Embed Size (px)
DESCRIPTION
BioPerf : An Open Benchmark Suite for Evaluating Computer Architecture on Bioinformatics and Life Science Applications. David A. Bader. Collaborators. Vipin Sachdeva (U New Mexico, Georgia Tech, IBM Austin) Tao Li (U Florida) Yue Li (U Florida) Virat Agrawal (IIT Delhi) - PowerPoint PPT Presentation
Citation preview

BioPerf: An Open Benchmark Suite for Evaluating Computer Architecture on Bioinformatics and Life Science ApplicationsDavid A. Bader

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Collaborators
• Vipin Sachdeva (U New Mexico, Georgia Tech, IBM Austin)
• Tao Li (U Florida)• Yue Li (U Florida)• Virat Agrawal (IIT Delhi)• Gaurav Goel (IIT Delhi)• Abhishek Narain Singh (IIT Delhi)• Ram Rajamony (IBM Austin)

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Acknowledgment of Support
• National Science Foundation – CAREER: High-Performance Algorithms for Scientific Applications (06-
11589; 00-93039)– ITR: Building the Tree of Life -- A National Resource for Phyloinformatics
and Computational Phylogenetics (EF/BIO 03-31654)– DEB: Ecosystem Studies: Self-Organization of Semi-Arid Landscapes:
Test of Optimality Principles (99-10123)– ITR/AP: Reconstructing Complex Evolutionary Histories (01-21377)– DEB Comparative Chloroplast Genomics: Integrating Computational
Methods, Molecular Evolution, and Phylogeny (01-20709)– ITR/AP(DEB): Computing Optimal Phylogenetic Trees under Genome
Rearrangement Metrics (01-13095)– DBI: Acquisition of a High Performance Shared-Memory Computer for
Computational Science and Engineering (04-20513).
• IBM PERCS / DARPA High Productivity Computing Systems (HPCS)– DARPA Contract NBCH30390004

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Contributions of this Work
• An open source, freely-available, freely-redistributable suite of applications and inputs, BioPerf, which spans a wide variety of bioinformatics application– www.bioperf.org
• Performance study on PowerPC G5, IBM Mambo simulator, and Alpha

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Outline
• Motivation • Bioinformatics Workload• BioPerf Suite• Performance Analysis on PowerPC G5
and Mambo• Conclusions and Future Work

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Motivation
• Improve performance on a wide range of bioinformatics applications– Heterogeneous in problems, algorithms,
applications• BioPerf workload assembled as a
representative set of bioinformatics applications important now and expected to increase in usage over the next 5—10 years
• Decide if this is YAW “yet another workload” or rather unique in its characteristics

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Related Work
• General benchmark suites: SPEC• Domain-specific benchmarks
– TPC, EEMBC, SPLASH, SPLASH-2• Few benchmark suites for bioinformatics
– Previous attempts have been incomplete: Analysis on old architectures (BioBench) [Albayraktaroglu et al., ISPASS 2005]
– Included proprietary codes in benchmark suite (BioInfoMark) [Li et al., MASCOTS 2005]
– Previous suites not available for download– Included several non-redistributable packages– Inputs not articulated and not included with
benchmark suite for similar comparisons

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Guiding Principles for BioPerf• Coverage: The packages must span the heterogeneity of
algorithms and biological and life science problems important today as well as (in our view) increasing in importance over the next 5-10 years.
• Popularity: Codes with larger numbers of users are preferred because these packages represent a greater percentage of the aggregate workloads used in this domain.
• Open Source: Open source code allows the scientific study of the application performance, the ability to place hooks into the code, and eases porting to new architectures.
• Licensing: Only packages for which their licensing allows free redistribution as open source are included. This requirement eliminated several popular packages, but was kept as a strict requirement to encourage the broadest use of this suite.
• Portability: Preference was given to packages that used standard programming languages and could easily be ported to new systems (both in sequential and parallel languages).
• Performance: We gave slight preference to packages whose performance is well-characterized in other studies. In addition, we strived for computationally-demanding packages and included parallel versions where available.

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
BioPerf Suite
• Pre-compiled binaries (PowerPC, x86, Alpha)• Scalable Input datasets with each code for
fair comparisons• Scripts for installation, running and
collecting outputs• Documentation for compiling and using the
suite• Parallel codes where available• Available for download from
www.bioperf.org

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
BioPerf workloadArea PackageExecutables
Sequence Homology Word-based BLAST blastp, blastn Profile-based HMMER
hmmpfam, hmmsearch Sequence Alignment Pairwise FASTA ssearch, fasta
Multiple CLUSTALW clustalw, clustalw_smp
Multiple TCOFFEE tcoffee
Phylogeny Parsimony/Likelihood PHYLIP
dnapenny, promlk
Gene Rearrangement GRAPPA grappa
Protein Structure Prediction PREDATOR predator
Gene Finding GLIMMERglimmer,glimmer-package
Molecular Dynamics CE ce

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Sequence Alignment
• Sequence Alignment is one of the most useful techniques in computational biology– Sequence Alignment : Stacking the
sequences against each other, with gaps if necessary, to expose similarity. ALIGNMENT
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
MATCH

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Sequence Alignment
• Sequence Alignment is one of the most useful techniques in computational biology– Sequence Alignment : Stacking the
sequences against each other, with gaps if necessary, to expose similarity. ALIGNMENT
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
MISMATCH

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Sequence Alignment
• Sequence Alignment is one of the most useful techniques in computational biology– Sequence Alignment : Stacking the
sequences against each other, with gaps if necessary, to expose similarity. ALIGNMENT
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
S1 : ACGCTGATATTA ACGCTGATAT---TA
S2 : AGTGTTATCCCTA AG--TGTTATCCCTA
“GAPS”

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Multiple Sequence Alignment
• Bring the greatest number of similar characters into same column.
• Provides much more information than pairwise alignment
V S N S
S
N
AA
S
V S N
—
S
S
—
N A —
A S— — —
Run-time of dynamic programming solution = O(2k nk)6 sequences of length 100 6.4X1013 calculationsHence heuristics employed

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Sequence Homology
• Find similar sequences (DNA/protein) to an unknown sequence (DNA/protein).
• Computationally expensive• Size of data is huge and grows exponentially every
year• Public databases available: Genbank, SwissProt, PDB
NCBI Genbank DNA sequences 5 million sequencesSwissprot Protein Sequences 160,000
sequencesPDB Protein Structure 32,000 structures
Problems with computational approach• Exact alignment is O(l2) dynamic programming
solution• Quicker but less accurate heuristics employed
BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Blast
• Basic Local Alignment Search Tool• Developed by NCBI• The most important bioinformatics
application for its popularity
Blastblastpblastn
The homo sapiens hereditary haemochromatosis proteinNon-redundant protein sequence nr developed by NCBI

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
FASTA
• Also performs pairwise sequence alignment
FASTAFasta34ssearch
The human LDL receptor precursor nr

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
ClustalW
• Multiple sequence alignment (MSA) program
ClustalW
ClustalwClustalw_smp
317 Ureaplasma’s gene sequences from NCBI Bacteria genomes database

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
T-Coffee
• A sequential MSA similar to ClustalW with higher accuracy and complexity
T-coffee
Tcoffee
50 sequences of average length 850 extracted from the Prefab database

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Hmmer
• Align multiple sequences by using hidden Markov models
Hmmer hmmsearchhmmpfam
Brine shrimp globin
HMM of 50 aligned globin sequences

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Phylogenetic Reconstruction
• Study the evolution of all sequences and all species
• Find the best among all possible trees.
• Given n taxa, number of possible trees (2n-3)!!
• 10 taxa 2 million trees
• Approaches like maximum parsimony, maximum likelihood, among others
The Tree of Life (10-100M organisms)

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Phylogeny Reconstruction: Phylip
• Collection of programs for inferring phylogenies
• Methods include– Maximum parsimony – Maximum likelihood – Distance based methods.
• Input: Aligned dataset of 92 cyclophilins proteins of eukaryotes each of length 220
BioPerf: an open bioinformatics and life sciences workload, David A. Bader
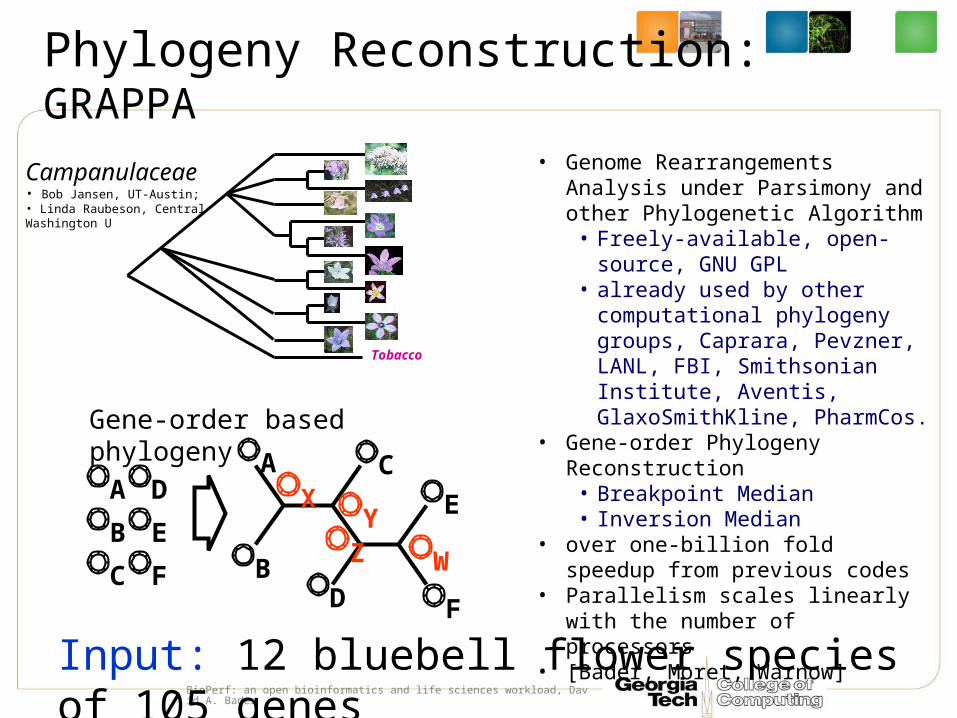
Phylogeny Reconstruction: GRAPPA
• Genome Rearrangements Analysis under Parsimony and other Phylogenetic Algorithm
• Freely-available, open-source, GNU GPL
• already used by other computational phylogeny groups, Caprara, Pevzner, LANL, FBI, Smithsonian Institute, Aventis, GlaxoSmithKline, PharmCos.
• Gene-order Phylogeny Reconstruction• Breakpoint Median• Inversion Median
• over one-billion fold speedup from previous codes
• Parallelism scales linearly with the number of processors
• [Bader, Moret, Warnow]
Tobacco
Campanulaceae• Bob Jansen, UT-Austin;• Linda Raubeson, Central Washington U
Input: 12 bluebell flower species of 105 genes
A
B
C
D
E
F
A
B
C
D
E
F
XYZ W
Gene-order based phylogeny

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Protein Structure Prediction
• Find the sequences, three dimensional structures and functions of all proteins and vice-versa– Why computationally?
• Experimental Techniques slow and expensive
– Problems with computational approach• Little understanding of how structure develops• Does function really follow structure ?

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Protein Structure : Predator
• Tool for finding protein structures• Relies on local alignments from
BLAST, FASTA• Input: 20 sequences from Swissprot
each of length about 7000 residues.

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
CE (Combinatorial Extension)
• Find structural similarities between the primary structures of pairs of proteins
CE ce
Two different types of hemoglobin which is used to transport oxygen

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Gene-Finding: Glimmer
• Gene-Finding: Find regions of genome which code for proteins
• Widely used gene finding tool for microbial DNA
• Input: Bacteria genome consisting of 9.2 million base pairs

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Pre-compiled binaries
• PowerPC• x86• Alpha

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
BioPerf Performance Studies
• Analysis at the instruction and memory level on PowerPC
• Livegraph data helps to visualize performance as it varies during phases of a run
• Identify bottlenecks of current processors and make inputs for better performance on future processors
• Ongoing work using Mambo simulator (IBM PERCS)
• Pre-compiled Alpha binaries for the majority of benchmarks for simulation
• In order to reduce the simulation time, we collect the simulation points for those benchmarks by using SimPoint

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Conclusions
• Bioinformatics is a rapidly evolving field of increasing importance to computing
• BioPerf is a first step to characterize bioinformatics workload: infrastructure to evaluate performance
• Performance data collected so far provides insight into the limitations of current architectures

BioPerf: an open bioinformatics and life sciences workload, David A. Bader
Related Publications• D.A. Bader, V. Sachdeva, A. Trehan, V. Agarwal, G. Gupta,
and A.N. Singh, “BioSPLASH: A sample workload from bioinformatics and computational biology for optimizing next-generation high-performance computer systems,” (Poster Session), 13th Annual International Conference on Intelligent Systems for Molecular Biology (ISMB 2005), Detroit, MI, June 25-29, 2005.
• D.A. Bader, V. Sachdeva, “BioSPLASH: Incorporating life sciences applications in the architectural optimizations of next-generation petaflop-system,”(Poster Session), The 4th IEEE Computational Systems Bioinformatics Conference (CSB 2005), Stanford University, CA, August 8-11, 2005
• D.A. Bader, Y. Li, T. Li, V. Sachdeva, “BioPerf: A Benchmark Suite to Evaluate High-Performance Computer Architecture on Bioinformatics Applications,” The IEEE International Symposium on Workload Characterization (IISWC 2005), Austin, TX, October 6-8, 2005





























