Embed Size (px)
Citation preview

Density Functionals that Recognize Covalent, Metallic, and Weak Bonds
Jianwei Sun,1,* Bing Xiao,1 Yuan Fang,2 Robin Haunschild,3 Pan Hao,2 Adrienn Ruzsinszky,1
Gabor I. Csonka,4 Gustavo E. Scuseria,3,5,6 and John P. Perdew1
1Department of Physics, Temple University, Philadelphia, Pennsylvania 19122, USA2Department of Physics and Engineering Physics and Quantum Theory Group, Tulane University,
New Orleans, Louisiana 70118, USA3Department of Chemistry, Rice University, Houston, Texas 77005, USA
4Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics,H-1521 Budapest, Hungary
5Department of Physics and Astronomy, Rice University, Houston, Texas 77005, USA6Chemistry Department, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
(Received 12 January 2013; published 4 September 2013)
Computationally efficient semilocal approximations of density functional theory at the level of the local
spin density approximation (LSDA) or generalized gradient approximation (GGA) poorly describe weak
interactions. We show improved descriptions for weak bonds (without loss of accuracy for strong ones)
from a newly developed semilocal meta-GGA (MGGA), by applying it to molecules, surfaces, and solids.
We argue that this improvement comes from using the right MGGA dimensionless ingredient to recognize
all types of orbital overlap.
DOI: 10.1103/PhysRevLett.111.106401 PACS numbers: 71.15.Mb, 31.15.E�, 73.22.Pr, 87.15.A�
Because of its computational efficiency and reasonableaccuracy, the Kohn-Sham density functional theory [1–3]with semilocal approximations to the exchange-correlationenergy, e.g., the local spin density approximation (LSDA)[4,5] and the standard Perdew-Burke-Ernzerhof (PBE)generalized gradient approximation (GGA) [6], is one ofthe most widely used electronic structure methods in ma-terials science, surface science, condensed matter physics,and chemistry. Semilocal approximations display a well-understood error cancellation between exchange andcorrelation in bonding regions. Thus, some intermediate-range correlation effects, important for strong and weakbonds, are carried by the exchange part of the approxima-tion. However, it is well known that these approximationscannot yield correct long-range asymptotic dispersionforces [7]. This raises doubts about the suitability ofsemilocal approximations for the description of weak inter-actions (including hydrogen bonds and van der Waalsinteractions), even near equilibrium where interestingproperties occur. These doubts are supported by the per-formance of LSDA and GGAs, which are not very usefulfor many important systems and properties (such as DNA,physisorption on surfaces, most biochemistry, etc.).
However, these doubts are challenged by recent devel-opments in semilocal meta-GGAs (MGGA) [8–14] (whichare useful by themselves and as ingredients of hybridfunctionals [14]). Compared to GGAs, which use thedensity nðrÞ and its gradient rn as inputs, MGGAs addi-tionally include the positive kinetic energy density� ¼ P
kjrc kj2=2 of the occupied orbitals c k. For simplic-ity, we suppress the spin here. By including training setsof noncovalent interactions, the molecule-oriented and
heavily parametrized Minnesota functional M06LMGGA was trained to capture medium-range exchangeand correlation energies that dominate equilibrium struc-tures of noncovalent complexes [9]. Madsen et al. showedthat the inclusion of the kinetic energy densities enablesMGGAs to discriminate between dispersive and covalentinteractions, which makes the M06L MGGA [9] suitablefor layered materials bonded by van der Waals interactions[15,16]. Besides improvement for noncovalent bonds,simultaneous improvement for metallic and covalent bondsis also an outstanding problem for semilocal functionals[17,18]. Reference [18] has shown that the revisedTao-Perdew-Staroverov-Scuseria (revTPSS) [10] MGGA,due to the inclusion of the kinetic energy density, simulta-neously predicts accurate results for the adsorption energyof CO on the Pt (111) surface and the lattice constant andsurface energy of the substrate, while GGA and LSDA [18]do not. These successful applications show the advantagesand flexibility brought by inputting the kinetic energydensity for MGGAs. The effect of the dependence on thekinetic energy density has also been studied recently inRefs. [11,14], leading to a new MGGA called MGGAmade simple (MGGA_MS2) [14]. However, importantquestions for the development of MGGAs and thus forunderstanding the results from MGGAs remain unan-swered: How should the kinetic energy density be builtinto MGGAs? Why? And what are the consequences?To answer these questions, let us look at the input,
the positive kinetic energy density, and how it is used inthe M06L, revTPSS, and MGGA_MS2 MGGAs. � can beexpressed explicitly in terms of the density nðrÞ for twokinds of paradigm systems. For one- and two-electron
PRL 111, 106401 (2013) P HY S I CA L R EV I EW LE T T E R Sweek ending
6 SEPTEMBER 2013
0031-9007=13=111(10)=106401(5) 106401-1 � 2013 American Physical Society
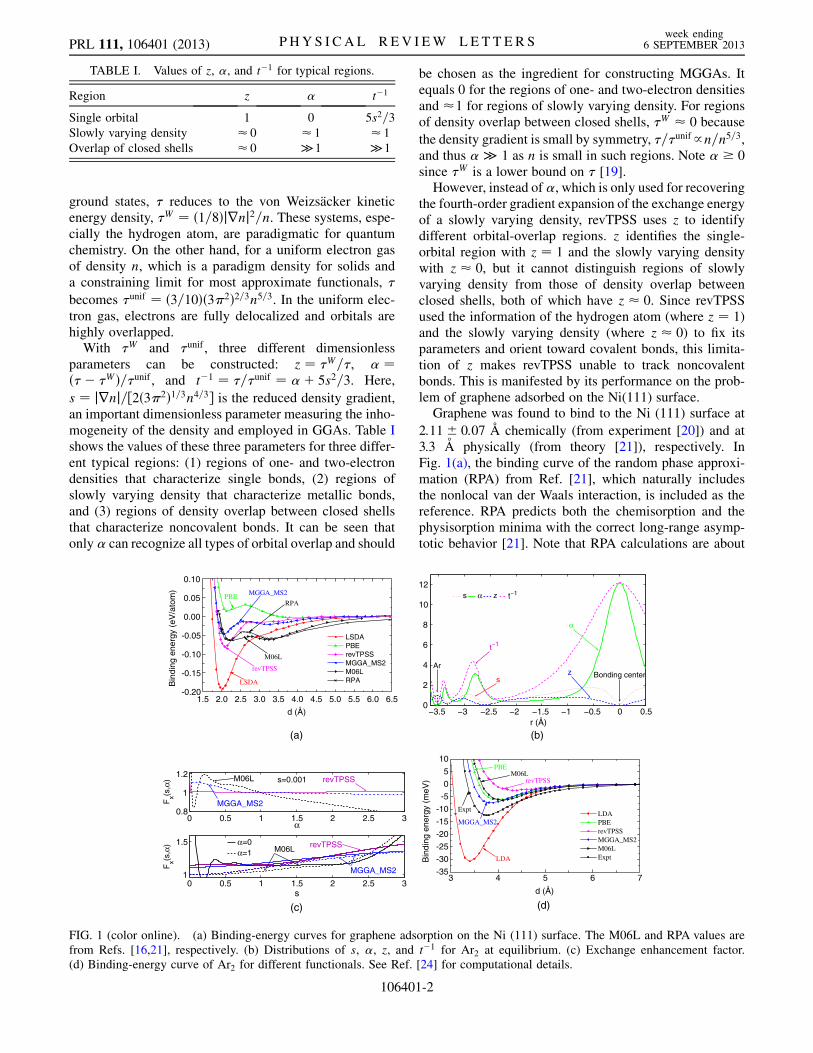
ground states, � reduces to the von Weizsacker kineticenergy density, �W ¼ ð1=8Þjrnj2=n. These systems, espe-cially the hydrogen atom, are paradigmatic for quantumchemistry. On the other hand, for a uniform electron gasof density n, which is a paradigm density for solids anda constraining limit for most approximate functionals, �
becomes �unif ¼ ð3=10Þð3�2Þ2=3n5=3. In the uniform elec-tron gas, electrons are fully delocalized and orbitals arehighly overlapped.
With �W and �unif , three different dimensionlessparameters can be constructed: z ¼ �W=�, � ¼ð�� �WÞ=�unif , and t�1 ¼ �=�unif ¼ �þ 5s2=3. Here,
s ¼ jrnj=½2ð3�2Þ1=3n4=3� is the reduced density gradient,an important dimensionless parameter measuring the inho-mogeneity of the density and employed in GGAs. Table Ishows the values of these three parameters for three differ-ent typical regions: (1) regions of one- and two-electrondensities that characterize single bonds, (2) regions ofslowly varying density that characterize metallic bonds,and (3) regions of density overlap between closed shellsthat characterize noncovalent bonds. It can be seen thatonly� can recognize all types of orbital overlap and should
be chosen as the ingredient for constructing MGGAs. Itequals 0 for the regions of one- and two-electron densitiesand �1 for regions of slowly varying density. For regionsof density overlap between closed shells, �W � 0 because
the density gradient is small by symmetry, �=�unif/n=n5=3,and thus � � 1 as n is small in such regions. Note � � 0since �W is a lower bound on � [19].However, instead of�, which is only used for recovering
the fourth-order gradient expansion of the exchange energyof a slowly varying density, revTPSS uses z to identifydifferent orbital-overlap regions. z identifies the single-orbital region with z ¼ 1 and the slowly varying densitywith z � 0, but it cannot distinguish regions of slowlyvarying density from those of density overlap betweenclosed shells, both of which have z � 0. Since revTPSSused the information of the hydrogen atom (where z ¼ 1)and the slowly varying density (where z � 0) to fix itsparameters and orient toward covalent bonds, this limita-tion of z makes revTPSS unable to track noncovalentbonds. This is manifested by its performance on the prob-lem of graphene adsorbed on the Ni(111) surface.Graphene was found to bind to the Ni (111) surface at
2:11� 0:07 �A chemically (from experiment [20]) and at3.3 A physically (from theory [21]), respectively. InFig. 1(a), the binding curve of the random phase approxi-mation (RPA) from Ref. [21], which naturally includesthe nonlocal van der Waals interaction, is included as thereference. RPA predicts both the chemisorption and thephysisorption minima with the correct long-range asymp-totic behavior [21]. Note that RPA calculations are about
TABLE I. Values of z, �, and t�1 for typical regions.
Region z � t�1
Single orbital 1 0 5s2=3Slowly varying density � 0 � 1 � 1Overlap of closed shells � 0 �1 �1
1.5 2.0 2.5 3.0 3.5 4.0 4.5 5.0 5.5 6.0 6.5-0.20
-0.15
-0.10
-0.05
0.00
0.05
0.10
revTPSS
M06L
RPA
Bin
ding
ene
rgy
(eV
/ato
m)
d (Å)r (Å)
LSDA PBE revTPSS MGGA_MS2 M06L RPALSDA
PBEMGGA_MS2
(a)
−3.5 −3 −2.5 −2 −1.5 −1 −0.5 0 0.50
2
4
6
8
10
12s α z t−1
ArBonding centerz
s
t−1
α
(b)
0 0.5 1 1.5 2 2.5 30.8
1
1.2
α
Fx(s
,α)
0 0.5 1 1.5 2 2.5 31
1.5
s
Fx(s
,α) α=0
α=1
revTPSS
revTPSSM06L
MGGA_MS2
M06L
MGGA_MS2
s=0.001
(c) (d)
3 4 5 6 7-35
-30
-25
-20
-15
-10
-5
0
5
10
Bin
ding
ene
rgy
(meV
)
d (Å)
LDA PBE revTPSS MGGA_MS2 M06L Expt
Expt
LDA
MGGA_MS2
M06LrevTPSS
PBE
FIG. 1 (color online). (a) Binding-energy curves for graphene adsorption on the Ni (111) surface. The M06L and RPA values arefrom Refs. [16,21], respectively. (b) Distributions of s, �, z, and t�1 for Ar2 at equilibrium. (c) Exchange enhancement factor.(d) Binding-energy curve of Ar2 for different functionals. See Ref. [24] for computational details.
PRL 111, 106401 (2013) P HY S I CA L R EV I EW LE T T E R Sweek ending
6 SEPTEMBER 2013
106401-2

2 orders of magnitude more time-consuming than thoseof semilocal functionals. Figure 1(a) shows that LSDApredicts the chemisorption minimum but misses thephysisorption one, while PBE misses the latter and almostmisses the former. Compared to the RPA results, revTPSSpredicts the chemisorption with a better binding energythan those of LSDA and PBE. But it still misses thephysisorption minimum, consistent with the result ofRef. [22], indicating its inability to capture noncovalentbonds. Although all semilocal functionals consideredhere yield wrong long-range asymptotic behaviors,MGGA_MS2 and M06L remarkably capture the doubleminima at accurate distances, demonstrating their ability todescribe both strong and weak bonds. If we use the experi-
mental and RPA binding distances (2:11� 0:07 �A, 3.3 A)as references for the chemisorption and physisorption,respectively, then MGGA_MS2 (2.09 A, 3.29 A) yields abetter agreement thanM06L (2.29 A, 3.25 A) [16]. In termsof the binding energies, M06L (64 meV, 64 meV) [16] isbetter than MGGA_MS2 (49 meV, 23 meV) when com-pared to RPA (67 meV, 60 meV) [21], with the first numberin the parentheses for the chemisorption and the second forthe physisorption.
In MGGA_MS2 [14], � is the only ingredient built from�, and it can recognize all types of orbital overlap.Although the two fitting parameters of MGGA_MS2 aredetermined by two data sets that do not involve weakinteractions [14], the ability of � to identify the overlapbetween closed shells helps MGGA_MS2 to captureweak interactions near equilibrium, which explains whyMGGA_MS2 captures both minima in Fig. 1(a).
M06L, on the other hand, uses t�1 to identify differentorbital overlap, with � only to make the one-electroncorrelation energy exact. Table I shows that t�1 � 1 forthe slowly varying density, and t�1 � 1 for the overlapbetween two closed shells. Madsen et al. have shown thatt�1 can discriminate between covalent and noncovalentinteractions [15], which then explains the ability ofM06L to capture the double minima in Fig. 1(a). Thisexample together with many other successful applicationsof M06L, including CO adsorbed on the Pt(111) surface[23] and layered solids [15], seems to suggest that t�1, inaddition to s, would be a good dimensionless parameterthat measures the inhomogeneity of the density. However,t�1 loses the ability to identify the single-orbital regions,where t�1 ¼ 5s2=3 provides no extra information in com-parison with s. It can be seen in Fig. 1(b) that, for the regionaround the nucleus of Ar where the 1s orbital dominates,t�1 resembles s in shape. To overcome the shortcomingof t�1 being unable to identify single-orbital regions,and to fit to a large number of data sets that includecovalent single bonds (� � 0), 35 fitting parameters hadto be introduced into M06L, causing an oscillation in theexchange enhancement factor near small � as shown inFig. 1(c). The exchange enhancement factor Fx character-izes the enhancement of the exchange energy density
with respect to its local approximation, defined by Ex½n� ¼Rd3rn�unifx ðnÞFxðs; �Þ, where �unifx ðnÞ ¼ �3ð3�2nÞ1=3=4�
is the exchange energy per electron of the uniformelectron gas.The oscillation in the M06L Fx can cause two major
problems. The first one is the lack of computational stabil-ity and the high requirements to converge calculations. Anexample is the oscillation of the binding curve of the Ardimer shown in Ref. [24]. The oscillation can be removedby using much higher computational settings for thecalculations, but even then M06L still yields a too-shallowminimum far away from the experimental binding dis-tance, as shown in Fig. 1(d). The performance of M06Lon the Ar dimer and its inability to bind the Kr2 and Xe2rare gas dimers [25], which are typical simple tests forvan der Waals interactions, contradict its good perform-ance on many other noncovalent systems. This also leads tothe second problem, the consistency in describing differentbonds in different chemical environments. To investigatethis, we study the performance of M06L on systems thatare far outside its training sets, e.g., lattice constants ofsolids. We choose the six ionic insulators and semiconduc-tors studied in Ref. [26]. Reference [26] showed that,although the covalent and ionic bonds dominate, the vander Waals interaction plays an essential role in determiningaccurate lattice constants. It found that the errors in latticeconstants of these six solids are reduced by a factor of 2,in comparison with experimental data, when the van derWaals interactions were included on top of the PBEfunctional.Table II shows that the local density approximation
(LDA) understimates the lattice constants of the 6 solidswhile PBE overestimates them. revTPSS improves thelattice constants over PBE slightly for ionic insulatorsbut significantly for semiconductors when compared toexperimental data. However, revTPSS still yields too-largelattice constants because revTPSS misses weak interac-tions in these solids. Therefore, the improvement ofrevTPSS over PBE should be ascribed to its betterdescription on covalent single bonds, by which those
TABLE II. Errors in lattice constants (A) of ionic insulatorsand semiconductors from different functionals. The zero-pointanharmonic expansion has been removed from the experimentallattice constants [27]. ME denotes mean error; MAE, meanabsolute error.
Solids LDA PBE M06L revTPSS MGGA_MS2 Expt.
C �0:022 0.014 �0:010 0.003 �0:007 3.555
Si �0:017 0.046 �0:010 0.017 0.005 5.422
Ge �0:013 0.124 0.137 0.038 0.005 5.644
GaAs �0:026 0.111 0.143 0.039 0.002 5.641
NaCl �0:098 0.130 0.117 0.102 0.029 5.565
MgO �0:018 0.073 0.012 0.052 0.019 4.188
ME �0:032 0.083 0.065 0.042 0.009
MAE 0.032 0.083 0.072 0.042 0.011
PRL 111, 106401 (2013) P HY S I CA L R EV I EW LE T T E R Sweek ending
6 SEPTEMBER 2013
106401-3
semiconductors are bound. Adding the van der Waalsinteractions on top of revTPSS would be expected tofurther improve the lattice constants. This is largely real-ized by MGGA_MS2 alone, which reduces the meanabsolute error from 0.042 A of revTPSS to a remarkable0.011 A. This demonstrates one more time thatMGGA_MS2 describes well both strong (covalent andionic) bonds and weak interactions because � can recog-nize all types of orbital overlap.
The pattern of the M06L performance on these 6 solids,however, is inconsistent. Compared to experimental data,M06L predicts accurate lattice constants for C and Si, but ityields too-large ones, even larger than those of PBE, forGe and GaAs, which have relatively large ionic radii andthus significant van der Waals interactions. Similar behav-ior is also observed for the two ionic solids. M06L yieldsan accurate lattice constant for MgO, but a too-large onefor NaCl. This random performance is likely related to theoscillation of the exchange enhancement factor near small� and small s as shown in Fig. 1(c), because small s is morerelevant to solids than to molecules. The inconsistent per-formance of M06L in this test set cautions against its use insolids, and is rooted in using t�1 to construct the functional.
The above discussions qualitatively explain the perform-ance of revTPSS, M06L, and MGGA_MS2, with a specialfocus on noncovalent interactions, whereas quantitativeanalysis using enhancement factors is a subtle issue.
Madsen et al. [15] argued that the inability of TPSS [8](whence also of revTPSS) to capture weak interactions isdue to the increase of its Fx at small s as � becomes large,as shown in Fig. 1(c) for revTPSS. That argument issupported by the fact that the MGGA_MS2 Fx at small sdecreases monotonically as � becomes large; see Fig. 1(c)of this work and Fig. 2 of Ref. [14]. The monotonicallydecreasing � dependence can be rationalized by the factthat the strength of different chemical bonds decreases withincreasing � (e.g., � ¼ 0 for single bonds, � � 1 formetallic bonds, and � � 1 for noncovalent bonds.) Notethat � is related to the electron localization function [28],� ¼ 1=ð1þ �2Þ, which has been used to establish arigorous topological classification of chemical bonds [29],but surprisingly was not recognized as a useful ingredientfor MGGAs. However, it should be stressed that theimprovement for weak interactions near equilibrium fromMGGA_MS2 results from the good balance between its sand � dependences [14,24].To show its potential impact on a wide range of research
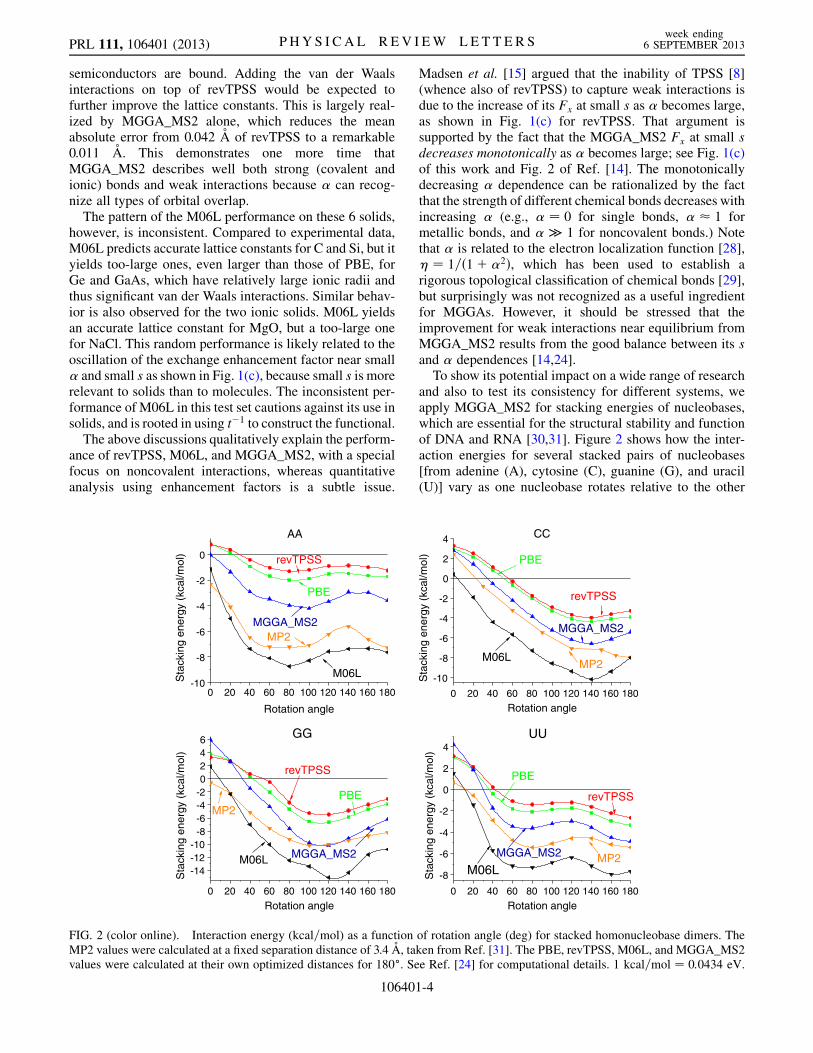
and also to test its consistency for different systems, weapply MGGA_MS2 for stacking energies of nucleobases,which are essential for the structural stability and functionof DNA and RNA [30,31]. Figure 2 shows how the inter-action energies for several stacked pairs of nucleobases[from adenine (A), cytosine (C), guanine (G), and uracil(U)] vary as one nucleobase rotates relative to the other
0 20 40 60 80 100 120 140 160 180
Sta
ckin
g en
ergy
(kc
al/m
ol)
Rotation angle
PBE
revTPSS
MGGA_MS2
M06L
MP2
AA
0 20 40 60 80 100 120 140 160 180
-10
-8
-6
-4
-2
0
2
4
Sta
ckin
g en
ergy
(kc
al/m
ol)
Rotation angle
PBE
revTPSS
MGGA_MS2
MP2
CC
M06L
0 20 40 60 80 100 120 140 160 180
-14-12-10
-8-6-4-20246
Sta
ckin
g en
ergy
(kc
al/m
ol)
Rotation angle
PBE
revTPSS
MGGA_MS2
MP2
GG
M06L
0 20 40 60 80 100 120 140 160 180
-8
-6
-4
-2
0
2
4
Sta
ckin
g en
ergy
(kc
al/m
ol)
Rotation angle
PBE
revTPSS
MGGA_MS2 MP2
UU
M06L
-10
-8
-6
-4
-2
0
FIG. 2 (color online). Interaction energy (kcal=mol) as a function of rotation angle (deg) for stacked homonucleobase dimers. TheMP2 values were calculated at a fixed separation distance of 3.4 A, taken from Ref. [31]. The PBE, revTPSS, M06L, and MGGA_MS2values were calculated at their own optimized distances for 180�. See Ref. [24] for computational details. 1 kcal=mol ¼ 0:0434 eV.
PRL 111, 106401 (2013) P HY S I CA L R EV I EW LE T T E R Sweek ending
6 SEPTEMBER 2013
106401-4

around their perpendicular axis. The reference point for therotation was defined using the method of Elstner et al. [32],in which the center of mass of each nucleobase was alignedwith the glycocidic bonds parallel. A counterclockwise(right-handed) rotation was then applied (see Fig. 2 ofRef. [31]). In each case, the nucleobase structure was fixedto that of the optimized isolated molecule. Except for theCC stacking structures, where MGGA_MS2 misses theminimum at 180� of MP2 (Møller-Plesset second-orderperturbation theory in the electron-electron interaction),MGGA_MS2 yields the stacking energies qualitativelyin agreement with the reference MP2 values [31]. Forall considered cases where binding between nucleobasesis expected, MGGA_MS2 significantly improves thestacking energies over PBE and revTPSS, but still under-estimates them about the same amount as M06L over-estimates, in comparison with the reference MP2 method.If this performance persists for other pairs, thenMGGA_MS2 can provide a better description for DNAand RNA conformations than PBE (and, when long-rangevan der Waals interactions are added [25], probably betterthan M06L). This could be an important step towardcomputer-assisted drug design since the computationalcost of the semilocal MGGA_MS2 is affordable for largebiomolecules.
The computationally efficient semilocal density func-tionals must fail for many stretched bonds [33], but wehave suggested that someMGGAs can be usefully accuratefor unstretched or modestly stretched ones, not just forstrong but even for weak bonds. Successful but simpleMGGAs employ only � ¼ ð�� �WÞ=�unif to recognizeall types of orbital overlap, and extrapolate monotonicallyfrom 0 � � & 1 to large �. This insight should guidethe construction of further-improved semiempirical andnonempirical density functionals.
This work was supported by NSF under GrantsNo. DMR08-54769 and No. CHE-1110884, by NSFCooperative Agreement No. EPS-1003897 with furthersupport from the Louisiana Board of Regents, by theWelch Foundation (C-0036), and by the DeutscheForschungsgemeinschaft (HA 5711/2-1). J. S. thanksJianmin Tao for helpful discussions. The computationswere made with the support of the Louisiana OpticalNetwork and the Tulane Center for ComputationalSciences.
*Corresponding [email protected]
[1] W. Kohn and L. J. Sham, Phys. Rev. 140, A1133 (1965).[2] R. G. Parr and W. Yang, Density Functional Theory of
Atoms and Molecules (Oxford University Press, Oxford,England, 1989).
[3] J. P. Perdew and S. Kurth, in A Primer in DensityFunctional Theory, Springer Lecture Notes in PhysicsVol. 620 (Springer, New York, 2003).
[4] J. P. Perdew and Y. Wang, Phys. Rev. B 45, 13 244 (1992).
[5] J. Sun, J. P. Perdew, and M. Seidl, Phys. Rev. B 81, 085123(2010).
[6] J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett.77, 3865 (1996).
[7] R. O. Jones and O. Gunnarsson, Rev. Mod. Phys. 61, 689(1989).
[8] J. Tao, J. P. Perdew, V.N. Staroverov, and G. E. Scuseria,Phys. Rev. Lett. 91, 146401 (2003).
[9] Y.ZhaoandD.G.Truhlar, J.Chem.Phys.125, 194101 (2006).[10] J. P. Perdew, A. Ruzsinszky, G. I. Csonka, L. A.
Constantin, and J. Sun, Phys. Rev. Lett. 103, 026403(2009); 106, 179902(E) (2011).
[11] J. Sun, B. Xiao, and A. Ruzsinszky, J. Chem. Phys. 137,051101 (2012).
[12] J.M. del Campo, J. L. Gazquez, S. B. Trickey, and A. Vela,Chem. Phys. Lett. 543, 179 (2012).
[13] P. Hao, J. Sun, B. Xiao, A. Ruzsinszky, G. I. Csonka,J. Tao, S. Glindmeyer, and J. P. Perdew, J. Chem. TheoryComput. 9, 355 (2013).
[14] J. Sun, R. Haunschild, B. Xiao, I.W. Bulik, G. E. Scuseria,and J. P. Perdew, J. Chem. Phys. 138, 044113 (2013).
[15] G. K.H. Madsen, L. Ferrighi, and B. Hammer, J. Phys.Chem. Lett. 1, 515 (2010).
[16] M. Andersen, L. Hornekaer, and B. Hammer, Phys. Rev. B86, 085405 (2012).
[17] L. Schimka, J. Harl, A. Stroppa, A. Grueneis, M.Marsman, F. Mittendorfer, and G. Kresse, Nat. Mater. 9,741 (2010).
[18] J. Sun, M. Marsman, A. Ruzsinszky, G. Kresse, andJ. P. Perdew, Phys. Rev. B 83, 121410(R) (2011).
[19] S. Kurth, J. P. Perdew, and P. Blaha, Int. J. Quantum Chem.75, 889 (1999).
[20] Y. Gamo, A. Nagashima, M. Wakabayashi, M. Terai, andC. Oshima, Surf. Sci. 374, 61 (1997).
[21] F. Mittendorfer, A. Garhofer, J. Redinger, J. Klimes, J.Harl, and G. Kresse, Phys. Rev. B 84, 201401(R) (2011).
[22] J. Wellendorff, K. T. Lundgaard, A. Møgelhøj, V. Petzold,D. D. Landis, J. K. Nørskov, T. Bligaard, and K.W.Jacobsen, Phys. Rev. B 85, 235149 (2012).
[23] S. Luo, Y. Zhao, and D.G. Truhlar, J. Phys. Chem. Lett. 3,2975 (2012).
[24] See Supplemental Material at http://link.aps.org/supplemental/10.1103/PhysRevLett.111.106401 forfurther information.
[25] L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys. 13,6670 (2011).
[26] G. X. Zhang, A. Tkatchenko, J. Paier, H. Appel, andM. Scheffler, Phys. Rev. Lett. 107, 245501 (2011).
[27] P. Hao, Y. Fang, J. Sun, G. I. Csonka, P. H. T. Philipsen,and J. P. Perdew, Phys. Rev. B 85, 014111 (2012).
[28] A. D. Becke and K. E. Edgecombe, J. Chem. Phys. 92,5397 (1990).
[29] B. Silvi and A. Savin, Nature (London) 371, 683 (1994).[30] P. Yakovchuk, E. Protozanova, and M.D. Frank-
Kamenetskii, Nucleic Acids Res. 34, 564 (2006).[31] V. R. Cooper, T. Thonhauser, and D. C. Langreth, J. Chem.
Phys. 128, 204102 (2008).[32] M. Elstner, P. Hobza, T. Frauenheim, S. Suhai, and E.
Kaxiras, J. Chem. Phys. 114, 5149 (2001).[33] A. Ruzsinszky, J. P. Perdew, G. I. Csonka, O. A. Vydrov,
and G. E. Scuseria, J. Chem. Phys. 125, 194112 (2006);126, 104102 (2007).
PRL 111, 106401 (2013) P HY S I CA L R EV I EW LE T T E R Sweek ending
6 SEPTEMBER 2013
106401-5





























