Embed Size (px)
Citation preview

Database searching

Purposes of similarity search
Function prediction by homology (Function prediction by homology (in silicoin silico annotation)annotation)
Search for identified gene in other organismsSearch for identified gene in other organisms Identifying regulatory elementsIdentifying regulatory elements Assisting in sequence assemblyAssisting in sequence assembly
ProblemsProblems Similar sequences can have different Similar sequences can have different
functionsfunctions Non-homologous sequences can have Non-homologous sequences can have
identical functionidentical function Feature space <> Sequence spaceFeature space <> Sequence space

Some databases
nrnr (GenBank nucleotide and protein) (GenBank nucleotide and protein) Month: monthly updateMonth: monthly update
swissprotswissprot (protein) (protein) ESTEST pdbpdb (proteins with 3D structures) (proteins with 3D structures) Various genome databases (human, Various genome databases (human,
mouse etc)mouse etc)

Main tools FASTAFASTA BLAST=Basic Local Alignment Search BLAST=Basic Local Alignment Search
ToolTool
ProcedureProcedure1.1. Choose scoring matrixChoose scoring matrix2.2. Find best local alignments using scoring Find best local alignments using scoring
matrixmatrix3.3. Determine statistical significance of resultDetermine statistical significance of result
List in decreasing order of significanceList in decreasing order of significance
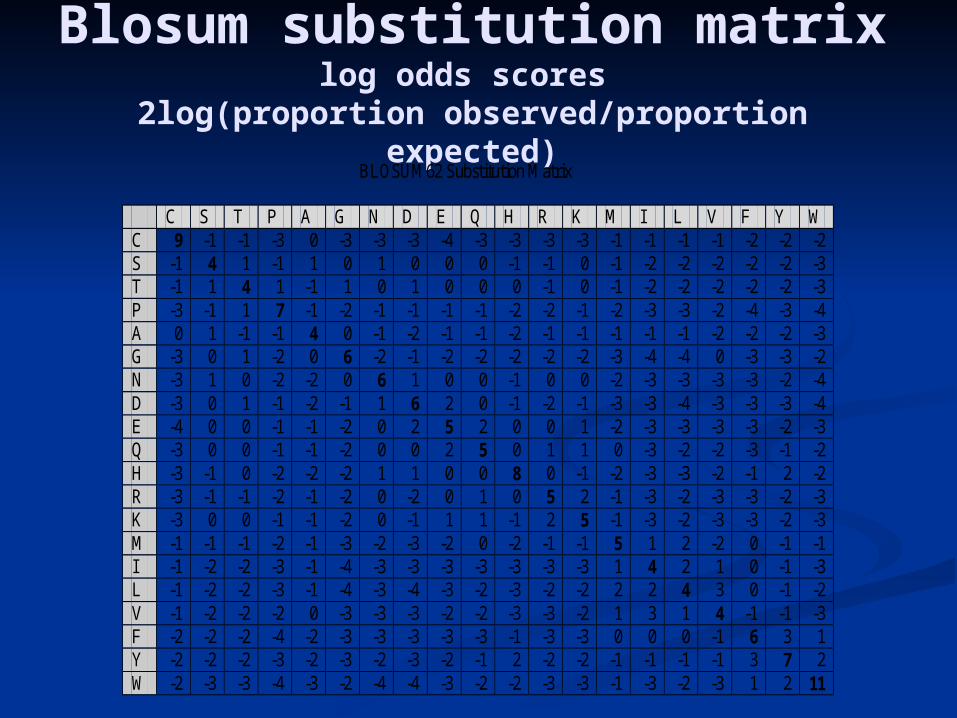
Blosum substitution matrixlog odds scores
2log(proportion observed/proportion expected)
BLOSUM62 Substitution Matrix
C S T P A G N D E Q H R K M I L V F Y W C 9 -1 -1 -3 0 -3 -3 -3 -4 -3 -3 -3 -3 -1 -1 -1 -1 -2 -2 -2 S -1 4 1 -1 1 0 1 0 0 0 -1 -1 0 -1 -2 -2 -2 -2 -2 -3 T -1 1 4 1 -1 1 0 1 0 0 0 -1 0 -1 -2 -2 -2 -2 -2 -3 P -3 -1 1 7 -1 -2 -1 -1 -1 -1 -2 -2 -1 -2 -3 -3 -2 -4 -3 -4 A 0 1 -1 -1 4 0 -1 -2 -1 -1 -2 -1 -1 -1 -1 -1 -2 -2 -2 -3 G -3 0 1 -2 0 6 -2 -1 -2 -2 -2 -2 -2 -3 -4 -4 0 -3 -3 -2 N -3 1 0 -2 -2 0 6 1 0 0 -1 0 0 -2 -3 -3 -3 -3 -2 -4 D -3 0 1 -1 -2 -1 1 6 2 0 -1 -2 -1 -3 -3 -4 -3 -3 -3 -4 E -4 0 0 -1 -1 -2 0 2 5 2 0 0 1 -2 -3 -3 -3 -3 -2 -3 Q -3 0 0 -1 -1 -2 0 0 2 5 0 1 1 0 -3 -2 -2 -3 -1 -2 H -3 -1 0 -2 -2 -2 1 1 0 0 8 0 -1 -2 -3 -3 -2 -1 2 -2 R -3 -1 -1 -2 -1 -2 0 -2 0 1 0 5 2 -1 -3 -2 -3 -3 -2 -3 K -3 0 0 -1 -1 -2 0 -1 1 1 -1 2 5 -1 -3 -2 -3 -3 -2 -3 M -1 -1 -1 -2 -1 -3 -2 -3 -2 0 -2 -1 -1 5 1 2 -2 0 -1 -1 I -1 -2 -2 -3 -1 -4 -3 -3 -3 -3 -3 -3 -3 1 4 2 1 0 -1 -3 L -1 -2 -2 -3 -1 -4 -3 -4 -3 -2 -3 -2 -2 2 2 4 3 0 -1 -2 V -1 -2 -2 -2 0 -3 -3 -3 -2 -2 -3 -3 -2 1 3 1 4 -1 -1 -3 F -2 -2 -2 -4 -2 -3 -3 -3 -3 -3 -1 -3 -3 0 0 0 -1 6 3 1 Y -2 -2 -2 -3 -2 -3 -2 -3 -2 -1 2 -2 -2 -1 -1 -1 -1 3 7 2 W -2 -3 -3 -4 -3 -2 -4 -4 -3 -2 -2 -3 -3 -1 -3 -2 -3 1 2 11

FASTA
Step 1 :Step 1 : Find hot-spots Find hot-spots (i.e. pairs of words of length k) that exactly match. (hashing)
Step 2Step 2: Locate best “diagonal : Locate best “diagonal runs”(sequences of consecutive hot spots runs”(sequences of consecutive hot spots on a diagonal) on a diagonal)
Step 3 : Step 3 : Combine sub-alignments Combine sub-alignments
form diagonal runs into form diagonal runs into
a longer alignmenta longer alignment
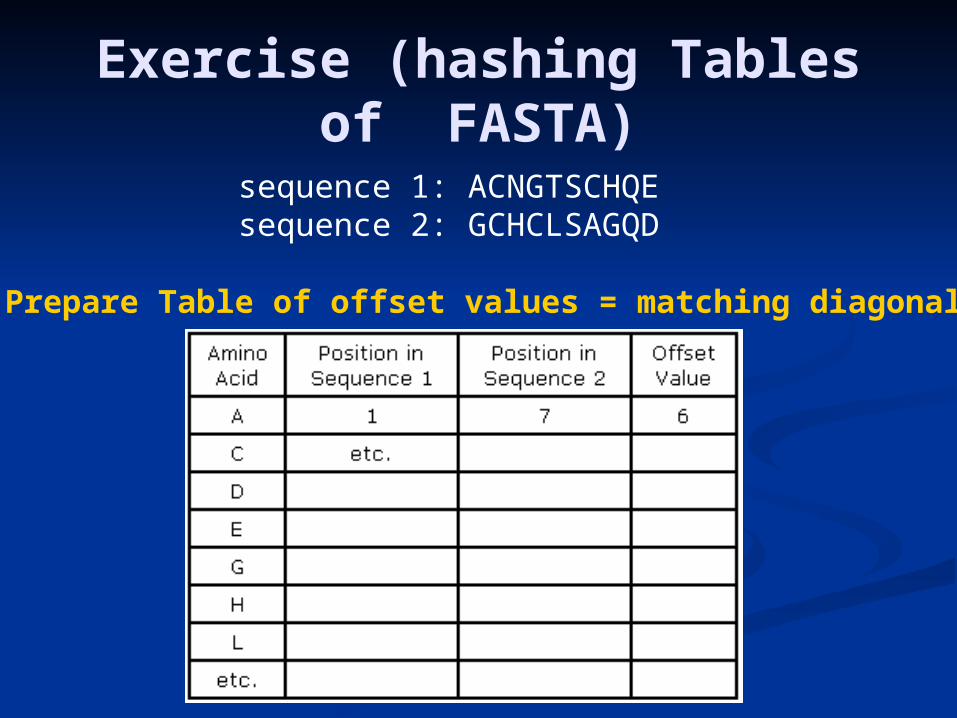
Exercise (hashing Tables of FASTA)
sequence 1: ACNGTSCHQEsequence 2: GCHCLSAGQD
Prepare Table of offset values = matching diagonals
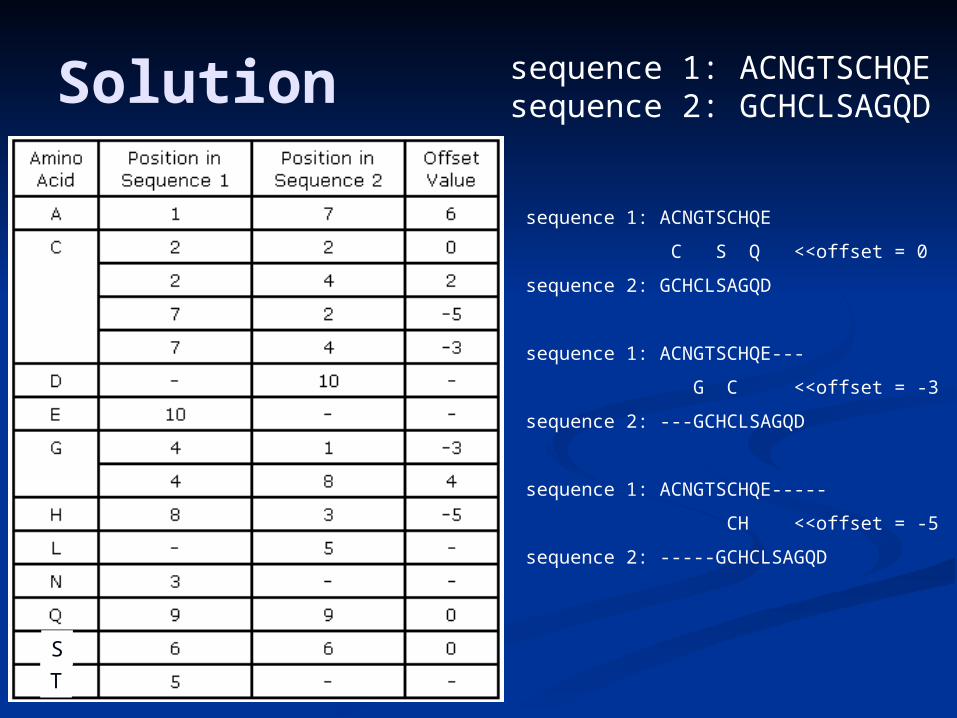
Solution sequence 1: ACNGTSCHQEsequence 2: GCHCLSAGQD
sequence 1: ACNGTSCHQE
C S Q <<offset = 0
sequence 2: GCHCLSAGQD
sequence 1: ACNGTSCHQE---
G C <<offset = -3
sequence 2: ---GCHCLSAGQD
sequence 1: ACNGTSCHQE-----
CH <<offset = -5
sequence 2: -----GCHCLSAGQD
S
T

The main steps of gapped BLAST
1.1. Specify word length (3 for proteins, 11 for Specify word length (3 for proteins, 11 for nucleotides)nucleotides)
2.2. Filtering for complexityFiltering for complexity
3.3. Make list of words to search forMake list of words to search for
4.4. Exact searchExact search
5.5. Join matches, and extend ungapped Join matches, and extend ungapped alignmentalignment
6.6. Calculate E-valuesCalculate E-values
7.7. Join high-scoring pairsJoin high-scoring pairs
8.8. Perform Smith-Waterman on best matchesPerform Smith-Waterman on best matches

Filtering sequences
sequencein typeof residues ofnumber
length sequence
)!/!(log/1 all
in
L
nLLK
i
iiN
Replacing sequence regions of low complexity K with X
Find K for sequence GGGG
and for sequence ATCG
L!= 4*3*2*1 = 24nG = 4, nC = 0, nT = 0, nA = 0ni! = 4! * 0! * 0! * 0! = 24K = ¼ log4 (24/24) = 0
L!= 4*3*2*1 = 24nG = 1, nC = 1, nT = 1, nA = 1ni! = 1! * 1! * 1! * 1! = 1K = ¼ log4 (24/1) = 0.573

The BLAST algorithm Break the search sequence into Break the search sequence into
wordswords WW = 3 for proteins, = 3 for proteins, WW = 12 for DNA = 12 for DNA
Include in the search all words that Include in the search all words that score above a certain value (T) for score above a certain value (T) for any search wordany search word
MCGPFILGTYC
MCG
CGP
MCG, CGP, GPF, PFI, FIL, ILG, LGT, GTY, TYC
MCG CGPMCT MGP …MCN CTP … …

The BLAST search algorithm

Search for the words in the database
Word locations can be precomputed and indexed
Searching for a short string in a long string
Searching the database

Search Significance Scores
A search will A search will alwaysalways return some return some hits.hits.
How can we determine how How can we determine how “unusual” a particular alignment “unusual” a particular alignment score is?score is? ORF’sORF’s
AssumptionsAssumptions
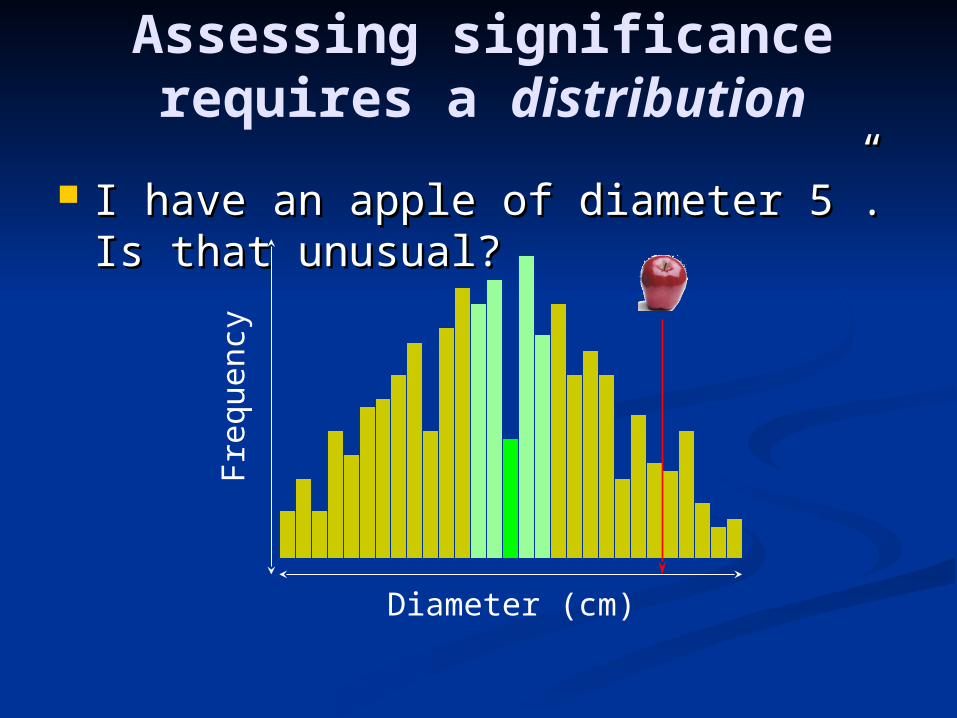
Assessing significance requires a distribution
I have an apple of diameter 5”. Is I have an apple of diameter 5”. Is that unusual?that unusual?
Diameter (cm)
Fre
quen
cy

Is a match significant?
Match scores for aligning my Match scores for aligning my sequence with sequence with random sequencesrandom sequences..
Depends on:Depends on: Scoring systemScoring system DatabaseDatabase Sequence to search forSequence to search for
LengthLength CompositionComposition
How do we determine the How do we determine the random random sequences?sequences?
Match score
Fre
quen
cy

Generating “random” sequences
Random uniform model:Random uniform model:P(G) = P(A) = P(C) = P(T) = 0.25P(G) = P(A) = P(C) = P(T) = 0.25
Doesn’t reflect natureDoesn’t reflect nature Use sequences from a databaseUse sequences from a database
Might have genuine homologyMight have genuine homology We want unrelated sequencesWe want unrelated sequences
Random shuffling of sequencesRandom shuffling of sequences Preserves compositionPreserves composition Removes true homologyRemoves true homology

What distribution do we expect to see?
The The meanmean of of nn random (i.i.d.) events random (i.i.d.) events tends towards a Gaussian tends towards a Gaussian distribution.distribution. Example: Throw Example: Throw nn dice and compute dice and compute
the mean.the mean. Distribution of means:Distribution of means:
n = 2 n = 1000

Determining significance of match
The score of an ungapped alignment is The score of an ungapped alignment is
S = sum s(S = sum s(xxii,y,yii).). The scores of individual sites are The scores of individual sites are
independent. independent. The distribution of the sum of independent The distribution of the sum of independent
random variables is a normal distribution random variables is a normal distribution (central limit theorem). (central limit theorem).

Determining significance of match
However, we don't select scores randomly. We take the However, we don't select scores randomly. We take the maximum extension of the initial word (HSP). maximum extension of the initial word (HSP).
The distribution of the maximum score of a large number The distribution of the maximum score of a large number N N of i.i.d. random variables is called the of i.i.d. random variables is called the extreme value extreme value distribution.distribution.
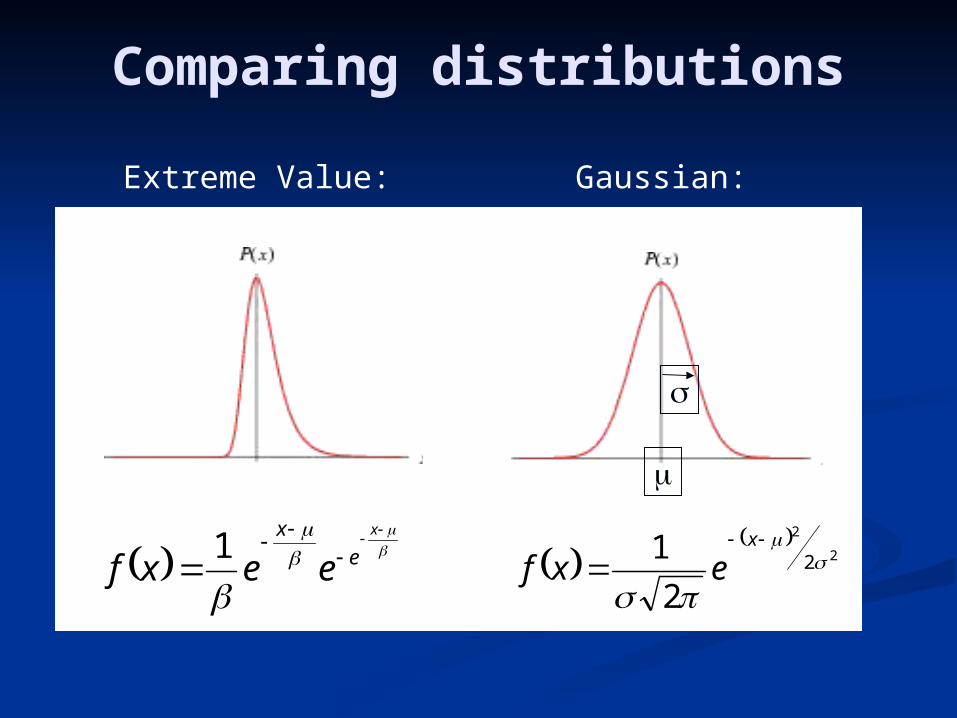
Comparing distributions
x
ex
eexf1
2
2
2
2
1
x
exf
Extreme Value: Gaussian:

Determining P-values If we can estimate If we can estimate and and , then we , then we
can determine, for a given match can determine, for a given match score score xx, the probability that a random , the probability that a random match with score match with score xx or greater would or greater would have occurred in the database.have occurred in the database.
For sequence matches, a scoring For sequence matches, a scoring system and database can be system and database can be parameterized by two parameters, parameterized by two parameters, KK and and , related to , related to and and .. It would be nice if we could compare hit It would be nice if we could compare hit
significance without regard to the significance without regard to the database and scoring system used!database and scoring system used!

)exp(1)( )( uxexSP
/)''log( nKmu
P(Score greater than P(Score greater than xx)=)=Probability of observing Probability of observing a score S > xa score S > x
m’m’ and and nn’ are effective query and database sequence ’ are effective query and database sequence lengths; lengths; KK and and are substitution matrix parameters. are substitution matrix parameters.
P -values

Determining significance of match
E-valueE-value = expected number of sequences = expected number of sequences scoring above scoring above SS in the given database in the given database
Low E-values => significant matchesLow E-values => significant matches When When EE < 0.01 < 0.01 PP-values and -values and EE-values are -values are
nearly identicalnearly identical
BIT-scoreBIT-score: Sum of scores for local : Sum of scores for local alignmentsalignments
DxSpeE )(1

Smith-Waterman local alignment

BLAST parameters Lowering the neighborhood word Lowering the neighborhood word
threshold (T) allows more distantly threshold (T) allows more distantly related sequences to be found, at the related sequences to be found, at the expense of increased noise in the expense of increased noise in the results set.results set.
Raising the segment extension cutoff Raising the segment extension cutoff (X) returns longer extensions for (X) returns longer extensions for each hit.each hit.
Changing the minimum Changing the minimum EE-value -value changes the threshold for reporting a changes the threshold for reporting a hit.hit.

Example – nodulation
Cloned sequence from Lotus japonicusCloned sequence from Lotus japonicus
Amino-acid level (Amino-acid level (BlastPBlastP))
LLANGNFVLRESGNKDQDGLVWQSFDFPTDTLLPQMKLGWDRKTGLNKILRLLANGNFVLRESGNKDQDGLVWQSFDFPTDTLLPQMKLGWDRKTGLNKILRSWKSPSDPSSGYYSYKLEFQGLPEYFLNNRDSPTHRSGPWDGIRFSGIPEKSWKSPSDPSSGYYSYKLEFQGLPEYFLNNRDSPTHRSGPWDGIRFSGIPEK
Nucleotide level (Nucleotide level (BlastNBlastN))cttctcgcta atggcaattt cgtgctaaga gagtctggca acaaagatca agatgggtta cttctcgcta atggcaattt cgtgctaaga gagtctggca acaaagatca agatgggtta gtgtggcaga gtttcgattt tcccactgac actttactcc cgcagatgaa actgggatgg gtgtggcaga gtttcgattt tcccactgac actttactcc cgcagatgaa actgggatgg gatcgcaaaa cagggcttaa caaaatcctc agatcctgga aaagcccaag gatcgcaaaa cagggcttaa caaaatcctc agatcctgga aaagcccaag tgatccgtcaagtgggtatt actcgtataa actcgaattt caagggctcc ctgagtattt tgatccgtcaagtgggtatt actcgtataa actcgaattt caagggctcc ctgagtattt tttaaacaac agagactcgc caactcaccg gagcggtccg tgggatggta tttaaacaac agagactcgc caactcaccg gagcggtccg tgggatggta tccgatttag tggtattccatccgatttag tggtattcca




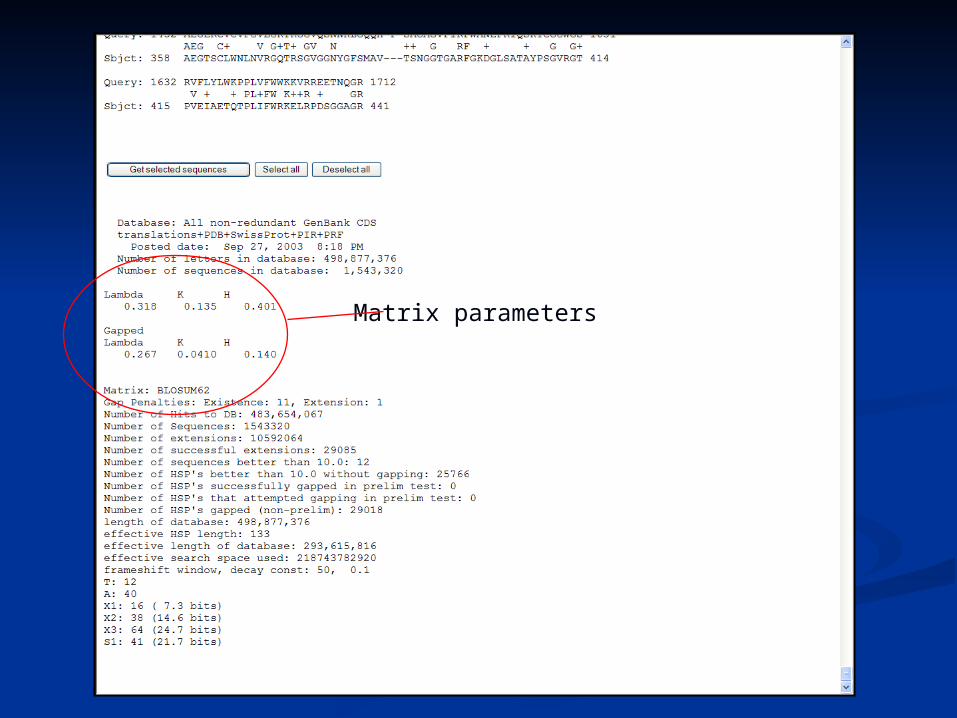
Matrix parameters
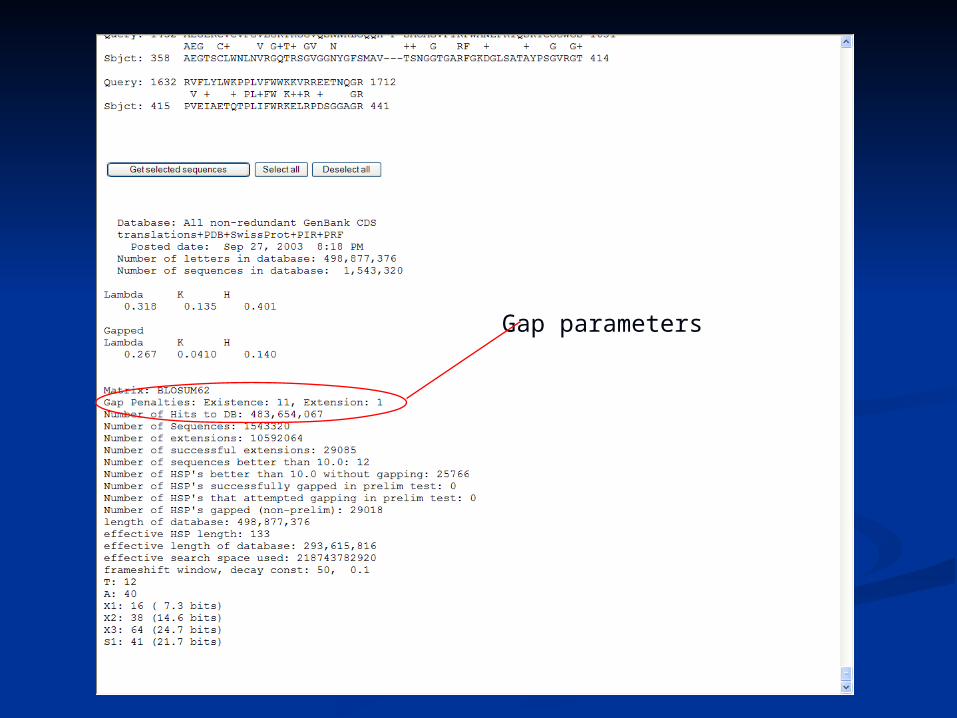
Gap parameters
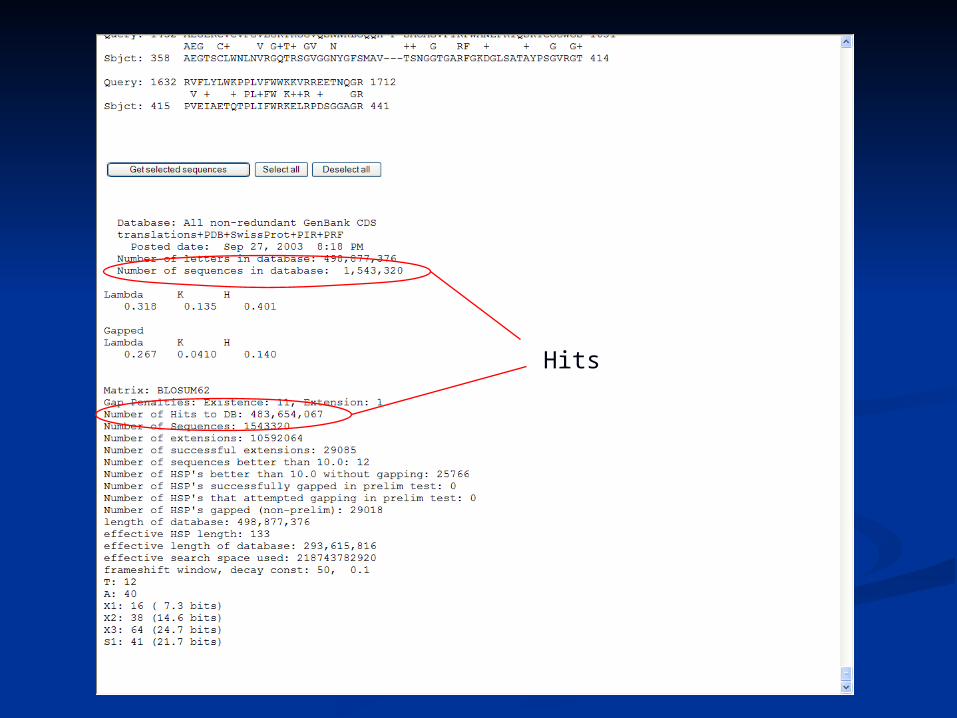
Hits

BLAST flavours
Basic flavoursBasic flavours BLASTP (proteins to protein database)BLASTP (proteins to protein database) BLASTN (nucleotides to nucleotide BLASTN (nucleotides to nucleotide
database)database) BLASTX (translated nucleotides to BLASTX (translated nucleotides to
protein database)protein database) TBLASTN (protein to translated TBLASTN (protein to translated
database)database) TBLASTX (translated nucleotides to TBLASTX (translated nucleotides to
translated database) - SLOWtranslated database) - SLOW
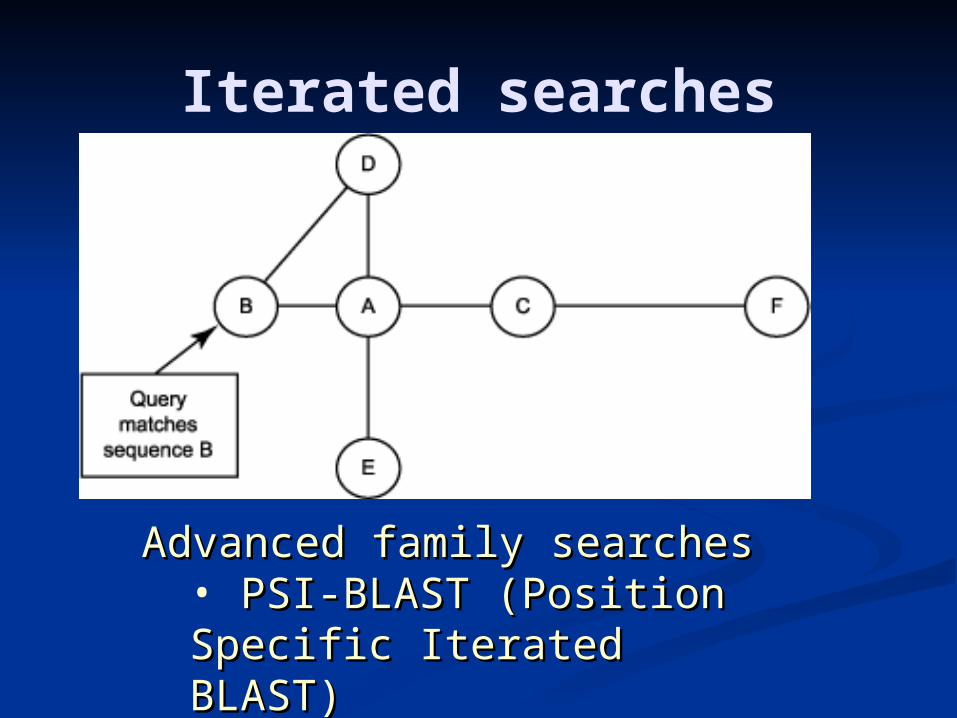
Iterated searches
Advanced family searchesAdvanced family searches• PSI-BLAST (Position PSI-BLAST (Position Specific Iterated BLAST)Specific Iterated BLAST)

PSI-blast
Search with BLAST using the given Search with BLAST using the given query. query.
while (there are new significant hits) while (there are new significant hits) combine all significant hits into a profile combine all significant hits into a profile search with BLAST using the profile search with BLAST using the profile
endend
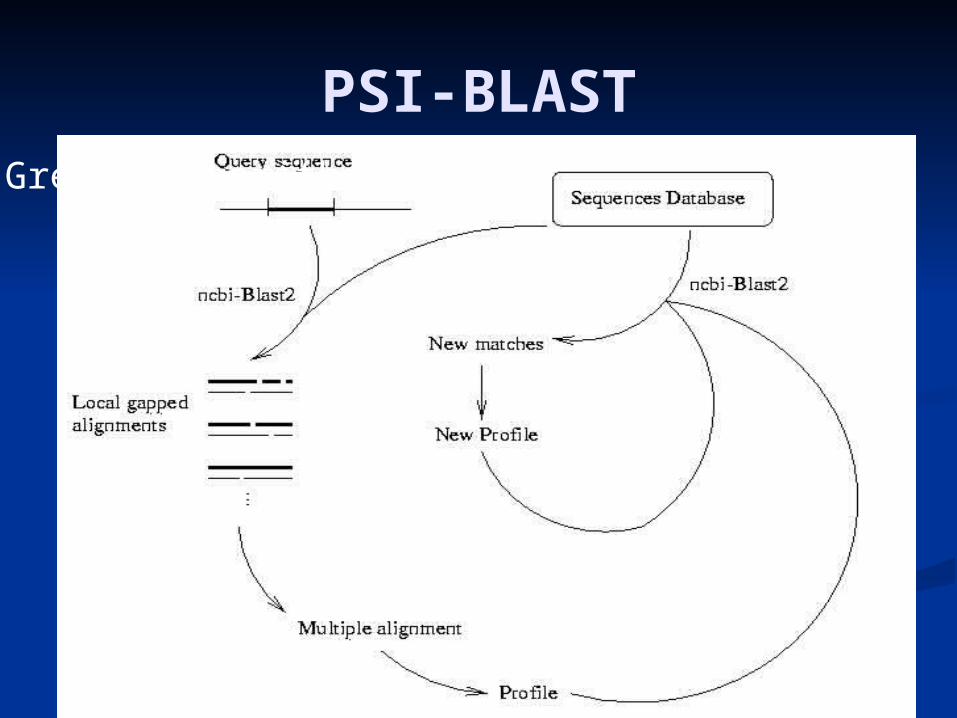
PSI-BLASTGreedy algorithm

Synteny Synteny between between the rat, the rat, mouse and mouse and human human genomes genomes (Nature (Nature 2004) 2004)
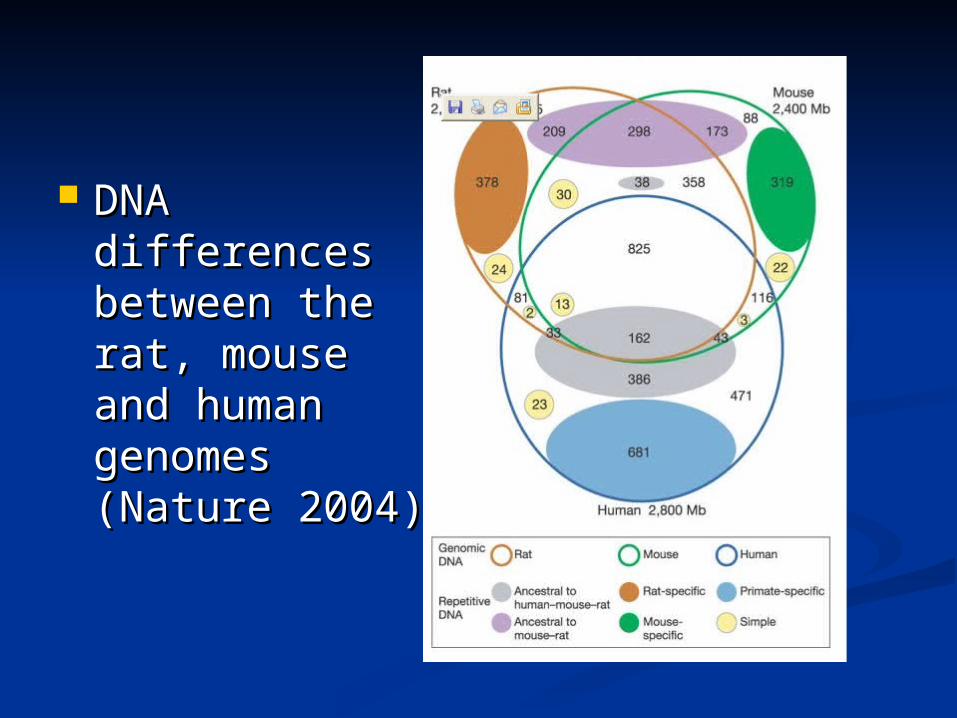
DNA DNA differences differences between the between the rat, mouse and rat, mouse and human human genomes genomes (Nature 2004)(Nature 2004)





























