Embed Size (px)
Citation preview

Universidad de Vigo
Departamento de Química Física
Grupo de Química Cuántica
Efectos electrónicos introducidos por sustitución,
asociación intermolecular y tautomería en
algunos procesos de interés químico o biológico.
Memoria presentada por M.Luisa Pita Ameneiros para optar al grado de Doctor por la Universidad de Vigo en el Programa de Doctorado en Ciencia y Tecnología Química. Vigo, Septiembre de 2015

Ricardo A. Mosquera Castro, Catedrático del Departamento de Química Física de la Universidad de Vigo
AUTORIZA:
a Dna. M.Luisa Pita Ameneiros a presentar para obtener el grado de Doctora por la Universidad de Vigo, en el programa interuniversitario de Ciencia y Tecnología Química, el trabajo titulado: “Efectos electrónicos introducidos por sustitución, asociación intermolecular y tautomería en algunos procesos de interés químico o biológico”, realizado en el Departamento de Química Física de la Universidad de Vigo bajo su dirección. Para que así conste firma la presente en Vigo a 7 de septiembre de 2015.
Fdo. Ricardo A. Mosquera Castro
Vº B. Rosana Álvarez Rodríguez
Coordinadora local del Programa de Doctorado
en Ciencia y Tecnología Química.
Universidade de Vigo

Indice:
1.- Resumen 4
2.-Introducción 6
3.- Objetivos 8
4.- Metodologia general 10
5.- Resultados y discusión 24
5.1.- Variaciación de la densidad electrónica ante reacciones de adicion electrófila en el anillo
piridinico 25
5.2.- Cambios en el comportamiento nucleofilo de la piridina por efecto de distintos grupos activantes
o desactivantes 34
5.3.- Estudio computacional de las preferencias conformacionales de los dimeros de glicina neitros,
protonados y desprotonados.. 52
5.4 Estudio de la Densidad Electrónica en Dímeros de Glicina Desprotonados. 83
5.5 La relación entre la distancia y la densidad del punto critico. Nueva concepción del orden de
enlace de H 104
5.6 Corrección counterpoise y optimización geométrica 123
5.7 Estudio QTAIM de aminas heterocíclicas (HCAs) 136
5.8 Estudio computacional de la actividad biológicade aminas heterocíclicas potencialmente
cancerígenas. 153
5.9 Estudio del equilibrio ceto –enólico de 3-etil 2,4 pentanodiona (epd) 168
5.10 Tautomeria imino-enamina en metabolitos de aminas heterociclicas con marcado potencial
mutagenico 180
6.- Conclusiones 202
7.- Bibliografia 204

1.- Resumen
En la presente tesis se han realizado varios estudios teóricos sobre determinados
fenómenos quimicos ampliamente conocidos como son la sustitución en anillos
aromáticos, la tautomeria o los enlaces de hidrógeno. Para llevar a cabo dichos análisis
se ha elegido en cada caso un grupo de compuesto de alto interés químico o biológico.
En primer lugar y usando la piridina como molécula modelo, analizamos los cambios
sufridos en el anillo piridínico ante adiciones electrófilas bien sea energéticos o de
población con la teoria cuántica de átomos en moléculas (QTAIM). Para ello se
optimizaron piridina y derivados sustituidos con los grupos funcionales : O-, CN, NO2,
CH3, F, OCH3. Las protonaciones en las posiciones 2,3 y 4 del anillo nos permitieron
analizar por una parte las variaciones en las afinidades protónicas, y por otra los
cambios de población global, así como sus componentes σ y π.
Seguidamente se realizo un estudio energético y de población electrónica minucioso
sobre los distintos enlaces de hidrógeno: O···H-O, N···H-N, O···H-N, O···H-C en el
dímero del aminoácido glicina. Para ello se optimizaron más de 200 geometrías distintas
de dímeros glicina neutra (en su forma zwitterionica y no iónica), protonada y
desprotonada .
Sobre dichos dímeros también fue llevado a cabo un análisis teorico-matemático sobre
las distintas relaciones que pueden tener lugar entre las densidades de los puntos críticos
implicados en el enlace de hidrógeno, así como con la distancia de la unidad X-H···Y.
El proceso de dimerización nos llevo a analizar y valorar la fiabilidad de las
optimizaciones geométricas que incluyen la corrección counterpoise como medida para
evitar el efecto BSSE. Para lo cual ampliamos el conjunto de dímeros de glicina
obtenidos, con dímeros más simples, modelo de un único enlace de hidrógeno.
La tercera via de estudio fue un análisis de las aminas heterocíclicas (HCAs) con
carácter mutagénico. De ellas estudiamos por una parte su papel en la formación de
aductos del ADN mediante un estudio QTAIM sobre la creación de los iones nitronio,
analizando el delicado equilibrio entre la activación metabolica de la 2-amino-1-metil-
6-fenilimidazo[4,5-b]piridina (PhIP) y su destoxificación.
Finalmente, hemos estudiado la tautomería ceto/enólica en la molécula de 3-etil-2,4-
pentanodiona (EPD) para poder compararla con la imino/enamino de las HCAs. Para
ello, hemos considerado las HCAs con carácter mutagénico más conocidas: 2-amino-1-

metil-6-fenilimidazo[4,5-b]piridina (PhIP), 3,4,8-trimetil-3H-imidazo[4,5-f]quinoxalin-
2-amina (4,8-diMeIQ) y 2-amino-3-metilimidazo(4,5-f)quinolina (IQ).

2.-Introducción
Desde hace varias decadas ha surgido un creciente interés por procesos
bioquímicos de sistemas heterociclicos en sustancias conocidas precursoras del cancer.
De las reacciones que sufran estos sistemas va a depender el que sean dichas sustancias
catalogadas con potencial mutagénico o no.
Es también conocido que su estructura y su capacidad de asociación es un factor clave a
la hora de encadenar reacciones que conlleven la formación de aductos del ADN. A
pesar de los numerosos estudios dedicados al efecto, todavía son escasos los estudios
teóricos de las reacciones biológicas en las que estos compuestos intervienen. Esto es
debido bien al tamaño de las moléculas en cuestión y al costoso tiempo computacional
que implicaría su estudio, o bien por los distintos tipos de reacciones a las que son
sometidos dichos compuestos a nivel biológico.
En la presente tesis nos proponemos analizar, gracias a la teoria cuántica de átomos en
moléculas (QTAIM) y a su estudio sobre la densidad electrónica por qué un
determinado tautómero o confórmero presenta una prevalencia para sufrir una
determinada reacción química sobre otros.
Así pues, un objetivo de esta tesis va a ser el estudio teórico de una serie de reacciones
de gran importancia biológica a través de sistemas de los que poseemos una precisa
información experimental o teórica como pueden ser la piridina y la glicina.
Las reacciones que van a ser estudiadas y las compuestos empleados para tal fin serán:
- Reacciones de sustitución en piridina
- Reacciones de asociación en glicina
- Reacciones de tautomerización:
a) cetoenolica en 3-etil-2,4-pentanodiona
b) enamina-imina en aminas heterocíclicas aromáticas (HCAs)
En primer lugar, para analizar los movimientos de densidades electrónicas ante un
proceso de adición electrófila se ha considerado la piridina. La glicina como aminoácido
esencial que es, nos permitirá observar la importancia y propiedades de los procesos de
asociación inter e intramoleculares como son los enlaces de hidrógeno. Finalmente,
estudiando la facilidad para formar tautómeros imino-enamino de las HCAs y por
comparación con la tautomeria ceto-enolica, podremos llegar a investigar cuando un

tautómero desembocará en reacciones de formación de aductos y cuando se
destoxificará.
Para calcular energías, frecuencias y densidades electrónicas de dichos sistemas hemos
empleado distintos niveles de teoria como la Teoria del Funcional de Densidad (DFT) o
el Método de Perturbación o incluso varios a la vez para el mismo sistema con el fin de
cotejar resultados. Los conjuntos base que hemos empleado más habitualmente en los
calculos han sido los 6-311G++(d,p) y 6-311G++(2d,2p), por considerarlos un
adecuado compromiso entre fiabilidad y rapidez. Por último, para el tratamiento de la
información hemos empleado la QTAIM desarrollada por Bader y col. que nos
proporciona un análisis topológico de la densidad electrónica.
La tesis está estructurada en 10 capítulos cada uno de los cuales permite el estudio de un
sistema o de una cualidad distinta del mismo.

3.- Objetivos
1.- Describir los cambios en las poblaciones electrónicas atómicas (globales y en sus
componentes σ y π) en sistemas heterocíclicos aromáticos del tipo de la piridina frente
a un ataque electrófilo.
2.- Estudiar las afinidades protónicas de sistemas heterocíclicos aromáticos frente a la
protonación en función de la activación/desactivación del anillo
3.- Evaluar la validez del modelo de resonancia aplicado a sistemas heterocíclicos
aromáticos a la hora de describir los cambios experimentados por la densidad
electrónica.
4.- Analizar los cambios de las poblaciones electrónicas atómicas global, σ y π en
sistemas heterocíclicos aromáticos frente a una ataque electrófilo según la
activación/desactivación del anillo.
5.- Predecir la viabilidad de zwitteriones en distintos medios: fase gas y disolución
acuosa.
6.- Realizar el análisis conformacional del dímero de glicina en fase gas y disolución
acuosa dependiendo del pH.
7.- Cuantificar la transferencia electrónica que tiene lugar entre los monómeros de los
diferentes dímeros neutros, protonados y desprotonados de glicina.
8.- Estudiar los distintos enlaces de hidrógeno N-H···N, N-H···O, O-H···O, C-H···O que
pueden ser obtenidos en dímeros de glicina (neutra, protonada y desprotonada).
9.- Analizar los cambios de la población electrónica atómica debidos a la formación de
los enlaces de hidrógeno.
10.- Investigar en la unidad X-H···Y, las posibles relaciones entre las densidades de los
puntos críticos de la unidad y/o con las longitudes de enlace.
11.- Precisar el concepto del orden de enlace de la unidad X-H···Y.
12.- Discutir la fiabilidad de los métodos de optimización que incluyen la corrección
counterpoise para el efecto BSSE considerando diferentes fragmentaciones.
13.- Estudiar la reactividad de los iones nitronio mediante el laplaciano de la densidad
electrónica.

14.- Valorar el efecto de la tautomeria ante la N-hidroxilación en las aminas
heterocíclicas aromáticas (HCAs) con potencial mutagénico.
15.- Estudiar las reacciones de activación metabólica y destoxificación de tres
importantes HCAs: 2-amino-1-metil-6-fenilimidazo[4,5-b]piridina (PhIP), 3,4,8-trimetil
-3H-imidazo[4,5-f]quinoxalin-2-amina (4,8-diMeIQ) y 2-amino-3-metilimidazo(4,5-
f)quinolina (IQ).
16.- Explicar la estabilidad de los tautómeros ceto/enol de 3-etil-2,4 pentanodiona y su
relación con el pH y la formación de enlaces de hidrógeno.
17.- Explicar la preferencia energética en la tautomeria imino/enamino de algunas
HCAs haciendo uso del método átomos cuánticos interactuantes (IQA) para desglosar la
energía de algunos compuestos modelo.

.
4.- Metodologia general
4.a) Método Hartree-Fock El objetivo de la Química Cuántica es interpretar y predecir tanto el
comportamiento estático como la reactividad quimica a nivel molecular. Para ello el
método Hartree-Fock (HF) es esencial pues nos permite obtener la energia y la función
de onda electrónica.
Así pues según su primer postulado el estado físico de un sistema está determinado por
una función de onda, Ψ, que es función de la posición y del tiempo y que está
relacionada con la energía (E) del sistema a través de la ecuación de Schrödinger
dependiente del tiempo. Para sistemas en estado estacionario, dicha ecuación se reduce
a la independiente del tiempo (1).
ψψ EH =∧
(1)
siendo∧
H el operador hamiltoniano suma del operador de energía cinética (∧
T ), que
incluye la debida al movimiento del núcleo y de los electrones, y del operador de
energía potencial (∧
V ), que habitualmente incluye sólo términos de tipo electrostático:
electrón-electrón , electrón-núcleo y núcleo-núcleo.
La resolución de esta ecuación es exacta para átomos hidrogénicos (un solo
electrón) cuando el hamiltoniano incluye exclusivamente términos electrostáticos y la
masa de las partículas es independiente de la velocidad, pero la situación se complica
con átomos polielectrónicos o moléculas debido a la interacción entre electrones. Una
simplificación habitual del problema consiste en introducir la aproximación de Born-
Oppenheiner [1] que implica suponer los núcleos fijos para poder resolver la ecuación
electrónica. En el contexto de esta aproximación es posible separar el hamiltoniano
molecular en un término nuclear y otro electrónico que depende paramétricamente de
las coordenadas nucleares. Para un conjunto de M núcleos (A, B, …, M) y N electrones
(i, j, …, N) e incluyendo exclusivamente términos electrostáticos en la energía
potencial, la forma del hamiltoniano electrónico está dada por la ecuación (2), en la que
el último término (repulsión internuclear) es constante para una geometría molecular

fija, rij es la distancia entre partículas y ZA la carga de los
núcleos.
∧
H electronico= ∑∑∑∑∑ ∑∑= >= == = >
+−+∇−M
A
M
AB AB
BAN
i
M
A iA
AN
i
N
i
N
ij
iR
ZZ
r
Z
rij 11 11 1
2 121
(2)
Aún así, utilizando la aproximación de Born-Oppenheimer, la ecuación de
Schrödinger dependiente del tiempo carece de solución exacta para un sistema
polielectrónico. Una opción para su resolución consiste en utilizar funciones
polielectrónicas de prueba formadas como producto de funciones monoelectrónicas iχ ,
llamadas espín-orbitales moleculares [2,3], que dependen de las coordenadas de
posición y espín del electrón que describen, i. Esto da lugar a las funciones
polielectrónicas tipo Hartree.
∏=
=ΨN
i
i iN1
)()...2,1( χ (3)
Además, la función polielectrónica tiene que cumplir la indiscernibilidad
electrónica y el principio de antisimetría (al intercambiar las coordenadas de dos
electrones, la función cambiará de signo). El determinante de los espín-orbitales
moleculares ortonormales [4] cumple dicho principio y es llamado determinante de
Slater [5], siendo !
1N
la constante de normalización.
)().....()(
.........................................
)2().........2()2(
)1(.).........1()1(
!1
)...2,1(
1
1
1
NNN
NN
Ni
Ni
Ni
χχχ
χχχ
χχχ
ψ = (4)
El valor medio de la energía para un determinante de Slater contiene dos
sumandos: uno incluye la energia de cada electron como si estuvieran solos en el campo
formado por los restantes núcleos, y otro se debe a la interacción entre todos los pares
de electrones, tal como expresa la ecuación (5) utilizando la notación de Dirac.
∑∑∑= >=
∧
−+==N
i
N
j
ijij
N
i
ii KJhHE1 11
)(ψψ (5)

ijJ es la integral de Coulomb que contiene los términos que implican la repulsión entre
las distribuciones de carga de los espín-orbitales. Siempre es positiva. ijK es la integral
de canje o intercambio consecuencia de la indiscernibilidad de los electrones . No tiene
equivalente clásico y siempre es positiva.
Al aplicar el teorema variacional, se busca encontrar los espín-orbitales que nos
conduzcan a un mínimo de energía. Este teorema garantiza que dicha energía nunca será
inferior a la real. La optimización se realiza imponiendo como restricciones la condición
matemática de ortonormalidad entre espín-orbitales moleculares.
jiij χχδ = (6)
Teniendo en cuenta esta condición se obtienen las ecuaciones de Hartree-Fock
enunciadas como sigue:
)1()1(1
j
N
j
ijif χλχ ∑=
∧
= i=1,2,…N (7)
La expresión (7) no corresponde a un sistema de ecuaciones de valores propios.
Sin embargo, es posible realizar una transformación unitaria que da lugar a las llamadas
ecuaciones canónicas de Hartree-Fock, que si son ecuaciones de valores propios.
)1()1( iiif χεχ ′=′∧
i=1,2,…,N (8)
∧
f representa en las ecuaciones (7) y (8) al operador de Fock, que es monoelectrónico, y
iε los valores propios de las energías orbitales.
Las ecuaciones deben resolverse de forma iterativa a partir de un conjunto iχ
inicial. Cuando converge el método, se ha alcanzado la autoconsistencia: los espín-
orbitales obtenidos son consistentes con el potencial creado por ellos mismos (Self
Consistent Field, (SCF).
Además la suma de todas las iε no se corresponde con la energía electrónica de
la molécula puesto que tendriamos repulsiones e intercambios repetidos. No obstante,
según el teorema de Koopmans, [6] se puede estimar el potencial de ionización como la
energía (cambiada de signo) del último espín-orbital ocupado. Debe indicarse que el
éxito de dicho teorema radica en una compensación de errores.
El procedimiento Hartree-Fock tiene en cuenta a las repulsiones interelectrónicas
de manera promediada. En consecuencia, permite un acercamiento mayor entre los
electrones de lo que realmente sucede e incluye repulsiones interelectrónicas superiores

a las reales. Esta limitación del método origina una diferencia entre la energía Hartree-
Fock y la que realmente correspondería al hamiltoniano utilizado. Dicha diferencia se
conoce como energía de correlación electrónica. Se han diseñado diferentes estrategias
tendentes a solucionar (de manera parcial) dicho problema. Tenemos así los métodos
DFT y los métodos post HF.
4.b) Teoria del funcional de densidad (DFT)
Los métodos DFT están basados en los teoremas de Hohenberg-Kohn. [7] Estos
indican que la energía, así como el resto de propiedades de una molécula en estado
fundamental están determinadas por su densidad electrónica, ρ. Es decir, la energía es
un funcional de ρ: E=E(ρ). Además, la densidad electrónica del estado fundamental será
aquella que minimiza la energía de dicho estado.
Estos teoremas no obtuvieron aplicación hasta que Kohn-Sham [8] introdujeron
un sistema de referencia ficticio de N electrones que no interaccionan entre sí y cuya
distribución de densidad electrónica, ρs(r), corresponde a la de un determinante de
Slater de espín-orbitales moleculares.
∑=
=N
j
js rr1
2
)()( χρ (9)
Para que la densidad de este sistema coincida con la del sistema real en el que si
hay interacciones, el funcional que relaciona ρ y E debe contener un término de
correlación-intercambio, Exc, no incluído en el método Hartree-Fock (HF). En dicho
término se incluyen tanto la energía de correlación como la interacción electrón-electrón
no clásica. Este término se estima con lo que deja de ser un método exacto pero su
inexactitud es distinta de la del método Hartree-Fock. En general, los funcionales
básicos del método DFT tienen una forma del tipo mostrado en la ecuación (10).
[ ]∑ ∫ ∫ ∫=
+−
++∇−=N
j
njjj Excrdrdrr
rrrdrVrE
121
21
212 )()(21
)()(21
][ ρρρ
ρχχρrr
rr
rrrrr
(10)
El primer sumando denominado Ts[ρ] corresponde a la energía cinética de los
electrones en un sistema que tiene la misma densidad que la del sistema real pero en el
que las interacciones electron-electron no estan incluidas, el segundo es el potencial de
interacción electron-nucleo, el tercero se corresponde con la interacción electrón-
electrón clásica y el cuarto corresponde al término de correlación-intercambio. Mientras
que HF nunca es exacto por no incluir la correlación, DFT podría serlo.

En general, los cálculos DFT proporcionan buenos resultados en sistemas
organometálicos y bioinorgánicos. Su principal inconveniente es que no pueden ser
mejorados sistemáticamente como HF y que es necesario realizar aproximaciones para
estimar el término Exc. Según las aproximaciones utilizadas para estimar este término
podemos distinguir:
a) Aproximación de la densidad local (LDA). En dicha aproximación el
funcional solo depende de ρ, la cual es tratada como la densidad de un gas uniforme de
electrones. Es una aproximación que consigue buenas geometrías y frecuencias pero que
sobreestima los enlaces débiles y las predicciones termoquímicas.
b) Aproximación de gradiente generalizado (GGA). Añade a la LDA
correcciones con basadas en el gradiente de ρ. Las aproximaciones pueden distinguir
entre el potencial de intercambio y el de correlación. Un ejemplo del primero es el
potencial de Becke [9], mientras que ejemplos del segundo son los potencial de Lee,
Parr y Yang (LYP) [10], o el P86 de Perdew [11].
c)Funcionales hibridos.que mezclan los anteriores e incluyen una parte de la
energía de intercambio HF. Uno de los más usados es el B3LYP [12] que presenta
diversas variantes. La más usado hoy en dia es la mostrada en la ecuación (11). LDA
corr
LYP
corr
B
correrccorr
LDA
ercambio
LYPB
xc EEEEEE )7.01(7.08.02.0)2.01( 880intint
3 −++++−= =+
λ (11)
Compuesto por las energías de correlación o intercambio de distintos autores. Los
coeficientes de dichas energías se obtienen ajustando valores para un conjunto de
átomos y moléculas de referencia.
4.c) Métodos post-HF
Son métodos que partiendo del método HF intentan incluir, a través de
correcciones, la energía de correlación electrónica. Podemos diferenciar
a) Interacción de configuraciones (CI). Es el método más directo para calcular la
energía de correlación pero computacionalmente costoso. Consiste en un método
variacional en el que la función de onda se expresa como una combinación lineal de
determinantes construidos a partir de los espín-orbitales HF ocupados y virtuales. De
hecho, se observa que incluso los resultados para el H2 mejoran cuando se tiene en
cuenta que los electrones no tienen porque estar descritos exclusivamente por el
determinante de Slater construido con los espín-orbitales de más baja energía. Por tanto,
considerando, además de dicho determinante, excitaciones simples (S) , dobles(D), …

resulta una función variacional como la mostrada en la ecuación (12), en la que el
coeficiente c0, y los conjuntos de coeficientes cS, cD, etc. son parámetros variacionales.
∑ +Ψ+Ψ+Ψ=ΨS
DDSS ccc .....00 (12)
Así pues al determinante HF, se le incorporan otros en los que se reemplaza 1 o
2 espín-orbitales ocupados por 1 o 2 virtuales respectivamente. Si la función llega a
incluir todas las excitaciones posibles se tiene el método "Full CI" y si, además, el
conjunto de funciones base fuera completo se tendría la energía exacta no relativista.
Dado que el número de determinantes en cuanto la molécula tiene 6 átomos es
del orden de 1019, en la práctica, se consideran solo algunas de todas las excitaciones
posibles y se habla de "CI truncado".
b) Métodos de perturbación de Møller-Plesset (MP). Están basados en que el
hamiltoniano que podemos resolver con el método HF y el del sistema verdadero se
diferencian en una perturbación, H’: ∧
H = ∧
H(0)+
∧
H ' (13)
Concretamente, los métodos MP utilizan como hamiltoniano imperturbado, ∧
H(0), la suma de los operadores de Fock.
Si se desarrolla la energía y la función de onda como una serie de Taylor, surgen
términos de corrección de órdenes sucesivos:
Ei=Ei(0) + Ei
(1) + Ei(2)+… (14)
Ψi= Ψi(0) + Ψi
(1) + Ψi(2) +… (15)
Truncando el desarrollo en el segundo orden surge el método MP2, si se hace en
el tercero el MP3, etc. Así pues, con Ψi(0) se obtiene una energía, Ei
(0), que es la suma
de las correspondientes a los orbitales moleculares HF ocupados en Ψi(0). En este caso,
estaríamos en el nivel MP0. La energía del nivel MP1 equivale exactamente a la HF.
Así, para incluir alguna corrección debida a la correlación electrónica debe como
mínimo utilizarse un nivel 2. Para dicho nivel la corrección a la energía sigue la
expresión (16).
H E '0
'00
20 ΨΨ= (16)
Este método presenta el inconveniente de que al no ser un método variacional no
hay garantía de que al disminuir la energía nos acerquemos al valor exacto pero presenta
menor coste computacional que CI.

c) Métodos Coupled Cluster (CC). Se caracterizan por desarrollar la función de
onda utilizando un operador de forma exponencial.
0ˆΨ=Ψ T
CC e (17)
T es el operador CC que puede expandirse en función de los operadores de excitación
....ˆˆˆˆ321 +++= TTTT (18)
Cada uno de estos operadores genera excitaciones simples, dobles, triples….
Hasta N electrones. Si emplearamos todas las excitaciones el tratamiento CC sería
equivalente a Full CI. Así pues CCSD emplea excitaciones simples y dobles. Es un
método no variacional y costoso computacionalmente hablando.
4.d) Teoria cuántica de Àtomos en Moléculas (QTAIM)
La función de onda electrónica, Ψ, no es medible experimentalmente dada su
característica de complejo. Por ello, se trabaja con ella como |ψ|2, que además tiene el
sentido físico de densidad de probabilidad de encontrar a cada uno de los electrones del
sistema descrito por Ψ en un determinado lugar del espacio. Integrando respecto a las
tres coordenadas espaciales y a la coordenada de espín de todos los electrones menos
uno y multiplicando por el número de electrones del sistema, obtenemos la densidad
eléctrónica, ρ(r), que es una función determinable experimentalmente y en cuyo análisis
se basa nuestro estudio
En los años 60 Hohenberg y Kohn [7] elaboraron un estudio sobre la densidad
electrónica de un gas no homogéneo de electrónes y sentaron las bases para el desarrollo
de los teoremas en los que se basa la Teoría del Funcional de Densidad (DFT). Gracias
a ello podemos calcular la densidad electrónica de un estado fundamental no
degenerado como aquella que minimiza la energía del estado fundamental y además
podemos escribir cualquier observable como un funcional de dicha densidad.
Posteriormente, Kohn y Sham [8] aplicaron esas ideas para desarrollar un método
autoconsistente similar al método Hartree-Fock que ya vimos al principio de este
epígrafe, y que corrige la correlación electrónica.
La DFT nos permite asimismo relacionar conceptos químicos como potencial
químico electrónico, dureza, blandura, con la densidad electrónica. Esta es una rama de
la DFT que se ha llamado “DFT conceptual” [13]. En cualquier caso, para asignar
valores concretos de estas u otras propiedades a átomos o grupos funcionales,

precisamos dividir nuestro sistema de estudio en subsistemas Aunque existen diversas
posibilidades para realizar dicha división, la QTAIM es la única que lo hace utilizando
exclusivamente conclusiones rigurosamente derivadas de los Postulados de la Mecánica
Cuántica [14].
La QTAIM, desarrollada por el grupo de investigación de Bader [15] puede
presentarse como un análisis topológico de la función de densidad electrónica. Esto
hace que sea una teoría excelente para describir los sistemas puesto que la densidad
electrónica es un observable que puede ser medido experimentalmente por difracción de
rayos X. El carácter escalar de ρ(r) hace que pueda representarse con mapas de
contorno o de relieve. ρ(r) es un campo escalar diferenciable, salvo en escasos puntos, y
por lo tanto existirá un gradiente asociado dado por la ecuación (19).
zk
yj
xir
δ
δρ
δ
δρ
δ
δρρ
rrrr++=∇ )( (19)
La representación del campo vectorial del gradiente de la densidad electrónica
origina las líneas de flujo. Las trayectorias descritas por dichas líneas tienen cuatro
características que originan su significado físico:
1.- No se cortan.
2.- En cada punto el vector gradiente es tangente a la línea de flujo.
3.- Cada trayectoria se origina o termina en un punto donde el gradiente se anula
o se hace infinito.
4.- En todo momento el vector gradiente es perpendicular a las superficies en las
que ρ(r) adopta un valor constante.
Las líneas de flujo se representan trazando trayectorias dirigidas en dirección
creciente de ρ(r) partiendo del infinito y terminando en los núcleos atómicos. El que el
gradiente se anule da lugar da lugar a la condición necesaria para la obtención de los
puntos críticos. Utilizando la matriz hessiana, pueden distinguirse cuatro tipos de puntos
[16]. Uno de ellos satisface la condición de máximo local y se corresponde muy
aproximadamente con las coordenadas nucleares. Otros tipos de punto son los de anillo
y los de caja. No obstante, son los puntos críticos llamados de enlace (BCP) los que
habitualmente presentan un mayor interés químico. En concreto sus propiedades se
relacionan con el carácter del enlace, a partir de ellos se trazan las líneas de máxima
densidad de carga (“bondpaths”) que suelen identificarse con los enlaces químicos y la

superficie perpendicular a dichas líneas en estos puntos permite delimitar los átomos en
las moléculas; eje fundamental de la Teoría QTAIM.
Si queremos hallar la derivada direccional de la función densidad electrónica en
el punto r ( )(rr
ρ∇ ) según la dirección del vector normal )(rnrr, realizaríamos el producto
escalar θρρ cos.)(.)()().( rnrrnrrrrrrr
∇=∇ [16] y para un ángulo determinado
obtendriamos líneas de flujo como la línea B de la Fig. 1.
En el caso particular de que el ángulo que formasen los dos vectores fuese de 90º
el coseno se anularía y tendríamos la condición: 0)().( =∇ rnrrrr
ρ . La superficie que
satisface esta condición es conocida como “superficie de flujo cero” y se corresponde
con la línea A de la Fig. 1.
Fig. 1. Isolineas de densidad electrónica y lineas de gradiente de densidad
La línea A de la Fig. 1 nos va a permitir establecer la condición cuántica de
subsistema ya que como la superficie de flujo cero no puede ser atravesada por otra
línea de gradiente ha dividido el espacio en dos cuencas. Cada uno de estos subsistemas
define al átomo conteniendo un nucleo y sus cuencas electrónicas.
Hay un punto especial en la línea A que es el BCP, ya anteriormente
mencionado y obtenido para ( ) 0=∇ rrρ . Dicho punto se encuentra entre dos núcleos y
la línea generada entre ellos que pasa por el BCP nos permite definir el camino del
enlace. El valor de su densidad electrónica nos permite medir la acumulación de carga
en ese punto y por tanto el orden de enlace. Esta es una condición necesaria pero no
suficiente ya que hemos de garantizar que no tenemos fuerzas sobre los núcleos

atómicos, de tal forma que necesitamos además estar en una situación molecular de
mínimo energético.
La caracterización del átomo como entidad fundamental gracias a las superficies
de flujo cero obtenidas del gradiente de densidad de carga, permite analizar
computacionalmente las propiedades atómicas y moleculares, proporcionando, entre
otros, datos termoquímicos e índices de reactividad.
De este modo, podremos obtener la población electrónica de un átomo, N(Ω),
por integración de la densidad electrónica en toda su cuenca.
∫Ω
=Ω rdrNrr
)()( ρ (20)
Para obtener la carga atómica, q(Ω), restaremos al número atómico del núcleo,
ZΩ la población obtenida en (20).
)()( Ω−=Ω Ω NZq (21)
Con respecto a la energía cinética, puede calcularse por dos vías [17]; o bien por
la energía cinética de Schrödinger:
[ ]∫ ∫Ω
Ψ∇Ψ+Ψ∇Ψ′−=Ω 2**2.2
)( τdrdNm
Krh
(22)
O bien por la energía cinética de gradiente;
[ ]∫ ∫Ω
Ψ∇Ψ∇′=Ω ..2
)( *τdrdNm
Grh
(23)
La diferencia entre ambas cantidades, que idealmente debieran ser
numéricamente iguales, permite definir una nueva propiedad atómica, L(Ω), como la
diferencia de (22) y (23) y que debería ser estrictamente cero cuando la determinación
de los límites atómicos y la integración numérica son exactos.
)()()( Ω−Ω=Ω GKL (24)

Es decir:
rdrm
Lrrh
)(4
)( 2ρ∇−
=Ω ∫Ω
(25)
La energía atómica total, E(Ω), se calcula utilizando la relación virial, γ, lo que
evita la integración numérica de términos bielectrónicos.
( ) )1)(( γ+Ω=Ω KE (26)
El laplaciano de la densidad electrónica, además de estar relacionado con el
cálculo de L(Ω), permite localizar las posiciones de máxima reactividad del sistema.
)(2 rr
ρ∇ representa a la suma de las segundas derivadas de la densidad electrónica en
un punto. Cuando esta suma es negativa el carácter de acumulación de carga (derivadas
segundas negativas) supera al de su dispersión (derivadas segundas positivas). En el
primer caso, 0)(2 <∇ rr
ρ , representa un máximo local de carga y recibe el nombre de
“valence shell charge concentration“, (VSCC) que se relaciona con el modelo de Lewis
de localización de pares de electrónes y el ampliamente usado modelo de repulsión de
pares de electrones de la capa de valencia (VSEPR) [18]. Así, por ejemplo, una reacción
ácido base se explica por la intersección de un máximo de la concentración local de
carga en la base con un mínimo en el ácido.
Otra propiedad analizada en este trabajo ha sido el tensor momento cuadrupolar,
Q(Ω), el cual informa de cómo se deforma la distribución esférica de la densidad
electrónica. Es especialmente significativo el elemento Qzz del tensor. Si Qzz(Ω) es
cero la distribución es esférica, si es positivo adopta forma de oblato y si es negativo de
prolato. En el primer caso la carga positiva se distribuye en un elipsoide aplastado y en
el segundo caso la carga positiva queda distribuida en un elipsoide alargado en
dirección del eje z. La comprobación de que tipo de momento cuadrupolar tiene cada
átomo de nuestro sistema permite predecir tipos de reactividad como pueden ser ante
ataques electrófilos o nucleófilos. El elemento Qzz del momento cuadrupolar atómico
[19] está relacionada con la densidad electrónica mediante la fórmula:

∫Ω
−−=Ω drrzeQzz )3()( 22ρ (27)
Por último, también podemos obtener otro índice de reactividad mediante las
Funciones Condensadas de Fukui. Dichas funciones llamadas también “índices de
Fukui” miden el cambio de la densidad electrónica en un punto cuando se incrementa
+
+
∂
∂=
)(
)(
rVN
rf
r
rρ
(28)
o disminuye
−
−
∂
∂=
)(
)(
rVN
rf
r
rρ
(29)
el número de electrónes manteniendo el potencial externo constante. +f representa el
cambio frente a un ataque nucleófilo (aumento de N) y −f representa el cambio frente a
un ataque electrófilo (disminución de N).
La integración de (28) y (29) al espacio particionado en átomos tal como lo
hemos realizado con QTAIM (núcleo más cuenca electrónica) nos dará los índices de
Fukui. El grupo de Química Cuántica de la Universidad de Vigo ha implementado un
nuevo método [20] para el cálculo de los índices de Fukui que resuelve las dificultades
encontradas en implementaciones previas, calculando una integración global según las
ecuaciónes (30) y (31):
[ ]∫Ω
++ −=
A
NNA rdrrfoo
rrr)()(1 ρρ (30)
[ ]∫Ω
−− −=
A
NNA rdrrfoo
rrr)()( 1ρρ (31)
En el contexto de la QTAIM se han definido índices de deslocalizacion
electrónica, δA,B, entre dos átomos A y B a partir de la densidad del agujero de Fermi
[21]. Se ha demostrado que los δA,B entre las correspondientes cuencas atómicas

QTAIM pueden calcularse a partir de las matrices atómicas de solapamiento orbital,
Sij(Ω) según la expresión (32).
( ) ( ) ( )∑∑ ΩΩ−=ΩΩi j
BijAijBA SS2,δ (32)
Considerando las simetrías de los orbitales moleculares, es posible distinguir
componentes σ y π en estos índices y aplicarlos para considerar el papel jugado en
diferentes procesos químicos por ambos tipos de deslocalización electrónica.
4.e) Partición energética IQA (Interacting Quantum Atoms)
Para el hamiltoniano electrónico no relativista podemos descomponer la energía
como la suma de los distintos términos que pueden ser observados en la ecuación (33).
ij
N
i
ne
M
jij
M
i
nn
M
ij
N
i ij
N
i
ee
N
iji
N
i ij
iM
j
M
i ij
jiM
ij
N
i ij
N
ij
N
i
i
VVVKr
Z
r
ZZ
rHE
∑∑∑∑∑ ∑∑∑∑
∑∑∑∑∑
−
==
−
=+==
−
=+===
−
=+=
−
=+==
∧
+++=Ψ−Ψ+
ΨΨ+ΨΨ+Ψ∇−Ψ=ΨΨ=
1
11
1
111
1
1111
1
11
1
111
2 121
(33)
Esto nos permite desglosar la energía en diferentes términos con un claro sentido físico.
Considerando las deficiencias de la aproximación HF, como la energía de correlación,
Ecorr, y que el término Vee puede ser divido en dos: aquel que proviene de la energia de
Coulomb clásica, VEE, (sin auto-interacción) y aquel potencial de intercambio debido a
la indiscernibilidad de las particulas, Vx, podemos escribir la ecuación (33) como:
corrHFneHFnnHFEEHFxHFcorr
ij
N
i
HFHF
M
j
ij
M
i
HFnnHF
M
ij
N
iijHFeeHF
N
iji
N
i
HFHF
EVVVVKEVne
VVKH
+++++=+ΨΨ+
ΨΨ+ΨΨ+ΨΨ=ΨΨ
∑∑
∑∑∑∑∑
==
−
=+=
−
=+==
∧
11
1
11
1
111
(34)
En el contexto de la QTAIM y utilizando la densidad electrónica de las cuencas
atómicas Ω, la ecuación (34) quedaría:
[ ] corr
HFZZ Z
nennEEx EZVZZVVVKE +
Ω++ΩΩ+ΩΩ+Ω= ∑∑ ∑∑∑∑∑ΩΩ≥ΩΩ f
''
),(),(),(),()( '''
(35)

Definiendo la energía potencial total monoatómica, VT(A,A), como (36) y la biatómica,
VT(A,B), como (37), se puede escribir la ecuación (35) como suma de energia cinética y
potencial mas un término de correlación (38)
( ) ( ) ( )AAneAAeeAAX VVVV ΩΩ+ΩΩ+ΩΩ= ,,, A)(A,T (36)
( ) ( ) ( ) ( ) ( )ABneBAneBAnnBAEEBAXT ZVZVZZVVVBAV Ω+Ω++ΩΩ+ΩΩ= ,,,,,),( (37)
corr
HFA BA
TT EBAVAAVKE +
++Ω= ∑ ∑∑∑≠Ω
),(21
),()( (38)
De esta forma la energía neta de un átomo A, Enet(ΩA), puede ser definida por la
ecuación (39). La energía electrónica molecular se obtiene sumando dichas energías
netas y las interacciones entre átomos distintos, ),( BAVT , como muestra la expresión
(40), y propusieron Blanco y colaboradores en la partición energética conocida como
IQA [22].
( ) ( ) ( )AAVKE TAAnet ,+Ω=Ω (39)
( ) ( ) corr
BA
TnetT EBAVEE +
+Ω= ∑ ∑∑Ω ≠
,21
(40)
Mientras que las energías netas atómicas se obtienen integrando en una cuenca atómica
la función densidad adecuada, los términos diatómicos, excluyendo Vnn(Z,Z'), requieren
una doble integración sobre los pares de cuencas atómicas (que habitualmente implica
un alto coste computacional). Esta fue llevada a cabo usando el programa
PROMOLDEN [22]. El término Ecorr es obtenido como diferencia entre la energía
obtenida al nivel CCSD (con una geometría optimizada con el mismo nivel) y la energía
HF puntual de esa misma geometría.

5.- Resultados y discusión

5.1.- Variaciación de la densidad electrónica ante reacciones
de adición electrófila en el anillo piridinico
Conocer la reactividad de los heterociclos es crucial para distintos procesos
industriales (procesado del petroleo, tintes, poliméros, etc), así como en un gran numero
de procesos biológicos. Además su papel como agentes químicos es realzado en su
interacción con sistemas electrófilos, siendo la protonación una de las reacciones más
comunes en las que un heterociclo puede participar. Esto se refleja en la existencia de
datos publicados de afinidades protónicas [1]. La piridina, en particular, presenta dos
características muy importantes de cara a su estudio en este tipo de reaccciones. De una
parte parte el interés que presenta para la industria en general y por otra parte su
relación con una sustancia ampliamente estudiada como es el benceno. Sin embargo, la
piridina muestra un carácter prácticamente inerte a la sustitución aromática electrófila.
Esto se explica por la elevada electronegatividad del nitrógeno que reduce la densidad
electrónica de los átomos de carbono del anillo.
Siguiendo el modelo de resonancia podriamos deducir que los electrófilos atacarian
preferentemente en las posiciones 3 y 5, mientras que los nucleófilos se dirigirian a las
posiciones 2, 4 y 6 [2]. Debido al N piridínico, las posiciones 3 y 5 resultan más
desactivadas que en el benceno.
Fig. 1.1. Formas resonantes de la piridina
En trabajos previos se ha destacado su facilidad para la formación de enlaces de
hidrogéno con agua o formación de aductos mediante dichos enlaces [3,4], pero los
procesos de metalación de piridinas por reacciones de sustitución electrófilia ha sido
considerado un desafio y se han elaborado diferentes métodos como la litiación dirigida
[5] para poderlos llevar a cabo. Algunos estudios teóricos van más allá y demuestran
que sulfuros de nitrilo ionizados reaccionan con piridina a través de reacciones de
sustitución electrófila mediante la formación del ión tiazolopiridinio [6]. Los lugares
favorecidos para la protonación en este ión son diferentes a los que muestra la piridina

neutra. Estudios de RMN demuestran que su protonación está favorecida en la posición
2 del anillo piridínico en contraposición con estudios teoricos [7].
Este trabajo pretende desarrollar un análisis QTAIM [8,9] de los efectos generados por
la protonación en diferentes posiciones del anillo piridinico. Nuestro grupo ya ha
estudiado el cálculo de la densidad electrónica de otro heterociclo nitrogenado usando la
teoria QTAIM para las reacciones de protonación [10-13], encontrándose en algunos
casos propiedades contrapuestas al modelo de resonancia (MR)[14].
Detalles computacionales
Se han realizado optimizaciones completas de la geometría molecular para todas
las moléculas estudiadas: piridina sin protonar y protonada en las posiciones 2, 3 y 4 del
anillo. En todos los casos se aplicaron cálculos DFT con el funcional B3LYP. También
se han realizado optimizaciones geometrícas al nivel MP2, que incluye la corrección de
perturbación de segundo orden, para cotejar los datos de energías moleculares obtenidos
en los dos niveles y garantizar su fiabilidad. En todos los casos se han utilizado
conjuntos “split valence” de funciones base, añadiendo funciones difusas y polarizadas,
en concreto: 6-311++G**. El análisis vibracional realizado ha mostrado que las
estructuras resultantes de la optimización son mínimos energéticos que no presentan
ninguna frecuencia imaginaria, que implicaría la obtención de un estado de transición.
Las Afinidades protónicas han sido calculadas de acuerdo con la ec. 1.1
( ) ( )NHCENHCEAP 5565 −= + (1.1)
Se ha usado el programa Gaussian03 [15] para computar las geometrías moleculares y
el paquete de programas AIMPAC [16] para obtener las propiedades atómicas QTAIM
y realizar el análisis topológico de las densidades eléctrónicas. En la integración de
densidades electrónicas se verificó que los valores de L(Ω) no superasen en valor
absoluto 2·10-3 au, lo cual se considera una condición necesaria para garantizar la
calidad de las propiedades QTAIM integradas. También se comprobó que las
poblaciones electrónicas moleculares eran suficientemente próximas a las obtenidas
como suma de las correspondientes propiedades atómicas, ΣN(Ω). La geometría plana
de la molecula de piridina nos ha permitido separar la población en σ y π. Dado que la

protonación en los carbonos deja los 2 hidrógenos en un plano perpendicular al anillo
piridínico, el programa ha interpretado su población electrónica como de tipo π. Esto ha
sido tenido en cuenta a la hora de analizar resultados.
Podemos obtener un índice de reactividad mediante las funciones condensadas de
Fukui. Dichas funciones, llamadas también “índices de Fukui”, miden el cambio de la
densidad electrónica en un punto cuando se incrementa o disminuye el número de
electrones, N, manteniendo el potencial externo constante. +f representa el cambio
frente a un ataque nucleófilo (aumento de N) y −f representa el cambio frente a un
ataque electrófilo (disminución de N).
En el grupo de Química Cuántica de la Universidad de Vigo se ha implementado un
nuevo método para el cálculo de los índices de Fukui que resuelve las dificultades
encontradas en implementaciones previas [17], calculando una integración global según
las ec. 1.2 y 1.3:
[ ]∫Ω
++ −=
A
NNA rdrrfoo
rrr)()(1 ρρ ( 1.2)
[ ]∫Ω
−− −=
A
NNA rdrrfoo
rrr)()( 1ρρ ( 1.3)
Otra propiedad analizada en este trabajo ha sido el tensor momento cuadrupolar, Q(Ω),
el cual informa de cómo se deforma la distribución esférica de la densidad electrónica.
Es especialmente significativo el elemento Qzz del tensor. Si Qzz(Ω) es cero la
distribución es esférica, si es positivo adopta forma de oblato y si es negativo de prolato.
El elemento Qzz del momento cuadrupolar atómico [18] se relaciona con la densidad
electrónica mediante la ec. 1.4
∫Ω
−−=Ω drrzeQzz )3()( 22ρ (1.4)
Resultados y discusión

a) Afinidades protónicas
Se han estudiado las especies resultantes de todas las protonaciones posibles de
la piridina (Fig. 1.2).
NC2
C3
C4
C5
C6
N1posición 2
posición 3
posición 4
H1
H2
H3
H4
H5
H6
(orto)
(meta)
(para) Fig. 1.2 Notación usada para las distintas protonaciones
En cada caso se ha calculado la afinidad protónica (AP), tendencia de la molécula a
captar un protón, como la diferencia de entalpía de la correspondiente reacción de
protonación. Para ello hemos restado las energías electrónicas moleculares una vez
corregidas con la energía de vibración en el cero Kelvin (ZPVE) y la corrección térmica
para la entalpía (CTE) a 298.15 K, de las formas protonadas y sin protonar (Tabla 1.1).
Tabla 1.1. Energías moleculares, E, (en au) y afinidades protónicas, AP, (en kJ mol-1) para las protonaciones sobre los distintos átomos del anillo piridínico. Tanto las energías moleculares como las afinidades protónicas incluyen las correspondientes correcciones por vibración en el punto cero (ZPVE) y la corrección térmica para la entalpía a 298.15 K (TCE). Se muestra también valor de APa experimental tomado de la referencia [1].
E AP a AP
N1 C2 C3 C4
C5H5N -248,3512 930,0 890,9 669,2 696,2 648,0
Tal y como puede ser predecible por el MR, las AP más elevadas corresponden
al N1. Este es la posición por donde la piridina puede atacar como nucleofilo al ser el
sitio más activado y además no pierde la aromaticidad del anillo. Le sigue el valor de
AP en la posición C3. Esto concuerda con las predicciones del modelo de resonancia, ya
que como muestra la Fig. 1.3, es la única C-protonación que no presenta una forma
resonante con una carga positiva sobre el nitrógeno.

Fig 1.3. Formas resonantes de la piridina bajo una protonación en posiciones orto, meta y para del anillo piridinico.
Podemos usar las funciones condensadas de Fukui como un parámetro de interpretación
de estas AP calculadas. Las funciones condensadas de Fukui (FFs) obtenidas mediante
QTAIM, son una herramienta muy usada para explicar la nucleofilia o electrofilia en
distintos sitios de la molécula. Se definen como la variación que experimenta la
densidad electrónica ante una variación del nunero de electrones a un potencial externo
constante. Esto puede entenderse bien como un aumento en el número de electrones
( +Af ) o una disminución ( −
Af ). El sitio más nucleofilico será aquel que sufra con mayor
probabilidad el ataque electrófilo. Así pues y como en nuestro caso el anillo perderá un
electrón al formar el enlace con el H+, nos interesa conocer −Af . De acuerdo con esa
definición el sitio más nucleofilo será aquel que tenga valores más positivos.
Comparando con las AP, observamos que sí obtenemos los valores de −Af más elevados
para el N1. Sin embargo, no obtenemos C3 como la siguiente posición de ataque sino
C4 (Tabla 1.2).
Tabla 1.2. Valores de −Af multiplicado por 102.
b) Variación de la población electrónica
La explicación que pueda aportarnos la teoria QTAIM pasa por el cálculo de la
densidad electrónica en cada una de las cuencas atómicas de la molécula. En la tabla 3
N1 C2 C3 C4
FFs 36,1 4,1 7 7,3

se muestra la variación de población del resultado de las distintas protonaciones en orto,
meta, para y en el nitrógeno con respecto a la piridina neutra. Dado el carácter plano de
la piridina hemos evaluado asimismo reparto de la población en σ/π proporcionado por
el AIMPAC.
Las variaciones de población fueron obtenidas como diferencia entre la población de la
molecula protonada menos la de la piridina sin protonar. Los resultados parecen marcar
una clara tendencia de los movimientos electrónicos atendiendo en donde se localice la
protonación. Antes de diseccionar las pérdidas o ganancias eléctrónicas de cada tipo de
átomos del ciclo hemos de decir que ante un ataque electrófilo, como puede ser una
protonación, los átomos más débiles en cuanto a su atracción por su densidad
electrónica serán aquellos que pierdan población en todas las distintas moléculas y en
todas los tipos de protonaciones. En el ciclo de la piridina esos son los hidrógenos que
pierden siempre población σ, como confirman todos los datos obtenidos.
Tabla 1.3. Incremento de población electrónica de la molecula protonada menos la molecula neutra. Resultados multiplicados por 102. Resultados dados en u.a.
Antes de explicar los movimientos electrónicos, es necesario comentar que el software
usado para la partición de la carga en σ/π consideró que los hidrógenos (el del propio
N1 C2 C3 C4 C5 C6 H1 H2 H3 H4 H5
∆Nσ -22,7 6,67 22,66 -3,52 21,71 -4,84 -49,38 -9,48 -8,65 -9,33 -8,27
∆Nπ -15,96 14,93 -27,48 -3,35 -26,6 -9,3 34,76 -1,38 -0,71 -1,51 -0,84 Protonación
en orto ∆NTOTAL -18,21 21,6 -4,82 -6,86 -4,88 -14,14 -14,62 -10,86 -9,37 -10,83 -9,12
∆Nσ -3,46 10,63 -1,2 15,38 -1,66 21,02 -9,34 -49,1 -9,04 -8,41 -9,74
∆Nπ -3,92 -24,54 3,79 -19,47 -5,13 -21,67 -1,19 36,2 -1,06 -0,84 -1,32 Protonación
en meta ∆NTOTAL -7,37 -13,91 2,59 -4,09 -6,78 -0,65 -10,53 -12,89 -10,11 -9,25 -11,06
∆Nσ 6,61 0,2 18,53 -2,99 18,52 0,2 -8,87 -9,54 -49,18 -9,54 -8,87
∆Nπ -22,25 -3,55 -23,26 7,98 -23,26 -3,56 -0,75 -1,21 34,54 -1,21 -0,75 Protonacion
en para ∆NTOTAL -15,63 -3,36 -4,72 5 -4,73 -3,37 -9,62 -10,75 -14,65 -10,75 -9,62
∆Nσ -22,53 5,38 -1,78 4,98 -1,79 5,38 -9,58 -7,81 -7,54 -7,81 -9,58
∆Nπ 28,04 -1,51 -6,31 -10,22 -6,31 -1,52 -0,62 -0,78 -0,81 -0,78 -0,62 Protonacion
en N1 ∆NTOTAL 5,51 3,87 -8,09 -5,23 -8,1 3,86 -10,2 -8,59 -8,36 -8,59 -10,2

anillo y el de la protonación), por estar uno encima del otro correspondían a una
distribución de población π en vez de σ. Este hecho se ha tenido en cuenta en todos los
cálculos para que los balances de población resultaran correctos.
Con los datos de la tabla 1.3 hemos elaborado un posible diagrama de reparto de
población que puede observarse en Fig. 1.4
Protonación en “C2” Protonación en “C3”
N
N
Protonación en “C4” Protonación en “N”
N
N
Fig. 1.4. Tabla representativa de los movimientos de población electrónica correspondientes a la generalidad de los casos y los aumentos o incrementos máximos y minimos de la población. La flecha de mayor tamaño representa el lugar de la protonación, las flechas exteriores al anillo los movimientos de población σ y las flechas interiores del anillo (en rojo) los movimientos π.
Analizando dicha representación podemos obtener las siguientes consecuencias:
1.- Como ya hemos apuntado los H de la molécula pierden población electrónica. En
general alrededor de 0,10 au cada H.
2.- Los movimientos de población σ y π no llevan la misma orientación. Mientras la
población π se dirige desde los átomos opuestos al lugar de la protonación, la población
σ no sigue un mismo patrón, sino que depende de la posición del N respecto a la
protonación. Podriamos decir que unicamente cuando la protonación es en orto respecto
al N, el C que soporta la protonación gana población σ. Esta población es cedida por el
propio N. En el resto de los casos donde el lugar de la protonación está alejado del N, el
carbono que sufre la protonación siempre pierde población σ.

3.- Los carbonos opuestos a la protonacion siempre ganan población σ. Esto está
motivado por su gran pérdida de población π.
4.- Los carbonos adyacents al carbono que sufre la protonación son los que más pérdida
de población π experimentan, por la ruptura de enlaces π del anillo.
5.- La protonación en el N no supone la pérdida de los dobles enlaces. Eso se observa en
las menores pérdidas de población π que experimentan los carbonos. Además,
observamos que es el único caso donde aumenta la población π en el anillo (∑∆Nπ)
incrementando la aromaticidad del anillo piridínico.
El siguiente parámetro a comparar es el momento cuadrupolar. De las componentes del
tensor momento nos interesa la que esta más alejada del anillo y esa es el momento
cuadrupolar en el eje z (Qzz). Esa distribución de densidad electrónica perpendicular al
anillo es llamada prolato y será el punto que más fácilmente será atacado por un
electrófilo. Esa forma prolato tiene valores de Qzz <0. Cuanto más negativo sea, mayor
densidad tiene esa componente y más sufrira el ataque electrófilo. Sus valores indican a
C3 como el más favorable ante un ataque electrófilo, coincidiendo con lo mostrado por
los valores de AP. Además presentan una correlacion (R2= 0,98) con la población del
carbono que sufre la protonación, N(C*).
Tabla 1.4. Valores de Qzz. Valores en au. C2 C3 C4
Qzz en MP2 -3,100 -3,360 -3,271
N(C*) 5,708 6,052 5,979
Conclusiones
Se ha estudiado la protonación del la piridina considerando distintos parámetros.
Los datos de afinidades protónicas obtenidos concuerdan con los datos experimentales
otorgando la posición favorable para la protonación al N1, seguida de la posición meta
a N1. La diferencia entre valores calculados y experimentales de AP es tan solo de 39
kJ/mol. Las funciones condensadas de Fukui siguen la misma idea en cuanto al N1
como sitio de protonación preferente pero otorgan el segundo lugar a la posición para.

Los valores de Qzz correlacionan con los de N(C*) y otorgan también al carbono en
meta como lugar favorable para la protonación.
Los datos de densidades electrónicas obtenidos por QTAIM nos permiten elaborar unas
representaciones de los movimientos de población σ/π para los distintos sitios de las
protonaciones. Así como la población π sigue el mismo sentido en dirección a la
protonación, los movimientos de la población σ dependen de si el N1 está directamente
enlazado con el C*.

5.2.- Cambios en el comportamiento nucleófilo de la piridina por efecto de distintos grupos activantes o desactivantes
Las reacciones de sustitución electrófila aromática en las que la piridina actúa
como nucleófilo son muy conocidas [1] y vienen siendo explicadas mediante el modelo
de resonancia (MR). Sin embargo dicho modelo no interpreta que dado que la piridina
es π deficiente dichas reacciones son mucho más lentas y requieren condiciones más
duras. Más aún cuando la reaccion de sustitución transcurre en piridinas sustituidas.
En ellas, el MR predice sitios más probables de sustitución electrófila considerando la
estabilidad de los iones carbonio intermedios. Así, la Fig. 2.1 muestra que hay tres
estructuras resonantes para la sustitución electrófila en las posiciones 2, 3 y 4. Sin
embargo, los carbocationes formados en las posiciones 2 y 4 presentan una forma
resonante que deja una carga positiva sobre el átomo de nitrógeno, por lo que estas
sustituciones seran menos estables que la originada por la formación del carbocatión en
3. Las posiciones favorecidas cambian considerablemente tras la sustitución con grupos
activantes, así pues el grupo activante en posición 2 y 4 dejan una carga negativa en el
N lo que estabilizará esas posiciones ante la sustitución. La sustitución en posición 3 no
deja esa forma resonante con carga negativa en el N.
Fig. 2.1. Formas resonantes de la piridina con grupo funcional activante y desactivante
Sin embargo, debemos tener en cuenta que el carácter activante-desactivante del grupo
competirá con el efecto inductivo del N piridínico provocando una nueva
reestructuración electrónica de la molécula. Nuestro estudio está dedicado a analizar los

efectos de la sustitución sobre la reactividad del anillo piridínico. Cabe esperar que estos
efectos dependan de la capacidad que tenga el grupo unido al anillo de la piridina para
estabilizar la carga positiva que se genere en el ión piridinio. Clásicamente, esto podrá
alcanzarse por dos vias: efecto inductivo o efecto resonante, predominando
normalmente este último. De aquí surge la siguiente clasificación: grupos activantes del
anillo piridinico por efectos inductivos (denominados +I) o por efectos mesómeros
(+R). Si lo que producen en el anillo piridinico es desactivación son denominados -I o -
R según el efecto sea inductor o mesómero. La conjugación de ambos efectos va a ser
clave a la hora de predecir cual es el sitio óptimo de reactividad de la piridina.
Numerosos estudios de nuestro departamento y de otros autores [2-17] han analizado
dichos efectos en otros sistemas. En este trabajo, estos cambios serán estudiados en
derivados de piridina monosustituidos con los grupos funcionales mostrados en la tabla
2.1 situados en posiciones 2, 3 y 4 del anillo. Para analizarlos vamos a utilizar la teoria
de átomos en moléculas (QTAIM) [18,19]. Dicha teoria permite evaluar la población
electrónica σ y π de los átomos del anillo [20], el momento cuadrupolar [20] y los
índices de Fukui [21], que nos van a servir como referente para conocer los sitios del
anillo piridinico más reactivos.
Tabla 2.1 . Sustituyentes considerados en este trabajo para el anillo piridínico
activante fuerte : O-
activante intermedio: OCH3
activante débil: CH3
desactivante fuerte: NO2
desactivante intermedio: CN
desactivante débil: F
Detalles computacionales
Se han realizado optimizaciones completas de la geometría molecular para todas
las moléculas estudiadas: piridina y sus derivados monosustituidos con: O-, OCH3, CH3,
F, CN, y NO2 en las posiciones 2, 3 y 4 del anillo, y el conjunto de todas las especies
protonadas de estos compuestos en cada uno de los seis átomos del anillo. La notación
empleada en la distintas protonaciones puede observarse en la Fig.2.2.

NC2
C3
C4
C5
C6
N1posición 2
posición 3
posición 4
H1
H2
H3
H4
H5
H6
(orto)
(meta)
(para) Fig. 2.2 Notación usada para las distintas protonaciones
El nivel de teoria B3LYP/6-311++G(2d,2p) ha sido empleado en todas las
optimizaciones geométricas. Asimismo, se ha usado el nivel MP2/6-311++G(2d,2p)
para garantizar las energias y geometrias conseguidas como mas estables. El análisis
vibracional realizado ha mostrado que las estructuras resultantes de la optimización son
mínimos energéticos que no presentan ninguna frecuencia imaginaria, que implicaría la
obtención de un estado de transición.
Se ha usado el programa Gaussian03 [22] para computar las geometrías moleculares y
el paquete de programas AIMPAC [23] para obtener las propiedades atómicas QTAIM
y realizar el análisis topológico de las densidades eléctrónicas. Todas Todas las
integraciones de las propiedades atómicas fueron conseguidas a un nivel de error de la
función L(Ω) más bajo en valor absoluto que 2·10-3 au, lo cual se considera una
condición necesaria para garantizar la calidad de las propiedades QTAIM integradas.
Ademas se comprobó que las energías y poblaciones electrónicas moleculares eran
suficientemente próximas a las obtenibles como suma de las correspondientes
propiedades atómicas, ΣN(Ω) y ΣE(Ω), respectivamente.
Las funciones integradas de Fukui se obtuvieron con un programa desarrollado en
nuestro grupo de investigación [24] y una versión modificada del paquete AIMPAC
[25]. Dichas funciones miden el cambio que experimenta la densidad electrónica ante
un cambio en el número de electrones bajo el mismo potencial externo. Dado que lo que
estudiamos es un ataque electrófilo, emplearemos −f que representa la función de

Fukui al disminuir el número de electrones y que nos servirá como uno de los posibles
índices de reactividad.
Otro índice empleado en este trabajo es el momento cuadrupolar en el eje z, Qzz(Ω). El
tensor momento cuadrupolar mide la deformación de la densidad a nivel atómico con
respecto a la forma esférica. Así pues la deformación de la carga positiva en el eje z
(prolato) tiene valor negativo según la ec. 2.1 y nos servirá para predecir las zonas de
ataque electrófilo [21]. Debe señalarse que los valores del momento cuadrupolar
utilizados en este trabajo fueron obtenidos con el nivel de cálculo MP2, dados los malos
resultados obtenidos al nivel B3LYP.
∫Ω
−−=Ω drrzeQzz )3()( 22ρ (2.1)
Por último, la separación de la densidad electrónica en componentes σ y π nos indicará
el impacto ante la activación o desactivación de la densidad del anillo piridínico.
En cada caso se ha calculado la afinidad protónica (AP), tendencia de la molécula a
captar un protón, definida como la entalpía de la correspondiente reacción de
protonación. Para ello hemos restado las energías electrónicas moleculares una vez
corregidas con la energía de vibración en el cero Kelvin (ZPVE) y la corrección térmica
para la entalpía (CTE) a 298.15 K, de las formas protonadas y sin protonar.
Resultados y discusión
a) Afinidades protónicas
Se han estudiado las especies correspondientes a todas las protonaciones
posibles de todas las piridinas monosustituidas. En lo que sigue cada una de estas
moléculas se idenfica mediante un término, cuyo primer dígito hace referencia al
sustituyente (1: piridina, 2: O-, 3: CN, 4: NO2, 5:CH3, 6: F, 7: OCH3), el segundo a la
posición del sustituyente (o: posición IUPAC 2, m: posición IUPAC 3, p: posición
IUPAC 4) y tercero y cuarto (sólo en las especies protonadas) al sitio de protonación:
(N1, C2, C3, C4, C5, C6). Las afinidades protónicas calculadas pueden observarse en
la tabla 2.
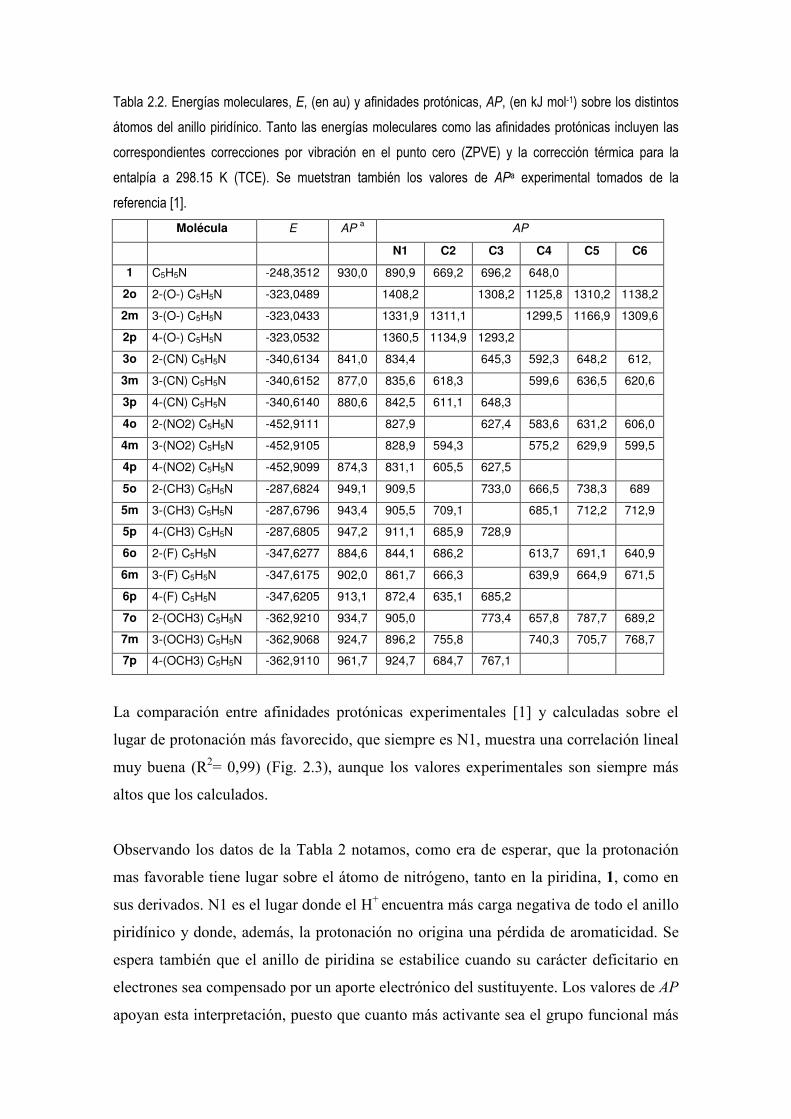
Tabla 2.2. Energías moleculares, E, (en au) y afinidades protónicas, AP, (en kJ mol-1) sobre los distintos átomos del anillo piridínico. Tanto las energías moleculares como las afinidades protónicas incluyen las correspondientes correcciones por vibración en el punto cero (ZPVE) y la corrección térmica para la entalpía a 298.15 K (TCE). Se muetstran también los valores de APa experimental tomados de la referencia [1].
Molécula E AP a AP
N1 C2 C3 C4 C5 C6
1 C5H5N -248,3512 930,0 890,9 669,2 696,2 648,0
2o 2-(O-) C5H5N -323,0489 1408,2 1308,2 1125,8 1310,2 1138,2
2m 3-(O-) C5H5N -323,0433 1331,9 1311,1 1299,5 1166,9 1309,6
2p 4-(O-) C5H5N -323,0532 1360,5 1134,9 1293,2
3o 2-(CN) C5H5N -340,6134 841,0 834,4 645,3 592,3 648,2 612,
3m 3-(CN) C5H5N -340,6152 877,0 835,6 618,3 599,6 636,5 620,6
3p 4-(CN) C5H5N -340,6140 880,6 842,5 611,1 648,3
4o 2-(NO2) C5H5N -452,9111 827,9 627,4 583,6 631,2 606,0
4m 3-(NO2) C5H5N -452,9105 828,9 594,3 575,2 629,9 599,5
4p 4-(NO2) C5H5N -452,9099 874,3 831,1 605,5 627,5
5o 2-(CH3) C5H5N -287,6824 949,1 909,5 733,0 666,5 738,3 689
5m 3-(CH3) C5H5N -287,6796 943,4 905,5 709,1 685,1 712,2 712,9
5p 4-(CH3) C5H5N -287,6805 947,2 911,1 685,9 728,9
6o 2-(F) C5H5N -347,6277 884,6 844,1 686,2 613,7 691,1 640,9
6m 3-(F) C5H5N -347,6175 902,0 861,7 666,3 639,9 664,9 671,5
6p 4-(F) C5H5N -347,6205 913,1 872,4 635,1 685,2
7o 2-(OCH3) C5H5N -362,9210 934,7 905,0 773,4 657,8 787,7 689,2
7m 3-(OCH3) C5H5N -362,9068 924,7 896,2 755,8 740,3 705,7 768,7
7p 4-(OCH3) C5H5N -362,9110 961,7 924,7 684,7 767,1
La comparación entre afinidades protónicas experimentales [1] y calculadas sobre el
lugar de protonación más favorecido, que siempre es N1, muestra una correlación lineal
muy buena (R2= 0,99) (Fig. 2.3), aunque los valores experimentales son siempre más
altos que los calculados.
Observando los datos de la Tabla 2 notamos, como era de esperar, que la protonación
mas favorable tiene lugar sobre el átomo de nitrógeno, tanto en la piridina, 1, como en
sus derivados. N1 es el lugar donde el H+ encuentra más carga negativa de todo el anillo
piridínico y donde, además, la protonación no origina una pérdida de aromaticidad. Se
espera también que el anillo de piridina se estabilice cuando su carácter deficitario en
electrones sea compensado por un aporte electrónico del sustituyente. Los valores de AP
apoyan esta interpretación, puesto que cuanto más activante sea el grupo funcional más

alta es AP. Observándose en la Tabla 2.2 la siguiente secuencia de afinidades
protónicas:
O- > OCH3 > CH3 > F > CN > NO2
R2 = 0,99
820
840
860
880
900
920
940
860 880 900 920 940 960 980
AP experimentales
AP
teó
rico
s
Fig.2.3. Correlación entre AP experimentales y teóricas (kJ/mol)
En la piridina no sustituida, los valores de AP calculada, indican que C3 es el lugar de
C-protonación preferido. Esto concuerda con las predicciones del modelo de resonancia,
ya que como muestra la Fig. 2.2, es la única C-protonación que no presenta una forma
resonante con una carga positiva sobre el nitrógeno. Sin embargo, cuando existen
grupos funcionales unidos al anillo, surgen ejemplos, como 5m, 6m ó 7m, donde las
predicciones del modelo de resonancia dejan de corresponderse con los valores de AP
mostrados en la Tabla 2.2 y C3 y C5 dejan de ser los lugares de C-protonación
preferidos.
Analizando ahora las posiciones de los grupos funcionales, observamos que con grupos
desactivantes, la posición que más facilita la protonación sobre el nitrógeno es la 4,
seguida por la 3 y por último la 2. Es decir, AP aumenta a medida que lo hace la
distancia al grupo desactivante, lo que facilita que el N pueda perder densidad
electrónica en el proceso de protonación. Este hecho no está de acuerdo con el modelo
de resonancia, ya que un sustituyende desactivante en 4, como el NO2, retiraría más
densidad electrónica de N1 que el mismo sustituyente en posición 3.

En el caso del grupo funcional activante la mayor AP corresponde a los isómeros en que
el grupo funcional se sitúa en las posiciones 2 ó 4, pero no en 3. El comportamiento de
los valores de AP sobre el N coincide con las predicciones del MR.
La Tabla 2.2 muestra los lugares preferidos para la C-protonación de derivados
sustituídos. Para su interpretación, debemos distinguir entre sustituyentes, Y, activantes
y desactivantes, así como considerar la posición ocupada por el sustituyente.
Tabla 2.2. Posición más favorable para la C-protonación exceptuando la protonación sobre el nitrógeno
Protonación C2 C3 C4 C5 C6
C5H6N x
2-O- x
3-O- x
4-O- x x
2-OCH3 x
3-OCH3 x
4-OCH3 x x
2-CH3 x
3-CH3 x
4-CH3 x x
2-F x
3-F x
4-F x x
2-CN x
3-CN x
4-CN x x
2-NO2 x
3-NO2 x
4-NO2 x x
I) Grupos desactivantes
En todos los casos deben considerarse dos efectos que retiran densidad
electrónica de los carbonos del anillo. Por una parte el efecto electronegativo del átomo
de nitrógeno, por otra el efecto desactivante del sustituyente Y. El primero, de acuerdo
con el modelo de resonancia, debe manifestarse de forma más atenuada sobre C3 y C5,
dando lugar, tal como hemos comentado en el caso de la molécula de piridina, a las
posiciones más afines a la C-protonación. El segundo se manifestará de manera

preferente en diferentes carbonos dependiendo de la posición del sustituyente. En
general serán las posiciones “meta” al grupo Y aquellas donde este efecto esté más
atenuado y tengan tendencia a una afinidad protónica más elevada. Es decir, en una
piridina sustituida están presentes dos efectos orientadores para la protonación: el
debido al nitrógeno y el del sustituyente. El primero es constante (C3 y C5) y el
segundo varía, de manera que podemos considerar tres casos (Ver Fig. 2.1):
a) “Y” en posición 2: que orientaría los protones hacia posiciones C4 y C6.
b) “Y” en posición 3: que orientaría los protones hacia las posiciones C5.
c) “Y” en posición 4: que orientaría los protones hacia las posiciones C2 y C6.
De acuerdo con la Tabla 2.3, que resume los resultados de la Tabla 2.2, en las 2- y 4-
sustituciones el efecto orientador del átomo de nitrógeno supera al del grupo funcional,
resultando favorecidas las protonaciones sobre C3 y C5. En cambio, en la 3-sustitución,
ambos efectos refuerzan la preferencia por la posición 5.
II) Grupos activantes
De manera semejante al caso anterior, consideraremos el efecto electronegativo
del nitrógeno, que retira densidad electronica de los carbonos del anilo, nuevamente, de
manera más atenuada en C3 y C5. Ahora, por el contrario, el efecto del sustituyente es
introducir densidad electrónica, de manera preferente en posiciones “orto” o “para” al
grupo Y. Nuevamente, el efecto orientador del sustituyente dependerá de su posición y
habrá que considerar tres casos (Ver Fig. 2.1):
a) “Y” en posición 2: que orientaría los protones hacia posiciones C3 y C5.
b) “Y” en posición 3: que orientaría los protones hacia las posiciones C2, C4 y
C6.
c) “Y” en posición 4: que orientaría los protones hacia las posiciones C3 y C5.
Los valores calculados para la afinidad protónica de los carbonos del anillo de piridina
(Tablas 2.2 y 2.3) indican que efectivamente, en derivados 2- y 4-sustituidos, ambos
efectos se suman y las mayores afinidadaes corresponden a C3 y C5. Por el contrario, en
los derivados 3-sustituidos, el efecto orientador del grupo Y supera al del átomo de
nitrógeno y las protonaciones más favorables corresponden a C2 (grupo O-) y C6
(grupos OCH3 y CH3).

En conclusión, la correlación entre las AP obtenidas cuando el mismo sustituyente Y se
encuentra en posiciones distintas, (Fig. 2.4 para las posiciones 2 (orto) y 3 (meta)),
muestra que a pesar de la buena correlación global (R2 = 0,93), deben diferenciarse dos
casos. Los compuestos con mayores afinidades protónicas (grupos O- y OCH3) dan
lugar a una dispersión de puntos que evidencia que el efecto de la posición puede ser
comparable al de la naturaleza del sustituyente. Por el contrario, los grupos de efecto
débil y los desactivantes fuertes, muestran una excelente correlación que indica que el
efecto de la posición es inferior al de la naturaleza del sustituyente. Son los casos en los
que el efecto orientador del nitrógeno supera al del grupo Y.
R2 = 0,93
400
600
800
1000
1200
1400
1600
400 600 800 1000 1200 1400 1600
protonaciones con GF en "orto"
pro
ton
ac
ion
es
co
n G
F e
n "
me
ta"
Fig. 2.4. Correlación entre AP en kJ/mol obtenidas con grupos funcionales en posición 2 y 3.
b) Interpretación electrónica de las afinidades protónicas
b.1) Criterios de poblaciones electrónicas
Con las energías de las moléculas protonadas en distintas posiciones del anillo y
con distintos grupos funcionales en posiciones “orto”, “meta” y “para” hemos obtenido
cuales eran las posiciones preferentes (Ver tabla 2.2). En esta sección compararemos
esos datos obtenidos con los de población electrónica (N(Ω)) que presenta la tabla 2.3.
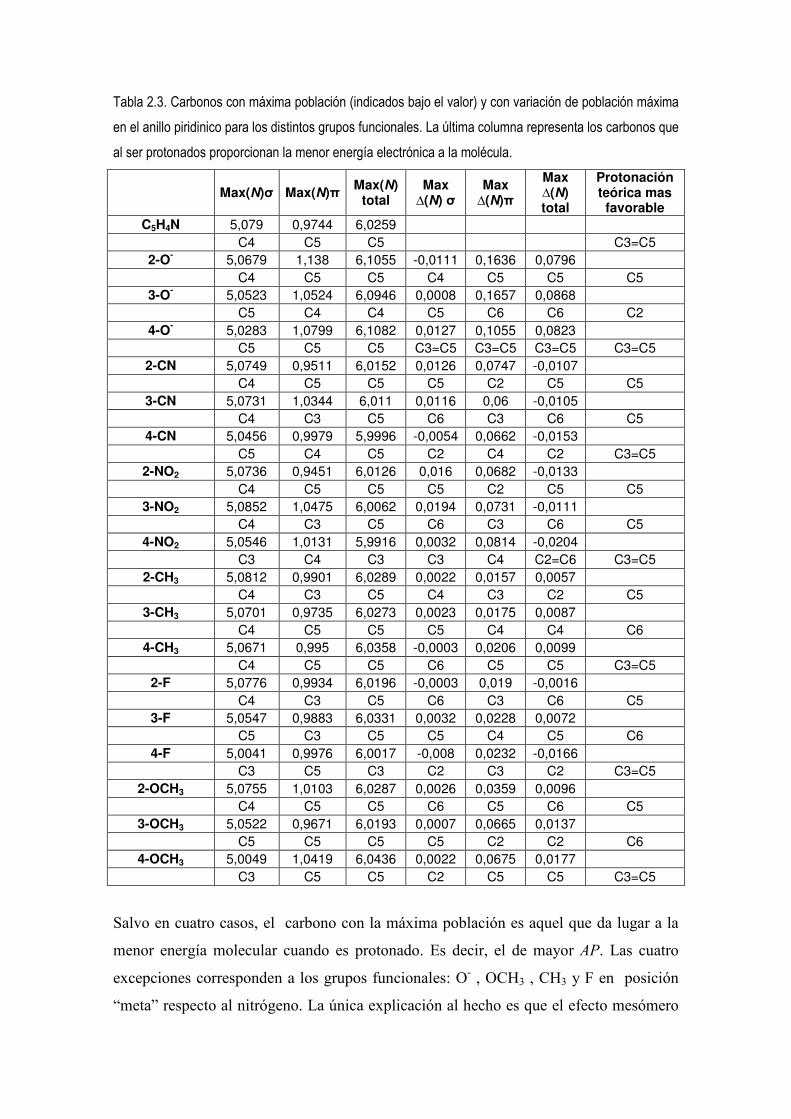
Tabla 2.3. Carbonos con máxima población (indicados bajo el valor) y con variación de población máxima en el anillo piridinico para los distintos grupos funcionales. La última columna representa los carbonos que al ser protonados proporcionan la menor energía electrónica a la molécula.
Max(N)σ Max(N)π Max(N)
total Max ∆(N) σ
Max ∆(N)π
Max ∆(N) total
Protonación teórica mas favorable
C5H4N 5,079 0,9744 6,0259 C4 C5 C5 C3=C5
2-O- 5,0679 1,138 6,1055 -0,0111 0,1636 0,0796
C4 C5 C5 C4 C5 C5 C5 3-O
- 5,0523 1,0524 6,0946 0,0008 0,1657 0,0868
C5 C4 C4 C5 C6 C6 C2 4-O
- 5,0283 1,0799 6,1082 0,0127 0,1055 0,0823
C5 C5 C5 C3=C5 C3=C5 C3=C5 C3=C5 2-CN 5,0749 0,9511 6,0152 0,0126 0,0747 -0,0107
C4 C5 C5 C5 C2 C5 C5 3-CN 5,0731 1,0344 6,011 0,0116 0,06 -0,0105
C4 C3 C5 C6 C3 C6 C5 4-CN 5,0456 0,9979 5,9996 -0,0054 0,0662 -0,0153
C5 C4 C5 C2 C4 C2 C3=C5 2-NO2 5,0736 0,9451 6,0126 0,016 0,0682 -0,0133
C4 C5 C5 C5 C2 C5 C5 3-NO2 5,0852 1,0475 6,0062 0,0194 0,0731 -0,0111
C4 C3 C5 C6 C3 C6 C5 4-NO2 5,0546 1,0131 5,9916 0,0032 0,0814 -0,0204
C3 C4 C3 C3 C4 C2=C6 C3=C5 2-CH3 5,0812 0,9901 6,0289 0,0022 0,0157 0,0057
C4 C3 C5 C4 C3 C2 C5 3-CH3 5,0701 0,9735 6,0273 0,0023 0,0175 0,0087
C4 C5 C5 C5 C4 C4 C6 4-CH3 5,0671 0,995 6,0358 -0,0003 0,0206 0,0099
C4 C5 C5 C6 C5 C5 C3=C5 2-F 5,0776 0,9934 6,0196 -0,0003 0,019 -0,0016
C4 C3 C5 C6 C3 C6 C5 3-F 5,0547 0,9883 6,0331 0,0032 0,0228 0,0072
C5 C3 C5 C5 C4 C5 C6 4-F 5,0041 0,9976 6,0017 -0,008 0,0232 -0,0166
C3 C5 C3 C2 C3 C2 C3=C5 2-OCH3 5,0755 1,0103 6,0287 0,0026 0,0359 0,0096
C4 C5 C5 C6 C5 C6 C5 3-OCH3 5,0522 0,9671 6,0193 0,0007 0,0665 0,0137
C5 C5 C5 C5 C2 C2 C6 4-OCH3 5,0049 1,0419 6,0436 0,0022 0,0675 0,0177
C3 C5 C5 C2 C5 C5 C3=C5
Salvo en cuatro casos, el carbono con la máxima población es aquel que da lugar a la
menor energía molecular cuando es protonado. Es decir, el de mayor AP. Las cuatro
excepciones corresponden a los grupos funcionales: O- , OCH3 , CH3 y F en posición
“meta” respecto al nitrógeno. La única explicación al hecho es que el efecto mesómero

se imponga al efecto inductivo cuando el grupo funcional esta en posición “meta”. Este
cambio sobre la dirección de protonación se observa experimentalmente con piridinas
activadas en posición meta donde la sustitución electrófila (SEAr) ocurre en la posición
2 [27]. Efectivamente, dado que O- y OCH3 ya son +R, F a pesar de ser desactivante
débil posee pares de electrones no enlazantes que pueden deslocalizarse por resonancia,
por tanto el efecto inductivo domina en la reactividad y en cambio el efecto de la
resonancia es el que predomina en la orientación. Con respecto a CH3 su efecto es +I
pero tiene un efecto mesómero por hiperconjugación al adoptar una hibridación sp2 y
conjugar así sus electrones con los del C al que esta enlazado. Trabajos anteriores
teóricos ya han advertido de este efecto [25].
Se calculó cómo variaba la densidad electrónica de cada átomo de la molécula respecto
a la de la piridina sin sustituyente. Asimismo, el balance global de la población de todo
el anillo se obtuvo como la diferencia del derivado piridínico menos el de la piridina sin
sustituyente (Tabla 2.4).
Tabla 2.4 .Diferencias entre la población electrónica (en au) multiplicada por 10 2 del anillo piridinico del derivado de la piridina menos la piridina sin sustituyente.
Molécula ∆N(Ω)σ ∆N(Ω)π ∆N(Ω)total
2-O- -58,1 32,4 -25,7
3-O- -55,8 30,1 -25,7
4-O- -57,4 31,9 -25,5
2-OCH3 -66,2 12,5 -53,7
3-OCH3 -64 10,2 -53,8
4-OCH3 -65,1 11,4 -53,7
2-CH3 -1,4 5,1 3,7
3-CH3 -1,5 4,2 2,7
4-CH3 -2,2 5,4 3,2
2-F -71,3 6,2 -65,1 3-F -69,7 6,7 -63 4-F -71 5,9 -65,1
2-CN -24 -1,8 -25,8 3-CN -24,3 -3,5 -27,7 4-CN -24,9 -2,3 -27,1
2-NO2 -44,3 -4 -48,3
3-NO2 -46,3 -7,3 -53,6
4-NO2 -45,9 -5,4 -51,3

En primer lugar se observa que las variaciones resultan poco afectadas por la posición
del grupo funcional. Así, las mayores variaciones de ∆N(Ω) total debidas a la posición
del sustituyente las presenta el grupo nitro, con una diferencia máxima de 0,05 au entre
posiciones 2 y 3. En general el anillo presenta una pérdida general de población que va
desde 0,25 au con O- hasta 0,53 au con F. De la norma anterior se exceptúa el grupo
metilo, con el cual se produce una ganancia neta de 0,03 au.
Analizando las componentes σ y π, vemos que las poblaciones π aumentan o
disminuyen pero en cuantías no similares a las de la población total. La variación de la
población σ es siempre negativa y resulta ser la contribución dominante a la pérdida de
población global. Es decir, la pérdida de población es mayoritariamente σ. Para explicar
esta pérdida de población del anillo hemos de estudiar la distribución de poblaciones
parciales de cada átomo (Anexo I). En ellas observamos dos tipos de comportamiento;
por un lado, con grupos activantes la perdida de densidad electrónica es llevada a cabo
exclusivamente por los carbonos y el nitrógeno del anillo. Por otro lado, para grupos
desactivantes, la pérdida de densidad electrónica tiene lugar en los hidrógenos. La
explicación es evidente. Para enlazar con el grupo funcional el anillo ha de donar
densidad electrónica y los átomos encargados de ello serán aquellos que tengan una
menor atracción por esa población y estos son los hidrógenos y su población σ. Pero eso
ocurrirá cuando no existe un aporte de población π al anillo, que es el caso de los
desactivantes. Cuando tenemos dicho aporte de población π los carbonos ganan
densidad electrónica hasta el punto de que ellos mismos podrán donar población σ. Es
necesario hacer notar que esa pérdida será mayor al aumentar la electronegatividad del
átomo con el que van a enlazar. Por ello, la mayor pérdida de población σ corresponde a
los derivados fluorados, seguidos por aquellos que contienen el grupo OCH3, luego el O-
. Es de notar como la pérdida es mayor en el OCH3 que en el O- debido a que la carga
negativa del oxígeno le hace atraer menos población. A continuación se encuentran los
derivados con NO2, luego con CN y por último con CH3.
Con respecto a la población π, ∆N(Ω)π presenta aumento o disminución dependiendo del
grupo funcional en cuestión de tal modo que para los grupos considerados como
activantes tenemos un aumento de población π. Es más, al aumentar el poder activante
del grupo mas aumentará la población π del anillo (O->OCH3>CH3). Para los grupos
desactivantes obtenemos la consecuencia lógica de una pérdida de población y esto lo

vemos en el grupo NO2 y en menor medida en el CN. El caso del F no se corresponde
con su carácter de desactivante débil, ya que según la Tabla 2.4 muestra un aumento de
densidad electrónica π, que es incluso mayor que el de los derivados metilados. Este
hecho se debe a que F tienes pares de electrones que puede compartir.
Veamos si la posición del sustituyente marca una pauta. Para la población σ las mayores
pérdidas las obtenemos estando en orto los grupos (O-, F,y OCH3). Estos grupos eran
los que mas perdían por estar enlazados a un átomo muy electronegativo. Además se
encuentran al lado del nitrógeno de la piridina con lo que la atracción de la población
será mayor. Para el resto, la mayor pérdida la obtenemos estando el grupo NO2 y CN en
“meta” y para el CH3 en posición “para”. Los desactivantes CN y NO2 pierden
población π en mayor medida cuando están en posición “meta”. Para los grupos
fuertemente activantes (O- y OCH3) la posición “orto” da lugar a las mayores
transferencias electrónicas π al anillo.
ACTIVANTES O- , OCH3 y CH3 DESACTIVANTES NO2 yCN FLUOR
N orto
Norto
Norto F
N
meta
N
meta
N
meta F
N
para
N
para
N
para F
Figura 2.5. Posible distribución electrónica para los grupos funcionales estudiados. Con flechas grandes y exteriores al anillo se representan los movimientos σ y con flechas pequeñas e interiores los π. Variaciones de población en a.u. La flecha grande indica la posición del grupo funcional.
Analizando la distribución σ y π de cada átomo por separado (Anexo II), hemos podido
realizar el siguiente esquema de distribución y posible movimiento de densidad
electrónica frente a la sustitución de un grupo funcional activante o desactivante
(Fig.2.5). En la figura 2.5 encontramos dos tipos de distribución conforme el grupo

funcional sea activante o desactivante y un caso particular intermedio: F. El fluor es
considerado generalmente como desactivante débil, como confirmarían las variaciones
de población que muestran los hidrógenos. Sin embargo las variaciones de población de
los carbonos se encuentran más próximas al comportamiento de los activantes que al de
los desactivantes. Por este motivo se describe el comportamiento del fluor como un
tercer caso con gráficas propias. No podemos garantizar el sentido de las flechas de
población en algunos casos puesto que se han construido teniendo en cuenta qué átomo
del anillo perdía población y quien lo ganaba y las flechas se han dibujado siguiendo esa
orientación. Las flechas interiores representan los movimientos de población π y con las
flechas exteriores se ha representado la distribución de población σ.
b.2) Funciones condensadas de Fukui
El hecho de que un átomo presente un índice −Af mas positivo significa que es
mayor la densidad electrónica del átomo en la especie neutra. Así, en una protonación,
el protón se dirigirá hacia dicho átomo. Por lo tanto los valores de −Af deberían
correlacionar con los valores de AP de las moléculas más estables. Sin embargo, como
se comentó en el capítulo 5.1. para la piridina no sustituida, esta correlación no se
encuentra. La tabla 2.5 muestra los índices −Af obtenidos en las piridinas sustituidas
que hemos estudiado.
Debe señalarse que en 14 de los 19 compuestos los carbonos con los valores más
positivos de −Af coinciden con las protonaciones más estables (resumidas en la Tabla
2.2). Las excepciones se deben a que las protonaciones más estables tienen lugar sobre
el átomo N1.
La Tabla 2.6 muestra las diferencias que experimenta el índice −Af con respecto a la
piridina no sustituida, que han sido calculadas con la ec. 2. En dicha tabla destaca el
valor negativo de esta variación para el grupo NO2. Dejando claro el poder desactivante
de este grupo.
[ ] −−=∆ ∫Ω
−−
tesustituyenA
NNA rdrrfoo
rrr )()( 1ρρ [ ]sustituidanoA
NN rdrroo
−Ω
−∫ −rrr )()( 1ρρ (2.2)

Tabla 2.5. Índices −Af multiplicados por 10 2 para los átomos del anillo de la piridina para cada
sustituyente y en distintas posiciones. Todo en au. −Af C2 C3 C4 C5 C6 N
piridina sin sustituyente
4,1 7 7,3 7 4,1 36,1
O- orto 0,8 11,7 3,9 15,8 4 13,4 meta 10,8 1,8 11,1 4,2 13,9 7,9 para 3 11,9 1 11,9 3 18,7
OCH3 orto 4,9 11,5 3,8 13,7 8,7 7,4 meta 10,5 8 6,1 6,1 11,9 8,3 para 3,2 11,4 6,4 6,9 4,1 17,5
CH3 orto 10,7 12,9 3,8 14,9 10,9 7,2 meta 12,2 13,2 4,2 10,1 13,4 7,9 para 4,3 6,4 5,9 6,7 4,4 34,2
F orto 10,2 13,7 4,3 15,5 11,7 8,2 meta 12,6 13 4,8 10,9 14 8,5 para 3,9 6,9 6 6,9 3,9 38,2
CN orto 9,5 10,8 3,7 13,3 10,1 6,3 meta 10,1 11,2 3,5 9,8 11,9 7,4 para 13,2 13,8 3,6 13,8 13,2 6,9
NO2 orto 1,4 4,4 5,9 6 4,2 22,2 meta 3,4 4,2 5,6 6,2 3,6 31,1 para 3,9 5,9 5,5 5,9 3,9 33,6
Tabla 2. 6. Variaciones de los Índices −Af respecto a la piridina sin sustituir multiplicado por 10 2.Todo en
au.
∆ −Af R C2 C3 C4 C5 C6 N
O- orto -3,3 4,7 -3,4 8,8 -0,1 -22,7 meta 6,7 -5,2 3,8 -2,8 9,8 -28,2 para -1,1 4,9 -6,3 4,9 -1,1 -17,4
OCH3 orto 0,8 4,5 -3,5 6,7 4,6 -28,7 meta 6,4 1 -1,2 -0,9 7,8 -27,8 para -0,9 4,4 -0,9 -0,1 0 -18,6
CH3 orto 6,6 5,9 -3,5 7,9 6,8 -28,9 meta 8,1 6,2 -3,1 3,1 9,3 -28,2 para 0,2 -0,6 -1,4 -0,3 0,3 -1,9
F orto 6,1 6,7 -3 8,5 7,6 -27,9 meta 8,5 6 -2,5 3,9 9,9 -27,6 para -0,2 -0,1 -1,3 -0,1 -0,2 2,1
CN orto 5,4 3,8 -3,6 6,3 6 -29,8 meta 6 4,2 -3,8 2,8 7,8 -28,7 para 9,1 6,8 -3,7 6,8 9,1 -29,2
NO2 orto -2,7 -2,6 -1,4 -1 0,1 -13,9 meta -0,7 -2,8 -1,7 -0,8 -0,5 -5 para -0,2 -1,1 -1,8 -1,1 -0,2 -2,5

Asimismo, la Tabla 2.6 muestra que la acumulación de densidad sobre el nitrógeno (que
siempre supera a la de los carbonos) es inferior en los NO2-derivados. No obstante, en
general tampoco es un buen índice de comparación pues desactivantes como el grupo
CN no dan lugar siempre a valores negativos.
b.3) El momento cuadrupolar Qzz
Para averiguar cual es el sitio favorecido en una protonación se ha utilizado un
criterio adicional basado en la componente Qzz del momento cuadrupolar [4]. Es fácil
imaginar que la protonación requiere sitios con alta densidad. Luego cuanto más alejada
del plano del anillo se mueva la distribución electrónica de un atomo intercíclico, más
posibilidades tendrá de reaccionar con el protón. Esta distorsión de la distribución
electrónica de un átomo está cuantificada por su momento cuadrupolar atómico (Q(Ω)).
Así pues podemos pensar que el carbono que presente mayor desviación de carga
respecto del anillo será el más recomendable para donarla. Los valores más negativos de
Qzz, dejan en torno al eje de distorsión la mayor concentración de carga negativa,
favoreciendo la sustitución eléctrófila.
La tabla 2.7 nos indica las variaciones del momento cuadrupolar en nuestras moléculas
para C*. Estos datos mantienen el mismo orden que el carácter activante. Esto es, los
valores más negativos de Qzz son aquellos de mayor concentración de carga y esos los
obtenemos para los grupos activantes (incluído F). Además, se encuentran valores
positivos para los grupos funcionales desactivantes. También se observa claramente que
de las tres posiciones del grupo funcional la más acumulativa de densidad electrónica es
la “para”, seguida de la “orto” Recordemos que esas tres posiciones las situábamos
respecto el nitrógeno del anillo piridínico.
Los valores obtenidos para la población electrónica del carbono más poblado del anillo
piridínico presentan una buena correlación con los datos de Qzz (Fig. 2.6). Esto indica
que ambos índices pueden ser utilizados para preveer cual va a ser la protonación más
estable de todas las del anillo. Finalmente, mostramos una correlación entre los valores
de Qzz(C*) para distintas posiciones del grupo funcional (Fig. 2.7), que permitirían
establecer un carácter específico a cada grupo, independientemente de su posición. No
obstante, esta correlación se observa únicamente con el nivel MP2, pero no con B3LYP.
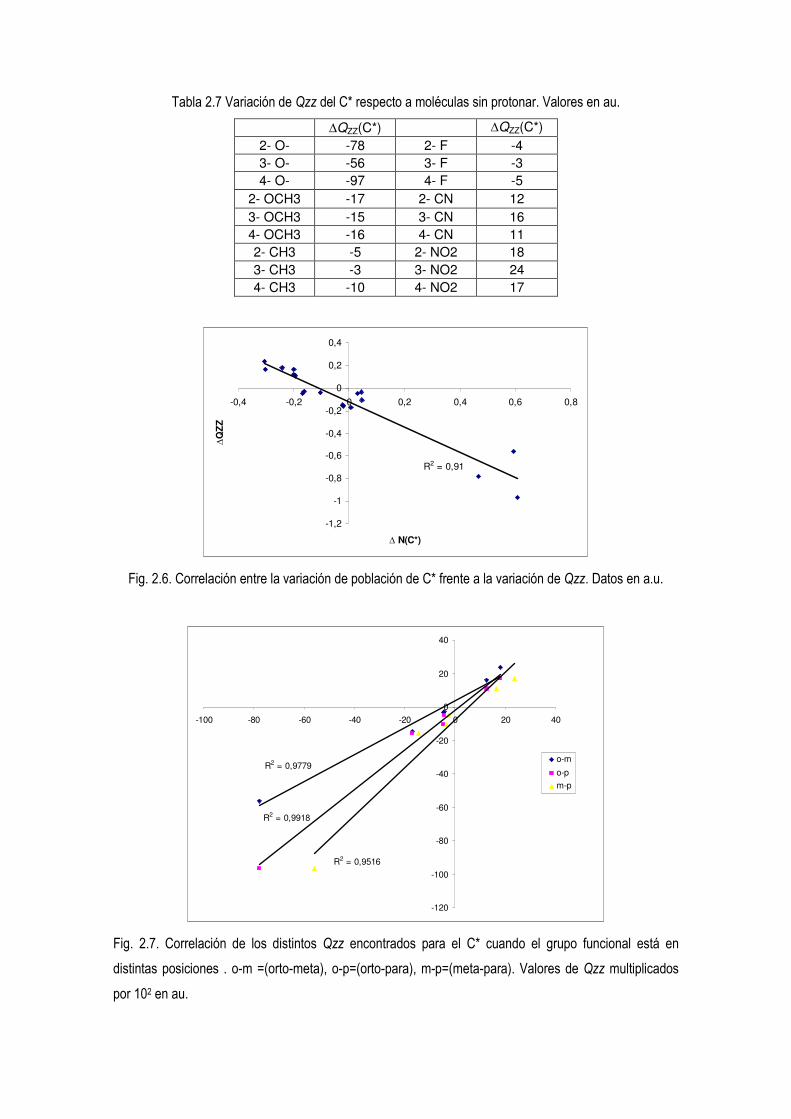
Tabla 2.7 Variación de Qzz del C* respecto a moléculas sin protonar. Valores en au.
∆QZZ(C*) ∆QZZ(C*) 2- O- -78 2- F -4 3- O- -56 3- F -3 4- O- -97 4- F -5
2- OCH3 -17 2- CN 12 3- OCH3 -15 3- CN 16 4- OCH3 -16 4- CN 11 2- CH3 -5 2- NO2 18 3- CH3 -3 3- NO2 24 4- CH3 -10 4- NO2 17
R2 = 0,91
-1,2
-1
-0,8
-0,6
-0,4
-0,2
0
0,2
0,4
-0,4 -0,2 0 0,2 0,4 0,6 0,8
∆ N(C*)
∆Q
ZZ
Fig. 2.6. Correlación entre la variación de población de C* frente a la variación de Qzz. Datos en a.u.
R2 = 0,9516
R2 = 0,9918
R2 = 0,9779
-120
-100
-80
-60
-40
-20
0
20
40
-100 -80 -60 -40 -20 0 20 40
o-m
o-p
m-p
Fig. 2.7. Correlación de los distintos Qzz encontrados para el C* cuando el grupo funcional está en distintas posiciones . o-m =(orto-meta), o-p=(orto-para), m-p=(meta-para). Valores de Qzz multiplicados por 102 en au.

Conclusiones
1.- Las afinidades electrónicas y la energia del protón añadido no muestran una única
correlación general ya que depende del grupo funcional en cuestion.
2.- A medida que aumentamos el carácter activante del grupo funcional, aumenta el
valor de las afinidades protónicas demostrando que aunque la piridina sea π deficitaria
si puede tomar parte en reacciones de adición electrófila cuando el grupo funcional sea
activante.
3.- En todos los compuestos estudiados encontramos la protonoción en el N como la
más favorecida.
4.- Existe una notable concordancia cualitativa entre las predicciones del modelo de
resonancia y las C-protonaciones preferidas para la piridina y sus derivados
monosusitutidos aquí estudiados. No obstante, este modelo no predice adecuadamente
la secuencia de AP que muestran el efecto de la posición del sustituyente sobre la N-
protonación.
5.- Las afinidades protónicas siguen la misma tendencia para una determinada posición
del sustituyente en todos los casos menos para los activantes fuertes (grupos O- y
OCH3) en los que el efecto de la posición es comparable al del sustituyente.
6.- La variación de la población σ del anillo es siempre negativa y resulta ser la
contribución dominante a la pérdida de población global. En los grupos considerados
como activantes aparece un aumento de población π. Esta tendencia se extiende al F, ya
que aunque es considerado un desactivante débil tiene pares de electrones que puede
compartir.
7.- No encontramos correlación entre AP e índices de Fukui.
8.- Los valores del momento cuadrupolar corroboran las conclusiones y tienen buenas
correlaciones con la densidad electrónica del C más susceptible de protonación.

5.3.- Estudio computacional de las preferencias
conformacionales de los dimeros de glicina neutros,
protonados y desprotonados.
La estructura molecular de los aminoácidos ha sido objeto de un gran número
de investigaciones teóricas e experimentales. Los dímeros de los aminoácidos y de otros
clusters de biomoléculas también han recibido una gran atención como modelos para
estudiar los enlaces de hidrógeno (HB), los cuales juegan un importante papel en la
estructura secundaria y terciaria de las proteínas [1-3], reconocimiento molecular [4], y
otros importantes procesos bioquimicos [5]. Más aún la abilidad de diversos dímeros de
aminoácidos para actuar como enlazadores moleculares ha propiciado el interés en su
estructura molecular y de como ésta es afectada por el pH [6].
Glicina, el aminoácido más pequeño y un modelo para compuestos más complejos,
recibió una atención considerable en estudios químicos estructurales que cubren una
amplia gama de propiedades y características. Un tema interesante de los aminoácidos
es como la interconversión entre las formas neutras y zwitteriónicas depende de
compuestos a los que está ligado. Así, mientras que los zwitterions de glicina no se han
encontrado aislados sea en monómeros o dímeros, si que fueron detectados en los
complejos de glicina-agua en fase gas. De hecho, Ramaekers señaló la existencia de
zwitteriones en complejos de agua-glicina n:1 en fase gas por espectroscopia IR [7]. Por
otra parte, la estabilización de zwitterions de glicina en fase gaseosa ha sido analizada
en varios estudios bien por microsolvatación con moléculas de agua [8] o mediante la
adición de iones metálicos [9]. Según Jensen y colaboradores, la complejación de
glicina aislada y no iónica por las moléculas de agua permite trazar una ruta de reacción
para producir glicina zwitteriónica. Este no es un problema sencillo si nos damos cuenta
el gran número de estructuras posibles para los complejos de glicina-agua. Así, una
amplia variedad de estructuras fue creada por Hong [10] para un complejo glicina-agua
(1:1). La complejidad aumenta considerablemente cuando se incluyen 5 moléculas de
agua para inducir la presencia de zwitterions [11].
Los dímeros de glicina neutra y protonada en fase gaseosa han sido objeto de trabajos
anteriores, mientras que no hemos encontrado ningún estudio estructural teórico o
experimental para los aniones. Con disociación radiactiva infrarroja de cuerpo negro

(BIRD) y con el objetivo de medir las energías de enlace de la disociación de
homodímeros de glicina protonada, Gly2H+, Price y colaboradores [12] encontraron que
su estructura más estable contiene un monómero no zwitteriónico que muestra un HB
tipo N-H•••N. En contraste, Raspopov y McMahon, combinando espectrometría de
masas de alta presión y cálculos de MPW1PW91/6-31G(d), informaron que la
estructura más estable de Gly2H+, muestra un HB N-H•••O [13]. La energía de esta
estructura está por debajo de los reportados por Price. Más tarde, Wu y McMahon
compararon los espectros infrarrojos calculados para las tres geometrías más estables de
Gly2H+, con el espectro experimental para establecer que la especie dominante está
formada por una estructura no zwitteriónica neutra y un catión enlazados a través de un
HB tipo H2N+-H···O [14]. Esta estructura también fue propuesta como la más estable
por Atkins y colaboradores, volviendo a analizar preferencias conformacionales de
Gly2H+, en fase gaseosa [15] y por Armentrout y colaboradores, mediante cálculos
MP2(full)/6-311+G(2d,2p)//B3LYP/6-311+G(d,p) [16]. Sin embargo, los autores de
este último, informan también de una nueva geometría de baja energía mostrando un
HB tipo N-H•••N, no totalmente convergente en el cálculo de la frecuencia.
Huang y colaboradores, estudiaron la estabilidad del dímero neutro glicina, Gly2, en
soluciones de agua sobresaturada concluyendo qué monómeros son preferidos en los
dímeros [17], de acuerdo con Hamad y su estudio ab initio de dinámica molecular [18].
Estudios experimentales y teóricos [15,19] permiten concluir que los dímeros Gly2 son
estabilizados por los cationes Na+, factibles en los sistemas biológicos. Análisis
conformacional y computacional para Gly2 se llevaron a cabo en distintos niveles
teóricos. La mayoría de ellos sólo considera fase gas con estructuras no zwitteriónicas
[20,21], mientras que los más recientes también consideran formas zwitteriónico y
solución acuosa [18,22].
El presente estudio amplía los anteriores, teniendo en cuenta dímeros neutros,
protonados y desprotonados, que pueden estar formados por monómeros no iónicos o
zwitteriónicos (ambos en fase gaseosa o en solución acuosa). En todos los casos, se ha
incluido una amplia gama de geometrías con diversos enlaces del hidrógeno para el
muestreo conformacional. De hecho, es crucial analizar un gran número de estructuras
iniciales cuando se trata con superficies con poca diferencia de energía molecular, como
la de los dímeros de glicina. Aunque describir las preferencias conformacionales de
estos sistemas es un objetivo primordial de este trabajo, nos proponemos analizar los
efectos proporcionados por la estructura de monómeros no iónicos o zwitteriónico en

varias propiedades, como son las características ácido/base del dímero de glicina, la
transferencia de electrón densidad entre monómeros, o la fuerza de los enlaces de
hidrógeno intermoleculares. Parte de estos análisis se llevaron a cabo con la teoría
cuántica de los átomos en moléculas (QTAIM) [23].
Detalles computacionales
Con el fin de tomar en cuenta las condiciones de pH diferentes hemos estudiado
dímeros de glicina monoprotonados, no protonados y desprotonados. Se pueden
construir uniendo monómeros diferentes: catión (COOH-CH2-NH3+), C, no iónico neutro
(COOH-CH2-NH2), N o zwitterion (COO--CH2-NH3+), Z y el anión (COO--CH2-NH2), A.
Específicamente, hemos considerado los siguientes dímeros: NC y ZC de especie
protonada; NN, ZZ y NZ de no protonados; y NA y ZA para dímeros desprotonados.
Para cada uno de estos dímeros, hemos construido un conjunto de unas 30 diferentes
geometrías basado en anteriores trabajos de Chocholousova y colaboradores., que
realizó una serie extensa de optimizaciones de MP2/6-31G(d,p) en fase gas, [20], así
como en Carvalho et al [21], y Friant-Michel et al., [22] pero los hemos ampliado
mediante la inclusión de nuevos tipos de enlaces del hidrógeno (HB) entre monómeros.
También hemos tomado en cuenta: i) diferentes posibilidades para formar
intramonomeros HB en N-H•••O = C; ii) rotaciones internas alrededor de los ángulos de
diedro N-C - C = O de cada monómero. Estas conformaciones pueden clasificarse
aproximadamente como: cíclica con mismas o diferentes tipos de hidrógeno
intermoleculares (HBs), lineales (con sólo un HB), apilados o staked (considerando
planos principales aproximadamente paralelos para los esqueletos de monómero, con el
apilamiento debido a HBs) y conformación gauche de monómeros (denotado a veces
como geometrías en forma de T).
Hemos utilizado el paquete gaussian 09 [24] para optimizar todo este tipo de
conformaciones iniciales. La optimización se llevaron a cabo utilizando el densidad
funcional (DFT) método B3LYP, el método DFT meta híbrido M06 y MP2. El conjunto
de base de 6-311++G(d,p) fue utilizado en todos los casos. Se incluyeron funciones de
polarización y difusas para permitir la mejor descripción de los HBs [25,26]. Más de
200 geometrías (sumando aquellos considerados en cada uno de los seis grupos de
dímeros) fueron optimizados para cada nivel computacional y para cada medio. Esto

difiere de lo que se hizo en un trabajo previo donde todas las geometrías empleadas
fueron optimizadas sólo en B3LYP/6-31+G(d,p) en fase gas excepto una geometría
lineal para el dímero ZZ [22]. Cuando las geometrías optimizadas con ambos métodos
son significativamente diferentes hemos recurrido a M06 para contrastar. Cabe destacar
que las optimizaciones de geometrías de varios dímeros, incluyendo uno con monómero
zwitteriónico, fueron especialmente complejas, obligándonos a emplear otros niveles
computacionales para refinar dichas geometrías. También, realizamos cálculos
puntuales MP3 sólo cuando las optimizaciones MP2 revierten las tendencias energéticas
observadas en otros niveles computacionales. Todos los valores energéticos que se
muestra en las tablas incluyen correcciones ZPVE y energía térmicas. El análisis
vibracional realizado ha demostrado que las estructuras optimizadas son mínimos con
ninguna frecuencia imaginaria.
Los dímeros en solución acuosa fueron optimizados haciendo uso del modelo continuo
polarizable (PCM) para tener en cuenta los efectos de la solvatación [27]. En todos los
casos, optimizaciones de PCM se realizaron utilizando las mismas geometrías iniciales
empleadas en cálculos de fase gas. Como no es suficientemente exacto el cálculo con
frecuencias PCM, no hemos estimado ZPVE y correcciones térmicas para la
optimización de las estructuras PCM [28]. Por otra parte, no hemos asumido que las
correcciones de fase de gas podrían ser transferidas en solución acuosa porque a veces
son totalmente diferentes las geometrías
El paquete AIMPAC [29] fue usado para obtener el análisis de densidad electrónica
QTAIM. Todas las propiedades atómicas fueron computadas con un error |L(Ω)| < 2·10-
3 au, lo que normalmente garantiza la conveniencia de las propiedades [30,31]. También
hemos corroborado que la población electronica y la energía son computadas en 10-3 au
and 2 kJ mol-1 respectivamente que las obtenidas por la su suma respectiva de la
población y energías globales ΣN(Ω) and ΣE(Ω). Nuestro estudio también abarca
densidad electrónica en el punto crítico ρb [23].
Resultados y discusión
a) Análisis conformacional.

A continuación se describen las estructuras más estables obtenidas para cada
fase (medio acuoso y fase gas) para el grupo de dímeros (NN, NZ, ZZ, NA, ZA, NC,
ZC) a nivel de MP2/6-311++G(d,p). Hay claras diferencias entre las estructuras e
optimizada en fase gas y las obtenidas en solución acuosa. Cálculos de MP2 y B3LYP
indican energías más negativas en los monómeros neutros de los dímeros en solución
acuosa para los zwitteriones que para estructuras no iónicas en todo el rango de pH,
mientras que la tendencia inversa se observa en fase gas. Por otra parte, las
optimizaciones MP2 y B3LYP generan las mismas geometrías más estables en todos los
casos excepto ZA en medio acuoso y NC en fase gas. Como era de esperar, enlaces del
hidrógeno tipo H2N···H-O y H2N···H-C no se observan, pero sorprendentemente
enlaces tipo H2N···H-N aparecen en varias de las estructuras más estables,
específicamente cuando el ion amonio está presente en medio acuoso, sin importar si
dicho ión amonio tiene un origen zwitteriónico o catiónico. Este tipo de enlace de
hidrógeno aparece, incluso, en la estructura de fase gas más estable para el dímero de
NC.
a.1) Dímeros neutros
Como aquellos previamente obtenidos [20-22] en fase gaseosa, el dímero más estable
está formado por dos monómeros no iónicos (NN) y presenta una estructura cíclica con
dos O-H···O HBs (Fig. 1). Las distintas posiciones de la dos restos -CH2-NH2 dan lugar
a distintos rotámeros cuyas energías relativas abarcan un rango de 8,9 kJ mol-1 de
acuerdo con cálculos de MP2 (10,2 a kJ mol-1 con B3LYP). A nivel computacional, el
rotámero más estable es el que ambos restos muestran el par solitario del N, Lp, en
disposición antiperiplanar al enlace C-C y el enlace N-C synperiplanar a la unidad C =
O. También se encontraron diversas estructuras como mínimos relativos que difieren de
éstos en el esqueleto principal. Incluyen dímeros donde se apilan los monómeros forma
un ciclo de ocho miembros (mostrando HBs N-H···O), dispuestos en forma de T (con
HBs O-H•••O y N-H•••O), formando un ciclo de ocho retorcido con HBs O-H•••O y C-
H•••O, etc.. La secuencia de la energía en ellos difiere significativamente de
optimizaciones B3LYP o MP2. En general, nuestros resultados confirman que los
dímeros cíclicos NN son más estables que arreglos apilados, contrariamente a lo que
había encontrado al explorar la superficie de energía potencial de este sistema [20] con
el potencial empírico de Cornell et al., [32].

La secuencia de estabilidades de los grupos de dímeros según B3LYP es NN> NZ> ZZ
(tabla 3.1). Aunque las optimizaciones MP2 revierten la estabilidad de NZ y ZZ,
cálculos puntuales MP3 de las estructuras MP2 proporcionan la misma secuencia de
energía obtenidos con B3LYP. Se observa que mientras que O-H•••O = C HBs aparecen
en las geometrías preferidas por dímeros de NN, N-H•••O = C y C-H•••O = C se
muestran en el zwitterion que contiene la estructuras más estable (NZ y ZZ) (Fig. 3.1).
Por otra parte, la geometría preferida se empaqueta progresivamente de NN (planar
cíclica) a ZZ (apiladas)
Tabla 3.1. Energías relativas entre los dimeros más estables para diversas zwitteriónicas/no-iónicas especies catiónicas , aniónicas y neutros, ∆1E. Todos los valores (en kJ mol-1) son relativos a la forma más estable no-iónica (NC, NN or NA, respectivamente). [a] valores ZZ-NN. [b] valores NZ-NN. [c] 61,8 kJ mol-1 corresponde a la optimización M06 de la misma estructura angular que la obtenida como más estable según optimizaciones MP2 y B3LYP. Sin embargo, el confórmero más estable de NZ según el estudio M06 es un dimero con estructura stacked, cuyas energia relativa NZ-NN es 59,0 kJ mol-1. [d] El valor positivo (1,0) corresponde a el mismo conformero obtenido como el más estable con B3LYP y MP2 (dispone de un HB N-H···N ). El dímero de NZ más estable obtenidos con M06 para solución acuosa muestra estructura apilada y su valor de NZ-NN es -11.5 kJ mol-1.
∆1E Fase gas Disolución acuosa
dimero catión neutra anión catión neutra anión
B3LYP 10,4 60,6a(47,2)b 7,8 1,7 -34,2a(-20,7)b -15,7
MP2 13,8 47,5a(55,2)b 12,7 -7,6 -34,1a(-19,6)b -23,6
M06 - 68,6a(61,8)b,c - -1,7 -29,8a(1,0)b,d -
MP3 - 85,3a(73,6)b - - - -
Debemos comentar varias diferencias con estudios anteriores, que afectan
exclusivamente a dímeros zwitteriónico (NZ y ZZ): i) la estuctura ZZ más estable en
fase gas siempre es staked, pero la estructura encontradas en este trabajo, ZZ-G1, (Fig.
3.1) es diferente a las obtenidas en trabajos anteriores [22]. De hecho, las segunda y
terceros de las estructuras más estables que se obtienen para este grupo, ZZ-G2 y ZZ-
G3, (Fig. 3.2) que son 1,7 y 6,1 kJ.mol-1 mayor en energía que ZZ-G1, son parecidas a
las geometrías propuestas en dichos trabajos.. Por otra parte, mientras que ZZ-G2 y ZZ-
G3 no presentan transferencia de densidad electrónica significativa entre monómeros,
un monómero retira del otro 4,2·10-3 au en ZZ-G1. Esta secuencia de energías
conformacionales se verifica a niveles B3LYP y MP2. Estructuras ZZ-G2 y ZZ- G3
muestran puntos críticos del enlace intermolecular, (BCPs), asociados al bond path
C•••C o O•••O no observados en ZZ-G1 que revelan una más estrecha proximidad
espacial entre ambos átomos que en las estructuras anteriores.
Fig. 3.1. Estructuras de energia más baja MP2 para dímeros neutros de glicina (ZZ, NZ, NN) en fase gas y solución acuosa. ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas.
78.8 1.45
7.1 2.68
23.7 2.08
74.2 1.50
42.0
ZZ-G1
45.4 1.67
45.2 1.67
0.0
ZZ-W1
51.8 1.74
85.3
NZ-W1
51.9 1.64
72.5 1.50
26.9 2.08
24.9
NZ-G1
43.1
43.1
1.70
1.70
0.0
NN-G1
35.1
5.6
7.3 7.5
11.9
5.3
1.90 2.28
2.87
3.27 2.77
2.67
0.0
NN-W1

+
Fig. 3.2. Geometrias más estables (segundo y tercer lugar) ZZ en fase gas y disolución acuosa (MP2). ρb
(en au multiplicado por 103) y sus correspondientes distancias interatómicas (en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas.
ZZ-G3

ii) la geometría del dímero NZ obtenido como los más estables a nivel MP2, NZ-G1,
difiere significativamente de la geometría staked estudiada previamente, NZ-GS, en el
mismo nivel computacional [22], que básicamente coincide con una de las estructuras
de B3LYP optimizado encontradas en este trabajo (Fig 3.3). Por otra parte, notamos que
todas nuestras pruebas para llevar a cabo optimizaciones MP2 para NZ estructuras
stacked en fase gas optimizaron como NN. Sin embargo, en el NZ nivel B3LYP se
pudieron optimizar las estructuras apiladas sin problemas, y el más estable de ellos sólo
es 10,7 kJ.mol-1 superior.
Fig 3.3. Estructuras B3LYP optimizadas como más estable para fase gas de dimeros NZ. La estructura preferida (basicamente equivalente a la NZ-G1 obtenida con MP2) está a la derecha, mientras NZ-GS es mostrada a la izquierda.
Los estudios computacionales en disolución acuosa de dímeros de glicina son escasos.
Friant-Michel y Ruiz López [22] estudiaron tres estructuras stacked ZZ, una estructura
lineal de NZ y dos NN cíclicas. Hamad y Catlow [18] estudiaron a su vez estructuras
cíclicas, con un doble HB en dimeros ZZ en solución acuosa mediante dinámica
molecular ab initio. En el presente trabajo, el conjunto completo de estructuras en fase
gaseosa fue vuelto a optimizar para medio acuoso con PCM, tanto a nivel B3LYP y
MP2. Se pueden destacar las siguientes normas generales:
i) La secuencia de energia cambia a: ZZ < NZ < NN (Tabla 3.1); ii) las geometrías
más estables optimizadas en PCM son diferentes de aquellas obtenidas en fase gas , por

tanto cabe resaltar que cálculos puntuales PCM basados en geometrías de fase gas no
pueden ser una buena aproximación para describir las preferencias conformacionales en
solución acuosa. La tendencia de este última se ejemplifica en las Fig. 3.1 y 3.2 en las
tres estructuras de energía más bajas en ZZ (las energías relativas ZZ-W2 y ZZ -W3
son, respectivamente, 2,5 y 3,0 kJ.mol-1 a nivel MP2). Las estructuras ZZ preferidas en
solución acuosa son en general más abiertas que las de la fase de gas. Notamos que
mantienen dos HBs N H•••O pero pierden un C-H••O y el intramonomero HB debido a
las interacciones intermoleculares con las moléculas de agua. Además, la geometría
preferida mantiene más densidad electrónica en los átomos más electronegativos.
En dímeros NZ, también observamos que las estructuras predominantes en fase gaseosa
son diferentes que las de medio acuoso. La Fig 3.1 muestra que estas geometrías son
totalmente distintas, incluso, tienen distinta clase de HBs. Así, la geometría más estable
de NZ en solución acuosa muestra un HB N-H•••N (que no se puede establecer entre
las dos unidades de NH3+ en ZZ). Este conformero mostrando este HB es
aproximadamente 4,4 kJ mol-1 más estable que la obtenida por la optimización PCM de
la geometría preferida de la fase de gas (Fig 3.4). Observamos que cada vez que un
dimero solvatado incluye una unidad NH3+, la estructura más estable muestra un HB
N+-H···N. Esto se observa incluso en dímeros de ZC en fase gas. Lo cual permite que
grupos carbonilo sean solvatados por las moléculas de agua, dando como resultado una
mayor estabilidad en el dímero
Varios dímeros NN, con muy distintas geometrías (apilados, cíclicos, lineales y en
forma de T) y hasta diferentes tipos de HBs, muestran energías muy similares de
solución acuosa. También observamos que las geometrías más estables son diferentes
de fase gas a la de solución acuosa (Fig. 3.1). Así, las estructuras apiladas son más
estables que las planas en solución acuosa. Esto puede explicarse teniendo en cuenta
que dejan grupos hidrofílicos para la solvatación con el agua.
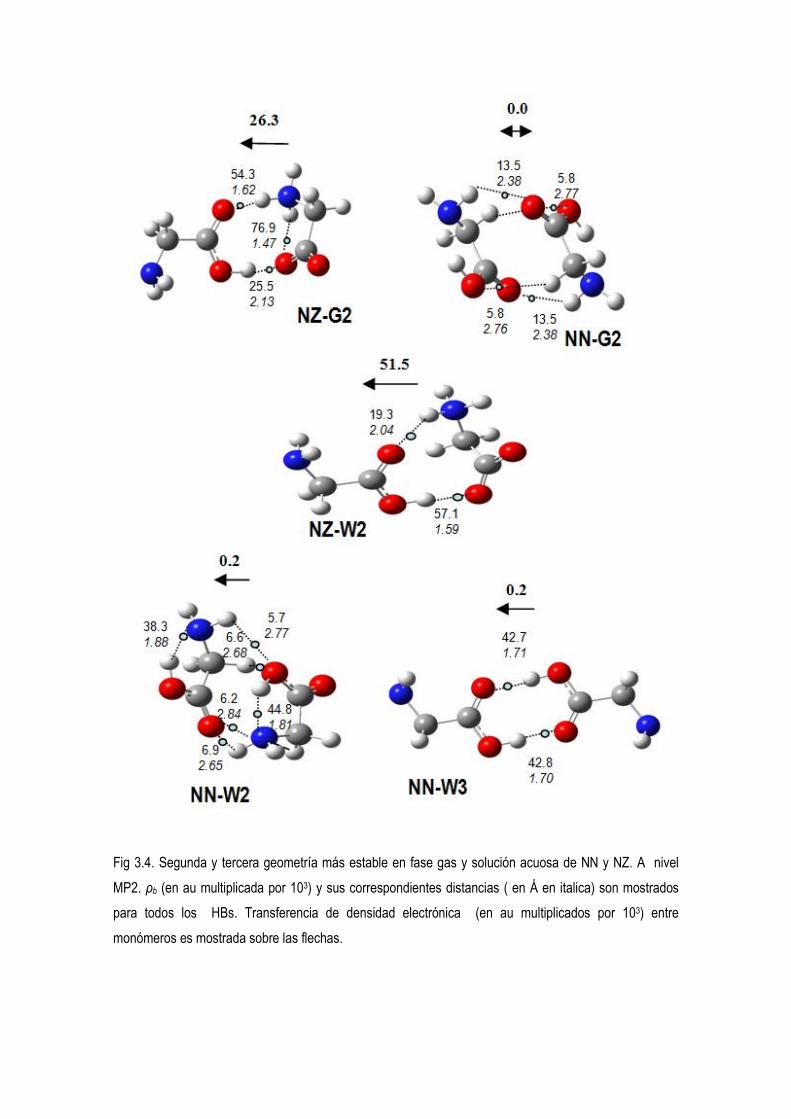
Fig 3.4. Segunda y tercera geometría más estable en fase gas y solución acuosa de NN y NZ. A nivel MP2. ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas.

a.2) Dímeros aniónicos
La primera desprotonation de formas neutras da lugar a dímeros de ZA o NA. En
fase gaseosa los dímeros de NA son preferibles sobre los ZA (tabla 3.1), mientras que
la tendencia inversa se observa en la disolución acuosa. En fase gaseosa, la estructura
más estable de NA muestra O-H•••O y N-H•••O HBs mientras que la estructura más
estable de ZA tiene dos HBs de conexión entre las unidades de NH3+ y OCO- (Fig 3.5).
La energía relativa de la siguiente forma NA estable supera 1 kJmol-1 y muestra un HB
C-H···O en lugar de uno N-H···O (Fig 3.6). Otro tipo de geometrías con el mismo HB
que el primero de ellos, son más lineales, menos estables y muestran una esructura
distorsionada. Esta distorsión es probablemente la razón de la menor estabilidad.
NA-G4 muestra solamente un HB O-H···O, donde el hidrógeno muestra distancias
internuclear absolutamente similares a ambos oxígenos (Fig 3.6). Este hecho, así como
otras propiedades de la densidad electrónica, sugiere que el H es compartido entre los
dos monómeros. Geometrías ZA-G1 y ZA-G2 (con dos N-H•••O HBs compartiendo el
mismo átomo de N) tienen algún tipo de primacía sobre aquellos donde dos tipos
diferentes de HB (N-H•••O y C-H•••O) son observados (ZA-G3 en la fig 6). ZA-G2 es
casi isoenergético con el conformero más estable, ya que sólo difieren en la estructura
alrededor de los enlaces C-C y C-N de un monómero mientras que mantienen el mismo
tipo de HBs. En general, rotaciones alrededor de los ángulos de diedro que mantenga
los mismos HBs no provocan diferencias grandes de la energía de estos dímeros.
Cuando estudiamos aniones en medio acuoso, volvemos a la situación descrita unas
líneas más arriba. Cuando existe el grupo NH3+ (dímero ZA) el HB N+-H···N vuelve a
estar presente en la geometría más estable. Cuando el grupo NH3+ desaparece (dímero
de NA), se establece solamente un O-H···O es la opción que permite una solvatación
más eficiente. En general, la presencia del grupo NH3+ gobierna el comportamiento de
la geometría sobre otros grupos, especialmente en solución acuosa.

Fig. 3.5. Estructuras de más bajas energías MP2 para dimeros aniónicos (ZA,NA) y cationicos (ZC,NC) de glicina ) en fase gas y solución acuosa. ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas.

Fig. 3.6. Otras geometrías importantes en fase gas a nivel MP2. ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas. En la zona del HB O···H···O, se encuentran dos BCPs en el dímero NA-G4. Los valores de ρb y su correspondiente distancia internuclear sugieren que el H es compartido por los dos monómeros. La transferencia de densidad electrónica NA-G4 es nula al compartir el H.

Fig. 3.7. Otras geometrías importantes en solución acuosa para dímeros NA a nivel MP2. ρb (en au
multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los
HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada
sobre las flechas.
ZA en medio acuoso es uno de los casos en donde optimizaciones B3LYP y MP2 no
conducen a las mismas geometrías preferidas. Según MP2 la estructura más estable,
ZA-W1, lleva un fuerte HB N+-H···N y una interacción débil de C-H···C, mientras que
ZA-W2, con un HB N+-H···O y C-H···C es más de 6 kJ mol-1 menos estable. La mayor
estabilidad de ZA-W1 sobre ZA-W2 puede asignarse a la disponibilidad de cuatro
átomos de oxígeno para establecer HBs con moléculas del disolvente, mientras que en
ZA-W2 hay sólo dos oxígenos y un NH3+ disponible para establecerlos. En contraste,
según B3LYP, ZA-W1 (teniendo HBs N+-H···N) son aproximadamente 4 kJ mol-1
menos estable que una estructura similar a ZA-W2, que obtenemos como la geometría

preferida. La descripción conformacional proporcionada por los cálculos M06 está de
acuerdo con MP2. Esto nos lleva a pensar que tal vez B3LYP no realiza una correcta
evaluación para HBs N+-H···N. La serie NA de dímeros (menos estable que ZA en
solución acuosa) contiene cuatro conformadores con energía muy parecida (Fig. 3.7).
Específicamente, NA-W1, NA-W2y NA-W3, sólo difieren en el ángulo diedro que
contiene el HB O-H···O.
a.3) Dímeros catiónicos
Como se detalla en la sección de introducción, estudios previos en dímeros de
glicina protonada (NC y ZC) indican la preferencia por formas NC en fase gaseosa [12-
16]. Aunque Price [12] señaló que la estructura más estable contiene un HB N-H•••N,
parece haberse establecido un consenso sobre la presencia de un HB N-H•••O en el
dímero más estable en los estudios restantes [13-16]. Por último, Armentrout. [19],
empleando MP2(full)/6-311+G(2d,2p) / B3LYP/6-311+G(d,p), informó sobre una
geometría que muestra una baja energía para dímeros NC (no totalmente convergente en
el cálculo de la frecuencia) con un HB N+-H···N, aunque la más estable de las geometría
muestra un HB N+-H···O. En nuestro conjunto de las 30 diferentes geometrías iniciales
consideradas para dímeros NC y ZC se incluyeron todas las geometrías consideradas en
estudios previos.
Recordamos que en cuanto a dímeros aniónicos, las geometrías más estable coinciden
en B3LYP y MP2, excepto cuando aparece el HB N+-H···N (Fig 3.5). Aquí también,
según el nivel computacional (de acuerdo con estudios previos), estructuras de NC son
más estables que ZC en fase gas. La estructura más estable para ZC contiene como HB
N+-H···O y C-H···O. La segunda geometría más estable de ZC es casi isoenergética con
la primera. Se obtiene haciendo girar el HB N+-H···O, lo cual refuerza dicho enlace,
mientras que el C-H···O se rompe. Los más estable de los dímeros NC tiene un HB N+-
H···N. La formación de dímeros de NC con otro HB distinto cuesta por lo menos 3.5 kJ
mol-1, según los cálculos de MP2. Así, observamos una segunda geometría NC con un
HB N+-H···O, (Fig 3.8). Los resultados de MP2 para dímeros de NC en fase gas

contradicen estudios previos experimentales y teóricos, pero nuestro estudio en B3LYP
está de acuerdo con ellos. De hecho la estructura con el HB N+-H···O está 8.2 kJ mol-1
por debajo de la de N+-H···N y la geometría del anterior coincide básicamente con el
ZC-G2, mientras que el de este último es sustancialmente igual a ZC-G1 (Fig. 3.5 y 3.8)
Fig. 3.8. Estructuras de más bajas energías MP2 para dimeros en fase gas calculations. ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas.
En solución acuosa, los resultados MP2 favorecen las formas ZC frente a NC, mientras
la tendencia inversa se observa con B3LYP. Aunque los resultados M06 coinciden en
geometrías con MP2, observamos que la diferencia de energía es ciertamente pequeña, y
teniendo en cuenta las limitaciones de los cálculos en PCM, así como la gran serie de
posibles conformadores NC y ZC, pensamos que no puede descartarse la presencia
simultánea de varios tipos de dímeros de ZC y NC en solución acuosa.

La geometría más estable según MP2 en medio acuoso, es una forma de ZC con un HB
N+-H···O, similar a la que se obtiene como el más estable dentro de la serie de ZC en
fase gas (Fig 3.5). El siguiente confórmero más estable (ZC-W2) resulta de una
distorsión del monómero Z que permite establecer un HB intramonomero N+-H···O. En
cambio, cuando el HBs C-H···O y N+-H···O se mantienen en la estructura solvatada con
agua, la energía aumenta 8 kJ mol-1 (ZC-W3), el cual es menos estable que el más
estable de dímeros de NC (NC-W1). Este conformero NC-W1, muestra un HB N+-H···N
(Fig 3.5). Otros conformeros NC mantienen estos HBs (por ejemplo NC-W2)
mostrando energías relativas muy similares, mientras que geometrías como ZC-W1 (asi
como NC-W3) conducen a energías más altas (al menos 15 kJ mol-1 ) (Fig 3.9).
Fig. 3.9. Estructuras de más bajas energías MP2 para dimeros en medio acuoso . ρb (en au multiplicada por 103) y sus correspondientes distancias ( en Å en italica) son mostrados para todos los HBs. Transferencia de densidad electrónica (en au multiplicados por 103) entre monómeros es mostrada sobre las flechas

b) Tendencias de energía
b.1). Zwitteriones versus monómeros no-iónicos
La relativa estabilidad de zwitteriones respecto a monómeros no iónicos es uno
de los temas más comunes tratados en estudios estructurales realizados en los
aminoácidos en las últimas décadas. La diferencia de energía entre el zwitterion más estable y la estructura no iónico, ∆1E, (Tabla 3.1), permite resumir la información
conformacional presentada en la sección anterior. Valores positivos de ∆1E significan
que la forma más estable es la no-iónica. Esto sucede en fase gas, de acuerdo con los
resultados experimentales. Esta tendencia se invierte en la solución acuosa, donde las
estructuras zwitteriónico se prefieren sobre los que contienen un monómero no iónico
(mostrado en la Tabla 3.1 por valores negativos de ∆1E).
Observamos una excepción. Corresponde a los dímeros catiónicos, donde las estructuras
solvatadas más estables NC y ZC muestran energías muy similares en todos los niveles
computacionales aquí empleados. También es notable que la diferencia de energía entre
dímeros que contienen monómeros de N y Z disminuye en valor absoluto, tanto en fase
gaseosa y en solución acuosa, de dímeros aniónicos y catiónicos en relación con el
neutro (Tabla 3.1).
b.2) Energías de solvatación
En primer lugar debemos mencionar que hemos optimizado la misma colección
de geometrías iniciales de fase gaseosa y en solución acuosa, pero la estructura más
estable no era siempre la misma. La diferencia entre la energía obtenida en solución
acuosa obtenida por PCM y la fase gas, es siempre es negativa, y está representada por
∆2E (Tabla 3.2). Observamos que el valor absolute de ∆2E es mayor en los dímeros
zwitteriónico que en los no iónicos. Esto significa que dímeros que contienen
zwitteriones están más estabilizados por la solucion acuosa que sus correspondientes
isómeros no iónicos. Por el contrario la diferencia de energía más baja, |∆2E|, es
observada en dímeros neutros, mientras que los cationes son más estabilizados en

solución acuosa que los aniones. Se espera que las energías de hidratación de los
cationes sean más negativas que las de aniones de tamaño similar [33]. La razón de esta
tendencia es que el número de enlaces de hidrógeno rotos cuando solvatamos un catión
en agua es menor que para solvatar un anión de tamaño similar. Sin embargo, la energía
en romper el HBs entre las moléculas del solvente no es tenida en cuenta por PCM
(incluso en el término de cavitación no electrostático [27]). Buscando una explicación,
hemos considerado los términos polarizarion y distorsión que se muestra en la tabla 3.
Esta repartición revela el protagonismo de la polarización. La mayor polarización se
muestra por las formas aniónicas (ZA) que también experimentan la mayor deformación
de la geometría. Como ambos efectos se contrarrestan mutuamente, la energía de
solvatación más grande corresponde a ZC, aún cuando los efectos de solvente no se
consideran explícitamente.
Tabla 3.2. Energias de solvatación, -∆2E, (kJ mol-1) obtenidas por la diferencia entre la energia de la estructura solvatada más estable menos la más estable de la fase gas .No se incluyen correciones térmicas.
B3LYP MP2 M06
NC 295,3 288,9
ZC 304,5 311,6
NN 55,4 59,4 13,2
NZ 134,5 134,7 81,7
ZZ 159,9 142,0 110,1
NA 251,3 241,1
ZA 271,9 279,3

Tabla 3.3. Términos de Deformación, Polarización , y no electrostático de la Energía de solvatación en MP2, en solución acuosa.. Valores en kJ mol-1. [a] Este término no se incluye en ∆2E de la tabla 3.2.
Deformación Polarización No
electrostaticoa
NC 65,2 -354,1 30,3
ZC 75,0 -386,6 31,1
NN 57,8 -117,2 34,3
NZ 117,2 -251,8 33,8
ZZ 100,3 -242,2 32,9
NA 57,5 -301,8 38,3
ZA 147,0 -426,3 33,3
La secuencia de energías de polarización (tabla 3.3) se puede explicar teniendo en
cuenta tres factores: i) la carga total de la molécula, la cual cuando se mantienen
constantes otros factores debería conducir a una secuencia que favorece aniones sobre
cationes sobre especies neutrales; II) la separación de carga dentro del sistema,
favoreciendo formas zwitteriónicas sobre los no iónicos; y iii) la geometría del dimero,
produciendo energías de polarización más grandes cuando la mayoría los grupos son
externos. De acuerdo con esto último los confórmeros con HBs N-H•••N son más
propensos a aumentar su energía de polarización que los que incluyen HBs O-H•••O.
Esto explica por qué la energía de polarización es para el conformero más estable de
dímeros NC más grande que la de NA.
Fig.3.10. Estructuras más estables obtenidas con PCM pra dímeros ZC (MP2 izquierda, B3LYP derecha).

Tabla 3.4. Energias de protonación y desprotonación (en kJ mol-1) para los confórmeros más estables de dímeros de glicina a nivel MP2 y B3LYP. (Energía anión o catión menos neutra)
MP2 B3LYP MP2 B3LYP
Proceso
Protonacion
Gas Gas Disol.
Ac.
Disol.
Ac..
NZ>CZ=ZC -991,7 -987,8 -1204,5 -1195,2
NZ>NC -1005,4 -998,2 -1196,9 -1196,8
ZZ>ZC -984,0 -1001,3 -1189,9 -1181,7
NN>NC -950,2 -951,0 -1221,5 -1232,5
Proceso
Desprotonacion
Gas Gas Disol.
Ac.
Disol.
Ac.
NZ>AZ=ZA 1320,4 1313,9 1211,4 1210,0
NZ>NA 1306,3 1306,1 1235,0 1225,7
ZZ>ZA 1328,1 1300,4 1226,0 1223,5
NN>NA 1362,9 1353,3 1210,3 1190,0
b.3) Habilidad para protonar y desprotonar.
Hemos comparado las habilidades de estructuras iónicas zwitteriónico y no
iónicas para ser protonadas o desprotonadas. Tabla 3.4 lista las energías de protonación
y desprotonación para la fase de gas y disolución acuosa computada a niveles MP2 y
B3LYP. Observamos que los zwitteriones neutros son más propensos a ser protonados o
desprotonados que formas no iónicos en fase gaseosa, mientras que la tendencia inversa
se observa en disolución acuosa. Esto se observa tanto cuando se compara la capacidad
de protonación/desprotonación de monómeros de N y Z en dímeros de NZ que cuando
la comparación se establece entre dímeros NN y ZZ. El único caso donde el nivel
computacional modifica los resultados es la protonación de dímeros NZ en disolución
acuosa. Esta excepción puede estar relacionada con las diferencias significativas

obtenidas entre optimizaciones B3LYP y MP2 para las estructuras más estables de la
formas de ZC (Fig. 3.10).
c) Análisis de la densidad electrónica
c.1) Propiedades de enlace
Todos los dímeros obtenidos como los más estables en cada serie se estabilizan a
través de HBs. Dependiendo de la serie y fase, se observan tres tipos de HB fuertes: O-
H•••O, N-H•••O o N-H•••N. En algunos casos se combinan con HBs débiles C-H•••O.
Todas ellas están representadas por los puntos críticos de enlace, BCPs, en los gráficos
moleculares QTAIM [23]. BCPs correspondiente al enlace C-H•••C y C•••O se
observan también en algunos casos particulares dímeros solvatados (ZA y ZC,
respectivamente). Además, bond paths asociados a enlaces del hidrógeno
intramoleculares N-H•••O y O-H•••N se observan en varios monomeros (Fig. 3.1-3.9).
Valores de densidades electrónicas en el BCP, ρb, generalmente son tomados como
indicadores relativos de la fuerza del enlace dentro de una serie de enlaces similares (se
muestran en la tabla 3.5 y 3.6). Notamos que HBs O-H•••O sólo están presentes en
dímeros que contienen un monómero no iónico neutro, N, y su ρb es más grande cuando
no hay otro HB en el dímero (NA-W1). Mas aún valores ρb para HBs O-H···O, decrecen
progresivamente cuando la fuerza de otro HB en el dímero aumenta. Así ρb para O-
H···O es 0,080 au en NA-G2, (acompañado por un HB C-H···O); 0,073 au en NZ-G1
(acompañado por N-H···O HB) y 0,040 au en NN-G1 (acompañado por otro HB O-
H···O). Este efecto puede estar relacionado con las distorsiones de la geometría de la HB
O-H•••O. Un buen ejemplo es proporcionado por la serie de dímeros de NA más
estables en solución acuosa: NA-W1, NA-W2 y NA-W3 donde la forma más estable
(con un ángulo diedro de 90º entre ambos grupos carboxílicos) , tiene el mayor valor ρb
y la distancia de enlace H···O más pequeña (Fig. 3.5 y 3.7). Rotaciones internas de los
grupos carboxílicos incrementan los valores del enlace H···O y reducen valores de ρb.
N-H···O es el HB más comunmente observado en la variedad de dímeros de glicina
estudiados. Sus valores de ρb van desde 0,081 au (similar al valor más grande de HB O-
H···O) en ZC-G1 y 0,013 au en NN-G2. Estos valores dependen de la longitud de
enlace H•••O y del ángulo de enlace N-H•••O. Así, los valores mínimos se observan en

geometrías apiladas. Un caso particular se observa para las estructuras más estables del
dimero ZA en fase gas (ZA-G1 y ZA- G2), donde están involucrados dos enlaces N-H
del monómero zwitteriónico con los dos oxígenos del anión (Fig. 3.5 y 3.6). Analizando
valores de ρb, observamos que la estructura preferida tiene valores intermedios (0,058 y
0,024 ua) entre las observadas para ZA-G3 (0,085 y 0,009 au). HBs tipo N-H•••N
muestran valores de ρb similares a los de la mayoría de los N-H•••O (alrededor de 0,06
ua, véase por ejemplo ZC-W1 y NC-W1) y están presentes en las estructuras más
estables del dimero de glicina conteniendo un (y sólo un) grupo NH3 +. Este HB se
observa más a menudo en solución acuosa ya que proporciona mayor separación entre
grupos COO - /COOH que permite solvataciones de ambos grupos de moléculas, junto
con un HB relativamente fuerte.
Algunos ejemplos impiden considerar una relación directa entre la forma preferida y los
valores de ρb. Así, en fase gaseosa, la más estable conformer ZZ muestra valores ρb
mayores que el NN, a pesar de que NN es la forma preferida en dímeros neutros en fase
gas (Tabla 3.1). En contraste, en solución de agua, los dímeros neutros preferidos (ZZ)
muestran valores ρb mayores que NN.
Los HB Intramonomérico (IMHB) están relacionados con la presencia de ciertos
arreglos coplanares. Puede ocurrir entre el carboxilato y uno de los enlaces N-H
perteneciente a un grupo NH3+ (el más usual IMHB en dímeros de glicina), o entre el
par solitario del nitrógeno y el grupo COOH (sólo observado en NC-G2 y NN-W1)
(Fig. 3.11).
Figura 3.11. Disposiciones moleculares favoritas de IMHBs (A and B) y conformaciones preferidas sin IMHB (C).
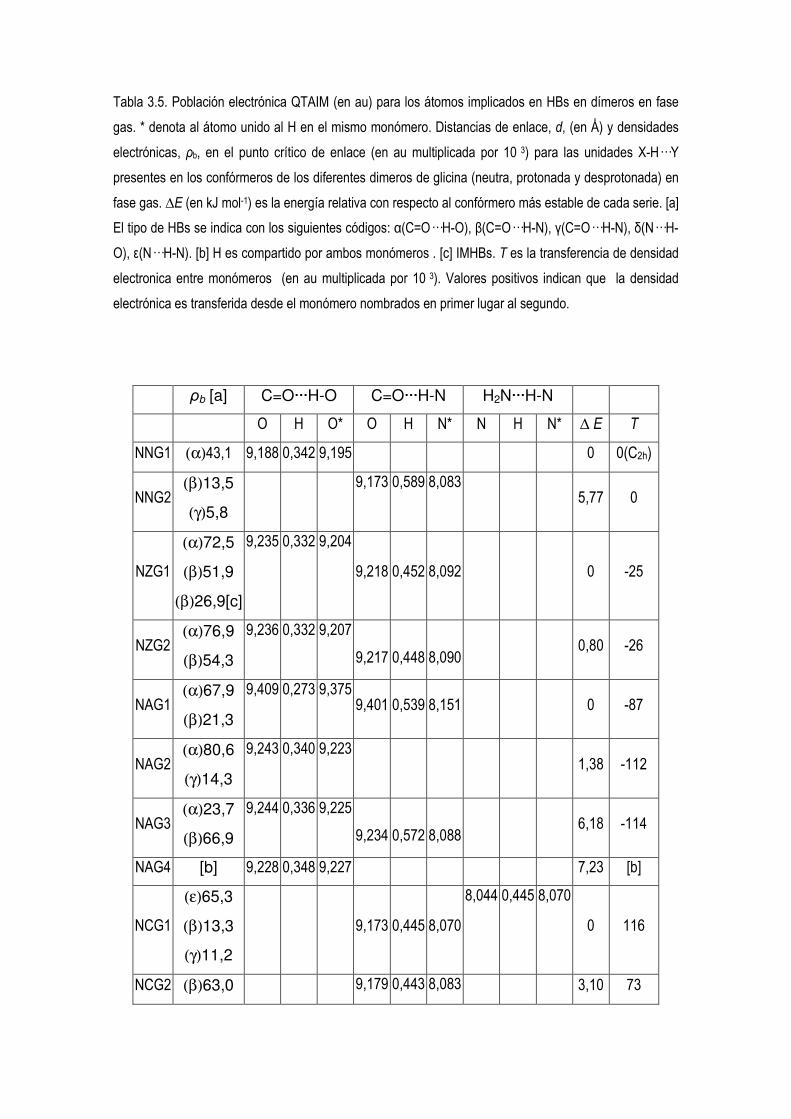
Tabla 3.5. Población electrónica QTAIM (en au) para los átomos implicados en HBs en dímeros en fase gas. * denota al átomo unido al H en el mismo monómero. Distancias de enlace, d, (en Å) y densidades electrónicas, ρb, en el punto crítico de enlace (en au multiplicada por 10 3) para las unidades X-H···Y presentes en los confórmeros de los diferentes dimeros de glicina (neutra, protonada y desprotonada) en fase gas. ∆E (en kJ mol-1) es la energía relativa con respecto al confórmero más estable de cada serie. [a] El tipo de HBs se indica con los siguientes códigos: α(C=O···H-O), β(C=O···H-N), γ(C=O···H-N), δ(N···H-O), ε(N···H-N). [b] H es compartido por ambos monómeros . [c] IMHBs. T es la transferencia de densidad electronica entre monómeros (en au multiplicada por 10 3). Valores positivos indican que la densidad electrónica es transferida desde el monómero nombrados en primer lugar al segundo.
ρb [a] C=O···H-O C=O···H-N H2N···H-N
O H O* O H N* N H N* ∆ E T
NNG1 (α)43,1 9,188 0,342 9,195 0 0(C2h)
NNG2 (β)13,5
(γ)5,8
9,173 0,589 8,083 5,77 0
NZG1
(α)72,5
(β)51,9
(β)26,9[c]
9,235 0,332 9,204
9,218 0,452 8,092 0 -25
NZG2 (α)76,9
(β)54,3
9,236 0,332 9,207 9,217
0,448
8,090
0,80 -26
NAG1 (α)67,9
(β)21,3
9,409 0,273 9,375 9,401 0,539 8,151 0 -87
NAG2 (α)80,6
(γ)14,3
9,243 0,340 9,223 1,38 -112
NAG3 (α)23,7
(β)66,9
9,244 0,336 9,225 9,234
0,572
8,088
6,18 -114
NAG4 [b] 9,228 0,348 9,227 7,23 [b]
NCG1
(ε)65,3
(β)13,3
(γ)11,2
9,173 0,445 8,070
8,044 0,445 8,070
0 116
NCG2 (β)63,0 9,179 0,443 8,083 3,10 73

(δ)48,0[c]
ZZG1
(β)78,8
(β)74,2
(γ)7,1
(β)23,7[c]
9,225 9,216
0,432 0,441
8,084 8,094
0 4
ZZG2 (β)81,2
9,223 0,438 8,085
1,70 0(Ci)
ZZG3
(β)70,4
(γ)7,2
(β)25,7[c]
9,223 0,439 8,094
6,13 0(Ci)
ZAG1
(β)57,8
(β)21,9
(β)4,43[c]
9,245 9,238
0,542 0,465
8,146 8,146
0 -106
ZAG2
(β)53,4
(β)24,3
(β)44,1[c]
9,245 9,240
0,533 0,471
8,149 8,149
0,57 -104
ZAG3
(β)84,5
(γ)8,5
(β)41,1[c]
9,249 0,432 8,114
12,91 -120
ZCG1
(β)81,3
(γ)12,5
(β)58,2[c]
9,194 0,427 8,088
0 114
ZCG2 (β)90,5
(β)60,4[c]
9,192 0,427 8,087 0,50 122

c.2) Transfererncia de densidad electrónica entre monómeros
El cálculo de las propiedades atómicas de QTAIM permite analizar la cesión de
densidad electrónica entre monómeros que puede tener lugar en la formación de
dímeros (Fig. 3.1-3.9). Poblaciones electrónicas atómicas, N(Ω), para los átomos
involucrados en enlaces de hidrógeno intermoleculares se muestran en las tablas 3.5 y
3.6, respectivamente, en fase gas y solución acuosa. Notamos que la mayoría de
dímeros neutros muestran cero o insignificante transferencia de densidad electrónica
(T) entre monómeros. En algunos casos de nula transferencia es debido a las estructuras
simétricas, como NN-G1.
Hay cuatro excepciones donde la transferencia es importante, y todos ellos han sido
encontrados en dímeros NZ en gas o en solución acuosa. En tres de ellas la densidad
electrónica va desde Z al monómero N (NZ-G1, NZ -G2 y NZ-W2), pero en otros casos
observamos que la es transferencia inversa (NZ-W1). En cualquier caso, mirando los
HBs establecidos, observamos que los dímeros donde la transferencia de electrones es
de Z a N comparten el mismo conjunto de HBs. Éstos muestran un HB H•••O-N donde
la H es donado por el grupo zwitteriónico NH3+ al oxígeno carboxílico y un O-H•••O
con monómeros neutros donando el H carboxílico. En contraste, NZ-W1 muestra una
interacción de N-H•••N. Comparando valores de ρb de HBs N-H•••O y O-H•••O en NZ-
G1 y NZ- G2, con transferencias electrónicas muy similares, indican que la distorsión
del HB O-H•••O tiene poco efecto sobre la transferencia de la densidad electrónica.
ZZ-G1 muestra una transferencia pequeña (0,004 ua) debido a la falta de simetría (Fig.
3.1), ya que la orientación de los grupos CH2 es diferente en cada monómero.

Tabla 3.6. Población electrónica QTAIM (en au) para los átomos implicados en HBs en dímeros en disolución acuosa. * denota al átomo unido al H en el mismo monómero. Distancias de enlace, d, (en Å) y densidades electrónicas, ρb, en el punto crítico de enlace (en au multiplicada por 10 3) para las unidades X-H···Y presentes en los confórmeros de los diferentes dimeros de glicina (neutra, protonada y desprotonada) en fase gas. ∆E (en kJ mol-1) es la energía relativa con respecto al confórmero más estable de cada serie. [a] El tipo de HB se indica con los siguientes códigos: α(C=O···H-O), β(C=O···H-N), γ(C=O···H-N), δ(N···H-O), ε(N···H-N). [b] H es compartido por ambos monómeros . [c] IMHBs. T es la trnsferencia de densidad electronica entre monómeros (en au multiplicada por 10 3). Valores positivos indican que la densidad electrónica es transferida desede el monómero nombrados en primer lugar al segundo
ρb[a] C=O···H-O C=O···H-N H2N···H-N
O H O* O H N* N H N* ∆ E T
NNW1
(β)11,9
(β)7,3
(γ)7,5
(γ)5,6
(δ)35,1[b]
9,336 9,371
0,602 0,584
8,170 8,207
0 0
NNW2
(β)6,9
(γ)6,2
(β)6,6
(β)38,3[b]
(β)44,8[b]
9,204
9,187
0,591
0,595
8,099
8,097 0,29 0
NNW3 (α)42,7 9,199 0,343 9,203 5,11 0(C2h)
NZW1 (ε)51,8 8,070 0,468 8,090 0 85
NZW2 (α)57,1
(β)19,3
9,258 0,345 9,216 9,215
0,500
8,059
4,41 -52
NAW1 (α)87,4 9,267 0,340 9,245 0 -109
NAW2 (α)83,6 9,258 0,339 9,244 1,48 -105
NAW3 (α)82,1 9,271 0,339 9,242 1,68 -99
NAW4 (α)66,3
(β)15,6
9,281 0,331 9,236 9,211
0,622
8,097
2,05 -72
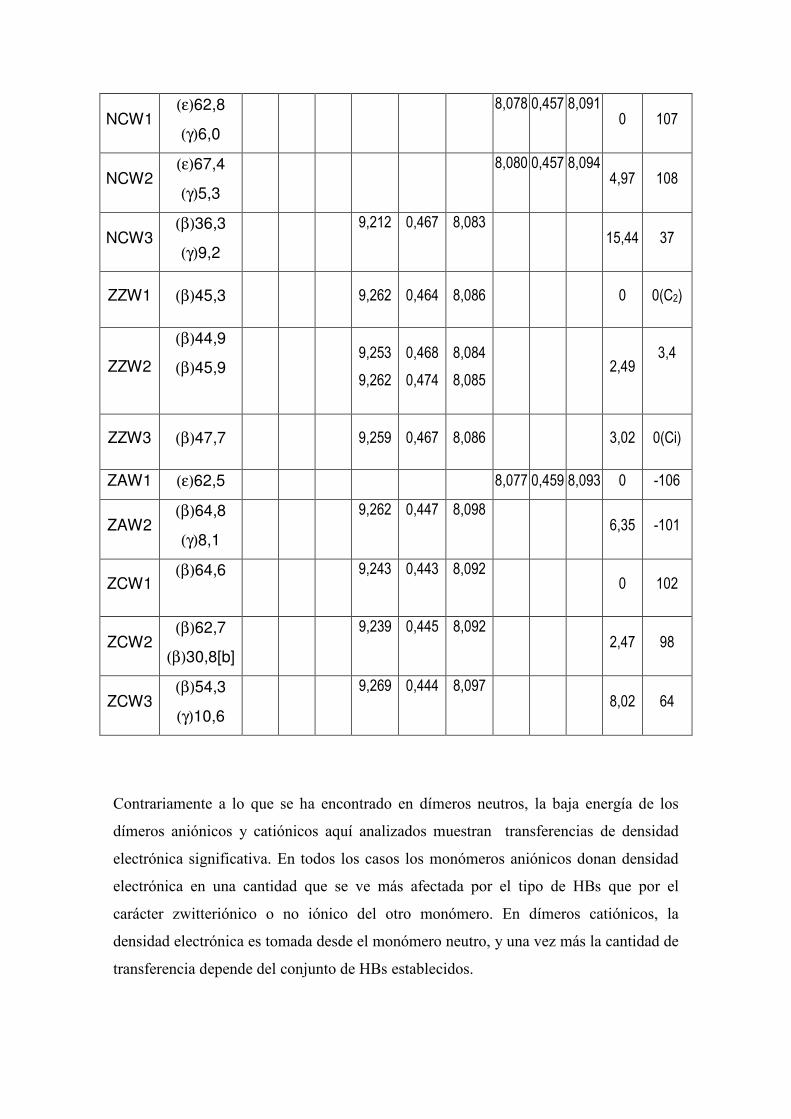
NCW1 (ε)62,8
(γ)6,0
8,078 0,457 8,091 0 107
NCW2 (ε)67,4
(γ)5,3
8,080 0,457 8,094 4,97 108
NCW3 (β)36,3
(γ)9,2
9,212 0,467 8,083 15,44 37
ZZW1 (β)45,3
9,262 0,464 8,086
0 0(C2)
ZZW2
(β)44,9
(β)45,9
9,253 9,262
0,468 0,474
8,084 8,085
2,49 3,4
ZZW3 (β)47,7
9,259 0,467 8,086
3,02 0(Ci)
ZAW1 (ε)62,5 8,077 0,459 8,093 0 -106
ZAW2 (β)64,8
(γ)8,1
9,262 0,447 8,098 6,35 -101
ZCW1 (β)64,6
9,243 0,443 8,092 0 102
ZCW2 (β)62,7
(β)30,8[b]
9,239 0,445 8,092 2,47 98
ZCW3 (β)54,3
(γ)10,6
9,269 0,444 8,097 8,02 64
Contrariamente a lo que se ha encontrado en dímeros neutros, la baja energía de los
dímeros aniónicos y catiónicos aquí analizados muestran transferencias de densidad
electrónica significativa. En todos los casos los monómeros aniónicos donan densidad
electrónica en una cantidad que se ve más afectada por el tipo de HBs que por el
carácter zwitteriónico o no iónico del otro monómero. En dímeros catiónicos, la
densidad electrónica es tomada desde el monómero neutro, y una vez más la cantidad de
transferencia depende del conjunto de HBs establecidos.

Diversos estudios de análisis conformacional realizados previamente en una serie de
complejos con HBs simples [34-36], concluyen que las energías relativas pueden
explicarse sobre la base de la suma de poblaciones electrónicas atómicas de hidrógenos.
Este no es el caso (Tabla 3.7), dada la disparidad de HBs (y otros factores) aquí
involucrados. Notamos que las geometrías más estables en fase gas muestran la mayor
suma de la población electrónica atómica de los hidrógenos. Sin embargo, esta
tendencia no se sigue en la solución acuosa, donde las formas más estables muestran los
valores N(O) más grandes. La suma de los valores de N(H) en el monómero neutro
muestra una clara dependencia con la carga del otro monómero adjunto. Son reforzados
por los aniones y debilitados por los cationes (Tabla 3.7).
Dos efectos debidos al medio afectan a todos los dímeros: a) La suma de N(H) en cada
dímero es siempre más grande en fase gas que en disolución acuosa; b) Al contrario, la
suma N(O) es más pequeña en fase gas. Todo esto es consecuencia directa de la
solvatación al ser favorecida al incrementar el valor de la carga atómica (Tabla 3.7).
Tabla 3.7. Población electrónica global de H (in au) en diversos dímeros en fase gas NG, y en disolución acuosa NA.
NN1 NN2 NN3 NZ1 NZ2 ZZ1 ZZ2 ZZ3
NG 7,072 6,978 6,982 6,983 6,884 6,918 6,850
NA 6,829 6,828 6,923 6,798 6,831 6,693 6,711 6,703
NA1 NA2 NA3 NA4 ZA1 ZA2 ZA3
NG 6,880 6,881 6,875 6,983 6,789 6,786 6,805
NA 6,735 6,738 6,698 6,737 6,587 6,621
NC1 NC2 NC3 ZC1 ZC2 ZC3
NG 7,125 7,071 7,023 7,064
NA 6,964 6,957 6,980 6,923 6,906 6,879

Conclusiones
Una gran colección de estructuras fueron optimizadas para diferentes series de
dímeros de glicina (neutros, aniónicos y catiónicos) con el fin de averiguar cuáles eran
los más estables. Se encuentran nuevas geometrías más estables, no descritas
previamente, para la serie ZZ en solución acuosa. La secuencia de estabilidad para
dímeros stacked, cíclicos y lineales depende de la carga total del dimero y de la fase.
Según los resultados de MP2, los dímeros más estables que incluyen un grupo NH3 +
muestran N•••H-N HBs. Esta preferencia conformacional no se confirma a nivel B3LYP
en todos los casos. Particularmente, en la serie del NC, optimizaciones de B3LYP,
indican que el dímero más estable muestra un HB O•••H-N, de acuerdo con todas las
interpretaciones publicadas de los espectros de IR en Gly2H + [12,14,16]. Los valores de
energía muestran: i) la dificultad de encontrar zwitterions en fase gaseosa; y ii) La
solvatación en agua es mayor cuando los dímeros contienen un monómero de Z que
para aquellos con monómeros N; iii) Z monómeros son más factibles a experimentar
procesos de protonación y desprotonación que N en gas fase, mientras que la tendencia
inversa se observa en la solución acuosa.
En general, HBs de O•••H-O muestran las mayores densidades electrónicas en el
correspondiente BCPs (hasta 0,08 au), mientras que esta cantidad es generalmente más
baja para HBs como O•••H-N (abarcando una amplia gama de valores dependiendo de
la presencia de otros HBs en el dímero) y N•••H-N (alrededor de 0,06 ua). HBs como
N•••H-O y N•••H-C no son encontrados en ninguna estructura optimizada, pero enlaces
O•••H-C se encuentran acompañando a otros HBs. También hemos observado que los
valores generales de ρc son generalmente más pequeños en disolución acuosa que en
fase gas, indicando que la solvatación debilita los HBs, como es observada en otros
trabajos [37]. En contraste, la población electrónica del hidrógeno que pertenece a este
HB aumenta en la solución acuosa con respecto a la de fase gas.
La mayor parte de los dímeros cargados muestran una transferencia electrónica
importante entre monómeros (alrededor de 0,1 ua). El monómero aniónico actúa como
donante de densidad del electrón (ZA o NA), mientras que el catión actúa como aceptor
(ZC o NC). En contraste, la mayor parte de los dímeros neutros muestran cero o
insignificante transferencia entre monómeros.

5.4 Estudio de la Densidad Electrónica en Dímeros de Glicina
Desprotonados.
Los dímeros de aminoácidos y otros grupos de biomoléculas han recibido gran
atención ya que son modelos simples para el estudio de enlaces de hidrógeno
intermoleculares y juegan un papel importante para estructuras de proteínas secundaria
y ternarias [1-3], reconocimiento molecular [4] y otros importantes procesos
bioquímicos [5]. Por otra parte, la capacidad de diversos dímeros de aminoácidos para
actuar como elementos de conexión moleculares ha motivado un alto interés por su
estructura molecular y cómo ésta es afectada por el pH [6].
Glicina, el aminoácido más pequeño y un modelo para compuestos más complejos,
recibió considerable atención en los estudios químicos estructurales que cubren una
amplia gama de propiedades y características [7-12]. Dímeros de glicina en fase gaseosa
neutros y protonados han sido objeto de trabajos previos [13-16], mientras que no
hemos encontrado ningún estudio estructural teórica o experimental para los aniones. Es
bien sabido que glicina no protonada se encuentra como zwitterion (Z) en solución
acuosa y una especie no iónica (N) en fase gas. Mientras que no se han encontrado
zwitteriones de glicina en monómeros aislados, se detectaron en complejos de glicina-
agua en fase gas. Por lo tanto, deben considerarse dos tipos de dímeros, dependiendo de
la naturaleza del monómero desprotonado: Z o N.
Teniendo en cuenta las suaves superficies de energía molecular que caracteriza a
dímeros de glicina sin carga [16], el presente estudio tendrá lugar en un conjunto amplio
de geometrías adecuadas para nuestro muestreo conformacional. Así, hemos
considerado diversos enlaces del hidrógeno entre monómeros, dando lugar a estructuras
lineales, cíclicas, apiladas y en forma de T, también, se realizaron rotaciones internas
alrededor de los ángulos de diedro N-C-C=O de cada monómero. Con el objetivo de
analizar el origen de las preferencias conformacionales para este sistema, analizar la
transferencia de electrón densidad entre monómeros y para evaluar la fuerza de los
enlaces intermoleculares de hidrógeno, se ha realizado un análisis de densidad
electrónica con la teoría cuántica de los átomos en moléculas (QTAIM) [17].

Detalles computacionales
Dímeros desprotonados de glicina pueden ser construidos por unión de un
monómero aniónico (COO--CH2-NH2), A, a uno no iónico neutro (COOH-CH2-NH2),
N, o a un zwitterion (COO--CH2-NH3+), Z. Así, podemos distinguir entre dímeros NA
y ZA, que fueron estudiados en fase gaseosa y en disolución acuosa, utilizando el
modelo continuo polarizado (PCM) [18]. Un conjunto diferente de conformaciones
iniciales fueron optimizadas para especies NA y ZA. Estos sistemas se basan en los
conformeros obtenidos en dímeros no protonados y protonados de trabajos previos [13,
14,16] y fueron ampliados mediante la inclusión de diversos tipos de enlaces del
hidrógeno (HB) entre monómeros
Se llevaron a cabo optimizaciones completas de geometría, en sistemas aislados y en
PCM, con el paquete Gaussian 09 [19] a nivel MP2 y con el método de funcional
densidad (DFT), B3LYP. Sin embargo, en aras de la concisión, sólo utilizaremos el
segundo nivel computacional cuando los resultados obtenidos son significativamente
diferentes. El conjunto base 6-311++G(d,p) fue utilizado en todos los casos. Se
incluyeron funciones difusas y de polarización para permitir la mejor descripción de
HBs [20, 21].
Los confórmeros son nombrados por acrónimos formados por letras latinas indicando la
disposición de los principales ángulos diedros en el monómero y letras griegas para
referirnos al tipo de HBs establecidos entre los monómeros. La primeroa letras del
acrónimo corrresponde a los mónomeros neutros (N o Z), luego seguido va el
descriptor del HB y finalmente la letra describiendo el ángulo diedro del monómero A.
La secuencia específica de los ángulos diedros para el monómero N son las unidades
Lp-N-C-C, N-C-C-O, y C-C-O···O* (con Lp=par solitario); y para el monómero A las
unidades Lp-N-C-C y N-C-C-O* (O* es el oxígeno implicado en el HB en A). Cuando
el primer monómero es Z , la primera letra se refierer a la unidad H*-N-C-C (H*es el H
implicado en el HB). En todos los casos, c, g, s, e, t, e’, s’ y g’ representan,
respectivamente, valores cercanos a 0, 60, 90, 120, 180, -120, -90, y -60 grados. El
código para los tipos de HB es: α= O-H···O, β= N-H···O, γ= C-H···O, δ= N-H···N.
Todos los valores de energía se mostrados en tablas para la fase de gas incluyen
correcciones térmicas a 298,15 K. El análisis vibracional realizado ha demostrado que

las estructuras optimizadas son mínimos con ninguna frecuencia imaginaria. Como no
es suficientemente exacto el cálculo con frecuencias PCM, no hemos estimado ZPVE
ocorrecciones térmicas para dicho cálculo [22]. Por otra parte, no hemos asumido que
las correcciones de fase de gas podrían ser transferidas en solución acuosa porque a
veces son geometrías totalmente diferentes.
El paquete AIMPAC [23] fue usado para realizar el análisis de la densidad electrónica
QTAIM. Todas las propiedades atómicas son computadas con |L(Ω)| < 2·10-3 au, lo cual
es una garantia de su calidad [24, 25]. También hemos comprobado que la población
electrónica y la energía estuviera en 10-3 au y 2 kJ mol-1, respectivamente, cuando
sumanos el global de poblaciones atómicas y energía: ΣN(Ω) y ΣE(Ω). Nuestro estudio
en el análisis de la población electrónica emplea propiedades atómicas y propiedades de
enlace QTAIM computadas en el punto crítico de enlace BCP, como son la densidad
electrónica, ρb, y el total de la energía electrónica.
Resultados y Discusión
a) Análisis Conformacional en fase gas
Como lo analizado en dímeros de glicina neutros y catiónicos [16], la mayoría
de confórmeros de NA son más estable que ZA en fase gas. Debido a la gran cantidad
de posibles mínimos nos referiremos sólo a las estructuras que son significativamente
diferentes. Particularmente, cuando la unidad N-C-C=O no es estrictamente plana pero
su ángulo diedro se desvía de planaridad un poco (digamos que por menos de 20º),
observamos la presencia de mínimos que sólo se diferencian en este ángulo diedro o son
casi enantiómeros. En aras de la claridad, todas estas estructuras están representadas en
este estudio por el más estable.
La Fig.4.1 enumera los confórmeros de energía más baja significativamente diferentes
aquí obtenidos para el dímero aniónico de glicina en fase gas. Los más estables son
dímeros de NA los cuales presentan cinco estructuras diferentes en fase gas. Todas estas
geometrías NA disponen de HBs O-H•••O. En el conformero más estable, este HB es
acompañado por un HB N-H•••O, así como en otros dos confórmeros. Los confórmeros
de NA sin HBs O-H•••O sólo se encuentran 50-60 kJ mol-1 por debajo del anterior.

∆E (kJ/mol) Dímero
0,0
NA-tgcαβtt
1,4
NA-ttcαγtt
6,2
NA-tccαβtt
8,5
NA-tttαtt
9,4
NA-tttαβtg'
11,0 ZA-e'cββtt
22,3
NA-cccββtc
27,0
ZA-e'cβγtt
40,0
ZA-ecβδgc
41,7
ZA-ecββtc
Fig. 4.1. Estructuras de los dímeros más destacables en fase gas. Diferencias de energía con respecto al dímero más estable en kJ mol-1.

La falta de HBs O-H•••O puede ser también estudiada como una razón para las altas
energías relativas encontrada en dímeros ZA (donde no existe ningún grupo O-H) en
fase gas. Por el contrario, Z monómeros son más adecuados para el establecimiento de
N-H•••O HB intramonomérico (intra-HB), ya que tienen dos oxígenos disponibles para
formarlos (Fig. 4.2a). Hay que destacar que bondpath intramoleculares tipo O-H•••N, no
se observan en dímeros de NA (Fig. 4.2b), en vez de ello se observan 2 HBs que
comparten el mismo O con 2 unidades H-N no planares, (generalmente asumidos como
intra-HBs) (Fig. 4.2 c).
Fig. 4.2. Principales disposiciones de monómeros de glicina neutra: (a) La forma Z con intra-HB N+-H···O no observado en el análisi QTAIM.; (b) La forma N con un intra-HB tipo O-H···N tampoco detectado como bondpath en el análisis QTAIM; y (c) La forma N con dos intra-HB N-H···O, sí detectados como bondpath en el análisis QTAIM.
(a)
(b)
(c)

Observando la geometría del confórmero más estable, podemos destacar que los
monómeros neutros adoptan un disposición oblicua poco convencional O = C-C-N
(tgc), que no corresponde a la mostrada por el conformero más estable de glicina aislado
(ttt). También notamos la presencia de dos HBs (O-H•••O y N-H•••O en este caso). Este
hecho es habitual entre los confórmero más estables, dando lugar a una preferencia por
estructuras cíclicas sobre las lineales. En estructuras cíclicas, notamos que las
longitudes de enlace de los dos HBs no son equivalentes. Estructuras apiladas en forma
de T se encuentran sólo en dímeros de alta energía.
Reemplazar la disposición tgc, infrecuente en el monómero de N, por uno de los
conformeros más estables de la glicina aislada (tcc) manteniendo el mismo conjunto de
HBs, hace aumentar la energía molecular 6,2 kJ mol-1, como se puede observar en NA-
tccαβtt
La estabilización de geometrías tgc puede explicarse como consecuencia de HBs más
fuertes con el monómero A. Así, la estructura más estable de la glicina aislado (ttt) solo
se acomoda en el confórmero NA-tttαtt, que solo establece un HB. Otra estructura
estable para la glicina N aislada (ttc) se muestra en un dímero de NA de baja energía
(1,4 kJ mol-1) donde el HB N-H···O se sustituye por uno débil C-H···O.
Un exhaustivo conjunto de dímeros NA ligados por un solo HB O-H···O, NA-tttαtt, ha
sido optimizado dado que va a ser la estructura más estable en solución acuosa. Sin
embargo en fase gas, esta estructura se encuentra 8 kJ mol-1 por debajo de la más estable
Es destacable que, en este conformero, los valores de las propiedades de enlace
QTAIM computados en el punto crítico de enlace (BCP) que rodean el hidroxilo H no
están en los rangos que analizados como característicos de un HB [26,27]. De hecho, el
átomo de hidrógeno se coloca casi en el centro entre ambos oxígenos. El enlace
covalente O-H en el monómero N se alarga hasta 1,151 Å, mientras que el "esperado"
HB H•••O se contrae a 1,262 Å.
Así que la unidad O-H•••O podría representarse mejor como formado por dos enlaces
covalentes. Por último, no podemos olvidar que dímeros donde cada monómero actúa
simultáneamente como HB donante y aceptor no se puede rechazar ya que son sólo 9,4
kJ mol-1 menos estable.
Los confórmeros ZA presentan un nuevo modelo de HB ligando ambos monómeros.La
presencia de NH3+ proporciona a la estructura más estable, ZA-e’cββtt, dos HBs N-
H···O . HBs los cuales provienen del mismo átomo de N. Esta geometría rígida obliga a un intra-HB establecido por el átomo de hidrógeno restante. Rotaciones internas que no

modifican el conjunto de HBs y el intra-HB causan variaciones de energía ligeras (0,7
kJ mol-1). Sin embargo, si reemplazamos un HB N-H•••O por un C-H•••O manteniendo
el intra-HB, ZA-e’cββtt, se incrementa la energía en 16 kJ mol-1. La desestabilización es
mayor cuando un N-H···O es reemplazado por un N-H···N (ZA-ecβδgc) (40 kJ mol-1).
Geometrías cíclicas de dímeros ZA donde ambos monómeros actúan simultáneamente
como donante y aceptor tienen una energía de unos 41 kJ mol-1 por encima de la del
conformero más estable (Fig. 4.1).
b) Análisis Conformacional en disolución acuosa
Como podía preverse, los dímeros más estables en medio acuoso son formas ZA,
pero las geometrías optimizadas (Fig. 4.3) son completamente diferentes de los
obtenidos para esta serie de confórmeros en fase gas. La optimización del ZA más
estable en gas (ZA-e’cββtt), pierde uno de los HBs N-H···O unidos por el mismo N,
dando lugar a una estructura más abierta
Una estructura 7 kJ mol-1 por debajo en energía, (ZA-ecδγgc), mantiene el N con esos
dos HBs pero reemplaza un N-H···O por un N-H···N. Esta estructura es similar a una de
las menos estables mostrada en fase gas (Fig. 4.1).
Es notable que en fase gas el HB más fuerte es N-H•••O, mientras que es el N-H•••N en
medio acuoso. De hecho, estos HBs juegan un papel importante como enlaces entre
monómeros en solución acuosa, siendo el único HB presente en la geometría más
estable (ZA-gcδgc). Esta estructura puede describirse como una pseudo-apilada con los
dos grupos carboxílicos en disposición trans. Esto permite una más eficaz solvatación
para ambas cargas negativas. La segunda estructura estable (ZA-gcβγtc) tiene un HB N-
H···O y un C-H···O, e incrementa su energía en más de 6 kJ mol-1. Esta es encontrada
como la más estable en solución acuosa con cálculos B3LYP, porque no hay
confórmero estable con HB tipo N H•••N. Rotaciones internas que mantienen esta
estructura básica llevan a confórmeros cuya energía relativa se diferencian por menos de
2 kJ mol-1. También es notable que el intra-HB (comunes en confórmeros en fase de
gas) no se muestren en los dímeros más estables de solución acuosa. Si forzamos la
formación de un intra-HB, como en ZA-tcβγtt, perdemos estabilidad (sobre unos 12,5 kJ
mol-1). Esto puede ser explicado porque la ausencia de intra-HB permite mejor
solvatación.
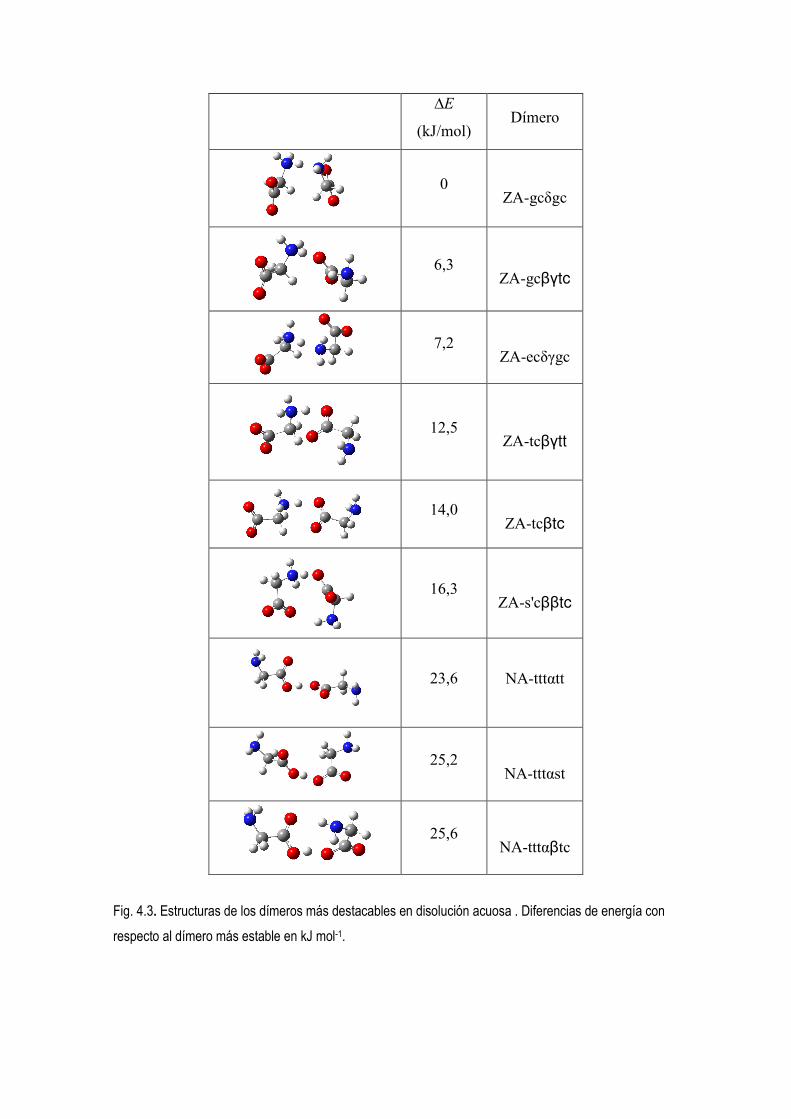
∆E
(kJ/mol) Dímero
0
ZA-gcδgc
6,3
ZA-gcβγtc
7,2
ZA-ecδγgc
12,5
ZA-tcβγtt
14,0
ZA-tcβtc
16,3
ZA-s'cββtc
23,6
NA-tttαtt
25,2
NA-tttαst
25,6
NA-tttαβtc
Fig. 4.3. Estructuras de los dímeros más destacables en disolución acuosa . Diferencias de energía con respecto al dímero más estable en kJ mol-1.

Con respecto a otras geometrías como las que se encuentran en fase de gas donde ambos
monómeros son aceptores/donantes, se encuentran 16,5 kJ mol-1 por encima de la más
estable, reduciendo sus energías relativas alrededor de 25 kJ mol-1 con respecto a la fase
gas, por tanto una posible presencia de estas geometrías en solución acuosa no debe ser
desechada.
En contraste, dímeros de NA en solución muestran solo un HB O-H···O en la geometría
más estable. Recordamos que en fase gas, esta geometría compartía el átomo de
hidrógeno entre los oxígenos en lugar de la tradicional unidad HB, que es la obtenida en
solución acuosa (1,052 Å y 1,444 Å, respectivamente, longitudes de enlace O-H y
O•••H ). Por otra parte, en solución acuosa, la geometría principal sitúa los monómeros
en un ángulo diedro de 90 º. Rotaciones internas alrededor de este HB elevan la energía
entre 1 y 5 kJ mol-1. Así, con HB tipo O-H···O y N-H···O, (como NA-tttαβtc) , se
encuentran por debajo de NA-tttαtt en estabilidad.
c) Análisis de la densidad electrónica
c.1) Explicación de las preferencias conformacionales en fase gas según el análisis
de la densidad electrónica
Como comentabamos anteriormente, el carácter zwitteriónico o no iónico del
monómero neutro da lugar a muy diferentes estructuras preferidas para el dímero en
fase gas (serie NA y ZA). Una de las características más llamativas del dimero más
estable en fase gaseosa, NA-tgcαβtt, es el ángulo de diedro O=C-C-N de 77º en el
monómero N. Curiosamente, este esqueleto es planar en los confórmeros más estables
del monómero de glicina [13]. Vamos a comparar las poblaciones electrónicas atómicas
de NA-tgcαβtt con las de NA-tccαβtt. Este último es el dímero de más baja energía
con el mismo HB que NA-tgcαβtt y con casi una estructura plana para el monómero N.
Observamos que la densidad electrónica transferida desde el monómero anión A a el
monómero neutro N en NA-tgcαβtt (0,119 au) refuerza básicamente la densidad
electrónica de la unidad C-CH2, (incrementa en 0,105 au con relación a N). La
población electrónica de esta unidad es 0,027 au más pequeña en NA-tccαβtt. Ésto está
en relación con la longitud de enlace C-C que aparece en este último (1,540 Å vs 1,531
Å). Cuando el enlace C-C se alarga la distancia entre grupos OH y NH2 aumenta lo
suficiente como para permitir dos HBs simultáneos con el anión. En general, mientras
que la fuerza de ambos HBs es bastante similar en los dos dímeros (O-H···O ligeramente
fuerte y N-H···O ligeramente débil en NA-tccαβtt), el enlace C-C es más fuerte en el

confórmero más estable. Observando los valores de ∆E(Ω) entre esos dos dímeros,
observamos que cada uno de esos carbonos se desestabiliza en 50 kJ mol-1 cuando el
esqueleto de N se hace plano (NA-tccαβtt).
Como tendencia general, observamos que ΣN(H) y ΣN(C) en fase gas ofrece los valores
más grandes. De este modo, la segunda estructura más estable en fase gas, NA-ttcαγtc,
reduce la densidad electrónica de los átomos más electronegativos con respecto a N y A
monómeros aislados. En este caso, esto es debido principalmente a la falta de HB en el
átomo de N. Rotaciones alrededor de los restos del aminoácido que no modifican el
conjunto de HB establecido entre los monómeros, tienen efectos insignificantes sobre
las poblaciones electronicas atómicas. Por ejemplo, valores ∆N(Ω) para la
interconversión NA-ttcαγtt→NA-ttcαγtc, no exceden de 0,003 au. En contraste el
proceso NA-tgcαβtt→NA-tccαβtt hace exceder 0,020 au para algunos átomos.
La similar estabilidad de NA-tgcαβtt y NA-ttcαγtt, resulta de un balance entre: i)
remplazar el HB N-H···O, por uno débil C-H···O y ii) reforzar el HB O-H···O (acortarlo
de 1,515 Å a 1,474 Å).
La alta energía relativa de NA-tttαtt (que dispone de un solo O-H···O) y NA-tttαβtg'
(donde ambos monómeros juegan el papel de aceptores y donantes) puede ser explicado
por la pérdida de densidad electrónica en los átomos atractores de densidad electrónica
(C, N y O) .A pesar de mostrar el HB O-H···O más fuerte (Tabla 4.1).
El esqueleto e’cββtt proporciona el primer ejemplo donde dímeros NA y ZA, teniendo
misma clase y número de HBs, pueden ser comparados. Teniendo en cuenta que
optimizaciones de monómeros Z en fase gas siempre acaban en monómeros N, es de
suponer que NA dímeros podrian ser las estructuras preferidas en fase gas frente a ZA.
Sin embargo, encontramos que hay estructuras ZA con menor energía que las NA con el
mismo tipo de HB. En ellas notamos que los HB son más cortos (con mayor ρb y más
fuertes [28]) para ZA (Tabla 4.1).
También, reemplazar en el dímero el hidrógeno unido al oxígeno en NA por otro unido
al nitrógeno en ZA, implica un factor de desestabilización y la población electrónica
atómica del hidrógeno aumenta en 0,205 au. Por otra parte, el anión se sitúa opuesto a la
región positiva del zwitterion en ZA, lo que permite una mayor transferencia electrónica
(Tabla 4.1). También remarcamos que los dos HBs N-H•••O en NA-e’cββtt, son
diferentes, estando uno de los oxigenos del anión en disposición syn en relación con el

grupo NH2. Aunque un intra-HB se asume generalmente para esta disposición, ningún
BCP asociado ha sido encontrado, incluso en monómeros aislados anionicos de glicina.
También destacamos que ZA-e’cββtt, es la estructura más estable entre los dímeros ZA.
Podría ser debido a los dos HBs N-H···O bifurcados que posee, convirtiéndose en el
único ZA con 3 de los 4 oxígenos implicados en HBs. El resto de las estructuras ZA
sólo presentan 2 oxígenos implicados en la formación de HBs presentando por tanto los
menores valores de ΣN(O)
c.2) Explicación de las preferencias conformacionales en solución acuosa según el
análisis de la densidad electrónica
Como ya hemos indicado, ZA-gcδgc es la estructura más estable en solución
acuosa. Presenta solo un HB N-H···N, tan fuerte como uno N-H···O (ρb entre 0,06 ay
0,07 au) y incluso como O-H···O (ρb es 0,07 au si está compartido y 0,09 au solo).
Cuando comparamos esta estructura con otra de las más estables, ZA-ecδγgc, que
dispone de dos fuertes HBs (N-H···N and N-H···O compartiendo el N), nos damos
cuenta que ΣN(O) es más alto en el anterior (por 0,036 au), mientras que los átomos
restante sufren ligeras variaciones. Ésto señala que la densidad electrónica adopta una
disposición más propensa a la solvatación de los oxígenos por moléculas de agua.
Observando las longitudes de los HBs, N-H···N es más grande que N-H···O (1,664 y
1,578 Å, respectivamente), lo cual está de acuerdo con estudios de difracción de
neutrones [29]. Observamos un decrecimiento de ΣN(H) and ΣN(C) a medida que la
estabilidad aumenta lo que nos conduce a pensar que en general, la estabilización de los
dímeros en solución acuosa sigue la tendencia opuesta observada en fase gas. De este
modo, el más estable de los dímeros en solución acuosa tiene el valor más bajo de
ΣN(H) y ΣN(C) y el mayor de ΣN(O) o ΣN(N). Este incremento en la población
electrónica de los átomos más electronegativos es más grande en ZA que en NA,
remarcando la carga negativa del grupo COO- en monómeros Z. La densidad electrónica
ganada por el grupo COO- proviene del los H del grupo metilo ya que su población
promedio es 0,917 au en dimeros ZA y 0,954 au en NA.
Las geometrías que mantienen un intra-HB incrementan la energía relativa a medida que
reducen las interaciones con el disolventes reduciendo por tanto la población de los
oxígenos. Ni estructuras basadas en HB N-H···O ni ciclos donante/donante incrementan
ΣN(O).
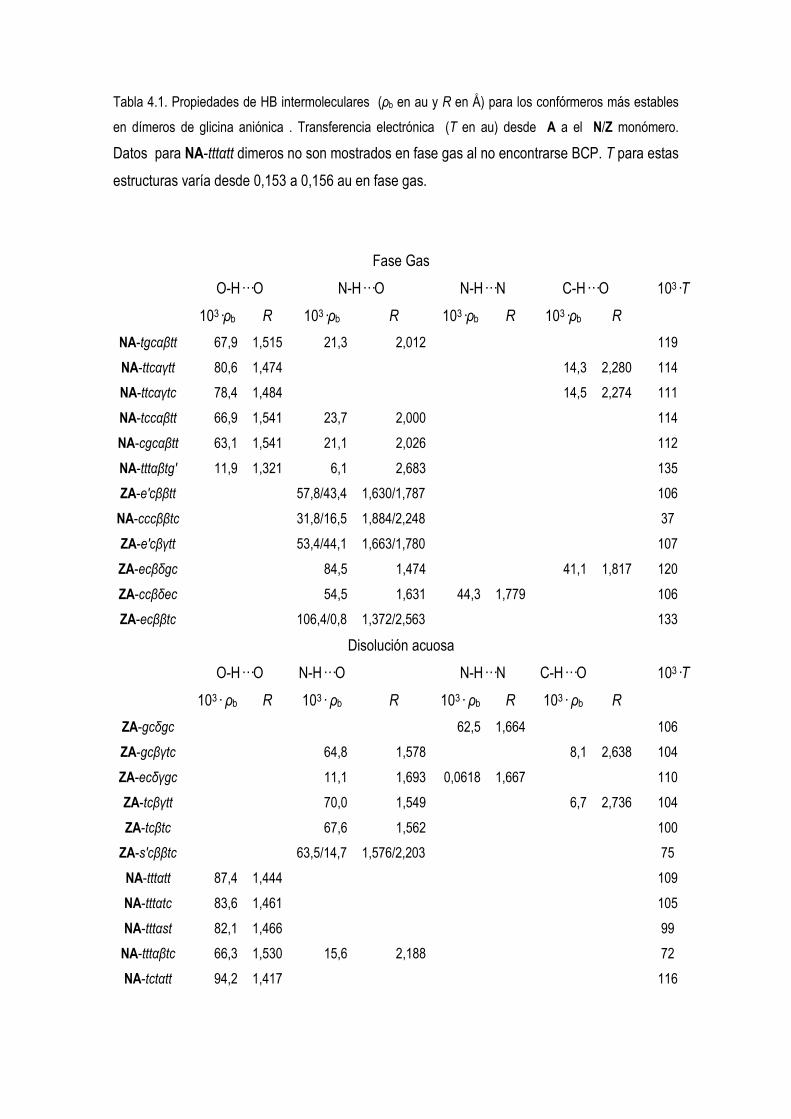
Tabla 4.1. Propiedades de HB intermoleculares (ρb en au y R en Å) para los confórmeros más estables en dímeros de glicina aniónica . Transferencia electrónica (T en au) desde A a el N/Z monómero.
Datos para NA-tttαtt dimeros no son mostrados en fase gas al no encontrarse BCP. T para estas estructuras varía desde 0,153 a 0,156 au en fase gas.
Fase Gas O-H···O N-H···O N-H···N C-H···O 103·T 103·ρb R 103·ρb R 103·ρb R 103·ρb R
NA-tgcαβtt 67,9 1,515 21,3 2,012 119 NA-ttcαγtt 80,6 1,474 14,3 2,280 114 NA-ttcαγtc 78,4 1,484 14,5 2,274 111 NA-tccαβtt 66,9 1,541 23,7 2,000 114 NA-cgcαβtt 63,1 1,541 21,1 2,026 112 NA-tttαβtg' 11,9 1,321 6,1 2,683 135 ZA-e'cββtt 57,8/43,4 1,630/1,787 106 NA-cccββtc 31,8/16,5 1,884/2,248 37 ZA-e'cβγtt 53,4/44,1 1,663/1,780 107 ZA-ecβδgc 84,5 1,474 41,1 1,817 120 ZA-ccβδec 54,5 1,631 44,3 1,779 106 ZA-ecββtc 106,4/0,8 1,372/2,563 133
Disolución acuosa O-H···O N-H···O N-H···N C-H···O 103·T 103· ρb R 103· ρb R 103· ρb R 103· ρb R
ZA-gcδgc 62,5 1,664 106 ZA-gcβγtc 64,8 1,578 8,1 2,638 104 ZA-ecδγgc 11,1 1,693 0,0618 1,667 110 ZA-tcβγtt 70,0 1,549 6,7 2,736 104 ZA-tcβtc 67,6 1,562 100 ZA-s'cββtc 63,5/14,7 1,576/2,203 75 NA-tttαtt 87,4 1,444 109 NA-tttαtc 83,6 1,461 105 NA-tttαst 82,1 1,466 99 NA-tttαβtc 66,3 1,530 15,6 2,188 72 NA-tctαtt 94,2 1,417 116

En NA dímeros, la situación es muy diferente. Mantenerlos grupos COOH obliga a la
geometría a formar al menos un O-H•••O sin excepciones. Por lo tanto, nos
encontramos con estructuras lineales que enfrentan átomos de O formando ángulo
diedro entre monómeros de 90 º, en la estructura más estable, a 180 º, en el menos
estable. Estas geometrias siguen la tendencia general siguiente: decrecer ΣN(H) y ΣN(C)
a medida que lo hace la estabilidad. Pensando en incrementar ΣN(O), hemos diseñado
un conjunto de geometrias con un HB O-H···O predominante y un N-H···O secundario.
Estas estructuras dan lugar a una gran reorganización electrónica decreciendo ΣN(H)
(por 0,037 au) y aumentando ΣN(O), la energia relativa es más grande que en dímeros
NA con un solo HB.
Un efecto claro de la solución acuosa tanto para ZA como para NA es la pérdida en
ΣN(H) que afecta a todos los monómeros implicados. (Fig. 4.4). De acuerdo con los
cálculos PCM, la energía decrece según la población electrónica de los átomos más
electronegativos crece. En otras palabras, PCM intensifica la diferencia de cargas.
También los dímeros ZA tienen menos ΣN(H) de NA en ambas fases mostrando que el
dipolo formado en estructuras zwitteriónico contribuye a un flujo de densidad de H a los
átomos electronegativos, especialmente hacia aquellos que están más cerca de la carga
positiva. Por otra parte, este efecto debe ser más intensa en el monómero de Z (que es el
conlleva la estructura dipolar en dímeros ZA) que en el anión adjunto. En cuanto a
características del HB O-H···O, observamos que básicamente obtenemos el mismo valor de ρb y longitudes de HB que las obtenidas en fase gas, incluso cuando este HB es
acompañado por C-H···O o uno muy débil N-H···O.
c.3) Transferencia electrónica entre monómeros
En todos los casos estudiados, encontramos que el monómero A transfiere
densidad electrónica al monómero neutro (N o Z) desde 0,037 hasta 0,135 au, aunque en
la mayoría de los casos son entre au 0,100 0,120. No hemos computado transferencia de
densidad del electrón, T, para dimeros NA-tttαtt, ya que el hidrógeno involucrado en el
enlace intermolecular tiene distancias de enlace O-H muy similares y no está claro que
podría ser exclusivamente asignado a cualquiera de ellos. No observamos ninguna
relación simple entre la T y la energía de la estabilidad. La tendencia general sólo parece
ser que los valores de T en fase gas son mayores que las observadas en solución acuosa.
Variaciones de T entre los confórmeros aquí considerados en la misma fase están
motivadas por un cambio en el tipo de HB formada, o por la modificación de

geometrías de dímero (Fig. 4.1). El mayor de los valores de T se observa en NA-tttαβtg’
en fase gas, mientras que una transferencia muy pequeña es encontrada en NA-cccββtc
en fase gas y para ZA-s’cββtc and NA-tttαβtc en solución acuosa.
Fig. 4.4. ΣN(H) (en au) para los monómeros de los dimeros ZA y NA en fase gas y disolución acuosa.
3.25
3.40
3.55
3.70
NA-tg
cαβtt
NA-tt
cαγtt
NA-tt
cαγtc
NA-tc
cαβtt
NA-tt
tαttI
NA-cg
cαβtt
NA-tt
tαttII
NA-tt
tαβtg
'
NA-tt
tαttII
I
ZA-e
'cββtt
I
ZA-e
'cββtt
II
ZA-e
'cββtt
III
ZA-e
'cββtt
IV
ZA-e
cβγg
c
ZA-cc
βδec
ZA-e
cββtc
ΣN(H)
neutral monomer
anionic monomer
GAS FASE
3.15
3.30
3.45
3.60
NA-tg
cαβtt
NA-tt
cαγtt
NA-tt
cαγtc
NA-tc
cαβtt
NA-tt
tαttI
NA-cg
cαβtt
NA-tt
tαttII
NA-tt
tαβtg
'
NA-tt
tαttII
I
ZA-e
'cββtt
I
ZA-e
'cββtt
II
ZA-e
'cββtt
III
ZA-e
'cββtt
IV
ZA-e
cβγg
c
ZA-cc
βδec
ZA-e
cββtc
ΣN(H)
neutral monomer
anionic monomer
DISOL. ACUOSA

c.4) Redistribución de la densidad electrónica de los dímeros de glicina con
respecto a los monómeros.
En las siguientes líneas vamos a analizar cómo los monómeros modifican sus
poblaciones atómicas de H, C, N y O durante el proceso de dimerización. Por brevedad,
sólo detallaremos la reorganización de la densidad electrónica que acompaña la
formación del dímero más estable en fase gaseosa y en solución acuosa. Hemos
dividido este proceso en dos pasos imaginarios: i) deformación del monómero, que
afecta principalmente al monómero N (∆DEF(N)); y ii) asociación monomérica
(∆ASOC(A) y ∆ASOC(N)).
La figura 4.5 muestra las variaciones experimentadas por las poblaciones electrónicas
atómicas en cada uno de estos pasos en fase gas. Observamos:
i) La rotación interna alrededor de los enlaces C-O y C C induce cambios significativos
en valores N(Ω) para el monómero N, aunque tiene efectos insignificantes en A (datos
no mostrados), así como su estructura optimizada para el monómero aislado es
básicamente la misma que la que aparece en el dimero.
ii) La densidad electrónica transferida por el anión procede de todo el monómero,
excepto uno de los hidrógenos de la amina y el carbono del carbonilo. Esta densidad
electrónica no mejorar N(H) de los hidrógenos implicados en HBs. De hecho, los
átomos que aumentan sus poblaciones atómicas son los más electronegativos en el
monómero N y los hidrógenos colocados más lejos en el anión. Esto es compatible con
un mecanismo donde el anión polariza el monómero de N reforzando la asociación entre
ambos monómeros con una contribución electrostática anión/dipolo inducido.
Las deformaciones experimentadas por los monómeros que dan lugar al dímero más
estable (ZA-gcδgc) en disolución acuosa son insignificantes. Por lo tanto, estamos sólo
analizando la reorganización electrónica referente a la asociación de monómeros,
(∆ASOC (A) y ∆ASOC(Z) , mostradas en la Fig. 4.6. Se puede observar que en todos los
átomos del monómero A, excepto el carbono metilénico, pierden población electrónica,
mientras que todos los átomos del monómero Z, excepto el carbono metilénico,
aumentan su N(Ω). Ambas excepciones pueden explicarse considerando el mismo
esquema general propuesto para la comprensión de valores ∆N(Ω) de las
protonatonaciones moleculares [30,31].

Fig..4.5. Población electrónica QTAIM (en au) para los confórmeros más estables de monómeros aislados en fase gas A y N, así como las variaciones experimentadas (en au multiplicadas 103) en los pasos imaginarios para la asociación , deformación del monomero N en NA-tgcαβtt:, ∆DEF(N), y asociación entre monómeros A y N , ∆ASOC(A) y ∆ASOC(N). Todas las poblaciones atómicas de las densidades electrónicas son computadas a nivel MP2/6-311++G(d,p) .
Según ésto, la donación de la población de electrónica entre los átomos ocurre a lo largo
del conjunto de los bondpaths. Así la cesión de densidad electrónica entre monómeros
tiene lugar a través del HB N-H•••N. Este hecho introduce la direccionalidad en la
transferencia electrónica y en los " nudos de acumulación electrónica". Estos últimos
pueden definirse como átomos que reciben densidad electrónica a través de varios
enlaces, pero sólo pueden donarla a otro átomo. Esto es lo que sucede en los carbonos
metilénicos. Del monómero A reciben densidad del electrón a través de tres enlaces y
106
11
-16-22 25
-19
-25 -19
-28
-24
3866
2
-94
-102
90-20-2860
0.6448.019
5.619
4.475
0.954
0.4019.134
9.1540.641
8.053 5.6854.350
0.695
1.026 9.264
9.261
1.023
42
-4-24
28
20
42-22
-53-17-11
∆DEF(N)
∆ASOC(A)
∆ASOC(N)
106
11
-16-22 25
-19
-25 -19
-28
-24
3866
2
-94
-102
90-20-2860
106
11
-16-22 25
-19
-25 -19
-28
-24
3866
2
-94
-102
90-20-2860
106
11
-16-22 25
-19
-25 -19
-28
-24
3866
2
-94
-102
90-20-2860
0.6448.019
5.619
4.475
0.954
0.4019.134
9.154
0.6448.019
5.619
4.475
0.954
0.4019.134
9.1540.641
8.053 5.6854.350
0.695
1.026 9.264
9.261
1.023
0.641
8.053 5.6854.350
0.695
1.026 9.264
9.261
1.023
42
-4-24
28
20
42-22
-53-17-11
42
-4-24
28
20
42-22
-53-17-11
42
-4-24
28
20
42-22
-53-17-11
-4-24
28
20
42-22
-53-17-11
∆DEF(N)∆DEF(N)
∆ASOC(A)∆ASOC(A)
∆ASOC(N)∆ASOC(N)
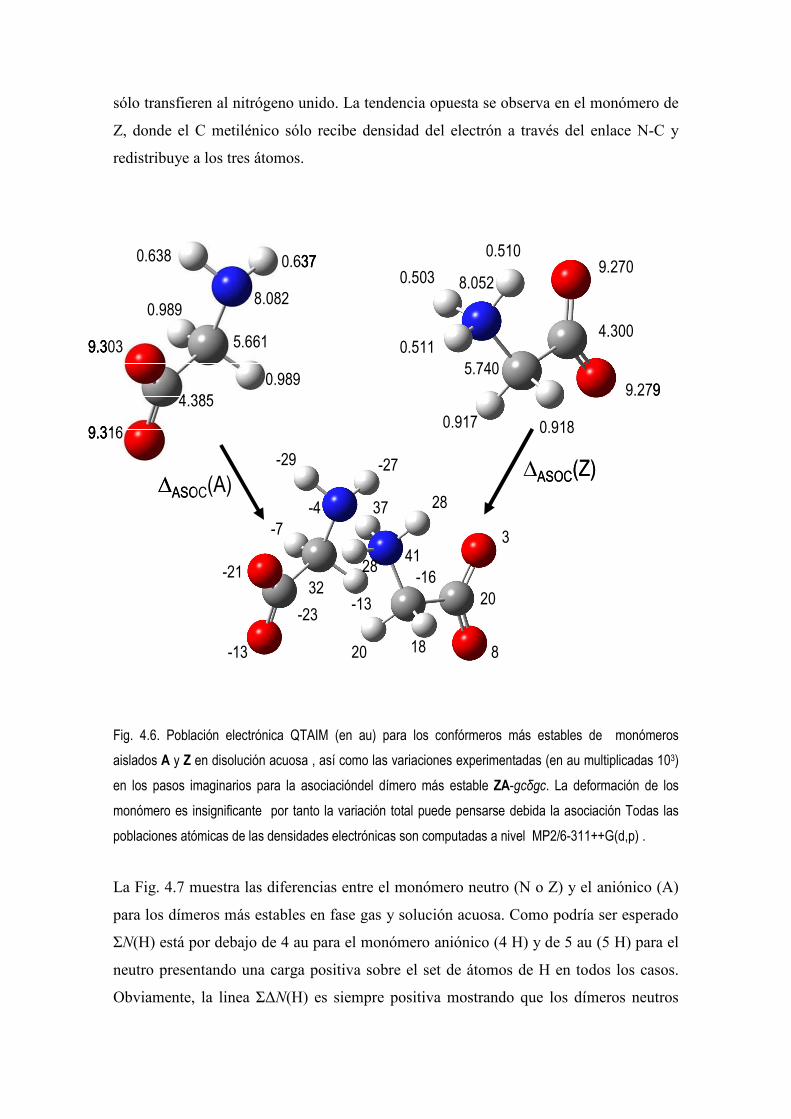
sólo transfieren al nitrógeno unido. La tendencia opuesta se observa en el monómero de
Z, donde el C metilénico sólo recibe densidad del electrón a través del enlace N-C y
redistribuye a los tres átomos.
Fig. 4.6. Población electrónica QTAIM (en au) para los confórmeros más estables de monómeros aislados A y Z en disolución acuosa , así como las variaciones experimentadas (en au multiplicadas 103) en los pasos imaginarios para la asociacióndel dímero más estable ZA-gcδgc. La deformación de los monómero es insignificante por tanto la variación total puede pensarse debida la asociación Todas las poblaciones atómicas de las densidades electrónicas son computadas a nivel MP2/6-311++G(d,p) .
La Fig. 4.7 muestra las diferencias entre el monómero neutro (N o Z) y el aniónico (A)
para los dímeros más estables en fase gas y solución acuosa. Como podría ser esperado
ΣN(H) está por debajo de 4 au para el monómero aniónico (4 H) y de 5 au (5 H) para el
neutro presentando una carga positiva sobre el set de átomos de H en todos los casos.
Obviamente, la linea Σ∆N(H) es siempre positiva mostrando que los dímeros neutros
0.638
9.303 5.661
4.385
0.637
0.989
9.316
8.082
0.989
0.503 9.270
4.300
5.740
0.918
0.510
9.279
8.052
0.917
0.511
373
20-16
18
28
8
41
20
28
-29
-2132
-23
-4-7
-13
-27
-13
∆ASOC(A)∆ASOC(Z)
0.638
9.303 5.661
4.385
0.637
0.989
9.316
8.082
0.989
0.638
9.303 5.661
4.385
0.637
0.989
9.316
8.082
0.989
0.503 9.270
4.300
5.740
0.918
0.510
9.279
8.052
0.917
0.511
373
20-16
18
28
8
41
20
28
-29
-2132
-23
-4-7
-13
-27
-13
∆ASOC(A)∆ASOC(Z)

tienen más población electrónica en los H que en los H del anión. Sin embargo esta
cantidad está entre 0,2 y 0,4 au, lejos de el esperado electrón. Esta tendencia es también
mostrada por los monómeros aislados.
Fig. 4.7. Diferencias entre la población electrónica total para cada átomo (X=C,H,O,N) entre el monómero
neutro (N or Z) y el aniónico Σx∈N/ZN(X)- Σx∈AN(X), (en au) para los monómeros de ZA y NA en fase
gas y disolución acuosa.
-0.50
-0.40
-0.30
-0.20
-0.10
0.00
0.10
0.20
0.30
0.40
NA-tg
cαβtt
NA-tt
cαγtt
NA-tt
cαγtc
NA-tc
cαβtt
NA-tt
tαttI
NA-cg
cαβtt
NA-tt
tαttII
NA-tt
tαβtg
'
NA-tt
tαttII
I
ZA-e
'cββtt
I
ZA-e
'cββtt
II
ZA-e
'cββtt
III
ZA-e
'cββtt
IV
ZA-e
cβγg
c
ZA-cc
βδec
ZA-e
cββtc N-ttt
N-tgc Z-
tt
Z-tg
ΣN(H) (N-A)
ΣN(N) (N-A)
ΣN(C) (N-A)
ΣN(O) (N-A)
FASE GAS
-0.50
-0.40
-0.30
-0.20
-0.10
0.00
0.10
0.20
0.30
0.40
NA-tt
tαtt
NA-tt
tαtc
NA-tt
tαst
NA-tt
tαβt
c
NA-tc
tαtt
NA-g
ttαtt
ZA-g
cδgc
ZA-g
cβγtc
ZA-e
cδγg
c
ZA-tc
βγttI
ZA-tc
βγttI
I
ZA-tc
βγttI
II
ZA-tc
βγtcI
ZA-tc
βγtcI
I
ZA-tc
βtc
ZA-s
'cββt
c
N-ttt
N-tg
c
Z-tt
Z-tg
ΣN(H) (N-A)
ΣN(N) (N-A)
ΣN(C) (N-A)
ΣN(O) (N-A)
DISOL. ACUOSA

Observando Σ∆N(C), vemos que es siempre menor que 0,1 au excepto en las
estructuras de cálculo puntual computado para los monómeros Z aislados en fase
gaseosa, que producian estructuras N cuando se dejaban optimizar. Las diferencias entre
dímeros NA y ZA, así como las ocasionadas por el medio, no parecen ser relevantes.
Sin embargo, es notable que la población electrónica de las cuencas de C es mayor en
monómeros N que en los de Z (no importa qué fase estamos hablando), mientras que la
tendencia inversa se observa para el respectivo monómero A comparando dímeros NA
y ZA.
También observamos que los valores de Σ∆N(N) siguen la tendencia opuesta que los
detallados para Σ∆N(C). Así, esta cantidad es más grande en monómeros Z que en N.
Este hecho puede ser explicado por la presencia de HB tipo N-H···O en dímeros ZA lo
cual incrementa N(N). Más aún, el monómero Z en ZA dímeros tiene más alta ΣN(N)
en fase gas que en solución acuosa debido a los dos HBs soportados por el mismo
nitrógeno en fase gas. Por la misma razón ΣN(N) es mayor en dímeros ZA que en NA.
La linea Σ∆N(O) indica que, en algunos casos, el oxígeno tiene una densidad electrónica
mayor en monómero Z que en N. Los oxígenos del monómero aniónico tienen una
población electrónica mayor que la del monómero neutro en fase gas, mientras que en
solución acuosa esta tendencia es seguida por los dímeros NA. En contraste, dos
oxígenos muy negativos en los monómeros Z son capaces de interactuar con moléculas
de agua en los dímeros ZA en disolución acuosa.
La linea Σ[N(O)+N(N)] versus Σ[N(H)+N(C)] (Figure 4.8) permite distinguir 3 grupos
de dímeros. Los valores más grandes del eje x corresponden con la más baja estabilidad.
Dímeros entre 26,68 au <Σ[N(H)+N(C)] <26,72 au son estructuras ZA en disolución
acuosa . Aquellos con 26,81 au < Σ[N(H)+N(C)] <26,89 au , coresponden a NA en
solución acuosa y a ZA en fase gas.
Este gráfico demuestra lo que habiamos comentado previamente sobre como el medio
acuoso incrementa Σ[N(O)+N(N)] disminuyendo Σ[N(H)+N(C)], ocurriendo justo lo
opuesto en fase gas . La razón está en que en fase gas, el incremento de Σ[N(H)+N(C)]
proviene del resto de O y N , que no interaccionan con el disolvente. Por tanto la
electronegatividad de los átomos implicados en HBs no es significativamente afectada
por lo que pueden maximizar la fuerza de los enlaces intermoleculares manteniendo
altos valores de ρb.
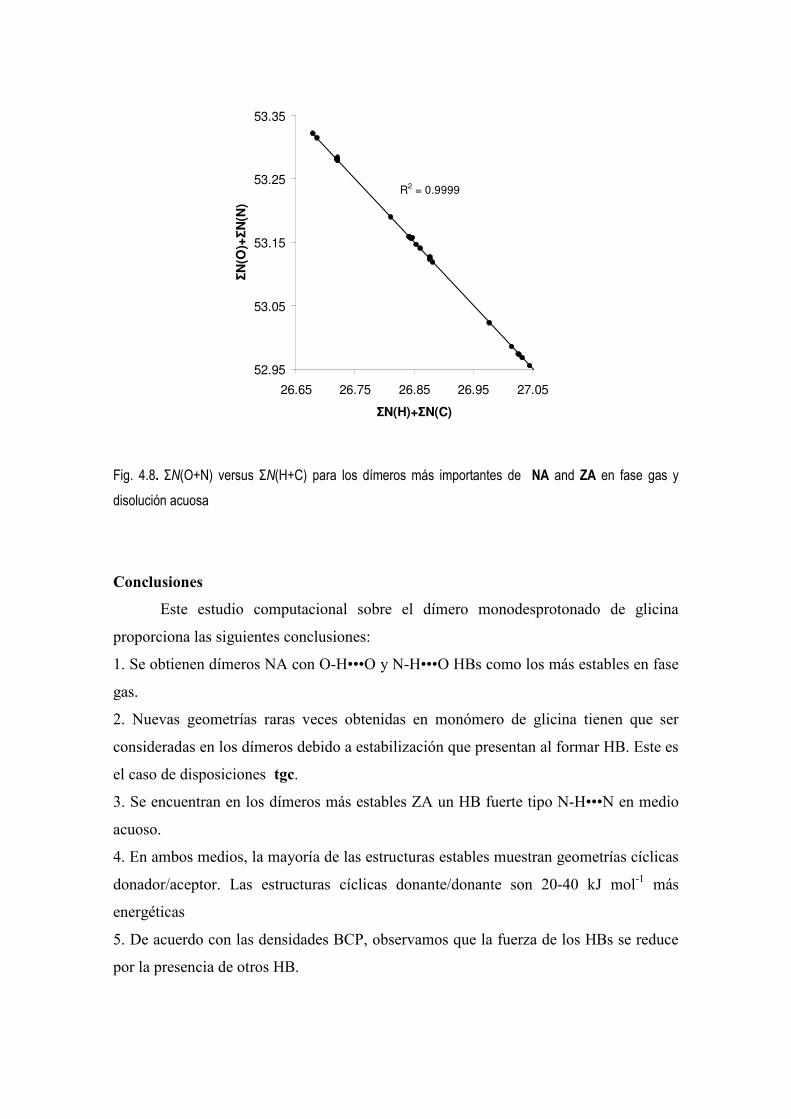
R2 = 0.9999
52.95
53.05
53.15
53.25
53.35
26.65 26.75 26.85 26.95 27.05
ΣN(H)+ΣN(C)
ΣN(O
)+ΣN
(N)
Fig. 4.8. ΣN(O+N) versus ΣN(H+C) para los dímeros más importantes de NA and ZA en fase gas y disolución acuosa
Conclusiones
Este estudio computacional sobre el dímero monodesprotonado de glicina
proporciona las siguientes conclusiones:
1. Se obtienen dímeros NA con O-H•••O y N-H•••O HBs como los más estables en fase
gas.
2. Nuevas geometrías raras veces obtenidas en monómero de glicina tienen que ser
consideradas en los dímeros debido a estabilización que presentan al formar HB. Este es
el caso de disposiciones tgc.
3. Se encuentran en los dímeros más estables ZA un HB fuerte tipo N-H•••N en medio
acuoso.
4. En ambos medios, la mayoría de las estructuras estables muestran geometrías cíclicas
donador/aceptor. Las estructuras cíclicas donante/donante son 20-40 kJ mol-1 más energéticas
5. De acuerdo con las densidades BCP, observamos que la fuerza de los HBs se reduce
por la presencia de otros HB.

6. En fase gas disminuyen las poblaciones de electrones de los átomos electronegativas
mientras medio acuoso hace lo contrario.
7. Intramonoméricos HBs aparece sólo en fase gas y en dímeros de ZA con dos HBs
dispuestos en el mismo nitrógeno.
8. Se observa una variación significativa de la población electrónica atómica en dímeros
con respecto a los correspondientes monómeros aislados. En fase gas el cambio de
densidad electrónica tiene dos orígenes: deformación de monómero y asociación. Los
efectos de asociación en fase gaseosa son compatibles con un anión – interacción dipolo
inducido.
9. La tranferencia de densidad electrónica desde el monómero aniónico al neutro oscila
entre 0,037 y 0,135 au, pero no se observan ninguna relación simple entre este hecho y
la estabilidad del dímero

5.5 La relación entre la distancia y la densidad del punto critico.
Nueva concepción del orden de enlace de H
El concepto de enlace químico es uno de los más discutidos desde hace decadas.
En parte esto es debido a la imprecisión que supone definir el átomo dentro de una zona
determinada del espacio. Esta definición se complica aún más cuando estudiamos
asociaciones moleculares debidas a fuerzas de largo alcance como las de Van der Waals
o los enlaces de hidrógeno (HB). Estos últimos presentan una importancia vital en
procesos químicos y biológicos, ya que su energía (que puede encontrarse entre 2 kJ
mol-1 y 170 kJ mol-1 dependiendo de los átomos implicados) es, a la vez,
suficientemente fuerte para estabilizar un cluster de moléculas y suficientemente débil
para experimentar procesos de disociación a temperatura ambiente.
La teoria cuantica de átomos en moléculas (QTAIM) [1] proporciona una definición
rigurosa, que permite calcular matemáticamente las propiedades de los átomos en las
moléculas y asociaciones moleculares, así como establecer sus fronteras. Todo ello se
hace a traves de una propiedad observable, la densidad electrónica ( ρ ). A partir de este
mismo observable, la QTAIM introduce también una definición matemática para el
enlace químico. Así, la presencia de un enlace químico entre dos átomos queda
vinculada a la existencia de un “bondpath”, con su correspondiente punto crítico de ρ,
en una cierta posición del espacio, cprr , entre dichos átomos. Incluso dicha teoría permite
calcular el orden de enlace relacionándolo con la densidad electrónica en el punto
crítico ( ( )cpbcp rrρρ = ) (ec. 5.1).
)( ba bcpen
−=
ρ (5.1)
Durante decadas, basándose en los trabajos de Pauling [2], se ha definido el orden de
enlace como una propiedad dependiente de la variación de la distancia interatómica (ec.
5.2). En dicha ecuación resulta n=1 si r∆ =0, situación que corresponde a la distancia
de enlace el equilibrio. En cambio n=0 cuando los átomos se separen a distancia
infinita. El parámetro c sería la distancia de enlace a la cual n=1/e. Sustituyendo n dada

por ec. (5.1) en la ec. (5.2), conseguimos una relación entre la densidad del punto crítico
y la distancia interatómica (ec. 5.3).
cren /∆−= (5.2)
bcpBAr ρ.−=∆ (5.3)
Trabajos anteriores [3] muestran como dicha ecuación 3 no se verifica en determinados
HBs y proponen en vez de una relación lineal entre r∆ y bcpρ , una relación logarítmica
(ec. 5.4).
bcpBAr ρln.−=∆ (5.4)
Para el caso específico de un HB dado, A-H···B (Fig. 5.1), podemos definir dos
distancias r1 y r2 y dos ordenes de enlace n1 y n2.
Fig 5.1. Representación esquemática de una unidad estructural con enlace de hidrógeno A-H···B y su nomenclatura.
Se ha establecido que estas dos distancias estas correlacionadas [4-6] ya que no puedn
variar independientemente. Según el modelo del enlace de valencia [7,8] la suma de los
dos órdenes ha de ser 1 para el caso especifico del H (ec. 5.5).
( )[ ] ( )[ ] 1//21
2010 =+=+ +−−+−− crrcrreenn (5.5)
Desarrollando esta ecuación podemos obtener fácilmente las ecuaciones 5.6 y 5.7, que
nos muestran la relación entre las dos distancias.
]ln[. //021
12 crcreecrrr ++=+ (5.6)
]1ln[ /)(02
10 crrecrr
−−−= (5.7)
A H B
r1 r2

Este estudio analiza el grado de validez de estas ecuaciones en distintas clases de
dimeros de glicina neutra, anionica y cationica. En ellos se compara el tipo y número de
HBs que presenta cada dímero.
Detalles computacionales
La geometria de los distintos dímeros fue optimizada con los niveles B3LYP/6-
311++G(d,p) y MP2/6-311++G(d,p) usando el paquete Gaussian 09 [9]. Se han incluido
funciones difusas y de polarización para obtener una mejor descripción del sistema con
HBs [10,11]. Las densidades electrónicas empleadas en el análisis topológico QTAIM
fueron obtenidas también con ambos niveles de cálculo y se ha usado el paquete
AIMPAC [12] para su interpretación.
El mismo análisis se ha hecho para fase gas y disolución acuosa usando el modelo PCM
para tener en cuenta los efectos de la solvatación [13].
En la integración de densidades electrónicas se verificó que los valores de L(Ω) no
superasen en valor absoluto 2·10-3 au, lo cual se considera una condición necesaria para
garantizar la calidad de las propiedades QTAIM integradas. También se comprobó que
las energías y poblaciones electrónicas moleculares eran suficientemente próximas a las
obtenibles como suma de las correspondientes propiedades atómicas, ΣN(Ω) y ΣE(Ω),
respectivamente. Los valores mostrados para energías moleculares incluyen las
correcciones ZPVE y térmica.
Resultados y discusión
La glicina, al ser el más pequeño de los aminoácidos, juega un papel importante
para modelizar y entender la reactividad de las biomoléculas entre si y con su entorno.
Se ha realizado una completa optimización de distintas geometrias de cada dímero y se
ha elegido los más estables de los que tienen los mismos HBs. Se ha tenido en cuenta
que los confórmeros de glicina pueden presentarse en forma de molécula neutra o de
zwitterion. Además, se ha realizado un análisis de los cambios que sufren dichos
dimeros con el pH, optimizándose por tanto los dímeros cationicos y aniónicos. La
nomenclatura empleada ha sido: NN=dimero formado por dos moléculas neutras, NZ=
dimero de molécula neutra con zwiterion, ZZ=dimero formado por dos zwiteriones,
NA=dimero de molécula neutra con anión, ZA=dimero de zwiterion con anion,
NC=dimero de molécula neutra con catión, ZC=dimero de zwiterion con catión. Dado

que las geometrias no tiene porque coincidir en fase gas y en medio acuoso, se ha
optimizado nuevamente en cada medio el conjunto completo de geometrías iniciales. El
confórmero resultante se nombra añadiendo la sigla G o W dependiendo del medio que
se haya considerado en que la optimización geométrica.
Las tablas 5.1 y 5.2 muestran las distancias de enlace y valores ρbcp de la unidad X-
H···Y obtenidas para fase gas y medio acuoso, respectivamente, en la serie de
confórmeros localizados para los diferentes dímeros de glicina.
En primer lugar se analiza la relación entre longitud del HB y su correspondiente ρbcp.
La Fig. 5.2 muestra una clara dependencia logaritmica tanto en fase gas como en
disolución acuosa. La correlación mostrada es independiente del tipo de HB, de la
geometria, del confórmero e incluso de la carga del dímero, aumentando el valor de r2 si
se analiza separadamente cada grupo, como ejemplifica la Fig. 5.3. También se observa
un mejor ajuste de los datos a la ec 5.4 frente a la ec 5.3. Es decir, observamos entre
d(H···X) y ρbcp una dependencia logaritmitca en lugar de lineal, como mostraron trabajos
de otros autores [3].
El estudio de esta dependencia en enlaces asociados al HB (X-H) se ha particularizando
para el caso de los dimeros aniónicos en fase gas. Se ha considerado el conjunto de
geometrías estudiadas para los aniones del dimero de glicina desprotonada. Los
resultandos muestran la misma tendencia no solo entre la distancia del HB y la densidad
de su punto crítico sino también entre la longitud del enlace asociado y la densidad de
su punto crítico (Fig. 5.3 y Fig. 5.4).
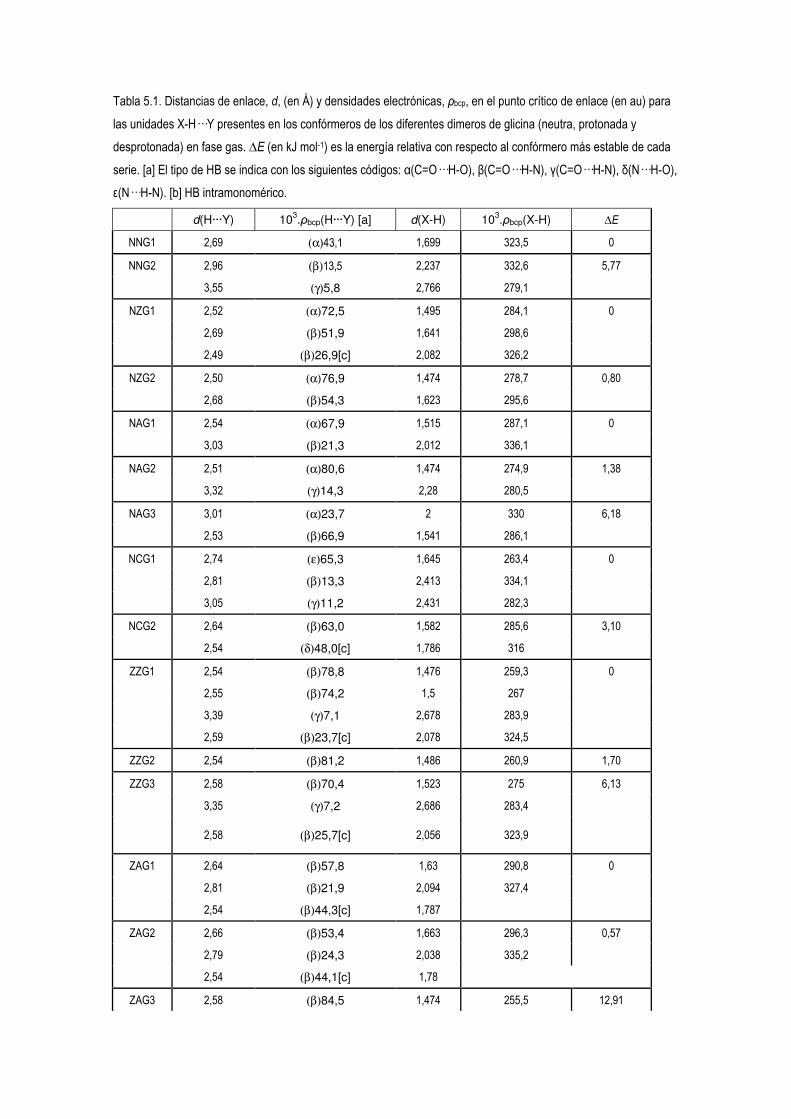
Tabla 5.1. Distancias de enlace, d, (en Å) y densidades electrónicas, ρbcp, en el punto crítico de enlace (en au) para las unidades X-H···Y presentes en los confórmeros de los diferentes dimeros de glicina (neutra, protonada y desprotonada) en fase gas. ∆E (en kJ mol-1) es la energía relativa con respecto al confórmero más estable de cada serie. [a] El tipo de HB se indica con los siguientes códigos: α(C=O···H-O), β(C=O···H-N), γ(C=O···H-N), δ(N···H-O), ε(N···H-N). [b] HB intramonomérico.
d(H···Y) 103.ρbcp(H···Y) [a] d(X-H) 103.ρbcp(X-H) ∆E
NNG1 2,69 (α)43,1 1,699 323,5 0
NNG2 2,96 (β)13,5 2,237 332,6 5,77
3,55 (γ)5,8 2,766 279,1
NZG1 2,52 (α)72,5 1,495 284,1 0
2,69 (β)51,9 1,641 298,6
2,49 (β)26,9[c] 2,082 326,2
NZG2 2,50 (α)76,9 1,474 278,7 0,80
2,68 (β)54,3 1,623 295,6
NAG1 2,54 (α)67,9 1,515 287,1 0
3,03 (β)21,3 2,012 336,1
NAG2 2,51 (α)80,6 1,474 274,9 1,38
3,32 (γ)14,3 2,28 280,5
NAG3 3,01 (α)23,7 2 330 6,18
2,53 (β)66,9 1,541 286,1
NCG1 2,74 (ε)65,3 1,645 263,4 0
2,81 (β)13,3 2,413 334,1
3,05 (γ)11,2 2,431 282,3
NCG2 2,64 (β)63,0 1,582 285,6 3,10
2,54 (δ)48,0[c] 1,786 316
ZZG1 2,54 (β)78,8 1,476 259,3 0
2,55 (β)74,2 1,5 267
3,39 (γ)7,1 2,678 283,9
2,59 (β)23,7[c] 2,078 324,5
ZZG2 2,54 (β)81,2 1,486 260,9 1,70
ZZG3 2,58 (β)70,4 1,523 275 6,13
3,35 (γ)7,2 2,686 283,4
2,58 (β)25,7[c] 2,056 323,9
ZAG1 2,64 (β)57,8 1,63 290,8 0
2,81 (β)21,9 2,094 327,4
2,54 (β)44,3[c] 1,787
ZAG2 2,66 (β)53,4 1,663 296,3 0,57
2,79 (β)24,3 2,038 335,2
2,54 (β)44,1[c] 1,78
ZAG3 2,58 (β)84,5 1,474 255,5 12,91

3,42 (γ)8,5 2,601 280,3
2,54 (β)41,1[c] 1,817
ZCG1 2,58 (β)81,3 1,485 261,3 0
3,25 (γ)12,5 2,359 280,9
2,48 (β)58,2[c] 1,652 286,7
ZCG2 2,56 (β)90,5 1,448 252,3 0,50
2,47 (β)60,4[c] 1,637 284,1
Tabla 5.2. Distancias de enlace, d, (en Å) y densidades electrónicas, ρbcp, en el punto crítico de enlace (en au) para las unidades X-H···Y presentes en los confórmeros de los diferentes dimeros de glicina (neutra, protonada y desprotonada) en disolución acuosa. ∆E (en kJ mol-1) es la energía relativa con respecto al confórmero más estable de cada serie. [a] El tipo de HB se indica con los siguientes códigos: α(C=O···H-O), β(C=O···H-N), γ(C=O···H-N), δ(N···H-O), ε(N···H-N). [b] HB intramonomérico.
d(H···Y) 103.ρbcp(H···Y) [a] d(X-H) 103.ρbcp(X-H) ∆E
NNW1 3,28 (β)11.9 2,28 343,7 0
3,11 (β)7.3 2,78 286,8
3,31 (γ)5.6 2,88 338,7
2,61 (δ)35.1[b] 1,90 327,1
NNW2 3,20 (β)6.9 2,65 329,5 0.29
3,19 (γ)6.2 2,84 277,8
3,59 (β)6.6 2,68 278,6
2,59 (β)38.3[b] 1,89 323,8
2,56 (β)44.8[b] 1,82 319,6
NNW3 2,70 (α)42.7 1,71 323,4 5.11
NZW1 2,79 (ε)51.8 1,74 293,6 0
NZW2 2,60 (α)57.1 1,59 305,8 4.41
2,88 (β)19.3 2,04 328,2
NAW1 2,49 (α)87.4 1,44 263,2 0
NAW2 2,51 (α)83.6 1,46 268,1 1.48
NAW3 2,50 (α)82.1 1,47 272,8 1.68
NAW4 2,53 (α)66.3 1,53 290,7 2.05
3,19 (β)15.6 2,19 274,7
NCW1 2,74 (ε)62.8 1,66 276,3 0
3,31 (γ)6.0 2,82 277,9
NCW2 2,72 (ε)67.4 1,63 269,9 4.97
3,50 (γ)5.3 2,78 277
NCW3 2,79 (β)36.3 1,76 315,1 15.44
3,47 (γ)9.2 2,44 279,4
ZZW1 2,70 (β)47.3 1,67 304,3 0

ZZW2 2,72 (β)44.9 1,70 305,8 2.49
2,68 (β)45.9 1,70 304,3
ZZW3 2,70 (β)47.7 1,67 303,1 3.02
ZAW1 2,74 (ε)62.5 1,66 278,3 0
ZAW2 2,65 (β)64.8 1,58 283 6.35
3,39 (γ)8.1 2,64 282,3
ZCW1 2,65 (β)64.6 1,58 280,7 0
ZCW2 2,66 (β)62.7 1,59 283,6 2.47
2,60 (β)30.8[b] 1,94 315,1
ZCW3 2,66 (β)54.3 1,61 296,2 8.02
3,39 (γ)10.6 2,35 283,4 0
dG= 0.4868Ln(ρ) + 0.2439R2 = 0.9888
dW = -0.4947Ln(ρ) + 0.2113R2 = 0.9788
1.3
1.8
2.3
2.8
3.3
0 0.02 0.04 0.06 0.08 0.1ρρρρ/au
d/A
Fig 5.2. Representacion de ρbcp frente a d(H···X) en fase gas (rombos azules) y en disolución acuosa (cuadrados rosa). Se muestran las ecuaciones de ajuste para ambos casos (dG y dW representan, respectivamente, fase gas y disolución acuosa)
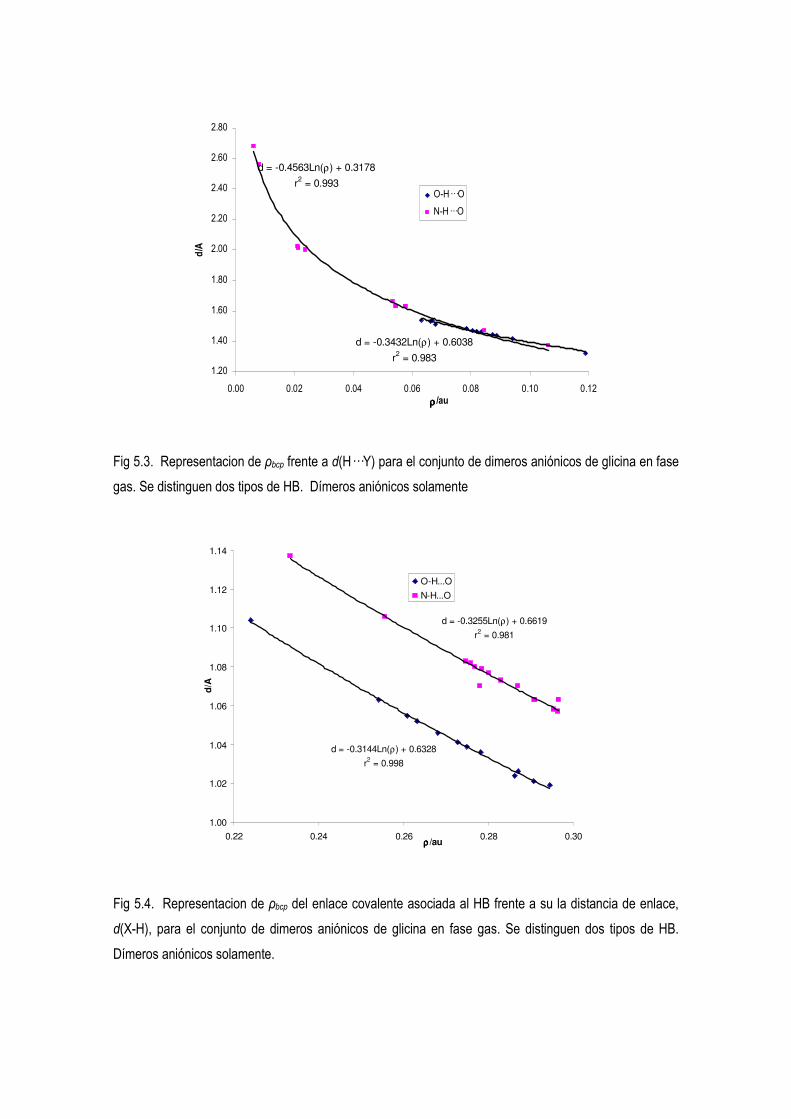
d = -0.3432Ln(ρ) + 0.6038
r2 = 0.983
d = -0.4563Ln(ρ) + 0.3178
r2 = 0.993
1.20
1.40
1.60
1.80
2.00
2.20
2.40
2.60
2.80
0.00 0.02 0.04 0.06 0.08 0.10 0.12ρρρρ /au
d/A
O-H···ON-H···O
Fig 5.3. Representacion de ρbcp frente a d(H···Y) para el conjunto de dimeros aniónicos de glicina en fase gas. Se distinguen dos tipos de HB. Dímeros aniónicos solamente
d = -0.3255Ln(ρ) + 0.6619
r2 = 0.981
d = -0.3144Ln(ρ) + 0.6328
r2 = 0.998
1.00
1.02
1.04
1.06
1.08
1.10
1.12
1.14
0.22 0.24 0.26 0.28 0.30ρρρρ /au
d/A
O-H...O
N-H...O
Fig 5.4. Representacion de ρbcp del enlace covalente asociada al HB frente a su la distancia de enlace, d(X-H), para el conjunto de dimeros aniónicos de glicina en fase gas. Se distinguen dos tipos de HB. Dímeros aniónicos solamente.

Seguidamente, analizamos si existe algún tipo de dependencia entre las densidades de
los puntos críticos del HB y su enlace covalente asociado. Teniendo en cuenta las
definiciones de la Fig. 5.1 podriamos escribir la ec. 5.7 como:
[ ]crrecrr
/)(01
021ln −−−−=− ( 5.8)
Y teniendo en cuenta la relación logaritmica de la ecuación 5.4 podriamos escribir ∆r en
función de la densidad electrónica del punto crítico del HB, ρbcp, y de su enlace
asociado, ρa.
[ ]cBA
abcpecBA
/)ln''(1lnln ρρ
−−−−=− (5.9)
Quedando entonces:
[ ]cBA
abcpe
B
c
B
A /)ln''(1lnln ρρ
−−−−= (5.10)
De la que podemos deducir:
c
B
bcpc
AA
c
A
c
B
a ee''
ρρ−
−= (5.11)
En la ec. 5.11 Fig.n varios parámetros cuyo valor debe calcularse. Se ha particularizado
su cálculo para dímeros aniónicos, por ser éstos los que presentan más diversidad de
geometrias con el mismo enlace de hidrógeno. De la Fig. 5.3 podemos observar que B'
es del orden de 0,3 tanto para el enlace de hidrógeno O-H···O como N-H···O. De la Fig.
5.4 podemos concluir que B es del mismo orden también para el enlace asociado.
El parámetro c está definido por la ec. 5.12 [14] a partir de la distancia mínina a la que
pueden estar los átomos pesados (X e Y) en el HB lineal, cuando la valencia a cada lado
del H es 1/2.

2ln22)( 0min2 rrr
c r −+= (5.12)
Sumando la ec. 5.8 para los valores de r1 y r2. Obtendríamos para el caso de que los
átomos pesados fueran el mismo (X=Y):
]1ln[2 /)(2102
21 crr
r ecrrrrr−−++−+=+ ( 5.13)
Definiendo las variables q2 = r1+r2 (distancia interatómica entre los átomos pesados ) y
q1 = r1-r2 (la posición relativa del H con respecto a la posición media) [4,5,14,15],
podemos calcular c ajustando la dependencia entre estos dos parámetros a la ec. 5.13.
Nótese que estos parámetros se definen con respecto a un enlace de H lineal. Con los
datos de las tablas 5.3 y 5.4 obtenidos en fase gas y disolución acuosa, hemos calculado
los parámetros q1 y q2 para los enlaces de H tipo O-H···O y N-H···O. La representación
gráfica de q2 frente a q1 puede observarse en Fig 5.5 (O-H···O) y Fig 5.6 (N-H···O).
Dicha gráfica ha sido construida incluyendo datos de fase gas y disolución acuosa.
Previamente, Steiner y colaboradores [4,5] estudiaron dicha correlación para el estado
sólido con datos de Cambridge Structural Database (CSD)
2.38
2.40
2.42
2.44
2.46
2.48
2.50
2.52
2.54
2.56
2.58
-0.6 -0.4 -0.2 0.0 0.2 0.4 0.6
q1=r1-r2
q2=
r1+
r2
Fig 5.5. Representación de q2 vs. q1 para los principales HBs del tipo O-H···O. Se incluyen datos para fase gas y medio acuoso.

Tabla 5.3. Densidades electrónicas en el punto crítico de enlace (en au) y distancias de enlace, d, (en Å) en la unidad X-H···Y para el conjunto de dimeros aniónicos de glicina en fase gas.
∆E(kJ/mol) ρa(O-H···O) da(O-H···O) ρbcp(O-H···O) d(O-H···O) ρa(N-H···O) da(N-H···O) ρbcp(N-H···O) d(N-H···O)
NA1 0,000 0,287 1,026 0,068 1,515 0,336 1,024 0,021 2,012
NA2 1,377 0,275 1,039 0,081 1,474
NA3 2,252 0,278 1,036 0,078 1,484
NA4 6,181 0,286 1,024 0,067 1,541 0,330 1,021 0,024 2,000
NA5 9,649 0,295 1,019 0,063 1,541 0,336 1,024 0,021 2,026
NA6 14,087 0,224 1,104 0,119 1,321 0,332 1,020 0,006 2,683
ZA1 0,000 0,291 1,063 0,058 1,630
ZA2 0,090 0,297 1,063 0,054 1,630
ZA3 0,092 0,291 1,063 0,058 1,630
ZA4 0,576 0,296 1,057 0,053 1,663
ZA5 12,900 0,256 1,106 0,085 1,474
ZA6 25,894 0,295 1,058 0,055 1,631
ZA7 27,592 0,233 1,137 0,106 1,372
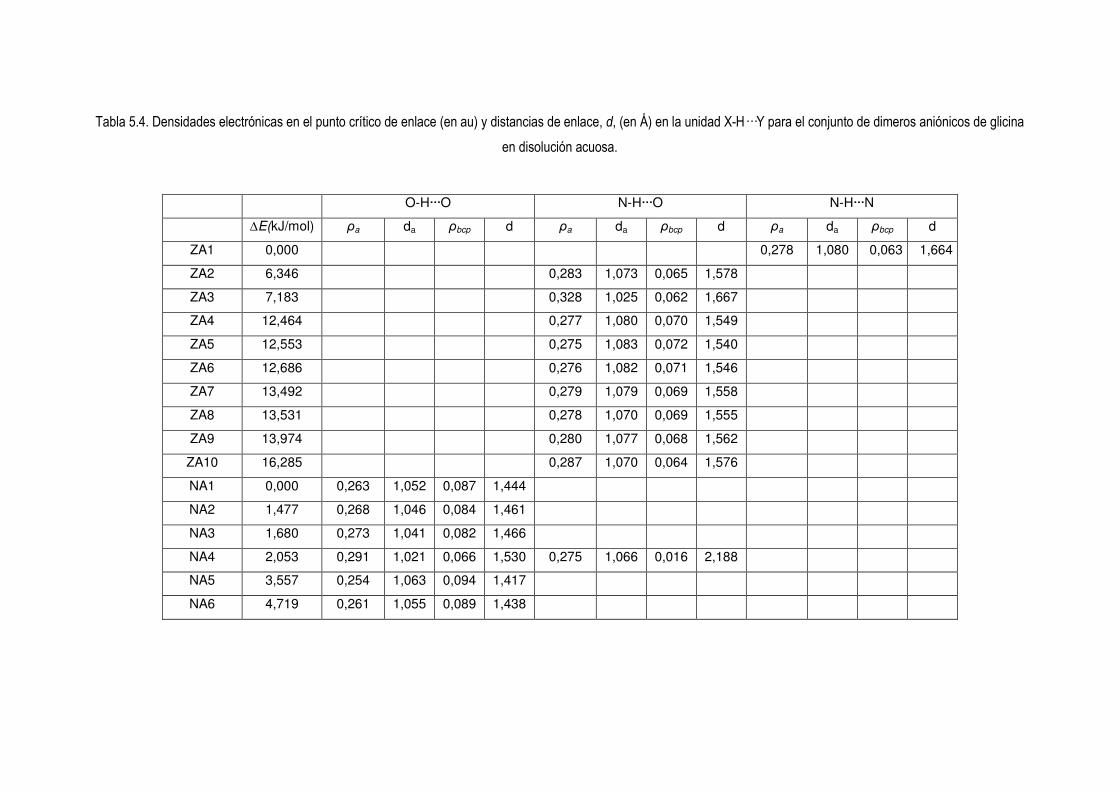
Tabla 5.4. Densidades electrónicas en el punto crítico de enlace (en au) y distancias de enlace, d, (en Å) en la unidad X-H···Y para el conjunto de dimeros aniónicos de glicina en disolución acuosa.
O-H···O N-H···O N-H···N
∆E(kJ/mol) ρa da ρbcp d ρa da ρbcp d ρa da ρbcp d
ZA1 0,000 0,278 1,080 0,063 1,664
ZA2 6,346 0,283 1,073 0,065 1,578
ZA3 7,183 0,328 1,025 0,062 1,667
ZA4 12,464 0,277 1,080 0,070 1,549
ZA5 12,553 0,275 1,083 0,072 1,540
ZA6 12,686 0,276 1,082 0,071 1,546
ZA7 13,492 0,279 1,079 0,069 1,558
ZA8 13,531 0,278 1,070 0,069 1,555
ZA9 13,974 0,280 1,077 0,068 1,562
ZA10 16,285 0,287 1,070 0,064 1,576
NA1 0,000 0,263 1,052 0,087 1,444
NA2 1,477 0,268 1,046 0,084 1,461
NA3 1,680 0,273 1,041 0,082 1,466
NA4 2,053 0,291 1,021 0,066 1,530 0,275 1,066 0,016 2,188
NA5 3,557 0,254 1,063 0,094 1,417
NA6 4,719 0,261 1,055 0,089 1,438

Ajustando los datos de Fig. 5.5 a la ec. 5.13, obtenemos para el enlace O-H···O los
parámetros de r0=0,961 Ǻ y c= 0,337.
En el caso de enlaces de hidrógeno tipo N-H···O, al tener r01 y r02 distintos para cada
enlace tendriamos la ec. 5.14. Con ello, la ec. 5.13 se transformaría en la ec. 5.15.
]/)210201(
1ln[022crrrr
ecrr+−−
++= (5.14)
]/)210201(
1ln[22102221crrrr
ecrrrrr+−−
++−+=+ (5.15)
La Fig 5.6 presenta el ajuste de los datos obtenidos para los enlaces N-H···O a la ec.
5.15. Los parámetros obtenidos son r01=1,042, r02= 1,021 y c=0,241. Los valores de c
encontrados en ambos enlaces están más próximos a los proporcionados por Steiner y
Benedict [4,14] que al valor 0,677 aportado por Pauling.
2.400
2.600
2.800
3.000
3.200
3.400
3.600
3.800
-2.000 -1.500 -1.000 -0.500 0.000 0.500 1.000 1.500 2.000
q1=r1-r2
q2=
r1+
r2
Fig 5.6. Representación de q2 vs. q1 para los principales HBs del tipo N-H···O. Se incluyen datos para fase gas y medio acuoso.

Al sustituir los valores de los parámetros c, B y B’ en la ec. 5.11 observamos que la
dependencia es cuasi-lineal. La Fig. 5.7 muestra como el conjunto de dímeros aniónicos
de glicina verifica una dependencia lineal.
d = -1.2502ρ + 0.3731
r2 = 0.991d = -1.2256ρ + 0.3624
r2 = 0.9947
0.22
0.23
0.24
0.25
0.26
0.27
0.28
0.29
0.30
0.05 0.06 0.07 0.08 0.09 0.10 0.11 0.12ρρρρ bcp/au
ρρ ρρa/a
u
O-H···O
N-H···O
Fig 5.7. Dependencia entre las densidades de los punto crítico de enlace del HB, ρbcp, y del enlace covalente asociado, ρa, para el conjunto de dimeros aniónicos de glicina en fase gas. Se distinguen dos tipos de HB.
Dado que algunos de los dimeros de glicina presentan varios HBs, hecho que podría
afectar a la dependencia entre ambos puntos críticos de enlace, se decidió extender el
estudio a una serie de aductos modelo formados por monómeros enlazados por un único
enlace O-H···O. Dicho conjunto y los datos correspondientes pueden observarse en la
Tabla 5.5. La Fig. 5.8 muestra la tendencia encontrada.

ρa = -1.3823 ρbcp + 0.3885
r2 = 0.985
0.23
0.25
0.27
0.29
0.31
0.33
0.35
0.37
0.00 0.02 0.04 0.06 0.08 0.10 0.12ρρρρbcp/au
ρρ ρρa// //au
Fig. 5.8. Dependencia entre las densidades de los punto crítico de enlace del HB, ρbcp, y del enlace covalente asociado, ρa, para el conjunto de aductos con enlace O-H···O mostrado en la Tabla 5. Se incluye para cada compuesto los datos obtenidos en fase gas y en disolución acuosa.
Se ha analizado si esta relación lineal entre densidades electrónica de los puntos críticos
se verfica también para geometrías distintas a la de los mínimos. Para ello, se han
considerado diferentes geometrías del dímero de agua. La Fig. 5.9 muestra que dicha
relación no es lineal, sino exponencial. Con lo cual, podemos pensar que la relación
lineal entre densidades electrónicas de puntos críticos solo se mantiene en el mínimo de
energía.
ρa = 0,005 ρbcp-1,2436
r2 = 0,9974
0.200.400.600.801.001.20
0.010 0.015 0.020 0.025 0.030 0.035 0.040 0.045
ρρρρ bcp/au
ρρ ρρa/au
Fig. 5.9. Dependencia entre las densidades de los punto crítico de enlace del HB, ρbcp, y del enlace covalente asociado, ρa, para una serie de geometrías del dímero de agua en fase gas.

Tabla 5.5. Densidades electrónicas en el punto crítico de enlace (en au) y distancias de enlace, d, (en Å) en la unidad X-H···Y para un conjunto de aductos con un único enlace O-H···O.
Fase gas Disolucón acuosa
Aducto ρbcp ρa d(H···O) d(O-H) E/au ρbcp ρa d(H···O) d(O-H) E/au
HO-H···OH2 0,022 0,356 1,949 0,966 -152,5067 0,027 0,349 1,877 0,970 -152,5729
(HO-H…OH)- 0,105 0,233 1,381 1,094 -151,9241 0,081 0,274 1,478 1,040 -152,0740
HO-H···OH-CH3 0,027 0,353 1,895 0,968 -191,6469 0,031 0,346 1,834 0,972 -191,7422
(HO-H···O-CH3)- 0,093 0,255 1,426 1,064 -191,0703 0,105 0,246 1,383 1,076 -191,2412
(HO-H···O-COH)- 0,023 0,348 2,022 0,972 -265,0572 0,043 0,328 1,723 0,987 -265,2092
HO-H···O(CH3)2 0,030 0,351 1,864 0,969 -230,7961 0,033 0,344 1,808 0,974 -230,9193
HO-H···OCHOH 0,019 0,356 2,060 0,965 -265,5741 0,024 0,352 1,929 0,968 -265,6554
CH3-O-H···OH2 0,024 0,359 1,944 0,964 -191,6462 0,029 0,353 1,867 0,969 -191,7417
(CH3O-H…OH)- 0,093 0,255 1,426 1,064 -191,0703 0,081 0,280 1,476 1,037 -191,2432
CH3O-H···OH-CH3 0,028 0,357 1,886 0,966 -230,7866 0,032 0,350 1,827 0,971 -230,9113
(CH3O-H···O-CH3)- 0,118 0,232 1,339 1,098 -230,2153 0,101 0,260 1,401 1,062 -230,4108
(CH3O-H···O-COH)- 0,045 0,327 1,702 0,991 -304,1961 0,045 0,332 1,708 0,986 -304,3708
CH3O-H···O(CH3)2 0,031 0,355 1,850 0,968 -269,9360 0,035 0,348 1,793 0,973 -270,0885
CH3O-H···OCHOH 0,020 0,360 2,061 0,964 -304,7137 0,025 0,356 1,917 0,967 -304,8244

Finalmente y dada la relación encontrada según la ecuación 5.11 entre las densidades
del punto crítico del HB y del enlace asociado, nos proponemos analizar las ec. 5.1 y
5.2 sobre el orden de enlace propuestas por Bader y Pauling respectivamente.
Dada la relación logarítmica encontrada para la densidad del punto crítico y la distancia
interatómica encontrada tanto para el enlace de H como para el enlace asociado (Fig.
5.2,5.3,5.4) , y sustituyendo dicha relación en la ecuación 5.2 quedaría:
c
BA
c
rn
]ln[ln
ρ−∆−=
∆−= (5.16)
Que nos conduciría a la ecuación (5.17)
c
B
PCn
=
0ρ
ρ (5.17)
Siendo PCρ la densidad electrónica en el punto critico de enlace de hidrógeno o enlace
asociado y 0ρ la densidad electrónica del enlace en el monómero aislado sin enlace de
hidrógeno, B=0,3144 y c=0,337 son parámetros obtenidos de la fig 5.4 y 5.5 para el
caso del enlace de hidrógeno: O-H···O
Dicha relación ha sido comprobada para el caso de los dímeros aniónicos de glicina,
usando los datos de las tablas 5.3 y 5.4 para el caso del enlace O-H···O tanto en gas
como en agua .Obtenemos unos ordenes de enlace coherentes que pueden analizarse en
la Tabla 5.6. n1 se refiere al HB y n2 al enlace asociado. Comprobamos que la suma de
ambos es muy próxima a 1 como corresponde al enlace de H según ec. 5.5.
Tabla 5.6. Órdenes de enlace calculados según ec. 5.5 para el conjunto de dimeros NA de la glicina.
n1 n2 n1+n2 NAG1 0,214 0,822 1,036 NAG2 0,251 0,789 1,040 NAG3 0,245 0,798 1,043 NAG4 0,211 0,819 1,030 NAG5 0,200 0,842 1,042 NAG6 0,361 0,652 1,013 NAW1 0,271 0,758 1,029 NAW2 0,260 0,771 1,031 NAW3 0,256 0,784 1,039 NAW4 0,209 0,831 1,041 NAW5 0,291 0,733 1,024 NAW6 0,275 0,751 1,027
Para explorar si esta fórmula puede extenderse con valor general, hemos considerado la
misma serie de aductos con enlace de hidrógeno O-H···O considerados anteriormente.
Para esta serie las constantes B y c toman, respectivamente los valores 0,3917 y 0,4859.
Se comprueba nuevamente que la suma de los ordenes de enlace obtenidos para los
enlaces O-H y H···O (Tabla 5.7) nunca difiere de la unidad en más de un 10%.
Tabla 5.7. Órdenes de enlace calculados para la unidad O-H···O con la expresión 16 a partir de densidades de punto crítico de enlace obtenidas al nivel B3LYP/6-311++G(d,p) para una serie de aductos de agua y metanol.
Aducto n1(H···O) n2(O-H) n1+n2 HO-H···OH2 0,113 0,965 1,078 (HO-H···OH)- 0,362 0,686 1,048 HO-H···OHCH3 0,129 0,959 1,088 (HO-H···O-CH3)
- 0,326 0,739 1,065 (HO-H···O=COH)- 0,108 0,949 1,058 HO-H···O(CH3)2 0,136 0,955 1,091 HO-H···O=CHOH 0,100 0,967 1,067 CH3-O-H···OH2 0,118 0,967 1,085 (CH3O-H···OH)- 0,328 0,734 1,063 CH3O-H···OH-CH3 0,132 0,962 1,093 (CH3O-H···O-CH3)
- 0,395 0,680 1,076 (CH3O-H···O=COH)- 0,183 0,895 1,078 CH3O-H···O(CH3)2 0,140 0,958 1,097 CH3O-H···O=CHOH 0,100 0,968 1,069

Conclusiones
1.- Nuestro trabajo corrobora la dependencia logaritmica entre la distancia del enlace de
H y su densidad electrónica en el punto crítico.
2.- Los valores de c encontrados se aproximan a los previamente reportados por otros
autores
3.- Basándonos en la relación anterior se ha encontrado una relación lineal entre las
densidades del punto crítico del HB y del anlace asociado. La validez de esta relación se
ha comprobado, tanto en fase gas como en disolución acuosa, en dimeros aniónicos de
glicina y en un conjunto de aductos modelo.
4.- La relación lineal a que se refiere la conclusión anterior solo tiene validez en las
estructuras que corresponden a un minimo energético, pero no así en estructuras
intermedias durante la formación del enlace.
5.- Se ha encontrado una relación nueva entre orden de enlace y densidad del punto
critico que difiere de las propuestas por Pauling y Bader y proporciona ordenes de
enlace en unidades X-H···Y que verifican una valencia total unidad para el átomo de
hidrógeno.

5.6 Corrección counterpoise y optimización geométrica
El cálculo de la energia de interacción de complejos moleculares ha abierto un
amplio campo de estudio, en particular cuando estos complejos estas unidos por fuerzas
de interacción débiles como son los enlaces de H de gran importancia en bioquímica.
Pero el estudio teórico de dichos sistemas conlleva al tan estudiado fenomeno de Basis
set superposition error (BSSE) resultando complejos artificialmente estabilizados al
considerar diferentes funciones bases para el cálculo del complejo y de los monómeros.
Este efecto es notable en procesos de dimerización al calcular la E dimerización
( ABE∆ ) como la ecuación 6.1.
))()(()()( BEAEABEABE BAABAB
βαβα +−=∆ ∪ (6.1) con α y β como conjuntos de
funciones base y A, B los momomeros en cuestión.
Entre los métodos aproximados usados para corregirlo, el método del counterpoise (CP)
[1] es el más habitual. El error se estima como diferencia entre la energía del monómero
calculada con su base y la calculada con la base usada para el dímero completo (6.2)
))()(()()( BEAEABEABE BAAB
CP
AB
βαβαβα ∪∪∪ +−=∆ (6.2)
Sin embargo esta ecuación no presupone un cambio en la geometria al pasar de los
monómeros al dímero. Por tanto se trataria ahora de calcular cuanto se benefician los
mómomeros en la geometria del dímero al usar las funciones base uno del otro pero sin
estar los átomos [2]. Así pues la estimación del BSSE supone realizar 4 cálculos
independientes (ec. 6.3)
)()()()( BEAEBEAEE II
B
II
A
I
AB
I
ABcorr −−+=∆ (6.3)
Donde I representaria la energía de cada monomero con su base en la geometria del
dímero y II representaría la energía de cada monómero con su base y la base del otro
monómero en las posiciones donde estarian los átomos de dicho monómero en el
dímero pero sin estos átomos. Es lo que se denominan "ghost orbitals".
Pero el debate continua hasta nuestros dias porque el problema del BSSE es
independiente del tipo de interacción para la formación del dímero y que además esta
cambia conforme los monómeros se acercan, diseñando por tanto una nueva superficie
de energia potencial (PES) [3], por ejemplo cuando especies de capa abierta
interaccionan con moleculas neutras. Por tanto el método CP nos genera dimeros con
grandes distancias entre monomeros menos estables, como por ejemplos enlaces de H,

debido a que BSSE no tiene en cuenta atracción fisica de éstos [4]. Se han ideado
mecanismos para construir superficies de energia potencial CP-corregidas [5]. Éste es
un método controvertido al estar basado en la idea de que dos geometrias distintas
originan una misma estructura en el mismo PES [6]. También ha sido propuesto un CP
a nivel atómico como suma de contribuciones atómicas [7]. Pero aún así subsiste otro
importante problema como es la elección de monómeros en la formación del dimero.
Así desde considerar dos o tres fragmentos [8], hasta considerar cada átomo como un
fragmento [9],
Nuestro trabajo está generado bajo la idea de que el BSSE no puede estar basado en la
elección de los fragmentos. De este modo, el método de counterpoise, como lo
entendemos, estaría invalidado. Ademas basándonos en los enlaces de H como fuerza de
atracción de gran distancia en procesos de dimerización y del estudio que la aplicación
del método CP hace de ellos al incrementar la distancia del enlace de H [10-11], hemos
aplicado el método del CP a un set de dímeros con enlace de H como son [NH3,H2O],
[NH4+,H2O], [NH3,OH-],[NH3,NH3],[H2O,H2O] de los cuales hemos calculado su
BSSE eligiendo dos tipos distintos de fragmentos para cada dímero. De todos ellos
hemos aplicado el método CP como un single-point (SP) en el dimero después de la
optimización y como un cálculo CP optimizando cada fragmento en el dimero
Detalles computacionales
Todos los cálculos han sido llevados a cabo usando 6-311++G(d,p) basis set
dentro de 4 niveles de teoria HF, B3LYP, MP2 y CCSD aplicandolos en el set de
programas de Gaussian-09 [12]. Funciones difusas y de polarización fueron usadas para
permitir una mejor descripción de los enlaces de H [13,14]. A todos los dimeros se les
ha realizado: i) una vez optimizados, cálculos single-point CP; ii) optimizationes de
geometria standard CP-corregidas en fase gas.
Todos los valores de energia mostradas en tablas incluyen correcciones térmicas a
298,15 K. El análisis vibracional realizado muestra que todas las estructuras
optimizadas son mínimos con cero frecuencias imaginarias.
Resultados y Discusion
Como hemos comentado en la introducción, han sido muchos los estudios
hechos a favor o en contra del método de CP para solucionar el problema del BSSE,

algunos de los cuales concluyen en mejorar computacionalmente hablando el basis set
[15], más que contrarrestar efectos del CP. Sin embargo y dado el coste computacional
que representa el aumentar el basis set al máximo tamaño, el método CP sigue siendo
muy usado en especial en dimeros con uniones débiles como enlaces de H para el
cálculo de la energía de interacción afectada por BSSE. Sabiendo además que en el
cálculo de la energía de interacción no se tienen en cuenta la correlación de los
electrones de un monómero con los del otro al aproximarse, lo que se ha denominado
BSCE (basis set correlation error), la correción CP sólo está dedicada entonces a
solucionar la descripción del sistema en parte [16].
Por otro lado, y quizá uno de los puntos más controvertidos a la hora de formarse una
supermolecula es predecir que 2 moléculas han sido sus predecesoras. Esto hace que el
cálculo de la energía de interacción dependa de la fragmentación elegida para el efecto.
El estudio de la hidratación del OH-, H3O+ y NH4+ ha sido estudiado con CP2 y CP3 (2
o 3 fragmentos) sin llegar a mostrar diferencias significativas en cuanto a la energía de
interacción [8]. Sin embargo otros estudios muestran diferencias significativas en el
cálculo de CP dependiendo de la elección de fragmentos en agregados HF···(HF)n···HF
[17].
En nuestro estudio nos proponemos analizar dímeros los cuales puedan provenir de la
interacción de dos tipos distintos de fragmentos y analizar que diferencias aportan en el
cálculo de la E de dimerización tanto la elección de fragmentos como su diferencia al
haber usado sus correspondientes CPs o no.
Se han optimizados al efecto, los monómeros y dimeros descritos a continuación
buscando la geometría más estable basándonos en trabajos anteriores [18-22] para los 4
niveles de cálculo mencionados.
Las optimizaciones de los monómeros y dimeros analizados en este trabajo así como
su nomenclatura y energia en kJ/mol para los cuatro niveles de cálculo pueden
observarse en tabla 1. Los datos de energía más bajos para los monómeros los adjudica
B3LYP. Para los dimeros el valor más elevado de la energía lo adjudica HF y el más
bajo B3LYP. Podemos intuir entonces que el tratamiento que hace B3LYP y HF sobre
los enlaces de H está tratado a la alza (HF) o a la baja (B3LYP) con respecto a métodos
más precisos como MP2 o CCSD. (En tabla 6.1 solo se muestran las diferencias
energéticas relativas)
Tabla 6.1 -Principales monómeros y dímeros analizados en este trabajo. Los datos de energia de los monómeros son la diferencia con respecto al monómero neutro. Los datos de energía de los dímeros son la diferencia con respecto al dímero de más alta energia.
HF/6-311++G(d,p) B3LYP/6-311++G(d,p) MP2/6-311++G(d,p) CCSD/6-311++G(d,p)
GAS GAS GAS GAS
molecula sin CP Nombre
∆E(kJ/mol) ∆E(kJ/mol) ∆E(kJ/mol) ∆E(kJ/mol)
NH3 m1a 0,00 0,00 0,00 0,00
NH4+ m1b -861,64 -846,57 -852,95 -860,10
NH2- m1c 1723,31 2530,72 1690,09 1708,02
H2O m2a 0,00 0,00 0,00 0,00
H3O+ m2b -696,61 -681,66 -687,36 -694,19
OH- m2c 1663,05 1622,20 1631,19 1651,35
H2N-H······O--H d1 -50507,66 -50657,98 -50601,11 -50559,74
H2N-H······N H3 d2 0,00 0,00 0,00 0,00
H3N+-H·····OH2 d3 -53055,41 -53144,01 -53103,94 -53089,13
HO-H·······NH3 d4 -52133,44 -52233,42 -52189,14 -52168,16
HO-H·······OH2 d5 -104252,97 -104445,63 -104359,04 -104318,88
d1f1 -50504,82 -50651,79 -50588,35 -50548,49
d1f2 -50501,45 -50648,91 -50574,96 -50536,26
d2f1 -16,32 2,08 4,51 4,03
d2f2 14,57 9,56 25,94 24,22
d3f1 -53052,49 -53140,50 -53095,99 -53081,88
d3f2 -53051,27 -53139,21 -53088,24 -53073,86
d4f1 -52131,43 -52230,27 -52182,54 -52162,23
d4f2 -52129,58 -52227,29 -52164,31 -52144,33
d5f1 -104250,63 -104442,26 -104352,20 -104312,77
Single-point CP
d5f2 -104249,38 -104439,61 -104334,07 -104295,06
d1f1 -50504,78 -50651,69 -50588,29 -50548,52
d1f2 -50501,99 -50649,21 -50575,35 -50536,70
d2f1 0,75 1,67 3,97 3,51
d2f2 6,23 8,81 25,64 23,26
d3f1 -53052,68 -53140,65 -53095,95 -53084,43
d3f2 -53051,44 -53139,44 -53088,30 -53073,96
d4f1 -52131,65 -52230,53 -52183,23 -52162,89
d4f2 -52129,71 -52224,75 -52164,70 -52144,84
d5f1 -104250,90 -104442,45 -104353,07 -104313,56
Optimizado CP
d5f2 -104249,56 -104439,65 -104334,60 -104295,95

El objetivo es ahora realizar dos diferentes fragmentaciones de cada dímero f1 y f2 que
estuvieran constituidas por monómeros distintos. Las fragmentaciones elegidas fueron
las siguientes (Tabla 6.2)
Tabla 6.2 Fragmentaciones de los dimeros f1 f2
d1 H2N-H······O--H H2N-······H-OH
d2 H2N-H······N H3 H2N-······ H-N+H3
d3 H3N+-H·····OH2 H3N······H-O+H2
d4 HO-H·······NH3 HO-······H-N+H3
d5 HO-H·······OH2 HO-······ H-O+H2
El mecanismo normal de incluir el método CP es añadirlo, después de optimizada la
molécula, como una corrección single-point CP o bien optimizar geometrias de un PES
que esté CP corregido (CP-optimazed). Ambos casos pueden observarse en la tabla
6.1. Analizando los datos obtenidos podemos observar que en todos los casos resulta
más negativa la energía de complejación considerando un procedimiento CP-optimized
que un single-point CP. La excepción en el d1f1 a nivel HF, B3LYP y MP2 pero su
diferencia es en torno a las centesimas en kJ/mol, lo cual no es significativo. La
diferencia en el resto no llega a 1 kJ/mol en B3LYP, MP2 y CCSD. Pensemos que un
single-point CP no deja de ser un artificio pues considera las geometrias de los
monómeros aislados las mismas que en el dímero.
También es destacable que las fragmentaciones tipo 1 (f1) proporcionan siempre
energías de complejación más negativas que las tipo 2. Para explicar esto consideremos
la tabla 6.3 donde se muestran los BSSE obtenidos con las dos fragmentaciones.
Podemos notar i) que el BSSE calculado siempre es mayor en la fragmentación 2 sin
importar ni dimero, ni tipo de cálculo ni método de optimización y ii) que siempre el
BSSE es siempre más grande cuando es calculado como un single-point.
Una posible explicación sería:
i) Dado que BSSE = [E(F1M)+E(F2M)] -[ E(F1D)+E(F2D)] = J-K, si el BSSE de f2 es
más grande significa que el término K es mas negativo lo que implica que los
monomeros con las bases completas del dimero se estabilizan más. Esto podría ser
debido a que la fragmentación f2 está formada por fragmentos más iónicos o iones
menos estables que con mayor numero de orbitales se estabiliza más. De ahí que al
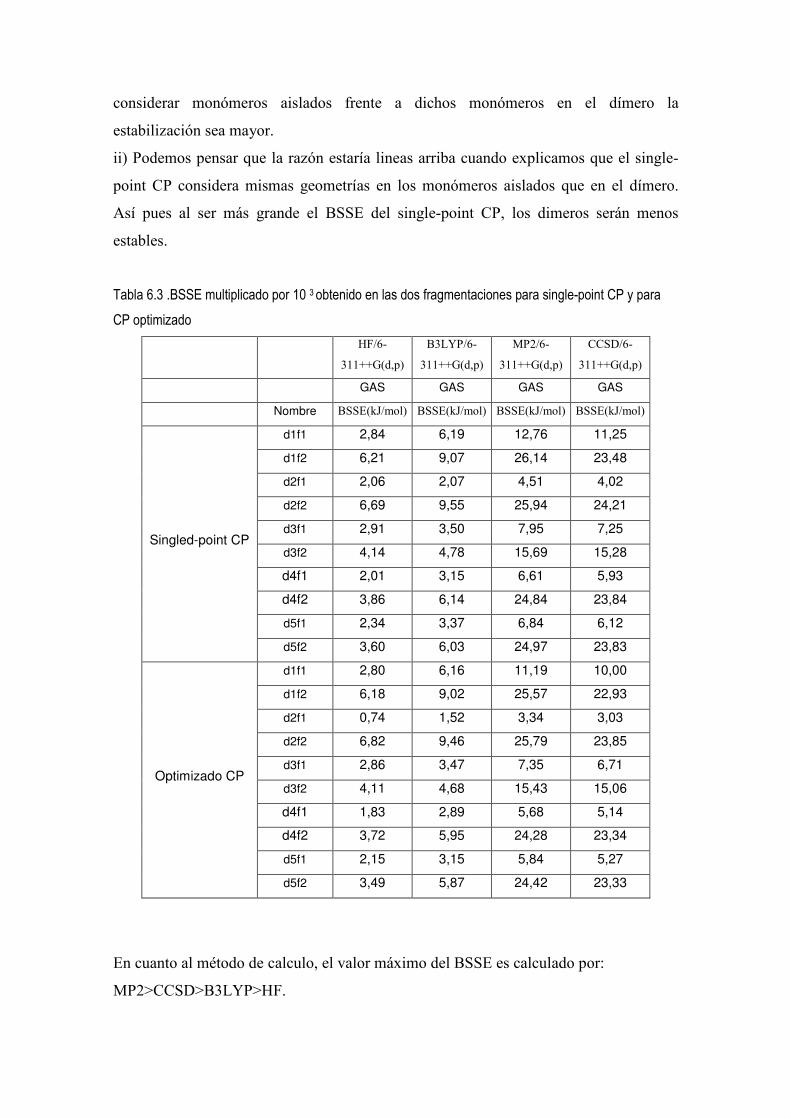
considerar monómeros aislados frente a dichos monómeros en el dímero la
estabilización sea mayor.
ii) Podemos pensar que la razón estaría lineas arriba cuando explicamos que el single-
point CP considera mismas geometrías en los monómeros aislados que en el dímero.
Así pues al ser más grande el BSSE del single-point CP, los dimeros serán menos
estables.
Tabla 6.3 .BSSE multiplicado por 10 3 obtenido en las dos fragmentaciones para single-point CP y para CP optimizado
HF/6-
311++G(d,p)
B3LYP/6-
311++G(d,p)
MP2/6-
311++G(d,p)
CCSD/6-
311++G(d,p)
GAS GAS GAS GAS
Nombre BSSE(kJ/mol) BSSE(kJ/mol) BSSE(kJ/mol) BSSE(kJ/mol)
d1f1 2,84 6,19 12,76 11,25
d1f2 6,21 9,07 26,14 23,48
d2f1 2,06 2,07 4,51 4,02
d2f2 6,69 9,55 25,94 24,21
d3f1 2,91 3,50 7,95 7,25
d3f2 4,14 4,78 15,69 15,28
d4f1 2,01 3,15 6,61 5,93
d4f2 3,86 6,14 24,84 23,84
d5f1 2,34 3,37 6,84 6,12
Singled-point CP
d5f2 3,60 6,03 24,97 23,83
d1f1 2,80 6,16 11,19 10,00
d1f2 6,18 9,02 25,57 22,93
d2f1 0,74 1,52 3,34 3,03
d2f2 6,82 9,46 25,79 23,85
d3f1 2,86 3,47 7,35 6,71
d3f2 4,11 4,68 15,43 15,06
d4f1 1,83 2,89 5,68 5,14
d4f2 3,72 5,95 24,28 23,34
d5f1 2,15 3,15 5,84 5,27
Optimizado CP
d5f2 3,49 5,87 24,42 23,33
En cuanto al método de calculo, el valor máximo del BSSE es calculado por:
MP2>CCSD>B3LYP>HF.

Consideremos ahora las energías de dimerización para los dos tipos de casos single-
point CP y CP-optimazed (Tabla 6.4). Los cálculos están realizados como diferencia
entre la energía CP corregida y sus correspondientes monómeros (ec. 6.4).
∆Edim(AB)=Edim(AB) -Emon (A) -Emon (B) . (6.4)
Tabla 6.4 .E de dimerización para distintas fragmentaciones y distintos cálculos en kJ/mol. HF/6-311++G(d,p) B3LYP/6-311++G(d,p) MP2/6-311++G(d,p) CCSD/6-311++G(d,p)
GAS GAS GAS GAS
∆Edim (kJ/mol) ∆Edim (kJ/mol) ∆Edim (kJ/mol) ∆Edim (kJ/mol) d1f1 -47,19 -61,03 -51,32 -50,25
d1f2 -104,08 -120,10 -96,84 -94,70
d2f1 8,71 -3,09 -2,74 -1,86
d2f2 -848,32 -833,19 -818,45 -829,60
d3f1 -70,18 -80,97 -74,83 -72,20
d3f2 -233,99 -244,61 -232,66 -230,08
d4f1 -10,75 -17,32 -14,32 -12,65
d4f2 -810,31 -789,96 -774,33 -786,00
d5f1 -8,05 -11,18 -8,52 -7,72
Singled-point CP
d5f2 -973,24 -949,07 -934,21 -947,16
d1f1 -47,15 -60,93 -51,27 -50,29
d1f2 -104,62 -120,40 -97,22 -95,14
d2f1 -0,47 -3,51 -3,28 -2,38
d2f2 -856,67 -833,93 -818,75 -830,56
d3f1 -70,36 -81,13 -74,78 -72,91
d3f2 -234,15 -244,83 -232,72 -230,18
d4f1 -10,98 -17,58 -15,01 -13,31
d4f2 -810,44 -790,42 -774,73 -786,51
d5f1 -8,32 -11,37 -9,39 -8,51
Optimizado CP
d5f2 -973,42 -949,11 -934,74 -948,05
Observamos que siempre es más negativa la de la fragmentación 2 dado que son
monómeros más iónicos o menos estables. Y que en general sale más negativo el
procedimiento optimizado con CP (alrededor de 1 kJ/mol la mayoria). Los valores
menos negativos de ∆Edim son otorgados por el cálculo MP2 y los valores mas negativos
los otorga HF. Hay un unico caso donde el valor menos negativo lo otorga CCSD y el
más negativo B3LYP. Este caso se refiere al dimero H2N-······H-OH (d1f2) y ocurre

tanto single-point como en CP optimizado debido a la valoración energética a la alza
que haces del ion NH2- ambos métodos respectivamente.
Para finalizar consideremos la geometria y como el enlace de H modifica su longitud en
los respectivos métodos (Tabla 6.5)
Analizando la tabla 6.5 observamos que, con cualquier nivel de cálculo, la geometría y
las frecuencias de vibración de los complejos obtenidos con el procedimiento CP-
optimized, no sólo difieren de los calculados en la optimización del complejo aislado,
como ya era conocido desde la introducción de este procedimiento [5], sino que son
dependientes de la fragmentación elegida. Teniendo el cuenta que el complejo obtenido
debe presentar la misma geometría con ambas fragmentaciones, resulta claro que el
procedimiento CP-optimized proporciona también artificios de cálculo.
Es un hecho conocido [5] que la distancia d(H···X) aumentan al usar un procedimiento
CP-optimized con B3LYP, MP2 y CCSD. (HF muestra dos casos que no cumplen esto)
El orden en cuanto a longitud del enlace de H según el método es:
B3LYP<MP2<CCSD<HF
Por tanto HF alarga mucho el enlace y B3LYP lo contrario. Tanto MP2 como CCSD
presentan mayor distancia en f1 que en f2 (fragmentación más iónica). Luego en parte,
estos cálculos tienen en cuenta la interacción de los monómeros al calcular CP.
Exactamente lo mismo ocurre con la frequencia de enlace covalente X-H
Por último con respecto a los ángulos, la fragmentación f1 siempre proporciona ángulos
más pequeños que la fragmentación f2 en los dimeros d1, d2, d3 en todos los niveles de
cálculo. Para los dímeros d4 y d5 sucede al revés puesto que son los casos donde la
fragmentación 2 deja grupos OH- como monómeros.
Basándonos en los datos de la tabla 5 podemos calcular para un enlace de H dado A-
H···B dos distancias r1 y r2. (Fig 6.1)
Fig. 6.1. Representación enlace de hidrógeno y su notación empleada
A H B
r1 r2

Tabla 6.5. Propiedades estructurales de los dimeros estudiados. Distancias en Ǻ. Frecuencias en cm-1. Angulos en grados.
d(X-H) d(H····Y) Freq X-H Freq H···Y Θ(X-H···Y)
HF B3LYP MP2 CCSD HF B3LYP MP2 CCSD HF B3LYP MP2 CCSD HF B3LYP MP2 CCSD HF B3LYP MP2 CCSD
d1 1,029 1,029 1,063 1,056 1,845 1,684 1,694 1,726 3292,5 3292,5 2777,6 2894,2 231.9 231.9 276.6 264,5 167.8 167,8 171,3 170,6
d2 1,003 1,020 1,018 1,051 2,461 2,255 2,263 2,304 3803,9 3424,2 3636,6 3025,6 116.0 148.3 147.2 282,8 167.8 168,8 165,8 179,9
d3 1,031 1,063 1,055 1,018 1,751 1,637 1,648 2,304 3282,2 2806,8 2953,8 3617,4 263.2 302.7 292.6 140,6 179.6 179,9 179,9 166,6
d4 0,949 0,977 0,972 0,968 2,129 1,962 1,973 2,014 4029,0 3562,0 3679,3 3753,1 153.4 181.2 173.2 167,1 173.6 171,6 170,9 172,1
Optimizado sin
CP
d5 0,946 0,970 0,965 0,963 2,056 1,933 1,948 1,979 4095,7 3706,7 3810,5 3974,7 148.4 197.7 171.6 166,8 179.0 175,0 178,2 179,2
d1f1 1,028 1,072 1,051 1,047 1,861 1,697 1,798 1,816 3310,5 2616,2 2989,9 3055,3 224.9 248.0 206.6 207,8 167.5 173,2 168,1 167,9
d1f2 1,029 1,072 1,067 1,056 1,847 1,699 1,723 1,757 3283,3 2612,5 2746,5 2872,5 231.5 257.7 226.0 234,1 168.2 173,2 170,6 170,1
d2f1 1,003 1,020 1,017 1,017 2,506 2,305 2,379 2,415 3806,9 3433,8 3505,0 3625,6 101.0 128.8 111.3 126,2 160.5 161,2 152,5 154,2
d2f2 1,003 1,020 1,018 1,022 2,484 2,276 2,388 2,395 3795,7 3420,2 3478,4 3588,8 90.6 124.5 130.9 129,4 169.6 167,7 167,2 169,4
d3f1 1,030 1,062 1,051 1,048 1,766 1,649 1,698 1,720 3209,5 2824,0 3026,9 3085,3 254.3 294.6 270.4 253,3 179.3 179,7 179,4 179,8
d3f2 1,031 1,063 1,059 1,055 1,757 1,649 1,665 1,686 3283,6 2815,6 2922,5 2991,4 270.2 302.5 291.0 285,2 179.6 179,8 179,7 180,0
d4f1 0,948 0,976 0,970 0,966 2,164 1,989 2,060 2,098 4037,9 3578,0 3721,1 3782,0 142.2 203.8 174.5 140 172.2 170,2 169,5 171,1
d4f2 0,949 0,977 0,977 0,972 2,114 1,973 1,991 2,020 4017,5 3564,7 3619,9 3688,9 155.6 185.2 179.0 172,5 171.2 167,3 168,9 169,1
d5f1 0,945 0,969 0,965 0,962 2,105 1,976 2,054 2,077 4223,0 3714,8 3977,0 3860,0 130.8 150.2 132.8 131,1 178.8 172,8 175,4 176,2
Optimizado CP
d5f2 0,946 0,970 0,970 0,968 2,051 1,946 1,974 1,996 4215,0 3885,8 3746,8 3792,4 161.6 170.0 162.5 159,8 177.1 171,4 173,3 173,2

Se ha establecido que estas dos distancias estas correlacionadas [23-25] ya que no
pueden variar independientemente. Conforme con el modelo del enlace de valencia
[26,27], la valencia para cada uno de esos enlaces seria: ( ec. 6.5y ec.6.6)
( )[ ]110 /1
brres
−= (6.5)
( )[ ]220 /2
brres
−= (6.6)
La suma de las dos nos tiene que dar el orden de enlace total, esto es la unidad en el
caso del H y aquí obtendríamos la ec. 6.7
( )[ ]020 /01 1ln brr
ebrr−
−−= (6.7)
Que considerando la suma de r1+r2 nos conduciria a la relación (ec. 6.8)
)1ln(2)(.2 /)(21021
21 brrebrrrrr
+−++−+=+ (6.8)
Relación válida en el caso que los atomos electronegativos que forman el enlace fuesen
del mismo tipo. En caso contrario deberiamos considerar un r01≠r02. Considerando
entonces la r1+r2 como la distancia interatómica entre los átomos pesados y 1/2(r1-r2 )
que significaria la posición relativa del H con respecto a la posición media. (Nótese que
esto sería cuando el enlace de H fuera lineal) encontramos una correlación exponencial
para cada nivel de cálculo de 0,9. Los datos pueden observarse en la Fig 6.2. En la
gráfica podemos visualizar la lógica consecuencia de que al aumentar la distancia entre
átomos pesados, el H se aleja más de la posición central. Por otro lado también
observamos como B3LYP y HF son los métodos con lo que se consiguen distancias más
pequeñas o más grandes respectivamente como ya habiamos anticipado al comienzo del
epígrafe.
También se aprecia los distintos enlaces de H repartidos en tres bloques. El primer
grupo con los valores más pequeños de r1-r2/2 lo constituye el enlace de H N-H···O
de los dímeros d3 y luego d1, el segundo grupo lo conforma el enlace de H, O-H···O del
dimero d5, y luego O-H····N del dímero d4 y por último tenemos el enlace N-H···N del
dimero d2. Esta claro que en primer lugar nos encontramos con d3 y d1 que son dimeros

anionicos. La carga hace que la distancia (r 1+r 2) sea más pequeña. El tener el oxígeno
cargado negativamente hace que el dimero d3 tenga todavía la distancia más pequeña
que d1.
El segundo grupo lo ocupan los dímeros con enlace O-H···X. En primer lugar nos
encontramos al dimero d5 (X=O) con una distancia (r 1+r 2) más pequeña que el
siguiente d4 (X=N) dado el carácter más electronegativo del O.
En último lugar nos encontramos el dimero d2 porque el enlace de H está formado por 2
átomos de N.
También es observable que los dimeros con la fragmentación f2 tienen menor el
parámetro r1-r2/2 con respecto a los de fragmentación f1. Recordemos que f2 era la
fragmentación con monómeros iónicos lo cual explica este hecho.
2,4
2,6
2,8
3,0
3,2
3,4
3,6
0,2 0,3 0,4 0,5 0,6 0,7 0,8
r1-r2/2
r1+r2
HF
B3LYP
MP2
CCSD
Fig 6.2. Representación relación r1+r2 versus r1-r2/2. En Ǻ
Con el análisis de frecuencia de la tabla 5 hemos realizado la Fig 6.3 en la que se puede
observar relación para cada tipo de cálculo entre la d(X···H) y la ν−
(X···H). Las
correlaciones van de 0,8 excepto HF que es 0,9. Las frecuencias siguen exactamente el
mismo orden que lo dicho en el epígrafe anterior. Para la menor distancia se encuentran
las frecuencias más altas y siguen el orden v(d3)>v(d1)>v(d4)>v(d5)>v(d2). Y también
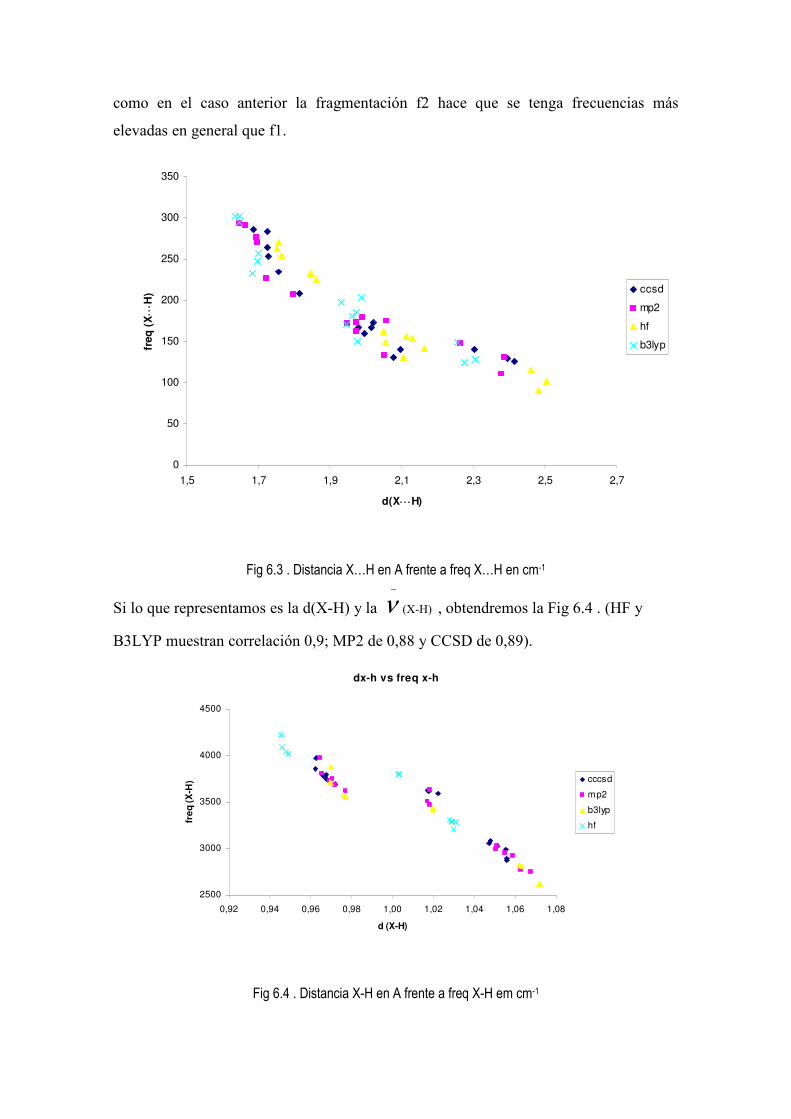
como en el caso anterior la fragmentación f2 hace que se tenga frecuencias más
elevadas en general que f1.
0
50
100
150
200
250
300
350
1,5 1,7 1,9 2,1 2,3 2,5 2,7
d(X···H)
freq
(X
···H
) ccsd
mp2
hf
b3lyp
Fig 6.3 . Distancia X…H en A frente a freq X…H en cm-1
Si lo que representamos es la d(X-H) y la ν−
(X-H) , obtendremos la Fig 6.4 . (HF y
B3LYP muestran correlación 0,9; MP2 de 0,88 y CCSD de 0,89).
dx-h vs freq x-h
2500
3000
3500
4000
4500
0,92 0,94 0,96 0,98 1,00 1,02 1,04 1,06 1,08
d (X-H)
fre
q (
X-H
) cccsd
mp2
b3lyp
hf
Fig 6.4 . Distancia X-H en A frente a freq X-H em cm-1

Conclusiones
1.- Se realizaron cálculos de energía en 4 niveles de cálculo distinto, HF, B3LYP,
CCSD y MP2. B3LYP genera la energía del sistema más elevada y HF la más pequeña.
2.- Las energías de complejación resultan distintas según se realice un single-point
cointerpoise o un counterpoise optimizado
3.- El BSSE con la fragmentación iónica es mayor en todos los niveles de cálculo por la
mayor estabilización de los fragmentos iónicos al aumentar el número de bases
4.- El BSSE depende del método elegido. Así la secuencia de mayor BSSE es:
MP2>CCSD>B3LYP>HF.
5.- La Energía de dimerización (∆Edim) es también obtenida más negativa para la
fragmentación iónica.
6.- El procedimiento CP-optimizado proporciona mayor estabilización al complejo
7.- Tanto la longitud como la frecuencia o el ángulo del enlace de H varian según el
procedimiento sea CP-optimizado o CP-single-point
8.- La teoria del enlace de valencia nos proporciona un enclave para estudiar la
variación de los parámetros mencionados en el punto anterior según el nivel de cálculo.
9.-Coincidencia del tratamiento del nivel de calculo en cuanto a propiedades de enlace
O-H···O, N-H···O, O-H···N, N-H···N

5.7 Estudio QTAIM de aminas heterocíclicas. (HCAs)
Desde la primera vez que se menciona la presencia de sustancias carcinogénicas
en la dieta humana [1], han sido numerosos los estudios epidemiológicos llevados a
cabo para determinarlas [2-7]. Entre dichas sustancias destaca el cada vez mas
numeroso grupo de las aminas heterociclicas (HCAs) formadas a partir de proteinas,
aminoácidos, creatinina, y carbohidratos bien sea por mecanismos a través de radicales
o por la reacción de Maillard [8, 9, 10]. Estas reacciones ocurren naturalmente al cocer
o freir carne o pescado según el procedimiento habitual. Dichas aminas han mostrado
tener potencial mutagénico [11,12] en roedores, simios y humanos, [13-16] según el test
desarrollado por Ames [17]. Sin embargo, la relación entre ingesta de HCAs y riesgo de
cancer no está del todo establecida. Por ejemplo, la dosis establecida para la indución de
cancer en roedores es muy elevada comparada con exposición diaria de los humanos a
las HCAs. Esto ha llevado a analizar que aductos de HCAs con aminoácidos o proteinas
pueden ser liberados en el proceso proteolítico de la digestión [18].
La activación metabólica de las HCAs [19-22] en el citocromo P-450 a través del
isoenzima CYP1A1/CYP1A2 requiere de un proceso previo de N-Oxidación que forma
el N-hidroxi metabolito. Este metabolito luego, por reacciones conjugadas de
acetilacion o sulfonacion con N acetiltransferasa o sulfotransferasa llega a formar un
ester inestable, el cual por escisión heterolítica dará lugar a un ión nitronio. La
estabilidad de este ión nitronio se ha considerado un factor clave a la hora de
determinar el potencial mutagéno de las HCAs por su ataque electrofílico al C8 de la 2'
deoxyguanosina del ADN [21]. (Fig.7.1)
AR NH2 AR NH
OH
AR NH
OR
AR N+
H
P450CYP1A1/CYP1A2 Sulfotrasferasa
N-Acetyltrasferasa-SO3
-COCH3
2-deoxyguanosine
DNA adducts
Aromatic amine N-Hidroxylamino-Ar Ar-Ester Ar-nitrenium ion Fig.7.1 Reacciones de formación del ión nitronium y aductos del ADN

Lógicamente, cuanta más estabilidad tengan dichos iones nitronio, más tiempo tendrán
para reaccionar selectivamente con el ADN [23]. Ha sido demostrado, mediante
estudios teóricos, que existe una relación entre la estabilidad del ión nitronio y la carga
del átomo de N exocíclico del catión nitronio [24,25], definida a partir de un estudio de
población natural (NPA). Asimismo, el logaritmo del potencial mutagénico (log MP)
está relacionado con la tasa de formación de los iones nitronio [26]. Este último estudio
expone ciertos factores críticos para la formación de aductos del ADN como son i) el
asentamiento geometrico de ArNH2 al enzima encargado de su activación, ii) la
facilidad de sustración del protón de la amina en el citocromo, iii) la facilidad de
disociación del ester aromático, iv) la reactividad del catión ArNH+ en contraposición
con su estabilidad. A todos estos factores debemos sumarle un balance entre la
comentada activación metabólica y la destoxificación, bien por formación de
metabolitos destoxificados por hidroxilación [27] o por glucuronidación [28-30](Fig.
7.2).
Ar NH2
4'-Hydroxylation
N-Oxidation
Glucuronidation
Ar NH
Ar NH2
Ar NHOH
O
OH
COOH
OH
OH
GlucuronidationAr N
O
OH
COOH
OH
OH
OH
OH
Glucuronidation
Ar NH2O
O
OH
COOH
OH
OH
DNA adducts
Fig.7.2 Reacciones de destoxificación de las aminas aromáticas heterocíclicas
Nuestro estudio va dirigido a profundizar el análisis de la estabilidad y reactividad del
catión ArNH+. Para ello, hemos usado un análisis de densidad electrónica llevado a
cabo con la Teoría Cuántica de Átomos en Moléculas (QTAIM) [31] .
La teoria QTAIM estudia topológicamente la densidad electrónica obtenida por cálculos
cuánticos. Su objetivo es caracterizar los átomos y su adaptación al entorno desde un
punto de vista matemático. El cálculo del gradiente de la densidad electrónica ∇ ρ nos
permite averiguar los puntos críticos de la función y el cálculo del laplaciano ∇ 2ρ nos
permite caracterizar topológicamente dichos puntos. Matemáticamente hablando la
derivada segunda de una función mide la curvatura local. Así pues, traduciendo su

interpretación a lenguaje químico, si el laplaciano de la densidad es mayor que cero
tendremos una zona de depresión de densidad electronica, mientras que si es negativo
nos encontramos con una zona donde se refuerza dicha propiedad. Por tanto, el análisis
de la distribución de∇ 2ρ es esencial para estudiar la reactividad química, identificando
zonas del sistema molecular sensibles al ataque nucleofilo o electrófilo.
En nuestro estudio hemos analizado, en una serie de HCAs con potencial mutagénico
conocido, la densidad electrónica ρ en el entorno del átomo de N exocíclico sobre el que
se coloca la carga positiva en la forma de Lewis. Es decir, el átomo que se considera
como encargado del ataque electrófilo. Además, hemos determinado donde se localizan
el par electrónico libre de ese átomo para estudiar cual es su papel en dicho ataque
electrófilo.
Detalles Computacionales
Se llevaron a cabo optimizaciones completas de la geometría en fase gas y en
medio acuoso usando el método PCM con el paquete Gaussian 09 [32] y usando el
método del funcional de densidad (DFT), B3LYP. El conjunto base utilizado fue 6-
311++G(d,p) en todos los casos. Se incluyeron funciones difusas y de polarización para
permitir una mejor descripción del enlace de hidrógeno [33,34].
Todos los valores de energías que se muestra en tablas para fase gas incluyen
correcciones térmicas a 298,15 K. El análisis vibracional realizado ha demostrado que
las estructuras optimizadas son mínimos, no presentan frecuencia imaginaria alguna.
Como no es suficientemente exacto el cálculo con frecuencias PCM, no hemos estimado
ZPVE y correcciones térmicas para las estructuras optimizadas en medio acuoso [35].
Por otra parte, no hemos asumido que las correcciones de fase de gas puedan ser
transferidas a disolución acuosa porque a veces ambas geometrías son muy diferentes.
El paquete AIMPAC [36] fue utilizado para realizar el análisis de densidad electrónica
QTAIM. Todas las propiedades atómicas se computaron con |L(Ω)| < 2·10-3 au, que
generalmente garantiza su calidad [37,38]. También comprobamos que la energía y la
población electrónica molecular se reproducen dentro de 10-3 au y 2 kJ mol-1,
respectivamente, por las obtenidas como suma de propiedades atómicas. Nuestra
discusión sobre el análisis de la densidad electrónica utiliza estas propiedades atómicas
y algunas propiedades de enlace QTAIM, computadas en puntos críticos de enlace,
BCP, como la densidad electrónica ρb y la densidad de energía total electrónica Eb y
∇ 2 ρ.

Resultados y Discusión
a) Relación densidad electrónica del N exociclico con la estabilidad del ión nitronio
Se ha elegido un grupo de aminas aromáticas previamente publicadas por
Debnath et al. [39] y caracterizadas por su potencial mutagénico (log MP) [40]. En este
conjunto de compuestos se pueden distinguir varias series que guardan cierta
proximidad estructural. Si bien en un estudio previo [30] se consideraron 6 series:
ab=aminobifenilos, ac=aminocarbonilos, af=aminofluorenos, f=estructura aromática por
anillos fusionados, m=aminas monocíclicas, aia=aminoimidazoazarenos. En el presente
trabajo hemos considerado conveniente realizar una clasificación más precisa. Para ello
los grupos más numerosos (m, f, y ac) han sido divididos en nuevos grupos. Así, en las
aminas monocíclicas (m) se ha distinguido la presencia de grupos funcionales activantes
y desactivantes. En la serie f se han separado aquellos derivados que contienen un grupo
aromático lineal de 3 anillos (tipo antraceno y fenazina) frente a los que contienen otras
estructuras no lineales o (tipo quinolina, fluoranteno, fenantreno, criseno, naftaleno, o
pireno). Por último, en la serie ac se han separado los derivados del piridoindol y los de
(2- ó) 3-aminopiridoimidazopiridina frente a los tetraazafluoranteno o 4-amino
piridoimidazopiridina.
Todas las HCA han sido optimizadas tanto en fase gas como en disolución acuosa
independientemente, presentando en cada fase distinta densidad electronica del N+ y
distinto valor del laplaciano∇ 2ρ.
La tabla 1 muestra, en fase gas y en disolución acuosa, los distintos tipos de aminas
aromáticas con la estabilidad relativa del catión medida con referencia a la anilina (∆E
en kcal/mol), calculada a través de la ecuación (7.1). Asimismo, se muestra su potencia
mutagénica, la población electrónica del N+ y el valor de∇ 2ρ asociado a su par solitario.
∆E= [E(ArNH+)-E(ArNH2)]- [E(C6H5NH+)-E(C6H5NH2)] (7.1)
Con los datos de dicha tabla se han elaborado las gráficas en disolución acuosa y fase
gas en las que se muestra la dependencia la población electrónica N(N+) y ∇ 2ρ frente a
la estabilidad relativa de ion nitronio correspondiente. Debido a cuestiones de espacio y
similaridad solo se muestran las graficas en disolución acuosa (Fig. 7.3 al 7.14). Se
observa que la estabilidad relativa presenta buenas correlaciones tanto con la población
electrónica del N+ como con el valor de ∇ 2ρ (del orden de 0,9). Esto sucede tanto en
disolución como en fase gas para todos los grupos. Todo ello corrobora a ∇ 2ρ como un
nuevo indicador de la estabilidad del catión.
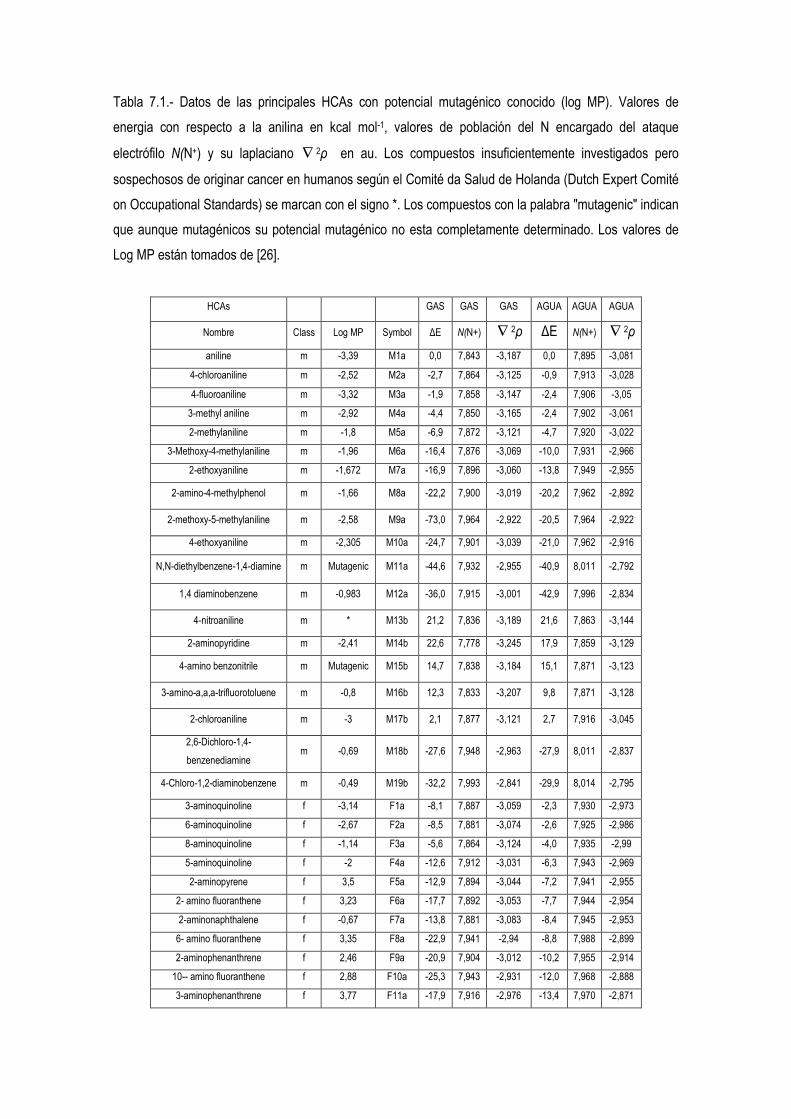
Tabla 7.1.- Datos de las principales HCAs con potencial mutagénico conocido (log MP). Valores de energia con respecto a la anilina en kcal mol-1, valores de población del N encargado del ataque
electrófilo N(N+) y su laplaciano ∇ 2ρ en au. Los compuestos insuficientemente investigados pero
sospechosos de originar cancer en humanos según el Comité da Salud de Holanda (Dutch Expert Comité on Occupational Standards) se marcan con el signo *. Los compuestos con la palabra "mutagenic" indican que aunque mutagénicos su potencial mutagénico no esta completamente determinado. Los valores de Log MP están tomados de [26].
HCAs GAS GAS GAS AGUA AGUA AGUA
Nombre Class Log MP Symbol ∆E N(N+) ∇ 2ρ ∆E N(N+) ∇ 2ρ
aniline m -3,39 M1a 0,0 7,843 -3,187 0,0 7,895 -3,081
4-chloroaniline m -2,52 M2a -2,7 7,864 -3,125 -0,9 7,913 -3,028
4-fluoroaniline m -3,32 M3a -1,9 7,858 -3,147 -2,4 7,906 -3,05
3-methyl aniline m -2,92 M4a -4,4 7,850 -3,165 -2,4 7,902 -3,061
2-methylaniline m -1,8 M5a -6,9 7,872 -3,121 -4,7 7,920 -3,022
3-Methoxy-4-methylaniline m -1,96 M6a -16,4 7,876 -3,069 -10,0 7,931 -2,966
2-ethoxyaniline m -1,672 M7a -16,9 7,896 -3,060 -13,8 7,949 -2,955
2-amino-4-methylphenol m -1,66 M8a -22,2 7,900 -3,019 -20,2 7,962 -2,892
2-methoxy-5-methylaniline m -2,58 M9a -73,0 7,964 -2,922 -20,5 7,964 -2,922
4-ethoxyaniline m -2,305 M10a -24,7 7,901 -3,039 -21,0 7,962 -2,916
N,N-diethylbenzene-1,4-diamine m Mutagenic M11a -44,6 7,932 -2,955 -40,9 8,011 -2,792
1,4 diaminobenzene m -0,983 M12a -36,0 7,915 -3,001 -42,9 7,996 -2,834
4-nitroaniline m * M13b 21,2 7,836 -3,189 21,6 7,863 -3,144
2-aminopyridine m -2,41 M14b 22,6 7,778 -3,245 17,9 7,859 -3,129
4-amino benzonitrile m Mutagenic M15b 14,7 7,838 -3,184 15,1 7,871 -3,123
3-amino-a,a,a-trifluorotoluene m -0,8 M16b 12,3 7,833 -3,207 9,8 7,871 -3,128
2-chloroaniline m -3 M17b 2,1 7,877 -3,121 2,7 7,916 -3,045
2,6-Dichloro-1,4-benzenediamine
m -0,69 M18b -27,6 7,948 -2,963 -27,9 8,011 -2,837
4-Chloro-1,2-diaminobenzene m -0,49 M19b -32,2 7,993 -2,841 -29,9 8,014 -2,795
3-aminoquinoline f -3,14 F1a -8,1 7,887 -3,059 -2,3 7,930 -2,973
6-aminoquinoline f -2,67 F2a -8,5 7,881 -3,074 -2,6 7,925 -2,986
8-aminoquinoline f -1,14 F3a -5,6 7,864 -3,124 -4,0 7,935 -2,99
5-aminoquinoline f -2 F4a -12,6 7,912 -3,031 -6,3 7,943 -2,969
2-aminopyrene f 3,5 F5a -12,9 7,894 -3,044 -7,2 7,941 -2,955
2- amino fluoranthene f 3,23 F6a -17,7 7,892 -3,053 -7,7 7,944 -2,954
2-aminonaphthalene f -0,67 F7a -13,8 7,881 -3,083 -8,4 7,945 -2,953
6- amino fluoranthene f 3,35 F8a -22,9 7,941 -2,94 -8,8 7,988 -2,899
2-aminophenanthrene f 2,46 F9a -20,9 7,904 -3,012 -10,2 7,955 -2,914
10-- amino fluoranthene f 2,88 F10a -25,3 7,943 -2,931 -12,0 7,968 -2,888
3-aminophenanthrene f 3,77 F11a -17,9 7,916 -2,976 -13,4 7,970 -2,871

1-aminonaphthalene f -0,6 F12a -20,5 7,926 -2,985 -14,1 7,971 -2,896
1-aminophenanthrene f 2,38 F13a -25,6 7,943 -2,945 -14,8 7,979 -2,878
9-aminophenanthrene f 2,98 F14a -24,7 7,930 -2,971 -15,0 7,976 -2,881
8- amino fluoranthene f 3,8 F15a -21,5 7,920 -2,983 -16,2 7,976 -2,875
4-aminopyrene f 3,16 F16a -27,2 7,939 -2,923 -16,5 7,991 -2,824
3- amino fluoranthene f 3,31 F17a -28,8 7,941 -2,949 -16,6 7,988 -2,857
6-aminochrysene f 1,83 F18a -33,3 7,950 -2,912 -20,2 7,999 -2,819
1,5-Diaminonaphthalene f -1,35 F19a -32,7 7,961 -2,875 -25,6 8,024 -2,739
2-aminopyrene f 1,43 F20a -38,9 7,968 -2,882 -26,8 8,016 -2,784
2-aminophenazine f 0,55 F21b -9,6 7,890 -3,03 1,8 7,935 -2,944
1,7-diaminophenazine f 0,75 F22b -14,6 7,898 -2,979 -1,0 7,955 -2,861
1,5-diaminophenazine f 1,12 F23b -15,9 7,893 -3,035 -1,3 7,937 -2,95
1-aminophenazine f -0,01 F24b -9,9 7,889 -3,053 -2,8 7,946 -2,952
1,9-diaminophenazine f 0,04 F25b -17,2 7,915 -2,947 -5,9 7,969 -2,855
1,6-diaminophenazine f 0,2 F26b -21,0 7,916 -2,964 -13,0 7,994 -2,816
2-aminoanthracene f 2,62 F27b -18,5 7,909 -2,981 -15,9 7,986 -2,822
2,7-diaminophenazine f 3,97 F28b -28,0 7,918 -2,968 -17,2 7,998 -2,8
1-Aminoanthracene f 1,18 F29b -32,1 7,981 -2,824 -21,8 8,021 -2,778
5-aminoanthracene f 0,87 F30b -40,1 8,01 -2,747 -24,9 8,043 -2,68
2-amine 5-phenyl pyridine ab 0,054 AB1 -3,0 7,865 -3,059 -0,4 7,964 -2,886
3,3'-diaminobiphenyl ab -1,3 AB2 -12,9 7,878 -3,019 -1,5 7,925 -2,952
4-amino-4′-nitrobiphenyl ab 1,04 AB3 -6,4 7,888 -3,057 -2,3 7,934 -2,975
2,4'-diaminobiphenyl ab -0,92 AB4 -15,7 7,877 -2,989 -3,7 7,933 -2,895
4-amino-biphenyl ab -0,14 AB5 -20,1 7,904 -3,014 -11,5 7,963 -2,902
3,4'-diaminobiphenyl ab 0,2 AB6 -24,6 7,910 -2,991 -13,3 7,971 -2,876
4,4'-diamino diphenyl disulfide ab -1,03 AB7 -29,9 7,927 -2,943 -14,1 7,982 -2,844
4,4'-diaminobiphenyl ab -0,39 AB8 -37,7 7,936 -2,922 -28,2 8,023 -2,740
3,3'-Dimethyl-4,4'-diaminobiphenyl
ab 0,01 AB9 -44,0 7,961 -2,898 -31,7 8,030 -2,749
4-[2-(4-aminophenyl)vinyl]aniline ab -2,15 AB10 -46,1 7,953 -2,877 -33,7 8,045 -2,681
4,4''-Diamino-3,3'-biphenyldiol ab 0,15 AB11 -49,3 7,957 -2,863 -35,4 8,034 -2,704
2-Amino-7-nitrofluorene af 3 AF1 -12,5 7,901 -3,031 -14,5 7,950 -2,939
2-Amino-7-bromofluorene af 2,62 AF2 -25,7 7,919 -2,977 -16,3 7,977 -2,87
2-Aminofluorene af 1,93 AF3 -27,0 7,916 -2,991 -17,8 7,977 -2,873
2-Amino-7-acetamidofluorene af 1,18 AF4 -33,1 7,931 -2,945 -22,4 7,998 -2,812
2-hydroxy-7-aminofluorene af 0,41 AF5 -34,2 7,930 -2,949 -24,5 8,001 -2,807
2,7-diaminofluorene af 0,48 AF6 -44,1 7,945 -2,906 -33,9 8,030 -2,726
Pyrido[3',2':4,5]imidazo[1,2-a]pyridin-3-amine
ac 2,26 AC1a -12,2 7,887 -3,069 -3,2 7,928 -2,999
4,6-Dimethylpyrido[3',2':4,5]imidazo[
1,2-a]pyridin-3-amine ac 4,83 AC2a -21,6 7,923 -3,018 -9,6 7,951 -2,965
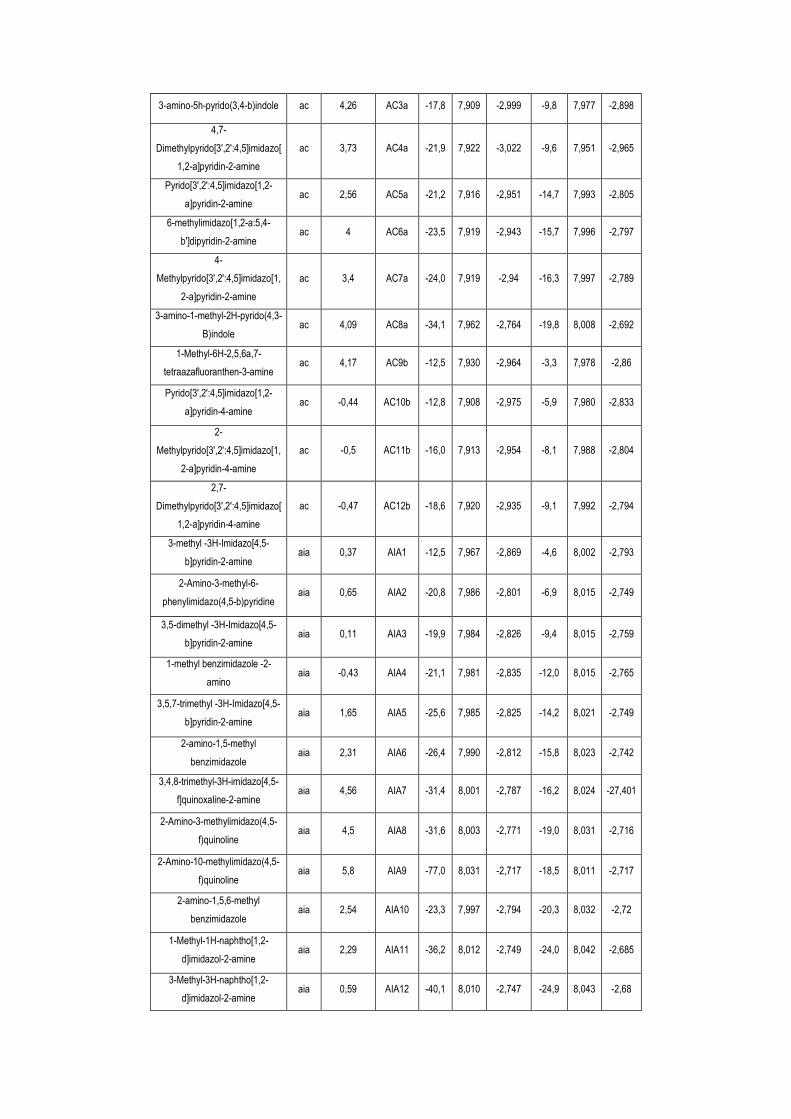
3-amino-5h-pyrido(3,4-b)indole ac 4,26 AC3a -17,8 7,909 -2,999 -9,8 7,977 -2,898
4,7-Dimethylpyrido[3',2':4,5]imidazo[
1,2-a]pyridin-2-amine ac 3,73 AC4a -21,9 7,922 -3,022 -9,6 7,951 -2,965
Pyrido[3',2':4,5]imidazo[1,2-a]pyridin-2-amine
ac 2,56 AC5a -21,2 7,916 -2,951 -14,7 7,993 -2,805
6-methylimidazo[1,2-a:5,4-b']dipyridin-2-amine
ac 4 AC6a -23,5 7,919 -2,943 -15,7 7,996 -2,797
4-Methylpyrido[3',2':4,5]imidazo[1,
2-a]pyridin-2-amine ac 3,4 AC7a -24,0 7,919 -2,94 -16,3 7,997 -2,789
3-amino-1-methyl-2H-pyrido(4,3-B)indole
ac 4,09 AC8a -34,1 7,962 -2,764 -19,8 8,008 -2,692
1-Methyl-6H-2,5,6a,7-tetraazafluoranthen-3-amine
ac 4,17 AC9b -12,5 7,930 -2,964 -3,3 7,978 -2,86
Pyrido[3',2':4,5]imidazo[1,2-a]pyridin-4-amine
ac -0,44 AC10b -12,8 7,908 -2,975 -5,9 7,980 -2,833
2-Methylpyrido[3',2':4,5]imidazo[1,
2-a]pyridin-4-amine ac -0,5 AC11b -16,0 7,913 -2,954 -8,1 7,988 -2,804
2,7-Dimethylpyrido[3',2':4,5]imidazo[
1,2-a]pyridin-4-amine ac -0,47 AC12b -18,6 7,920 -2,935 -9,1 7,992 -2,794
3-methyl -3H-Imidazo[4,5-b]pyridin-2-amine
aia 0,37 AIA1 -12,5 7,967 -2,869 -4,6 8,002 -2,793
2-Amino-3-methyl-6-phenylimidazo(4,5-b)pyridine
aia 0,65 AIA2 -20,8 7,986 -2,801 -6,9 8,015 -2,749
3,5-dimethyl -3H-Imidazo[4,5-b]pyridin-2-amine
aia 0,11 AIA3 -19,9 7,984 -2,826 -9,4 8,015 -2,759
1-methyl benzimidazole -2-amino
aia -0,43 AIA4 -21,1 7,981 -2,835 -12,0 8,015 -2,765
3,5,7-trimethyl -3H-Imidazo[4,5-b]pyridin-2-amine
aia 1,65 AIA5 -25,6 7,985 -2,825 -14,2 8,021 -2,749
2-amino-1,5-methyl benzimidazole
aia 2,31 AIA6 -26,4 7,990 -2,812 -15,8 8,023 -2,742
3,4,8-trimethyl-3H-imidazo[4,5-f]quinoxaline-2-amine
aia 4,56 AIA7 -31,4 8,001 -2,787 -16,2 8,024 -27,401
2-Amino-3-methylimidazo(4,5-f)quinoline
aia 4,5 AIA8 -31,6 8,003 -2,771 -19,0 8,031 -2,716
2-Amino-10-methylimidazo(4,5-f)quinoline
aia 5,8 AIA9 -77,0 8,031 -2,717 -18,5 8,011 -2,717
2-amino-1,5,6-methyl benzimidazole
aia 2,54 AIA10 -23,3 7,997 -2,794 -20,3 8,032 -2,72
1-Methyl-1H-naphtho[1,2-d]imidazol-2-amine
aia 2,29 AIA11 -36,2 8,012 -2,749 -24,0 8,042 -2,685
3-Methyl-3H-naphtho[1,2-d]imidazol-2-amine
aia 0,59 AIA12 -40,1 8,010 -2,747 -24,9 8,043 -2,68

M1aM2a
M3aM4a
M5aM6a
M7a M8aM9a
M10aM11a
M12a
M13b
M15b
M16b M17b
M18bM19b
R2 = 0.9524
R2 = 0.9828
-60
-40
-20
0
20
40
7.86 7.88 7.9 7.92 7.94 7.96 7.98 8 8.02N(N+)
∆E
Fig.7.3: ∆E (kcal/mol) N(N+) para monocyclic. Terminaciones en "a" representa que el anillo tiene un
grupo activante y en "b" desactivante.
Fig.7.4: ∆E (kcal/mol) vs. ∇ 2ρ (au) para monocyclic. Terminaciones en "a" representa que el anillo tiene
un grupo activante y en "b" desactivante
M12a M11aM10aM9a
M8aM7aM6a
M5aM4a M3aM2aM1a
M19bM18b
M17bM16b
M15bM14b
M13b
R2 = 0.9345
R2 = 0.978
-50
-30
-10
10
30
-3.15 -3.05 -2.95 -2.85 -2.75∇2ρ
∆E

Fig.7.5: ∆E (kcal/mol) N(N+) para fused. Terminaciones en "a" representa ciclos de 2 , 3 y 4 fusionados, y
en "b" ciclos de 3 lineales
Fig.7.6: ∆E (kcal/mol) vs. ∇ 2 ρ para fused. Terminaciones en "a" representa ciclos de 2 , 3 y 4
fusionados, y en "b" ciclos de 3 lineales
F1a
F2aF3a
F4aF5a
F6a F7aF8aF9a
F10aF11aF12a
F13aF14a
F15aF16aF17a F18a
F19aF20aF30b
F29b
F28bF27b
F26b
F25bF24bF23b
F22bF21b
R2 = 0.9678
R2 = 0.9493
-30
-25
-20
-15
-10
-5
0
5
7.92 7.94 7.96 7.98 8 8.02 8.04 8.06
N(N+)
∆E
F1a
F2aF3a
F4aF5a
F6a F7aF8aF9a
F10aF11a
F12aF13a
F14a
F15aF16aF17a F18a
F19aF20aF30b
F29b
F28bF27b
F26b
F25bF24bF23b F22b
F21b
R2 = 0.9678
R2 = 0.9493
-30
-25
-20
-15
-10
-5
0
5
7.92 7.94 7.96 7.98 8 8.02 8.04 8.06
∆E
∇2ρ

Fig.7.7: ∆E (kcal/mol) N(N+) para aminobyphenyls
Fig.7.8: ∆E (kcal/mol) vs. ∇ 2 ρ (au) para aminobyphenyls
Fig.7.9: ∆E (kcal/mol) vs. N(N+) (au) para aminofluorenes
AB10AB9AB8
AB7AB6AB5AB4
AB3AB2 AB1
R2 = 0.9267
-35
-30
-25
-20
-15
-10
-5
0
5
7.92 7.94 7.96 7.98 8 8.02 8.04
N(N+)
∆E
AB11AB10AB9
AB8
AB7AB6AB5
AB4AB3
AB2 AB1
R2 = 0.9069
-40
-35
-30
-25
-20
-15
-10
-5
0
-3 -2.95 -2.9 -2.85 -2.8 -2.75 -2.7 -2.65
∆E
∇2ρ
AF6
AF5
AF4AF3
AF2AF1
R2 = 0.9239
-35
-30
-25
-20
-15
-10
7.94 7.96 7.98 8 8.02 8.04
N(N+)
∆E

Fig.7.10: ∆E (kcal/mol) vs. ∇ 2ρ (au) para aminofluorenes
Fig.7.11: ∆E (kcal/mol) vs. N(N+) para aminocarboniles
Fig.7.12: ∆E (kcal/mol) vs.∇ 2 ρ para aminocarboniles
AF6
AF5AF4
AF3
AF2
AF1
R2 = 0.9326
-35
-30
-25
-20
-15
-10
-2.95 -2.9 -2.85 -2.8 -2.75 -2.7
∇2ρ
∆E
AC8aAC7a
AC6aAC5a
AC4a AC3aAC2a
AC1a
AC12bAC11bAC10b
AC9b
R2 = 0.9019
R2 = 0.907
-20
-15
-10
-5
0
7.92 7.94 7.96 7.98 8
N(N+)
∆E
AC1a
AC2a AC3a
AC4aAC5aAC6aAC7a AC8a
AC12bAC11b
AC10bAC9b
R2 = 0.9006
R2 = 0.9972
-20
-15
-10
-5
0
-3 -2.95 -2.9 -2.85 -2.8 -2.75 -2.7 -2.65
∆E
∇2ρ
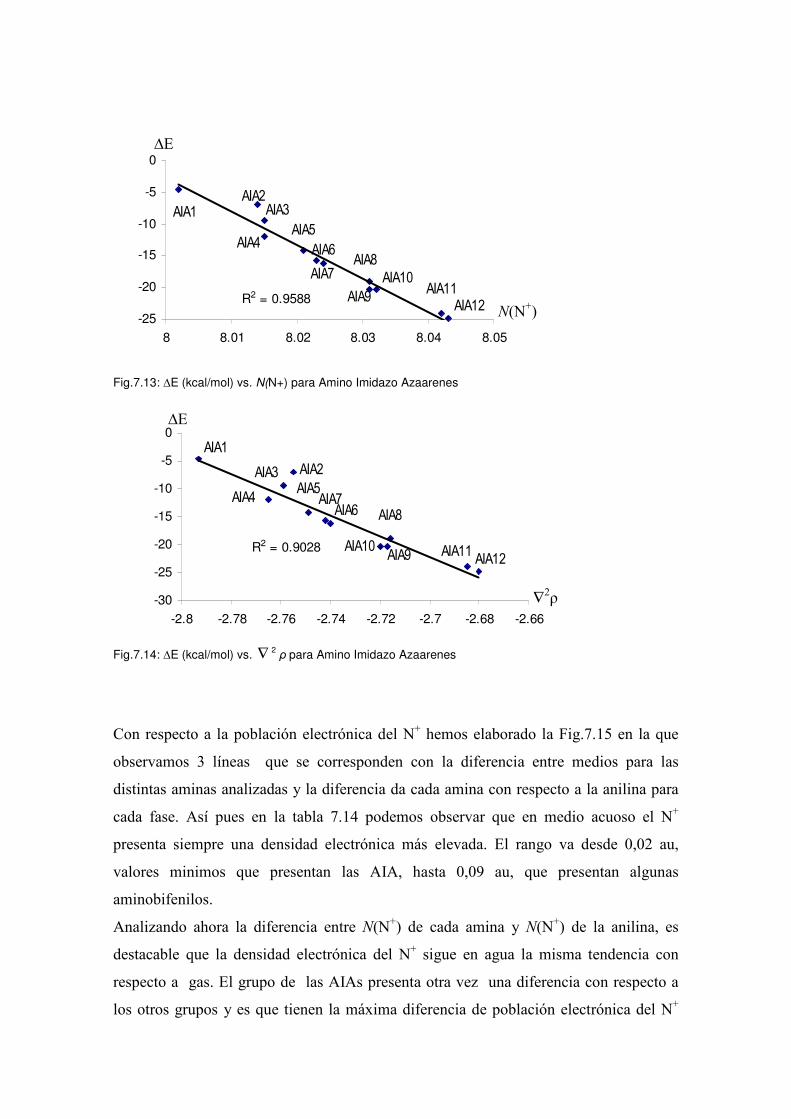
Fig.7.13: ∆E (kcal/mol) vs. N(N+) para Amino Imidazo Azaarenes
Fig.7.14: ∆E (kcal/mol) vs. ∇ 2 ρ para Amino Imidazo Azaarenes
Con respecto a la población electrónica del N+ hemos elaborado la Fig.7.15 en la que
observamos 3 líneas que se corresponden con la diferencia entre medios para las
distintas aminas analizadas y la diferencia da cada amina con respecto a la anilina para
cada fase. Así pues en la tabla 7.14 podemos observar que en medio acuoso el N+
presenta siempre una densidad electrónica más elevada. El rango va desde 0,02 au,
valores minimos que presentan las AIA, hasta 0,09 au, que presentan algunas
aminobifenilos.
Analizando ahora la diferencia entre N(N+) de cada amina y N(N+) de la anilina, es
destacable que la densidad electrónica del N+ sigue en agua la misma tendencia con
respecto a gas. El grupo de las AIAs presenta otra vez una diferencia con respecto a
los otros grupos y es que tienen la máxima diferencia de población electrónica del N+
AIA1AIA2
AIA3
AIA4AIA5
AIA6AIA7
AIA8
AIA9AIA10
AIA11AIA12R2 = 0.9588
-25
-20
-15
-10
-5
0
8 8.01 8.02 8.03 8.04 8.05
N(N+)
∆E
AIA12AIA11AIA10 AIA9
AIA8AIA7
AIA6AIA5AIA4
AIA3 AIA2AIA1
R2 = 0.9028
-30
-25
-20
-15
-10
-5
0
-2.8 -2.78 -2.76 -2.74 -2.72 -2.7 -2.68 -2.66
∆E
∇2ρ

con respecto a la anilina. Aspecto éste que se nota mucho más en gas, siendo además la
única serie en la que todas las moléculas tienen valores superiores en gas que en agua
con respecto a la anilina. Esto se debe a que al contrario que la piridina, los imidazoles
presentan un carácter PI excedente. Por tanto el ion N+ tiene mayor densidad electrónica
que la mayoria de las aminas.
También destacan valores negativos (relativos a la anilina) de N(N+) en aquellas
aminas monocíclicas que poseen sustituyantes desactivantes del anillo aromático.
-0,100
-0,050
0,000
0,050
0,100
0,150
0,200
M1a
M5a
M9a
M13
bM
17b
F2a F6aF10
aF14
aF18
aF22
bF26
bF30
bAB4
AB8AF1
AF5AC3a
AC7a
AC11b
AIA3
AIA7
AIA11
INCR MEDIO
Incr anilina gas
incr anilina agua
Fig.7.15 . Diferencia de N(N+) (linea azul diferencia agua-gas sale todos positivos, linea amarilla Incremento de N(N+) respecto a la anilina en fase acuosa y linea rosa mismo incremento en fase gas.
b) Relación de ∇ 2 ρ del N exociclico con la estabilidad del ión nitronio.
Dentro de los múltiples factores que pueden dotar de significativa potencia
mutagénica a un HCA, la localización de la densidad electrónica del par electrónico del
N exocíclico podría ser un valioso indicativo de su reactividad.
Tal y como hemos dicho en la introducción, el laplaciano representa una medida de la
localización o concentración de carga. Los valores obtenidos pueden verse en tabla 7.1.
Nuestra meta es encontrar si existe algún tipo de relación entre dicha localización y la
estabilidad del ión nitronio.
A priori debería existir dicha relación porque si extrapolamos la densidad de carga por
una función exponencial decreciente en r (distancia al nucleo). [42].
reNr ..)( ξρ −= (7.2)
su derivada segunda seria :
∇ 2 )(rρ = )/.2.(. 2. reN r ξξξ −− (7.3)

Y como resultado podriamos preveer entonces entre ambas magnitudes una
dependencia.
Se han buscado los valores de laplaciano más negativo (concentración de carga) y que
esté más alejado del nucleo (región de localización del par solitario) y se han
representado frente a la densidad electrónica.
La Fig 7.16 muestran la extrecha relación lineal (R2= 0,94 y 0,96 en gas y en agua
respectivamente) entre datos densidad electronica y laplaciano en medio acuoso para
todo el set de aminas aromáticas
Fig. 7.16 N(N+) vs. Laplaciano de Ρ en medio acuoso. (∇ 2 ρ)
Por tanto si hemos comprobado la existencia de una dependencia entre la densidad del
N exocíclico con la estabilidad del ión nitronio, también existirá algún tipo de relación
con respecto al laplaciano.
De las Fig. 7.16 podemos extraer las siguientes conclusiones:
a) No importa a que grupo de HCAs pertenezcan, siempre correlacionan
b) Los monociclos son los que tienen el par más localizado y la menor densidad
electrónica de todas las aminas.
Una explicación cualitativa se basa en considerar que el N extrae menos
densidad electrónica de un monociclo que de un sistema aromático mayor. En
consecuencia la población electrónica del N+ es menor en los monociclos y la repulsión
interelectrónica también, esto contribuye a que el par solitario esté más localizado en los
monociclos al reducirse las repulsiones. Esto concuerda con el hecho de que cuando el
-3,2
-3,1
-3
-2,9
-2,8
-2,7
-2,6 7,8 7,85 7,9 7,95 8 8,05 8,1
N(N+)
∇ 2 ρ
M F AB
AF
AC AIA

ciclo es piridínico (más deficiente de carga que el benceno), tiene la menor densidad
electrónica de todo el grupo y presentan menor localización del par.
Siguiendo esta argumentación luego tenemos los monociclos con grupos desactivantes
fuertes (tipo NO2) o activantes débiles. Por último tenemos los grupos activantes del
anillo.
Llama la atención la diferencia gas y agua. En gas la densidad electrónica de N
exociclico perteneciente al ciclo piridínico (m14b, no mostrado), se encuentra bastante
alejado de los monociclos con grupo -NO2 y -CF3. Y en agua se encuentran en la
misma zona debido a la solvatación de estos grupos por moléculas del solvente.
También observamos que las aminas monociclicas con –OH, grupo activante, tienen
una relación N(N+)/∇ 2 ρ semejante a la presentada por algunos fused. Mientras que la
de aminas monocíclicas con -NH2 muestran una relación próxima a la mayoria de los
AC. Es decir incluir estos grupos propociona un aporte de densidad electronica
semejante al suministrado por otro anillo fusionado.
c) Los fused siguen la misma tendencia comentada al principio de los monociclos; los
de mayor localización son los biciclos en el que uno de los anillos es piridinico. Les
siguen los triciclos tipo phenazine debido a poseer un ciclo tipo pyrazine, dos N
intraciclicos en el medio. Recordemos que pyrazine es menos básica que la piridina por
el efecto inductivo que presenta uno de los N sobre el otro, ademas de la resonancia
entre esos dos nitrogenos. Este razonamiento podria explicar que se encuentren por
encima de los monociclos piridinicos en cuanto a carga del N exociclico.
Por último los compuestos formados por 3 o 4 ciclos tipo fluorantene, pyrene,
phenanthrene, chrysene serian los que presentan una menor localización del par y
aumento de carga del N exociclico. En fase gas y agua, el compuesto fused con mayor
carga en N exocíclico sería el 5 -amino anthracene (F29b). Por lo que podemos decir
que el efecto mesómero predomina frente al efecto inductivo.
d) Observando los aminobiphenyls (AB), aquellos con más carga son los que pertenecen
a la serie benzidine con los grupos aminos en posición para (4,4'-benzidine). Uno de los
que mas población electrónica tiene (en agua y gas) es AB10 con lo que la existencia
del doble enlace interbencenos mantiene el efecto de resonancia que le confiere
densidad al N exocíclico

Estos derivados de 4,4'-diaminobyphenyl pueden tener tanta densidad de carga sobre el
N exociclico como los AIAs. Y se diferencian clarísimamente del resto de los biphenyls
con los grupos aminos en otras posiciones. Como en los grupos anteriores, el que menos
carga tiene en fase gas, es cuando uno de los benzenos es una piridina.
e) En el grupo de los AC, resulta clave la orientación del N del ion amonio con respecto
al N contenido en el ciclo. Tanto en agua como en gas la mayor población electrónica
corresponde a AC8a, cuyo N intracíclico está protonado, por lo que éste sustrae menos
densidad electrónica del anillo. Esto genera que el par electronico del ion nitronio pueda
estar menos localizado. En el otro extremo se encuentra AC1a con la menor población
electrónica, ya que presenta el ion nitronio en meta respecto al N intracíclico. Esta
posición se ve desfavorecida con respecto a orto y para por los efectos resonantes
ilustrados en la Fig. 7.17. Con la unidad -NH+ situada en posición "meta", el N
intracíclico nunca quedará cargado positivamente en sus formas resonantes con lo cual
dicha unidad podrá retirar menos densidad del anillo piridínico.
N
NH+
N
NH
N
NH
N
NH+
N
NH
N
NH
N NH+
N NH N NH
+
+
+
+
+
+
Fig 7.17. Formas resonantes de la piridina sustituida por grupo amino en posiciones 2, 3 y 4
f) El grupo de los aminefluorenes (AF) presenta una correlacion de 0,999. Su estructura
es similar a los amino carboniles (AC) excepto por el hecho de que no presentan N
intracíclicos. Este hecho provoca que se encuentren valores de N(N+) y∇ 2 ρ en el rango
de los AC. Pero podemos observar como aquellos con grupos activantes como -NH2, -
NHOR, -OH presentan una menor localización del par electrónico del N+ frente a
aquellos con grupos desactivantes. Este efecto se nota pese a la gran distancia entre
dicho N y el grupo funcional.

g) Por último, mencionar que uno de los grupos estructuralmente más variados con un
mayor potencial mutagénico, AIAs, presenta correlaciones de 0,9 tanto en agua como en
gas. Todos ellos tienen en común que el N del ión nitronio se encuentra emplazado en el
medio de los N de un anillo imidazol, lo que le confiere ciertas peculariedades tanto a la
carga como a la localización del par electrónico del N del catión nitronio. Tal es así que
presentan la densidad electrónica del N exacíclico más alta, así como la menor
localización del par de dicho átomo. En agua la densidad electrónica del N supera las 8
ua. Por tanto, y de acuerdo con los trabajos de Colvin y colaboradores [43] sobre la
estabilidad de los cationes iminio frente a los nitronio, podemos concluir un efecto de
conjugacion entre los 3 nitrógenos que daría lugar a una mayor deslocalización del par
electrónico con el consecuente aumento de densidad electrónica del N del catión
iminio.(Fig 7.18)
N
N NH
+
CH3
Fig.7.18. Representación efecto de conjugación en el ión nitronio
De toda la serie de los AIAs, los que tienen el par electrónico menos localizado son los
triciclicos (AIA11 y AIA12) y los de par más localizado son los biciclicos.
Conclusiones
Se ha analizado un conjunto de aminas aromáticas precursoras de aductos con
ADN, cuyo potencial mutagénico ha sido determinado o es esperable. El análisis
consistió en relacionar la estabilidad del ión nitronio que forman estas aminas, con la
población electrónica del átomo N+ y con la localización de su par solitario (∇ 2 ρ ). Este
último elemento se ha tenido como factor estimativo de la reactividad química de un
atacante electrófilo. Nuestros resultados muestran correlaciones de alrededor de 0,9 en
todos los casos. También ha sido encontrada una buena correlación lineal entre N(N+) y
∇ 2 ρ tanto en medio acuoso como en fase gas. Estas correlaciones permiten analizar el
comportamiento de las distintos tipos de aminas aromáticas en términos de localización
del par solitario: mayor en las aminas monociclicas y menor en el grupo de las AIAs.

5.8 Estudio computacional de la actividad biológica de aminas
heterocíclicas potencialmente cancerígenas
La preocupación por los sustancias tomadas en la ingesta diaria que se sabe o se
suponen carcinogénicas ha hecho que aparezcan cada dia nuevos estudios para
determinarlas [1-6]. Entre estas sustancias se encuentran algunas aminas heterociclicas
(HCA) formadas naturalmente en la elaboración de los alimentos que ingerimos.
Incluso se han elaborado tests que adjudican a cada HCA un determinado potencial
mutagénico [7].
Su carácter mutagénico pasa por la formación de aductos con el ADN [8]. Pero
para ello la amina ha de activarse metabólicamente según un proceso (Fig 8.1) [9,11],
en que se forman iones nitronio, que son los responsables de la formación de dichos
aductos.
AR NH2 AR NH
OH
AR NH
OR
AR N+
H
P450CYP1A1/CYP1A2 Sulfotrasferasa
N-Acetyltrasferasa-SO3
-COCH3
2-deoxyguanosine
DNA adducts
Aromatic amine N-Hidroxylamino-Ar Ar-Ester Ar-nitrenium ion Fig.8.1. Camino de reacción para la formación del ión nitronio.
Además solo un porcentaje de estas aminas llegará a formar aductos porque este
proceso compite con la formación de metabolitos destoxificados bien sea por
hidroxilación [12] o por glucuronidación [13-15]( Fig 8.2)
Ar NH2
4'-Hydroxylation
N-Oxidation
Glucuronidation-1
Ar NH
Ar NH2
Ar NHOH
O
OH
COOH
OH
OH
Glucuronidation-2Ar N
O
OH
COOH
OH
OH
OH
OH Ar NH2O
O
OH
COOH
OH
OH
DNA adductsGlucuronidation-3
Fig.8.2 . Reacciones de destoxificación de aminas aromáticas con capacidad para formar aductos del ADN.

Los estudios computacionales sobre estas reacciones son escasos, y se concentran en la
estabilidad de los iones nitronio [16-18] como factor clave a la hora de formación de
aductos del ADN.
En este trabajo se presenta un estudio estructural y de análisis de la densidad electrónica
mediante la Teoría Cuántica de Átomos en Moléculas (QTAIM) [19] de las distintas
reacciones de activación y destoxificación de una HCA. Dada la complejidad que
presenta elaborarlo para todas las aminas con potencial mutagénico conocido, hemos
escogido como modelo la molecula de 2-amino-1-metil-6-fenilimidazo[4,5-b]piridina
(PhIP). Esta elección se debe a que es una de las aminas más estudiadas [20-31], tiene
una potencia mutagénica elevada log MP=2,75 [18], y es una de las más abundantes
HCAs que se crean durante el proceso de cocinado de alimentos. Así, se ha encontrado
a PhIP en concentraciones que incluso alcanzan los 182 ng/g, mientras que para las
restantes HCAs no se detectan concentraciones superiores a 24 ng/g [5].
Detalles Computacionales
Se llevaron a cabo completas optimizaciones de geometría para fase gas y para
medio acuoso usando el método PCM con el paquete Gaussian 09 [32] y usando el
método del funcional de densidad (DFT), B3LYP. La función base utilizada fue 6-
311++G(d,p)en todos los casos. Se añadieron funciones difusas y de polarización para
permitir una mejor descripción del enlace de hidrógeno [33,34].
Todos los valores de energías que se muestra en tablas para la fase de gas incluyen
correcciones térmicas a 298,15 K. El análisis vibracional realizado ha demostrado que
las estructuras optimizadas son mínimos con ninguna frecuencia imaginaria. Como no
es suficientemente exacto el cálculo con frecuencias PCM, no hemos estimado ZPVE y
correcciones térmicas para PCM estructuras optimizadas [35].
El paquete AIMPAC [36] fue utilizado para realizar el análisis de densidad electrónica
QTAIM. Todas las propiedades atómicas se computaron con |L(Ω)| < 2·10-3 au, que
generalmente garantiza su calidad [37,38]. También comprobamos que la energía y la
población electrónica molecular fueron reproducidos dentro de 10-3 au y 2 kJ mol-1,
respectivamente, por los obtenidos como suma de las propiedades atómicas ΣN(Ω) and
ΣE(Ω). .
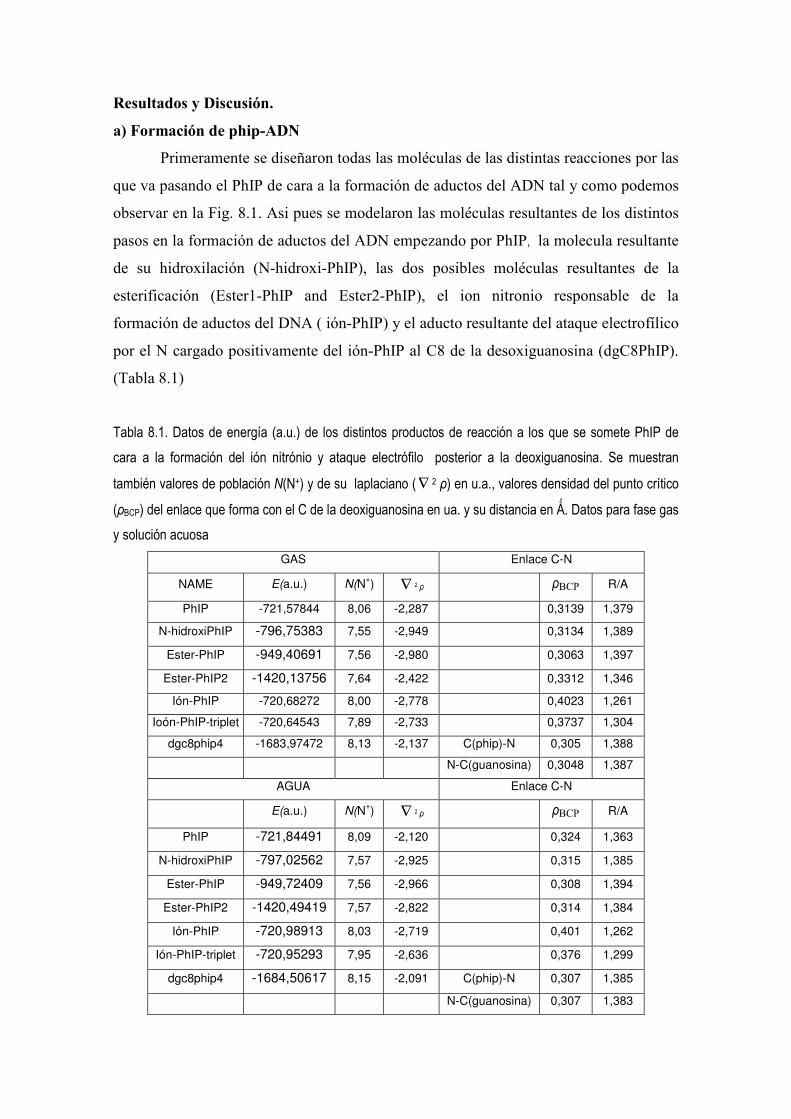
Resultados y Discusión.
a) Formación de phip-ADN
Primeramente se diseñaron todas las moléculas de las distintas reacciones por las
que va pasando el PhIP de cara a la formación de aductos del ADN tal y como podemos
observar en la Fig. 8.1. Asi pues se modelaron las moléculas resultantes de los distintos
pasos en la formación de aductos del ADN empezando por PhIP, la molecula resultante
de su hidroxilación (N-hidroxi-PhIP), las dos posibles moléculas resultantes de la
esterificación (Ester1-PhIP and Ester2-PhIP), el ion nitronio responsable de la
formación de aductos del DNA ( ión-PhIP) y el aducto resultante del ataque electrofílico
por el N cargado positivamente del ión-PhIP al C8 de la desoxiguanosina (dgC8PhIP).
(Tabla 8.1)
Tabla 8.1. Datos de energía (a.u.) de los distintos productos de reacción a los que se somete PhIP de cara a la formación del ión nitrónio y ataque electrófilo posterior a la deoxiguanosina. Se muestran
también valores de población N(N+) y de su laplaciano ( ∇ 2 ρ) en u.a., valores densidad del punto crítico
(ρBCP) del enlace que forma con el C de la deoxiguanosina en ua. y su distancia en Ǻ. Datos para fase gas y solución acuosa
GAS Enlace C-N
NAME E(a.u.) N(N+) ∇ 2 ρ ρBCP R/A
PhIP -721,57844 8,06 -2,287 0,3139 1,379
N-hidroxiPhIP -796,75383 7,55 -2,949 0,3134 1,389
Ester-PhIP -949,40691 7,56 -2,980 0,3063 1,397
Ester-PhIP2 -1420,13756 7,64 -2,422 0,3312 1,346
Ión-PhIP -720,68272 8,00 -2,778 0,4023 1,261
Ioón-PhIP-triplet -720,64543 7,89 -2,733 0,3737 1,304
dgc8phip4 -1683,97472 8,13 -2,137 C(phip)-N 0,305 1,388
N-C(guanosina) 0,3048 1,387
AGUA Enlace C-N
E(a.u.) N(N+) ∇ 2 ρ ρBCP R/A
PhIP -721,84491 8,09 -2,120 0,324 1,363
N-hidroxiPhIP -797,02562 7,57 -2,925 0,315 1,385
Ester-PhIP -949,72409 7,56 -2,966 0,308 1,394
Ester-PhIP2 -1420,49419 7,57 -2,822 0,314 1,384
Ión-PhIP -720,98913 8,03 -2,719 0,401 1,262
Ión-PhIP-triplet -720,95293 7,95 -2,636 0,376 1,299
dgc8phip4 -1684,50617 8,15 -2,091 C(phip)-N 0,307 1,385
N-C(guanosina) 0,307 1,383

Varios conformeros del PhIP (según la posición del anillo bencenico y la posición de
los H del grupo amino) fueron optimizados tanto en disolución acuosa y en fase gas
para encontrar la molecula más estable. Todas las moleculas convergieron en una
esqueleto plano para el biciclo nitrogenado que forma un ángulo diedro de 41.4 º con el
anillo bencénico. (Fig.8.3)
Fig 8.3. PhIP
La estructura mostrada en la Fig. 8.3 para PhIP podría convertirse a través de un
proceso tautomérico en 2-imino-1-metil-6-fenilimidazo[4,5-b]piridina. Esta molécula
presenta isomería Z/E (imino-PhIP1 o imino-PhIP2) dependiendo de que el H del grupo
imino se encuentre, respectivamente, en posición cis o trans respecto al grupo metílico,
(Fig 8.4).
Fig.8.4. imino-PhIP2
Viendo los datos de la tabla 8.2 observamos que el más estable es el PhIP con grupo
amino. El análisis de población nos muestra que el PhIP es más estable por tener una
ganancia densidad electrónica neta en los N e H frente al esqueleto carbonado.
Especificamente ganan densidad los N que soportan el grupo metilo y el del anillo
piridinico

Tabla 8.2.Diferencias de energía entre los principales tautómeros del PhIP (kJ.mol-1) e incrementos de población electrónica global en u.a. 103
∆N (imino-amino PhIP). Datos para medio acuoso ∆E(kJ/mol) ∆N(C) ∆N(H) ∆N(N)
PhIP 0
imine-PhIP1 26.5
imine-PhIP2 24.6 -97 35 62
Sin embargo vamos a observar más adelante que el N-hidroxiimino va a ser más estable
que el N-hidroxiphip que es la molécula resultante de la hidroxilación del PhIP del que
hablaremos seguidamente
Se optimizó N-hidroxi-PhIP del cual podriamos tener dos posibilidades: la posición en
cis (N-hidroxi-PhIP) o trans (N-hidroxi-PhIP2) del grupo hidroxilo con respecto al
grupo metilo del anillo imidazol . Los resultados de estabilidad nos muestran como más
estable la posición en cis por 6,13 kJ/mol. El análisis de población nos muestra que con
esta disposición pierden densidad electronica la mayoria de los carbonos, y los N que
no soportan ningún sustituyente de los anillos (Tabla 8.3). Concretamente, aumenta la
densidad electrónica el O, el N que soporta el grupo metilo y el N exocíclico. El análisis
de la densidad electrónica nos muestran que no se encuentra un bondpath entre un H del
grupo metilo con el O del grupo hidroxilo pero la interacción electrostática favorece la
disposición alternada de los H del metilo respecto al H hidroxílico (interacción también
encontrada en dimeros de glicina [39,40]. (Fig 8.5). Sin embargo, los datos
computacionales revelan la existencia de un enlace N21···H-O de 0,02 ua a una
distancia de 2.15 Ǻ, en la N-Hidroxi-PhIP2 tanto en fase gas como en medio acuoso.
Tabla 8.3. Diferencias de energía entre los principales tautómeros aminos del N-hidroxi-PhIP (kJ.mol-1) y incrementos de población electrónica global (N-hidroxi-PhIP)-( N-hidroxi-PhIP2) en u.a. Datos para medio
acuoso ∆E(kj/mol) -6,1
103·∆N(Ctotal) -13
103·∆N(Htotal) 9
103·∆N(Ntotal) -4
103·∆N(O) 8

Fig 8.5. N-hidroxi-PhIP
Si comparamos datos de población atómica del N-hidroxi-PhIP con el PhIP,
observamos que con excepción del anillo bencénico que está muy alejado como para
sufrir cambios apreciables, la mayoría de los átomos pierden población en este proceso.
Sólo es de notar un aumento de población en el anillo imidazol frente a la gran pérdida
que experimenta el N del grupo amino en 0,65 ua. (Tabla 8.1).
Esta molécula, igual que PhIP, puede tautomerizar transformándose en una molécula
con dos isómeros Z/E (considerando la disposición del grupo imino respecto al
hidroxilo): N-hidroxi-iminoPhIP (trans del hidroxilo con respecto al grupo metilo. (Fig.
8.6) y N-hidroxi-iminoPhIP2 (posición cis del hidroxilo con respecto al grupo metilo).
Sorprende el hecho de que encontramos un tautomero más estable que el N-hidroxi-
PhIP por 11 kJ/mol (Tabla 8.4). La posición trans del imino es la que resulta favorecida
(Tabla 8.4).
Por tanto dos hechos llaman nuestra atención:
a) Que el N-hidroxi-iminoPhIP adopte como más estable la posición trans frente a la cis
que era la que estabilizaba a N-hidroxi-PhIP.
b) Que el N-hidroxi-iminoPhIP sea más estable que la más estable de las posiciones del
N-hidroxi-PhIP que se sabe resultado de la hidroxilación del PhIP [11].

El primer hecho es fácilmente explicable porque al existir un doble enlace C=N la
distancia se acorta en 0,098 Ǻ, lo que hace que el O en cis se encuentre tan cercano al
grupo metilo aumentando el impedimento estérico.
Sobre el segundo hecho debe comentarse que N-hidroxi-iminoPhIP pierde la
aromaticidad del ciclo imidazol y el O del hidroxilo no forma enlace de H con el H del
grupo imidazol (estan a 2,389 A). El recuento de densidad electrónica indica que esta
disposición imino permite una mayor población en el conjunto de los N y del O, es decir
de las átomos más electronegativos (Tabla 8.4). Vamos a comprobar en el capítulo 10
de la presente tesis que optimizadas las moléculas via MP2 el tautómero más estable en
disolución acuosa será el que tiene el grupo amino. Por tanto la preferencia tautomérica
depende del medio y del método de cálculo
Tabla 8.4. Diferencias de energía entre los principales tautómeros (iminos y aminos) del N-Hidroxi-PhIP (kJ.mol-1) e incrementos de población electrónica global en u.a. 103
∆N ((hidroxi-iminoPhIP)-(hidroxi-aminoPhIP)). Datos de densidad del punto critico en u.a. Longitud de enlace en Ǻ
∆E(kJ/mol)) ∆N(Ctotal) ∆N(Htotal) ∆N(Ntotal) ∆N(Ototal)
ρBCP
C-N
R/A
C-N
ρBCP
N-O
R/A
N-O
N-hidroxiPhIP 0 0,314 1,39 0,288 1,437
N-hidroxi-
iminoPhIP -11 -115 -25 91 49 0,382 1,292 0,284 1,439
N-hidroxi-
iminoPhIP2 8,8
Fig 8.6. N-hidroxi-imino-PhIP

Con respecto al siguiente proceso de esterificación, también se construyeron varios
conformeros de los cuales mostramos los más estables. (Fig 7).
Ester-PhIP1 Ester-PhIP2
Fig. 8.7. Productos más estables de las posibles esterificaciones del PhIP
Así como al hidroxilar el ciclo imidazol ganaba densidad, al esterificar este ciclo la
pierde densidad tanto en Ester-PhIP1 como en PhIP2. Es de notar las grandes
diferencias en la reorganización de la población según el tipo de esterificación.
Podemos observar como pierde más población el O del hidroxilo al enlazar con un S,
notándose menos su efecto a lo largo de los ciclos diferenciándose así del proceso de
esterificación 1 (Tabla 8.5)
Tabla 8.5. Diferencias de energía entre los principales esteres del PhIP (kJ.mol-1) e incrementos de población electrónica global en u.a. 103
∆N ((Ester-PhIP)- (N-HidroxiPhIP )). Datos en medio acuoso ∆N(Ctotal) ∆N(Htotal) ∆N(Ntotal) ∆N(Ototal) ∆N(anillo imidazol)
Ester-PhIP1 9 -16 -29 -6 -22
Ester-PhiIP2 13 14 -21 -87 -32
De la tabla 8.1 también podemos observar la pérdida de población del N responsable de
la formación de aductos comparando la población en el hidroxi, con la población en la
esterificación 1 (pérdida de 1,15 au) o la esterificación 2 (pérdida de 1,07 au).
Con el siguiente paso de formación del ionPhIP, (Fig 8.8), incluso el ciclo bencénico
extremo que no se veía afectado hasta el momento va a perder población. A pesar de
tener formalmente una carga positiva el N, según su estructura de Lewis, el grupo amino
tiene mayor población que cuando estaba esterificado o hidroxilado tanto en fase agua
como en gas (tabla 8.1)

Fig 8.8. Ión PhIP
Pensemos ahora en la formación del ión phip. En primer lugar el estado singulete o
triplete en el cual se encuentran los iones phip es un hecho discutido desde hace tiempo
[41]. Hemos optimizado tanto el estado singulete como el triplete, y el resultado nos
muestra como más estable el estado singulete. (Tabla 8.1). Nuestros resultados
concuerdan con otros obtenidos [42,43]. Como podemos observar en la tabla 8.6, el
singulete deja mayor población en N e H a costas de los C y deja una mayor población
en el N cargado positivamente, encargado del ataque electrófilo.
Tabla 8.6. Incrementos de población electrónica global en u.a. 103∆N. Datos para medio acuoso
∆N(Ctotal) ∆N(Htotal) ∆N(Ntotal)
ión-PhIP triplete -ión-PhIP singulete 144 -75 -70
Por último se han optimizado varios conformeros del aducto dgC8PhIP y de ellos ha
resultado ser el más estable el que se muestra en la Fig 8.9. Este confórmero presenta 3
enlaces de hidrogeno, cuya densidad puede ser observada en la Fig. 8.9
El análisis de población del ataque, (Tabla 8.7), nos muestra una pérdida de población
neta en la deoxiguanosina. De los cárbonos solamente dos son los que pierden; aquel
que sufre el ataque (-0,417 ua casi medio e-) y otro del anillo imidazol. Respecto a los
dos que se mantienen, el resto sufre pérdida de población. De los N los que más pierden
son los del anillo imidazol (-0,02 cada uno). Y por último de los O, uno se mantiene y
los demás ganan población. En resumen los 0,46 que pierde el anillo imidazol de la
deoxiguanosina es ganado por el anillo imidazol del PhIP.

Fig 8.9. dgC8PhIP
También observar que el N atacante del ión PhIP gana 0,16 ua más de la población que
la que tenía en el ión. El enlace que se forma tiene una densidad electrónica en el punto
crítico comparable al de una esterificación (0,307 ua y una longitud también parecida
1,38 A). Ver Tabla 8.1
Tabla 8.7. Diferencias de población electrónica entre (dgC8PhIP- deoxiguanosina) o (dgC8PhIP-ión-PhIP) en u.a. 103
∆N. Datos en medio acuoso. Destacar que las sumas de población electrónica no dan cero porque la deoxiguanosina pierde un H. Por tanto si le sumamos ese H sí queda una suma de 0,001 de error.
∆N(Ctotal) ∆N(Htotal) ∆N(Ntotal) ∆N(Ototal) ∆N(anillo imidazol)
dgC8PhIP-deoxiguanosina -394 -37 -47 8 -464
dgC8PhIP-ión-PhIP 719 291 337 456
0.008 a.u.
0,003 a.u. 0,009 a.u.

b) Metabolismo del PhIP
Aparte de la estabilidad de los iones nitronio formados en las HCAs, otro factor
clave de cara a la formación de aductos del ADN es el proceso de destoxificación del
PhIP. En la medida que el PhIP siga los procesos de destoxificación, no formará parte
de reacciones para la formación de aductos con el ADN.
La detoxificación del PhIP puede llevarse a cabo de 2 formas via hidroxilación o
glucuronidación [15].
El proceso de hidroxilación puede llevar a varios productos dependiendo de la posición
del átomo que sufre el ataque nucleofílico. En el caso concreto del PhIP, el único lugar
desencadenante del proceso mutagénico es si la hidroxilación ocurre en el N del grupo
amino, pero han sido observados otro tipos de metabolitos [29] con hidroxilaciones en
otras posiciones, (en concreto 2-OH-PhIP el cual en vez del grupo amino tiene un OH y
se desconoce se potencial carcinogeno o el 4'-hidroxi-PhIP que no es carcinogeno [12]).
La tasa entre productos genotóxicos y destoxificados depende de la metabolización via
los enzimas CYP1A1, CYP1B1, CYP3A4, CYP2C9, CYP2A3 intra o
extrahepaticos.[27].
Se han optimizado todos los posibles metabolitos procedentes de la hidroxilación y los
resultados pueden observarse en la tabla 8.8. Recordar que del metabolito hidroxilado
responsable de la formación de aductos ya hablamos.
El camino para la destoxificación pasa por una hidroxilación en un carbono del anillo
benzénico extremo. Resultado de esto podemos obtener 4'-hidroxi-PhIP , 3'-hidroxi-
PhIP y 2'-hidroxi-PhIP. De ellos el más estable resulta ser el 3'-hidroxi-PhIP por 0,6 y
7,5 kJ/mol respecto al 4' y al 2'- hidroxi-PhIP respectivamente. Con la hidroxilación en
3' el anillo benzénico gana 0,01 ua de densidad electronica a cambio de perderla los N
intracíclicos. (Fig 8.10).
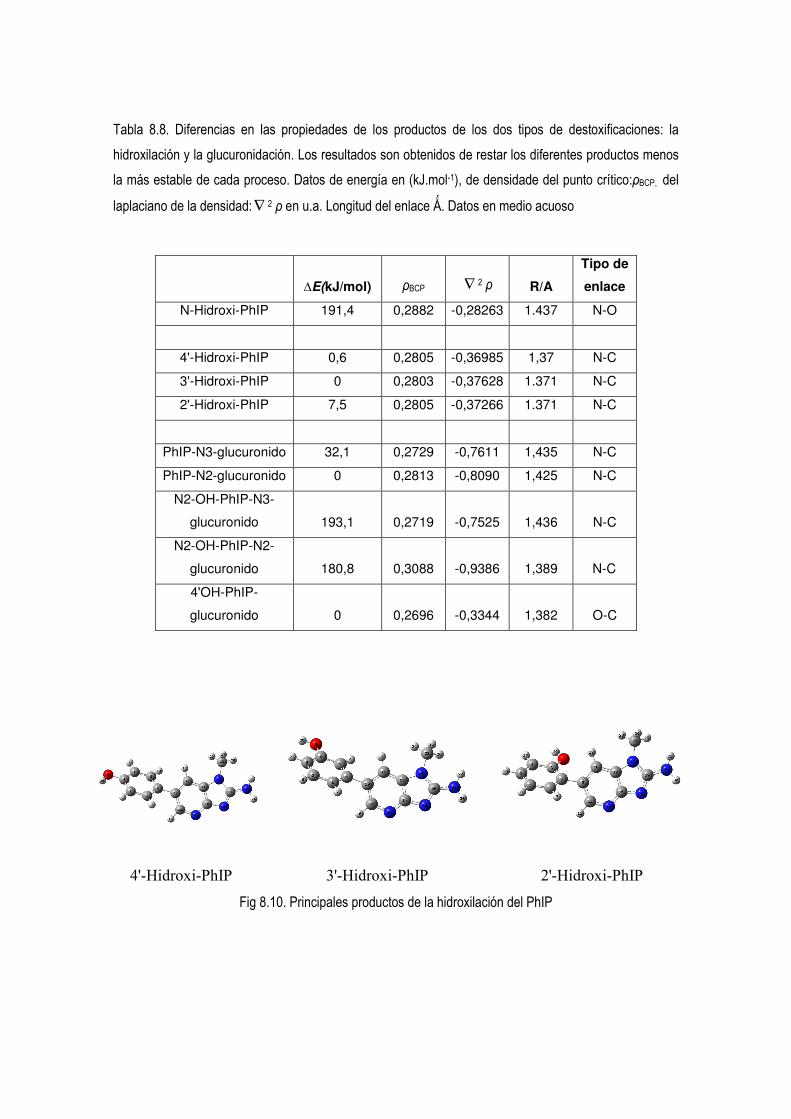
Tabla 8.8. Diferencias en las propiedades de los productos de los dos tipos de destoxificaciones: la hidroxilación y la glucuronidación. Los resultados son obtenidos de restar los diferentes productos menos la más estable de cada proceso. Datos de energía en (kJ.mol-1), de densidade del punto crítico:ρBCP, del
laplaciano de la densidad:∇ 2 ρ en u.a. Longitud del enlace Ǻ. Datos en medio acuoso
4'-Hidroxi-PhIP 3'-Hidroxi-PhIP 2'-Hidroxi-PhIP
Fig 8.10. Principales productos de la hidroxilación del PhIP
∆E(kJ/mol) ρBCP ∇ 2 ρ R/A
Tipo de
enlace
N-Hidroxi-PhIP 191,4 0,2882 -0,28263 1.437 N-O
4'-Hidroxi-PhIP 0,6 0,2805 -0,36985 1,37 N-C
3'-Hidroxi-PhIP 0 0,2803 -0,37628 1.371 N-C
2'-Hidroxi-PhIP 7,5 0,2805 -0,37266 1.371 N-C
PhIP-N3-glucuronido 32,1 0,2729 -0,7611 1,435 N-C
PhIP-N2-glucuronido 0 0,2813 -0,8090 1,425 N-C
N2-OH-PhIP-N3-
glucuronido 193,1 0,2719 -0,7525 1,436 N-C
N2-OH-PhIP-N2-
glucuronido 180,8 0,3088 -0,9386 1,389 N-C
4'OH-PhIP-
glucuronido 0 0,2696 -0,3344 1,382 O-C

Curiosamente este tipo de hidroxilación en el anillo bencénico genera moléculas más
estables que la N-hidroxilación por unas 191,4 kJ/mol. Viendo resultados sobre
densidad electrónica es fácil evidenciar que con la hidroxilación en el anillo bencénico
ganan densidad todos los átomos más electronegativos N y O a costa de C e H. Por
tanto un gran porcentaje del PhIP sigue el proceso de destoxificación según 4'-Hidroxi-
PhIP
Otro tipo de metabolismo del PhIP que compite con la hidroxilacion es la
glucuronidación, el cual es el proceso de transferir el ácido glucurónico a un sustrato
con el fin de mejorar su solubilidad y así poder ser eliminado por el organismo via orina
o heces. Tal y como vemos en la fig 8.2 existen varias formas de glucuronidar al PhIP
que hemos denominado glucuronidación-1, glucuronidación-2 y glucuronidación-3.
Además y dado el proceso de conjugación la glucuronidation 1 y 2 pueda dar lugar a
dos glucuronidos. (Fig. 8.11)
De los dos glucuronidos analizados en el proceso de glucuronidación-1 se encuentra
como más estable la glucuronidación en el N2 por 32 kJ/mol teniendo además una
mayor fortaleza en el enlace N-C creado con el ácido glucurónico (valores de ρBCP más
elevado, tabla 8.8). Ya mencionamos anteriormente que el imino-PhIP es más estable
que el amino-PhIP por lo que la glucuronidación en el N2 del grupo imino se verá
desfavorecida. La misma explicación y tendencia es válida para el proceso de
glucuronidación-2. Observamos 180 kJ/mol menos estable que 4'OH-PhIP-glucuronido
y con unos valores de ρBCP del enlace N-C implicado en la glucuronidation mas elevados
para el N2-glucuronido. Haciendo el desglose de densidades electrónicas de los átomos,
podemos observar que en la glucuronidación en N2 pierden densidad los atomos más
electronegativos y los hidrógenos a favor de los carbonos de los anillos.

PhIP-N3-glucuronido PhIP-N2-glucuronido
N2-OH-PhIP-N3-glucuronido N2-OH-PhIP-N2-glucuronido 4'-OH-PhIP-glucuronido Fig. 8.11. Principales productos de la glucuronidación del PhIP
Conclusiones.
1.- El tautomero imino de phip es más de 20 kJ/mol menos estable que el amino ya que
posee mayor población los nitrógenos
2.- Sin embargo tras el proceso de N-hidroxilación el tautómero imino es más estable
que el amino según el método B3LYP, ya que está estructura permite mayor población
en los átomos más electronegativos
3.- El proceso de N-hidroxilación permite aumentar la densidad electrónica al ciclo
imidazol
4.- El ión nitronio formado es más estable como singulete que en estado triplete, al dejar
mayor población en el N+.
5.- El proceso de ataque electrófilo de los iones nitronio ocurre mediante una pérdida de
medio electrón en la deoxiguanosina
6.- La densidade del punto crítico de formación del enlace N+ al C de la guanosina es
comparable al de una esterificación N···O

7.- El proceso de N-hidroxilación genera metabolitos más inestables que los metabolitos
provenientes de la hidroxilación en el anillo bencénico por lo que la mayor parte del
PhIP es destoxificado.
8.- En la glucuronidación de PhIP los productos más estables son aquellos en los que se
glucuronida en el anillo imidazol y no en el grupo amino del PhIP

5.9 Estudio del equilibrio ceto –enólico de 3-etil-2,4-
pentanodiona (epd)
Ha sido experimentalmente estudiado el efecto del solvente en las β-dicetonas
[1] comprobando que pueden existir en solución como compuesto puro en tres formas
tautomericas:
R
CH3 R1
O
OHO
R
CH3 R1
O O
R
CH3
R1
O
Form a Form b Form c
Fig 9.1. Formas tautoméricas de las dicetonas
Mientras que la forma c es observada en raros ocasiones, el equilibrio tautomérico entre
la forma a y b ha sido ampliamente analizado por espectroscopia RMN [2]
manteniendo una K = [enol]/[diceto] distinta según el disolvente. En el caso de la
acetilacetona [1] dichos valores son:
Disolvente K (RMN) fracción molar(enol) (cmol/mol)
1,4 dioxano 4,8 83
Fase gas 11,7 92
Agua 0,23 19
Tabla 9.1. Propiedades del equilibrio tautomérico diceto/enol
Se observa un mayor porcentaje de forma enol en disolvente apolares que en
disolvente polares. Esto es debido a que la forma enol es la menos polar de los dos
tautómeros ya que el puente de H intramolecular ayuda a reducir la repulsión dipolo
dipolo de los grupos carbonilicos. Además dicho puente de H intramolecular será mas
favorable cuando el disolvente no compite en formar puentes de H. Así pues si la
acetilcetona es diluida con disolventes apolares el contenido de enol aumenta y si es con
disolventes polares disminuye. Así por tanto, el predominio de la forma diceto en
disolventes polares tiene su origen en la estabilización que presentan los grupos
cetónicos al formar puentes de hidrógeno con las moléculas del disolvente. Estudios
actuales analizan también el efecto de los sustituyentes [3] por espectrometria RMN.
Nosotros nos proponemos analizar el equilibrio en un derivado de la acetilcetona, 3-etil-

2,4-pentanodiona (EPD) pero usando calculos teóricos de optimización de la
modelización molecular y posterior cálculo de la densidad electrónica haciendo uso de
la Teoría de Átomos en Moléculas (QTAIM), la cual realiza un estudio topológico de
dicha densidad junto con un cálculo de propiedades atómicas.
Estudios ab initio ya han sido llevados acabo para analizar la relativa estabilidad de
varios enoles de la acetilcetona en fase gas y calculo de la K equilibrio keto/enol en
solución acuosa [4], [5], [6].
Los tautómeros de la forma enólica son evidentes cuando los restos R y R’ son
distintos (Fig. 9.2) pero en nuestro caso que tenemos dos metilos, podemos pensar en
un equilibrio parecido ya sugerido por otros autores [7].
R
CH3 R1
O
O
HO
R
CH3 R1
O
Form 1 Form 2
R1
R OO H
CH3
R
CH3 R1
O
O H
Form 3 Form 4
Fig 9.2. Tautomeros ceto/enol
Adaptándonos a nuestra molécula en particular EPD hemos llevado modelizado todos
los posibles confórmeros de la reacción de enolización por una parte y de la reacción de
acificación en medio acuoso por otra. Una vez optimizadas y encontrado el confórmero
más estable hemos analizado su población para poder interpretar su estabilidad.
Detalles computacionales
Se han realizado optimizaciones completas de la geometría molecular para todas
las moléculas estudiadas. Para el cálculo de las geometrías, frecuencias y densidades
electrónicas de los sistemas se ha empleado la Teoría de Funcionales de la Densidad
(DFT) y más concretamente el método B3LYP. En todos los casos se han utilizado
conjuntos “split valence” de funciones de base, añadiendo funciones difusas y
polarizadas, en concreto: 6-311++g(2d,2p), elegida por la fiabilidad, flexibilidad y
operatividad que confiere a los cálculos. Pero a pesar de ser una base bastante fiable y
para poder cotejar con los datos experimentales también se han realizado
optimizaciones geometrícas al nivel MP2, que incluye la correción de perturbación de
segundo orden. El análisis vibracional realizado ha mostrado que las estructuras

resultantes de la optimización son mínimos energéticos que no presentan ninguna
frecuencia imaginaria, que implicaría la obtención de un estado de transición.
Estas optimizaciones han sido llevadas a cabo tanto en fase gas como en medio acuoso
empleando el modelo Polarizable Continuum Model (PCM) [8] que aproxima el efecto
del solvente mediante esferas centradas en las posiciones atómicas.
Se ha usado el programa Gaussian03 [9] para computar las geometrías moleculares y el
paquete de programas AIMPAC [10] para obtener las propiedades atómicas QTAIM y
realizar el análisis topológico de las densidades eléctrónicas. En la integración de
densidades electrónicas se verificó que los valores de L(Ω) no superasen en valor
absoluto 2·10-3 au, lo cual se considera una condición necesaria para garantizar la
calidad de las propiedades QTAIM integradas. También se comprobó que las energías y
poblaciones electrónicas moleculares eran suficientemente próximas a las obtenibles
como suma de las correspondientes propiedades atómicas, ΣN(Ω) y ΣE(Ω),
respectivamente. Los valores mostrados para energías moleculares incluyen las
correcciones ZPVE y térmica.
Resultados y discusión.
El conjunto de moléculas estudiadas puede observarse en la Fig.9.3.
La primera reacción que estudiamos correspondería con el equilibrio dicetona↔enol en
medio acuoso neutro. Para cada uno de estos casos construimos tres moleculas con el
etilo en syn, anti y gauche. Y para cada una de ellas hicimos un scan de los diedros ω 2,
ω 3 (Fig 9.4) de la posición de los carbonilos girandolos hasta 360º
La reacción sería:
EDP1EDP2
EDP3EDP4EDP5
Reacción nº1
La optimización de todas las geometrias concluyo con la mínima energía en la
geometria la molécula diceto EDP2 frente EPD1, con el etilo en syn y los grupos
cetónicos formando un diedro de 121,77º.
Ademas se hizo otro scan a la molécula cetoenólica EDP3, la más estable de su grupo,
sobre el diedro ω 4 cuyos resultados pueden ser observados en la Fig 9.4 en la que los
mínimos corresponden a posiciones semejantes. Dado que el scan presentaba algún
punto discordante tanto en fase agua como en fase gas se hizo un scaneo más preciso de
un intervalo alrededor de esos puntos no encontrando ninguna anomalía.(Fig 9.5)

Medio acuoso neutro
CH3 CH3
O O
C
H H
CH3
H
CH3 CH3
O O
C
CH3 H
H
H
EPD1(KKcca) EPD2(KKccg)
CH3 CH3
O
C
H H
CH3O
H
CH3 CH3
O
C
H CH3
HOH
CH3 CH3
O
C
CH3 H
HOH
EPD3(EKcca) EPD4(EKccm) EPD5(EKccg)
H2C CH3C
H H
CH3
OH
OH
CH3 CH3
O O
C
H H
CH3
H
H
EPD12(EEcca) EPD13(EEcca)
Medio acuoso ácido
CH3 CH3
O
C
CH3 H
HO+H
H
CH3 CH3
O
C
H CH3
HO+H
H
CH3 CH3
O
C
H H
CH3O+
H
H
EPD6(EKccg) EPD7(EKccm) EPD8(EKcca)
CH3 CH3C
H H
CH3O+
HO
H
CH3 CH3C
H CH3
HO+H
OH
CH3 CH3C
CH3 H
HO+H
OH
EPD9(EEcca) EPD10(EEccm) EPD11(EEccg)
Fig.9.3. Moléculas EPD estudiadas con distinta geometria y a distinto pH. Nomenclatura: mayúsculas, enol=E, ceto=K; minúsculas (ver nomenclatura diedro Fig 4.), cc= diedro ω2 y ω3 son 0º ambos, g si ω4=60º, a si ω4=180º y m si ω4=-60º.
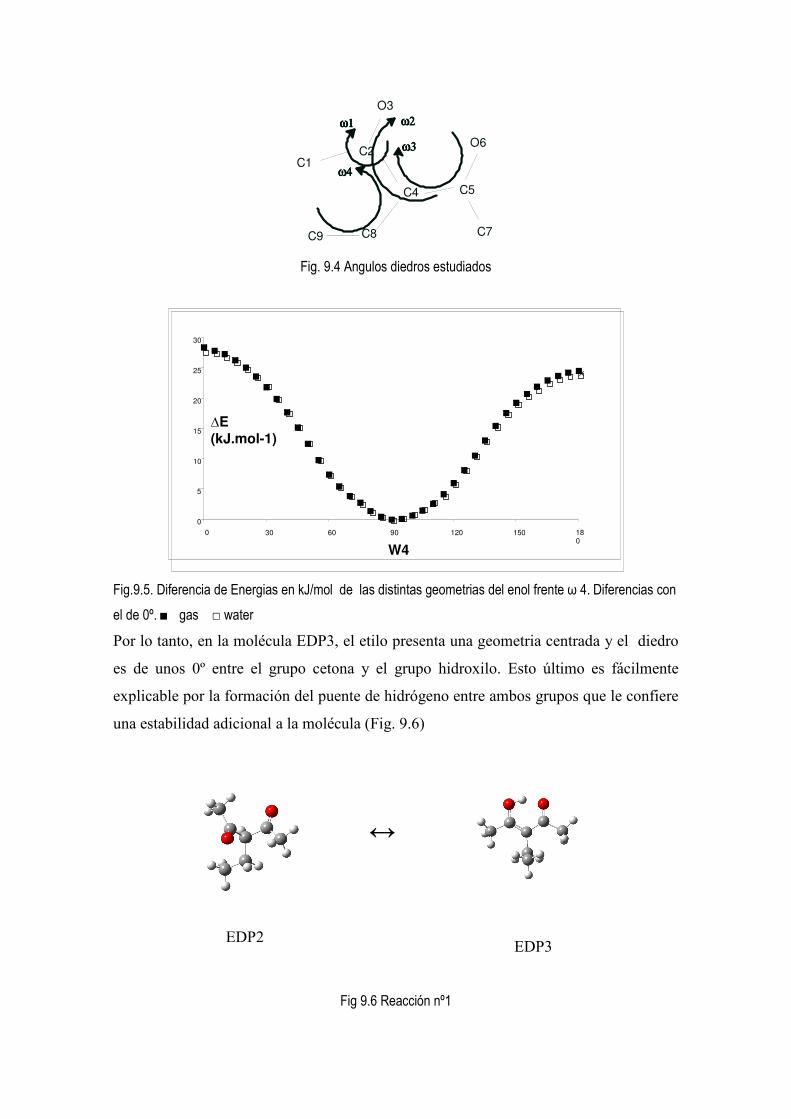
C2
O3
C1
C4 C5
C8C9
O6
C7
ω1ω1ω1ω1 ω2ω2ω2ω2
ω3ω3ω3ω3
ω4ω4ω4ω4
Fig. 9.4 Angulos diedros estudiados
Fig.9.5. Diferencia de Energias en kJ/mol de las distintas geometrias del enol frente ω 4. Diferencias con el de 0º. gas water
Por lo tanto, en la molécula EDP3, el etilo presenta una geometria centrada y el diedro
es de unos 0º entre el grupo cetona y el grupo hidroxilo. Esto último es fácilmente
explicable por la formación del puente de hidrógeno entre ambos grupos que le confiere
una estabilidad adicional a la molécula (Fig. 9.6)
EDP2
↔
EDP3
Fig 9.6 Reacción nº1
0
5
10
15
20
25
30
0 30 60 90 120 150 180
W4
∆E (kJ.mol-1)

La segunda reacción sería la acidificación de la reacción anterior. La protonación del
enol podría dar lugar a dos nuevas moléculas el dienol o la pérdida del doble enlace.
EDP6EDP7EDP8
EDP9EDP10EDP11
EDP3
Reacción nº2
Los resultados de energía nos muestran que del primer grupo de la reacción 2 la
geometria más favorable es la del EPD6 y del segundo grupo de los dienoles la
molécula EPD9 (Tabla 9.2). Entre ambas estructuras prevalece la menor energía en
EPD9 por lo que ante acidificación se formará más favorablemente el dienol también
esto puede explicarse por la estabilidad que le confiere el puente de hidrógeno y la
resonancia que presentan los dobles enlaces. Por lo que la reacción podriamos
considerarla como la de la Fig. 9.7
EDP3
↔
EDP9
Fig. 9.7. Reacción nº2
Al igual que para EPD3 se ha hecho un Scan al diedro ω 4 de EPD9 de 360 º para ver
si encontrabamos algún confórmero pero obtuvimos dos mínimos a 90º y 270º que
representan las posiciones simétricas del etilo. (Fig.9.8)
Los datos obtenidos por ambos métodos tanto B3LYP como MP2 de energía total
pueden observarse en la tabla 9.2 mostrando las geometrías más estables de cada grupo
de moléculas de la Fig. 9.3.
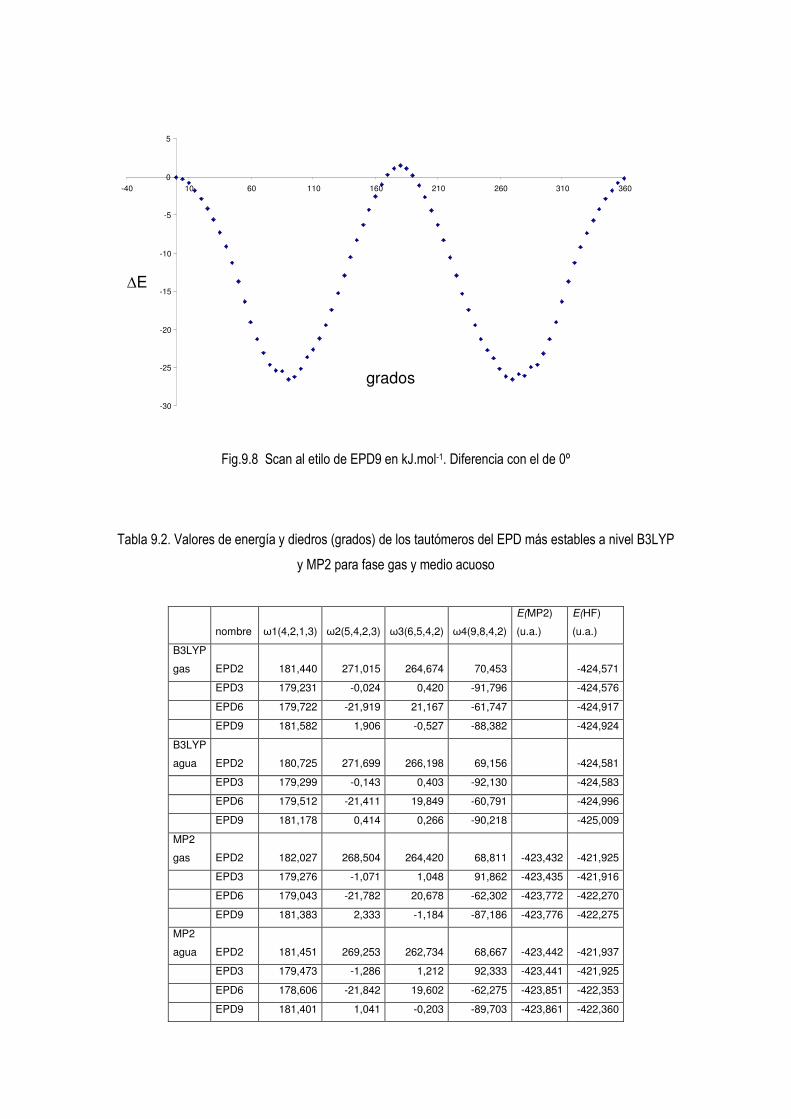
Fig.9.8 Scan al etilo de EPD9 en kJ.mol-1. Diferencia con el de 0º
Tabla 9.2. Valores de energía y diedros (grados) de los tautómeros del EPD más estables a nivel B3LYP y MP2 para fase gas y medio acuoso
nombre ω1(4,2,1,3) ω2(5,4,2,3) ω3(6,5,4,2) ω4(9,8,4,2)
E(MP2)
(u.a.)
E(HF)
(u.a.)
B3LYP
gas EPD2 181,440 271,015 264,674 70,453 -424,571
EPD3 179,231 -0,024 0,420 -91,796 -424,576
EPD6 179,722 -21,919 21,167 -61,747 -424,917
EPD9 181,582 1,906 -0,527 -88,382 -424,924
B3LYP
agua EPD2 180,725 271,699 266,198 69,156 -424,581
EPD3 179,299 -0,143 0,403 -92,130 -424,583
EPD6 179,512 -21,411 19,849 -60,791 -424,996
EPD9 181,178 0,414 0,266 -90,218 -425,009
MP2
gas EPD2 182,027 268,504 264,420 68,811 -423,432 -421,925
EPD3 179,276 -1,071 1,048 91,862 -423,435 -421,916
EPD6 179,043 -21,782 20,678 -62,302 -423,772 -422,270
EPD9 181,383 2,333 -1,184 -87,186 -423,776 -422,275
MP2
agua EPD2 181,451 269,253 262,734 68,667 -423,442 -421,937
EPD3 179,473 -1,286 1,212 92,333 -423,441 -421,925
EPD6 178,606 -21,842 19,602 -62,275 -423,851 -422,353
EPD9 181,401 1,041 -0,203 -89,703 -423,861 -422,360
-30
-25
-20
-15
-10
-5
0
5
-40 10 60 110 160 210 260 310 360
grados
∆E

Tabla 9.3. Valores de energía de solvatación y deformación (kJ.mol-1) de los tautómeros del EPD más estables a nivel B3LYP y MP2 para fase gas y medio acuoso
Energía de
solvatatacion
y
deformación
(kJ/mol)
Reacción
nº 1
Reacción
nº 2
Comparación
MP2-B3LYP
Comparación
AGUA-GAS
Comparación
EDP3-EDP2
Comparativa
EDP9-EDP3
B3LYP gas EPD2
EPD3 -13,630
EPD6
EPD9 -877,438
B3LYP agua EPD2 -27,732
EPD3 -19,688 -5,586
EPD6 -207,847
EPD9 -223,140 -1080,889
MP2 gas EPD2 2993,803
EPD3 3003,279 -4,154
EPD6 3010,826
EPD9 3019,442 -861,275
MP2 agua EPD2 2997,368 -24,167
EPD3 3005,194 -17,774 2,240
EPD6 3012,820 -205,853
EPD9 3019,702 -222,880 -1066,381
Observando la tabla 9.3 podemos extraer las siguientes conclusiones; en primer lugar
observamos un aumento de 3000 kj/mol a favor de la metodología B3LYP con respecto
a MP2 en todas las moléculas. Las energías de solvatación son parecidas, teniendo una
diferencia de 2 kJ aproximadamente entre un método y otro. Siendo más bajas las
energías de solvatación adjudicadas por el método B3LYP, que implica una mayor
estabilización a la molécula en medio acuoso.
Observando la tabla 9.3 también podemos analizar hacia donde estan desplazadas las
reacciones tanto por un método como por otro. Para la reacción nº1 tanto un método
como otro proporciona mayor estabilidad a la forma enol frente a la cetona. Este
resultado es el esperado en fase gas ya que confirma los resultados experimentales de la
tabla 1 con respecto a la acetilcetona, en caso de B3LYP el enol se encuentra a unos

13,630 kJ/mol más bajo de energía y en el caso de MP2 a -4,154 kJ/mol. Sin embargo,
en fase agua los resultados de B3LYP no coinciden con los datos experimentales ya que
según la tabla 9.1 y para la acetilcetona predomina la cetona al estabilizar los grupos
cetónicos con moléculas de agua y este método vuelve a darnos predominancia del enol
en 5,586 kJ/mol más bajo de energía. Para obviar que fuera un problema particular de
nuestra molécula EPD se optimizaró la acetilcetona y los resultados tampoco
concordaron a nivel B3LYP con los resultados experimentales. De ahí que
emplearamos otro algoritmo mas exacto como el MP2 para llegar a resultados correctos.
Con esta metodología ya observamos la predominancia de la cetona con 2,240 kJ/mol
mas estable.
Con respecto a la reacción nº 2 ambos métodos muestran un claro desplazamiento del
equilibrio hacia la formación del dienol en unos 800 kJ/mol en fase gas y alrededor de
1000 kJ/mol en fase agua por ambos métodos. Esto nos esclarece que en la reacción nº 1
hay un equilibrio de los dos compuestos puesto que la diferencia energética es muy
pequeña pero en la reacción nº2 esta totalmente desplazada a la formación del enol.
Una vez encontrado el método idóneo de optimización calculamos la K equilibrio para
ver dicha concordancia con los resultados experimentales. Calculada como keq=e-∆G/RT
también calculamos las fracciones molares del enol (Tabla 9.4) reacción nº1 cuyos
resultados concuerdan aproximadamente con los experimentales de la tabla1.
Seguidamente efectuamos un analisis DFT de la población y energía atómicas para la
reacción nº1 pudiendose observarse en la Fig. 9.9 los resultados obtenidos con MP2
para fase gas y disolución acuosa. Para los cálculos se estimo la población y energía
atómica de la cetona más estable y del enol más estable.
Tabla 9.4. Datos de fracciones molares y constante de equilibrio obtenidos para la reacción 1
EDP3
Diceto
(% Fracción molar)
cetoenol
(% Fraccción molar) keq=kenol/kcetona
Fase agua 71 29 0,41
Fase gas 16 84 5,34

AGUA
CH3
CH3CH3
OH
O
-0,6020,014
-0,010
0,0210,021
0,017
0,133
0,034
0,37
CH3
CH3CH3
OH
O
-3,642
17,071
-18,307
104,642
-183,785
-5,092
-628,834
751,930
-32,672
GAS
CH3
CH3CH3
OH
O
-0,6310,036
-0,009
0,0300,022
0,015
0,123
0,047
0,365
CH3
CH3CH3
OH
O
-21,003
11,966
-15,205-35,469
78,337
-179,884
-11,983
-619,888
790,853
Fig.9.9. Diagrama de flujo de población (izquierda) y diagrama de flujo de energía (derecha). Estas gráficas representan la variacion de población (izquierda) y de energía en kJ/mol (derecha) de EDP3-EDP2. En la variación de población las flechas representan un posible camino para el desplazamiento de la población al pasar de EDP2 a EDP3. En la variación de energía las flechas hacia arriba representan un aumento de la energía y las flechas hacia abajo una disminución de la misma al pasar de EDP2 a EDP3
Analizando la variación de población MP2, (ver Fig 9.4 y considerar O6 como el
oxígeno del hidroxilo), podemos considerar en primer lugar que los movimientos de
densidad electrónica son parecidos tanto en fase gas como en disolución acuosa. La
formación del enol desde la cetona implica un aumento de población del C5>C2>C4 y
de toda la estructura en general. Todo este aumento de población proviene de que H
unido al O, pierde mas de ½ electrón y muy ligeramente del grupo metilo próximo al
grupo hidroxilo. Es decir existe un desplazamiento de la carga hacia los oxígenos y el
etilo. La pérdida de medio electrón no permanece en el O6 al que se va unir sino que se
distribuye: en primer lugar al C5 (de ahí su gran aumento mas de ¼ de e-) y lo que resta
a los carbonos centrales. Todo el 0,64 que pierde el H al unirse al O6 no va via
esqueleto carbonado. Mirando la Fig. 9.9 vemos que tanto el C1 como el C2 como el C4
ganan densidad, por tanto el O3 del grupo cetónico unido al C2 tiene que ganar esos

0,02 u.a. de densidad electrónica mediante un enlace de H con el grupo hidroxilo.
Aspectos de densidad de población nos indican pues, la presencia de enlace por puente
de hidrógeno tal y como nos indican los criterios de Koch y Popelier [11] y aspectos
geometricos nos confirman esta presencia como un enlace de H de fuerza moderada
[12], con distancia OH---O de 1,57 A y ángulo O-H – O de 151,2º.
Respecto a la energía, las flechas hacia abajo indican las parte que se estabilizan y arriba
desestabilizan. Logicamente el más estabilizado resulta ser C5 en el que está situado el
grupo hidroxilo porque gana la población que le quitaba un enlace π con el O. Un dato
curioso que llama la atención resulta ser el O del grupo cetona aunque gana algo de
carga debida al puente se desestabiliza. Podriamos pensar en una explicación basada en
la distancia de enlace. La distancia de enlace en el enol es 1,26 Ǻ, la distancia de enlace
en la cetona es 1,21 Ǻ. Esto indica que al formar el enlace de H la longitud de enlace se
estira perdiendo por tanto el O algo de su poder de atracción por la carga del C. La
densidad de carga π del doble enlace se estira.
Con respecto a la reacción II de acidificación ya comentamos que la molécula más
estable pasaba a ser EPD9, el dienol. Los resultados de variación de población en fase
acuosa pueden observarse en la Fig. 9.10
CH3
CH3CH3
OH
OH
-0,041
-0,055
-0,066
-0,1170,192
-0,067
-0,093 CH3
CH3CH3
OH
OH
-140,67
-11,56
-5,98
100,78-366,50
31,74
143,33
-0,631
--0,019
760,51
-140,68
Fig.9.10 . Diagrama de flujo de población (izquierda) y diagrama de flujo de energía (derecha). Estas gráficas representan la variacion de población (izquierda) y de energía en kJ/mol (derecha) de EPD9-EPD4 . En la variación de población las flechas representan un posible camino para el desplazamiento de la población al pasar de EPD4 a EPD9. En la variación de energía las flechas hacia arriba representan un aumento de la energía y las flechas hacia abajo una disminución de la misma al pasar de EPD4 a EPD9. Datos en medio acuoso. Balances incompletos porque no se muestran datos del protón añadido para formar el dienol. Datos MP2.

Solo aumenta de población el C2 al perder el enlace π que le unia al Oxígeno. En global
toda la mlécula pierde población al enlazarse con el nuevo H y formar el dienol. Pero
este nuevo grupo enol provoca disminución de energía en los dos oxígenos, en el etilo y
en el C2 (-366,50 Kj/mol) al formarse el doble enlace ahí C2=C4.
CONCLUSIONES
Hemos realizado un análisis energético de la geometrica de la molécula de EPD
a la búsqueda de conformeros para dos posibles reacciones en medio acuoso neutro y
ácido. Para la primera reacción se ha encontrado dos conformeros del EPD una dicetona
y un dienol con una geometria especifica dependiendo del medio tratado y en un
equilibrio quimico con la reacción parcialmente desplazada a la formación del enol en
medio gas y la reacción parcialmente desplazada a la formación de la dicetona en medio
acuoso neutro. Se han usado dos metodologías distintas para el cálculo de la energía, en
una primer momento se ha usado B3LYP y como las K equilibrio no coincidian con los
datos experimentales se ha utilizado metodología MP2. Para la segunda reacción el
equilibrio se encuentra desplazado a la formación del dienol tanto en medio gas como
en medio acuoso ácido. Por último se ha realizado un análisis de población para poder
explicar las reaccciónes estudiadas.

5.10 Tautomeria imino-enamina en metabolitos de aminas
heterociclicas con marcado potencial mutagenico
En los últimos años las aminas heterocíclicas (HCA) han sido objeto de atención
debido a estar consideradas como sustancias con carácter mutágenico capaces de
provocar distintos tipos de cáncer [1]. Entre las HCA consideradas como mutagénicas
destacan, por su número, las que presentan un ciclo imidazol fusionado con otros anillos
y con un grupo amino en C2. [2](Fig10.1).
N
N
CH3
1
2
34
5
1-methyl imidazole Fig. 10.1. Numeración IUPAC para el 1-metil imidazol
Dicho grupo amino va a ser el responsable de la formación de aductos con el DNA tras
un proceso que incluye hidroxilación, esterificación y formación del nitrenium ion [3]
(Fig. 10.2).
Ar NH2 Ar NH
OH
Ar NH
OR
Ar N+
H
P450CYP1A1/CYP1A2 Sulfotrasferase
N-Acetyltrasferase-SO3
-COCH3
2'-deoxyguanosine
DNA adducts
Aromatic amine N-Hydroxylamino-Ar Ar-Ester Ar-nitrenium ion
Fig. 10.2 Esquema resumiendo los procesos seguido por las HCAs para formar aductos del ADN. Entre los grupos más comunes aril, Ar, de HCAs estan: policlorobifenilos, carbolinas, fluorenes, azaarenes de imidazo y otros anillos fusionados.
Además dichas reacciones compiten con la destoxificación de la HCAs mediante
hidroxilaciones en otras posiciones [4] o mediante glucuronidation [5]. Tanto las
especies Ar-NH2 como Ar-NHOH son susceptibles de experimentar procesos de
tautomerización que dan lugar a las correspondientes formas imino. Estas siguen otras
rutas metabólicas que no originan el aducto con el ADN. Es decir, la tautomería de las

aminas e hidroxilaminas aromáticas puede ser un factor decisivo tanto para la formación
de aductos, así como en la destoxificación.
Así como la tautomeria ceto-enolica ha sido muy estudiada, la tautomeria amina-
enamina lo ha sido menos. En el equilibrio amino enamino entre acetaldimide (CH3-
CH=NH) y vinylamine (CH2=CH-NH2) nos encontramos con una reaccion reversible
en la cual la E activación es baja. En dicho equilibrio se favore la formación de la imina
a veces sin importar si el sustituyente central (X) es aceptor o donante de densidad
electrónica (equilibrio CH2=CX-NH2 ↔ CH3-CX=NH) [6], pero otras veces efecto de
los sustituyentes pueden variar el producto más estable [7]. Habitualmente se ha
razonado que un enlace π C=N es mas fuerte que uno C=C, y por ello la formación de la
imina esta termodinámicamente favorecida, al igual que ocurre con la mayor estabilidad
de una cetona frente a un enol. Sin embargo, esta tendencia se invierte en compuestos
anulares o cuando el grupo amino es un sustituyente de ciclos aromaticos. Así, por
ejemplo, se ha demostrado teórica y experimentalmente que el tautómero amino es más
estable que el imino en adenines [8] y 2-aminothiazole [9]. No obstante, los estudios
realizados sobre distintos tipos de ciclos no arrojan un patrón común. [10]. Así, otros
factores, como temperatura [11], medio [12,13] o los enlaces de H que se puedan formar
[14,15] pueden determinar cuál es el tautomero más estable.
Aunque la tautomerización en ciclos con N intraanular ha sido estudiada desde hace
varias décadas [16,17], ha estado particularizada en las bases nitrogenadas del ADN
[18-21]. Dado el interes que presentan las aminas aromáticas (HCAs) con demostrado
potencial mutagenico [22], nos parece primordial ahondar en las reacciones que dichas
sustancias sufren antes de formar los aductos con el ADN, así como cuál de los
productos de dichas reacciones podemos considerar como tautómero más estable y por
qué. En particular, consideramos 3 moleculas con alta potencia mutagénica y que
poseen ese anillo imidazol en su estructura. Nos referimos a 2-amino-1-metil-6-
fenilimidazo[4,5-b]piridina (PhIP), 3,4,8-trimetil-3H-imidazo[4,5-f]quinoxalin-2-amino
(4,8-diMeIQ) y 2-Amino-3-metilimidazo(4,5-f)quinolina (IQ) (Fig. 10.3).

Ia (PhIP) IIa (4,8-diMeIQ) IIIa (IQ)
Fig. 10.3. Principales HCAs objeto de nuestro estudio
Además estudiaremos compuestos modelo para analizar el efecto de diversos factores
como puede ser la hidroxilación sobre el equilibrio tautomérico.
Detalles Computacionales
Partiendo del anillo imidazol y teniendo en cuenta trabajos anteriores
caracterizados por la posible no-planaridad del grupo amino situado en C2 [23, 24],
todas los tautómeros estudiados han sido optimizados a nivel MP2/6-311++G(d,p)
utilizando el programa Gaussian 09 [25]. Cuando así se ha requerido también se han
hecho optimizaciones de las mismas moléculas a nivel HF, B3LYP y CCSD utilizando
el mismo conjunto base. En todos los casos se ha garantizado el carácter de mínimo de
la geometría analizada mediante el cálculo de frecuencias. En los casos necesarios se
han optimizado los diferentes confórmeros de cada tautómero, aunque sólo se muestran
las diferencias de energía entre los confórmeros más estables de cada tautómero. Para el
cálculo en medio acuoso, las moléculas han sido optimizadas con el modelo PCM [26]
pero no se ha utilizado el cálculo de frecuencias con este método dada su escasa
fiabilidad [27].
Hemos realizado el análisis topológico de la densidad electrónica QTAIM con el
programa AIMPAC [28] que mediante la integración de la función densidad nos
proporciona la correspondiente población electrónica atómica N(Ω). Dicha propiedad
han sido calculada con valores de L(Ω) menores que 1 x 10-3 au, lo que habitualmente
representa una buena fiabilidad en N(Ω) [28]. La partición de energía proporcionada por
método "Interacting Quantum Atoms" (IQA) [29] fue empleado para analizar el origen

de las especies tautoméricas en compuestos modelo empleando el programa
PROMOLDEN [30]. De acuerdo con esta partición, la energía electrónica molecular, E,
se desglosa en términos monoatómicos (energías atómicas netas), Enet(Ω), y biatómicos,
energías de interacción, Vint(Ω,Ω’) (ec. 10.1).
( ) ( )∑ ∑Ω Ω′<Ω
Ω′Ω+Ω= ,intVEE net (10.1)
Enet(Ω) contiene todos los terminus de energía que afectan exclusivamente a la cuenca
atómica. Refiriéndonos a la densidad electrónica de una cierta cuenca atómica, Ω, sería
su energía cinética, T(Ω), su atracción por el núcleo ,ω, Vn,e(ω,Ω), y la correspondiente
repulsión interelectrónica, Ve,e(Ω,Ω) (ec. 10.2).
( ) ( ) ( ) ( )ΩΩ+Ω+Ω=Ω ,, ,, eeennet VVTE ω (10.2)
Vint(Ω,Ω’) contiene todas las interacciones entre y su densidad electrónica da amabas
cuencas atómicas (ec. 10.3).
( ) ( ) ( ) ( ) ( )Ω′Ω+Ω′+Ω′+′=Ω′Ω ,,,,, ,,,,int eeenennn VVVVV ωωωω (10.3)
Resultados y Discusión
a) Tautomerismo en HCAs.
Dado que las moléculas de PhIP, 4,8-diMeIQ o IQ necesitan de una activación
metabólica [31-34] en el citocromo P-450 a través del isoenzima CYP1A1/CYP1A2
para formar el N-hidroxi metabolito, se optimizaron los distintos tautómeros de la amina
y del producto resultante de su N-hidroxilación. La diferencia de energía entre los
confórmeros más estables de cada tautómero (obtenida como resta imino – amina), ∆tE,
confirma la preferencia por la forma amina aromática (Tabla 10.1). Esta tendencia se
rompe en los compuestos N-hidroxilados, donde se observan valores de ∆tE negativos
(indicativos de una preferencia por la forma imino) para algunos compuestos y niveles
de cálculo. Se observa que los valores de ∆tE obtenidos con MP2 favorecen más la
forma amina que los B3LYP. Este efecto es prácticamente constante en magnitud (20±6
kJ mol-1). Aunque el efecto de incluir la disolución acuosa a través del modelo PCM no
altera los valores de ∆tE de manera tan constante, si se observa que siempre lo hace
favoreciendo la forma amina respecto al valor obtenido en fase gas. Esto conduce a que
las formas amino resulten preferidas frente a las imino en los cálculos MP2 para
disolución acuosa. Debe notarse que la N-hidroxilación para la formación de aductos
con el ADN ocurriría sobre el tautómero amino de los compuestos Ia, IIa y IIIa. Por lo

que los valores negativos de ∆tE, que implicaría una mayor estabilidad del tautomero
imino, podrían indicar que su potencial mutagénico sería reducido.
Tabla 10.1. Diferencia energeticas, ∆tE (en kJ/mol) del tautomero imino menos el tautomero amino. * Dado la cantidad de memoria precisada en el cálculo la contribución térmica para fase gas, ha sido obtenida de molécula precursora: a=molécula VIIb -*Dadas las necesidades computacionales precisadas la no importancia de la información generada, no se ha optimizado IXb en fase gas con MP2.
B3LYP/6-311++G(d,p) MP2/6-311++G(d,p)
HCAs GAS
∆E(kJ/mol)
WATER
∆E(kJ/mol)
GAS
∆E(kJ/mol)
WATER
∆E(kJ/mol)
Ia 3,71 26,48 17,87 36,98
IIa 33,12 35,32 49,18 51,07
IIIa 14,93 30,29 32,64 47,03
Ib -28,97 -11,00 -0,15 16,73
IIb 3,93 9,62 26,50 34,29
IIIb -14,91 4,04 9,85a 29,12
IVb -42,41 -36,19 -27,48 -26,37
Vb 14,93 20,13 39,81 45,61
VIb -24,69 -4,90 -2,70 16,34
VIIb -9,77 -1,47 11,91 20,57
VIIIb -8,05 0,64 12,07 22,09
IXb -21,00 -4,87 * 19,87
Para analizar el origen del efecto del nivel de cálculo sobre los valores de ∆tE se ha
considerado una serie de compuestos N-hidroxilados más simples (IVb-IXb) (Fig. 10.4),
incluyendo un cluster con solvatación explicita por 2 moléculas de agua (IXb). Todos
ellos incluyen un ciclo imidazol y muestran tendencias similares a las descritas para los
compuestos I-III, tanto al variar el nivel de cálculo como al incluir el disolvente. Así
pues el efecto del cambio de estabilidad cometido por B3LYP es independiente del
tamaño de la molécula incluso del algoritmo de cálculo del PCM puesto que el cluster
formado con la molécula I con dos moléculas de agua arroja los mismos resultados.
Esto es una preferencia, incluso en el cluster de B3LYP por el tautómero imino (-4,87
kJ/mol), en clara diferencia con MP2 (19,87 kJ/mol mas estable el tautomero amino).

IVb Vb VIb
VIIb VIIIb IXb
Fig. 10.4. Compuestos modelo utilizados en la comparación con HCAs hidroxiladas.
Podemos analizar cuando exactamente el B3LYP cambia de opción en cuanto al
tautomero favorecido en la N-hidroxilación observando el comportamiento de las
moléculas modelo Vb-VIIIb. La Vb no cambia de preferencia tautomerica, sigue
favoreciendo al tautomero amino tanto para MP2 como B3LYP. Pero al añadir un
segundo ciclo fusionado ya es cuando las preferencias cambian. Además depende del
ciclo fusionado:
a) Si dicho ciclo es tipo piridínico (VIb), B3LYP, tanto medio acuoso como gas, y MP2
en gas, favorecen la estabilidad del tautómero imino.
b) Si el ciclo es bencénico B3LYP sigue manteniendo la preferencia por tautomero
imino en medio acuoso y gas.
c) Si aportamos un grupo activante al ciclo bencénico, ya solo el B3LYP mantiene dicha
preferencia en medio acuoso.

Podemos entonces concluir que B3LYP con anillos π desactivantes contrarrestan el
efecto de la aromaticidad que favorecía la estabilidad del tautomero amino. A medida
que activamos el ciclo imidazol vuelve a predominar el efecto de la aromaticidad.
El análisis de población y energia molecular hecho al respecto para el compuesto I entre
el tautómero imino menos el amino, nos muestra exactamente lo que podiamos intuir
del B3LYP. Los atomos que forman el esqueleto anillo piridinico pierden 43 u.a. de
población en global y unos 100 kJ/mol en estabilización para aumentar la población y la
estabilidad del grupo hidroxi-imino.(Fig. 10.5)
-2.6-18.78
48.9-33.14
-0.2-0.49
0.4-1.26
0.4-0.76
0.4-1.37
0.3-0.28
-0.30.55 -2.7
3.75
1.614.26
32.4-43.70
-23.039.28
-41.883.66
-60.6136.57
-15.922.85
0.20.05
0.4-0.34
0.4-0.32
0.3-0.34
0.3-0.43
2.6-3.98
-2.53.84
10.8-376.25
11.7-9.68
2.4-1.89
-48.583.80
-5.511.90
-15.260.64
88.9-1.69
22.9-342.66
Fig. 10.5. Tautomero N-hydroxi-PhIP forma imino menos forma amino. Variaciones de población electrónica atómica (en au multiplicado por 103) y variaciones de energia atómica QTAIM (en cursiva y kJ/mol).
b) Evolución estructural de la preferencia tautomérica.
Volviendo a la tabla 10.1 observamos que el compuesto IVb tiene un valor
negativo de ∆tE en ambas fases y niveles de cálculo. Se trata del análogo no aromático
de Vb (cuyos valores de ∆tE son, en cambio, siempre claramente positivos) por
dihidrogenación del enlace C=C. Por lo cual podemos inducir que la aromaticidad juega
un papel importante en la estabilidad del tautomero amino. Por ello, decidimos
optimizar una nueva serie de compuestos (X-XIV) (Tabla 10.2).

Tabla 10.2. Diferencias de E entre la forma imino menos la forma amino. En kJ/mol. Hemos usado PCM para la optimización en medio acuoso.
GAS MEDIO ACUOSO
Sin Etérmica Con Etérmica Sin Etérmica
HF/6-
311++G(d,p) B3LYP/6-
311++G(d,p
MP2/6-
311++G(d,p)
HF/6-
311++G(d,p)
B3LYP/6-
311++G(d,p
MP2/6-
311++G(d,p)
HF/6-
311++G(d,p) B3LYP/6-
311++G(d,p
MP2/6-
311++G(d,p)
1,83 9,79 17,29 1,98 9,25 16,45 -4,55
-2,44
12,15
2-Imidazolidinimine (Xa)
46,71 41,09 61,12 44,12 38,9 52,49 41,74 37,79 55,64
2-aminoimidazole (XIa)
-15,24 -7,38 -10,98 -15,73 -7,98 -11,95 -22,14 -13
-14,98
1-Cyclopenten-1-amine (XIIa)
-13,09 3,88 3,47 -12,15 3,95 3,98 -14,88 3,52
4,03
1,3-Cyclopentadien-1-amine
(XIIIa)
-19,37 -24,01 -15,72 -21,26 -24,80 -16,81 -24,23 -36,71
-18,24
N-Hydroxy-2-imidazolidinimine
(Xb)
32,30 21,39 43,51 27,73 18,23 40,73 25,27 21,34
46,31
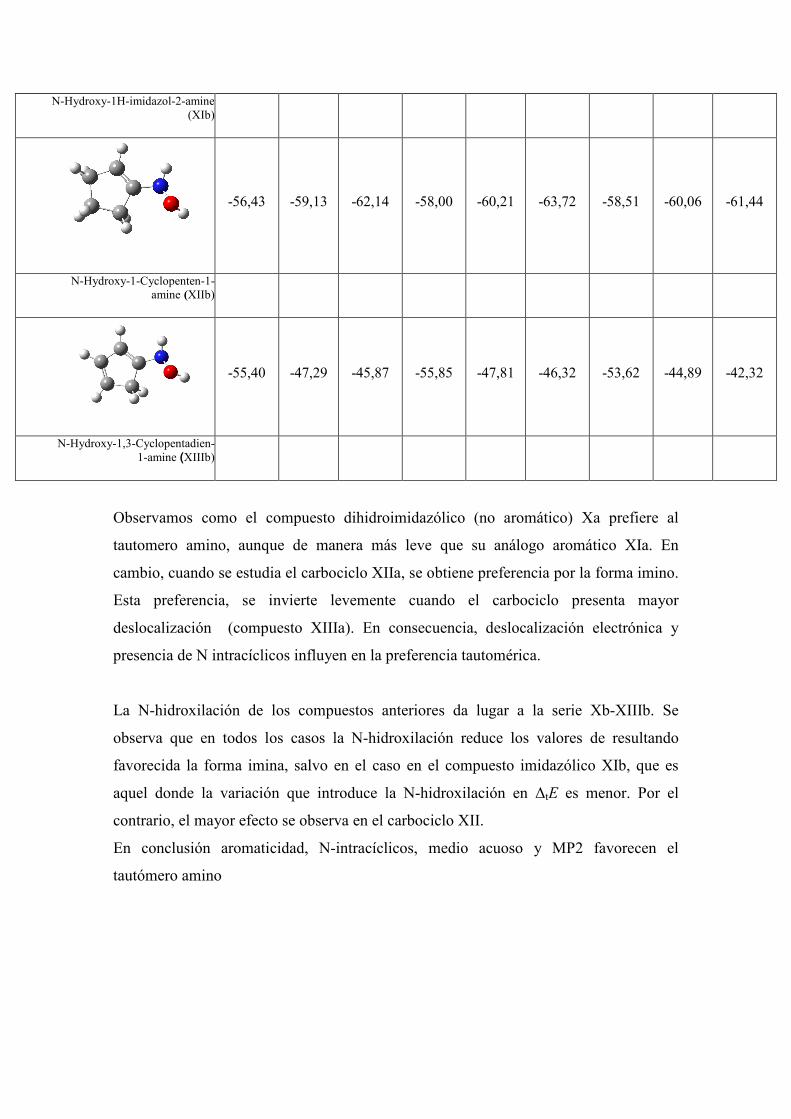
N-Hydroxy-1H-imidazol-2-amine (XIb)
-56,43 -59,13 -62,14 -58,00 -60,21 -63,72 -58,51 -60,06 -61,44
N-Hydroxy-1-Cyclopenten-1-amine (XIIb)
-55,40 -47,29
-45,87
-55,85 -47,81 -46,32 -53,62 -44,89 -42,32
N-Hydroxy-1,3-Cyclopentadien-1-amine (XIIIb)
Observamos como el compuesto dihidroimidazólico (no aromático) Xa prefiere al
tautomero amino, aunque de manera más leve que su análogo aromático XIa. En
cambio, cuando se estudia el carbociclo XIIa, se obtiene preferencia por la forma imino.
Esta preferencia, se invierte levemente cuando el carbociclo presenta mayor
deslocalización (compuesto XIIIa). En consecuencia, deslocalización electrónica y
presencia de N intracíclicos influyen en la preferencia tautomérica.
La N-hidroxilación de los compuestos anteriores da lugar a la serie Xb-XIIIb. Se
observa que en todos los casos la N-hidroxilación reduce los valores de resultando
favorecida la forma imina, salvo en el caso en el compuesto imidazólico XIb, que es
aquel donde la variación que introduce la N-hidroxilación en ∆tE es menor. Por el
contrario, el mayor efecto se observa en el carbociclo XII.
En conclusión aromaticidad, N-intracíclicos, medio acuoso y MP2 favorecen el
tautómero amino
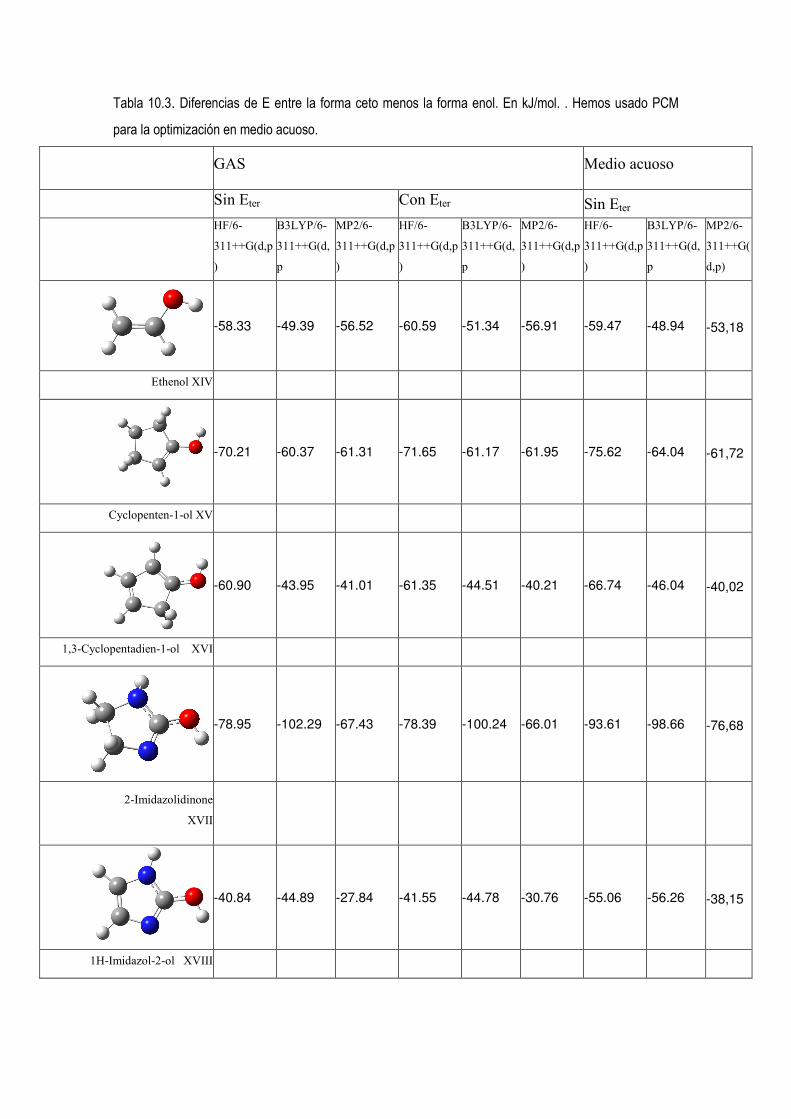
Tabla 10.3. Diferencias de E entre la forma ceto menos la forma enol. En kJ/mol. . Hemos usado PCM
para la optimización en medio acuoso.
GAS Medio acuoso
Sin Eter Con Eter Sin Eter
HF/6-
311++G(d,p
)
B3LYP/6-
311++G(d,
p
MP2/6-
311++G(d,p
)
HF/6-
311++G(d,p
)
B3LYP/6-
311++G(d,
p
MP2/6-
311++G(d,p
)
HF/6-
311++G(d,p
)
B3LYP/6-
311++G(d,
p
MP2/6-
311++G(
d,p)
-58.33 -49.39 -56.52 -60.59 -51.34 -56.91 -59.47 -48.94
-53,18
Ethenol XIV
-70.21 -60.37 -61.31 -71.65 -61.17 -61.95 -75.62 -64.04
-61,72
Cyclopenten-1-ol XV
-60.90 -43.95 -41.01 -61.35 -44.51 -40.21 -66.74 -46.04
-40,02
1,3-Cyclopentadien-1-ol XVI
-78.95 -102.29 -67.43 -78.39 -100.24 -66.01 -93.61 -98.66
-76,68
2-Imidazolidinone
XVII
-40.84 -44.89 -27.84 -41.55 -44.78 -30.76 -55.06 -56.26
-38,15
1H-Imidazol-2-ol XVIII
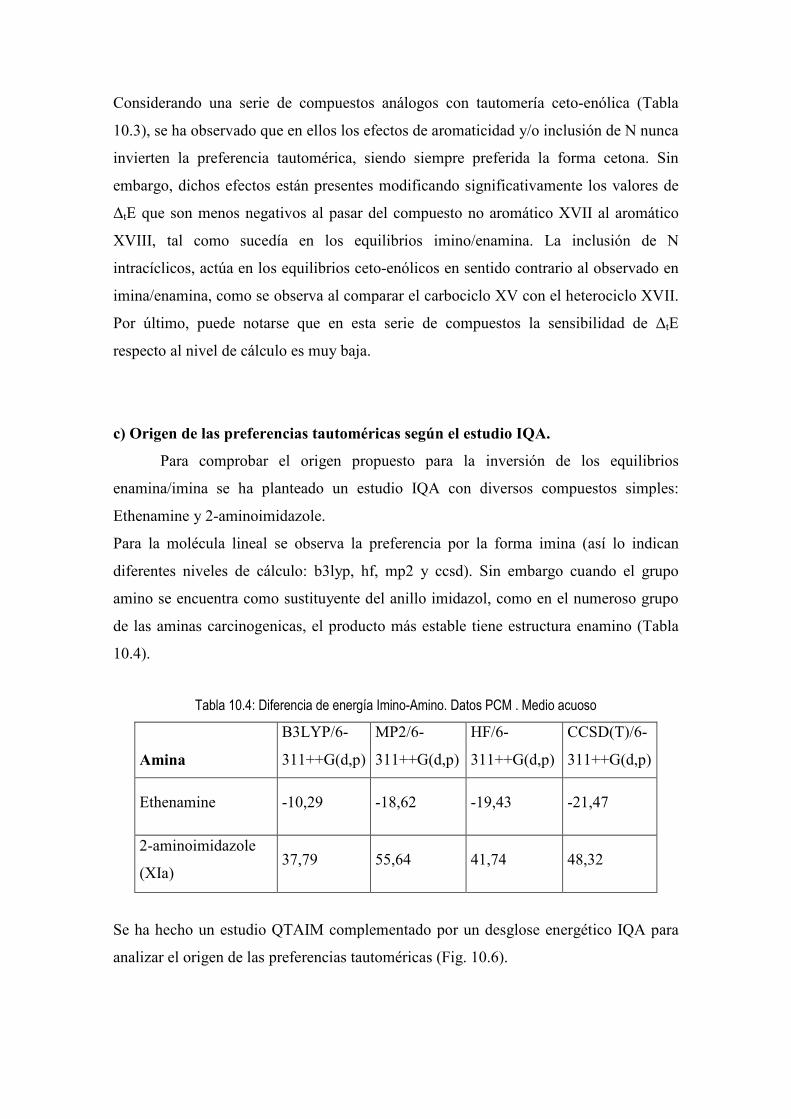
Considerando una serie de compuestos análogos con tautomería ceto-enólica (Tabla
10.3), se ha observado que en ellos los efectos de aromaticidad y/o inclusión de N nunca
invierten la preferencia tautomérica, siendo siempre preferida la forma cetona. Sin
embargo, dichos efectos están presentes modificando significativamente los valores de
∆tE que son menos negativos al pasar del compuesto no aromático XVII al aromático
XVIII, tal como sucedía en los equilibrios imino/enamina. La inclusión de N
intracíclicos, actúa en los equilibrios ceto-enólicos en sentido contrario al observado en
imina/enamina, como se observa al comparar el carbociclo XV con el heterociclo XVII.
Por último, puede notarse que en esta serie de compuestos la sensibilidad de ∆tE
respecto al nivel de cálculo es muy baja.
c) Origen de las preferencias tautoméricas según el estudio IQA.
Para comprobar el origen propuesto para la inversión de los equilibrios
enamina/imina se ha planteado un estudio IQA con diversos compuestos simples:
Ethenamine y 2-aminoimidazole.
Para la molécula lineal se observa la preferencia por la forma imina (así lo indican
diferentes niveles de cálculo: b3lyp, hf, mp2 y ccsd). Sin embargo cuando el grupo
amino se encuentra como sustituyente del anillo imidazol, como en el numeroso grupo
de las aminas carcinogenicas, el producto más estable tiene estructura enamino (Tabla
10.4).
Tabla 10.4: Diferencia de energía Imino-Amino. Datos PCM . Medio acuoso
Amina
B3LYP/6-
311++G(d,p)
MP2/6-
311++G(d,p)
HF/6-
311++G(d,p)
CCSD(T)/6-
311++G(d,p)
Ethenamine -10,29 -18,62 -19,43 -21,47
2-aminoimidazole
(XIa) 37,79 55,64 41,74 48,32
Se ha hecho un estudio QTAIM complementado por un desglose energético IQA para
analizar el origen de las preferencias tautoméricas (Fig. 10.6).

8
6
3
1
2
4
7
5
350.2-186.4
352,76.2
-789.0524.4
58.5241.5 48.2
-21.2
8.4-1.3
-12.81.6
-16.4-1.6
12
3
45
6
7 8
Fig. 10.6a Ethenamine (1E)-Ethanimine
2
9
8
5
4
1
3
7
6
11
10
72.2-28.6
111.4100.5
-45.9207.0
-44.6-0.7
6.4-131.8
-20.5143.6
0.833.2
-13.26.0
-45.9-57.6
20.634.1
-44.81.5
2
1
7
11
6
3
10
4
8
5
9
Fig. 10.6b 2-aminoimidazole (XIa) 2-imineimidazole
Fig 10.6 a y b. Diferencias de población electrónica atómica entre tautómeros imino-amino (en a.u .multiplicado por 103) y diferencia correspondiente de energías atómicas netas IQA, ∆Enet(Ω) (en cursiva y kJ/mol)
En la molecula lineal Fig 10.6a, observamos la gran pérdida de población del C unido al
grupo imino -0,789 au y la gran desestabilización que esto le supone (524.4 kJ mol-1).
Aunque el N de dicho grupo gana 58,5 a.u. en población electrónica, también se
inestabiliza en 241,5 kJ mol-1. Por tanto, otros deben de ser los factores de estabilización
del grupo imino.
En la tabla 10.5 del desglose IQA aportado para la energía de interacción, observamos
como la estabilidad relativa del doble enlace C1=N3 frente a C1-N3 (1402,9 kJ/mol)

compensa la perdida por el enlace C1-C2 frente al C1=C2 (501,3 kJ/mol). También en
la misma tabla podemos observar, la menor incidencia del proceso de formación y
rotura de los enlaces del H7 desde el N3 al C2. Por tanto podriamos concluir que la
formación del un enlace π C=N, reemplazando un C=C menos fuerte, es la causa de la
estabilidad del imino frente a la amina.
Tabla 10.5.- Diferencias de energías interacción IQA (en kJ mol-1) entre tautómero imino menos enamino en el compuesto lineal de la Fig 10.6a.
E. Interaction C1 C2 N3 H4 H5 H6 H7
C1
C2 501,3
N3 -1402,9 24,5
H4 4,6 -5,4 2,0
H5 3,2 6,7 9,4 1,1
H6 8,1 8,5 -6,3 2,0 -3,6
H7 -197,6 -680,6 1150,1 -4,6 -11,9 -3,2
H8 140,5 -7,2 44,3 -7,0 -2,5 1,4 -156,0
Sin embargo, en la molecula 2-aminoimidazol pasa lo contrario siendo más estable el
grupo amino que el imino. La fig. 10.6b muestra que los cambios de energía o de
población no son tan drásticos como en la molécula lineal, a pesar de que el N del grupo
imino gana más población que su correspondiente en la molécula lineal. Ese mismo N
vuelve a ser más estable, como sucedía en el compuesto lineal, en la amina. El C de
dicho grupo sigue siendo más estable en la amina pero no sufre una pérdida de
población tan elevada como en la correspondiente molécula lineal. También el H que se
transpone sufre unos cambios más moderados. Sin embargo, el N3 que lo soporta es
mucho más estable en la amina.
En este caso el proceso es cambiar un enlace C=N de dentro del anillo por el mismo
enlace que esta fuera de él. El enlace C2=N1 ha desaparecido en la molecula iminica lo
que supone una desestabilización de 99,4 kJ/mol de Vint (Tabla 10.6). Todas las
interacciones del interior del ciclo, excepto C4···C2 y C5···N3, son más estables en la

molecula amínica. Esto podría relacionarse con la aromaticidad del grupo imidazol
cuando la molecula tiene un grupo amino; y su pérdida cuando el grupo es imino.
Además, la formación del enlace imino no es tan estable como en la lineal.
Estamos hablando de -836,1 frente a -1402,9 kJ/mol. Dada la diferencia entre estos dos
últimos valores y pensando en que tipo de influencia representaba en el ciclo, se
analizaron los desgloses IQA del carbociclo 1-Cyclopenten-1-amine (XIIa) y su
correspondiente tautómero imínico.
Tabla 10.6. Diferencias de energías de interacción IQA (en kJ mol-1) (tautómero imino-tautómero enamino) en el compuesto imidazólico de la Fig. 10.6b.
E. Interaction N1 C2 N3 C4 C5 N6 H7 H8 H9 H10
N1
C2 99,4
N3 -7,4 270,0
C4 118,1 -156,9 101,9
C5 -18,9 113,1 -107,8 -17,5
N6 75,9 -836,1 122,1 12,6 -74,2
H7 5,1 14,6 0,2 -23,7 19,3 -21,3
H8 -17,9 23,7 -28,2 13,0 15,3 -14,2 4,5
H9 -8,5 11,9 -9,0 -3,5 14,2 -7,9 3,4 1,2
H10 -33,4 63,6 -987,7 128,8 45,9 849,3 -0,5 12,9 4,9
H11 37,2 -5,5 36,5 -24,8 2,5 34,7 -10,3 2,2 0,7 -103,0
Los datos de energía del 1-Cyclopenten-1-amine (XIIa) y su correspondiente tautómero
imínico se muestran en la tabla 10.7. Como hemos observado en la tabla 10.2, el
equilibrio tautomérico está desplazado hacia la formación del tautómero imino como en
la molécula lineal
Los datos de E interacción para el enlace C=N imino (-1444,8 kJ/mol), se aproximan a
los de la molécula lineal. Asi como también son similares los datos de destrucción del
doble enlace C2=C1 intracíclico en el tautomero imino (499,2 kJ/mol), transposición del
H14 al C2 (-673,2 kJ/mol) y su correspondiente pérdida en el grupo amino (1172,3
kJ/mol). Por todo ello concluimos que el ciclo, al no tener aromaticidad, no estabiliza la
forma amina y su comportamiento es parecido al de la molécula lineal.

Tabla 10.7 Diferencias de energías de interacción IQA (en kJ mol-1) (tautómero imino-tautómero enamino) en el carbociclo XIIa, mostrado en la tabla 10.2.
Interaction
energies
1 2 3 4 5 6 7 8 9 10 11 12 13 14
1
2 499,2
3 40,1 16,6
4 38,4 1,8 -13,6
5 44,9 -5,1 -8,7 -13,8
6 -1444,8 5,4 7,3 -3,8 15,9
7 17,0 13,0 -0,6 1,1 1,9 -17,6
8 -3,0 -0,2 0,6 1,5 0,9 -3,6 -0,3
9 -8,9 -0,9 -3,0 -0,6 -0,4 5,2 -1,7 0,2
10 -11,3 -2,3 0,7 1,1 1,3 4,2 -0,6 0,5 -0,6
11 -4,5 -0,5 1,4 3,0 1,6 -2,5 -0,3 -0,2 0,1 0,1
12 5,5 2,2 1,9 3,5 6,3 -20,8 -0,2 -0,7 -0,4 -0,2 -0,5
13 -1,5 2,9 -0,2 -0,1 2,4 -8,5 0,1 -0,1 -0,7 -0,5 -0,2 0,7
14 -205,3 -673,2 -35,5 -20,8 -25,3 1172,3 -3,6 4,8 4,7 5,9 4,9 8,5 4,4
15 139,8 -2,9 -5,6 -3,4 -14,2 39,0 3,6 1,9 -0,1 0,6 1,3 7,8 1,3 -157,4
La variación de los índices de deslocalización (DI) entre átomos enlazados de los
tautómeros, indica como evoluciona la fortaleza de ese enlace entre ambas formas.
Asimismo, si el DI considerado corresponde a átomos del ciclo que no están enlazados,
su variación es un estimador de cómo se modifica la deslocalización electrónica dentro
del anillo. Incluso se ha demostrado una correlación entre dichos índices y la de otros
indices de aromaticidad ( Harmonic Oscillator Model of Aromaticity (HOMA) que
describe la contribución de la geometría y la energía de la molécula en la aromaticidad)
[35,36]. Por tanto, podemos utilizar dicha variación como indicador de la variación de
efectos resonantes. Así, la Fig. 10.7 muestra la variación que experimentan los DIs en
los tautómeroa amino/imino de 2-aminoimidazole (XIa) y de 1-Cyclopenten-1-amine
(XIIa). En ambas moléculas la mayoria de los átomos que generan el ciclo presentan
mayores DIs en el tautómero amino (datos de ∆DI negativos) ya que éste conlleva la
formación de un doble enlace intracíclico.

Los datos positivos de DIs (C4,C5) se refieren a que el doble enlace es más fuerte en el
tautómero imino. Sin embargo, en 2-aminoimidazole (XIa) ∆tDI (C4,C5) es mayor a
consecuencia de la pérdida de aromaticidad al pasar del tautomero amino al imino.
Dado que ninguno de los tautómeros de 1-Cyclopenten-1-amine (XIIa) es aromático,
solo observamos un ∆tDI significativo para el par C2,X3 (-0,743 au) que representa al
doble enlace perdido. Analizando los datos entre átomos intraciclicos no enlazados y
basándonos en el tautómero amino, si que observamos una cierta deslocalización solo
entre los átomos en α al doble enlace (C4 y X1) en XIIa. En el caso del XIa esta
deslocalización alcanza hasta C5, demostrándonos la conjugación del sistema de
electrones π
En cuanto al enlace exterior al anillo, lógicamente ambas presentan más deslocalización
en el tautomero imino en el enlace iminico (∆tDI positivos).
C4X3
C2
X1C5
N6
H*
H
-377-743
-122-30
858
-318
-56-29
408548
a
b
c
-14-27a= -57
-17b= -44-7c=
Fig 10.7. Diferencia de Indices de deslocalización (DI) de los principales átomos en el tautomero imino menos el tautómero amino, de los compuestos 2-aminoimidazole y 1-Cyclopenten-1-amine (en cursiva). Resultados en au multiplicados por 103 . X=C en 1-Cyclopenten-1-amine o X=N en 2-aminoimidazole. H* = Hidrogeno que se transpondra al enlazarse al átomo X3.

Como ejemplo de cambios en población y energia para las HCAs estudiadas, hemos
considerado datos de población electrónica y energias atómicas QTAIM del estudiado
PhIP (Fig 10.8). Dado el coste computacional de un cálculo IQA para estos sistemas se
obtuvieron unicamente las energías QTAIM de cada átomo.
-1.10.26
-0.71.17
-0.50.62
-0.70.19
-0.5-1.14
-5.85.61
-0.613.71
-0.81.35 24.1
-31.77 -45.379.39
-29.0130.34
-0.11.07
-0.80.76
-0.80.64
-0.80.75
-1.01.18
-4.54.78
-7.28.37
26.6-401.87
61.0172.33
-29.934.12
-6.19.06
19.7-16.84
10.9-9.71
-20.022.25
-35.453.81
76.2-94.19
-12.37.47
-13.034.61
Fig 10.8. Tautomero PhIP forma imino menos el tautomero PhIP forma amino. Variaciones de población electrónica atómica (en au multiplicado por 103) y variaciones de energia atómica QTAIM (en cursiva y kJ/mol).
La Fig. 10.8 nos muestra el tautómero amino del PhIP, el H del grupo amino en cis con
el grupo metilo pasará a enlazar con el N que no soporta al metilo del grupo imidazol en
el tautómero imino. En la Fig. 10.8 ganan población en el tautómero amino todos los
ciclos a costa de perderla el N del propio grupo amino y el N anteriormente
mencionado. Esto supone a nivel energético que en conjunto los ciclos son más estables.
También es observable como el N del ciclo imidazol que no soporta el metilo, tiene
26,6 au más de población en el tautomero imino gracias a estar enlazado con el H
procedente del grupo NH2
Llama la atención que el N del grupo amino pese a tener menor población que en el
tautómero imino, se estabiliza en 172.33 kJ/mol. Podemos concluir que en el tautomero
amino la distribución de población genera un compuesto con mayor densidad
electrónica en los ciclos, fortaleciendo el grupo amino.
.

d) Equilibrio tautomérico en la glucurodination de aminas arómaticas.
La glucuronidation juega un papel muy importante en la detoxificación de las
HCAs. Va a ser el mecanismo natural que tiene el organismo para deshacerse de toxinas
peligrosas para los seres vivos. Durante el proceso el acido glucuronido se conjuga con
las aminas aromaticas formando compuestos mas solubles que son eliminados en la
orina. Esto puede ocurrir via hepática o extrahepatica [37].
De las 3 HCAs que estamos estudiando hemos elegido al PhIP como molécula base para
formar sus posibles glucuronidos, por ser de las 3 la HCAs más abundante en la ingesta
diaria [38]. Así pues el N-hydroxy-PhIP-N2-glucuronide es el glucuronido más
numeroso de los encontrados en la orina después de haber administrado PhIP [39], o al
haber consumido carne muy cocinada [40] .
Pero no es el único. En la Fig. 10.9 podemos ver los dos procesos de glucuronidación
(glucuronidation 1 y 2), en los cuales va a estar implicado el PhIP. La glucuronidación 1
va a dar lugar a dos posibles tautómeros: PhIP-N2-glucuronide y PhIP-N3-glucuronide,
dependiendo si el ácido glucuronico es enlazado en posición N2 o N3 del PhIP. Y la
glucuronidación 2 ocurrirá después de que el PhIP se bioactive mediante el proceso de
hidroxilación, que dará también lugar a dos posibles tautómeros : N2-OH-PhIP-N2
glucuronide y N2-OH-PhIP-N3-glucuronide. [5]
Para nuestro estudio hemos optimizado los 4 posibles tautómeros y los resultados en
cuanto a energia pueden analizarse en la tabla 10.8. Sin importar el medio siempre
resulta más estable el tautómero amino.
Tabla 10.8. Diferencia de energias entre el tautómero imino menos el amino en kJ/mol. Las E en gas llevan añadida la correción térmica pero sin ella los resultados siguen la misma tendencia. Resultados dados en B3LYP unicamente dado el coste computacional que representa MP2 y que los resultados concuerdan con los obtenidos experimentalmente [39].
∆E(kJ/mol)
B3LYP/6-311++G(d,p)
GAS WATER
PhIP-N2-glucuronide - -
PhIP-N3-glucuronide 13.95 32.01
N2-OH-PhIP-N2-glucuronide - -
N2-OH-PhIP-N3-glucuronide 2.46 12.28

Ar NH2
N-Oxidation
Glucuronidation-1
Ar NHOH
Glucuronidation-2
DNA adducts N
NH
N
N
OH O
OH
COOH
OH
OH
N
NH
N
N
OH
O
OH COOH
OHOH
N
NH
N
N
H O
OH
COOH
OH
OH
N
NH
N
N
H
O
OH COOH
OHOH
PhIP- N2-glucuronide
PhIP- N3-glucuronide
N2-OH-PhIP- N3-glucuronide
N2-OH-PhIP- N2-glucuronide
Fig.10.9 . Procesos de glucuronidación.

Efectuamos ahora el análisis de población de los glucuronidos implicados. Dada la
magnitud de las moleculas, efectuaremos un análisis sobre los grupos de átomos o
átomo según la terminología de la fig 10.10
N
N1
C2
N3
N4
G6
G4
O
OH
COOH
OH
OH
G3
G2 G1 G5
Fig 10.10. Terminología empleada para el reparto de poblaciones atóminas
Los resultados podemos observarlos en la tabla 10.9. En ambas glucuronidaciones, la
unión al ácido glucurónico podrá ser en N3 o N4 observando valores de dicha tabla
vemos una mayor población de dichos N en el tautomero imino. N4 tiene mayor
población que N3 porque es el N del grupo imino. Por la misma razón en el tautómero
imino G6 sea N o grupo OH gana población (69,5 gana si es un único H y 40,1 gana si
es un OH).
Sin embargo sabemos que es más estable el tautómero amino. Porque observando los
ciclos G1, G2 y G3 notamos que tienen más población y por tanto se estabilizan más.
Observamos asimismo, que en la glucuronidation 2 el benceno final, G3, practicamente
no se siente afectado.
Es de analizar que el acido glucuronico G5 también tiene más población en el tautomero
amino en general pero de forma distinta. Mientras en glucuronidación 1 son los H los
que mantienen más población en el tautómero amino; en la glucuronidación 2 son los C.
Recordemos que en la glucuronidación 1, el ácido glucurónico enlaza en un grupo NH y
en el segundo enlaza en un NOH En ambos casos los O del ácido glucurónico
mantienen prácticamente la misma población.

Tabla 10.9. Datos de población de grupos o atomos más relevantes de los glucuronidos. Datos obtenidos restando el tautómero imino menos el amino. Los resultados están multiplicados por 103 en ua. G4 es siempre un CH3. G6 =H en glucuronidación 1 y OH en glucuronidación 2. Resultados en agua utilizando método PCM.
Glucuronidación 1 Glucuronidación 2
∆N(Ω) ∆N(H) ∆N(C) ∆N(Ω) ∆N(H) ∆N(C)
G1 -67,9 -83,3
G2 -64,6 -12,1 -44,7 -24,8 -5,7 -15,4
G3 -12,2 -5,8 -5,7 0,2 0,3 -0,1
G4 4,8 14,6 -9,8 6,4 -8,5 14,9
G5 -19,6 -33,8 13,9 -9,2 17,5 -29,8
G6 69,5 40,1 -1,9
N1 -12,1 -24,4
C2 -17,2 -19,8
N3 13,3 5,1
N4 35,2 28,4
Conclusiones
Dada la importancia que tienen hoy en dia las HCAs y los numerosos estudios
experimentales que se estan haciendo al efecto en la búsqueda, determinación y análisis
de su potencial carcinogénico, hemos realizado un estudio teórico de estabilidad de los
distintos tautómeros en los que puede encontrarse dichas HCAs.
El conocimiento de la forma más estable es clave para decidir si una determinada HCA
seguirá la via de destoxificación o bien la formación de aductos del DNA dado que no
todos los tautómeros son precursores de formación de aductos.
Basándonos en 3 de las más importantes cancerigenas HCAs, hemos demostrado la
estabilidad de su forma amina a nivel de distintos métodos de cálculo. Todos
coincidentes excepto cuando la amina sufre la bioactivación mediante un proceso de
hidroxilación. En dicho caso B3LYP (agua y gas) e incluso MP2 gas, nos proporcionan
el tautómero imino como más estable.
Para obtener una explicación satisfactoria a este hecho y analizar estas preferencias
tautonómicas, hemos realizado un estudio IQA y QTAIM de la tautomeria imino-
enamino lineal y ciclica.

Las energia de interacción nos muestran la mayor estabilidad del enlace C=N frente al
C=C en la molécula lineal, pero no así las energias de interacción de los mismos enlaces
en 2-aminoimidazole. La comparación de la tautomeria ceto-enolica con la amino-imino
en el estudio de estabilidad de compuestos de ciclo de 5 átomos, nos ha llevado a la
conclusión que la aromaticidad favorece el tautomero amino.
Por último hemos realizado un estudio energético y QTAIM de los posibles tautómeros
resultantes de la glucuronidation confirmando la estabilidad del tautómero amino.

6.- Conclusiones generales
1.- Las posiciones de protonación que predicen las diferentes afinidades protónicas
calculadas para la piridina y sus derivados coinciden con el comportamiento observado
experimentalmente.
2.- Se han obtenido estquemas representativos de los movimientos de población
electrónica atómica σ y π que acompañan a las protonaciones del heterocíclo de
piridina en esta molécula y en los derivados aquí estudiados.
3.- En los heterociclos piridínicos, la posición de protonación que predice como más
favorable el elemento zz del tensor momento cuadrupolar atómico coincide con la
prevista utilizando un criterio de población electrónica atómica.
4.- La inclusión de un sustiyente activante permite que tengan lugar reacción de
sustitución electrófila aromática en un heterociclos deficitarios de densidad electrónica,
como es el caso de la piridina.
5.- Las predicciones del modelo de resonancia en derivados de la piridina presentan
discrepancias con los resultados del análisis QTAIM.
6.- La formas zwitteriónicas de la glicina y de su dimero son inestables en fase gas,
incluso cuando se adoptan geometrías que permiten formar enlaces de hidrógeno entre
átomos con carga formal.
7.- Los confómeros más estables de los dimeros de glicina que contienen un monómero
zwiteriónico presentan enlaces de tipo N•••H-N.
8.- Los dimeros de glicina en disolución acuosa presenta valores de la densidad
electrónica en el punto crítico más bajos que en fase gas. Esto se debe a la estabilización
de los átomos más electrónegativos por interacciones con el disolvente, y da lugar
geometrias más abiertas en disolución acuosa
9.- Los dimeros de glicina neutra presentan transferencias electrónicas muy bajas entres
sus monómeros. En cambio, en los dimeros iónicos, se obtienen transferencias que
alcanzan 0,1 au.
10.- En los procesos de dimerización el análisis QTAIM revela que los átomos más
electronegativos pierden más población electrónica en fase gas que en disolución
acuosa.
11.- Se ha obtenido y comprobado computacionalmete una nueva relación matemática
entre las densidades electrónicas de los puntos críticos del enlace de hidrógeno y de su
enlace covalente asociado.

12.- Se propone una nueva definición de orden de enlace para los enlaces de hidrógeno
a partir de la densidad de su punto crítico.
13.- Se ha demostrado que la combinación de la optimización geométrica con la
corrección counterpoise al BSSE da lugar a geometrías dependientes de la
fragmentación utilizada.
14.- Tanto la población electrónica del átomo de N+, del heterociclo de las aminas
arómaticas con potencial mutagénico, como el valor de ρ2∇ asociable a su par
solitario, presentan buenas correlaciones con la estabilidad relativa de los
correspondientes iones nitronio. Por ello, pueden ser considerados como factores
estimativos de su reactividad química.
15.- Así como el tautómero imino del PhIP (2-amino-1-metil-6-fenilimidazo[4,5-
b]piridina) es menos estable que el amino, la N-hidroxilación revierte esta tendencia.
Ello indica que otros factores han de ser los responsables de la formación del N-
hidroxiamino-PhIP y sus consiguientes y peligrosos iones nitronio.
16.-La N-hidroxilación es una ruta de reaccción que genera metabolitos más inestables
que las hidroxilaciones en el anillo bencénico del PhIP. Esto indica también que la
mayoria del PhIP no reacciona para formar iones nitronio.
17.- Los glucuronidos más estables del PhIP se obtienen cuando el ácido glucurónico
ataca al N1 del anillo imidazólico del PhIP.
18.- Los procesos tautoméricos en el EPD (3-etil-2,4-pentanodiona) dependen del
medio y del pH, en cuanto a la formación de tautómero más estable. En la reacción en
fase gaseosa la forma predominante es el tautómero enol (mostrando estabilidad por
formación de HB) y en medio acuoso neutro la forma dicetónica ( en este caso la
estabilidad es debida a interacciones con el disolvente). En medio acuoso ácido el
equilibrio tautomérico está desplazado a la formación del dienol.
19.- Estudios IQA sobre desglose energético de tautómeros imino-enaminos en modelo
lineales y ciclicos, con o sin aromaticidad nos revelan la aromaticidad como la
responsable de la estabilidad del tautómero amino.

7.- Bibliografia
a) Para el punto 4 referente a la metodología empleada: [1] M. Born, J. R. Oppenheimer, Ann. Physik, 1927,84, 457,.
[3] F. Hund, Z. Phys., 1928,51, 759,.
[3] R. S.Mulliken, Phys. Rev., 1928,32, 761,.
[4] C. C. J.Roothaan, Rev. Mod. Phys. 1951,23, 69,.
[5] J. C. Slater, Phys. Rev., 1931,38, 1109, .
[6] A Szabo, N. S. Ostlund, Modern Quantum Chemistry: Introduction to Advanced
Electronic Structure Theory; Dover Publications Inc: Mineola, New York, 1996.
[7] P. Hohenberg, W. Kohn, Phys. Rev., 1964,136, B864,.
[8] W. Kohn, L. J. Sham, Phys. Rev., 1965,140, A1133,.
[9] A. D. Becke, J. Chem. Phys., 1993,98, 1372,.
[10] C. Lee, W. Yang, R. G. Parr, Phys. Rev. B, 1988,37, 785,.
[11] J. P. Perdew, Phys. Rev. B, 1986,33, 8822,.
[12] A. D. Becke, D Axel., J. Chem. Phys. 1993,98, 5648,.
[13] R. G. Parr, W. Yang, “Density Functional Theory of Atoms and
Molecules”, Oxford University Press, New York, 1989
[14] R. F. W. Bader, Pure Appl. Chem., 1988,60, 145,.
[15] R. F. W. Bader, “Atoms in Molecules: A Quantum Theory”, Oxford
University Press, Oxford, U.K. 1990.
[16] A. M. Pendás, ”Análisis de la densidad electrónica”, 2006.
[17] N. Otero, “Estudio QTAIM de las protonaciones del indol y derivados”, Memoria
de grado, Universidad de Vigo, 2007.
[18] R. F. W. Bader, R.J.Gillespie, P.J.MacDougall, J.Am.Chem.Soc., 1988,110,.
[19] R. F. W. Bader, “Properties of Atoms in Molecules: Electrofilic Aromatic
Substitution” , J. Chem. Phys., 1989,93,.
[20] N. Otero, M. Mandado, R. A. Mosquera, J. Chem. Phys., 2007,126, 234108.
[21] R. F. W. Bader, A. Streitwieser, A. Neuhaus, K. E. Laidig, P. Speers, J. Am. Chem.
Soc., 1996,118, 4959,.

[22] M. A. Blanco, A. Martín Pendás, E. Francisco, J. Chem. Theory. Comput., 2005,1,
1096,.
b) Especificamente y para cada apartado la bibliografia a ala que nos hemos referido
es la siguiente::
1. Variaciación de la densidad electrónica ante reacciones de adicion
electrófila en el anillo piridinico
[1] http.://webbook.nist.gov/chemistry/
[2] I.L.Finar. Organic Chemistry , Longman group limited, 1973.
[3] Y. Ibrahim, R. Mabrouki, M. Meot-Ner, M. Samy El-Shall. J. Phys. Chem. A 2007,
111, 1006
[4] D. L. Wertz, A.a Winters, T. Craft, J. Holloway. Energy & Fuels 2006, 20, 239
[5] J. A. Turner. J. Org. Chem. 1983,48, 3401-3408
[6] P. Gerbaux, Y. Van Haverbeke, R. Flammang. J Am Soc Mass Spectrom 2003, 14, 241
[7] H.Seel , H. Giinther*. J. Am. Chem. SOC. 1980, 102, 7051
[8] R.F.W. Bader, Chem. Rev.,1991, 91893.
[9]R. F. W. Bader, “Atoms in Molecules: A Quantum Theory”, Oxford
University Press, Oxford, U.K. 1990
[10] A. M. Graña, R. A. Mosquera, Chem. Phys., 1999,243, 17.
[11] A. Vila, R. A. Mosquera, Chem. Phys. Lett., 2000,332, 474.
[12]A. Vila, R. A. Mosquera, Tetrahedron, 2001,57, 9415.
[13]A. Vila, R. A. Mosquera, Chem. Phys. Lett., 2003,371, 540.
[14] M. J. González Moa, R. A. Mosquera, J. Phys. Chem. A, 2005,109, 3682.
[15]GaussianR 03 program Gaussian, Inc. 340 Quinnipiac St., Bldg. 40, Wallingford
CT 06492
[16] R. F. W.. Bader, AIMPAC: A suite of programs for the AIM theory; Mc Master
University, Hamilton, Ontario, Canada L8S 4M1.
[17] N. Otero, M. Mandado, R. A. Mosquera, J. Chem. Phys., 2007, 126, 234108
[18] R. F. W. Bader, “Properties of Atoms in Molecules: Electrofilic Aromatic
Substitution” , J. Chem. Phys., 1989,93,

2. Cambios en el comportamiento nucleofilo de la piridina por efecto de
distintos grupos activantes o desactivante
[1] http.://webbook.nist.gov/chemistry/
[2] K.B. Wiberg, K.E. Laidig, J. Am. Chem. Soc.1987 ,109, 5935.
[3] K.B. Wiberg, C. M. Breneman, J. Am. Chem. Soc., 1992,114, 831.
[4] K. E. Laidig, L. M. Cameron, J. Am. Chem. Soc., 1996,118, 1737.
[5] R. Glaser, G. S. K. Choy, J. Am. Chem. Soc., 1993,115, 2340.
[6] P. Speers, K. E. Laidig, A. Streitwieser, J. Am. Chem. Soc., 1994,116, 9257.
[7] A. M. Graña, R. A. Mosquera, Chem. Phys., 1999,243, 17.
[8] R. A. Mosquera, A. M. Graña, Rec. Res. Devel. Chem. Phys., 2001,2, 23.
[9] A. Vila, R. A. Mosquera, J. Phys. Chem. A, 2000,104, 12006.
[10] A. Vila, R. A. Mosquera, Chem. Phys. Lett., 2000,332, 474.
[11] A. Vila, R. A. Mosquera, Chem. Phys. Lett., 2003,371, 540.
[12] A. Vila, R. A. Mosquera, Tetrahedron,2001, 57, 9415.
[13] M. J. González Moa, R. A. Mosquera, J. Phys. Chem. A, 2003,107, 5361.
[14] M. J. González Moa, R. A. Mosquera, J. Phys. Chem. A, 2005,109, 3682.
[15] M. Mandado, C. Van Alsenoy, R. A. Mosquera, J. Phys. Chem. A, 2004,108, 7050.
[16] M. Mandado, R. A. Mosquera, A. M. Graña, Chem. Phys. Lett., 2004,386, 454.
[17] M. Mandado, C. Van Alsenoy, R. A. Mosquera, Chem. Phys. Lett., 2005,405, 10.
[18] R.F.W. Bader, Chem. Rev. 1991, 91, 893.
[19]R. F. W. Bader, “Atoms in Molecules: A Quantum Theory”, Oxford University
Press, Oxford, U.K. 1990
[20] R.F.W. Bader, C.J. Chang J.Phys. Chem. 1989, 93, 2946
[21] J. Cioslowski, M. Martinov, S.T. Mixon. J.Phys. Chem. 1993, 97, 10948
[22] Gaussian 03 program Gaussian, Inc. 340 Quinnipiac St., Bldg. 40, Wallingford CT
06492
[23] Bader, R. F. W.. AIMPAC: A suite of programs for the AIM theory; Mc Master
University, Hamilton, Ontario, Canada L8S 4M1.
[24] N. Otero, M. Mandado, R. A. Mosquera, J. Chem. Phys., 2007,126, 234108.
[25] A. Vila, R. A. Mosquera, J. Comput. Chem., 2007,28, 1516
[27] Geissman T.A. Principios de química orgánica, ed reverté,1973

3. Estudio computacional de las preferencias conformacionales de dímeros
de glicina neutros, protonados y desprotonados.
[1] W. Saenger, Principles of Nucleic Acid Structure; Springer-Verlag: New York,
1984.
[2] G.E. Schulz, R.H. Schirmer, Principles of Protein Structure; Springer-Verlag: New
York, 1979.
[3] L. Rodríguez-Santiago, M. Noguera, J. Bertran, M. Sodupe, In Quantum
Biochemistry. chapter 7. 219-243. Wiley-VCH. Weinheim, 2010.
[4] F. Pichierri, M. Aida, M. M. Gromiha, A. Sarai, J. Am. Chem. Soc. 1999, 121,
6152.
[5] C. Desfrançois, S. Carles, J. P. Schermann, Chem. Rev. 2000, 100, 3943.
[6] W. Ni, R. A. Mosquera, J. Pérez-Juste, L. M. Liz-Marzán, J. Chem. Phys. Lett.
2010, 1, 1181.
[7] R. Ramaekers, J. Pajak, B. Lambie, G. Maes, J. Chem. Phys. 2004, 120, 4182.
[8] J. H. Jensen, M. S. Gordon, J. Am. Chem. Soc. 1995, 117, 8159.
[9] C. Kapota, J. Lemaire, P. Maitre, G. Ohanessian, J. Am. Chem. Soc. 2004, 126,
1836.
[10] K. HongWei, R. Li, X. Xin, Y. YiJing, Sci. Chin. Ser. B. 2010, 53, 383.
[11] S. Xu, J. M. Nilles, K. H. Bowen, Jr., J. Chem. Phys. 2003, 119, 10698.
[12] W. D. Price, P. D. Schnier, E. R. Williams, J. Phys. Chem. B. 1997, 101, 664.
[13] S. A. Raspopov, T. B. McMahon, J. Mass. Spectrom. 2005, 40, 1536.
[14] R. Wu, T. B. McMahon, J. Am. Chem. Soc. 2007, 129, 4864.
[15] C. G. Atkins, K. Rajabi, E. A. L. Gillis, T. D. Fridgen, J. Phys. Chem. A 2008,
11, 10220.
[16] P. B. Armentrout, A. L. Heaton, S. J. Ye, J. Phys. Chem. A 2011, 115. 11144.
[17] J. Huang, T. C. Stringfellow, L. Yu, J. Am. Chem. Soc. 2008, 130, 13973.
[18] S. Hamad, C. Richard, A. Catlow, CrystEngComm. 2011, 13, 4391.
[19] M. G. Campo, J. Chem. Phys. 2006, 125, 114511.

[20] J. Chocholousová, J. Vacek, F. Huisken, O. Werhahn, P. Hobza, J. Phys. Chem. A.
2002, 106, 11540.
[21] M. F. de Carvalho, R. A. Mosquera, R. Rivelino, Chem. Phys. Lett. 2007, 445, 117.
[22] P. Friant-Michel, M. F. Ruiz-López, Chem.Phys.Chem. 2010, 11, 3499.
[23] R. F. W. Bader, Atoms in Molecules. A Quantum Theory; Oxford Univer-sity Press:
New York, 1990.
[24] Gaussian 09. Revision E.01. M.J. Frisch et al. Gaussian Inc. Wallingford CT
(2004)
[25] M. Frisch., J. Pople, J. Bene, J. Phys. Chem. 1985, 89, 3664.
[26] J. E. Del Bene, M. J. T. Jordan, J. Mol.Struct. 2001, 573, 11.
[27] J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 2005, 105, 2999.
[28] E. Rezabal, J. M. Mercero, X. Lopez, J. M. Ugalde, J. Inorg. Biochem. 2007, 101,
1192.
[29] F. Biegler-Konig, J. Schonbohm, R. Derdau, D. Bayles, R. F. W. Bader. AIM 2000.
Version 1; Bielefeld. Germany. 2000
[30] A. M. Graña, R. A. Mosquera, J. Chem. Phys. 1999, 110, 6606.
[31] F. Aicken, P. L. A. Popelier, Can. J. Chem. 2000, 78, 415.
[32] W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, Jr., D. M.
Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, P. A. Kollman, J. Am. Chem.
Soc. 1995, 117, 5179.
[33] J. O. M. Bockris, B. E. Conway, R. E. White, "Modern aspects of
electrochemistry". New York: Plenum Press. 1996.
[34] N. C. J. Stutchbury, D. L. Cooper, J. Chem. Phys. 1983, 79, 4967.
[35] M. J. González Moa, M. Mandado, R. A. Mosquera, Chem. Phys. Lett. 2006, 428,
255.
[36] N. Otero, L.Estévez, M. Mandado, R. A. Mosquera, Eur. J. Org. Chem. 2012, 2403.
[37] L. Estévez, R. A. Mosquera, J. Phys. Chem. A 2008, 112, 10614.
4. Estudio de la densidad electrónica en dímeros de glicina desprotonados
[1] W. Saenger, Principles of Nucleic Acid Structure; Springer-Verlag: New York.
1984.
[2] G.E. Schulz, R.H. Schirmer. Principles of Protein Structure; Springer-Verlag: New
York. 1979.

[3] L. Rodríguez-Santiago, M. Noguera, J. Bertran, M. Sodupe. In Quantum
Biochemistry. chapter 7, 219-243. Wiley-VCH. Weinheim. 2010.
[4] F. Pichierri, M. Aida, M. M. Gromiha, A. Sarai J. Am. Chem. Soc. 1999, 121, 6152-
6157.
[5] C. Desfrançois, S. Carles, J. P. Schermann Chem. Rev. 2000, 100, 3943-3962.
[6] W. Ni, R. A. Mosquera, J. Pérez-Juste, L. M. Liz-Marzán J. Chem. Phys. Lett.
2010, 1, 1181-1185
[7] J. H. Jensen, M. S. Gordon J. Am. Chem. Soc. 1995, 117, 8159-8170.
[8] W. D. Price, P. D. Schnier, E. R. Williams J. Phys. Chem. B. 1997, 101, 664-673.
[9] S. A. Raspopov, T. B. McMahon J. Mass. Spectrom. 2005, 40, 1536-1545.
[10] R. Wu, T. B. McMahon J. Am. Chem. Soc. 2007, 129, 4864-4865.
[11] J. Huang, T. C. Stringfellow, L. Yu J. Am. Chem. Soc. 2008, 130, 13973-13980.
[12] S. Hamad, C. Richard, A. Catlow CrystEngComm. 2011, 13, 4391-4399.
[13] J. Chocholousová, J. Vacek, F. Huisken, O. Werhahn, P. Hobza J. Phys.Chem. A .
2002, 106, 11540-11549.
[14] M. F. de Carvalho, R. A. Mosquera, Roberto Rivelino Chem. Phys. Lett. 2007,
445, 117-124.
[15] C. G. Atkins, K. Rajabi, E. A. L. Gillis, T. D. Fridgen J. Phys. Chem. A 2008, 11,
10220-10225.
[16] P. Friant-Michel, M. F. Ruiz-López Chem.Phys.Chem. 2010, 11, 3499-3504.
[17] R. F. W. Bader, Atoms in Molecules. A Quantum Theory; Oxford Univer-sity
Press: New York. 1990.
[18] J. Tomasi, B. Mennucci, R. Cammi Chem. Rev. 2005, 105, 2999-3093.
[19] Gaussian 09, Revision E.01, M.J. Frisch et al. Gaussian Inc. Wallingford CT
(2004).
[20] M. Frisch, J. Pople, J. del Bene J. Phys. Chem. 1985, 89, 3664-3669.
[21] J. E. del Bene, M. J. T. Jordan J. Mol.Struct. 2001, 573, 11-23.
[22] E. Rezabal, J. M. Mercero, X. Lopez, J. M. Ugalde J. Inorg. Biochem. 2007, 101,
1192-1200.
[23] F. Biegler-König, J. Schönbohm, D. Bayles J. Comput. Chem. 2001, 22, 545-559.
[24] A. M. Graña, R. A. Mosquera J. Chem. Phys. 1999, 110, 6606-6616.
[25] F. Aicken, P. L. A. Popelier Can. J. Chem. 2000, 78, 415-426.
[26] M. T. Carroll, R. F. W. Bader Mol. Phys. 1988, 65, 695-722.
[27] U. Koch, P. L. A. Popelier J. Phys. Chem. 1995, 99, 9747-9754.

[28] E. Espinosa, E. Molins, C. Lecomte Chem. Phys. Lett. 1998, 285, 170-173.
[29] T. Steiner J. Chem. Soc. Chem. Commun. 1995, 1331-1332
[30] M. J. González Moa, M. Mandado, R. A. Mosquera Chem. Phys. Lett. 2006, 428,
255-261.
[31] D. Ferro-Costas, R. A. Mosquera J. Phys.Chem. A 2013, 117, 257-265.
5. La relación entre la distancia y la densidad del punto crítico. Nueva
concepción en el oreden de enlace de H.
[1] R. F. W. Bader, Atoms in Molecules. A Quantum Theory; Oxford
University Press: New York, 1990.
[2] L. Pauling, J. Am. Chem. Soc. 1947, 69, 542. (b) Brown, I. D.
Acta Crystallogr. 1992, B48, 553.
[3] I.Alkorta, I. Rozas, J. Elguero. Structural Chemistry, Vol. 9, No. 4, 1998
[4] Th.; Steiner, W. Saenger, Acta Crystallogr. 1994, B50, 348.
[5] Th. Steiner, J. Chem. Soc., Chem. Commun. 1995, 1331.
[6] P. Gilli,.; V. Bertolasi,.; V. Ferretti, G. Gilli,. J. Am. Chem. Soc. 1994, 116, 909.
[7] L., Pauling, Atomic radii and interatomic distances in metals, J. Am. Chem. Soc.,
1947,69, 542.
[8] I. D., Brown, Chemical and steric constraints in inorganic solids, Acta Cryst.,
1992,B48, 553.
[9] Gaussian 09, Revision E.01, M.J. Frisch et al. Gaussian Inc. Wallingford CT ,
2004
[10] M., Frisch, J., Pople, J .E.., del Bene J. Phys. Chem., 1985, 89, 3664.
[11] J.E.Del Bene , M. J. T. Jordan, J. Mol.Struct., 2001, 573, 11.
[12] R. F. W.. Bader, AIMPAC: A suite of programs for the AIM theory; Mc Master
University, Hamilton, Ontario, Canada L8S 4M1.
[13] J. Tomasi, B. Mennucci, R. Cammi, Chem. Rev. 2005, 105, 2999.
[14] H. Benedict, H.Limbach, M. Wehlan, W.Fehlhammer,N.S. Golubev, R. Janoschek.
J. Am. Chem. Soc. 1998, 120, 2939.
[15] O. Picazo, I. Alkorta, J.Elguero. J. Org. Chem. 2003, 68, 7485.
6. Correción counterpoise y optimización geométrica.

[1] S.F. Boys, F. Bernardi, Mol. Phys. 1970, 10 553.
[2] J. Emsley, O.P.A. Hoyte, R.E. Overill.,J Am Chem Soc 1978,100, 3303
[3] N. Kobko, J.J. Dannenberg J Phys Chem A 2001,105,1944.
[4] D. Hugas, S. Simon , M. Duran. Chemical Physics Letters 2004,386 , 373.
[5] S. Simon , M. Duran, J. J. Dannenberg. J. Chem. Phys. 1996; 105, 11024 .
[6] C. Barrientos · J. A. Sordo. Theor Chem Account ,2007, 118,733.
[7] F. Jensen, J. Chem. Theory Comput. 2010, 6, 100–10
[8] P. Salvador ,M.Duran, J. J. Dannenberg, J. Phys. Chem. A 2002, 106, 6883.
[9] A. Galano, J. R.Alvarez–Idaboy. J Comput Chem 2006,27,1203.
[10] S. Simon , M. Duran, J. J. Dannenberg, J. Phys. Chem. A 1999, 103, 1640-1643
[11] S. Simon, J. Bertran, and M. Sodupe,J. Phys. Chem. A 2001, 105, 4359-4364
[12] Gaussian 09. Revision E.01. M.J. Frisch et al. Gaussian Inc. Wallingford CT,
2004.
[13] M. Frisch. J. Pople. J. Bene. J. Phys. Chem. 1985, 89, 3664.
[14] J. E. Del Bene. M. J. T. Jordan. J. Mol.Struct. 2001, 573, 11.
[15] D.W. Schwenke, D.G. Truhlar, J Chem Phys, 1985,82,2418.
[16] J. R. Alvarez-Idaboy , A. Galano. Theor Chem Acc ,2010, 126,75,
[17] J.J. Dannenberg, J Phys Chem ,1993, 97,2488,
[18] F. B. van Duijneveldt,J. G. C. M. van Duijneveldt-van de Rijdt, J. H. van Lenthe.
Chem. Rev. 1994, 94, 1873.
[19] J. C. Jiang, H.-C. Chang, Y. T. Lee, and S. H. Lin,J. Phys. Chem. A 1999, 103,
3123.
[20] A. Bende, M. Knapp-Mohhammady, S. Suhai., J Quantum Chem, 2003,92,152.
[21] S. Abdel Azeim ,G.van der Rest.J. Phys. Chem. A 2005, 109, 2505.
[22] A. Kumar Srivastava ,N.Misra.J. Comput. Methods Mol. Des., 2014, 4 ,19.
[23] Th. Steiner, W. Saenger, Acta Crystallogr. 1994, B50, 348.
[24] Th. Steiner, J. Chem. Soc., Chem. Commun. 1995, 1331.
[25] P.;Gilli, V Bertolasi,.; V. Ferretti, G. Gilli, J. Am. Chem. Soc. 1994, 116, 909.
[26] L.Pauling, Atomic radii and interatomic distances in metals, J. Am. Chem. Soc.,
1947, 69, 542.
[27] I. D., Brown, Chemical and steric constraints in inorganic solids, Acta Cryst., 1992
,B48, 553.

7. Estudio QTAIM de aminas heterocíclicas (HCAs)
[1] E. M. P., Widmark Nature, 1939, 143, 984.
[2] K. Skog, Food Chem. Toxicol, 2002, 40, 1197
[3] L.Dongxin, K. R Kaderlik, R. J Turesky., D.W Miller., J O Lay Jr., F. F Kadlubar.,
Chem. Res Toxicol, 1992, 5, 691
[4] J.S Felton. , M.G Knize., N. G. Shen, P.R Lewis., B.D Andresen., J Happe., F.T..
Hatch , Carcinogenesis, 1986, 7, 1081
[5] E.L Jamin., D Arquier., C Canlet., E Rathahao., J.Tulliez, L Debrauwer., J Am Soc
Mass Spectrom, 2007, 18, 2107
[6] H. Kataoka, M Miyake., S Nishioka., T. Matsumoto, K Saito., K Mitani., Mol.Nutr.
Food Res. 2010,54, 1039
[7] M Murkovic., J.Chromatogr. B. Analyt. Technol. Biomed. Life Sci., 2004, 802, 3
[8] E. G Snyderwine., M Venugopal., M Yu. Mutatation Research. 2002, 506, 145.
[9] S. Zöchling, M. Murkovic, Food Chem, 2002, 79, 125.
[10] M. Murkovic, J. Chromatogr. B. 2004, 802, 3.
[11] J.S Felton., M.G Knize., C.P Salmon., M.A Malfatti., K.S Kulp.. Environ. Mol.
Mutagen, 2002, 39, 112
[12] M. S Alaejos,., V González,., A. M Afonso,., 2008, 25, 2.
[13] C Magagnotti., F.Orsi, R Bagnati., N Celli., D Rotilio., R Fanelli. , L Airoldi., Int.
J. Cancer, 2000, 88, 1
[14] W. W Huber., L.P McDaniel., K.R Kaderlik., C. H Teitel., N. P Lang., F.F
Kadlubar ., Mutation Research ,1997,376,115
[15] S Shibutani., A Fernandes., N Suzuki., L Zhou., F Johnson, A.P Grollman., The
Journal of biological chemistry., 1999 ,274,27433,
[16] K. H Dingley., K. D Curtis. , S Nowell., S.S Felton., N.P Lang., K.N Turteltaub..
Cancer Epidemiology, Biomarkers & Prevention, 1999, 8, 507.
[17] B. N Ames., J McCann., E Yamasaki. , Mutation Research , 1975, 31, 347.
[18] F.L., Martin, K.J Cole., D.H Phillips., P.H Grover., Biochemical and Biophysical
Research Communications, 2002, 293, 1497
[19] M.S Connors., M.A. Malfatti , J.S Felton., Chemico-Biological Interactions, ,1995,
96 ,185.

[20] V.M Arlt ., R Singh ., M Stiborová ., G Gamboa da Costa ., E Frei ., J.D Evans
., P.B Farmer ., C.R Wolf ., C.J Henderson ., D.H Phillips .. Drug. Metab.
Dispos, 2011 , 39 , 2169
[21] R Singha., V.M Arlt., C.J Hendersond., D.H Phillips., P.B Farmera., Gamboa da
Costa G. , Journal of Chromatography B, 2010, 878, 2155.
[22] L Peng., R.J Turesky., Chem Res Toxicol., 2011, 24, 2004.
[23] E.J Kim., A.M Matuszek., B. Yu, J Reynisson., Aust. J. Chem., 2011, 64, 910
[24] G. L Borosky,., Chem. Res. Toxicol., 2007, 20, 171.
[25] G. L Borosky,., Journal of Molecular Graphics and Modelling, 2008, 27, 459.
[26] I Shamovsky,., L Ripa, N Blomberg., L.A Eriksson., P Hansen., C Mee., C
Tyrchan ., M O'Donovan., P Sjö., Chem. Res. Toxicol., 2012, 25, 2236
[27] J.F Han., X.Y, He J.S Herrington., L.A White., J.F Zhang., J.Y Hong., Drug
Metabolism and disposition, 2008, 36,745.
[28] M.A Malfatti., K.S Kulp., M.G Knize., C., Davis, J.P Massengi., S Williams.,
S., Nowell, S MacLeod., K.H Dingley., K.W Turteltaub.,. N.P Lang., J.S Felton,
Carcinogenesis, 1999 , 20 ,705.
[29] M.A Malfatti., E.A Ubick., J.S Felton., Carcinogenesis, 2005, 26, 2019.
[30] N.J Goodderham., S Murray., A.M Lynch., M Yadollahi-Farsani., K Zhao., A.R
Boobis ., D.S Davies., Drug Metabolism and Disposition, 2001, 29,529.
[31] R. F. W Bader., Atoms in Molecules. A Quantum Theory; Oxford University Press:
New York. 1990
[32] Gaussian 09, Revision E.01, M.J. Frisch et al. Gaussian Inc. Wallingford CT ,
2004.
[33] M Frisch., J Pople., J .E del Bene.., J. Phys. Chem., 1985, 89, 3664.
[34] J.E Del Bene., M. J. T Jordan. , J. Mol.Struct., 2001, 573, 11.
[35] E Rezabal., J. M Mercero., X Lopez., J. M Ugalde, J. Inorg. Biochem, 2007, 101,
1192.
[36] F Biegler-König., J Schönbohm., D Bayles., J. Comput. Chem., 2001, 22, 545.
[37] A.M Graña., R.A Mosquera., J. Chem. Phys., 1999, 110, 6606.
[38] F Aicken., P. L. A Popelier., Can. J. Chem., 2000, 78, 415.
[39] A.K Debnath., G Debnath., A.J Shusterman., C Hansch., Environ. Mol. Mutagen.,
1992,19, 37.
[40] CG Cash , Mutat.Res ,2001,491, 31.

[41] F.T.,Hatch, M.E Colvin., E.T Seidl., Environmental and Molecular Mutagenesis,
1996, 27,314-330.
[42] A.M Pendás., Análisis de la densidad electrónica, 2006.
[43] M.E Colvin., E.T Seid., I.M.B Nielsen. , L Le Bui. , F.T Hatch., Chemico-
Biological Interactions , 1997,108, 39.
8. Estudio computacional de la actividad biológica de aminas heterocíclicas
potencialmente cancerígenas
[1] K. Skog, Food Chem. Toxicol, 2002, 40, 1197
[2] L.Dongxin, K. R Kaderlik, R. J Turesky., D.W Miller., J O Lay Jr., F. F Kadlubar.,
Chem. Res Toxicol, 1992, 5, 691
[3] J.S Felton. , M.G Knize., N. G. Shen, P.R Lewis., B.D Andresen., J Happe., F.T..
Hatch , Carcinogenesis, 1986, 7, 1081
[4] E.L Jamin., D Arquier., C Canlet., E Rathahao., J.Tulliez, L Debrauwer., J Am Soc
Mass Spectrom, 2007, 18, 2107
[5] H. Kataoka, M Miyake., S Nishioka., T. Matsumoto, K Saito., K Mitani., Mol.Nutr.
Food Res. 2010,54, 1039
[6] M Murkovic., J.Chromatogr. B. Analyt. Technol. Biomed. Life Sci., 2004, 802, 3
[7] B. N Ames., J McCann., E Yamasaki. , Mutation Research , 1975, 31, 347.
[8] R Singha., V.M Arlt., C.J Hendersond., D.H Phillips., P.B Farmera., Gamboa da
Costa G. , Journal of Chromatography B, 2010, 878, 2155.
[9] M.S Connors., M.A. Malfatti , J.S Felton., Chemico-Biological Interactions, ,1995,
96 ,185.
[10] V.M Arlt ., R Singh ., M Stiborová ., G Gamboa da Costa ., E Frei ., J.D Evans
., P.B Farmer ., C.R Wolf ., C.J Henderson ., D.H Phillips .. Drug. Metab.
Dispos, 2011 , 39 , 2169
[11] L Peng., R.J Turesky., Chem Res Toxicol., 2011, 24, 2004.
[12] J.F Han., X.Y, He J.S Herrington., L.A White., J.F Zhang., J.Y Hong., Drug
Metabolism and disposition, 2008, 36,745
[13] M.A Malfatti., K.S Kulp., M.G Knize., C., Davis, J.P Massengi., S Williams.,
S., Nowell, S MacLeod., K.H Dingley., K.W Turteltaub.,. N.P Lang., J.S Felton,
Carcinogenesis, 1999 , 20 ,705.

[14]. M.A Malfatti., E.A Ubick., J.S Felton., Carcinogenesis, 2005, 26, 2019.
[15] N.J Goodderham., S Murray., A.M Lynch., M Yadollahi-Farsani., K Zhao., A.R
Boobis ., D.S Davies., Drug Metabolism and Disposition, 2001, 29,529
[16] G. L Borosky,., Chem. Res. Toxicol., 2007, 20, 171.
[17] G. L Borosky,., Journal of Molecular Graphics and Modelling, 2008, 27, 459.
[18] I Shamovsky,., L Ripa, N Blomberg., L.A Eriksson., P Hansen., C Mee., C
Tyrchan ., M O'Donovan., P Sjö., Chem. Res. Toxicol., 2012, 25, 2236.
[19] R. F. W Bader., Atoms in Molecules. A Quantum Theory; Oxford University Press:
New York. 1990
[20] G Nauwelaers., E. E Bessette., D Gu., Y Tang., J Rageul., V Fessard., J Yuan., M.
C., Yu, S Langouët., Turesky R. J., Chem Res Toxicol. 2011 ,24, 913.
[21] J.F Han., X.Y He, J.S Herrington., L.A White., J.F Zhang., J.Y Hong., Drug
Metabolism and disposition, 2008, 36,745.
[22] R. S Kinga.,. C. H Teitela, J. G Shaddockb.,. D. A Cascianob, F. F Kadlubara.,
Cancer Letters, 1999, 143, 167.
[23] S Langouët., A Paehler., D. H Welti., N Kerriguy., A Guillouzo., R. J Turesky..
Carcinogénesis, 2002, 23, 115.
[24] D Lin., K. R Kaderlik., R. J Turesky., D.W Miller., J. O Lay Jr,., F. F.. Kadlubar,
Chem. Res. Toxicol. 1992, 5, 691
[25] M. A., Malfatti, J. S Felton., Chemical Research in Toxicology, 2004.
[26] M.A Malfatti., K.S Kulp., M.G Knize., C., Davis, J.P Massengi., S Williams.,
S., Nowell, S MacLeod., K.H Dingley., K.W Turteltaub.,. N.P Lang., J.S Felton,
Carcinogenesis, 1999 , 20 ,705.
[27] M.A Malfatti., E.A Ubick., J.S Felton., Carcinogenesis, 2005, 26, 2019.
[28] F.L Martin., K.J., G.H Cole Muir.,. G.G Kooiman,. J.A Williams, R.A Sherwood.,
P.L Grover., D.H Phillips., Prostate Cancer and Prostatic Diseases, 2002, 5,
96.
[29] F. G Crofts, P. T., Strickland, C. L Hayes., T. R Sutter.. Carcinogenesis, 1997, 18,
1793.
[30] T Nguyen., M Novak., J. Org. Chem., 2007, 72, 4698.
[31] S Zöchling., M Murkovic., Food Chemistry, 2002, 79, 125.
[32] Gaussian 09, Revision E.01, M.J. Frisch et al. Gaussian Inc. Wallingford CT,
2004.
[33] M Frisch., J Pople., J .E del Bene.., J. Phys. Chem., 1985, 89, 3664.

[34] J.E Del Bene., M. J. T Jordan. , J. Mol.Struct., 2001, 573, 11.
[35] E Rezabal., J. M Mercero., X Lopez., J. M Ugalde, J. Inorg. Biochem, 2007, 101,
1192.
[36] F Biegler-König., J Schönbohm., D Bayles., J. Comput. Chem., 2001, 22, 545.
[37] A.M Graña., R.A Mosquera., J. Chem. Phys., 1999, 110, 6606.
[38] F Aicken., P. L. A Popelier., Can. J. Chem., 2000, 78, 415.
[39] J Chocholousová., J Vacek., F Huisken., O Werhahn., P Hobza., J. Phys.
Chem. A., 2002, 106, 11540.
[40] P Friant-Michel., M. F Ruiz-López., Chem.Phys.Chem., 2010, 11, 3499.
[41] G.D Hartman., H.B Schlegel.. Chem. Biol. Interactions, 1981, 36, 319-330.
[42] M. B Sullivan ., K Brown., C. J Cramer ., D. G Truhlar .. J. Am. Chem.
Soc., 1998, 120, 11778.
[43] C Gonzalez ., A Restrepo-Cossio ., M Márquez ., K. B Wiberg ., De Rosa M.. J.
Phys. Chem. A, 1998, 102 , 2732.
9. Estudio del equilibrio ceto –enólico de la 3-etil 2,4 pentanodiona (epd)
[1] C.Reichardt , Solvents and Solvent effects in Organic Chemistry , Ed.Wiley VCH,
2011.
[2] R. M Claramunt,.; C Lopez,.; M. D. S Maria,.; D Sanz,.; J Elguero,.
Prog. Nucl. Magn. Reson. Spectrosc. 2006, 49, 169.
[3] D. Bal, A. Kraska-Dziadecka, A. Gryff-Keller.; J. Org. Chem. 2009,74, 8625.
[4] J.J. Dannenberg , R. Ríos , J.Phys.Chem. 1994, 98 6714.
[5]M.Karelson,U Maran, A.R. Katritzky, Tetrahedron 1996,52, 11325.
[6] S Moon,.; Y Kwon,. Magn. Reson. Chem. 2001, 39, 89.
[7] F. Toda, R. Bishop, Separations and reactions in organic supremolecular chemistry,
2004,8
[8] S. Miertuš, E. Scrocco, J. Tomasi, Chem. Phys., 1981, 55 ,117.
[9] Gaussian(R) 03 program Gaussian, Inc. 340 Quinnipiac St., Bldg. 40, Wallingford
CT 06492
[10] R. F. W.. Bader, AIMPAC: A suite of programs for the AIM theory; Mc Master
University, Hamilton, Ontario, Canada L8S 4M1.
[11] U. Koch, P.L.A. Popelier, J. Phys. Chem., 1995,99, 9747.

[12] I. Alkorta, I. Rozas, J. Elguero, Chem. Soc. Rev. 1998, 27, 163.
10. Tautomeria imino-enamina en metabolitos de aminas heterociclicas con
marcado potencial mutagenico
[1] K. I. Skog, M. A. E. Johansson, M. I. Jägerstad, Food and Chemical Toxicology,
1998, 36, 879.
[2] M. Sanz Alaejos, J.H. Ayala, V. González, A.M. Afonso, Journal of
Chromatography B, 2008, 862, 15.
[3] H.A.J.Schut, E. G.Snyderwine, Carcinogenesis, 1999, 20, 353.
[4] J. Han, X. He, J. S. Herrington, L. A. White, J. Zhang, J. Hong, Drug metabolism
and disposition, 2008, 36,745.
[5] N. J. Gooderham, S. Murray, A. M. Lynch, M. Yadollahi-farsani, K. Zhao,A. R.
Boobis, D. S. Davies, Drug metabolism and disposition, 2001, 29,529.
[6] P. Perez, A. Toro-Labbé .Theor.. Chem. Acc. 2001, 105, 422.
[7] J. Lin, Ch. Wu, M.Lien, J. Phys. Chem., 1995, 99, 16903.
[8] B.Pullman, H.Berthod, M. Dreyfus, Theoret.chim.Acta, 1969,15, 265.
[9] X. Chen, Y. Hu., J. Gao, Journal of Molecular Structure , 2013, 1049, 362.
[10] I. ShcherbakovA, J. Elguero, A.R. Katritzky, Adv. Heterocycl. Chem., 2000, 77,
51.
[11] S.Süleyman, M. Genc, Murat, S. Saydam, . Chin. J. Chem, 2012, 30, 400.
[12] El D. Raczyńska , B. Kamińska. J Mol Model , 2013, 19,3947.
[13] A. Laxer, D. T. Major, H. E. Gottlieb, . B. Fischer, J. Org. Chem, 2001, 66, 5463.
[14] A. A. Hasanein, S. A. Senior. J Quantum Chem, 2011, 111, 3993.
[15] Z. S. Safi. The Islamic University Journal , 2009, 17, 29.
[16] S. J. Angyal, C. L. Angyal. J. Chem. Soc., 1952, 1461.
[17] M. Dreyfus, 0. Bensaude, G. Dodin, J. E. Dubois, Journal of the American
Chemical Society , 1976, 98, 6338.
[18]A. R. Katritzky, M. Karelsons, J. Am. Chem. Soc. 1991, 113, 1561-1566.
[19]T.-K. Ha, M.J. Keller, R. Gunde’, H.H. Gunthard*. Journal of Molecular Structure
(Theochem), 1996, 364, 161- 181.
[20]K. Tomic, J. Tatchen, C. M. Marian. J. Phys. Chem. A, 2005, 109, 8410-8418.
[21] O. O. Brovarets,. D. M. Hovorun, Phys. Chem. Chem. Phys., 2013, 15, 20091-
20104-

[22] I.Shamovsky, L. Ripa, N. Blomberg, L. A. Eriksson, P. Hansen, C. Mee, C.
Tyrchan, M. O'Donovan, P. Sjö, Chem. Res. Toxicol. 2012, 25, 2236−2252
[23] D.M. Hovorun, L.Gorb, J. LeszczynskI, International Journal of Quantum
Chemistry, 1999, 75, 245-253.
[24]N. Otero, L. Estévez, M. Mandado, R.A. Mosquera, Eur. J. Org. Chem. 2012,
2403–2413
[25] Gaussian 09, Revision E.01, M.J. Frisch et al. Gaussian Inc. Wallingford CT ,
2004.
[26] J. Tomasi, B. Mennucci, R. Cammi Chem. Rev. 2005, 105, 2999-3093.
[27] E. Rezabal, J. M. Mercero, X. Lopez, J. M. Ugalde J. Inorg. Biochem. 2007, 101,
1192-1200.
[28] R. F. W. Bader, Atoms in Molecules. A Quantum Theory; Oxford University Press:
New York. 1990.
[28] A. M. Graña, R. A. Mosquera, J.Chem. Phys. 1999, 110, 6606-6616.
[29] M. A. Blanco, A. M. Pendás and E. Francisco, J. Chem. Theory Comput. 2005, 1,
1096–1109.
[30] A. Martín Pendás, E. Francisco, Promolden, A QTAIM/IQA .
[31] M.S.Connors, M.A. Malfatti, J.S. Felton, Chemico-Biological Interactions, ,1995,
96 ,185-202.
[32] V.M Arlt ., R. Singh , M. Stiborová, G. Gamboa da Costa , E. Frei , J.D.
Evans, P.B. Farmer, C.R. Wolf , C.J. Henderson , D.H. Phillips . Drug. Metab. Dispos,
2011 , 39 , 2169-2173.
[33] R. Singha, V.M. Arlt , C.J. Hendersond , D.H. Phillips , P.B. Farmera , G. Gamboa
da Costa, Journal of Chromatography B, 2010, 878, 2155–2162.
[34]L. Peng , R.J. Turesky, Chem Res Toxicol., 2011, 24, 2004–2017.
[35]J. Poater, X. Fradera, M. Duran, M.Sola, Chem. Eur. J. , 2003, 9, 400-406.
[36] J. Poater, M. Duran, M. Sola, B.S ilvi, Chem. Rev. 2005, 105, 3911-3947.
[37]M. A. Malfatti, J. S. Felton , Chemical Research in Toxicology, 2004.
[38] H. Kataoka, M. Miyake, S. Nishioka, T. Matsumoto, K. Saito, K. Mitani,
Mol.Nutr. Food Res. 2010,54, 1039-1048
[39] M.A Malfatti, K.S. Kulp, M.G. Knize, C. Davis, J.P. Massengill, S. Williams, S.
Nowell, S. MacLeod, K.H. Dingley, K.W. Turteltaub, N.P. Lang, J.S. Felton,
Carcinogenesis, 1999, 20, 705–713.

[40] K.S. Kulp, M.G. Knize, M.A. Malfatti, C.P. Salmon , J.S. Felton, Carcinogenesis,
2000, 21, 2065–2072.
Anexo I Gráficas de ∆N=variación de la población σ/π para los distintos grupos funcionales , en posiciones orto, meta y para respecto del N de la piridina en moléculas sin protonar.
∆N GF=(O-)
en orto meta y para sin protonar
-1
-0,8
-0,6
-0,4
-0,2
0
0,2
0,4
C2 C4 C6 H3 H5 C3 C5 H1 H4 N C2 C4 C6 H2 H5
∆sigma
∆pi
∆total
∆N GF=(OCH3)
en orto, meta y para sin protonar
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
0
0,1
0,2
C2 C4 C6 H3 H5 C3 C5 H1 H4 N C2 C4 C6 H2 H5
var sigma
var pi
var total

∆N GF=(CH3)
en orto, meta y para sin protonar
-0,1
0
0,1
C2 C3 C4 C5 C6 H2 H3 H4 H5 N C2 C3 C4 C5 C6 H1 H3 H4 H5 N C2 C3 C4 C5 C6 H1 H2 H4 H5 N
var sigma
var pi
var total
∆N GF=(F)
en orto, meta y para sin protonar
-0,6
-0,5
-0,4
-0,3
-0,2
-0,1
0
0,1
C2 C4 C6 H3 H5 C3 C5 H1 H4 N C2 C4 C6 H2 H5
var sigma
var pi
var total
∆N GF=(CN)
en orto, meta y para sin protonar
-0,2
-0,1
0
0,1
C2 C4 C6 H3 H5 C3 C5 H1 H4 N C2 C4 C6 H2 H5
var sigma
var pi
var total
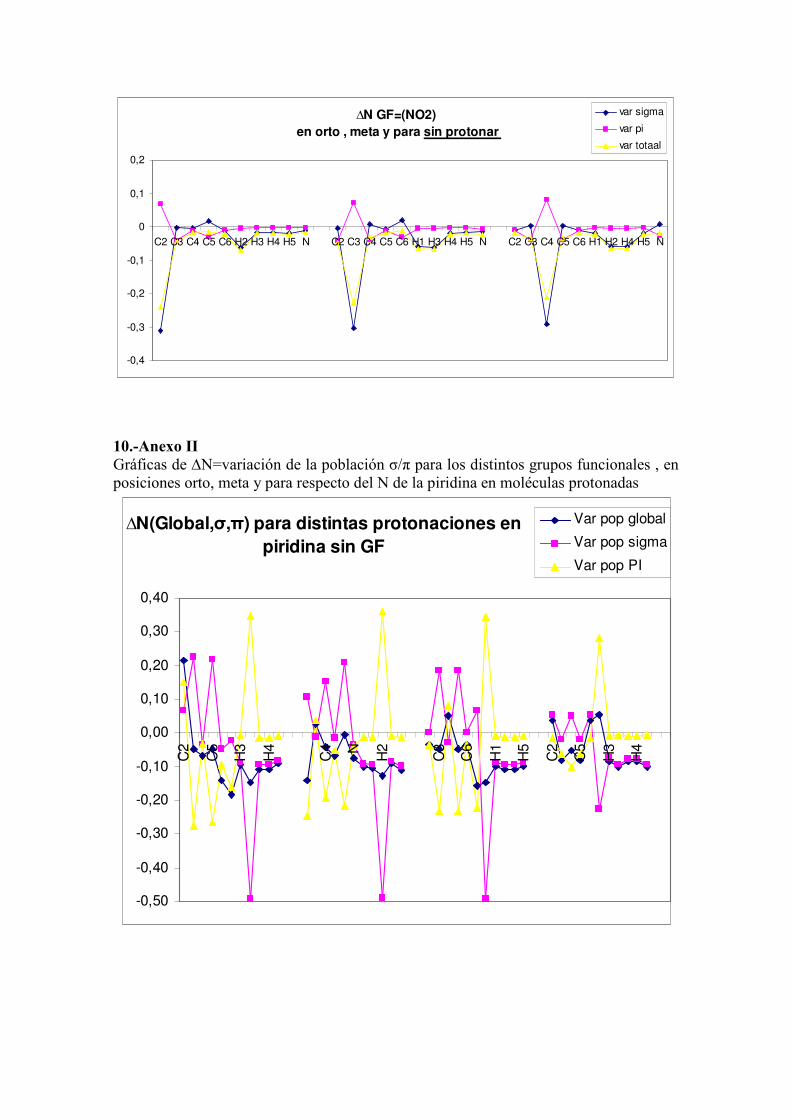
∆N GF=(NO2)
en orto , meta y para sin protonar
-0,4
-0,3
-0,2
-0,1
0
0,1
0,2
C2 C3 C4 C5 C6 H2 H3 H4 H5 N C2 C3 C4 C5 C6 H1 H3 H4 H5 N C2 C3 C4 C5 C6 H1 H2 H4 H5 N
var sigma
var pi
var totaal
10.-Anexo II Gráficas de ∆N=variación de la población σ/π para los distintos grupos funcionales , en posiciones orto, meta y para respecto del N de la piridina en moléculas protonadas
∆N(Global,σ,π) para distintas protonaciones en
piridina sin GF
-0,50
-0,40
-0,30
-0,20
-0,10
0,00
0,10
0,20
0,30
0,40
C2
C5
H3
H4
C4 N H2
C3
C6
H1
H5
C2
C5
H3
H4
Var pop global
Var pop sigma
Var pop PI

∆N(Global,σ,π) para distintas protonaciones en
piridina GF= O- (orto)
-0,8
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C6 H5 C5 H4 C4 H3xO
11 C3 H2 N C2 C6 H5
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= O- (meta)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C6 H5 C5 H4 C4 H3xO
11 C3 H1 N C2 C6 H5
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= O- (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2
C4
C6
H2
H5
xO11 C3
C5
H1
H4 N C2
C4
C6
H2
H5
xO11
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CN (orto)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C6 H5 C4 H3xC
11 C2 C6 H5 C4 H3xC
11 C2 C6 H5
var sigma
var pi
var total

∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CN (meta)
-0,6
-0,4
-0,2
0
0,2
0,4
C2
C6
H5
C4
H3
xC11 C2
C6
H5
C4
H3
xC11 C2
C6
H5
Var sigma
Var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CN (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C5 H2 N C3 C6 H4xC
11 C4 H1 H5xN
12
var sigma
var pi
var total

∆N(Global,σ,π) para distintas protonaciones en
piridina GF= NO2 (orto)
-1
-0,8
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
0,8
1
C2
C6
H5
xO13 C3
H2 N C4
H3
xN11 C5
H4
xO12 C2
C6
H5
xO13
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= NO2 (meta)
-1
-0,8
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
0,8
1
C2
C6
H5
xO13 C3
H1 N C4
H3
xN11 C5
H4
xO12 C2
C6
H5
xO13
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= NO2 (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C5 H2 NxO
13 C2 C5 H2 NxO
13 C2 C5 H2 NxO
13
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CH3 (orto)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 H2xC
11 C6 N C5 H5xH
14 C4 H4xH
13 C3 H3xH
12
var sigma
var pi
var total

∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CH3(meta)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2
C6
H5
xH13 C2
C6
H5
xH13 C2
C6
H5
xH13 C2
C6
H5
xH13 C2
C6
H5
xH13
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= CH3 (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C5 H2 NxH
13 C4 H1 H5xH
12 C3 C6 H4xC
11xH
14
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= F (orto)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C6 H5 C5 H4 C4 H3xF
11 C3 H2 N C2 C6 H5
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= F (meta)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2
C6
H5
C5
H4
C4
H3
xF11 C3
H1 N C2
C6
H5
var sigma
var pi
var total
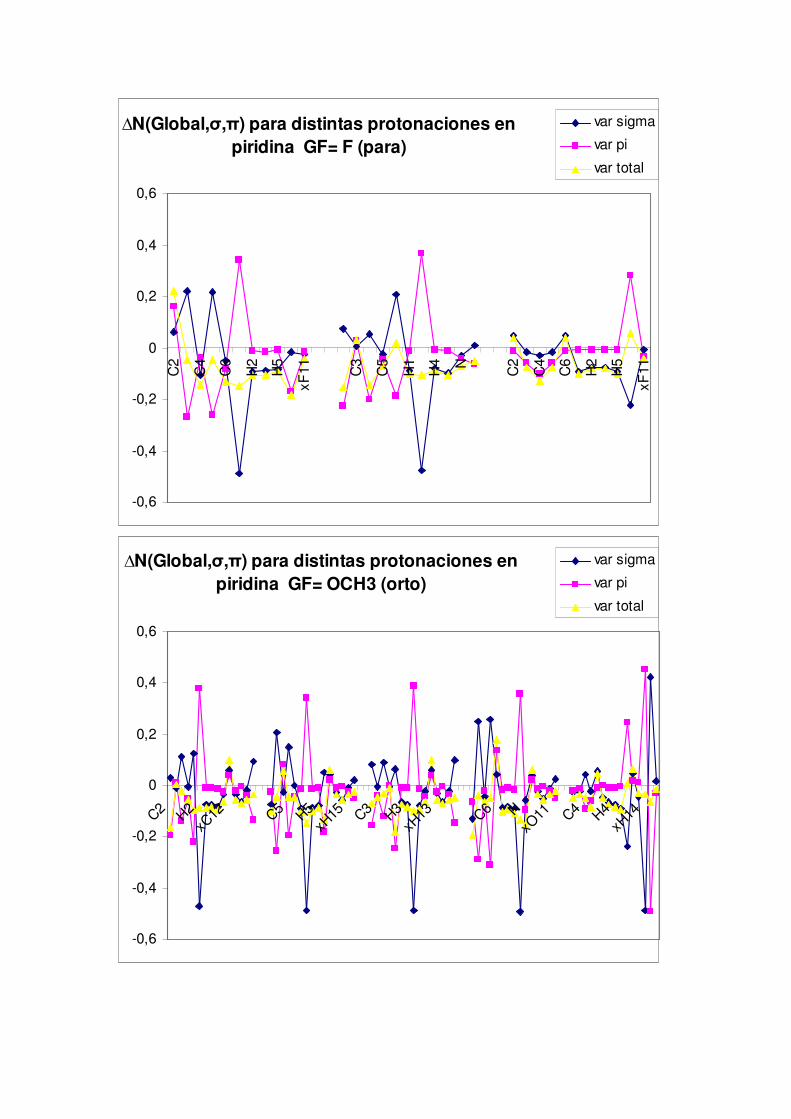
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= F (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6C
2
C4
C6
H2
H5
xF11 C3
C5
H1
H4 N C2
C4
C6
H2
H5
xF11
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= OCH3 (orto)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 H2xC
12 C5 H5xH
15 C3 H3xH
13 C6 NxO
11 C4 H4xH
14
var sigma
var pi
var total
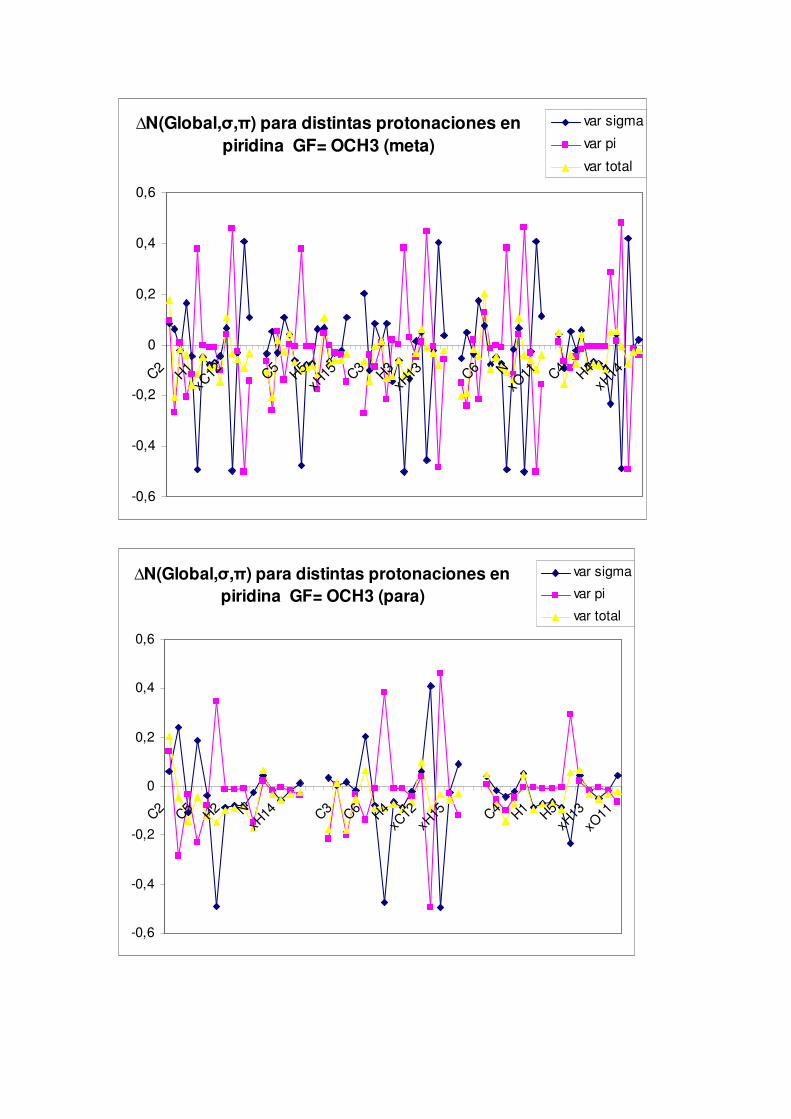
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= OCH3 (meta)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 H1xC
12 C5 H5xH
15 C3 H3xH
13 C6 NxO
11 C4 H4xH
14
var sigma
var pi
var total
∆N(Global,σ,π) para distintas protonaciones en
piridina GF= OCH3 (para)
-0,6
-0,4
-0,2
0
0,2
0,4
0,6
C2 C5 H2 NxH
14 C3 C6 H4xC
12xH
15 C4 H1 H5xH
13xO
11
var sigma
var pi
var total

Anexo II Variación de la población de los carbonos y del nitrógeno de la piridina estando el grupo funcional en orto respecto a la piridina sin sustituyente. Notación 02=O-, 03=CN, 04=NO2, 05=CH3, 06=F y 07=OCH3.Todo en a.u. orto C2 C3 C4
sigma pi total sigma pi total sigma pi total
02o -0,5900 -0,1337 -0,7237 -0,0319 0,0870 0,0551 -0,0111 0,0522 0,0412
07o -0,5139 -0,0312 -0,5452 -0,0542 0,0321 -0,0221 -0,0035 -0,0025 -0,0059
05o 0,0007 0,0050 0,0057 -0,0137 0,0157 0,0020 0,0022 -0,0006 0,0017
06o -0,5477 0,0005 -0,5472 -0,0606 0,0190 -0,0416 -0,0014 -0,0106 -0,0118
03o -0,1339 0,0747 -0,0592 -0,0044 -0,0244 -0,0288 -0,0041 -0,0098 -0,0138
04o -0,3101 0,0682 -0,2419 -0,0021 -0,0421 -0,0442 -0,0054 -0,0122 -0,0175
C5 C6 N
sigma pi total sigma pi total sigma pi total
02o -0,0840 0,1636 0,0796 -0,0176 0,0226 0,0050 -0,0763 0,0912 0,0150
07o -0,0331 0,0359 0,0028 0,0026 0,0071 0,0096 -0,0420 0,0793 0,0375
05o -0,0090 0,0120 0,0030 -0,0023 -0,0024 -0,0047 -0,0069 0,0192 0,0124
06o -0,0206 0,0143 -0,0063 -0,0003 -0,0013 -0,0016 -0,0274 0,0433 0,0160
03o 0,0126 -0,0233 -0,0107 -0,0073 -0,0139 -0,0213 -0,0328 -0,0130 -0,0458
04o 0,0160 -0,0293 -0,0133 -0,0114 -0,0099 -0,0214 -0,0132 -0,0029 -0,0160 Variación de la población de los carbonos y del nitrógeno de la piridina estando el grupo funcional en meta respecto de la piridina sin sustituyente. Notación 02=O-, 03=CN, 04=NO2, 05=CH3, 06=F y 07=OCH3. Todo en a.u. meta C2 C3 C4
sigma pi total sigma pi total sigma pi total
02m -0,0267 0,0829 0,0562 -0,6598 -0,1919 -0,8517 -0,0368 0,1207 0,084
07m -0,0528 0,0665 0,0137 -0,4859 -0,0282 -0,5141 -0,0446 0,0342 -0,0102
05m -0,0120 0,0173 0,0053 -0,0117 -0,0049 -0,0166 -0,0089 0,0175 0,0087
06m -0,0552 0,0196 -0,0356 -0,4983 0,0139 -0,4844 -0,0465 0,0228 -0,0236
03m -0,0172 -0,0269 -0,0441 -0,1371 0,06 -0,0771 -0,0059 -0,0202 -0,026
04m -0,0049 -0,0425 -0,0474 -0,3031 0,0731 -0,23 0,0062 -0,0388 -0,0324
C5 C6 N
sigma pi total sigma pi total sigma pi total
02m 0,0008 0,0384 0,0392 -0,0789 0,1657 0,0868 0,0349 0,0428 0,0778
07m 0,0007 -0,0073 -0,0066 -0,0345 0,0274 -0,0071 -0,0031 0,0022 -0,0007
05m 0,0023 -0,0009 0,0014 -0,0118 0,0091 -0,0028 0,0057 0,0023 0,0081
06m 0,0032 0,004 0,0072 -0,0197 0,0143 -0,0054 -0,0086 -0,0043 -0,0128
03m -0,0037 -0,0112 -0,0149 0,0116 -0,0221 -0,0105 -0,0047 -0,0055 -0,0101
04m -0,0081 -0,0116 -0,0197 0,0194 -0,0305 -0,0111 -0,0149 -0,0081 -0,0229

Variación de la población de los carbonos y del nitrógeno de la piridina estando el grupo funcional en para respecto de la piridina sin sustituyente. Notación 02=O-, 03=CN, 04=NO2, 05=CH3, 06=F y 07=OCH3. Todo en a.u. para C2 C3 C4
sigma pi total sigma pi total sigma pi total
02p 0,0127 0,0581 0,0708 -0,0233 0,1054 0,0821 -0,6849 -0,1762 -0,8611
07p 0,0022 0,0022 0,0044 -0,0465 0,0371 -0,0094 -0,4964 -0,0226 -0,5189
05p -0,0003 0,0006 0,0002 -0,0111 0,0195 0,0084 -0,0119 -0,0031 -0,0149
06p -0,008 -0,0087 -0,0166 -0,0473 0,0232 -0,0241 -0,5025 0,0195 -0,483
03p -0,0054 -0,0099 -0,0153 -0,0061 -0,0204 -0,0265 -0,1419 0,0662 -0,0756
04p -0,0101 -0,0102 -0,0204 0,0032 -0,0374 -0,0342 -0,2924 0,0814 -0,2109
C5 C6 N
sigma pi total sigma pi total sigma pi total
02p -0,0232 0,1055 0,0823 0,0127 0,0579 0,0706 -0,0813 0,1342 0,053
07p -0,0498 0,0675 0,0177 -0,0137 -0,0076 -0,0213 -0,0268 0,0312 0,0045
05p -0,0107 0,0206 0,0099 -0,0003 0,0006 0,0003 -0,0063 0,0111 0,0049
06p -0,0478 0,0232 -0,0247 -0,008 -0,0087 -0,0167 -0,0185 0,0148 -0,0036
03p -0,0059 -0,0204 -0,0263 -0,0057 -0,01 -0,0157 0,0018 -0,0196 -0,0176
04p 0,003 -0,0374 -0,0343 -0,0101 -0,0103 -0,0204 0,0065 -0,026 -0,0194





























