protection agencies.
• The definition of activities of data protection supervisors for which the Regulation provides for increased responsibility and accountability in the context of processing personal data.
• Organisations will only have to deal with a single national data protection authority in the EU country where they have their main establishment. Likewise, people can refer to the data protection authority in their country, even when a company is based outside the EU processes their data.
• Wherever consent is required for data to be processed, it is clarified that this has to be given explicitly, rather than assumed.
• People will have easier access to their own data and have to be able to transfer personal data from one service provider to another more easily (right to data portability). This will improve competition among services. A ‘right to be forgotten’ will help people better manage data protection risks online as people

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 33 of 96
will have to be able to delete their data if there are no legitimate grounds for retaining it.
• EU rules must apply if personal data is handled abroad by companies that are active in the EU market and offer their services to EU citizens.
• Independent national data protection authorities will be strengthened so they can better enforce the EU rules at home. Data protection authorities will be empowered to fine companies that violate EU data protection rules [35].
Informed consent
Consent is the main instrument through which the principle of self-determination is expressed (Directive 95/46/EC) [32]. Consent must be considered a prerequisite and an essential provision to any treatment of data, even more when the processing modalities through which it is carried out result in the creation of risks and potential problems to the security and integrity of the data itself. Although consent is regulated in partially different ways across the various legal framework, however, its need is consistently acknowledged in all the countries, and must satisfy four criteria:
• consent must be a clear and unambiguous indication of wishes;
• consent must be freely given;
• consent must be specific;
• consent must be informed.
From a technical point of view, consent to the processing of health data must generally be made in writing (Directive 95/46/EC art .2). This formality, even if it is easily manageable through traditional paper-based interactions at the time of the first contact between the patient and the health care body that provides the health service, may, however, be a critical point to solve if not properly managed also from a digital point of view.
Most relevantly, the Article 29 of Working Party, which was established under the Directive 95/46/EC of the European Parliament and of the Council of 24 October 1995 for the protection of individuals with regard to the processing of personal data, published an "Opinion 15/2011 on the definition of consent, Adopted on 13 July 2011" [37], states that informed consent has to be taken into account in the wording of any security, privacy & contractual disclaimer.
Italy
The Italian transposition of the rules established at the European level is one of the most restrictive ones.
Legislative Decree 30 June 2003, n. 196
The reference in Italy is the Legislative Decree 30 June 2003, n. 196 “Codice in materia di protezione dei dati personali” (Italian Data Protection Code - IDPC) [38]

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 34 of 96
that implemented the relevant European Directives. Articles 4 defines what sensitive data are and Article 3 introduces the Data minimization principle.
• Art 4, par. 1
o (d) “sensitive data” are: “personal data allowing the disclosure of racial or ethnic origin, religious, philosophical or other beliefs, political opinions, membership of parties, trade unions, associations or organizations of a religious, philosophical, political or trade-unionist character, as well as personal data disclosing health and sex life”.
• Art. 3 “Data minimization principle”: information systems and software shall be configured by minimizing the use of personal data and identification data, in such a way as to rule out their processing if the purposes sought in the individual cases can be achieved by using either anonymous data or suitable arrangements to allow identifying data subjects only in cases of necessity, respectively.
This principle imposes data controllers to adopt organizational measures to minimize the use of personal and identification data. That point can be reached by using anonymous data or suitable arrangements to allow identifying data subjects only in cases of necessity.
The security measures provided in the IDPC are classified as:
• Suitable and preventative security measures (Title V, Chapter I) and
• Minimum-security measures (Title V, Chapter II).
The regulation is contained in articles 31 and following, within the “Technical Specifications Concerning Minimum Security Measures (Annex B)”, and in article 3 on “Data Minimization Principle”.
• Art 31 “Security Requirements” states: “Personal data undergoing processing shall be kept and controlled, also in consideration of technological innovations, of their nature and the specific features of the processing, in such a way as to minimize, by means of suitable preventive security measures, the risk of their destruction or loss, whether by accident or not, of unauthorized access to the data or of processing operations that are either unlawful or inconsistent with the purposes for which the data have been collected”.
• Art. 33 “Minimum Security Measures” states: “Within the framework of the more general security requirements referred to in Section 31, or else provided for by specific regulations, data controllers shall be required in any case to adopt the minimum security measures pursuant to this Chapter in order to ensure a minimum level of personal data protection”.
• Art 34 Processing personal data by electronic means shall only be allowed if the minimum security measures referred to below are adopted in accordance with the arrangements laid down in the technical specifications as per Annex B specifically:
o Computer-based authentication;
o Management procedures for implementation of authentication credentials;

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 35 of 96
o Use of an authorization system, that can allow the user to access specific resources to pinpoint the authorization profile;
o Regular update of the specifications concerning the scope of the processing operations that may be performed by the individual entities in charge of managing and/or maintenance of electronic means;
o Protection of electronic means and data against unlawful data processing operations, unauthorized access and specific software;
o Implementation of procedures for safe keeping of backup copies and restoring data and system availability;
o Implementation of encryption techniques or identification codes for specific processing operations performed by health care bodies in respect of data disclosing health and sexual life.
Art 37, par. 1
o (a) a notification to the Data Protection Authority is mandatory if the treatment involves: “genetic data, biometric data, or other data disclosing geographic location of individuals or objects by means of an electronic communications network”.
The notification of processing operations shall have to be submitted to the Authority in advance of the processing and only once, regardless of the number of operations to be performed and the duration of the processing, and may concern one or more processing operations for related purposes. A notification shall only be effective if it is transmitted via the Data Protection Authority’s Website by using the ad-hoc form, which shall contain the request to provide all the pieces of information listed in article 38, par. 2.
The Code provides also a specific regulation on the treatment of health data in Part II, Title V “Processing of personal data in the health care sector”, Articles 75-94.
Informed consent
Processing of personal data shall only be allowed if the person gives his or her express consent, as stated in the Italian Data Protection Code (IDPC) [38] and reported below.
Art 23, par. 1. Consent has to be given “freely and specifically with regard to a clearly identified processing operation, if it is documented in writing, and if the data subject has been provided with the information referred to in Article 13”
Art 13. The consent shall always be accompanied by the specific Information Notice, reporting: the terms of service, the voluntary nature, the right to data access set by art. 7 of IDPC, the processing operations carried out for scientific purposes.
Art 26, par. 1. sensitive data may only be processed with the data subject’s written consent and with the Data Protection Authority’s prior authorization, by complying with the prerequisites and limitations set out in this Code as well as in laws and regulations.
Art 76, par. 1. Health professionals and public health care bodies may process personal data disclosing health, also within the framework of activities in the

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 36 of 96
substantial public interest pursuant to Article 85 (Tasks of the National Health Service),
a) With the data subject’s consent, also without being authorized by the “Garante” (the supervising authority), if the processing concerns data and operations that are indispensable to safeguard the data subject’s bodily integrity and health;
b) Without the data subject’s consent, based on the Garante’s prior authorization, if the purposes referred to under a) concern either a third party or the community as a whole”.
• Art 81. Providing One’s Consent
o The consent to process the sensitive data (disclosing health) of the individual can be expressed in a unique declaration that can be oral or written.
o In the case of an oral declaration, the healthcare professional or public health care authority takes note of the consent expressed and of the delivery to the interested person of the General Privacy Informative Note.
Art 82. The Information Notice and the consent on the processing of the personal data of an individual, can take place after the delivery of the healthcare treatment, without delay, in the following cases:
o Emergencies or cases involving public hygiene.
o In cases of physical impediment, lack of legal capacity, or unable to distinguish right and wrong, when the consent cannot be obtained from the entity legally representing the data subject, or else a next of kin, a family member, a person cohabiting with the data subject or, failing these, the manager of the institution where the data subject is hosted.
o In case of impending and irretrievable danger for the data subject’s health or bodily integrity.
o In cases when the delivery of the necessary medical treatment were to suffer in terms of its timeliness or effectiveness - by the need to obtain the data subject’s prior consent.
Greece
In Greece an independent administrative agency, the Data Protection Authority, was funded and operated since November 1997, according to the Law 2472/97 [39]. The fundamental principle of the Authority is that “every citizen should always be able to know who, where, when, how and why processes his/her personal data”.
The management and protection of personal data of the visitor/user of a website and of online services are subject to relevant provisions of Greek Law (Law 2472/1997) on the protection of individuals and the protection of personal data as supplemented by the decisions of the Chairman of the Commission for Personal Data Protection, P.A. 207/1998 and 79/2000 and article 8 of Law 2819/2000 and Law 2774/1999 and European law (Directives 95/46/EP and 97/66/EP).

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 37 of 96
Greek Constitution
Besides the Data Protection Law, a further article 9(a) was added to the Greek Constitution during the Constitutional Revision of 2001 and it provides for the protection of personal data and the establishment of the relevant independent authority, i.e. the Greek Data Protection Authority. The new Article reads as follows: “All persons have the right to be protected from the collection, processing and use, especially by electronic means, of their personal data, as specified by law. The protection of personal data is ensured by an independent authority, which is established and operates as specified by law”. Generally speaking, the Greek data protection law is quite similar to the European directive.
Greek Data Protection Authority and permissions
The only person who has access to the patient information is only the patient himself/herself for whom are the data, or a legal representative, i.e. parent, authorized lawyer, jurist supporter etc. Apart from these persons, a third person can ask for access to the patient information stored by the hospital. In such circumstance, the third person should clarify the reasons why they ask for the patient’s information. Following this, the hospital should ask permission from the Greek Data Protection Authority. This is described in the article 7 of the Greek Law 2472/1997.
The Greek Data Protection Authority decides regularly that the conduct of research is an obligation mainly for the Hellenic Republic according to the article 16 paragraph 1 of the Greek Constitution. Access to the files of a Hospital etc. need the authorization from the Greek Data Protection Authority. A researcher may have access legally to the information within these files. In this case, the Data Protection Authority defines the rules and the pre-requisites.
In general terms, according to the Greek Data Protection Authority, the sensitive personal data including health-related information, are collected by doctors and health institutions or hospitals in order to provide healthcare and services. The collection of data in a file (medical file article 14 law number 3418/2005) that is carried on by medical/health institutions, should receive the permission of the Greek Data Protection Authority in order to be carried on (law number 2472/1997). In contrast, private doctors do not have the above obligation when they create and keep such kind of data.
Any other process that is involving medical data, other than the one that has to deal with healthcare uses, should be reported to the Greek Data Protection Authority and the Authority should investigate and then approve or reject its use.
Informed consent
According to Greek laws, patients should not be treated primarily as consumers and health data should be strictly protected against unlawful processing [40]. In Greece, so far, while the ePrescription system has been developed not equally significant steps have not taken place as regards the Electronic Health Records (HER) [41]. For the operation of the ePrescription system, special permission by the Greek Data Protection Authority has been given [42]. The protection of health data takes place in the light of legislation on medical secrecy and in the light of legislation on the protection of personal data [43].

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 38 of 96
Thus, all health data are protected by Article 14 of the Code of Medical Ethics (law 3418/2005), which states, under the title “observance of medical records”, the requirements for electronic record keeping by doctors, clinics and hospitals. Article 371 of the Greek Penal Code states that professional secrecy is also applied to health data.
Moreover, the provisions of the law 2472/1997 for the data protection are applied, including Articles 7 and 7A of which contain provisions similar to those in Directive 95/46/EU.
As regards the protection of personal data in the ePrescription system, in particular, there are adequate provisions in the law 3892/2010, under which access to health data kept in the central electronic prescription system is regulated:
• The insured have access to and knowledge of their data, which are registered in the system (Article 6 par. 6).
• Physicians have access to data that has been registered by them or by other doctors, provided that the express and specific consent of the patient has been given (Article 3 par. 8).
• Pharmacists have access to prescriptions performed by themselves (Article 4 par.9).
• Social insurers have access to data only for specific reasons and with the requirement of a specific consent (Article 7 par.1).
• Health service units have access to health referrals (Article 5 par.8).
A further legal constraint that has been imposed by the legal framework regards the Protection of Privacy of patient as described in article 47 in the law 2071/1992 [44] regarding requirements for security policy of HealthCare Organizations and the time extent for health records maintenance for Hospital as from the law 1258/1981.
Germany
Federal Data Protection Act
In Germany, the reference law is the Federal Data Protection Act (Bundesdatenschutzgesetz, BDSG) whose most important provisions are illustrated below:
• Art 4. Admissibility of data processing and use:
o Processing and use of personal data shall be admissible only if the BDSG, or any other legal provision, permits or prescribes them or if the data subject has consented to its use.
o When consent is obtained from the data subject, he or she shall be informed of the purpose of storage and of any envisaged communication of his data and, at his or her request, of the consequences of withholding consent. Consent shall be given in writing unless special circumstances warrant any other form. If consent is to be given together with other written declarations, the declaration of consent shall be made distinguishable in its appearance.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 39 of 96
o In the field of scientific research, a special circumstance shall also be deemed to exist where the defined purpose of research would be impaired considerably if consent were obtained in writing. In such a case the information to be collected as well as the reasons from which considerable impairment of the defined purpose of research would arise, shall be recorded in writing.
• Art 5. Confidentiality, states that persons engaged in data processing shall not process or use personal data without authorization (confidentiality). On taking up their duties, including whenever they work for private bodies, they shall be required to give an explicit commitment to maintain such confidentiality. This undertaking shall continue to be valid after termination of their activity.
• Art 6 Inalienable rights of the data subject:
o The person’s right to be informed (articles 19, 34) and to be able to correct, erase or block (articles 20, 35) which may not be excluded or restricted by a legal transaction.
o If the data are stored in a data file that can be stored by several bodies and if it is not possible to identify the controller of the data file, the person may approach any of these bodies. Such body is obliged to forward the request to the controller of the data file. The person shall be informed of the forwarding of the request and of the controller of the data file. Public prosecution and police authorities, as well as public finance authorities may inform the Federal Commissioner for Data Protection instead of the person as long as they store the personal data to perform their legal duties within the area of application of the Fiscal Code for monitoring and control purposes.
• Art 7. Compensation by public bodies:
o Where a public body causes harm to the person through automated processing of his or her personal data that is inadmissible or incorrect under the provisions of BDSG or other data protection provisions, such body is obliged to compensate the person for the harm thus caused, irrespective of any fault.
o In grave cases of violation of privacy, the person shall receive adequate pecuniary compensation for the immaterial harm caused.
o If, in the case of a data file, several bodies are entitled to store the data and the injured person is unable to ascertain the controller of the data file, each body shall be liable.
o Where several parties are responsible, they shall be jointly and severally liable.
• Art 8. Compensation by private bodies: if a person asserts a claim against a private body for compensation because of automated data processing that is inadmissible or incorrect under the BDSG or other data protection provisions, and if it is disputed whether the harm caused results from a circumstance for which the controller of the data file is responsible, the burden of proof shall rest with the controller of the data file.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 40 of 96
• Art. 9 Technical and organizational measures: public and private bodies processing personal data either on their own behalf or on behalf of others shall take the technical and organizational measures necessary to ensure the implementation of the provisions of the BDSG.
• Art. 11. Commissioned processing or use of personal data:
o Where other bodies are commissioned to process or use personal data, the responsibility for compliance with the provisions of BDSG and with other data protection provisions shall rest with the principal.
o The agent shall be carefully selected, with particular regard for the suitability of the technical and organizational measures taken by them. The commissioning shall be stated in writing, specifying the processing and use of the data, the technical and organizational measures and any sub-contracting.
o The agent may process or use the data only as instructed by the principal. If the agent thinks that an instruction of the principal infringes the BDSG or any other data protection provisions, he or she shall point this out to the principal without delay.
The second part of the Federal Data Protection Act specifically addresses the issue of data processing by public bodies, with the first chapter focusing on the establishment of the legal basis for data processing, as follows.
• Art 12 Scope:
o The provisions shall apply to public bodies of the Federation as far as they do not participate in public-law enterprises.
o Where data protection is not governed by Land legislation, articles 12 to 17, 19 and 20 shall also apply to public bodies of the Länder in so far as they:
§ execute federal law and do not participate in competition as public-law enterprises or,
§ act as bodies of the judicature and are not dealing with administrative matters.
• Art 13. Collection of data:
o Collection of personal data shall be admissible if knowledge of them is needed to perform the duties of the bodies collecting them.
o Personal data shall be collected from the person. They may be collected without his or her participation only if:
§ A legal provision prescribes or peremptorily presupposes such collection or,
§ The nature of the administrative duty to be performed necessitates collection of the data from other persons or bodies or,
§ Collection of the data from the data subject would necessitate disproportionate effort and there are no indications that overriding legitimate interests of the data subject are impaired.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 41 of 96
o If personal data are collected from the data subject with his or her knowledge, he or she shall be informed of the purpose of collection. If information is collected from the data subject pursuant to a legal provision that makes the supply of particulars obligatory or if such supply is the prerequisite for the granting of legal benefits, the person shall be informed that such supply is obligatory or voluntary, as the case may be. At his or her request, he or she shall be informed of the legal provision and of the consequences of withholding details.
o Where personal data are collected from a private body and not from a person, such body shall be informed of the legal provisions requiring the supply of details or that such supply is voluntary, as the case may be.
• Art 14. Storage, modification and use of data
o The storage, modification or use of personal data shall be admissible where it is necessary for the performance of the duties of the controller of the data and if it serves the purposes for which the data were collected. If there has been no preceding collection, the data may be modified or used only for the purposes for which they were stored.
o Storage, modification or use for other purposes shall be admissible only in the following cases:
§ A legal provision prescribes or peremptorily presupposes this.
§ The person has consented.
§ It is evident that this is in the interest of the data subject and there is no reason to assume that he or she would withhold consent if he or she knew of such other purpose.
§ Details supplied by the person have to be checked because there are actual indications that they are incorrect.
§ Data can be taken from generally accessible sources or the controller of the data file would be entitled to publish them, unless the data subject clearly has an overriding legitimate interest in excluding the change of purpose.
§ It is necessary for immediate threat to public safety.
§ It is necessary to prosecute criminal or administrative offences, to implement sentences or measures as defined by the Penal Code or to execute decisions imposing administrative fines.
§ It is necessary to avoid a grave infringement of another person's rights.
§ It is necessary for the conduct of scientific research when scientific interest in conducting the research project substantially outweighs the interest of the person in excluding the change of purpose, and the research purpose cannot be achieved by other means or can be achieved only with disproportionate effort.
o Processing or use for other purposes shall not be deemed to occur if this serves the exercise of powers of supervision or control, the execution of auditing or the conduct of organizational studies for the controller of the

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 42 of 96
data file. This shall also apply to processing or use for training and examination purposes by the controller of the data file, unless the data subject has overriding legitimate interests.
o Personal data stored exclusively for the purpose of monitoring data protection, safeguarding data or ensuring proper operation of a data processing system may be used exclusively for such purposes.
• Art 15. Communication of data to public bodies:
o Communication of personal data to private bodies shall be admissible if:
§ It is necessary for the performance of the duties of the communicating body and the requirements of article 14 are met or;
§ The recipient credibly proves a justified interest in knowledge of the data to be communicated and the person does not have a legitimate interest in excluding their communication.
o Responsibility for the admissibility of communication shall rest with the communicating body.
o The communicating body shall inform the person of the communication of his data. This shall not apply if it can be assumed that he or she will acquire knowledge of such communication in another manner or if such information would jeopardize public safety or otherwise be detrimental to the Federation or a to Land.
o The recipient may process or use the communicated data only for the purpose for which they were communicated to him or her. The communicating body shall point this out to the recipient. Processing or use for other purposes shall be admissible if communication above would be admissible and the communicating body has consented.
• Art 17. Communication of data to bodies outside the area of application of the BDSG:
o Article 16, in conjunction with the relevant laws and agreements as well as article 16, shall apply to the communication of personal data to bodies outside the area of application of BDSG and to supranational or international bodies.
o Communication shall not occur where there is reason to assume that this would be incompatible with the purpose of a German law.
o Responsibility for the admissibility of communication shall rest with the communicating body.
o It shall be pointed out to the recipient that the communicated data may be processed or used only for the purpose for which they were communicated to him.
The second chapter of the BDSG focuses on personal rights, in particular the following articles apply:
• Art 19 Provision of information to the person:
o The person shall, at his or her request, be provided with information on:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 43 of 96
§ Stored data concerning him or her, including any reference in them to their origin or recipient, and
§ The purpose of storage.
o The request should specify the type of personal data on which information is to be provided. If the personal data are stored in records, information shall be provided only in so far as the data subject supplies details making it possible to locate the data and the effort needed to provide the information is not out of proportion to the interest in such information expressed by the person. The controller of the data file shall exercise due discretion in determining the procedure for providing such information and, in particular, the form in which it is provided.
o The paragraph above shall not apply to personal data that are stored merely because they may not be erased due to legal, statutory or contractual provisions on their preservation or exclusively serve purposes of data security or data protection control.
o If the provision of information relates to the communication of personal data to authorities for the protection of the constitution, to the Federal Intelligence Service, the Federal Armed Forces Counterintelligence Office and, where the security of the Federation is concerned, other authorities of the Federal Ministry of Defence, it shall be admissible only with the consent of such bodies.
o Information shall not be provided if:
§ This would be prejudicial to the proper performance of the duties of the controller of the data file,
§ This would impair public safety or order or otherwise be detrimental to the Federation or a Land or,
§ The data or the fact that they are being stored must be kept secret in accordance with a legal provision or by virtue of their nature, in particular on account of an overriding justified interest of a third party, and for this reason, the interest of the data subject in the provision of information must be subordinated.
o If no information is provided to the person, it shall, at his or her request be supplied to the Federal Commissioner for Data Protection, unless the relevant supreme federal authority determines in a particular case that this would jeopardize the security of the Federation or a Land. The communication from the Federal Commissioner to the person must not allow any conclusions to be drawn in terms of information at the disposal of the controller of the data file, unless the latter consents to more extensive information being provided.
o Information shall be provided free of charge.
• Art 20. Correction, erasure and blocking of data:
o Incorrect personal data shall be corrected. If it is discovered that personal data in records are incorrect or if the person disputes that they

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 44 of 96
are correct, a note to this effect shall be made in the record or it shall be recorded by some other means.
o Personal data in data files shall be erased if:
§ Their storage is inadmissible or,
§ Knowledge of them is no longer required by the controller of the data file for the performance of his or her duties.
o Instead of erasure, personal data shall be blocked in so far as:
§ Preservation periods prescribed by law, statutes or contracts rule out any erasure,
§ There is reason to assume that erasure would impair legitimate interests of the data subject or,
§ Erasure is not possible or is only possible with disproportionate effort due to the specific type of storage.
o Personal data in data files shall also be blocked if the data subject disputes that they are correct and it cannot be ascertained whether they are correct or incorrect.
o Personal data in records shall be blocked if the authority determines the particular case that, without blocking, legitimate interests of the person would be impaired and the data are no longer required for the performance of the authority's duties.
o Blocked data may be communicated or used without the consent of the data subject only if:
§ This is indispensable for scientific purposes, for use as evidence or for other reasons in the overriding interests of the controller of the data file or a third party and
§ Communication or use of the data for this purpose would be admissible if they were not blocked.
o If necessary to protect legitimate interests of the person, the correction of incorrect data, the blocking of disputed data and the erasure or blocking of data due to inadmissible storage shall be notified to the bodies to which these data are transmitted for storage within the framework of regular data communication.
Lastly, the last part of the BDSG addresses special provisions, with the following article.
• Art 40. Processing and use of personal data by research institutes:
o Personal data collected or stored for scientific research purposes may be processed or used only for such purposes.
o Communication of personal data to other than public bodies for scientific research purposes shall be admissible only if these undertake not to process or use the communicated data for other purposes and to comply with the provisions of the following paragraph.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 45 of 96
o Personal data shall be anonymised as soon as the research purpose permits this. Until then, the characteristics enabling information concerning personal or material circumstances to be attributed to an identified or identifiable individual shall be stored separately. They may be combined with the information only to the extent required by the research purpose.
o The bodies conducting scientific research may publish personal data only if:
§ The data subject has consented or,
§ This is indispensable for the presentation of research findings.
Informed consent
Art 34 of Federal Data Protection Act (Bundesdatenschutzgesetz, BDSG) on informed consent:
o The data subject may request information on:
§ Stored data concerning him or her, including any reference in them to their origin and recipient.
§ The purpose of storage and.
§ Persons and bodies to whom his or her data are regularly communicated if his or her data are processed automatically.
o If the personal data are stored in the normal course of business for the purpose of communication, the data subject may request information on their origin and recipient only if he or she has well-founded doubts about the correctness of the data. In such case, information on the origin and recipient shall be provided even if these particulars are not stored.
o In the case of bodies that store personal data in the normal course of business for the purpose of supplying information, the data subject may request information on his or her personal data even if they are not stored in a data file. The data subject may request information on their origin and recipient only if he or she proves that he has well-founded doubts about the correctness of the data.
o Information shall be provided in writing unless special circumstances warrant any other form.
o Information shall be provided free of charge. However, if the personal data are stored in the normal course of business for the purpose of communication, a fee may be charged if the data subject can use the information vis-à-vis third parties for commercial purposes. The fee shall not exceed the costs directly attributable to the provision of information. No fee may be charged in cases where special circumstances give rise to the assumption that stored personal data are incorrect or that their storage was inadmissible, or where the information has revealed that the personal data have to be corrected or have to be erased.
o Where information is not provided free of charge, the data subject shall be given the possibility to acquire personal knowledge of the data and, in

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 46 of 96
particular, concerning him or her within the framework of his entitlement to information. This shall be pointed out to him or her in a suitable manner.
Romania Romanian Constitution
The Romanian Constitution adopted in 1991 recognizes under Title II (Fundamental Rights, Freedoms and Duties) the rights of privacy, inviolability of domicile, freedom of conscience and expression. In particular, article 26 states that public authorities shall respect and protect intimacy, family and private life.
Government Decision regarding the protection of the employ/office classified information
Another relevant act is the Government Decision 781 regarding the protection of sensitive information within working environments [45], which was published in the official gazette no. 575 dated 5 August 2002.
The national standards for the protection of classified information in Romania, approved by Government Decision no. 585/2002, shall apply to sensitive employment-related information. The decision specifies:
• Classification, declassification and minimum protection measures.
• General rules of evidence, preparation, storage, processing, copying, handling, transportation, transmission and destruction.
• Obligations and responsibilities of public authorities and institutions managers, companies and other legal entities.
Law on patient rights
According to Law no. 46 of 21.01.2003 on patient rights published on Official Journal no. 51 of 29.01.2003 [46], the following principles apply:
• Art. 2. Patients have the right to the highest quality care that the society can provide in accordance with human financial and materials resources.
• Art. 3. The patient has the right to be respected as a human person, without any discrimination.
• Art. 20. The patient cannot be photographed or filmed in a medical unit without his/her consent, unless those images are necessary for diagnosis or treatment and to avoid suspicion of a medical fault.
The right to information confidentiality and privacy of patient are detailed in Chapter IV within the following articles.
• Art. 21. All information on patient conditions, results of investigations, diagnosis, prognosis, treatment, personal data are confidential even after death.
• Art. 22. Confidential information can be provided only if the patient explicitly gives its consent or if expressly required by law.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 47 of 96
• Art. 24. The patient has access to personal medical data.
• Art. 25. Any interference in the private and family life of the patient is prohibited, with the exception where this interference positive influence diagnosis, treatment or care given and only with patient consent. Are considered exceptions the cases when a patient is dangerous to himself or to public health.
Law on the Protection of Individuals
A further relevant law is No. 677/2001 on the Protection of Individuals with Regard to the Processing of Personal Data and the Free Movement of Such Data [47], amended and completed, published in the Official Journal of Romania, Part I, No. 790/12 December 2001, adopted by the Chamber of Deputies in the session of the 22 October 2001, in accordance with the provisions of Article 74 paragraph (2) of the Romanian Constitution.
The Law No 677/2001 transposes into domestic legislation the Directive 95/46/EC and it establishes the fundamental legal framework for the protection of individuals with regard to processing of personal data in Romania. The purpose of this law is to guarantee and protect the individual’s fundamental rights and freedoms, especially the right to personal, family and private life, with regard to the processing of personal data. The present law applies to personal data processing, performed, totally or partially, through automatic means, as well as to the processing through means other than automatic, which are part of, or destined to, a personal data filing system.
For the purposes of this law, the following terms are defined:
• Personal data: any information referring to an identified or identifiable person. In turn, an identifiable person is a person that can be identified, directly or indirectly, particularly with reference to an identification number or to one or more specific factors of his physical, physiological, psychological, economic, cultural or social identity.
• Personal data processing: any operation or set of operations that is performed upon personal data, by automatic or non-automatic means, such as collecting, recording, organizing, storing, adapting or modifying, retrieval, consultation, use, disclosure to third parties by transmission, dissemination or by any other means, combination, alignment, blocking, deletion or destruction.
• Storage: keeping the collected personal data on any type of storage support.
• Personal data filing systems: any organized personal data structure that may be accessed according to some specific criteria, regardless of the fact that this structure is distributed according to functional or geographical criteria.
• Data controller: any natural or legal person, including public authorities, institutions and their legal bodies, that establishes the means and purpose of the personal data processing; if the purpose and means of the personal data processing is set out or based on a legal provision, the data controller shall be the natural or legal person assigned as data controller by that specific legal provision.
• Data processor: a natural or legal person, of private or public law, including public authorities, institutions and their legal bodies, which processes personal data on the data controller’s behalf.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 48 of 96
• Third party: any natural or legal person, of private or public law, including public authorities, institutions and their local bodies other than the data subject, than the controller, or the processor who, under direct authority of the controller or of his processor, is authorized to process data.
• Recipient: any natural or legal person, of private or public law, including public authorities, institutions and their local bodies, to whom the data are disclosed, regardless of the fact that it is a third party or not. The public authorities, which receive data in accordance with a special type of inquiry competence, will not be considered consignees.
• Anonymous data: data that, due to its specific origin or specific manner of processing, cannot be associated to an identified or identifiable person.
The most relevant articles of Law No 677/2001 are:
• Art 4. Personal data to be processed must be:
o Processed fairly and in accordance with the existing legal provisions.
o Collected for specific, explicit and legitimate purposes. Further processing of personal data for statistical, historical or scientific research, will not be considered incompatible with the purpose they were initially collected for, if it is carried out according to the provisions of the law, including those referring to the notification submitted to the supervisory authority, as well as according to the guarantees regarding personal data processing, set out by the legal provisions on statistics’ activity or the historical or scientific research.
o Adequate, pertinent and non-excessive in relation to the purpose for which they are collected and further processed.
o Accurate and, if necessary, updated. For this purpose, appropriate measures shall be taken in order to erase and/or rectify inaccurate or incomplete data, from the point of view of the purpose for which they were collected and later processed.
o Stored in such a manner that allows the identification of the data subject only for the time limit required to fulfil the purposes for which they are collected and later processed. The storage of data for a longer period of time than the one mentioned, for statistical, historical or scientific research purposes, shall be carried out in accordance with the guarantees regarding personal data processing, provided in the relevant legal framework, and only for the period of time required to achieve these purposes.
• Art 8. Processing of personal data with an identification function
o The processing of the personal identification number or of other personal data with a general identification function may be carried out only if:
§ The data subject has given his/her express and unequivocal consent; or
§ The processing is expressly stated by a legal provision.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 49 of 96
o The supervisory authority may establish other situations in which the processing of data may be carried out, only after adequate guarantees have been provided in order to observe the data subject’s rights.
• Art 9. Processing personal data regarding the state of health:
o The processing of health data may be carried out only by, or under the supervision of, medical staff who are under a pledge of professional confidentiality, except for the cases when the data subject has given, in writing, his/her unequivocal consent and as long as the consent has not been withdrawn, as well as except for the cases when the data processing is necessary for the prevention of an imminent danger, the prevention of a criminal offence or the prevention of the result of such an action or for the removal of the damaging results of such an action.
o The medical staff, health institutions and their staff may process personal health data without the authorization of the supervisory authority only when the data processing is required in order to protect the data subject’s life, physical integrity or health. When the mentioned purposes refer to other people or to the general public and the data subject has not given his/her written and unequivocal consent, the preliminary authorization of the supervisory authority must first be demanded and obtained. The processing of personal data is forbidden beyond the limits of the authorization.
o Except for emergency reasons, the authorization may be given only after consulting the Romanian Medical College.
o Personal health data may only be collected from the data subjects themselves. Exceptionally, these data can be collected from other sources only when it is required in order not to compromise, the processing’s purpose, and when the data subject cannot or does not wish to provide them.
The rights of the data subject in the context of personal data processing are further analysed in Chapter IV of the law and, in particular, in the following articles:
• Art 12. Informing the Data Subject.
• Art 13. The Right of Access to Data.
• Art 14. The Right of Intervention upon the Data.
• Art 15. The Right to Object.
• Art 17. The Right Not to be Subject to an Individual Decision.
• Art 18. The Right to Refer to a Court of Law.
• Art 19. Confidentiality of Data Processing.
• Art 20. Security of Data Processing.
o It is the data controller’s obligation to apply adequate technical and organizational measures in order to protect the data against accidental or unlawful destruction, loss, alteration, disclosure or unauthorized access, notably if the respective processing involves the data’s transmission within a network, as well as against any other form of illegal processing.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 50 of 96
o These measures shall ensure, depending on the state of the art techniques employed and the costs, adequate security against processing risks as well as observing the nature of the data that must be protected.
o When appointing a data processor, the data controller has the obligation to assign a person who presents sufficient guarantees regarding technical security and the organizational measures concerning the data to be processed, as well as the obligation to ensure that the assigned person complies with these measures.
o The supervisory authority may decide, in individual cases, that the data controller should adopt additional security measures, except such measures that regard the guaranteed security of telecommunication services.
o Data processing performed by an appointed data processor shall be initiated following a written contract which should necessarily contain the following:
§ The processor’s obligation to act strictly in accordance with the instructions received from the data controller;
§ The fact that accomplishing the obligations also applies to the data processor. The minimum-security requirements shall be issued by the supervisory authority and shall be periodically updated, according to the technological progress and the accumulated experience.
o Any person who acts under the authority of the data controller or of the data processor, including the data processor, who has access to personal data, may process them only in accordance with the data controller’s specific instructions, except when the above-mentioned person’s actions are based on a legal obligation.
Ratification of the Convention on the protection of individuals
Law no. 682 / 28th November 2001 ratifies the Convention on the protection of individuals with regard to automatic processing of personal data [48], with its annex addressing the protection of individuals with regard to automated processing of personal data.
Order on the minimum safety requirements for personal data processing
It is also worth mentioning the order no. 52 dated April 18, 2002 which stated the minimum safety requirements for personal data processing [49], as published in the Official Journal no. 383 of June 5, 2002.
These minimum safety requirements for personal data processing set the grounds for controllers to adopt and implement appropriate technical and organisational measures to ensure the confidentiality and integrity of personal data. Accordingly, the controllers shall set their own safety procedures and policies.
The minimum safety requirements for personal data processing cover several aspects, the most important being the following:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 51 of 96
• User's identification and authentication. A user is any person that acts under the controller’s, or the authorised representative's authority, with an acknowledged access right to personal databases.
• Access type. The users shall access only personal data necessary to fulfil their job related tasks. In order to do so, the controllers shall set the access types according to the functionality (such as: administration, entering, processing, saving etc.), and according to the actions to be performed on the personal data (such as: writing, reading, deleting etc.), and shall set out the procedures for these access types.
• Data collection. The controller designates authorised users for personal data collection and input within an IT system.
• Back-ups. The controller shall set the time frames for carrying on backups of personal data, and of the software used for computer processing.
• Access to files. The controller shall take measures to ensure that any access to databases containing personal information is registered either in an log file –in case of automatic processing- or in a registry for non-automatic processing of personal data, as determined by the controller.
• Telecommunication Systems. The controller shall periodically check the authentication and the access types in order to detect any malfunctions regarding the use of the telecommunication systems.
• Employees’ Training. During the users' training courses, the controller shall inform them on the provisions of Law 677/2001 regarding the protection of individuals with regard to personal data processing and the free flow of such data, on the minimum safety requirements for personal data processing, and on the risks involved by personal data processing, according to the user's specific activity.
Law on the processing of personal data and the protection of privacy in electronic communications sector
The law no 506/2004 on the processing of personal data and the protection of privacy in the electronic communications sector [50], which was published in the Official Journal of Romania, Part I, no. 1101 of November 25 2004, closely follows the Directive 2002/58/CE of the European Parliament and the Council on personal data processing and privacy protection in the electronic communication sector.
The law establishes the specific conditions for safeguarding the right to privacy with respect to the processing of personal data in the electronic communications sector. The provisions of this law apply to the providers of public electronic communications networks and of publicly available electronic communications services, as well as to the providers of value added services and of directories of subscribers who, in the frame of their commercial activity, are processing personal data.
The most important topics addressed by the law regard:
• Security measures. In case of a particular risk of a breach of the security of the network, the provider of a publicly available electronic communications service must:
o Inform the subscribers of such risk and of the possible consequences;

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 52 of 96
o Inform the subscribers of any possible remedy;
o Inform the subscribers of the likely costs involved to eliminate the risk.
• Confidentiality of the communications. Confidentiality of communications and the related traffic data by means of public electronic communications networks and publicly available electronic communications services must be guaranteed. Listening, tapping, storage or other kinds of interception or surveillance of communications and the related traffic data are prohibited, except for the following cases:
o These operations are carried out by the users who participate in that communication;
o The users who participate in that communication have previously given their written consent;
o These operations are carried out by the competent authorities, under the conditions set out by the legal provisions in force.
The provisions of previous paragraphs shall not prevent technical storage, which is necessary for the conveyance of a communication without prejudice to the principle of confidentiality.
The use of an electronic communications network to store information or to gain access to information stored in the terminal equipment of a subscriber or user is only allowed on condition that:
• The subscriber or user concerned was provided with clear and comprehensive information in accordance with Art. 12 of Law no. 677/2001, inter alia about the purposes of the storage or access to information stored.
• The subscriber or user concerned was offered the possibility to refuse such storage or access to information stored.
The previous provisions shall not prevent the technical storage or access in the following cases:
• When these operations are performed for the sole purpose of carrying out or facilitating the transmission of a communication over an electronic communications network.
• When these operations are strictly necessary for the provision of an information society service explicitly requested by the subscriber or user.
Anti-corruption law
According to the anti-corruption law 161/2003 [51], as published in the Official Journal on 21/04/2003: Title III, the following provisions are defined in order to help preventing and fighting cyber-crime:
• “Data on the users” are represented by any information that can lead to identifying a user, including the type of communication and the serviced used, the post address, geographic address, IP address, telephone numbers or any other access numbers and the payment means for the respective service as well as any other data that can lead to identifying the user.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 53 of 96
• ”Security measures” refer to the use of certain procedures, devices or specialised computer programmes by means of which the access to a computer system is restricted or forbidden for certain categories of users.
The following offences against the confidentiality and integrity of data and computer systems are identified:
• Illegal access to a computer system.
• Illegal interception of any transmission of computer data that is not published to, from or within a computer system.
• Illegal alteration, deletion or deterioration of computer data.
• Unauthorised data transfer from a computer.
• Unauthorised data transfer by means of an information data storing means.
• Serious hindering, without right, of a computer system operation, by the introduction, transmission, altering, deleting or deteriorating computer data or by restricting the access to these data.
According to the law no. 677/2001, location data is defined as any data processed in an electronic communications network, indicating the geographic position of the terminal equipment of a user of a publicly available electronic communications service. With regard to location data and other traffic data, the following provisions apply:
• Where location data other than traffic data, relating to users or subscribers of public electronic communications networks or publicly available electronic communications services, can be processed, such data may only be processed in the following situations:
o The data concerned are made anonymous;
o With the prior express consent of the user or subscriber to whom that data relate, to the extent and for the duration necessary for the provision of a value added service;
o When the value added service with user location function is intended for one-way undifferentiated transmission of information to users.
• The provider of the publicly available electronic communications service must make available to the user or subscriber, prior to obtaining his/her consent, information on:
o The users or subscribers giving their consent for the processing of data shall have the right to withdraw their consent for the processing of data at any time or to temporarily refuse the processing of each connection to the network or for each transmission of a communication. The provider of the publicly available electronic communications service must make available to users or subscribers a simple means, free of charge, to exercise these rights.
o Processing of location data other than traffic data may only be carried out by the persons acting under the authority of the provider of the public electronic communications network or publicly available communications service or of the third party providing value added

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 54 of 96
services, and is allowed only to the extent it is necessary for the purposes of providing the value added service.
Informed consent
According to law No.677/2001 [47], all physical persons must be informed if personal data is to be collected. Statistical and anonymous data is not subject to laws and regulations.
When collecting personal data, users must be informed about their rights and must provide a written consent for data usage. Afterwards, users can intervene on their data or request the interruption of their processing, although that request has no backwards effect.
The Civil Code of July 17th, 2009 [52], republished by the Law 287/2009, published in Official Gazette no. 505 of July 15th, 2011, includes Article 71 on the right to privacy, which states that:
• Every person has the right to have respect for his private life.
• No one shall be subjected to any interference in private life, personal, family life or in inhabitation, residence or his correspondence, without his consent.
• It is also forbidden to use in any manner correspondence, manuscripts or other personal documents as well as private information, without explicit consent.
Slovenia Personal data protection act of the republic of Slovenia [53]
The following main articles of the act are worth highlighting:
• Art. 6: Meaning of terms. Terms used in this Act shall have the following meanings:
1. Personal data - is any data relating to an individual, irrespective of the form in which it is expressed.
2. Individual - is an identified or identifiable natural person to whom personal data relates; an identifiable natural person is one who can be identified, directly or indirectly, in particular by reference to an identification number or to one or more factors specific to his physical, physiological, mental, economic, cultural or social identity, where the method of identification does not incur large costs or disproportionate effort or require a large amount of time.
3. Processing of personal data - means any operation or set of operations performed in connection with personal data that are subject to automated processing or which in manual processing are part of a filing system or which are intended for inclusion in a filing system, such as in particular collection, acquisition, recording, organisation, storage, adaptation or alteration, retrieval, consultation, use, disclosure by transmission, communication, dissemination or otherwise making available, alignment or connecting, blocking, anonymising, erasure or destruction; processing may be performed manually or by using automated technology (means of processing).

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 55 of 96
4. Automated processing – is the processing of personal data using information technology means.
5. Filing system – is any structured set of data containing at least one piece of personal data, which is accessible according to criteria enabling the use or combination of the data, irrespective of whether the set is centralised, decentralised or dispersed on a functional or geographical basis; a structured set of data is a set of data organised in such a manner as to identify or enable identification of an individual.
6. Data controller - is a natural person or legal person or other public or private sector person which alone or jointly with others determines the purposes and means of the processing of personal data or a person provided by statute that also determines the purposes and means of processing.
7. Data processor - is a natural person or legal person that processes personal data on behalf and for the account of the data controller.
8. Data recipient – is a natural or legal person or other private or public sector person to whom personal data are supplied or disclosed.
9. Supply of personal data – is the supply or disclosure of personal data.
10. Foreign recipient and foreign data controller – is a recipient of personal data in a third country and a data controller in a third country.
11. Third country - is a country that is not a Member State of the European Union or a part of the European Economic Area.
12. Filing system catalogue - is a description of a filing system.
13. Register of Filing Systems - is a register containing data from filing system catalogues.
14. Personal consent of an individual – is a voluntary statement of the will of an individual that his personal data may be processed for a specific purpose, and this is given on the basis of information that must be provided to such individual by the data controller pursuant to this Act; personal consent of an individual may be written, oral or some other appropriate consent of the individual.
15. Written consent of the individual - is the signed consent of the individual having the form of a document, the provision of a contract, the provision of an order, an appendix to an application or other form in accordance with statute; a signature shall also mean on the basis of a statute a form equivalent to a signature given by means of telecommunication and a form equivalent by statute to a signature given by an individual who does not know how to write or is unable to write.
16. Oral or other appropriate consent of the individual - is consent given orally or by means of telecommunication or other appropriate means or in some other appropriate manner from which it can be concluded unambiguously that the individual has given his consent.
17. Blocking - is such labelling of personal data that restricts or prevents their further processing.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 56 of 96
18. Anonymising - is such alteration to the form of personal data such that they can no longer be linked to the individual or where such link can only be made with disproportionate efforts, expense or use of time.
19. Sensitive personal data - are data on racial, national or ethnic origin, political, religious or philosophical beliefs, trade-union membership, health status, sexual life, the entry in or removal from criminal record or records of minor offences that are kept on the basis of a statute that regulates minor offences (hereinafter: minor offence records); biometric characteristics are also sensitive personal data if their use makes it possible to identify an individual in connection with any of the aforementioned circumstances.
20. Same connecting codes - are the personal identification number and other uniform identification numbers defined by statute relating to an individual that can be used to obtain or retrieve personal data from filing systems in which the same connecting codes are also processed.
21. Biometric characteristics - are such physical, physiological and behavioural characteristics which all individuals have but which are unique and permanent for each individual specifically and which can be used to identify an individual, in particular by the use of fingerprint, recording of papillary ridges of the finger, iris scan, retinal scan, recording of facial characteristics, recording of an ear, DNA scan and characteristic gait.
22. Public sector - are state bodies, bodies of self-governing local communities, holders of public powers, public agencies, public funds, public institutes, universities, independent institutions of higher education and self-governing communities of nationalities.
23. Private sector - means legal or natural persons performing an activity in accordance with the statute regulating commercial companies or a commercial public service or craft, and persons of private law; public commercial institutes, public companies and commercial companies, irrespective of the share or influence held by the state, self-governing local communities or self-governing communities of nationalities, are a part of the private sector.
• Art. 13: Processing of sensitive personal data. Sensitive personal data may only be processed in the following cases:
1. If the individual has given explicit personal consent for this, such consent as a rule being in writing, and in the public sector provided by statute.
2. If the processing is necessary in order to fulfil the obligations and special rights of a data controller in the area of employment in accordance with statute, which also provides appropriate guarantees for the rights of the individual.
3. If the processing is necessarily required to protect the life or body of an individual to whom the personal data relate, or of another person, where the individual to whom the personal data relate is physically or contractually incapable of giving his consent pursuant to subparagraph 1 of this Article.
4. If sensitive personal data are processed for the purposes of lawful activities by institutions, societies, associations, religious communities, trade unions or other non-profit organisations with political, philosophical, religious or trade-union aim, but only if the processing concerns their members or individuals in regular

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 57 of 96
contact with them in connection with such aims, and if they do not supply such data to other individuals or persons of public or private sector without the written consent of the individual to whom they relate.
5. If the individual to whom the sensitive personal data relate publicly announces them without any evident or explicit purpose of restricting their use.
6. If sensitive personal data is processed by health-care workers and health-care staff in compliance with statute for the purposes of protecting the health of the public and individuals and the management or operation of health services.
7. If this is necessary in order to assert or oppose a legal claim.
8. If so provided by another statute in order to implement the public interest.
• Art. 14: Protection of sensitive personal data.
1. Sensitive personal data, during processing, must be specially marked and protected, such that access to them by unauthorised persons is prevented, except in instances from subparagraph 5 of Article 13 of this Act.
2. In the transmission of sensitive personal data over telecommunications networks, data shall be considered as suitably protected if they are sent with the use of cryptographic methods and electronic signatures such that their illegibility or non-recognition is ensured during transmission.
• Art. 15: Automated decision-making. Automated data processing, in the context of which a decision may be taken regarding an individual that could have legal effect in relation to him, or substantive influence on him, and which is based solely on automated data processing intended for the evaluation of certain personal aspects relating to him, such as in particular his success at work, credit rating, reliability, handling or compliance with conditions required, shall only be permitted if the decision:
1. is taken during the conclusion or implementation of a contract, provided that the request to conclude or implement a contract submitted by the individual to whom the personal data relate has been fulfilled or that there exist appropriate measures to protect his lawful interests, such as in particular agreements enabling him to object to such decision or to express his position;
2. is provided by statute which also provides measures to protect the lawful interests of the individual to whom the personal data relate, particularly the possibility of legal remedy against such decision.
• Art. 16. Purpose of collection, and further processing. Personal data may only be collected for specific and lawful purposes, and may not be further processed in such a manner that their processing would be counter to these purposes, unless otherwise provided by statute.
• Art. 17: Processing for historical, statistical and scientific-research purposes.
1. Irrespective of the initial purpose of collection, personal data may be further processed for historical, statistical and scientific-research purposes.
2. Personal data shall be supplied to the data recipient for the purpose of processing from the previous paragraph in an anonymised form, unless otherwise provided by statute or if the individual to whom the personal data

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 58 of 96
relate gave prior written consent for the data to be processed without anonymising.
3. Personal data supplied to data recipient in accordance with the previous paragraph shall on completion of processing be destroyed, unless otherwise provided by statute. The data recipient shall be obliged without delay after destruction of the data to inform the data controller who supplied him the personal data in writing when and how he destroyed them.
4. Results of processing from the first paragraph of this Article shall be published in anonymised form, unless otherwise provided by statute or unless the individual to whom the personal data relate gave written consent for publication in a non-anonymised form or unless written consent for such publication has been given by the heirs to the deceased person under this Act.
• Art. 19: Informing the individual of the processing of personal data.
1. If personal data are collected directly from the individual to whom they relate, the data controller or his representative must communicate to the individual the following information, if the individual is not yet acquainted with them:
o data on the data controller and his possible representative (personal name, title or official name respectively and address or seat respectively),
o the purpose of the processing of personal data.
2. If in view of the special circumstances of collecting personal data from the previous paragraph, there is a need to ensure lawful and fair processing of personal data of the individual, the person from the previous paragraph must also communicate to the individual the additional information, if the individual is not yet acquainted with them, and in particular:
o a declaration as to the data recipient or the type of data recipients of his personal data;
o a declaration of whether the collection of personal data is compulsory or voluntary, and the possible consequences if the individual will not provide data voluntarily;
o information on the right to consult, transcribe, copy, supplement, correct, block and erase personal data that relate to him.
3. If personal data were not collected directly from the individual to whom they relate, the data controller or his representative must communicate to the individual the following information no later than on the recording or supply of personal data to the data recipient:
o information on the data controller and his possible representative (personal name, title or official name respectively and address or seat respectively),
o the purpose of the processing of personal data.
4. If in view of the special circumstances of collecting personal data from the previous paragraph, there is a need to ensure lawful and fair processing of personal data of the individual, the person from the previous paragraph must also communicate to the individual additional information, and in particular:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 59 of 96
o information on the type of personal data collected,
o a declaration as to the data recipient or the type of data recipients of his personal data,
o information on the right to consult, transcribe, copy, supplement, correct, block and erase personal data that relate to him.
5. Information from the third and fourth paragraphs of this Article shall not need to be ensured if in order to process personal data for historical, statistical or scientific-research purposes it would be impossible or would incur large costs or disproportionate effort or would require a large amount of time, or if the recording or supply of personal data is expressly provided by statute.
• Art. 21. Duration of storage of personal data.
1. Personal data may only be stored for as long as necessary to achieve the purpose for which they were collected or further processed.
2. On completion of the purpose of processing, personal data shall be erased, destroyed, blocked or anonymised, unless pursuant to the statute governing archive materials and archives they are defined as archive material, or unless a statute otherwise provides for an individual type of personal data.
• Art. 23: Protection of personal data of deceased individuals.
1. Data controller may supply data on a deceased individual only to those data recipients authorised to process personal data by statute.
2. Irrespective of the previous paragraph, data controller shall supply data on a deceased individual to the person who under the statute governing inheritance is the deceased person’s legal heir of the first or second order, if they demonstrate a lawful interest in the use of personal data and the deceased individual did not prohibit in writing the supply of such personal data.
3. Unless otherwise provided by statute, a data controller may also supply data from the previous paragraph to any other person intending to use such data for historical, statistical or scientific-research purposes if the deceased individual did not prohibit in writing the supply of such personal data.
4. If the deceased individual did not issue a prohibition from the previous paragraph, persons who under the statute governing inheritance are his legal heirs of the first or second order may prohibit in writing the supply of his data, unless otherwise provided by statute.
• Art. 30: Right of the individual to information.
1. Data controller shall on request of the individual be obliged:
o To enable consultation of the filing system catalogue;
o To certify whether data relating to the individual are being processed or not, and to enable each individual to consult personal data contained in filing system that relate to him/her, and to transcribe or copy them.
o To supply the individual with an extract of personal data contained in filing system that relate to him/her.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 60 of 96
o To provide a list of data recipients to whom personal data were supplied, when, on what basis and for what purpose.
o To provide information on the sources on which records contained about the individual in a filing system are based, and on the method of processing.
o To provide information on the purpose of processing and the type of personal data being processed, and all necessary explanations in this connection.
o To explain technical and logical-technical procedures of decision-making, if the controller is performing automated decision-making through the processing of personal data of an individual.
2. The extract from subparagraph 3 of the previous paragraph may not replace the document or certificate under the regulations on administrative or other procedures and this shall be indicated on the extract.
• Art. 32: Right to supplement, correct, block, erase and to object.
1) On the request of an individual to whom personal data relate, the data controller must supplement, correct, block or erase personal data which the individual proves as being incomplete, inaccurate or not up to date, or that they were collected or processed contrary to statute.
2) At the request of the individual, the data controller must inform all data recipients and data processors to whom the controller has supplied the personal data of the individual, before the measures (as stated in the previous paragraph) have been carried out, of their supplementation, correction, blocking or erasure pursuant to the previous paragraph. Exceptionally, the data controller shall not need to do this if it would incur large costs, disproportionate efforts or would require a large amount of time.
3) Individuals whose personal data are processed in accordance with the fourth paragraph of Article 9 or the third paragraph of Article 10 of this Act shall have the right through objection at any time to demand the cessation of their processing. The data controller shall grant the objection if the individual demonstrates that the conditions for processing have not been fulfilled pursuant to the fourth paragraph of Article 9 or the third paragraph of Article 10 of this Act. In this case, the personal data of the individual may no longer be processed.
4) If the data controller does not grant the objection from the previous paragraph, the individual that lodged the objection may request that the National Supervisory Body for Personal Data Protection decides on whether the processing is in accordance with the fourth paragraph of Article 9 or the third paragraph of Article 10 of the Act. The individual may lodge such request within seven days of delivery of the decision regarding on objection.
5) The National Supervisory Body for Personal Data Protection shall decide on the request from the previous paragraph within two months of receipt of the request. The lodging of a request shall withhold the processing of personal data of the individual in respect of which the request was lodged.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 61 of 96
6) The costs of all actions of the data controller from the previous paragraphs shall be borne by the data controller.
The general rule for collecting patient’s health data is that either patient’s consent is required or that collection of certain data is prescribed by the law. In Slovenia, the data is mostly collected on the basis of legal provisions as the law prescribes which health data is collected and in such case patient consent is not required [54].
The protection of personal health data in Slovenia is a constitutionally guaranteed right. The Slovenian constitution declares the protection of personal data and it prohibits their application in the manner, which is different from legal purposes declared during their collection. In Slovenia there is no particular law only dedicated to the regulation of personal health data protection. Therefore, the regulation of personal health data protection is determined by the Personal Data Protection Act [53] (National Assembly of the Republic of Slovenia, Ur.l. RS, št. 113/2005-ZInfP, 51/2007-ZUstS-A, 67/2007, 94/2007-UPB1).
The Act regulates the rights, obligations and principles to prevent unconstitutional, illegal and unjustified intrusion into the privacy and dignity of an individual as related to the personal data processing. In this Act, health data is considered and therefore regulated as “sensitive personal data”.
Section II of this Act (Personal Data Processing) regulates the personal data acquisition, update, storage and processing procedures. According to art. 8, personal data processing may be performed only if determined by the law or upon previously obtained consent of the personal data subject. According to art. 13/6, sensitive personal data may be processed if it is needed for the purposes of medical prevention, diagnosis and treatment, and management or the provision of healthcare services handled by healthcare professionals and other medical staff in accordance with the Act.
Another Act related to personal health data in Slovenia is the Healthcare Databases Act (National Assembly of the Republic of Slovenia, Ur.l. RS, št. 65/2000), which regulates the collection, processing and transfer of databases, used and exchanged between legal and natural persons engaged in medical activities.
Macedonia
The Law on Personal Data Protection [56] ("Official Gazette of the Republic of Macedonia” n.7/05, 103/08, 124/10 and 135/11) regulates the protection of personal data as fundamental freedoms and rights of the natural persons, and especially the rights to privacy as related to the personal data processing.
For the purposes of this law, the following terms are defined:
1) “Personal data” shall be any information pertaining to an identified or identifiable natural person, the identifiable entity being an entity whose identity can be determined directly or indirectly, especially as according to the personal identification number of the citizen or on the basis of one or more characteristics, specific for his/her physical, mental, economic, cultural or social identity.
2) “Personal data processing” shall be every operation or a sum of operations performed on personal data, automatically or otherwise, such as: collection, recording, organizing, storing, adjusting, or altering, withdrawing, consulting,

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 62 of 96
using, revealing through transmitting, publishing or making them otherwise available, aligning, combining, blocking, deleting or destroying.
3) “Personal Data Collection” shall be a structured group of personal data available in accordance to specific criteria, regardless whether it is centralized, decentralized or dispersed on a functional or a geographical basis.
4) “Personal Data Subject” shall be any natural person to whom the processed data refer.
5) “Controller of the Personal Data Collection” shall be any natural person or legal entity, a state administration body or other body, who, independently or together with others, determines the purposes and the ways of personal data processing (hereinafter: controller). When the purposes and the ways of personal data processing are determined by law or any other regulation, the same law, i.e. regulation determines the controller or the special criteria for his/her selection.
6) “Personal Data Collection Processor” shall be a natural person or a legal entity or a legally authorized state administration body processing the personal data on the behalf of the controller.
7) “Third Party”, shall be any natural person or legal entity, a state administration body or other body, which is not a personal data subject, a controller, a Personal Data Collection Processor or any person who, under a direct authorization by the controller or by the Personal Data collection processor is authorized to process the data.
8) “User” shall be any natural person or a legal entity, a state administration body or other body, to whom the data are disclosed.
9) “Consent of the personal data subject” shall be freely and explicitly given statement of will, of the personal data subject whereby (s)he agrees to the processing of his/her personal data for previously determined purposes.
10) “Special categories of personal data” shall be personal data revealing the racial or ethnic origin, the political views, religious or other beliefs, membership in a trade union and data relating to the health condition of the people, including genetic data, biometric data or data referring to the sexual life.
11) “Third country” shall be country not being European Union member or not being member of the European Economic Space.
The most relevant articles of this Law are:
• Art. 3, according to which this law shall be applied:
o to entirely or partly automated personal data processing and,
o to other processing of the personal data, which are part of an existing collection of personal data or are intended to be part of a collection of a personal data.
• Art. 3-a, which states that the personal data protection shall be guaranteed without discrimination based on his/her nationality, race, colour of the skin, religious conviction, ethnical belonging, sex, language, political or other convictions, material status, birth background, education, social background, citizenship, place or type of residence or other personal circumstances.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 63 of 96
The Entire Section II of this law is regarding the personal data processing but more relevant articles are:
• Art. 5, according to which, personal data shall be:
o Processed justly and pursuant to law.
o Collected for specific, clear and legally determined purposes and processed in a manner pursuant to those purposes. Further data processing for historic, scientific or statistical research shall not be considered as not being in compliance with the primary purposes for the data collection, provided that the appropriate protection measures have been undertaken in accordance with law.
o Appropriate, relevant and not too extensive in relation to the purposes for collecting and processing.
o Accurate, complete and, where necessary, updated, whereby all proper measures for deleting and correcting the inaccurate or incomplete data shall be undertaken, considering the purposes for which they have been collected or processed and,
o Stored in a form, which enables identification of the personal data subject, not longer than necessary to meet the purposes for which the data have been collected for further processing.
After expiration of the preservation period, the personal data may only be processed for historical, scientific and statistic purposes. The policy for protection of the privacy, personal and family life of the personal data subject from their unauthorized use, shall be applied when personal data are used for the purposes referred to in paragraph 2 of this Article, and in as short term as possible the data shall be made anonymous. The controller shall be responsible for the quality of the personal data in accordance with paragraph 1 of this Article.
• Art. 6, according to which personal data processing may also be performed:
o upon previously obtained consent of the personal data subject;
o for executing the agreement where the personal data subject is contracting party or upon the request of the personal data subject prior to his/her accepting of the agreement;
o for fulfilling the legal obligation of the controller;
o for protection of the life or the essential interests of the personal data subject;
o for exercising activities of public interest or an official authorization of the controller or data being revealed to a third party or
o fulfilment of the legitimate rights of the controller, of a third party or a person to whom the data have been disclosed, unless the freedom and the rights of the personal data subject shall prevail such interests.
The controller shall prove the existence of the consent of the personal data subject, referred to in paragraph 1 line 1 of this Article.
Processing of special categories of personal data is regulated in the Section III of this law. In particular,

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 64 of 96
• Art. 8 of this law says that processing of special categories of personal data shall be forbidden. As an exception to paragraph 1 of this Article the processing of special categories of personal data may be performed:
o On the basis of an explicit consent of the personal data subject given for processing such data, unless a law envisages that the prohibition for processing such data may not be withdrawn by a written consent of the personal data subject.
o If it is necessary for carrying out specific rights and obligations of the controller in the field of labour law, to the extent and with adequate guarantees determined by the laws in this area.
o When it is necessary for the protection of the essential interests of the personal data subject or of other person physically disabled to give consent or lacking the capacity to give consent.
o If the processing is carried out in the framework of the activities of institutions, associations or any non-profit institutions for political, religious, trade-union or other purpose, provided that the data processing refers exclusively to their members or natural persons with whom regular contact regarding their aims are held, such data, as well, shall not be disclosed to third parties without the consent of the personal data subject.
o When the processing refers to data, which the personal data subject has publicly disclosed.
o When it is necessary for the purpose of determining or meeting individual legal interests.
o When it is necessary for the purpose of acquiring, exercising and protecting the rights of the personal data subject in a procedure with competent bodies.
o If it is needed for the purposes of medical prevention, diagnosis, treatment or management of a public health institution and is carried out by a person whose profession is to provide medical protection under oath of secrecy to the data revealed to him/her during the performance of his/her profession.
• Art. 31 states that the personal data transfer to other countries may be carried out only if the other country provides adequate degree of personal data protection.
The Section IV of the Law on Personal Data Protection is defining the rights of the personal data subject. In particular, the following articles are worth underlying.
• Art. 10 states that when data is collected from the personal data subject, the latter must be informed on:
o the identity of the controller and of its authorized representative in the Republic of Macedonia, if any;
o the purposes of the processing;
o the users or categories of users of personal data;
o the compulsoriness of responding to questions;

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 65 of 96
o possible consequences of not responding and
o existence of the right to access and the right to correct his/her personal data.
The controller shall not inform the personal data subject if (s)he has already been introduced to the matters listed in paragraph 1 lines 1 to 6 of this Article.
• Art. 11 says that, when the data are not collected from the personal data subject, the controller shall, at the time of recording the personal data or if disclosure of the personal data to a third party is envisaged, no later than the time when the data are firstly disclosed, inform the personal data subject on:
o the identity of the controller and of his/her authorized representative in the Republic of Macedonia, if any;
o the purposes for the processing;
o the data categories;
o the users or categories of users of the personal data and
o the existence of the right to access and the right to correct the data referring to the personal data subject.
The controller shall not inform the personal data subject if (s)he has already been introduced to the matters referred to in paragraph 1 lines 1 to 5 of this Article.
As an exception to Paragraph 1 of this Article, the controller shall not have an obligation to inform the personal data subject about the processing of personal data for historical, scientific and statistic purposes, if:
• the same is impossible or quests for disproportionate effort or costs or
• the collection or disclosure of the personal data has been determined by law.
According to Art. 12, the personal data subject may request from the controller to inform him/her:
• whether his/her personal data are being processed;
• on the purposes and legal base for personal data processing and the users or categories of users to whom the personal data are being disclosed;
• the logic of automatic processing, in case a decision has been made on the automatic processing affecting the personal data subject.
The controller shall be obliged to respond to the personal data subject referred to in paragraph 1 of this Article, within 15 days as of the day of accepting the request.
Should the controller have responded to the request of the personal data subject referred to in paragraph 1 of this Article, (s)he shall not have the obligation to respond again to a same or similar request of the said subject if, in the meantime, changes in his/her personal data have not occurred, except if six months have passed from the day of submitting the prior request to the new request.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 66 of 96
When the personal data are being processed in accordance with Article 6 paragraph 1 lines 5 and 6 of this Law, the personal data subject shall have the right to request for freezing his/her personal data processing.
• Art. 14 states that upon the request of the personal data subject, the controller is obliged to supplement, amend, delete or prevent the use of the personal data, if they are incomplete, incorrect or not updated and if their processing is not in conformity with the provisions of this Law.
In case when the controller shall determine that the personal data are incomplete, incorrect or not updated, (s)he is obliged to supplement, amend or delete them, regardless of whether the personal data subject has submitted a request for amending the personal data.
For the performed supplement, amendment or deletion of personal data, as pursuant to the paragraph 2 of this Article, the controller shall be obliged within 30 days from the day of accepting the request, to inform in written the personal data subject, the personal data users or third parties to whom the personal data have been disclosed to, unless it is not possible or it quests for disproportional effort or costs.
• According to Art. 23, in order to provide secrecy and protection of the processing of the subject’s personal data, the controller and processor have to apply proper technical and organizational measures for protection of accidental or illegal damaging of the personal data, or their accidental loss, change, unauthorized disclosing or approach, especially when the processing includes transmission of data over a network and protection of any kind of illegal forms of processing.
The personal data referred to in Article 8 and 9 of this Law, may be transferred via electronic telecommunications network only if specially protected by proper methods, therefore not being readable in the transfer process.
The measures referred to in paragraph 1 of this Article have to provide degree of protection of the personal data appropriate to the risk during the processing and the nature of the data being processed.
The controlled and processor shall be obliged to adopt and apply documentation containing description of the technical and organizational measures for providing secrecy and protection of the personal data processing.
• According to Art. 24, only the person with authorization from the controller or processor, including the processors themselves, may provide personal data processing. It is mandatory that the persona referred to in paragraph 1 of this Article shall:
o be introduced with the principles for personal data protection prior to accessing the personal data;
o perform personal data processing in accordance with the directions received from the controlled, unless otherwise regulated and
o preserve the personal data as confidential, as well as the measures for their protection.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 67 of 96
9. Medical devices
According to the Directive 2007/47/EC amending Council Directive 93/42/EEC [3] “medical device means any instrument, apparatus, appliance, software, material or other article, whether used alone or in combination, including the software intended by its manufacturer to be used specifically for diagnostic and/or therapeutic purposes and necessary for its proper application, intended by the manufacturer to be used for human beings for the purpose of:
• Diagnosis, prevention, monitoring, treatment or alleviation of disease,
• Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,
• Investigation, replacement or modification of the anatomy or of a physiological process,
• Control of conception,
and which does not achieve its principal intended action in or on the human body by pharmacological, immunological or metabolic means, but which may be assisted in its function by such means”.
Certification The CE marking indicates a product’s compliance with EU legislation and so enables the free movement of products within the European market. It is mandatory according to the Directive 2007/47/EC amending Council Directive 93/42/EEC [3]. This Directive identifies the steps of the procedure for EC type-examination of medical devices in terms of: 1) Classification of the device; 2) Verification of the compliance with the essential requirements and 3) CE marking.
1) Classification of the device. The classification is the first action that the manufacturer has to make in order to identify the class of the device. Devices shall be divided into Classes I, IIa, IIb and III. Classification shall be carried out in accordance with Annex IX.
2) Verification of the compliance with the essential requirements. To receive the CE marking, Medical Devices must meet the essential requirements sent out in Annex I. As reported in this Annex, “the devices must be designed and manufactured in such a way that, when used under the conditions and for the purposes intended, they will not compromise the clinical condition or the safety of patients, or the safety and health of users or, where applicable, other persons, provided that any risks which may be associated with their intended use constitute acceptable risks when weighed against the benefits to the patient and are compatible with a high level of protection of health and safety. This shall include:
• reducing, as far as possible, the risk of use error due to the ergonomic features of the device and the environment in which the device is intended to be used (design for patient safety), and
• consideration of the technical knowledge, experience, education and training and where applicable the medical and physical conditions of intended users (design for lay, professional, disabled or other users).”

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 68 of 96
According to article 5, “Member States shall presume compliance with the essential requirements referred to Article 3 in respect of devices which are in conformity with the relevant national standards adopted pursuant to the harmonized standards the references of which have been publishes in Official Journal of the European Communities; Member States shall publish the references of such publish the references of such national standards”.
The compliance with the harmonized standards is not mandatory but is presumption of conformity with the essential requirements of the directive. The harmonized standards are emitted by the European Committee for Standardization (CEN) and by the European Committee for Electrotechnical Standardization (CENELEC).
3) CE marking. According to the article 11:
• “in a case of devices falling within Class III, other than devices which are custom-made or intended for clinical investigations, the manufacturer shall, in order to affix the CE marking, either:
o follow the procedure relating to the EC declaration of conformity set out in Annex II (full quality assurance); or
o follow the procedure relating to the EC type-examination set out in Annex III, coupled with:
§ the procedure relating to the EC verification set out in Annex IV; or
§ the procedure relating to the EC declaration of conformity set out in Annex V (production quality assurance).
• In a case of devices falling within Class IIa, other than devices which are custom-made or intended for clinical investigations, the manufacturer shall, in order to affix the CE marking, follow the procedure relating to the EC declaration of conformity set out in Annex VII, coupled with either:
o the procedure relating to the EC verification set out in Annex IV; or
o the procedure relating to the EC declaration of conformity set out in Annex V (production quality assurance); or
o the procedure relating to the EC declaration of conformity set out in Annex VI (product quality assurance).
Instead of applying these procedures, the manufacturer may also follow the procedure referred to in paragraph 3 (a).
• In the case of devices falling within Class IIb, other than devices which are custom-made or intended for clinical investigations, the manufacturer shall, in order to affix the CE marking, either:
o follow the procedure relating to the EC declaration of conformity set out in Annex II (full quality assurance); in this case, point 4 of Annex II is not applicable; or
o follow the procedure relating to the EC type-examination set out in Annex III, coupled with:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 69 of 96
§ the procedure relating to the EC verification set out in Annex IV; or
§ the procedure relating to the EC declaration of conformity set out in Annex V (production quality assurance); or
§ the procedure relating to the EC declaration of conformity set out in Annex VI (product quality assurance).
• In the case of devices falling within Class I, other than devices, which are custom-made or intended for clinical investigations, the manufacturer shall, in order to affix the CE marking, follow the procedure referred to in Annex VII and draw up the EC declaration of conformity required before placing the device on the market.
• In the case of custom-made devices, the manufacturer shall follow the procedure referred to in Annex VIII and draw up the statement set out in that Annex before placing each device on the market.”
10. Other requirements
Electromagnetic compatibility requirements The usage of radio spectrum is regulated at the national, European and international level. All the technologies using radiofrequency transmission to be used in UNCAP operate in the so-called ISM (Industrial, Scientific and Medical) bands.
The ISM bands are portions of the radio spectrum reserved internationally for the use of RF energy for industrial, scientific and medical purposes. Large portions of the ISM bands are unlicensed, i.e., no license is required to operate a device transmitting in such a band. Which implies, conversely, that devices may be operating in a harsh environment characterised by high levels of interference.
The ISM bands are defined by the ITU-R in 5.138, 5.150, and 5.280 of the Radio Regulations [59]. Individual countries' use of the bands designated in these sections may differ due to variations in national radio regulations.
In the EU, low power wireless devices are generally referred to as short-range devices (SRD). The allocation of frequency bands and their use in the EU are based on recommendations by the Electronic Communications Committee (ECC), which is part of the European Conference of Postal and Telecommunication Administration (CEPT). The ECC document covering SRD is ERC/REC 70-03. The 45 member countries of the CEPT must then adopt these recommendations into law for them to be binding, so there are occasionally differences between the member countries.
Electromagnetic Compatibility (EMC) refers to the ability of a device to operate properly in its intended environment without producing excessive interference to other devices. All electronic devices must meet certain regulations regarding EMC. These regulations cover both intentional (for example, transmission signals) and non-intentional (electrical noise) radiation.
Other potential regulatory issues are induced radiation and RF exposure. Induced radiation refers to how well a device withstands unintentional radiation from an external source (e.g. high voltage line or microwave oven). RF exposure regulations, on the other hand, determine if the device emits radiation that is harmful to human

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 70 of 96
beings. This is normally only a concern for high-power transmission devices, but there have been some concerns (yet to be proven) that long-term exposure to even low-levels of electromagnetic radiation could potentially result in cancer and other health problems. For devices that may be positioned within 20 cm of a human body, SAR (Specific Absorption Rate) testing is required to ensure radiation levels are below a certain limit. In Europe, compliance in terms of RF exposure for the kind of devices used within UNCAP is standardized as CENELEC EN 62479:2010, which has been made part of 2006/95/EC directive.
Usability and Inclusiveness
Web accessibility initiative (WAI) The Web Accessibility Initiative (WAI) [60] is an effort to improve the accessibility of the World Wide Web for people with disabilities.
People with disabilities (visual, auditory, physical, speech, cognitive, and neurological) may encounter difficulties when using computers generally, but also on the Web. Since people with disabilities often require non-standard devices and browsers, making websites more accessible also benefits a wide range of user agents and devices, including mobile devices, which have limited resources. WAI develops a series of accessibility standards and guidelines.
The Web is an increasingly important resource in many aspects of life: education, employment, government, recreation, and more. It is essential that the Web be accessible in order to provide equal access and equal opportunity to people with disabilities. An accessible Web can also help people with disabilities more actively participate in society.
Much of the focus on Web accessibility has been on the responsibilities of Web developers. Software needs to help developers produce and evaluate accessible Web sites, and be usable by people with disabilities.
WAI is composed by a series of different accessibility standards and guidelines (WCAG, UAAG, ATAG, WAI-ARIA) [60-64].
Web pages or web-based interfaces WCAG (Web Content Accessibility Guidelines) have been developed with a goal of proving a single shared standard for Web content accessibility that meets the needs of individuals, organizations, and governments internationally. Guidelines addresses the information in a Web site, including text, images, forms, sounds etc.
The guidelines are organized around the following four principles:
• Perceivable - Information and user interface components must be presentable to users in ways they can perceive.
• Operable - User interface components and navigation must be operable.
• Understandable - Information and the operation of user interface must be understandable.
• Robust - Content must be robust enough that it can be interpreted reliably by a wide variety of user agents.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 71 of 96
The WCAG documents explain how to make Web content more accessible to people with disabilities. Web "content" generally refers to the information in a Web page or Web application, including:
• Natural information such as text, images, and sounds, etc.
• Code or mark-up that defines structure, presentation, etc.
WCAG is primarily intended for:
• Web content developers (page authors, site designers, etc.).
• Web tool developers.
• Web accessibility evaluation tool developers.
• Others who want or need a standard for Web accessibility.
The following is a list of guidelines from the Web Content Accessibility Guidelines 2.0 (WCAG 2.0), part of the W3C recommendation.
• Text Alternatives, i.e. to provide text alternatives for any non-text content so that it can be changed into forms people need (large print, braille, speech). It is include short equivalents for images (including icons, buttons, and graphics), description of data represented von charts and diagrams and illustrations or brief descriptions of non-text content such as audio and video files.
• Time-based Media, i.e. to provide alternatives for time-based media. People who cannot hear audio or see video need alternatives. Examples of alternatives for audio and video include text transcripts and captions of audio content, such as recordings of people speaking; audio descriptions, which are narrations to describe important visual details in a video; sign language interpretation of audio content.
• Adaptable, i.e. to create content that can be presented in different ways (for example simpler layout) without losing information or structure. Headings, lists, tables, and other structures in the content are marked-up properly. This allows content to be correctly read aloud, enlarged, or adapted to meet the needs and preferences of the user. For instance, it can be presented using custom colours combinations, text size, or other styling to facilitate reading.
• Distinguishable, i.e. to make it easier for users to see and hear content including separating foreground from background and not used colours as the only way of conveying information or identifying content. If any audio on a Web page plays automatically for more than 3 seconds, a mechanism is available to pause, either stop or change the audio volume. Another point is that captions and images of text and text can be resized without assistive technology up to 200% without loss of content or functionality.
• Keyboard accessible, i.e. all functionalities that are available by mouse have to be made available by keyboard and all functionality available from a keyboard have to made available without requiring specific timings for individual keystrokes.
• Enough Time, i.e. to provide users enough time to read and use content and consent the re-authenticate when a session expires without losing data.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 72 of 96
• Seizures, i.e. not to design content in a way that is known to cause seizures. Content that flashes at certain rates or patterns can cause photo-sensitive reactions, including seizures. Flashing content is ideally avoided entirely, or only used in a way that does not cause known risks.
• Navigable, i.e. to provide ways to help users navigate, find content, and determine where they are. Information about the user's location within a set of Web pages is available and a mechanism is available to allow the purpose of each link to be identified from link text alone. The keyboard focus is visible and the focus order follows a meaningful sequence.
• Readable, to make text content readable and understandable. One example is that the default human language of each Web page can be programmatically determined and use the clearest and simplest language possible, or providing simplified versions. Providing definitions for any unusual words, phrases, idioms, and abbreviations.
• Predictable, to make web pages appear and operate in predictable ways. Many people rely on predictable user interfaces and are disoriented or distracted by inconsistent appearance or behaviour. When any component receives focus, it does not initiate a change of context. Navigational mechanisms that are repeated on multiple Web pages within a set of Web pages occur in the sane relative order each time they are repeated, unless a change is initiated by the user.
• Input Assistance, to help users avoid and correct mistakes. If an input error is automatically detected, the item that is in error is identified and the error is described to the user in text. It helps people who do not understand the functionality, are disoriented or confused, forget, or make mistakes using forms and interaction for any other reason.
• Compatible, to maximize compatibility with current and future user agents (browsers, assistive technologies, and other user agents).
The User Agent Accessibility Guidelines (UAAG) explains how to make user agents accessible to people with disabilities, particularly to increase accessibility to the Web. User agents include Web browsers, media players, and assistive technologies, which are software that some people with disabilities use in interacting with computers. UAAG is primarily for developers of Web browsers, media players, assistive technologies, and other user agents.
Here is a list with a selection of guidelines from the UAAG 2.0, part of the W3C recommendation.
• Render Alternative Content: The user should be able to choose to render any type of recognized alternative content that is present for a content element. This is the case where a person with low vision could find some image painful (e.g. high contrast).
• Text Size, Colour and Font (by Element and/or Globally): The user should be able to set all of the following characteristics of visually rendered text content.
• Speech Rate, Volume, and Voice: If synthesized speech is produced, the user should be able to specify the volume, (independently of other sources of audio).

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 73 of 96
• Allow Zoom: The user should be able to rescale content within top-level graphical view (zoom in at least 500% respect the default size and zoom out less the 10% respect the default size)
• Follow Text Keyboard Conventions: The interface should adopt keyboard conventions for the operating environment.
• Sequential Navigation Between Elements: The user should be able to move the keyboard focus backwards and forwards through all recognized enabled elements in the current view.
• Provide text search: The user should be able to perform a search within rendered content, including rendered text alternatives and rendered generated content, for any sequence of printing characters from the document character set.
• Provide structural navigation: Users should be able to view, navigate, and configure the elements used in navigating hierarchy.
• Ensure that the user interface is understandable: Users should be able to turn off non-essential messages from the author or user-agent.
• Help users avoid and correct mistakes: Users should be able to access submission forms requiring confirmation, to go back after navigating, to have their text checked for spelling errors, undo text entry, to avoid or undo settings changes, and to receive indications of progress activity.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 74 of 96
11. Conclusions
This document is a collection of regulation constraints for:
• Design and development of a system compliant with the current European and national privacy and security directives.
• Implementation of clinical investigations with human subjects designed according to ethics principles.
• Using devices in clinical experimentation in a safe and controlled way
• Obtaining EU certification for UNCAP intended as a medical device.
• Creating a safe device to be used by professional and laypeople.
Ethics
Experimentation with UNCAP should be configured as a clinical investigation with humans using medical devices.
Clinical trials should be compliant with Helsinki declaration and EU directive 2001/20/EC. This directive concerns essentially clinical trials with drugs.
According to Section 2.2 Annex X of the Directive, experimentations with Medical devices must be performed according to the Helsinki declaration. In particular, these prescription apply for UNCAP:
• The primary purpose of medical research involving human subjects is to understand the causes, development and effects of diseases and improve preventive, diagnostic and therapeutic interventions (methods, procedures and treatments). Even the best-proven interventions must be evaluated continually through research for their safety, effectiveness, efficiency, accessibility and quality.
• Some groups and individuals are particularly vulnerable and may have an increased likelihood of being hurt or of incurring additional harm. For this reason, all vulnerable groups and individuals should receive specifically considered protection.
• A clinical trial may be initiated only if the Ethics Committee and/or the competent authority comes to the conclusion that the anticipated therapeutic and public health benefits justify the risks and may be continued only if compliance with this requirement is permanently monitored
• the trial subject or, when the person is not able to give informed consent, his legal representative must give his written consent after being informed of the nature, significance, implications and risks of the clinical trial
• Clinical investigations must be performed based on an appropriate plan of investigation reflecting the latest scientific and technical knowledge and defined in such a way as to confirm or refute the manufacturer's claims for the device; these investigations must include an adequate number of observations to guarantee the scientific validity of the conclusions.
• Clinical investigations must be performed in circumstances similar to the normal conditions of use of the device.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 75 of 96
• The investigations must be performed under the responsibility of a medical practitioner or another authorized qualified person in an appropriate environment. The medical practitioner or other authorized person must have access to the technical and clinical data regarding the device.
• All the appropriate features, including those involving the safety and performances of the device, and its effect on patients must be examined.
• In the case of devices intended for clinical investigations, the manufacturer or the authorised representative, established in the Community, shall notify the competent authorities of the Member States in which the investigations are to be conducted
All the participant countries, including Republic of Macedonia, have adopted the European Directive with no meaningful changes.
Privacy and security
After an in-depth analysis of European and national regulations regarding privacy and data protection, the following conclusions can be drawn:
• The data subject must be informed about aims and modalities of personal data treatment and her/his written consent must be collected in advance.
• Collected data must be used only for the purposes stated in the informed consent
• a notification to the Data Protection Authority is mandatory if the treatment involves: “genetic data, biometric data, or other data disclosing geographic location of individuals or objects by means of an electronic communications network”
• Personal data shall be processed in a way that guarantees a level of security appropriate to the risks presented by the processing and the nature of the data
• In the case of collection, storage and management of sensitive data, in particular data able to disclose the health status of a person (even the location of a person inside a specialist’s room can fall into this category), technical solutions compliant to a minimum common set, extracted from the European and national regulations, should be implemented, in particular:
o Computerized authentication;
o Use of an authorization system, that can allow the user to access to specific resource to pinpoint the authorization profile;
o Regular update of the specifications concerning scope of the processing operations that may be performed by the individual entities in charge of managing and/or maintenance of electronic means;
o Protection of electronic means and data against unlawful data processing operations, unauthorized access and specific software;
o Implementation of procedures for safe keeping backup copies and restoring data and system availability (i.e. back-up copies);

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 76 of 96
o Encryption techniques or identification codes for specific processing operations performed by health care bodies in respect of data disclosing health and sex life.
Medical devices
According to the Directive 2007/47/EC UNCAP is a medical device since it is a “… instrument, apparatus, appliance, software, material or other article, …..intended by the manufacturer to be used for human beings for the purpose of:
• Diagnosis, prevention, monitoring, treatment or alleviation of disease,
• Diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap,…”
To be commercialized, UNCAP needs to receive the CE marking. Steps and regulations to obtain the certification depend on the class of the medical device. The identification of the device class should be identified as soon as possible in order to design the system in accordance with the state of the art represented by the harmonized standards, as reported in chapter 10.
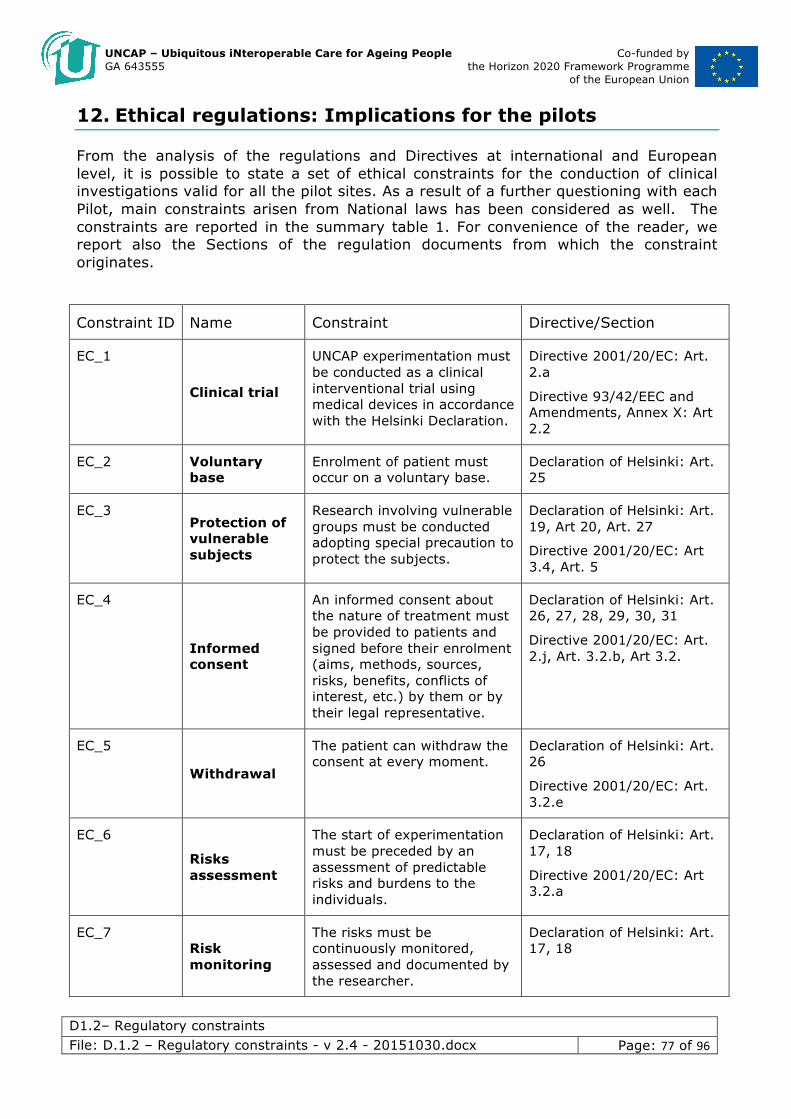
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 77 of 96
12. Ethical regulations: Implications for the pilots
From the analysis of the regulations and Directives at international and European level, it is possible to state a set of ethical constraints for the conduction of clinical investigations valid for all the pilot sites. As a result of a further questioning with each Pilot, main constraints arisen from National laws has been considered as well. The constraints are reported in the summary table 1. For convenience of the reader, we report also the Sections of the regulation documents from which the constraint originates.
Constraint ID Name Constraint Directive/Section
EC_1
Clinical trial
UNCAP experimentation must be conducted as a clinical interventional trial using medical devices in accordance with the Helsinki Declaration.
Directive 2001/20/EC: Art. 2.a
Directive 93/42/EEC and Amendments, Annex X: Art 2.2
EC_2 Voluntary base
Enrolment of patient must occur on a voluntary base.
Declaration of Helsinki: Art. 25
EC_3 Protection of vulnerable subjects
Research involving vulnerable groups must be conducted adopting special precaution to protect the subjects.
Declaration of Helsinki: Art. 19, Art 20, Art. 27
Directive 2001/20/EC: Art 3.4, Art. 5
EC_4
Informed consent
An informed consent about the nature of treatment must be provided to patients and signed before their enrolment (aims, methods, sources, risks, benefits, conflicts of interest, etc.) by them or by their legal representative.
Declaration of Helsinki: Art. 26, 27, 28, 29, 30, 31
Directive 2001/20/EC: Art. 2.j, Art. 3.2.b, Art 3.2.
EC_5
Withdrawal
The patient can withdraw the consent at every moment.
Declaration of Helsinki: Art. 26
Directive 2001/20/EC: Art. 3.2.e
EC_6
Risks assessment
The start of experimentation must be preceded by an assessment of predictable risks and burdens to the individuals.
Declaration of Helsinki: Art. 17, 18
Directive 2001/20/EC: Art 3.2.a
EC_7 Risk monitoring
The risks must be continuously monitored, assessed and documented by the researcher.
Declaration of Helsinki: Art. 17, 18
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 78 of 96
EC_8
Research protocol
The design and performance of the research study must be clearly described and justified in a research protocol.
Declaration of Helsinki: Art. 21, 22
Directive 93/42/EEC and Amendments, Annex X: Art. 2.3.1
EC_9
Ethical committee approval
The research protocol must be submitted for consideration, comment, guidance and approval to the concerned research ethics committee before the study begins.
Declaration of Helsinki: Art. 23
Directive 2001/20/EC: Art 3.2.a, Art. 7
EC_9
Right to privacy
The privacy of the research subjects must be protected and personal information must be kept confidential.
Declaration of Helsinki: Art. 24
Directive 2001/20/EC: Art. 3.2.c
EC_10 Medical device
The UNCAP box shall be treated as a medical device.
Directive 93/42/EEC end Amendments: Art1.a
EC_11
Notification to the competent authority
The manufacturer or the authorised representative, should notify the competent authorities of the Member States in which the investigations are to be conducted.
Directive 93/42/EEC and Amendments: Art. 15
EC_12
Adverse events
All serious adverse events must be fully recorded and immediately notified to the ethical committee all competent authorities of the Member States in which the clinical investigation is being performed.
Declaration of Helsinki: Art. 23
Directive 93/42/EEC and Amendments, Annex X: Art. 2.3.5
EC_13
Responsibility
The investigations must be performed under the responsibility of a medical practitioner or another authorized qualified person in an appropriate environment.
A further specific insurance that covers patients’ risk associated to the clinical investigation is not request, with the exception of Italy.
Directive 93/42/EEC and Amendments, Annex X: Art. 2.3.6
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 79 of 96
EC_14
Continuous Evaluation
Interventions must be evaluated continually through research for their safety, effectiveness, efficiency, accessibility, quality and adverse events.
Declaration of Helsinki: Art. 23
Directive 93/42/EEC and Amendments, Annex X: Art. 2.3.4, Art. 2.3.5
13. Privacy and security regulations: Implications for the architecture and the pilots
As a result of the previous collection of the relevant European and National laws and regulations, the main constraints that UNCAP project may consider are listed below.
System constraints
Constraint ID
Name Constraint Directive/Section
PR_1
Data minimisation/
Proporzionality
The UNCAP system must be designed to minimize the use of personal data and identification data, in such a way as to rule out their processing if the purposes sought in the individual cases can be achieved by using either anonymous data or suitable arrangements to allow identifying data subjects only in cases of necessity
Directive 95/46/EC: Art. 6(1)(b)
PR_2
Data loss/destruction
The UNCAP system must implement preventive security measures in order to minimize the risk of their destruction or loss, whether by accident or not.
Directive 95/46/EC:
Art.17
PR_3
Unauthorized access/processing
The UNCAP system must implement preventive security measures in order to minimize the risk of unauthorized access or processing operations that are either unlawful or
Directive 95/46/EC:
Art.7
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 80 of 96
inconsistent with the data collection purposes.
PR_4
‘Data controllers’ and ‘Data processors’
The UNCAP system shall identify data controllers’ and ‘data processors’, namely the actors who process personal data. In particular, the data controller must take technical and organizational security measures appropriate to the risk presented by the processing.
Directive 95/46/EC: Art. 2 and art.17.
PR _5
Need for encryption
The UNCAP system should use strong encryption to protect sensitive data. In particular, the transfer of the latter should be in encrypted form.
There has been an implementation of encryption techniques or identification codes for specific processing operations performed by health care bodies in respect of data disclosing health and sexual life.
Directive 2002/58/EC; WP29 Opinion 5/2014
PR _6
Encryption key management
The UNCAP system shouldUse an encryption key management solution to protect keys and provide different keys for different data protection needs.
WP29 Opinion 05/2014
PR _7
Appointment of a Data Protection Officer (DPO)
The appointment of a Data Protection Officer (DPO) is not usually required.
The only exception is represented by Germany; a DPO should be appointed in case of more than nine persons
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 81 of 96
permanently engaged in automated data processing (Federal Data Protection Act 1990, as amended (BDSG)).
PR _8
Breach notification
There is not a mandatory requirement to report data security breaches or losses.
Nevertheless, there are new obligations both in Germany and Italy (along with the coming regulation).
Directive 95/46/EC
WP29 Opinion 03/2014
Minimal security measures The UNCAP system shall implement the following security measures:
PR_9
Authentication credentials/access control
The UNCAP system must implement an authentication procedure in such a way to allow the processing of personal data only to persons provided with authentication credentials and after the completion of the procedure.
The minimal authentication credentials must consist of an ID code and a secret password.
The use of an authentication device or a biometric feature that may be associated with either an ID code or a password (strong authentication) is preferred.
PR_10 Management of authentication credentials
The UNCAP system shall implement controls in order to ensure that the password has a length of be at least eight
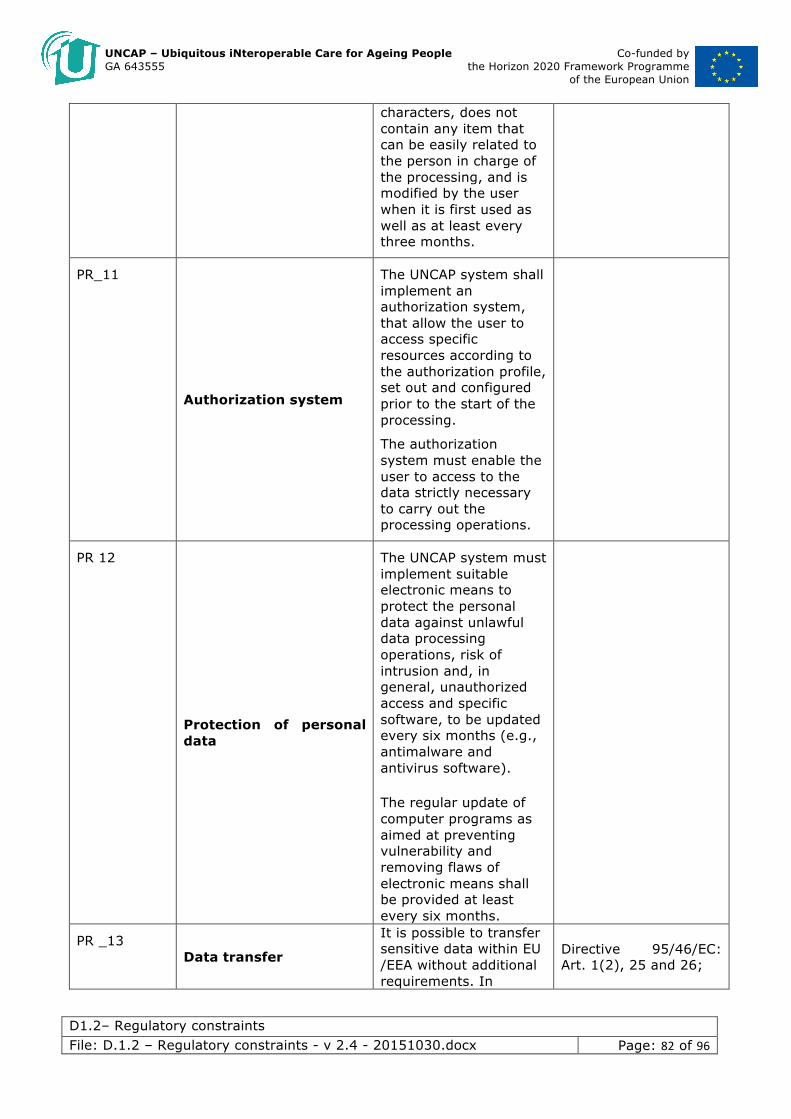
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 82 of 96
characters, does not contain any item that can be easily related to the person in charge of the processing, and is modified by the user when it is first used as well as at least every three months.
PR_11
Authorization system
The UNCAP system shall implement an authorization system, that allow the user to access specific resources according to the authorization profile, set out and configured prior to the start of the processing.
The authorization system must enable the user to access to the data strictly necessary to carry out the processing operations.
PR 12
Protection of personal data
The UNCAP system must implement suitable electronic means to protect the personal data against unlawful data processing operations, risk of intrusion and, in general, unauthorized access and specific software, to be updated every six months (e.g., antimalware and antivirus software).
The regular update of computer programs as aimed at preventing vulnerability and removing flaws of electronic means shall be provided at least every six months.
PR _13 Data transfer
It is possible to transfer sensitive data within EU /EEA without additional requirements. In
Directive 95/46/EC: Art. 1(2), 25 and 26;
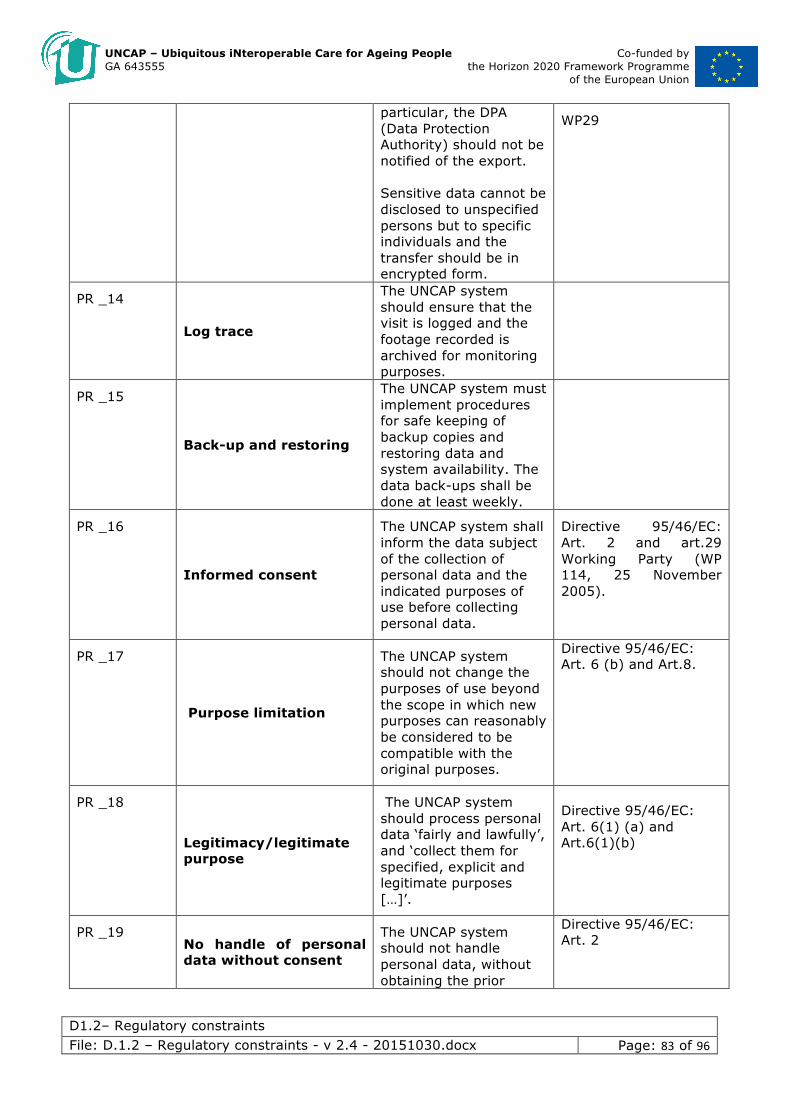
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 83 of 96
particular, the DPA (Data Protection Authority) should not be notified of the export. Sensitive data cannot be disclosed to unspecified persons but to specific individuals and the transfer should be in encrypted form.
WP29
PR _14
Log trace
The UNCAP system should ensure that the visit is logged and the footage recorded is archived for monitoring purposes.
PR _15
Back-up and restoring
The UNCAP system must implement procedures for safe keeping of backup copies and restoring data and system availability. The data back-ups shall be done at least weekly.
PR _16
Informed consent
The UNCAP system shall inform the data subject of the collection of personal data and the indicated purposes of use before collecting personal data.
Directive 95/46/EC: Art. 2 and art.29 Working Party (WP 114, 25 November 2005).
PR _17
Purpose limitation
The UNCAP system should not change the purposes of use beyond the scope in which new purposes can reasonably be considered to be compatible with the original purposes.
Directive 95/46/EC: Art. 6 (b) and Art.8.
PR _18
Legitimacy/legitimate purpose
The UNCAP system should process personal data ‘fairly and lawfully’, and ‘collect them for specified, explicit and legitimate purposes […]’.
Directive 95/46/EC: Art. 6(1) (a) and Art.6(1)(b)
PR _19 No handle of personal data without consent
The UNCAP system should not handle personal data, without obtaining the prior
Directive 95/46/EC: Art. 2
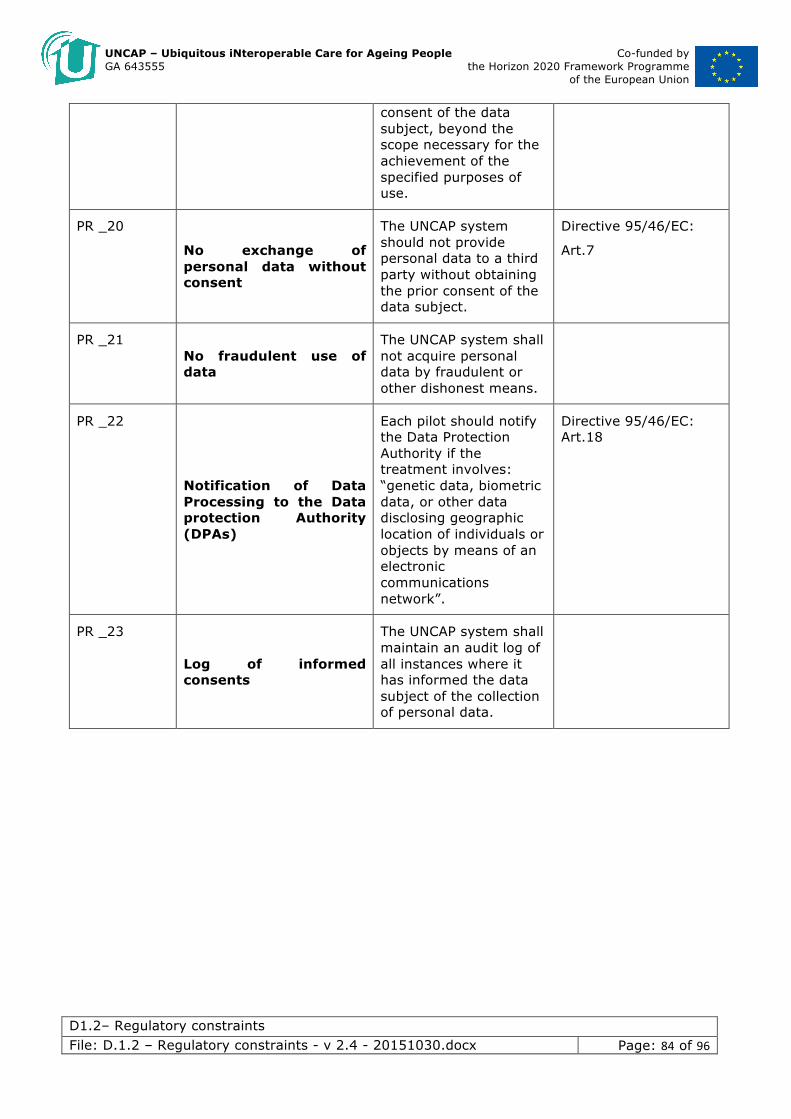
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 84 of 96
consent of the data subject, beyond the scope necessary for the achievement of the specified purposes of use.
PR _20
No exchange of personal data without consent
The UNCAP system should not provide personal data to a third party without obtaining the prior consent of the data subject.
Directive 95/46/EC:
Art.7
PR _21 No fraudulent use of data
The UNCAP system shall not acquire personal data by fraudulent or other dishonest means.
PR _22
Notification of Data Processing to the Data protection Authority (DPAs)
Each pilot should notify the Data Protection Authority if the treatment involves: “genetic data, biometric data, or other data disclosing geographic location of individuals or objects by means of an electronic communications network”.
Directive 95/46/EC: Art.18
PR _23
Log of informed consents
The UNCAP system shall maintain an audit log of all instances where it has informed the data subject of the collection of personal data.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 85 of 96
16. Analysis of the very local regulatory constraints and requirements
As a result of a more detailed analysis of all relevant laws at national and at very local level, viz. restriction at pilot site level, the main constraints that UNCAP system and each Pilot may consider are listed below.
List of Pilots:
• Germany (DE): Höhenkirchen-S. (Bavaria) • Greece (GR): Athens, Thessaloniki • Italy (IT): Pergine (Trentino-Alto Adige), Tarzo (Veneto), Health district “Ovest
Vicentino” (Veneto), Città della Pieve (Umbria) • Macedonia (MK): Skopje • Romania (RO): Baia Sprie (Transylvania), Simleu Silvaniei (Transylvania) • Slovenia (SI): Maribor (Lower Styria)
Data protection, privacy and data sharing: implications for the pilots
The EU Data protection Directive 95/46/EC represents the legislative framework regarding the protection of personal data. The directive has been implemented at national/local levels and some differences may are likely to persist, may slow down UNCAP’s pilot tests.
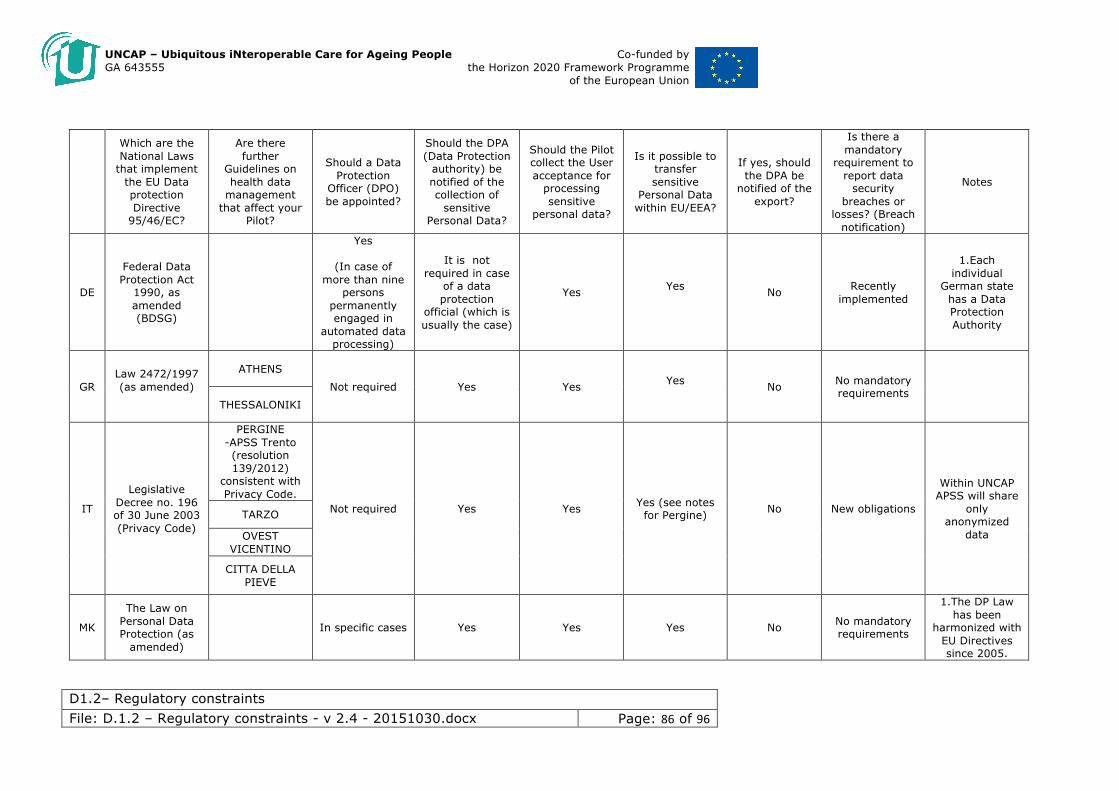
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 86 of 96
Which are the National Laws
that implement the EU Data protection Directive
95/46/EC?
Are there further
Guidelines on health data
management that affect your
Pilot?
Should a Data Protection
Officer (DPO) be appointed?
Should the DPA (Data Protection
authority) be notified of the collection of
sensitive Personal Data?
Should the Pilot collect the User acceptance for
processing sensitive
personal data?
Is it possible to transfer sensitive
Personal Data within EU/EEA?
If yes, should the DPA be
notified of the export?
Is there a mandatory
requirement to report data
security breaches or
losses? (Breach notification)
Notes
DE
Federal Data Protection Act
1990, as amended (BDSG)
Yes
(In case of more than nine
persons permanently engaged in
automated data processing)
It is not required in case
of a data protection
official (which is usually the case)
Yes Yes No Recently
implemented
1.Each individual
German state has a Data Protection Authority
GR Law 2472/1997 (as amended)
ATHENS
Not required Yes Yes Yes No No mandatory
requirements
THESSALONIKI
IT
Legislative Decree no. 196 of 30 June 2003 (Privacy Code)
PERGINE -APSS Trento (resolution 139/2012)
consistent with Privacy Code.
Not required Yes Yes Yes (see notes for Pergine) No New obligations
Within UNCAP APSS will share
only anonymized
data
TARZO
OVEST VICENTINO
CITTA DELLA PIEVE
MK
The Law on Personal Data Protection (as
amended)
In specific cases Yes Yes Yes No No mandatory requirements
1.The DP Law has been
harmonized with EU Directives since 2005.
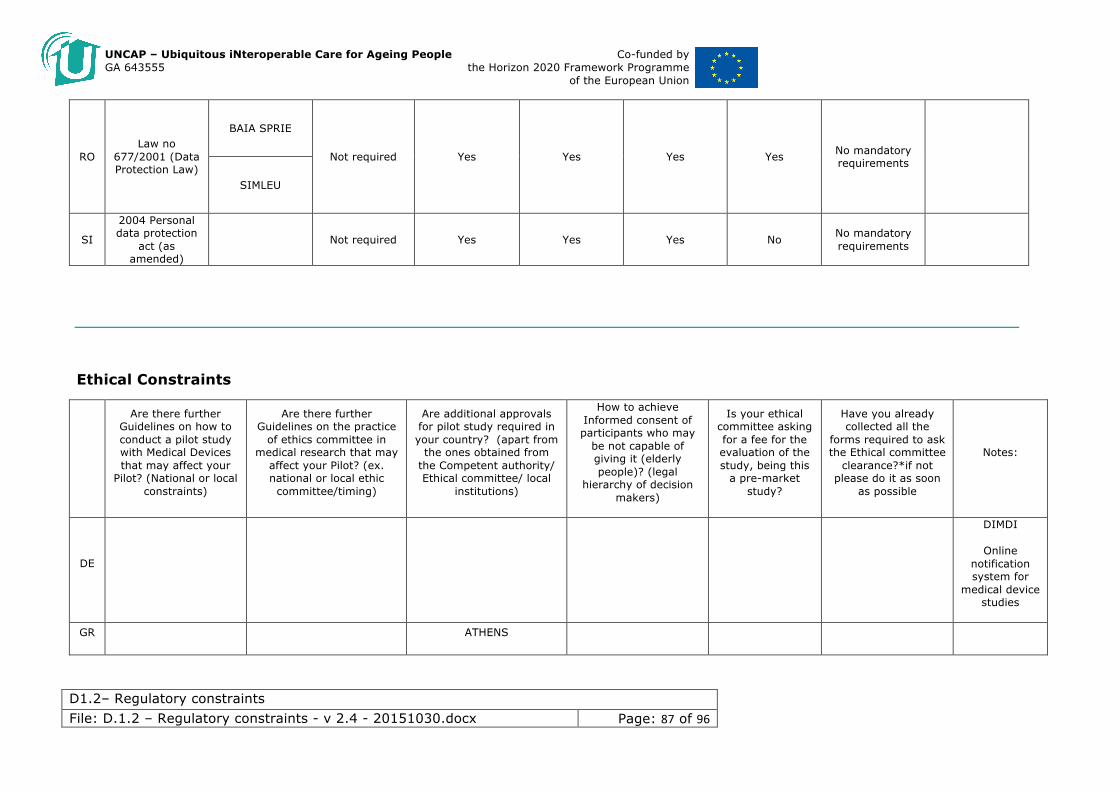
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 87 of 96
RO Law no
677/2001 (Data Protection Law)
BAIA SPRIE
Not required Yes Yes Yes Yes No mandatory requirements
SIMLEU
SI
2004 Personal data protection
act (as amended)
Not required Yes Yes Yes No No mandatory requirements
Ethical Constraints
Are there further Guidelines on how to conduct a pilot study with Medical Devices that may affect your
Pilot? (National or local constraints)
Are there further Guidelines on the practice
of ethics committee in medical research that may
affect your Pilot? (ex. national or local ethic committee/timing)
Are additional approvals for pilot study required in your country? (apart from
the ones obtained from the Competent authority/ Ethical committee/ local
institutions)
How to achieve Informed consent of participants who may
be not capable of giving it (elderly people)? (legal
hierarchy of decision makers)
Is your ethical committee asking for a fee for the evaluation of the study, being this
a pre-market study?
Have you already collected all the
forms required to ask the Ethical committee
clearance?*if not please do it as soon
as possible
Notes:
DE
DIMDI
Online notification system for
medical device studies
GR ATHENS
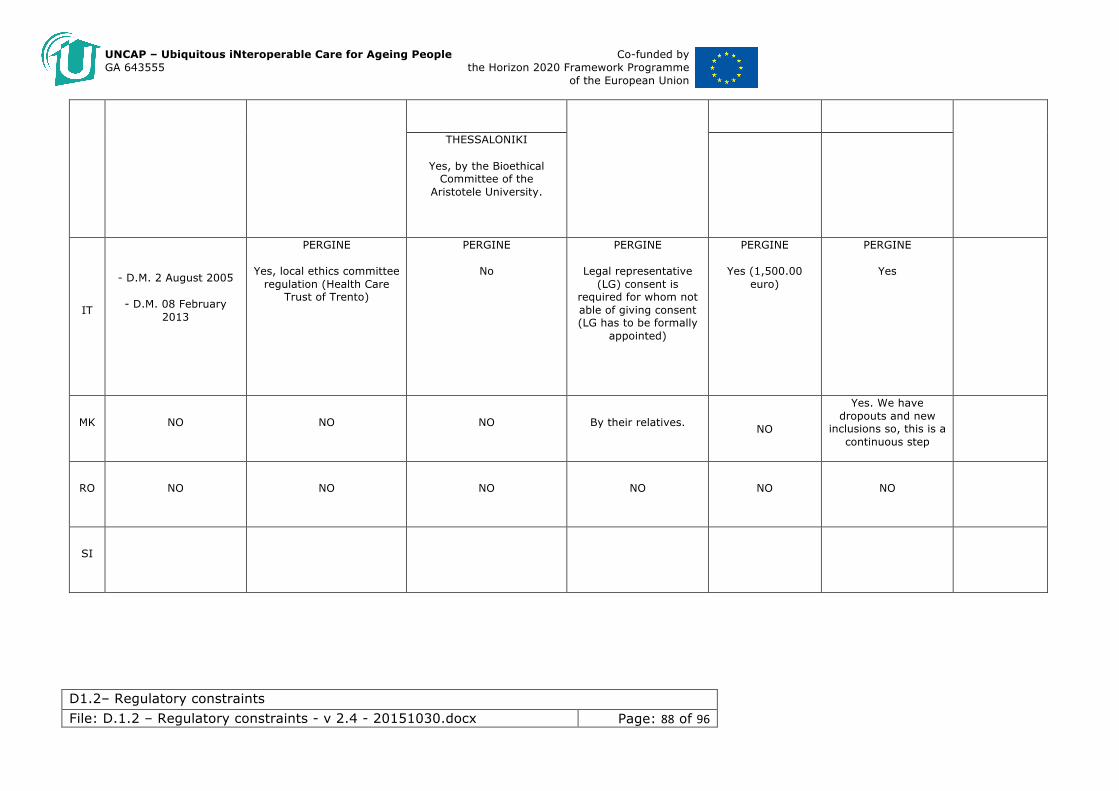
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 88 of 96
THESSALONIKI
Yes, by the Bioethical Committee of the
Aristotele University.
IT
- D.M. 2 August 2005
- D.M. 08 February 2013
PERGINE
Yes, local ethics committee regulation (Health Care
Trust of Trento)
PERGINE
No
PERGINE
Legal representative (LG) consent is
required for whom not able of giving consent (LG has to be formally
appointed)
PERGINE
Yes (1,500.00 euro)
PERGINE
Yes
MK NO NO NO By their relatives.
NO
Yes. We have dropouts and new
inclusions so, this is a continuous step
RO NO NO NO NO NO NO
SI
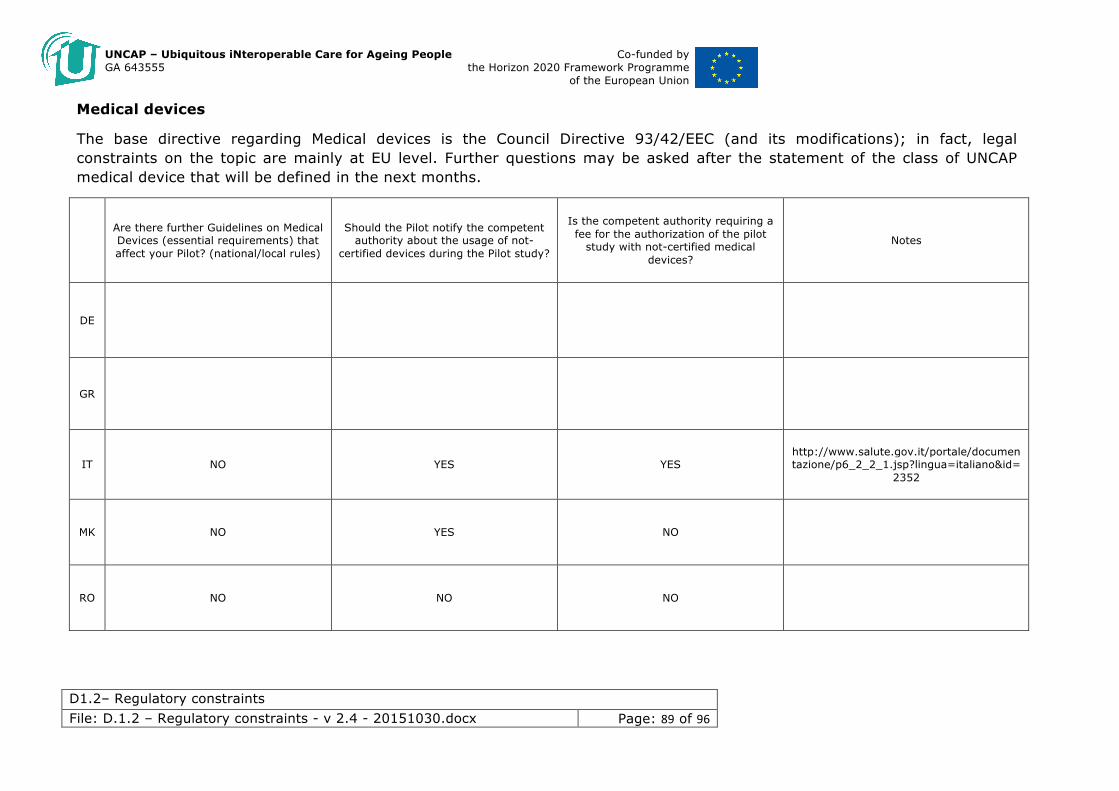
UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 89 of 96
Medical devices
The base directive regarding Medical devices is the Council Directive 93/42/EEC (and its modifications); in fact, legal constraints on the topic are mainly at EU level. Further questions may be asked after the statement of the class of UNCAP medical device that will be defined in the next months.
Are there further Guidelines on Medical Devices (essential requirements) that affect your Pilot? (national/local rules)
Should the Pilot notify the competent authority about the usage of not-
certified devices during the Pilot study?
Is the competent authority requiring a fee for the authorization of the pilot
study with not-certified medical devices?
Notes
DE
GR
IT NO YES YES http://www.salute.gov.it/portale/documentazione/p6_2_2_1.jsp?lingua=italiano&id=
2352
MK NO YES NO
RO NO NO NO

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 90 of 96
SI

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 91 of 96
Annex I Conducting Clinical investigations in Italy
The Department of Planning and Organization of the National Health Service (Il Dipartimento della programmazione e dell'ordinamento del Servizio Sanitario Nazionale) is the regulatory agency within the Italian Ministry of Health that deals with regulating pharmaceuticals, medical devices and other health products. According to the constitutional law n. 3 of 18 October 2001, the department develops and monitors quality assurance systems, coordinates health policies, authorizes and controls medicines, medical devices and other health products, as well as legal medical activities.
Within it, the Directorate General for Medical Devices, Pharmaceutical Services and Safety in Healthcare (La Direzione generale dei dispositivi medici, del servizio farmaceutico e della sicurezza delle cure) deals with the implementation of medical device regulations, preventing and dealing with clinical risk, regulating the advertising of medical products and providing medical-legal consultancy to state institutions.
Italian regulation for clinical investigation on medical devices is based on these main Italian decrees:
• D.Lgs. 507/92 “Attuazione della direttiva 90/385/CEE concernente il ravvicinamento delle legislazioni degli Stati membri relative ai dispositivi medici impiantabili attivi”. Article 7 and Annex 6 and 7, subsequently amended by D.Lgs 37/2010
• D.Lgs. 46/97 “Attuazione della direttiva 93/42/CEE concernente i dispositivi medici”. Article 14 and Annex VIII and X subsequently amended by D.Lgs 37/2010
• D.M. 2 August 2005 “Modalità di presentazione della documentazione per notifica di indagine clinica con dispositivi medici”
• D.M. 12 maggio 2006 “Requisiti minimi per l’istituzione, l’organizzazione, il funzionamento dei Comitati etici per le sperimentazioni cliniche dei medicinali
• Informative Note 27 luglio 2004 on Adverse Event Reporting
• D.M. 15 novembre 2005 “Approvazione dei modelli di schede di segnalazione di incidenti o mancati incidenti”
In Italy, the Clinical evaluation is a general prerequisite to show the compliance of the medical device to the essential requirements (conformity assessment) (D.Lgs. 46/97 and D.M. 37/2010 as transposition of the 2007/47/CE). Since the introduction of the D.Lgs. 37/2010, the clinical evaluation that was mandatory only for Class III devices, is now required also for Class IIa and Class IIb devices.
The aim of the clinical investigation are defined in Attachment X of the D.Lgs. 46/97 and Attachment 7 of the D. Lgs 507/92, and subsequently modified as reported in D.Lgs 25.01.2010 in accordance with Annex X of directive 93/42/EEC .
Article 7 and 14 of the D.Lgs. 46/97 and D. Lgs 507/92, and subsequent amendments (D.Lgs 25.01.2010) regulate:

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 92 of 96
• the procedure for the notification to the Italian Competent Authority (Ministero della Salute),
• the time before starting the clinical investigation,
• the institutions (Hospitals, health care structures) where the clinical investigation can be carried out,
• the communications to be provided to the Competent Authority,
• the costs associated to the notification to the Competent Authority,
• the clinical investigation that do not require a notification to the competent authority,
• the requirements for the Ethical Comittees,
The following clinical investigations must provide a notification to the Italian Competent Authority (Ministero della Salute):
• clinical investigations on medical device without CE marking,
• clinical investigations on medical device with CE marking but subjected to modification of characteristics and/or used out with the indication for use.
For Class III devices, active implantable devices and long term implantable devices in class IIa and IIb, the clinical investigation can start 60 days after the notification to the Italian Competent Authority (Ministero della Salute), providing that within this time frame the Ministero della Salute did not communicate a rejection of the study.
The 60 day period start since the notification has been received from the Ministero della Salute.
Clinical investigation on medical devices different from those mentioned above can start before in case the approval from the Ethical committee has been already obtained.
The Ethical committee interrogation can be made before or contextually to the notification to the Ministero della Salute.
The notification should include a declaration about the execution of the Ethical interrogation and a copy of the Ethical approval should be submitted as soon as available.
Whether a Member state deny or suspend the approval of a clinical investigation, that decision and the motivations is communicated to all EU Member States and to the EU Commission (2007/47/CE Communication among Member States).
After the 60 days time frame is completed, the starting date of the clinical investigation (enrolment of the first patient) should be communicated to the Ministero della Salute by certified or ordinary mail. The clock is stopped if the competent authority requires additional documentations or clarification about the investigation.
The notification is not valid if the payment as defined in the D.M.26 January 2005 (Article 2,item 2,D.M.2 August 2005) has not been performed. Currently the cost for the notification submission is €2160.45.
Similarly, at the conclusion of the study (last patient enrolled), the date of study conclusion should be communicated to the Ministero della Salute by certified or ordinary mail.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 93 of 96
Adverse events (AE), Serious Adverse Events (SAE) and Unanticipated Serious Adverse Events (USADE) related or not related to the investigational device as defined in ISO/FDIS 14155:2011 and in MEDDEV 2.7/3 December 2010 must be notified to the Ministero della salute by the sponsor of the clinical investigation (informative note 27 July 2004 and D. Lgs 15 November 2005 “approvazione dei modelli di schede di segnalazione di incidenti o mancati incidenti”).
Clinical investigations concerning Class III medical devices without CE-mark, implantable and long-term invasive medical devices in Classes IIa and IIb (medical devices to high risk class), can be made in the health-care structures listed at Article 2 of the D.M. 12 March, 2013. The clinical investigations on medical devices without CE-mark in Class I, IIa and IIb, other than implantable devices and long-term invasive devices (low risk class), can be carried out in the facilities listed in Article 3 of DM 12 March, 2013. The clinical investigations relating to active implantable medical devices without CE-mark can be made in the structures listed in Article 2, paragraphs 1 and 2 of the DM 25 June, 2014.
Contents of the notification to the Ministero della Salute
Declaration of the sponsor stating that:
• the sponsor assume the responsibility of the clinical investigation,
• the investigation is conducted according to a specific clinical protocol,
• the investigation is conducted respecting the ethical principles of the Helsinky Declaration, the Good Clinical Practices, the norm ISO 14155:2011,
• the medical device conform to the essential requirements,
• during the clinical investigation, any adverse event will be notified to the Ministero della salute and to the competent Ethical Committee,
• the cost related to the clinical investigation charge on the sponsor and neither on the National Health system, nor on the patient,
• a specific insurance will be stipulated to cover the patients’ risk associated to the clinical investigation,
• a final report, realized by the investigator, will be provided to the Italian Ministry of Health and the competent Ethical committee.
Attachment to the notification (according to D.M. 2 August 2005 “Modalità di presentazione della documentazione per notifica di indagine clinica con dispositivi medici”)
• Clinical Investigator’s Brochure ( according to ICH/GCP ,UNI EN ISO 14155),
• Risk analysis (UNI CEI EN ISO 14971:2004 or similar),
• Indication for the use of the medical device for the investigator (in Italian),
• Essential literature and available scientifi evidences regarding the medical device and the use intended in the clinical investigation. Critical review of the literature signed from the scientific responsible of the sponsor,
• Clinical protocol (according to ICH/GCP,UNI EN ISO 14155) dated and signed by sponsor and clinical investigator,

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 94 of 96
• Copy of the Ethical committee approval, including the listo f the committee members and details of the study evaluation,
• List of the Italian/European and extra-european centers involved in the study, qualification of the clinical investigator, expected timeline of the investigation
• Document designation of the agent to act in the name and on behalf of the manufacturer
• Invoice of the payment of € 2,160.45 for covering the cost of the evaluation by the competent authority;
Ethical principles required for conducting a clinical investigation in Italy:
• Helsinky declaration
• Oviedo European convention
• Good Clinical practice
• Medical Deontology code
Specific notes to the management of the clinical investigation:
• Non CE-marked medical devices provided for clinical investigation are not required to be delivered to the pharmaceutical department but can be delivered directly to the clinical investigator.
• Traceability of the devices should be documented (provide a Device Accountability Log).
• Provide a separated storage of investigational devices from similar devices used for routine diagnosis or patients treatment.
• Possible difficulties in returning failed device.
• Malfunctioning evaluation can be influences by the operator skill and the organizational context.
Procedure of notification to the Italian ministry of health for conducting pre-market clinical investigations
Source: Italian Ministry of Health website: http://www.salute.gov.it/ Accessed at 13 April 2015
Who should submit the notification and ask for the Italian Competent authority evaluation
Manufacturers, authorized representatives or their delegates
What is needed to request the evaluation
1. Notification of the clinical investigation with medical device / active implantable medical device;
2. Invoice of the payment of € 2,160.45;
3. documentation related to the legal representative of the manufacturer (sponsor) or authorized representative (Section 2.6 of the circular of 2 August 2011);

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 95 of 96
4. Declaration of the manufacturer;
5. “procura” in case of delegates of the manufacturer;
6. copy of the ethics committee opinion or declaration of submission of the study to the ethical evaluation;
7. Form for the request of evaluation of clinical investigations with medical devices;
8. Instruction of use of the investigational device with the indication "intended exclusively for clinical investigation;
9. Clinical protocol;
10. Curriculum vitae of the principal investigator with information on clinical trials and clinical experience conducted on the use of medical devices of the same type and class of the device being notified;
11. Declaration of the responsible of the Department which will host the trial concerning the activities of clinical trials reporting any previous clinical activity using medical devices of the same type and class of the device being notified.
For Class III devices, and implantable devices including active and long-term invasive devices in Classes IIa and IIb, the following documentation should also be transmitted:
12. Clinical Investigator's Brochure;
13. Risk analysis document and list of the applied standards, as well as descriptions of the solutions adopted to meet the essential requirements;
For devices in Class I, Class IIa and IIb other than the above, the following document should also be transmitted:
14. Declaration proving the successful evaluation of the risks and the risk minimization measures.
The complete list of documents and forms to be sent are available at http://www.salute.gov.it/portale/ministro/p4_8_0.jsp?lingua=italiano&label=servizionline&idMat=DM&idAmb=SC&idSrv=ICPRE&flag=P
The notification and requested documents should be sent:
• by mail to: Direzione Generale dei Dispositivi Medici, del servizio Farmaceutico e della Sicurezza delle Cure (DGDFSC) - Uff.06 Sperimentazione clinica dei dispositivi medici ex DGFDM. Viale G. Ribotta, 5 00144 Roma
• or by certified e-mail to: [email protected], with subject “DM-SC-ICPRE”
• or by e-mail to: [email protected], with subject “DM-SC-ICPRE” providing digital signature.
How long it takes
Within 60 days from submission of valid notification with complete documentation.

UNCAP – Ubiquitous iNteroperable Care for Ageing People Co-funded by GA 643555 the Horizon 2020 Framework Programme
of the European Union
D1.2– Regulatory constraints File: D.1.2 – Regulatory constraints - v 2.4 - 20151030.docx Page: 96 of 96
The term of 60 days foreseen for the evaluation of the notification by the Ministry of Health is interrupted in case of a request by the Office of supplementary documentation (administrative, technical and scientific) and begins to run again upon receipt of the documentation. The non-receipt of documentation or clarification requested within 90 days will be deemed as a waiver by the sponsor to conduct the clinical investigation.
The authorization for the clinical investigation is communicated to the sponsor with a written note. However tacit assent at the expiration of 60 days will apply if no there is no communication from the Ministry to the sponsor.
Terms of appeals
Against the measures of denial, it is allowed a hierarchical appeal within 30 days to the Director General of the Directorate General of the Ministry of Health or a judicial review by the Regional Administrative Court (TAR) within 60 days.
Payment methods
The payment of the administrative fee for handling the notification should be made by:
• Bank Transfer:
o Account holder Tesoreria Provinciale di Viterbo;
o IBAN: IT24F0760114500000060413416;
o Reason: ref. Article. 5, paragraph 12, L. 407/90; - Request for authorization for clinical investigation with medical device, specifying the title of the study; - Ministry of Health
• Or by post payment
o Account n°: 60413416
o Account holder Tesoreria Provinciale di Viterbo;
o Reason: ref. Article. 5, paragraph 12, L. 407/90; - Request for authorization for clinical investigation with medical device, specifying the title of the study; - Ministry of Health
The results of the notification will be communicated by e-mail or certified e-mail. No publication of the outcome is provided.





























