Embed Size (px)
Citation preview

Linköping Studies in Science and Technology
Dissertation No. 1436
Design and Synthesis of Inhibitors Targeting
BACE-1, an Aspartic Protease Involved in the
Pathogenesis of Alzheimer’s Disease
Veronica Sandgren
Department of Physics, Chemistry and Biology
Linköping University, Sweden
Linköping 2012

© Copyright 2012 Veronica Sandgren, unless otherwise noted
Veronica Sandgren
Design and Synthesis of Inhibitors Targeting BACE-1, an Aspartic Protease
Involved in the Pathogenesis of Alzheimer’s Disease
ISBN: 978-91-7519-933-7
ISSN: 0345-7524
Linköping Studies in Science and Technology, Dissertation No. 1436
Electronic publication: http://www.ep.liu.se
Printed in Sweden by LiU-Tryck, 2012.

"Anyone who has never made a mistake has never tried anything new."
Albert Einstein (1879-1955)


v
Abstract
Alzheimer’s disease (AD) is the most common form of dementia, occurring in
an estimated 24 million people worldwide. Accumulation of amyloid-β peptides
leads to development of plaques in the brain, which eventually stimulates
hyperphosphorylation of tau proteins leading to tangles. This is believed to play
a crucial role in the pathology of AD. The amyloid-β peptides are formed when
the amyloid precursor protein (APP) is cleaved first by the human aspartic
protease BACE-1 and then by the protease γ-secretase. BACE-1 catalyzes the
rate-limiting step in this sequence, and hence it has emerged as an important
therapeutic drug target.
The research reported in this thesis is focused on the design and synthesis of
BACE-1 inhibitors, where the synthetic work involves development of both
acyclic and cyclic inhibitors. Initially, a series of linear inhibitors incorporating
substituted cyclopentanes in the P2 position were synthesized and evaluated in
an attempt to find a replacement for the widely used isophthalamide moiety, and
this endeavor generated an inhibitor with activity in the nanomolar range. In the
second study, a series of hydroxyethylene-based inhibitors with extended P1
substituents was synthesized and evaluated, which resulted in several truncated
inhibitors also with activities in the nanomolar range. The third investigation
targeted a series of P1-P3-linked hydroxyethylamine-based macrocyclic
inhibitors and provided several highly potent compounds, however it did not
deliver high cell permeability inhibitors. In addition, two inhibitors were co-
crystallized with BACE-1 to provide X-ray crystal structures, which enabled
analysis of the binding properties of these inhibitors. In the final study, the
P2/P3 macrocyclic amide moiety and the P1-P3 ether oxygen bridge from the
previous work were replaced with a keto functionality and a carbon, respectively,
in an attempt to improve the permeability properties whilst maintaining the
beneficial potencies of this class of macrocyclic inhibitors. The compounds
synthesized did indeed display enhanced cell permeability properties, but this
approach resulted in decreased potency.
In short, this thesis presents several novel BACE-1 inhibitors, discusses the
synthetic strategies, and reports biological data on the target compounds.


vii
Populärvetenskaplig sammanfattning
Alzheimers sjukdom är den vanligaste demenssjukdomen och över 24 miljoner
individer världen över anses vara drabbade. Sjukdomen drabbar främst personer
över 65 år och hög ålder anses vara en stor riskfaktor. Gradvis nedbrytning av
hjärnvävnaden hos den drabbade ger upphov till en rad olika symptom varav
minnesstörningar är ett vanligt första sådant. Allteftersom sjukdomen fortskrider
framträder andra symptom såsom förvirring, humörsvängningar,
språksvårigheter och försämrade kroppsliga funktioner. De behandlingar som
finns tillgängliga idag kan lindra symptomen och bromsa sjukdomsförloppet
något, men de stoppar inte sjukdomens progress. Med en åldrande befolkning
finns det ett stort behov av att finna ett läkemedel som kan bromsa
sjukdomsförloppet mer effektivt och i bästa fall bota Alzheimers sjukdom. Den
bakomliggande orsaken till sjukdomen är inte helt kartlagd, men förekomst av så
kallade plack i hjärnan är ett diagnostiskt kännetecken. Plack består av olösliga
peptider (korta proteiner), vilka klibbar ihop sig och ger upphov till stora
ansamlingar. Dessa klibbar sig ofta fast på hjärnans nervändar, vilket stör
kommunikationen i hjärnan och ger med tiden upphov till celldöd. Peptiderna
bildas då ett större protein klyvs av två olika proteaser. Ett proteas är ett enzym
som katalyserar (snabbar på) klyvningen av proteiner. En vanlig strategi inom
läkemedelsforskningen är att utveckla ett läkemedel som hämmar, dvs.
blockerar ett sådant enzym vars funktion ger upphov till en sjukdom. En
inriktning inom Alzheimersforskningen är att försöka hämma enzymet (BACE-1)
som klyver det initiala stora proteinet. Detta görs genom att försöka identifiera
en kemisk förening (inhibitor/hämmare) som binder till enzymets så kallade
aktiva säte vilket är den yta på enzymet där proteinet binder in innan det klyvs.
Om en inhibitor hämmar BACE-1 skulle detta förhindra klyvningen av proteinet
och detta skulle i sin tur minska uppkomsten av de peptider som ger upphov till
placken i hjärnan och förhoppningsvis fördröja eller stoppa sjukdomsförloppet
hos Alzheimerssjuka.
Arbetet i denna avhandling bygger på att försöka designa och syntetisera
inhibitorer som hämmar BACE-1-proteaset. Det finns många svårigheter att
överkomma när man försöker designa en inhibitor. Först måste man ta reda på
vilka delar i en molekyl som är fördelaktiga för inbindning till det intressanta
enzymet. Detta har vi undersökt i arbete I och II. Föreningen ska också vara

selektiv mot andra enzymer, så att den inte bromsar någon annan livsviktig
funktion i kroppen. Inhibitorn får heller inte vara för proteinlik eftersom det då
finns stor risk att andra enzymer i bl.a. mag-tarmkanalen hinner klyva
molekylen innan den når enzymet man vill hämma. I arbete III har vi
syntetiserat hämmare med minskad proteinkaraktär. Vi fann flera inhibitorer
som effektivt hämmade BACE-1-enzymet. Tyvärr fann vi också att dessa
föreningar hade låg membranpermeabilitet, vilket leder in på en fjärde svårighet.
När man försöker hämna ett enzym som finns i hjärnan (som vid Alzheimers
sjukdom) måste molekylen ta sig igenom den så kallade blod-hjärnbarriären.
Detta är ett svårgenomträngligt membran mellan blodbanan och hjärnvävnaden
som skyddar hjärnan från främmande och eventuellt skadliga substanser. I denna
barriär sitter också proteiner som kan ”slänga ut” substanser som lyckats ta sig
igenom membranet. Dessa två faktorer försvårar designen av BACE-1
inhibitorer ytterligare. I arbete IV har vi försökt att modifiera och förbättra
hämmarnas egenskaper från arbete III för att öka deras möjlighet att ta sig
igenom blod-hjärnbarriären och undvika de proteiner som vill transportera ut
främmande substanser från hjärnan. Vi lyckades öka permeabiliteten hos
inhibitorerna, men tyvärr hade dessa hämmare då en minskad effekt mot BACE-
1. Avslutningsvis så visar denna avhandling hur svårt det är att designa
hämmare mot en sjukdom som Alzheimers, där molekylen inte bara ska vara
aktiv utan dessutom måste inneha ett flertal andra viktiga egenskaper som
behövs för att det ska kunna bli ett säkert och effektivt läkemedel.

ix
List of papers
This thesis is based on the following papers, which are referred to in the text by
their Roman numerals (I–IV):
I Sandgren, V., Kvarnström, I., Dahlgren, A. Exploration of the active site of BACE-1: Design and synthesis of
inhibitors incorporating substituted cyclopentanes in the P2 position Submitted
II Sandgren, V., Bäck, M., Kvarnström, I., Dahlgren, A. Design and synthesis of hydroxyethylene-based BACE-1 inhibitors
incorporating extended P1 substituents Submitted
III Sandgren, V., Agback, T., Johansson, P. O., Lindberg, J., Kvarnström,
I., Samuelsson, B., Belda, O., Dahlgren, A.
Highly potent macrocyclic BACE-1 inhibitors incorporating a
hydroxyethylamine core: Design, synthesis and X-ray crystal structures
of enzyme inhibitor complexes Submitted
IV Sandgren, V., Belda, O., Johansson, P. O., Kvarnström, I., Dahlgren,
A., Samuelsson, B. Design and synthesis of novel macrocyclic BACE-1 inhibitors Manuscript


xi
Abbreviations
ACE angiotensin-converting enzyme
AD Alzheimer’s disease
APP amyloid precursor protein
Asp aspartic acid
Aβ amyloid-β protein
BACE-1 β-site APP cleaving enzyme-1
BBB blood-brain barrier
BnBr benzyl bromide
Boc tert-butyloxycarbonyl
Boc2O di-tert-butyl dicarbonate
Cath D cathepsin D
CM cross-metathesis
CNS central nervous system
Cy cyclohexyl
DCE 1,2-dichloroethane
DCM dichloromethane
DDQ 2,3-dichloro-5,6-dicyanobenzoquinone
DIAD diisopropyl azodicarboxylate
DIPEA N,N-diisopropylethylamine
DMAP 4-dimethylaminopyridine
DMF N,N-dimethylformamide
DMSO dimethyl sulfoxide
DPP IV dipeptidyl peptidase IV
DPPA diphenylphosphoryl azide
EDC N-(3-dimethylaminopropyl)-N -ethylcarbodiimide
Gln glutamine
Gly glycine
HATU O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate
HCTU O-(6-chlorobenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate
HCV hepatitis C virus
HE hydroxyethylene

HEA hydroxyethylamine
HIV human immunodeficiency virus
HOBt 1-hydroxybenzotriazole
IC50 inhibitor concentration causing a 50% decrease in the
enzyme activity
Ile isoleucine
LDA lithium diisopropylamide
MeI methyl iodide
Mes 2,4,6-trimethylphenyl
MsCl methanesulfonyl chloride
NBS N-bromosuccinimide
NMDA N-methyl-D-aspartate
NMR nuclear magnetic resonance
NOESY nuclear overhauser effect spectroscopy
papp apparent permeability coefficient
PCy3 tricyclohexylphosphine
PDC pyridinium dichromate
PEPPSI pyridine, enhanced, precatalyst, preparation, stabilization,
and initiation
PPh3 triphenylphosphine
PS presenilin
p-TsCl para-toluenesulfonyl chloride
RCAM ring-closing alkyne metathesis
RCM ring-closing metathesis
ROM ring-opening metathesis
S3sp S3 subpocket
TBAB tetrabutylammonium bromide
TBTA tert-butyl-2,2,2-trichloroacetimidate
Tf2O triflic anhydride
TFA trifluoroacetic acid
THF tetrahydrofuran
Tyr tyrosine
Val valine

xiii
Contents
PREFACE............................................................................................................ 1
1. INTRODUCTION .......................................................................................... 3
1.1 PROTEASES.................................................................................................. 3 1.1.1 Aspartic Proteases............................................................................................. 4
1.2 PROTEASE INHIBITORS................................................................................. 5 1.2.1 Design Strategies............................................................................................... 5
1.3 ALZHEIMER’S DISEASE................................................................................ 6 1.3.1 APP and Aβ....................................................................................................... 7 1.3.2 The Amyloid Cascade Hypothesis.................................................................... 8 1.3.3 BACE-1............................................................................................................. 8 1.3.4 Therapeutic Strategies for AD .......................................................................... 9 1.3.5 BACE-1 Inhibitors .......................................................................................... 10
2. AIMS .............................................................................................................. 13
3. DESIGN AND SYNTHESIS OF ACYCLIC BACE-1 INHIBITORS
(PAPERS I AND II) .......................................................................................... 15
3.1 DESIGN OF BACE-1 INHIBITORS INCORPORATING SUBSTITUTED
CYCLOPENTANES IN THE P2 POSITION (PAPER I) ............................................ 15 3.1.1 Synthesis of Cyclopentane-Based Inhibitors .................................................. 16 3.1.2 Biological Data and Structure-Activity Relationships (SAR) of the
Cyclopentane-Containing Inhibitors ........................................................................ 21 3.1.3 Conclusions..................................................................................................... 25
3.2 DESIGN OF HYDROXYETHYLENE-BASED BACE-1 INHIBITORS
INCORPORATING EXTENDED P1 SUBSTITUENTS (PAPER II)............................. 25 3.2.1 Synthesis of Building Blocks.......................................................................... 26 3.2.2 Synthesis of the Final Products....................................................................... 29 3.2.3 Biological Data and SAR of the HE-Based Inhibitors.................................... 30 3.2.4 Conclusions..................................................................................................... 33
4. DESIGN AND SYNTHESIS OF CYCLIC BACE-1 INHIBITORS
(APPENDIX I, PAPERS III AND IV) ............................................................ 35
4.1 STRATEGY AND RETROSYNTHETIC ANALYSIS USED TO OBTAIN
MACROCYCLIC BACE-1 INHIBITORS (APPENDIX I) ........................................ 35 4.1.1 Synthesis of Alkyne Derivatives..................................................................... 36

4.1.2 Conclusions..................................................................................................... 38 4.2 DESIGN OF MACROCYCLIC BACE-1 INHIBITORS INCORPORATING A HEA
CORE (PAPER III)............................................................................................. 38 4.2.1 Ring-Closing Metathesis (RCM) .................................................................... 39 4.2.2 Synthesis of Building Blocks.......................................................................... 41 4.2.3 Synthesis of the Final Products....................................................................... 44 4.2.4 Biological Data and SAR of the Macrocyclic Inhibitors ................................ 46 4.2.5 X-ray Crystal Structure Results ...................................................................... 49 4.2.6 Conclusions..................................................................................................... 51
4.3 DESIGN OF MACROCYCLIC BACE-1 INHIBITORS WITH IMPROVED
PERMEABILITY (PAPER IV).............................................................................. 51 4.3.1 Synthesis of HEA Scaffolds............................................................................ 53 4.3.2 Synthesis of the Final Products....................................................................... 55 4.3.3 Biological Data on the Macrocyclic Inhibitors ............................................... 57 4.3.4 Conclusions..................................................................................................... 59
5. SUMMARY ................................................................................................... 61
APPENDIX........................................................................................................ 63
ACKNOWLEDGEMENTS ............................................................................. 67
REFERENCES.................................................................................................. 71

1
Preface
________________________________________________________________
This thesis is focused on the synthesis of compounds designed to inhibit the
aspartic protease BACE-1, which has emerged as an important drug target for
Alzheimer’s disease. The introduction is intended to provide the reader with a
short background on the enzymes known as proteases, to explain why some of
these are interesting in the context of drug development, and to outline the
aspects that must be considered when attempting to design a protease inhibitor.
This chapter also gives a general description of Alzheimer’s disease and
considers what is known about this condition and the progress research has
made towards finding a treatment. The subsequent chapters summarize and
discuss the work reported in the four appended papers (designated I–IV).
The work underlying this thesis was performed at the Department of Physics,
Chemistry and Biology at Linköping University, Linköping, Sweden, in
collaboration with the pharmaceutical company Medivir AB in Huddinge,
Sweden. During my doctoral studies in organic chemistry, I was also introduced
to the area of medicinal chemistry, and scientists in several different fields
helped me in various ways. Altogether, this has been a challenging and
educational journey that I have enjoyed very much.

2

3
1. Introduction
________________________________________________________________
1.1 Proteases
Proteases are enzymes that catalyze the hydrolytic cleavage of peptide bonds,
and they are found in both mammalian and non-mammalian species. The
proteases play important roles in the human body, where they are involved in
numerous processes including the synthesis and function of proteins. They
regulate physiological mechanisms such as digestion, fertilization,
immunological defense, and wound healing.1 Proteases can also cause
physiological changes that can lead to a number of pathologies, for instance
cardiovascular and inflammatory diseases, osteoporosis, and neurodegenerative
disorders.2 Accordingly, it can be of great importance to find inhibitors of those
proteases in the search for treatments of various diseases. In the 1960s,
Schechter and Berger3 introduced the nomenclature used to describe the
interactions between an enzyme and its substrate/inhibitor (Figure 1). The active
site of an enzyme is divided into subsites that are designated S or S´ depending
on which side of the scissile bond they are located; the non-prime subsites are on
the left side and the prime subsites on the right. The different parts of the
substrate are designated P or P´ and have the same numbering as the subsites
they occupy.

4
Figure 1. Standard nomenclature for the enzyme-substrate complex described by Schechter and Berger.3
There are currently 570 human proteases listed in the human Degradome
database,4 and these are classified according to their catalytic mechanism as
being aspartic, metallo, cysteine, serine, or threonine proteases.5 Each of these
five classes is further divided into subclasses based on the structural
characteristics of the enzymes. The present research focused on the first class of
these enzymes, namely, the aspartic proteases.
1.1.1 Aspartic Proteases
To date, 21 human aspartic proteases have been identified, and they constitute
the smallest of the five classes of proteases.6 Despite being few in number, the
aspartic proteases include several important drug targets such as β-secretase,
renin, and HIV-1 protease. Typically, the members of this group bind 6–10
peptide residues in their active site, and these oligopeptides are cleaved by
hydrolysis involving a catalytic dyad consisting of two aspartic acids.7 Several
aspartic proteases also have one or more flaps that close down on the substrate
to complete the active site. The most widely accepted mechanism for aspartic
proteases is a general acid-base mechanism in which an activated water
molecule acts as a nucleophile on the scissile amide bond (Figure 2). The water
molecule is activated by a deprotonated aspartic acid and then attacks the
scissile amide bond. A second aspartic acid donates a proton to the amide
nitrogen and thereby generates a zwitterionic tetrahedral intermediate, which in
turn collapses into the cleaved residues.1,8

5
Figure 2. The general acid-base mechanism of aspartic proteases.
1.2 Protease Inhibitors
As mentioned above, inhibition of proteases can be of interest for the treatment
of various diseases, including hypertension, coagulation disorders, diabetes,
parasitic and viral infections, and neurodegenerative disorders.1 Drugs that are
commercially available today target a number of different proteases, for
example, angiotensin-converting enzyme (ACE; regulates blood pressure),
thrombin (regulates blood clotting), HIV-1, HCV NS3, and dipeptidyl peptidase
IV (DPP IV; controls blood glucose levels). There are also numerous drug
candidates undergoing clinical or preclinical studies for possible use in treatment
of conditions such as Alzheimer’s disease, HCV infection, and diabetes.6 To
regulate a specific protease, an inhibitor has to compete with the natural
substrate, and therefore it must have specific properties that allow it to interact
with and retard or prevent the activity of the target protease.
1.2.1 Design Strategies
Traditionally, protease inhibitors have been developed through screening of
natural products or large libraries of compounds. If a peptide-like lead
compound is found, in most cases it must be modified, for example, by
truncation to obtain a shorter peptidomimetic and/or by replacing the scissile
bond with a non-cleavable isostere. Such optimization of the structure was
previously achieved through trial and error. However, this procedure has been
facilitated by the introduction of computer-assisted structure-based drug design,
and thus important information can now be acquired through computer modeling,
NMR spectroscopy, and X-ray crystallography.1,9
There are many aspects that have to be taken into consideration when designing
a protease inhibitor. To become potential drug candidates, the compounds must

6
have appropriate pharmacodynamics and pharmacokinetics properties.1 They
should be potent, but they must also show selectivity towards other proteases in
order to reduce the risk of side effects. Furthermore, the molecules should be
minimally peptidic in character, because, within the human body, peptides often
exhibit low stability, low bioavailability, and generally poor pharmacokinetic
profiles. It is also preferable that drugs are orally bioavailable and have a
clearance rate that allows convenient administration once or twice a day. In this
regard, Lipinski’s rule of five is often mentioned and can be used as a guideline
to achieve an orally active compound.10 Other properties discussed in relation to
oral bioavailability are the number of rotatable bonds and the polar surface
area,11 and further aspects of importance are low toxicity and a high therapeutic
index.12 When targeting a CNS disorder (e.g., Alzheimer’s disease), additional
issues have to be considered, such as the necessity of crossing the blood-brain
barrier (BBB). To be able to penetrate the BBB, an inhibitor should have low
molecular weight (< 400 Da), low polarity, and intermediate lipophilicity, or
alternatively it can enter the brain through some kind of active transport.13
Moreover, the molecule should not be a substrate for efflux transporters such as
P-glycoproteins.14 Thus it is clear that developing a protease inhibitor, especially
one that is intended to be active within the CNS, is a challenging and time-
consuming task.
1.3 Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder that was first
described by Alois Alzheimer in 1906. It is the most common form of dementia,
and the major risk factor is aging, as illustrated by a doubling of the incidence
every fifth year in people between the ages of 65 and 85 years.15 AD is
characterized by a decline in cognitive functions. The disease progresses slowly,
but once a diagnosis is made, death occurs on average nine years later.16
Extracellular plaques and intracellular tangles are two pathological features
observed in the brain of an AD patient. Plaques consist mainly of amyloid-β (Aβ)
peptides, whereas tangles are made up of hyperphosphorylated tau protein.17
There are two classes of this disease: the rare form designated early-onset AD
(EOAD) and the main form called late-onset AD (LOAD). EOAD generally
arises before the age of 65,18 and it has a strong genetic correlation. Genetic
analysis has identified several mutations that have been shown to increase levels
of the presumably most dangerous form of Aβ (designated Aβ42), and therefore

7
it is assumed that this polypeptide plays a role in the progression of AD.19,20
These findings support what is referred to as the amyloid hypothesis, which is
discussed in section 1.3.2. LOAD, also called sporadic AD, usually has its onset
at the age of 65 or older and shows less pronounced genetic inheritance.
Although EOAD and LOAD differ initially, they have comparable final
pathology and are therefore considered to be analogous diseases that might be
treated in similar ways.18 At present, no treatments are available that target the
underlying pathology of AD. Indeed, the two AD therapies that are now in use
merely ameliorate the symptoms of the disease; these treatments consist of
acetylcholinesterase inhibitors and/or memantine.18,21 Acetylcholinesterase
inhibitors prolong the life of acetylcholine in the synaptic cleft and thereby
improve cognitive function.22 Memantine is an N-methyl-D-aspartate (NMDA)
antagonist that prevents overstimulation of the NMDA receptor.
Overstimulation can contribute to high levels of glutamate in the brain, which
can cause calcium overload in the cells. This excess of calcium damages and
eventually kills the nerve cells, and is assumed to play a role in
neurodegenerative disorders such as AD.23 Sadly, these standard treatments do
not affect the underlying pathogenesis of AD, and thus there is an enormous
medical need for new therapies.
1.3.1 APP and Aβ
Amyloid precursor protein (APP) is a transmembrane protein that is found in
tissues throughout the body but is expressed at the highest levels in the CNS.24
The physiological function of APP is not yet fully understood,18 although it is
known that this protein is processed along two different pathways (Figure 3). In
the major pathway, APP is first cleaved by the enzyme α-secretase to release the
soluble fragment α-sAPP into the extracellular space. The remaining membrane-
bound C83 fragment is then further processed by γ-secretase to liberate a p3
peptide, which is considered to be non-amyloidogenic. The minor pathway gives
rise to Aβ. In this route, β-secretase cleaves APP to give the fragments β-sAPP
and C99. Thereafter, γ-secretase cleaves the C99 peptide at several positions,
which generates different Aβ peptides16,25 that range in size from 37 to 43
residues,26 with the major species being Aβ40 and Aβ42. Aβ40 makes up 80–
90% of all Aβ peptides, whereas Aβ42 constitutes only 5–10%. Aggregation of
the more hydrophobic Aβ42 leads to formation of oligomers, which eventually

8
results in development of plaques.18,27 Recent research suggests that it may be
the oligomers, rather than the plaques, that cause the disease.28
Figure 3. Schematic overview of the proteolytic processing of APP.
1.3.2 The Amyloid Cascade Hypothesis
As mentioned above, several gene mutations that are associated with EOAD
have been shown to increase the production of Aβ42. These mutations are found
in the genes coding for APP, presenilin-1 (PS1), and presenilin-2 (PS2),19,20,29
and both these presenilins are components of the γ-secretase complex.30 The
augmented production of Aβ42 is believed to play an early and crucial role in
the progression of AD. According to the amyloid cascade hypothesis, an
imbalance between Aβ production and Aβ clearance in the brain initiates a
neurotoxic cascade.31-33 This imbalance leads to accumulation and aggregation
of Aβ peptides and ultimately gives rise to amyloid plaques and neorofibrillary
tangles, which results in neuronal cell death and dementia. It should be
mentioned that even though this hypothesis is supported by both genetic and
pathological studies,18,32 it is not universally accepted.
1.3.3 BACE-1
In 1999, five different research groups34-38 identified the enzyme β-secretase or
BACE-1 (also called Asp2 or memapsin 2) as a transmembrane protein that
belongs to the class of aspartic proteases. Like other members of this class of

9
proteases, BACE-1 has an active site that contains two aspartic acids (Asp32
and Asp228) and a flap that closes down on the substrate. The active site
consists of a long cleft with eight main subsites spanning from S4 to S4´.39 It has
also been shown that the active site of BACE-1 is more open and less
hydrophobic compared to the active sites in other human aspartic proteases.40
The expression of BACE-1 is highest in the CNS, although lower levels are
found in other tissues as well.18 The homologue BACE-2 is also a
transmembrane protein,41 but it is only expressed at very low levels in the brain
and is not thought to be involved in Aβ production.42
1.3.4 Therapeutic Strategies for AD
As already mentioned, there are currently no drugs that actually target the
underlying pathology of AD. However, in theory, there are several possible
targets, which can be divided into two groups of general disease-modifying
strategies: anti-amyloid approaches and neuroprotective/neurorestorative
approaches.43 The former group includes methods such as vaccination and use of
secretase inhibitors and anti-fibrillization and anti-aggregation agents; example
of neuroprotective/neurorestorative approaches are treatments using antioxidants
and anti-inflammatory agents, and tau-related therapies.44
In anti-amyloid therapy, the two main strategies have been to reduce the
production or increase the clearance of Aβ42, with the former being considered
to be the most direct approach.31 This leads to three possible strategies:
stimulation of α-secretase or inhibition of either BACE-1 or γ-secretase.
Inasmuch as α-secretase normally processes most of the APP, it is plausible that
stimulation of α-secretase will increase the risk of side effects, because such
activation requires changes in the metabolism of all α-secretase substrates,
including APP. Consequently, most resources have been focused on finding a
secretase inhibitor. It has been shown that γ-secretase has several substrates
other than APP,45 including the Notch receptors, which are important for cell-
cell communication and processes such as cell development.46 This raises
concern about an increased risk of side effects, although some γ-secretase
inhibitors have entered clinical trials.47 Based on current knowledge, it appears
that BACE-1 has significantly fewer substrates compared to γ-secretase.31 Also,
in experiments using BACE-1 knockout mice,48-50 no Aβ production or side

10
effects on health or fertility were observed, and the animals showed only minor
behavioral changes. These findings, together with the knowledge that BACE-1
catalyzes the first step of Aβ production, renders BACE-1 an attractive
therapeutic drug target.
1.3.5 BACE-1 Inhibitors
A common strategy in the design of enzyme inhibitors is to replace the scissile
amide bond with a non-cleavable transition-state isostere. This approach has
been used to design inhibitors against other aspartic proteases such as HIV
protease and renin,1,51 and it has also been applied in the search for BACE-1
inhibitors. Widely used scaffolds include the statine-based, norstatine,
hydroxyethylene (HE), and hydroxyethylamine (HEA) isosteres (Figure 4),
which differ from each other by the functionality alpha and beta to the hydroxyl
group.52 Note that the HEA isostere requires inversion of configuration of the
hydroxyl group to achieve the best interaction with BACE-1.53,54
Figure 4. Examples of isosteres used in the design of BACE-1 inhibitors. The earliest published BACE-1 inhibitors were substrate analogues based on the
Swedish mutant APP, which differs from the natural APP in the P1 and P2
position.55 In the design of the peptidomimetic inhibitor P10-P4´StatVal, the
scissile bond was replaced with a non-cleavable statine residue and Asp was
replaced with Val in the P1´ position.34,56 Shortly thereafter, Tang and
coworkers40 published inhibitor A (OM99-2, Figure 5) in complex with BACE-1,
and this inhibitor incorporated a HE isostere. The X-ray structure of A
complexed with BACE-1 gave useful information, for instance, showing that
there was no specific interaction between the inhibitor and the S3´-S4´ part of
BACE-1. These observations were used in further optimizations that resulted in
inhibitor B.57 Peptidic inhibitor C, which was synthesized by Kimura et al.,58
incorporated a norstatine motif and exhibited good enzyme and cellular activity,

11
but, most interestingly, it led to reduction of Aβ in both wild-type and transgenic
mice after direct injection into the hippocampus.59 These peptidomimetic
inhibitors provided critical biological information about BACE-1, although it
was necessary to design smaller, non-peptidic inhibitors to produce viable drug
candidates.
Figure 5. Examples of BACE-1 inhibitors reported in the literature. Many research groups have replaced the peptide backbone with a substituted
isophthalamide moiety in an attempt to generate more drug-like inhibitors.
Merck research laboratories indentified the HEA inhibitor D, which showed
good cellular activity and selectivity against cathepsin D and renin.53 Another
HEA inhibitor (GSK188909, E) synthesized by Beswick et al.60 also exhibited
selectivity over other aspartic proteases, but, most significantly, it was the first
BACE-1 inhibitor reported to be orally active in transgenic mice.61 Other

12
researchers62 described the HE transition-state mimic F, which contained the
same isophthalamide as inhibitor D; F offered good enzymatic and cellular
inhibitory potency, and it was also efficacious in vivo upon intraperitoneal
injection in transgenic (Tg2576) mice. The macrocyclic inhibitor G reported by
Merck was the result of further optimization of inhibitor D;63 this macrocycle
displayed increased permeability and improved cellular potency compared to D,
and it also possessed good in vivo properties when tested in a murine model.
Compound CTS-21166 (structure not yet disclosed) was the first BACE-1
peptidomimetic inhibitor to enter phase I clinical trials,64 with several other
inhibitors in preclinical stage.6 In November 2011, Merck announced that they
plan to initiate a phase II clinical trial in 2012 for their BACE-1 inhibitor MK-
8931. Therefore, clinical efficacy data are still awaiting to be publicly disclosed.

13
2. Aims
________________________________________________________________
The general aim of the present work was to design and synthesize novel
compounds, and to evaluate the inhibitory effect of these molecules on the
human aspartic protease BACE-1, which is an attractive therapeutic target in
Alzheimer’s disease.
The specific objectives of the research were as follows:
• To synthesize a series of novel compounds to determine whether the
widely used isophthalamide P2 moiety could be replaced with a
substituted cyclopentane.
• To synthesize a series of hydroxyethylene-based inhibitors with
extended P1 substituents in order to explore possible interactions with
the S1-S3 pocket.
• To reduce the peptidic character of the molecules, which was done by
synthesizing a series of P1-P3-linked hydroxyethylamine-based
macrocyclic inhibitors.

14

15
3. Design and Synthesis of Acyclic BACE-1
Inhibitors (Papers I and II)
________________________________________________________________
3.1 Design of BACE-1 Inhibitors Incorporating Substituted
Cyclopentanes in the P2 Position (Paper I)
Previous studies conducted by our research group provided inhibitors containing
a novel HE motif with a methoxy group in the P1´ position. This series
identified highly potent BACE-1 inhibitors that also showed selectivity over
cathepsin D, as exemplified by inhibitor I (Figure 6).65
Figure 6. Inhibitor I previously reported by our research group. The substituted isophthalamide II (Figure 7) has been widely used as a P2
moiety in the search for BACE-1 inhibitors.66 However, many of these inhibitors
have proven to be substrates for P-gp efflux, and thus they cannot reach a
significant concentration in the CNS.67 Therefore, we were interested in finding
an alternative P2 scaffold. The substituted cyclopentane III (Figure 7) had
previously been used in our laboratories,68 and modeling and overlays had

16
suggested that it could replace the isophthalamide in the P2 position. Due to the
new stereocenters in the cyclopentane moiety, we studied diverse configurations.
In addition, we explored different substituents in various positions, and we also
evaluated two macrocycles containing the cyclopentane scaffold.
Figure 7. The widely used isophthalamide (II) and the substituted cyclopentane moiety (III) employed in our series of target molecules.
3.1.1 Synthesis of Cyclopentane-Based Inhibitors
Figure 8 outlines the general structure of the cyclopentane scaffold used in the
different BACE-1 inhibitors, together with the different R substituents that were R
O
HN
O
HNR1 R2
NH2
NH2
R1-NH2
NH2
F
NH2
NH2
NH2
NH2
E1
A1
C1
B1
D1
F1
H1
MeO
NH2
NH2
Cl
NH2
NH2
I1
K1
NH2
G1
J1
L1
N1
H2NHN
OH
H2NHN
NH
O
OH OMe
O
O
F F
H2N
OH
NH
O
O
HN
O
OH
O
P1
Q1
R1
S1
R2-NH2
H2NHN
NH
O
OH OMe
O
O
R
H
H2NHN
NH
O
OH OMe
O
O
F
F
T1
O1
NS
O O
NS
Bn
O O
M1
Figure 8. A summary of the different R-substituents connected or coupled to the cyclopentane scaffold.

17
connected or coupled to it. Amines A1–L1 were all commercially available,
whereas P1–S1 were synthesized as described in the literature.53,65,69 Amine T1
was prepared as described in Paper I.
Synthesis of the target compound trans-3 is outlined in Scheme 1. Commercially
available trans-DL-1,2-cyclopentanedicarboxylic acid was mono-benzylated
using 4-dimethylaminopyridine (DMAP), benzyl alcohol, and N-(3-
dimethylaminopropyl)-N -ethylcarbodiimide (EDC) in DCM to give a racemic
mixture of compound 1 (59%).70 The afforded carboxylic acid was coupled with
amine A1 by use of O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium
hexafluorophosphate (HATU) and N,N-diisopropylethylamine (DIPEA) in DMF
to yield 2 (96%) as a diastereomeric mixture (1:1). Attempts to separate this
mixture were unsuccessful. Hydrogenolysis with Pd-C in ethanol provided the
corresponding carboxylic acid, which was subsequently coupled with amine Q1
to furnish the target compound trans-3 in 76% yield over two steps. The
diastereomeric mixture (1:1) of trans-3 could not be separated, and therefore
measurements of the enzyme activity against BACE-1 were performed with this
mixture. Target compound trans-4 (Table 1) was synthesized in similar yields
using amine B1 instead of A1 (Figure 8). Compounds cis-5 and cis-6 (Table 1)
were synthesized from commercially available (±)-cis-cyclopentane-1,2-
dicarboxylic acid in the same manner as trans-3 and trans-4 and in similar
yields. All final products were tested as diastereomeric mixtures.
O
HO
O
OH
O
HO
O
O
O
HN
O
O
HN
HN
OHN O
O
OH OMe
O
NH
O
F F
a b
c, d
trans-DL 1 2
trans-3
96%
76%
59%
Scheme 1. Synthesis of target compound trans-3. Reagents and conditions: (a) DMAP, BnOH, EDC, DCM, 0 °C to rt; (b) A1, DIPEA, HATU, DMF, rt; (c) Pd-C, H2, EtOH, rt; (d) Q1, DIPEA, HATU, DMF, rt. Synthesis of the cyclopentane scaffold with a sulfonamide functionality in the 4-
position is outlined in Scheme 2. The bicyclic lactone 7 was available from an
earlier project in our laboratories68 and was here protected with tert-butyl-2,2,2-

18
trichloroacetimidate (TBTA) to furnish the tert-butyl ester 8 in 76% yield.71
Initially, di-tert-butyl dicarbonate (Boc2O) and DMAP were used in the same
way as in our previous project,68 but, due to low yields, the new strategy was
developed. The lactone was opened using K2CO3 in MeOH, which gave the
cyclopentane 9 in 99% yield.72 Next, Mitsunobu conditions using
triphenylphosphine (PPh3), diisopropyl azodicarboxylate (DIAD), and
diphenylphosphoryl azide (DPPA) in THF provided the azide 10 (93%) with
inversion of configuration.73 Reduction of the azide with PPh3 and H2O in
MeOH gave the amine 11 in 96% yield. The amine was then reacted with either
methanesulfonyl chloride (MsCl) or phenylmethanesulfonyl chloride in pyridine
and DCM, and then subjected to N-alkylation with methyl iodide (MeI) and
NaH in DMF to furnish compounds 12 and 13 (71% and 56%, respectively, over
two steps).
Scheme 2. Synthesis of cyclopentane scaffolds 12 and 13. Reagents and conditions: (a) TBTA, DCM, rt; (b) K2CO3, MeOH, 0 °C; (c) PPh3, DIAD, DPPA, THF, 0 °C; (d) PPh3, MeOH, H2O, rt; (e) MsCl or phenylmethanesulfonyl chloride, pyridine, DCM, 0 °C to rt; (f) NaH, MeI, DMF, rt. An additional bicyclic lactone (Scheme 3) was prepared to synthesize the
enantiomer of compound 12. Compound (+)-14 was prepared according to a
procedure described in the literature,74 and the ketone was selectively reduced to
the alcohol 15 (97%) using sodium borohydride (NaBH4) in MeOH.75 Thereafter,
hydrolysis of the diester with NaOH in MeOH gave a dicarboxylic acid, which
was treated with acetic anhydride in pyridine76 to provide the bicyclic lactone 16
in 94% yield over two steps. Compound 16 was subsequently used to prepare
compound 19 (Scheme 5) according to the synthetic route of 12 (Scheme 2).

19
Scheme 3. Reagents and conditions: (a) NaBH4, MeOH, 0 °C; (b) NaOH (aq), MeOH, rt; (c) Ac2O, pyridine, rt. Two slightly different methods were employed to synthesize the different linear
target compounds. General method I (described in Scheme 4) was used to obtain
the final products 18, 22, 25, and 29–43 (Tables 1–4). The tert-butyl ester
functionality in compound 12 was removed by use of triethylsilane (Et3SiH) and
trifluoroacetic acid (TFA) in DCM,77 and the resulting acid was coupled with
amine A1 using triethylamine (TEA), EDC, and 1-hydroxybenzotriazole (HOBt)
in DMF to give compound 17 (92% over two steps). The subsequent methyl
ester hydrolysis was achieved with LiOH (aq.) in dioxane, and the
corresponding acid was coupled with amine Q1 using HATU and DIPEA in
DMF to furnish target compound 18 in 55% yield over two steps. The other
products synthesized according to this method were prepared by varying the
amines coupled to the cyclopentane scaffold. Target 42 (Table 4) was obtained
by performing an additional methyl ester hydrolysis.
NS
O O
O
O
O
O
NS
O O
O
O
HN
O
HN
HN
OO
OH OMe
O
NH
O
NSOO
OHN
F F
a, b c, d
12 17
18
92% 55%
Scheme 4. General method I. Reagents and conditions: (a) Et3SiH, TFA, DCM, rt; (b) A1, TEA, HOBt, EDC, DMF, 0 °C; (c) LiOH (aq), dioxane, rt; (d) Q1, DIPEA, HATU, DMF, rt. Synthesis of target molecule 21 was achieved using general method II as
outlined in Scheme 5, and the same route was employed to prepare target
compounds 26–28 (Table 1). First, the methyl ester functionality in compound

20
19 was hydrolyzed, and the carboxylic acid obtained was subjected to a peptide
coupling with amine A1 using TEA, EDC, and HOBt to afford compound 20 in
73% yield over two steps. Subsequently, the tert-butyl ester functionality was
removed, and lastly the corresponding carboxylic acid was coupled with amine
Q1 using HATU and DIPEA in DMF to generate target product 21 (46% over
two steps).
Scheme 5. General method II. Reagents and conditions: (a) LiOH (aq), MeOH, rt; (b) A1, TEA, HOBt, EDC, DMF, 0 °C to rt; (c) Et3SiH, TFA, DCM, rt; (d) Q1, DIPEA, HATU, DMF, rt. Preparation of the two macrocyclic molecules 23 and 24 is described in Scheme
6. Compound 22 was generated according to general method I (Scheme 4) using
amines L1 and T1. Ring-closing metathesis (RCM, described in section 4.2.1)
using Hoveyda-Grubbs 2nd generation catalyst78 in refluxing DCE gave a
diastereomeric mixture (2:3) of the 16-membered macrocycle 23 (14%). The
double bond in the macrocyclic ring was then hydrogenated to afford the
saturated compound 24 in 82% yield.
Scheme 6. Synthesis of the macrocycles 23 and 24. Reagents and conditions: (a) Hoveyda-Grubbs 2nd generation catalyst, DCE (dry), N2, reflux; (b) Pd/C, EtOH (95%), H2, rt.

21
3.1.2 Biological Data and Structure-Activity Relationships (SAR) of
the Cyclopentane-Containing Inhibitors
All inhibitors were screened against BACE-1 (IC50), and the structures are
presented in Tables 1–4, along with biological data. The different stereocenters
of the cyclopentane scaffold were evaluated by synthesizing the final products
listed in Table 1. The two trans-isomers 3 and 4 showed better activity against
BACE-1 than the cis-isomers 5 and 6. These results indicate that the trans-
isomer produces the best fit to the active site of BACE-1, an observation that is
supported by modeling and overlay. Therefore, we kept the trans-configuration
when synthesizing other target compounds.
Replacing the isophthalamide moiety (II ) in inhibitor I with a trisubstituted
cyclopentane scaffold rendered inhibitor 25, which was similar to the two trans-
isomers 3 and 4 with respect to activity against BACE-1. Earlier studies have
shown a strong stereochemical preference for the (R)-configuration at the methyl
group in the P3 position,62,79 and thus we synthesized inhibitor 18 to ascertain
whether this was also true for our series. Interestingly, 18 with (S)-
stereochemistry at the methyl group showed increased activity compared to 25,
with an IC50 value of 0.26 µM, and it also exhibited selectivity over cathepsin D
and renin. Computer modeling and overlays were used to compare compounds
18 and 25, but it was difficult to explain why the (S)-configuration was favored.
Molecular modeling can be helpful when trying to explain the positions of
substituents and their interactions with an active site, although that approach is
rather hypothetical. Therefore, we sent compound 18 for co-crystallizations with
BACE-1 to determine what kind of information X-ray analysis could provide,
but unfortunately these co-crystallization attempts were not successful. Further
examinations were performed to explore the different configurations of the
cyclopentane substituents in positions 1, 2, and 4, which rendered compounds 21
and 26–30. All these inhibitors showed decreased activity, demonstrating that
the initial configurations at these positions were more favorable.

22
Table 1. Biological data for target compounds 3–6, 18, 21, and 25–31
Cpd. R IC 50 (µµµµM)
BACE-1 Cpd. R
IC 50 (µµµµM)
BACE-1
trans-3
4.5 26
3.9
trans-4
4.5 27
1.6
cis-5
> 10 28
> 10
cis-6
> 10 29
> 10
18a
0.26 30
> 10
21
> 10 31
3.5
25
4.9
aInhibition of cathepsin D (Ki > 5 µM) and renin (Ki > 5 µM).

23
Exploration of the P3 position while keeping the rest of inhibitor 18 constant led
to the synthesis of final products 32–40 (Table 2). These inhibitors were all
significantly less potent than compound 18. Surprisingly, compound 37 with a
fluorine in the para position of the P3 site proved to be completely inactive,
which is in contrast to previous results showing an increase in activity when this
approach was used for other BACE-1 inhibitors.53,79 This finding suggests that
inhibitor 18 can not achieve optimal interaction with the S3 position of the
active site.
Table 2. Biological data for target compounds 32–40
HN
HN
NH
O
OH OMe
O
O
F F
O
NSOO
OHN
R
Cpd. R IC 50 (µµµµM)
BACE-1 Cpd. R
IC 50 (µµµµM)
BACE-1
32
5.1 37
> 10
33
3.4 38
5.3
34 > 10 39
6.6
35
> 10 40
> 10
36
10
Macrocycles 23 and 24 were synthesized to investigate the additional free space
that had been revealed in the BACE-1 active site by X-ray crystallographic data
in a study performed by Stachel et al.63 These 16-membered macrocycles proved
to be inactive, and it is plausible that this ring-size was not ideal for this purpose.
24
Table 3. Biological data for target compounds 22–24
Compound R IC 50 (µµµµM) BACE-1
22
> 10
23
> 10
24
> 10
In a final attempt to evaluate the cyclopentane scaffold as an alternative P2
scaffold, we used the cyclopentane together with other isosteres and side chains
that had previously been used to produce active BACE-1 inhibitors.53,65,69 This
furnished compounds 41–43 (Table 4), all of which proved to be inactive against
BACE-1.
Table 4. Biological data for target compounds 41–43
Compound Structure IC 50 (µµµµM)
BACE-1
41
> 10
42
> 10

25
43
> 10
3.1.3 Conclusions
Compounds incorporating a substituted cyclopentane P2 moiety were
synthesized and evaluated against BACE-1. Also, different stereoisomers of the
cyclopentane part were studied and various substituents were evaluated. The
results showed that (S)-configuration of the methyl group in the
methylbenzylamine substituent in the P3 position was necessary for the activity
of this type of inhibitors. The most active inhibitor (product 18) had an IC50
value of 260 nM, and it showed selectivity against cathepsin D and renin.
Attempts were made to co-crystallize this compound with BACE-1 but were not
successful.
3.2 Design of Hydroxyethylene-Based BACE-1 Inhibitors
Incorporating Extended P1 Substituents (Paper II)
Earlier studies of aspartic proteases such as the malaria plasmepsins80 and
human renin81 have shown that the S1 pocket of these enzymes can
accommodate large P1 residues, many of which can extend into the S3 pocket.
Previous research in our laboratories explored the possibility of moving further
into the S1 pocket of BACE-1 by employing statine-based inhibitors with
phenyloxymethyl residues (IV) in the P1 position (Figure 9).69 Some of these
inhibitors displayed substantial activity against BACE-1 in the enzyme assay but
were unfortunately inactive in the cell-based assay. Accordingly, we wanted to
investigate the possibility of improving the cell permeability of our inhibitors
and also to study potential interactions with the S1-S3 binding pocket.
Carboxylic acid functionalities were found to be essential for high activity in the
statine series, which proved to be an unfavorable feature in relation to cell
permeability. Therefore, we abandoned the statine-based isostere in favor of a
HE central core that had previously provided successful results in our
laboratories.65 Another valuable quality of this HE isostere is that it contains one
less amide linkage than found in the statine core, which also decreases the
peptidic character of the inhibitors, an attribute that is generally viewed as

26
beneficial in terms of permeability. Various P1´ residues were incorporated into
the HE isostere (V) with the aim of improving the central core (Figure 9). The
purpose of including the X substituent on the P1 residue was to get the inhibitor
to protrude even deeper into the S1 pocket, and the Y substituent was
incorporated to extend into the S3 pocket.
Figure 9. Statine-based central cores (IV) that were employed in a previous study of potent BACE-1 inhibitors69 and a general illustration of the hydroxyethylene (HE) isostere (V) with different substituents used in the present work.
3.2.1 Synthesis of Building Blocks
A general illustration of the HE central core together with the different
substituents is presented in Figure 10. However, it is worth mentioning that most
of the target compounds were evaluated as free amines, i.e., without the R
carboxylic acid substituents. Amines A2–D2 were synthesized from
commercially available precursors, whereas phenols E2 and F2 were
commercially available. Sulfonamide K2 and L2 were generated according to
published procedures.53,82

27
Figure 10. A general illustration of the hydroxyethylene central core and the different R substituents that were used. The P1 residues G2 and H2 were obtained as their corresponding benzyl ethers
as described in Scheme 7. An electrophilic aromatic substitution was performed
on 3-benzyloxy-phenol (E2) using N-bromosuccinimide (NBS) in DCM at –15
°C to generate the aryl halide 44 in 71% yield.83 Next, compound 44 was
subjected to Mitsunobu conditions with 3-methoxy-1-propanol, PPh3, and DIAD
in THF to give compound 45 (90%). Microwave-assisted palladium coupling
conditions using 4-fluorophenylboronic acid or phenylboronic acid, K3PO4, and
PEPPSITM-IPr catalyst in EtOH, H2O, and DMF furnished compounds 46 and 47
in 70% and 81% yield, respectively. The benzyl ether of compound I2 was
synthesized from E2 by applying the same conditions utilized to prepare
compound 45, and compound J2 was obtained from commercially available 4-
bromophenol using the same Suzuki protocol employed to synthesize compound
46.

28
Scheme 7. Reagents and conditions: (a) NBS, DCM, –15 °C; (b) 3-methoxy-1-propanol, PPh3, DIAD, THF, 0 °C to rt; (c) 4-fluorophenylboronic acid or phenylboronic acid, K3PO4, PEPPSI™-IPr catalyst, DMF, EtOH, H2O, 125 °C (microwave heating). The synthesis of lactones 55a–d is depicted in Scheme 8. Compound 48 was
prepared from 1,2:5,6-di-O-isopropylidene-α-D-glucofuranose over five steps,
as described in the literature.84-87 Methylation of the hydroxyl group using MeI
and Ag2O in DMF furnished compound 49 in 89% yield. Hydrolysis under
acidic conditions gave the hemiacetal 50a (85%), which was subsequently
oxidized to the lactone 51a (77%) using pyridinium dichromate (PDC) in
DCM.65 Lactone 50b was synthesized according to a procedure reported in the
literature85 and was used as a precursor for synthesis of compounds 51b–d.
Compound 50b was alkylated at the C2 position with ethyl bromide, 2-
iodopropane, or benzyl bromide to furnish 51b–d in 32–63% yield. NOESY
spectra of the three products confirmed the R configuration at C2, where H2
correlates with H3β, and H3α correlates with H4 (Figure 11).
Figure 11. A general illustration of the NOE correlations observed in the NOESY spectra of compounds 51b–d. The benzyl groups were removed by catalytic hydrogenolysis to give the diols
52a–d (62–98%). These were converted into their corresponding tin acetals by
use of dibutyltin oxide (Bu2SnO) in refluxing toluene, and subsequent treatment
with tetrabutylammonium bromide (TBAB) and 4-methoxybenzyl bromide
furnished the primary alkylated compounds 53a–d in 70–92% yield over two

29
steps.88 A Mitsunobu protocol using PPh3, DIAD, and DPPA provided the
azides 54a–d (57–99%), with inversion of configuration at C5. Finally, an
oxidative removal of the p-methoxybenzyl group using 2,3-dichloro-5,6-
dicyanobenzoquinone (DDQ) in H2O and DCM gave compounds 55a–d (91–
100%).89
O
BnO
BnO
O
R
O
PMBO
HO
O
O
PMBO
O
R
N3O
HO
O
R
N3
R
O
BnO
BnO
O
OHO
BnO
BnO
O
OO
BnO
BnO
OH
O
48 49 (89%) 50a (85%)
51a R = OMe, 77%51b R = Et, 32%51c R = Bn, 63%51d R = iso-Pr, 41%
52a R = OMe, 83%52b R = Et, 98%52c R = Bn, 84%52d R = iso-Pr, 62%
54a R = OMe, 99%54b R = Et, 73%54c R = Bn, 74%54d R = iso-Pr, 57%
55a R = OMe, 97 %55b R = Et, 94%55c R = Bn, 91%55d R = iso-Pr, quant.
O
HO
HO
O
R
53a R = OMe, 90%53b R = Et, 70%53c R = Bn, 88%53d R = iso-Pr, 92%
a b
c
f, g
h
i
O
BnO
BnO
Ode
50b
Scheme 8. Compound 50a was used to synthesize 51a, and compound 50b was employed to prepare 51b–d. Reagents and conditions: (a) MeI, Ag2O, DMF; (b) H2SO4, dioxane; (c) PDC, 4 Å molecular sieves; (d) (i) LDA, tripyrrolidinophosphine oxide, EtBr or 2-iodopropane, THF (dry), –68 °C or (ii) LDA, BnBr, THF (dry), –78 °C; (e) (i) H2/Pd(OH)2-C, AcOH, EtOH or (ii) H2/Pd-C, EtOH; (f) Bu2SnO, toluene, reflux; (g) TBAB, 4-OMeBnBr, toluene, 90 °C; (h) PPh3,
DIAD, DPPA, THF (dry); (i) DDQ, H2O, DCM.
3.2.2 Synthesis of the Final Products
The synthetic route to target compounds 58a and 59a (shown in Scheme 9)
represents the general method used to obtain all HE-based inhibitors. Note that
most of the final products were evaluated as free amines, thus the final peptide
coupling was performed only for compounds 59a, 61, 62, 67, and 68 (Table 5).
Compound 46 was converted into the phenol G2 by hydrogenolysis using
H2/Pd-C in EtOH. Initially, we applied Mitsunobu conditions to react G2 with
compound 55a, but we encountered elimination problems with that approach
and hence a new strategy was applied. The alcohol 55a was first converted into
the corresponding triflate by use of triflic anhydride (Tf2O) and pyridine in

30
DCM and was then reacted with the phenol G2 in a substitution reaction using
Cs2CO3 in DCM containing 4 Å molecular sieves, which gave compound 56a in
47% yield over two steps. The elimination problems could not be fully avoided
with this new approach, but they were minimized by use of fresh Cs2CO3 in dry
DCM and by letting the reaction proceed for a short time at 40 °C under N2. The
lactone 56a was opened with amine C2 in DIPEA and DMF to furnish
compound 57a in 83% yield. Reduction of the azide using PPh3 and H2O in
MeOH gave the target compound 58a (84%). The amine was also coupled with
amine K2 using HATU and DIPEA, which provided the final target 59a in 61%
yield. The other target compounds were synthesized in similar yields.
Scheme 9. Reagents and conditions: (a) triflic anhydride, pyridine (dry), DCM (dry); (b), H2/Pd-C, EtOH; (c) Cs2CO3, 4 Å molecular sieves, DCM (dry), 40 °C; (d) C2, 2-hydroxypyridine, DIPEA, DMF, 75 °C; (e) PPh3, MeOH, H2O; (f) K2, DIPEA, HATU, DMF.
3.2.3 Biological Data and SAR of the HE-Based Inhib itors
All target compounds were screened against BACE-1 (IC50), and they are
summarized in Table 5 together with biological data. The inhibitors were
synthesized from compounds 55a–d according to Scheme 9 using appropriate R
substituents (shown in Figure 10). In addition, cell percent inhibition of BACE-1
at the concentration of 1 µM was determined for the four most potent inhibitors
(compounds 58c and 68–70).

31
Target compound 61 incorporated a m-benzyloxy P1 substituent, and 62
contained a p-methoxy group, and both these products showed modest activity
against BACE-1 (2.2 and 4.6 µM, respectively). Despite the somewhat limited
efficacy of these two compounds, the results were promising and suggested that
more advanced P1 substituents could be tolerated and potentially provide
inhibitors with increased activity. Further development led to inhibitor 63, which
lacked the P2 substituent and contained a p-chloro substituent in the P3´ part of
the molecule. Intriguingly, 63 showed activity similar to that of product 61 (2.7
vs. 2.2 µM). Compound 58a had an IC50 value of 1.2 µM, indicating that
isoleucine is preferable to valine in the P2´ position. The activity of this class of
inhibitors depended on interactions from both the para and meta groups in the
P1 position, as observed for products 65 and 66 (IC50 values > 10 µM) lacking
the para and the meta substituent, respectively. Unfortunately, introduction of
the P2 sulfonamide K2 into 58a and 64 yielded compounds that were highly
hydrophobic in character; this caused precipitation problems during the activity
measurements, and thus no inhibition data could be obtained for compounds 59a
and 67. Introducing the larger P2-P3 sulfonamide L2 provided inhibitor 68 with
an IC50 value of 69 nM. This result is interesting, since it may indicate that the
active site can accommodate large compounds containing both a P3 substituent
and a sizable P1 residue. Further exploration of the P1´ and P3´ positions was
done to determine whether a more drug-like compound could be found, with the
P2-P3 substituent excluded. Compound 58b with an ethyl group in the P1´
position proved to be equipotent with 58a. Also, product 58d with a P1´
isopropyl group showed activity similar to that of 58a and 58b. Introducing the
larger benzyl P1´ substituent gave the inhibitor 58c (IC50 = 470 nM), which was
found to be somewhat more active than inhibitor 58a. Final investigations were
performed to examine whether the amide-bond between the P2´ and P3´
positions contributes to biological activity, and this effort resulted in inhibitors
69 and 70 (300 and 540 nM, respectively). Interestingly, both these products
showed activities similar to those of compounds 58c and 58d, which implies that
the P2´-P3´ amide was not required for BACE-1 activity in our series of
inhibitors.

32
Table 5. Target compounds and inhibition data
Cpd. R R1 R1´ R´ IC 50 (µµµµM)
BACE-1
60 H2N
> 10
61
2.2
62
4.6
63 H2N
2.7
58a H2N
1.2
64 H2N
2.5
65 H2N
> 10
66 H2N
> 10
59a
NDa

33
67
NDa
68
0.069
58b H2N
1.1
58c H2N
0.47
58d H2N
0.81
69 H2N
0.30
70 H2N
0.54
aND = not determined The inhibitors 58c, 68, 69, and 70 displayed 19%, 3%, 14%, and 28% inhibition
of BACE-1, respectively, in a cell-based assay at a concentration of 1 µM. As
expected, decreased size and less peptidic character leads to increased inhibition.
3.2.4 Conclusions
In summary, a new series of HE-based BACE-1 inhibitors incorporating
extended P1 substituents was synthesized. Other positions in the inhibitors were
also varied and studied, and most of the final products were evaluated as free
amines. The truncated compounds 58c, 58d, 69, and 70 are interesting, because,
even though they lack a P2 substituent, they show promising activity against

34
BACE-1. Moreover, compounds 69 and 70 possess only one amide bond, which
makes them more drug-like compared to other inhibitors described in the
literature. Unfortunately, all attempts to co-crystallize inhibitor 58a with BACE-
1 failed. It is plausible that X-ray data could have produced valuable information
on how these inhibitors interact with the active site of BACE-1, which might
have provided new ideas for additional modifications.

35
4. Design and Synthesis of Cyclic BACE-1 Inhibitors
(Appendix I, Papers III and IV)
________________________________________________________________
4.1 Strategy and Retrosynthetic Analysis Used to Obtain
Macrocyclic BACE-1 Inhibitors (Appendix I)
Development of low molecular weight, brain-penetrating BACE-1 inhibitors has
proven to be a challenging task.66,90 Nevertheless, it has been shown that the
pharmacokinetics of standard linear inhibitors can be significantly improved by
cyclization,91 and this approach has indeed been applied in the search for AD
therapies. RCM has been widely used to obtain such macrocycles (RCM is
discussed further in section 4.2.1).63,92,93 Considering that RCM tends to give
mixtures of both geometrical isomers (E/Z) at the newly formed double bond,
we were interested in finding a method that would offer adequate regiochemistry
control. We also wanted to examine the possibility of evaluating alkyne
derivatives that could be used to prepare cycloalkenes and cycloalkanes.
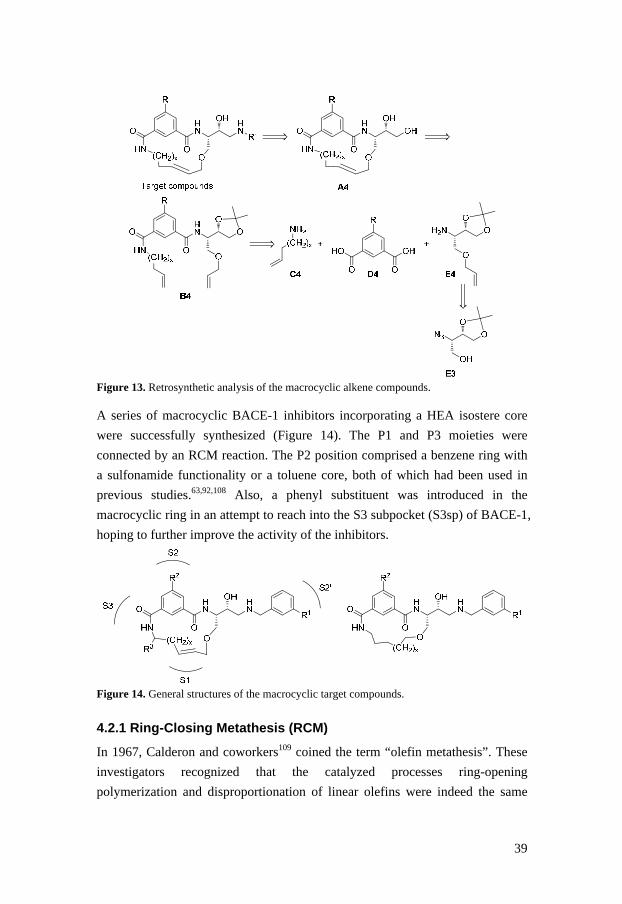
A retrosynthetic analysis was set up as illustrated in Figure 12. Tentatively, the
target alkyne compounds could be synthesized from the diol A3 using either of
two ways: by making an epoxide from the diol, which could be opened using
different amines; or by selectively tosylating the primary hydroxyl group and
then reacting it with appropriate amines. Hopefully, it would be possible to
prepare the alkyne macrocycle by applying ring-closing alkyne metathesis
(RCAM)94 to the acyclic diyne B3, which could be obtained by subjecting

36
amines C3 and D3 and commercially available 3-methoxycarbonyl-5-
methylbenzoic acid to standard peptide coupling procedures. The amine C3
could be synthesized from commercially available 3-pentyn-1-ol by a three step
protocol reported in the literature.95 Furthermore, the amine D3 could be
prepared by alkylation of E3 and reduction of the azide functionality. E3 could
be synthesized from commercially available (-)-diethyl D-tartrate in a four-step
procedure according to previously published reports.96-98
Figure 12. Retrosynthetic analysis of the macrocyclic alkyne compounds.
4.1.1 Synthesis of Alkyne Derivatives
The synthesized alcohol E396-98 was alkylated using NaH and 1-bromo-2-butyne
in DMF, which gave compound 71 in 74% yield (Scheme 10). Reduction of the
azide with 1,3-propanedithiol and TEA in MeOH generated the amine 72 in
68% yield.99 We initially tried to reduce the azide with PPh3 and H2O, but this
strategy provided no product.

37
Scheme 10. Synthesis of alkyne compound 72. Reagents and conditions: (a) NaH, 1-bromo-2-butyne, DMF, rt; (b) 1,3-propanedithiol, TEA, MeOH, rt. As described in Scheme 11, the carboxylic acid 73 was coupled with the
synthesized amine C395 by use of EDC, HOBt, and TEA in DMF to furnish the
alkyne derivative 74 in 32% yield. Treating compound 74 with LiOH in
dioxane/H2O afforded the corresponding carboxylic acid, which was
subsequently coupled with amine 72 using O-(6-chlorobenzotriazol-1-yl)-
N,N,N′,N′-tetramethyluronium hexafluorophosphate (HCTU) and DIPEA in
DMF to provide the dialkyne 75 in 79% yield over two steps. The crucial
RCAM reaction was performed in a glove box and was first tested using the
Schrock alkylidyne complex ((tBuO)3W≡CCMe3)100 in dry toluene101 under N2
atmosphere at 85 °C, but this approach was unsuccessful. Further attempts were
Scheme 11. Synthetic route for preparing compound 76. Reagents and conditions: (a) C3, EDC, HOBt, TEA, DMF, 0 °C; (b) LiOH (aq.), dioxane/H2O (2:1), rt; (c) 72, HCTU, DIPEA, DMF, rt; (d) (tBuO)3W≡CCMe3, toluene (dry) or chlorobenzene (dry), N2 or Ar, 80 °C or 85 °C, glove box. made using different conditions achieved by varying the solvent (THF94 and
chlorobenzene102) and also by replacing the N2 atmosphere with Ar, but still no
product was obtained. Some additional investigations were performed using the
reference substance 7794 as shown in Scheme 12. The alkyne macrocycle 78 was

38
produced in 57% yield when the reaction was performed in chlorobenzene under
Ar. Encouraged by this result, we repeated the reaction with compound 75 under
these conditions, but this merely resulted in decomposition of the dialkyne. It is
plausible that failure of this ring-closing alkyne metathesis was due to the
rigidity of the molecule.
Scheme 12. An overview of the different investigations performed using the RCAM reaction by varying the solvent. Reagents and conditions: (1) Schrock alkylidyne complex, toluene (dry), Ar, 80 °C; (2) Schrock alkylidyne complex, THF (dry), Ar, 80 °C; (3) Schrock alkylidyne complex, chlorobenzene, Ar, 80 °C.
4.1.2 Conclusions
The strategy we tested was to synthesize and evaluate different alkyne
macrocycles that could subsequently be used to prepare the corresponding
cycloalkenes and cycloalkanes. Unfortunately, our attempts to perform the
RCAM reactions were unsuccessful, and therefore we shifted our focus to
synthesizing alkene structures.
4.2 Design of Macrocyclic BACE-1 Inhibitors Incorpo rating a
HEA Core (Paper III)
A new retrosynthetic route was developed based on the knowledge gathered in
the alkyne project described above (Figure 13). In AD research, various
methodologies for synthesizing macrocyclic inhibitors have been used, such as
RCM,63,92,93,103,104 reductive amination,105 peptide coupling,63,106 and
macrolactonization.107 We employed an RCM protocol to generate the different
macrocyclic compounds designated A4. The diene B4 could be produced by
performing peptide couplings between different amines (C4, E4) and different
acids (D4), and the amine E4 could be synthesized from E3 used in the alkyne
project (see section 4.1).
39
Figure 13. Retrosynthetic analysis of the macrocyclic alkene compounds. A series of macrocyclic BACE-1 inhibitors incorporating a HEA isostere core
were successfully synthesized (Figure 14). The P1 and P3 moieties were
connected by an RCM reaction. The P2 position comprised a benzene ring with
a sulfonamide functionality or a toluene core, both of which had been used in
previous studies.63,92,108 Also, a phenyl substituent was introduced in the
macrocyclic ring in an attempt to reach into the S3 subpocket (S3sp) of BACE-1,
hoping to further improve the activity of the inhibitors.
Figure 14. General structures of the macrocyclic target compounds.
4.2.1 Ring-Closing Metathesis (RCM)
In 1967, Calderon and coworkers109 coined the term “olefin metathesis”. These
investigators recognized that the catalyzed processes ring-opening
polymerization and disproportionation of linear olefins were indeed the same

40
reaction.110,111 RCM, ring-opening metathesis (ROM), and cross-metathesis (CM)
are all examples of olefin metathesis reactions that provide a route to
unsaturated molecules. The underlying mechanism was correctly described in
1971 by Chauvin,112 and this had a marked impact on metathesis research,
because it provided important clues for developing catalysts. However, it was
not until the 1990s that reports on well-defined molybdenum and ruthenium
based catalysts started to appear. The molybdenum-based Schrock’s catalyst113
(Figure 15) was shown to be highly reactive, but it has disadvantages of being
sensitive to air and incompatible with a number of polar functional groups.
Grubbs and colleagues focused on ruthenium instead of molybdenum, which
significantly improved the tolerance to functional groups and led to the
discovery of the 1st generation Grubbs’ catalyst (Figure 15).114 Despite the
greater stability and capacity to tolerate functional groups, the Ru-based
catalysts were significantly less active than Schrock’s Mo catalysts. However, in
the late 1990s, Grubbs and other researchers found that replacing one phosphine
with a N-heterocyclic carbene ligand in Grubbs’ first catalyst drastically
increased the activity, and this resulted in development of the 2nd generation
Grubbs’ catalyst (Figure 15).115 In 2000, Garber et al.78 described the Hoveyda-
Grubbs 2nd generation catalyst, which incorporated a styrenyl ether moiety and
was found to be particularly stable and reactive. Over the years, various
modified versions of this catalyst have been synthesized, one example of which
is the Zhan catalyst-1B.116
Mo
N Ph
iPr iPr
O
OF3CF3C
F3CCF3
Schrock's catalyst
Ru
PCy3
PCy3
Cl
Cl
Ph
Grubbs' catalyst,1st generation
Ru
PCy3
Cl
Cl
Ph
NMesMesN
Grubbs' catalyst,2nd generation
Ru
OCl
Cl
NMesMesN
Hoveyda-Grubbs catalyst,2nd generation
Ru
OCl
Cl
NMesMesN
S NMe2
O
O
Zhan catalyst-1B
Cy = cyclohexylMes = 2,4,6-trimethylphenyl
Figure 15. Example of olefin metathesis catalysts.

41
As a result of the advent of well-defined molybdenum and ruthenium catalysts
in the 1990s and 2000s, RCM has emerged as an efficient way to transform
acyclic dienes into unsaturated cyclic compounds. These catalysts should in fact
be referred to as initiators, because they must first be converted into the actual
catalytically active metal-carbene complex (Figure 16). The RCM mechanism
with Ru-based catalysts starts with dissociation of the ligand L (PCy3 or loss of
coordination of the isopropoxide moiety). Coordination of the intermediate VII
with a diolefin generates VIII , and a [2+2] addition takes place to form the
metallacyclobutane complex (IX). This intermediate decomposes into complex
X with the release of styrene. An intramolecular [2+2] cycloaddition occurs to
furnish XI , which decomposes into the methylidene intermediate catalyst XII
and the cyclized target compound. The very active compound XII reacts with
another diolefin, and the metallacyclobutane XIII that is generated decomposes,
which liberates ethene and creates an additional intermediate X. This step is
irreversible because ethene evaporates, and it is the driving force of the
metathesis reaction.
Figure 16. General mechanism of the ring-closing metathesis reaction with Ru-based catalysts.
4.2.2 Synthesis of Building Blocks
Synthesis of key building block 80, which was used to produce all the target
compounds, is outlined in Scheme 13. The alcohol E3 was allylated with allyl
bromide and Ag2O in toluene to give compound 79 in 78% yield. Attempts to
reduce the azide group in compound 79 were initially performed using PPh3 and

42
H2O in MeOH, but these experiments were all unsuccessful. Therefore, we
employed 1,3-propanedithiol and TEA in MeOH to provide the corresponding
amine 80 in 73% yield.
Scheme 13. Synthesis of compound 80, which was employed as a building block for synthesis of all the target compounds. Reagents and conditions: (a) allyl bromide, Ag2O, toluene, rt; (b) 1,3-propanedithiol, TEA, MeOH, rt. The building blocks (R)-86, (S)-86, (R)-87, and (S)-87 (Scheme 14) were
synthesized to prepare the target compounds incorporating a phenyl substituent.
The epoxide functionality in commercially available (R)-(+)-styrene oxide was
opened with lithium acetylide ethylenediamine complex in DMSO to furnish
compound (S)-81 in 95% yield.117 Mitsunobu conditions utilizing DPPA gave
the azide (R)-82 (97%) with inversion of configuration. The reduction of the
azide group using 1,3-propandithiol and TEA in MeOH proved to be slow, but,
after two days of stirring at room temperature, the corresponding amine (R)-83
could be collected in 92% yield. The enantiomer (S)-83 was synthesized in the
same manner from commercially available (S)-(-)-styrene oxide. Thereafter,
HCTU and DIPEA in DMF were used to couple the two amines (R)-83 and (S)-
83 with either the acid 73 (Scheme 11) or compound 92 (Scheme 16),
synthesized as previously reported,53 which furnished the amides (R)-84, (S)-84,
(R)-85, and (S)-85 in 73–89% yield. Finally, alkyne reduction by use of
Lindlar’s catalyst afforded the four building blocks (R)-86, (S)-86, (R)-87, and
(S)-87 (84–99%), and these were used to synthesize the final products 109, 110,
113, and 114.

43
R
O
O
HN
Oa
OHb
N3
cNH2
d
O
R
O
O
HN
e
O
* * * *
* *
(R)(S)
(S)-81 (95%)(R)-81 (quant.)
(R)-82 (97%)(S)-82 (76%)
(R)-83 (92%)(S)-83 (87%)
(R)-84 R = Me (89%)
(R)-85 R = N(Me)SO2Me (89%)
(S)-84 R = Me (73%)
(S)-85 R = N(Me)SO2Me (89%)
(R)-86 R = Me (99%)
(R)-87 R = N(Me)SO2Me (84%)
(S)-86 R = Me (99%)
(S)-87 R = N(Me)SO2Me (92%) Scheme 14. Synthesis of building blocks 86–87. Reagents and conditions: (a) lithium acetylide ethylenediamine complex, DMSO (dry), Ar, rt; (b) PPh3, DIAD, DPPA, THF, –12 °C to rt; (c) 1,3-propanedithiol, TEA, MeOH, rt; (d) compound 73 or 88, HCTU, DIPEA, DMF, rt; (e) Lindlar’s catalyst, quinoline, MeOH, H2, rt. Synthesis of amines 91a and 91b is described in Scheme 15. Compound 89a
was prepared from commercially available 3-tert-butylphenol according to a
two-step procedure reported in the literature.118 The 3-tert-butylphenol was
reacted with trifluoromethanesulfonic anhydride (Tf2O) in pyridine to generate
the triflate 88a in 89% yield. Negishi conditions utilizing zinc cyanide (Zn(CN)2)
and tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) in DMF at 85 °C
furnished compound 89a in 76% yield. Attempts to reduce the nitrile group in
89a were first made using lithium aluminium hydride (LiAlH4), but that
approach was not successful in our hands. Therefore, a new strategy was
developed in which sodium borohydride (NaBH4) and catalytic nickel chloride
(NiCl2·6H2O) in MeOH were used, and the generated amino group was
protected in situ with Boc2O to give compound 90a (64%).119 By trapping the
produced amine, the risk of dimerization between the amine and an intermediate
imine was limited. The Boc group in 90a was subsequently removed using
Et3SiH and TFA in DCM to give 3-tert-butylbenzylamine in quantitative yield.
In the same manner, 3-isopropylbenzylamine was synthesized from
commercially available 3-isopropylphenol. These two amines were used in the
final step in the synthesis of the target compounds 98, 103, 104, 106–110, and
112–114.

44
Scheme 15. Synthesis of amines 91a and 91b. Reagents and conditions: (a) Tf2O, pyridine, rt; (b) Zn(CN)2, Pd(PPh3)4, DMF, 80 °C, Ar; (c) Boc2O, NiCl2·6H2O, NaBH4, TEA, MeOH, 0 °C to rt; (d) Et3SiH, TFA, DCM, rt.
4.2.3 Synthesis of the Final Products
The target compound 98 was synthesized as described in Scheme 16. Compound
93a was produced in 83% yield by coupling compound 9253 with commercially
available 3-butenylamine hydrochloride utilizing EDC, HOBt, and TEA in DMF.
Methylation of the amide nitrogen in 93a using MeI and NaH in DMF gave
compound 93b (100%), which was used in the synthesis of target compound 104
(Table 6). The methyl ester functionality in compound 93a was hydrolyzed
(LiOH), and the corresponding carboxylic acid was coupled with amine 80
(HATU, DIPEA) to afford diene 94 in 90% yield over two steps. An RCM
reaction was preformed on 94 using Hoveyda-Grubbs 2nd generation catalyst in
DCM, but the corresponding macrocycle was produced in very low yield.
Therefore, we decided to remove the acetal in compound 94 before the RCM
step by using 70% AcOH (aq.), which resulted in the diol 95 in quantitative
yield. Next, we tested performing RCM reaction on 95 with Hoveyda-Grubbs
2nd generation catalyst78 in refluxing DCM, and this approach was successful
and furnished the desired 15-membered macrocycle 96 in 98% yield. The
cyclization resulted in exclusive formation of the trans isomer, as confirmed by 1H-NMR analysis. It is not known why use of the diol 95 instead of the
protected compound 94 in the RCM step gave a much higher yield, although it is
possible that the more flexible diol allowed the intramolecular RCM reaction to
proceed more freely. The primary hydroxyl group in 96 was selectively
tosylated using p-TsCl, Bu2SnO, and TEA in dry DCM to generate compound
97 in 64% yield.120 Finally, the tosylate 97 was reacted with 3-
isopropylbenzylamine in refluxing EtOH to generate target compound 98 in

45
37% yield. The final products 102–104 and 108–114 (Table 6) were synthesized
by the same route and with similar yields (benzylamine was used instead of 3-
isopropylbenzylamine in the final step for compounds 102 and 111, and 3-tert-
butylbenzylamine was used for compound 103). The 16-membered macrocycle
104 was synthesized using 4-pentenylamine hydrochloride instead of 3-
butenylamine hydrochloride in the peptide-coupling step. Attempts were also
made to synthesize the corresponding 14-membered macrocycle, but these RCM
reactions were not successful, perhaps due to the shorter olefin linker, which
possibly would result in a macrocycle with too much ring strain. The
macrocycle 108 was synthesized as a mixture that could be separated into the cis
isomer (regiochemistry at the macrocyclic double bond) and traces of a related
compound that could be tentatively identified as the trans isomer by NMR
analysis.
Scheme 16. Synthesis of target molecule 98. Reagents and conditions: (a) 3-butenylamine hydrochloride, TEA, HOBt, EDC, DMF, 0 °C to rt; (b) NaH, MeI, DMF, rt; (c) i) LiOH, H2O/dioxane 1:2, ii) compound 80, DIPEA, HATU, DMF, rt; (d) AcOH/H2O, 60 °C; (e) Hoveyda-Grubbs 2nd generation catalyst, DCM (dry), N2, reflux; (f) p-TsCl, Bu2SnO, TEA, DCM (dry), rt; (g) 3-isopropylbenzylamine, EtOH 99.5%, reflux.

46
The three saturated macrocyclic compounds 105–107 were synthesized from
compounds 100 and 101 (Scheme 17). The 16-membered macrocycle 99 was
synthesized in the same manner as compound 96, but using 4-pentenylamine
hydrochloride instead of 3-butenylamine hydrochloride in the peptide-coupling
step (Scheme 16). Hydrogenation of the double bond in compounds 96 and 99
was achieved in the presence of Pd-C in MeOH to furnish 100 and 101 in 95%
and 78% yield, respectively. To obtain the final products 105–107, the same
synthetic route was used as for compound 98, except that benzylamine was used
instead of 3-isopropylbenzylamine in the final step for compound 105.
Scheme 17. Synthesis of the saturated compounds 100 and 101 used to produce target compounds 105–107. Reagents and conditions: (a) Pd-C 10%, H2, MeOH, rt.
4.2.4 Biological Data and SAR of the Macrocyclic Inhibitors
Table 6 presents the structures of all the synthesized target compounds along
with biological data. All the macrocycles were 15-membered, except 104 and
107, which were 16-membered. The inhibitors were all collected and tested as
trans isomers, except for compound 108, which was tested as the cis isomer.
Encouraged by the biological results for compound 102 (IC50(enzyme) = 270 nM
and IC50(cell) = 560 nM), we decided that further investigations would be of
interest. Compounds 98 and 103 were synthesized to reach further into the S2´
pocket of BACE-1. These two products showed 20-fold more potent enzymatic
activity compared to compound 102, and 98 had a notable IC50 value of 2.3 nM
in the cell-based assay. To investigate the effect of ring-size on biological
activity, we synthesized the 16-membered macrocycle 104, and this resulted in
loss of both enzymatic and cell activities. The somewhat more flexible saturated
macrocycles 105–107 were prepared to elucidate the impact of the double bond
on potency. In general, compounds 105 and 106 showed decreased activity
compared to their unsaturated analogues. Interestingly, the 16-membered
macrocycle 107 displayed higher activity than compound 104, which might be

47
explained by the enhanced flexibility of the macrocycle. When a phenyl
substituent was introduced in the macrocyclic ring, the (R)-isomer 110 proved to
achieve the best interaction with BACE-1. The (R)-isomer showed slightly
improved enzymatic activity compared to inhibitor 98 (3.2 vs. 14 nM), and it
also had an impressive cell activity of 0.15 nM. In contrast, the (S)-isomer 109
exhibited a substantial drop in activity, indicating an unfavorable interaction
with the active site of BACE-1. Although some of the sulfonamide-based
inhibitors did display good potency, they all had poor cell-based (Caco-2)
permeability. Since other BACE-1 inhibitors with a sulfonamide group in the P2
position have been shown to be substrates for P-gp efflux, which results in low
permeability, we replaced the sulfonamide group with a less polar methyl
substituent, and this provided the inhibitors 111–114. Unfortunately, this
approach led to decreased potency and no improvement in permeability.
Table 6. BACE-1 inhibition data for compounds 98 and 102–114
Compound Structure IC 50 enzyme
(nM)
IC 50 cell
(nM)
102
270 560
98
14 2.3
103
N
HN
O
O
HN
S
HN
O
O O
OH
15 4.6
104a
180 73
105
5300 > 10000

48
106
16 49
107a
18 7.7
108
7.5 6.9
109
400 260
110
3.2 0.15
111
> 10000 > 10000
112
500 220
113
4900 2300
114
78 160
a16-membered macrocycle

49
4.2.5 X-ray Crystal Structure Results
Inhibitors 109 and 114 were co-crystallized with BACE-1 to obtain X-ray
crystal structures (PDB ids 4DPF and 4DPI, respectively; Figures 17 and 19).
The crystal structures showed that the general binding mode of the P1-P3
macrocyclic BACE-1 inhibitors was as anticipated for HEA-based
inhibitors.60,121-123 The hydroxyl and the protonated amino groups form hydrogen
bonds with the catalytic dyad (Asp32 and Asp228), and the carbonyl of Gly34
forms a hydrogen bond with the ionized amine. Moreover, the P2´ benzyl is
sandwiched between Tyr198 and the peptide backbone of the flap, and the
isopropyl extension is clamped in between Ile126 (S2´) and Val69 (flap).
Similar to P1-P3 groups of linear HEA analogues, the P1-P3 macrocycle is
coordinated between the flap and active site by a set of hydrogen bonds. One
additional hydrogen bond was observed between the ether oxygen of the
Figure 17. Compound 114 bound to the active site of BACE-1 (PDB id: 4DPI). macrocycle and the backbone carbonyl of the flap residue, Gln73 bridged by
structural water (Figure 18). Also, the P3-amide of compound 114 was observed
in two conformations that were staggered and equiplanar with respect to the P2-

50
phenyl plane. In solution, the low energy state of the P3-amide is in staggered
conformation and does not interact with the protein. However, the equiplanar
conformation is predominant in the X-ray structure. In this conformation, the P3
NH forms a hydrogen bond with the backbone CO of Gly230, and the amide CO
is in range for an electrostatic interaction to the side chain of Gln73 in the flap.
Figure 18. The hydrogen bond coordination of the macrocycle of compound 114 between residues of S1-S3 and the flap. The figure also shows the two conformational isomers of the compound (equiplanar, yellow; staggered, light blue) and the two accompanying rotamers of Thr232. The structural overlay of compounds 109 and 114 is illustrated in Figure 19,
which visualizes the preference for the (R)- over the (S)-isomer. In the case of
the (R)-isomer, the phenyl substituent protrudes into the amphiphilic S3sp
gaining hydrophobic interactions, whereas for the (S)-isomer the macrocycle and
phenyl group adopt an unfavorable binding conformation.

51
Figure 19. Structural overlay of compounds 109 (pink) and 114 (yellow) showing the preference of the (R)-isomer for S3sp interactions.
4.2.6 Conclusions
A series of macrocyclic BACE-1 inhibitors containing a HEA isostere scaffold
were synthesized and evaluated, and some of the inhibitors showed promising
low nanomolar BACE-1 activities. In general, the compounds displayed good
selectivity over cathepsin D, but, disappointingly, they suffered from poor cell-
based (Caco-2) permeability. X-ray crystallography studies demonstrated that
the macrocycle-attached phenyl substituent did not extend fully into the S3
subpocket of BACE-1, which indicates that further optimization is possible.
4.3 Design of Macrocyclic BACE-1 Inhibitors with Improved
Permeability (Paper IV)
As described above, we have previously synthesized several macrocyclic
BACE-1 inhibitors (e.g., compound XIV; Figure 20) and found that they
exhibited good enzymatic and cell activity, but, unfortunately, they also had low
permeability. Earlier studies had shown that the P2/P3 amide functionality in
inhibitors is not critical for BACE-1 activity.63,124 Furthermore, it has been
reported that peptides, even those as small as di- or tripeptides, can have poor

52
BBB permeability.125 Therefore, in an attempt to improve the permeability, we
designed and synthesized a series of macrocycles (XV ) in which the P2/P3
amide was replaced with an acetophenone type ketone moiety (Figure 20). Also,
in several of the prepared inhibitors, the ether oxygen in the macrocyclic ring
was replaced with carbon in an additional attempt to improve pharmacokinetic
properties. Moreover, in place of the benzyl P2´ moiety a tert-butyl oxazole
moiety was examined.
Figure 20. Lead compound XIV together with a general structure of the target keto macrocycles (XV ). Inasmuch as the substitution reaction in the final step in the synthetic route
described in Scheme 16 had given low yields, a new synthetic strategy was
developed (Figure 21) in which the RCM reaction was to be performed in the
final step of the reaction sequence. To produce the diene A5, the two
synthesized building blocks B5 and C5 were reacted using a standard peptide-
coupling procedure. R
OHN
OY
R
OHN
O(CH2)x Y
HN
OH
R'
HN
OH
R
O
O
OH+
H2NHN
OH
Y
Target compoundsA5
C5
(CH2)x
B5
R'
(CH2)x
R'
Figure 21. Retrosynthetic analysis of the new macrocyclic compounds.

53
4.3.1 Synthesis of HEA Scaffolds
Synthesis of HEA scaffold 120, which was used to prepare the ether
macrocycles, is described in Scheme 18. The acetal in the previously produced
compound 79 (Scheme 13) was hydrolyzed with aqueous AcOH to give the diol
115 in 78% yield. The primary hydroxyl group in 115 was selectively tosylated
using p-TsCl, Bu2SnO, and TEA in dry DCM to generate tosylate 116 (74%)
which was used to furnish the epoxide 117 in 84% yield, by use of K2CO3 in
MeOH. Next, the epoxide was opened with 3-isopropylbenzylamine to give
compound 118 in 56% yield. For purification reasons, the prepared amine was
protected using Boc2O and TEA in MeOH to give compound 119 (81%). Finally,
the azide group in 119 was reduced with 1,3-propanedithiol and TEA in MeOH
to generate amine 120 in 76% yield.
N3
O
O
O N3
O
OH
OH N3
O
OH
OTs
N3
O
ON3
O
OHHN R
O
OH
N
Boc
119 R = N3 (81%)
120 R = NH2 (76%)
b c
d e
f
a
79 115 (78%) 116 (74%)
117 (84%) 118 (56%)
Scheme 18. Synthesis of building block 120. Reagents and conditions: (a) AcOH (aq), 60 °C; (b) TEA, Bu2SnO, p-TsCl, dry DCM, rt; (c) K2CO3, MeOH, 0 °C to rt; (d) 3-isopropylbenzylamine, TEA, MeOH, 30 °C; (e) Boc2O, TEA, MeOH, rt; (f) 1,3-propanedithiol, TEA, Aliquat 336, MeOH, rt. The carbon analogue scaffolds 130a, 130b, and 131 were obtained as outlined in
Scheme 19. Synthesis of 2,3-di-O-isopropylidene D-glyceraldehyde was
performed according to a procedure reported in the literature,126 and freshly
prepared Grignard reagent, pent-4-enylmagnesium bromide, or hex-5-
enylmagnesium bromide, was added to produce compounds 121a and 121b in
diastereomeric mixtures that could be separated. The hydroxyl group of (S,R)-
121a was inverted using Mitsunobu conditions (i.e., PPh3, DIAD, and p-
nitrobenzoic acid) followed by treatment with NaOMe in MeOH to furnish the
alcohol 123 in 62% yield over two steps. Another Mitsunobu reaction utilizing
DPPA generated the azide 124a in 98% yield. Azide 124b (88%) was

54
synthesized directly from (R,R)-121b using the same Mitsunobu conditions as
for compound 124a. The acetal in 124a was hydrolyzed (74%), and the primary
alcohol was tosylated to generate compound 126a in 84% yield. The epoxide
127a was then prepared in 90% yield using Na2CO3 in MeOH. Epoxide opening
by 3-isopropylbenzylamine and TEA in MeOH rendered compound 128a (61%),
and compound 128b was synthesized in the same manner from the azide 124b.
For purification reasons, the amine functionality in compound 128a was
protected using Boc2O and TEA to give compound 129 in 97% yield. Finally,
reduction of the azide group in compounds 128a, 128b, and 129 generated
amines 130a, 130b, and 131.
Scheme 19. Synthesis of building blocks 130a-b and 131. Reagents and conditions: (a) pent-4-enylmagnesium bromide or hex-5-enylmagnesium bromide in THF, Et2O (dry), –30 °C to rt; (b) p-nitrobenzoic acid, DIAD, PPh3, THF, rt; (c) NaOMe, MeOH, rt; (d) DPPA, PPh3, DIAD, THF (dry), –30 °C to rt; (e) 70% AcOH (aq), 60 °C; (f) TEA, Bu2SnO, p-TsCl, DCM, DMF, rt; (g) Na2CO3, MeOH, 0 °C to rt; (h) 3-isopropylbenzylamine, TEA, MeOH, 30 °C; (i) Boc2O, TEA, MeOH, rt; (j) 1,3-dipropanedithiol, TEA, Aliquat 336, MeOH, rt.

55
4.3.2 Synthesis of the Final Products
The synthesis of target compounds 139a and 139b is depicted in Scheme 20.
The carboxylic acid in compound 9253 was selectively reduced with borane
dimethyl sulfide complex (BH3·SMe2) in THF to give the alcohol 132a in 95%
yield. A Parikh-Doering oxidation127 generated aldehyde 133a (84%), and
subsequent addition of freshly prepared Grignard reagent pent-4-enylmagnesium
bromide furnished alcohol 134a in 65% yield. Oxidation of the alcohol using
Dess-Martin periodinane in DCM gave ketone 135a (76%).128 The methyl ester
in 135a was hydrolyzed, and the corresponding carboxylic acid was coupled
(HATU/DIPEA) with compound 131 to generate 136a in 89% yield over two
steps. Compound 136b was synthesized in the same manner as 136a but starting
from compound 73 (Scheme 11) instead of 92, and using amine 130a instead of
131 in the peptide-coupling step. The amino group in compound 136b was
protected by use of Boc2O and TEA, which gave compound 137 in 65% yield.
Protection of the amino group was done because we had discovered that that
approach allowed the purification of the macrocycles after the RCM reaction
step to proceed much more smoothly. An RCM reaction was performed on the
dienes 136a and 137 using Hoveyda-Grubbs 2nd generation catalyst in refluxing
DCM to furnish macrocycles 138a and 138b in 21% and 47% yield, respectively.
The cyclization led to exclusive formation of the trans isomers, as confirmed by 1H-NMR analysis. Finally, deprotection of the Boc-group was achieved using
Et3SiH and TFA in DCM to produce the target macrocycles 139a and 139b
(91% in both cases). In summary, the RCM reaction generated three additional
macrocycles in 95%, 63%, and 70% yield, respectively, and these were used to
prepare target compounds 143, 144, and 146 (Table 7). Attempts to synthesize
the 14-membered macrocycle corresponding to 138b by use of either Hoveyda-
Grubbs 2nd generation catalyst or Zhan catalyst-1B were unsuccessful, possibly
because the shorter olefin linker resulted in a too rigid molecule.

56
R1
HO
O
O
O
R1
HO O
O
R1
O O
O
R1
HO O
O
R1
O O
O
R1
OHN
O
N
OH R2
R1
OHN
O
N
OH Boc
R1
OHN
O
HN
OH
a b c
d e
g h
92 R1 = N(Me)SO2Me
73 R1 = Me
132a R1 = N(Me)SO2Me
(95%)
132b R1 = Me
(69%)
133a R1 = N(Me)SO2Me
(84%)
133b R1 = Me
(73%)
134a R1 = N(Me)SO2Me
(65%)
134b R1 = Me
(79%)
135a R1 = N(Me)SO2Me
(76%)
135b R1 = Me
(89%)
136a R1 = N(Me)SO2Me
R2 = Boc (89%)
136b R1 = Me, R2 = H
(85%)
137 R1 = Me, R2 = Boc
(65%)
f
138a R1 = N(Me)SO2Me
(21%)
138b R1 = Me
(47%)
139a R1 = N(Me)SO2Me
(91%)
139b R1 = Me
(91%) Scheme 20. Synthesis of target compounds 139a and 139b. Reagents and conditions: (a) BH3·SMe2, THF (dry), Ar, 0 °C to rt; (b) SO3-pyridine, TEA, DMSO (dry), rt; (c) pent-4-enylmagnesium bromide in THF, Et2O (dry), 0 °C; (d) Dess-Martin periodinane, DCM, rt; (e) (i) LiOH (aq), dioxane, rt: (ii) compound 131 or 130a, HATU, DIPEA, DMF, 0 °C to rt; (f) Boc2O, TEA, MeOH, rt; (g) Hoveyda-Grubbs 2nd generation catalyst, DCM (dry), reflux; (h) Et3SiH, TFA, DCM, rt. The amine 140 (Scheme 21) was synthesized from the epoxide 127a using the
synthetic route to compound 130a (Scheme 19) but with 2-(5-(tert-butyl)oxazol-
2-yl)propan-2-amine instead of 3-isopropylbenzylamine. The RCM reaction
with compound 141 was unsuccessful. The acyclic 141 could not be regenerated,
and the color of the reaction mixture indicated that the catalyst had decomposed
early in the reaction process. Therefore, we synthesized compound 145 by the
same synthetic route as we had used to obtain compound 98 in our previous
project (Scheme 16, described in section 4.2.3).

57
Scheme 21. Proposed synthetic route to compound 142. Reagents and conditions: (a) the carboxylic acid of 135a, HATU, DIPEA, DMF, 0 °C to rt; (b) Hoveyda-Grubbs 2nd generation catalyst, DCM (dry), reflux.
4.3.3 Biological Data on the Macrocyclic Inhibitors
The structures of all the synthesized target compounds are outlined in Table 7
along with BACE-1 inhibition data and Caco-2 permeability values for selected
products. All the macrocycles were 15-membered, except for 146, which was
16-membered.
Replacement of the P2/P3 amide functionality in the lead compound XIV with a
keto group rendered target compound 143. Encouragingly, 143 exhibited
activity similar to that of the lead compound, but, disappointingly, it did not
show increased cell permeability. Introducing the less polar methyl group in the
P2 position furnished compound 144, which as expected had lower enzyme and
cell activity but significantly improved permeability (papp = 13 x 10–6 cm/s).
Compared to 144, compound 145 showed additional improvement in cell
permeability (papp = 32 x 10-6 cm/s), likely due to increased shielding of the
amine moiety from the gem methyl groups of the P2´ substituent resulting in
decreased hydrogen bonding to the solvent, whereas the cell-based activity was
decreased likely due to the same effect, vide supra, i.e., decreased hydrogen
bonding to the BACE-1 active site. Compound 139a, the carba analogue of 143,
was similar to 143 with regard to enzyme and cell activity, and it also had an
inadequate Caco-2 value of < 1. Not surprisingly all activity was lost in the Boc-
protected compound 138b. Compound 139b had cell permeability similar to that

58
of its ether analogue 144 and also showed improved enzyme activity, although,
compared to compound 139a, it had 30-fold lower enzyme activity and 320
times lower activity in the cell-based assay. These data substantiate earlier
findings that the sulfonamide functionality is important for the activity of these
kinds of inhibitor compounds, although it at the same time renders inadequate
cell permeability. Other BACE-1 inhibitors incorporating a sulfonamide
substituent in the P2 position have been found to be substrates for P-gp efflux,67
which might explain why compounds 139a and 143 exhibited poor permeability.
The 16-membered macrocycle 146 had activities similar to those of the
corresponding 15-membered compound 139b but also showed a small drop in
cell permeability.
Table 7. BACE-1 inhibition data and cell permeability values for compounds 138b, 139, and 143–146
Cpd. Structure IC 50 enzyme
(nM)
IC 50 cell
(nM)
Caco-2 papp
(*10-6 cm/s)
143
12 4.6 < 1
144
1100 1400 13
145
1500 3300 32
139a
8.9 3 < 1
138b
> 10000 > 10000 ND
139b
280 970 13
146a
260 1000 7.3
a16-membered macrocycle

59
4.3.4 Conclusions
A series of macrocyclic BACE-1 inhibitors incorporating a P2/P3 keto
functionality was developed, and some of these compounds showed highly
promising low nanomolar activity against BACE-1. Moreover, several inhibitors
displayed improved cell-based permeability compared to other inhibitors
previously synthesized by our group, but not unexpectedly these target
compounds incorporating a toluene P2 core displayed decreased potency. As
previously shown, the sulfonamide functionality in the P2 position was
important for the activity of this type of compounds, but it also resulted in lower
cell-based permeability. In summary, our attempts to improve the permeability
compared to earlier published inhibitors from our group show promising results.
Further work on this series is of interest where focus will aim to explore
inhibitors where the sulfonamide functionality in the P2-position is replaced by
small lipophilic groups and where potency-enhancing substituents as shown in
Paper III are introduced aiming at the S3 subpocket.

60

61
5. Summary
________________________________________________________________
Novel series of BACE-1 inhibitors were designed and synthesized with
gradually less peptidic character. Initially, a novel hydroxyethylene transition
state isostere previously produced by our group was further evaluated by
introducing a substituted cyclopentane moiety in the P2 position, instead of the
widely used isophthalamide structure (Paper I). Different stereoisomers of the
cyclopentane scaffold and a selection of P3 substituents were evaluated.
Surprisingly, (S) stereochemistry at the methyl group in the most favorable
substituent in the P3 position was found to be significantly better than (R). The
most active inhibitor had an IC50 value of 260 nM, and it also proved to be
selective towards cathepsin D and renin.
Another series of hydroxyethylene-based inhibitors with various P1´ groups and
extended P1 substituents were synthesized from chiral carbohydrate precursors
(Paper II). Various P2´-P3´ substituents were also evaluated. Truncated
inhibitors with enzyme potency in the nanomolar range were identified, and two
compounds possessed only one amide bond, which made them more drug-like.
Unfortunately, they exhibited low inhibition in a cell-based assay.
The next step in the current research was to synthesize and evaluate a series of
macrocyclic inhibitors that contained a hydroxyethylamine isostere and had the
P1 and P3 moieties connected by ring-closing metathesis (Paper III). Several of
these inhibitors displayed potent low nanomolar activity in both the enzyme and

62
cell-based assays. A phenyl substituent was also introduced in the macrocyclic
ring to protrude into the S3 subpocket, which rendered an inhibitor with IC50
values of 3.2 and 0.15 nM in the enzyme and the cell-based assay, respectively.
Two targets from this series were co-crystallized with BACE-1, and the X-ray
crystal structures provided valuable information about the binding mode of this
kind of compounds. For instance, it was disclosed that the macrocycle-attached
phenyl substituent did not extend fully into the S3 subpocket, which suggests
that further optimization is possible. In general, the compounds in this series
showed good selectivity towards cathepsin D, but displayed low membrane
permeability.
In an attempt to improve the permeability, another series of macrocyclic
inhibitors were synthesized in which the P2/P3 amide functionality present in
the molecules reported in Paper III was replaced with a keto group (Paper IV).
In addition, the ether oxygen in the macrocyclic ring was replaced with a carbon.
Inhibitors containing a sulfonamide functionality in the P2 position showed
activity against BACE-1 in the low nanomolar range in both the enzyme and
cell-based assays, but not unexpectedly they still exhibited low cell permeability.
However, replacement of the sulfonamide, a known P-gp efflux substrate moiety
in these types of inhibitors, with a less polar methyl group furnished inhibitors
with improved permeability properties, the best with a Caco-2 value of 32 x 10–6
cm/s.
In conclusion, a wide range of novel BACE-1 inhibitors were synthesized, some
of which have provided information that makes them interesting for future
research. Furthermore, synthetic strategies were designed and evaluated. This
thesis illustrates the difficulties that are encountered when designing inhibitors
that are not only required to be potent, but must also possess adequate
permeability and selectivity properties. Together, the results of the current
research can hopefully constitute useful additions to the vast body of data
needed to design drugs for successful treatment of Alzheimer’s disease.

63
Appendix
________________________________________________________________
I. Experimental data for unpublished compounds in section 4.1
(S)-4-((R)-1-Azido-2-(but-2-ynyloxy)ethyl)-2,2-dimethyl-1,3-dioxolane (71).
NaH (60%, 0.099 g, 2.48 mmol) was added to a solution of E3 (0.309 g, 1.65
mmol) in dry DMF (10 mL). After 10 minutes of stirring at room temperature,
1-bromo-2-butyne (0.30 mL, 3.31 mmol) was added, and the mixture was left to
stir for 22 hours at room temperature. The solution was diluted with brine and
water and extracted with EtOAc. The combined organic extracts were dried,
filtered, and co-evaporated with xylene. Purification using flash column
chromatography (toluene/EtOAc 39:1) gave compound 71 (0.29 g, 74%) as a
yellow oil. 1H-NMR (CDCl3, 300 MHz): δ 1.34 (s, 3H), 1.44 (s, 3H), 1.85 (t, J =
2.5 Hz, 3H), 3.57 (dd, J = 9.6, 6.3 Hz, 1H), 3.68 (dd, J = 6.3, 3.6 Hz, 1H), 3.75
(dd, J = 9.6, 3.6 Hz, 1H), 3.91 (dd, J = 8.3, 5.8 Hz, 1H), 4.01–4.10 (m, 2H), 4.14
(q, J = 2.5 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ 4.1, 25.8, 27.0, 59.8, 63.3,
66.9, 69.8, 75.1, 75.7, 83.7, 110.2.
(S)-2-(But-2-yn-1-yloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethanamine
(72). 1,3-Propanedithiol (0.61 mL, 6.10 mmol) and TEA (0.85 mL, 6.10 mmol)
were added to a solution of 71 (0.29 g, 1.22 mmol) in MeOH (5 mL). The
mixture was stirred at room temperature for 22 hours. The solution was diluted
with H2O and MeOH, acidified with 1M HCl, and washed with Et2O. The water
phase was made basic with 1M NaOH and extracted with Et2O. The combined
organic extracts were dried, filtered, and evaporated to give compound 72 (0.18

64
g, 68%) as a colorless oil. 1H-NMR (CDCl3, 300 MHz): δ 1.27 (s, 3H), 1.34 (s,
3H), 1.78 (t, J = 2.2 Hz, 3H), 2.37 (bs, 2H), 3.01–3.06 (m, 1H), 3.37 (dd, J = 9.3,
6.6 Hz, 1H), 3.58 (dd, J = 9.3, 3.6 Hz, 1H), 3.82 (dd, J = 10.7, 9.1 Hz, 1H),
3.94–4.04 (m, 2H), 4.05 (q, J = 2.2 Hz, 2H); 13C-NMR (CDCl3, 75.5 MHz): δ
3.7, 25.4, 26.7, 53.3, 59.3, 66.5, 71.5, 75.2, 77.8, 82.8, 109.1.
Methyl 3-methyl-5-(pent-3-yn-1-ylcarbamoyl)benzoate (74). Compound 73
(1.38 g, 7.09 mmol), HOBt (1.21 g, 8.94 mmol), and TEA (2.5 mL, 17.8 mmol)
were added to a solution of compound C3 (20 mmol) in DMF (14 mL). The
mixture was cooled to 0 °C and EDC (1.67 g, 8.69 mmol) was added. The
reaction mixture was left to reach room temperature, and was stirred for 19
hours. The mixture was co-evaporated with xylene and purified using flash
column chromatography (toluene/EtOAc 9:1) to give compound 75 (0.58 g, 32%)
as white crystals. 1H-NMR (CD3OD/CDCl3, 300 MHz): δ 1.74 (s, 3H), 2.42 (t, J
= 7.1 Hz, 2H), 2.43 (s, 3H), 3.46 (t, J = 7.1 Hz, 2H), 3.90 (s, 3H), 7.84 (d, J =
1.7 Hz, 1H), 7.95 (d, J = 1.7 Hz, 1H), 8.24 (d, J = 1.7 Hz, 1H); 13C-NMR
(CD3OD/CDCl3, 75.5 MHz): δ 3.2, 20.0, 21.3, 40.4, 52.7, 76.7, 77.5, 126.4,
131.4, 133.2, 133.6, 135.9, 139.9, 167.8, 169.0.
N1-((S)-2-(But-2-yn-1-yloxy)-1-((S)-2,2-dimethyl-1,3-dioxolan-4-yl)ethyl)-5-
methyl-N 3-(pent-3-yn-1-yl)isophthalamide (75). Aqueous LiOH (40 mg/mL,
1.1 mL, 1.87 mmol) was added to a solution of 74 (0.243 g, 0.94 mmol) in H2O
(6 mL) and dioxane (12 mL). The solution was stirred at room temperature for 3
hours and co-evaporated with toluene to give the corresponding carboxylic acid.
The carboxylic acid (0.045 g, 0.18 mmol) was dissolved in DMF (15 mL).
Compound 72 (0.040 g, 0.19 mmol), HCTU (0.084 g, 0.20 mmol), and DIPEA
(0.095 mL, 0.55 mmol) were added. The mixture was stirred at room
temperature for 4 hours. The solution was diluted with brine and H2O and
extracted with EtOAc. The combined organic extracts were dried, filtered, and
co-evaporated with toluene. Purification using flash column chromatography
(toluene/EtOAc 9:1, 3:1, 1:1) gave compound 75 (0.064 g, 79%) as a colorless
oil. 1H-NMR (CD3OD/CDCl3, 300 MHz): δ 1.33 (s, 3H), 1.42 (s, 3H), 1.74 (t, J
= 2.5 Hz, 3H), 1.76 (t, J = 2.5 Hz, 3H), 2.43 (s, 3H), 2.42 (m, 2H), 3.47 (t, J =
7.1 Hz, 2H), 3.76 (d, J = 4.4 Hz, 2H), 3.90 (dd, J = 8.5, 5.2 Hz, 1H), 4.05 (dd, J
= 8.5, 5.8 Hz, 1H), 4.23–4.34 (m, 2H), 4.10–4.15 (m, 2H), 7.76 (s, 2H), 7.99 (s,
1H); 13C-NMR (CD3OD/CDCl3, 75.5 MHz): δ 2.8, 19.3, 20.8, 25.0, 26.4, 39.7,

65
47.6, 52.3, 59.0, 67.2, 68.6, 74.8, 75.1, 76.0, 78.2, 82.8, 110.0, 123.2, 131.0,
131.1, 134.7, 134.9, 139.1, 168.3, 168.3.
1,6dioxa-Cyclododec-9-yne-2,5-dione (78). Compound 77 (0.0268g, 0.107
mmol) and Schrock alkylidyne complex (5 mg, 0.01 mmol) were dissolved in
chlorobenzene (5.0 mL) under Ar atmosphere in a glove box. The solution was
stirred at 80 °C for 6 hours and the solvent was evaporated. The crude product
was purified using flash column chromatography (toluene/EtOAc 19:1) and
gave 79 (0.012 g, 57%) as colorless crystals. 1H-NMR (CDCl3, 300 MHz): δ
2.46 (t, J = 5.8 Hz, 4H), 2.69 (s, 4H), 4.26 (t, J = 5.6 Hz, 4H); 13C-NMR (CDCl3,
75.5 MHz): δ 19.8, 30.1, 61.6, 78.9, 171.8.
I I. Experimental procedures for unpublished compounds in section 4.3
(2R,3S)-3-Amino-1-((2-(5-(tert-butyl)oxazol-2-yl)propan-2-yl)amino)oct-7-
en-2-ol (140). Compound 140 was synthesized in the same manner as compound
130a, but using 2-(5-(tert-butyl)oxazol-2-yl)propan-2-amine instead of 3-
isopropylbenzylamine to open the epoxide. 1H-NMR (CD3OD, 300 MHz): δ
2.16-2.62 (m, 4H), 2.29 (s, 9H), 2.49 (s, 6H), 2.99 (m, 1H), 3.35 (dd, J = 8.2,
11.5 Hz, 1H), 3.54 (dd, J = 3.5, 11.5 Hz, 1H), 3.71-3.79 (m, 1H), 4.28-4.32 (m,
1H), 4.45-4.52 (m, 1H), 5.89-6.04 (m, 2H), 6.73-6.87 (m, 1H), 7.66 (s, 1H); 13C-
NMR (CD3OD, 75.5 MHz): δ 25.9, 26.6, 27.3, 28.9, 32.4, 32.7, 34.9, 46.3, 55.4,
55.8, 74.1, 115.2, 119.7, 139.7, 162.5, 168.7.
N-((2R,3S)-1-((2-(5-(tert-Butyl)oxazol-2-yl)propan-2-yl)amino)-2-
hydroxyoct-7-en-3-yl)-3-(hex-5-enoyl)-5-(N-methylmethylsulfonamido)
benzamide (141). Compound 141 was synthesized using a standard peptide
coupling procedure in 99% yield. 1H-NMR (CDCl3, 300 MHz): δ 1.16 (s, 9H),
1.30-1.58 (m, 11H), 1.45 (s, 6H), 1.72-1.83 (m, 1H), 1.95-2.13 (m, 2H), 2.55 (dd,
J = 5.3, 12.3 Hz, 1H), 2.67 (dd, J = 3.5, 8.8 Hz, 1H), 2.82 (s, 3H), 2.94 (t, J =
7.6 Hz, 2H), 3.31 (s, 3H), 3.62-3.68 (m, 1H), 4.12-4.23 (m, 1H), 4.83-5.03 (m,
4H), 6.50 (s, 1H), 7.88 (d, J = 8.8 Hz, 1H), 7.98-8.03 (m, 2H), 8.23-8.28 (m, 1H); 13C-NMR (CDCl3, 75.5 MHz): δ 23.1, 25.6, 26.8, 26.9, 28.6, 30.4, 31.4, 33.0,
33.6, 35.5, 38.0, 45.4, 54.0, 54.4, 71.6, 114.9, 115.5, 118.8, 125.0, 128.4, 128.8,
136.4, 137.8, 138.2, 138.4, 142.4, 160.9, 165.8, 167.0, 198.9.

66

67
Acknowledgements
________________________________________________________________
Jag vill tacka alla er som på olika sätt har bidragit till att göra mina år i Linköping till en oförglömlig, lärorik och otroligt rolig resa. Ett speciellt tack riktas till:
Ingemar Kvarnström, min handledare. Tack för ditt förtroende, stöd och ständigt öppna dörr.
Bertil Samuelsson, min biträdande handledare. Tack för att du har delat med dig av dina oändliga kemikunskaper.
Åsa Rosenquist, för att du tog dig tid för mig och mina projekt trots ditt fullbokade schema.
Oscar Belda, för att du har orkat med alla mina frågor och delat med dig av dina kunskaper.
Fredrik Thorstensson, min handledare under ex-jobbet. Tack för att du tog hand om mig när jag var ny och vilsen. Tack för att du tidigt lärde mig att kemins vägar är krokiga och att finna charmen i detta.
Anders Dahlgren, för all din hjälp med texter, korrekturläsning och alla timmar ute på labbet.
Per-Ola Johansson, för trevligt ressällskap i Boston och New York.
Marcus Bäck, för korrekturläsning och alla trevliga samtal gällande både kemi och livet. Tack för alla roliga konferensresor och sightseeingturer.
Fredrik Wångsell, för att du var som en liten bonushandledare åt mig min första tid på labbet och för alla roliga stunder på labb och utanför.

68
Bisse, för allt skratt du delade med dig av på labbet. Det blev väldigt tyst och tomt när du flyttade till Göteborg.
Alla mina ex-jobbare och stipendiater: Björn Malm, Tobias Sundelin, Sofia Sauke, Robin Hertzberg, Malin Sandberg, Sandra Sjögren, Emelie Svensson, Jonas Garmert, Mattias Tengdelius och Åsa Einarsson, för att ni har bidragit med stort och smått i projekt både i och utanför denna avhandling.
Alla andra som genom åren har bidragit till en härlig atmosfär på labbet.
Gamla och nuvarande ”grabbar i korridoren”: Andreas, Hamid, Jeff, Johan, Marcus H, Mattias, Mattias, Peter, Roger och Stefan. Ett extra tack till Timmy för att du har ”suttit i båten” med mig och varit en välbehövlig ventilationskälla.
Alma och Lan, för alla roliga stunder och för att ni hjälpte till med att utöka antal X-kromosomer i korridoren. Nu på sistone även Michi.
Leffe, för att du hjälpte mig att hålla koll på alla siffror. Tack för trevligt cykelsällskap, det kommer att bli många fler timmar framöver.
”Stockholmstjejerna” Catarina och Jenny för att ni har förgyllt alla möten genom åren.
Alla medförfattare och personal på Medivir som har hjälpt mig på olika sätt.
Patricia Ödman för eminent språklig granskning.
Medivir AB för finansiellt stöd.
Susanne, för att du har sådant tålamod när det kommer till pappersarbete. Tack för att du alltid har en minut över för en lägesrapport gällande arbetet och livet.
Alla gamla och nuvarande medlemmar av ”runda bordet”, speciellt: Anna-Lena, Annica, Alexandra, Daniel K, Ina, Jutta, Karin M, Linda, Maria J, Patrik, Sofie och Therese, ni har förgyllt mina luncher och fikapauser. Tack för allt skratt och alla trevliga samtalsämnen mellan himmel och jord. Ett extra tack till: Cissi för att du alltid gör mig glad, Maria L för långa samtal, Madhan for your never-ending laughter och Sara för allt idrottssnack.
Alla andra på kemiavdelningen som har bidragit till en trevlig atmosfär alla dessa år.
”Borneo babes:en” Anngelica och Karin, för alla underbara korta och långa resor. Tack för galna upptåg, skratt och djupa samtal.
Patricia, för att du alltid finns där och delar med dig av din värme.

69
Tjejgänget under studietiden, för alla lugna spelkvällar och mindre lugna utekvällar. Ett speciellt tack till min labbpartner Linda - vi kompletterade varandra perfekt och Camilla - sju lika val i ”sten, sax, påse” är otäckt.
Alla mina underbara vänner utanför kemivärlden - ni vet vilka ni är. Tack för underbara middagar, sällskap till, under och från olika motionslopp, fjällresor och allt annat roligt vi hittar på.
Min familj: mamma AnnBritt, pappa Kenneth, bonusföräldrar Yvonne och Mats, brorsan Patrik, syster Tessan, bonussyskon Sara och Peder, för att ni alltid finns där och stöttar när det behövs. Tack även ”familjen Nilsson” för att ni tog emot mig med öppna armar.
Min älskade lilla familj: Daniel, för att du alltid finns där och tror på mig. Tack för att du påminner mig om vad som är viktigt. Du är bäst. Fabian, min underbara lilla solstråle. Tack för att jag får återupptäcka världen tillsammans med dig. – Jag älskar er!
Linköping 2012

70

71
References
________________________________________________________________
(1) Leung, D.; Abbenante, G.; Fairlie, D. P. J Med Chem 2000, 43, 305-41. (2) Turk, B. Nat Rev Drug Discov 2006, 5, 785-99. (3) Schechter, I.; Berger, A. Biochem Biophys Res Commun 1967, 27, 157-
62. (4) Quesada, V.; Ordonez, G. R.; Sanchez, L. M.; Puente, X. S.; Lopez-
Otin, C. Nucleic Acids Res 2009, 37, D239-43. (5) Puente, X. S.; Sanchez, L. M.; Overall, C. M.; Lopez-Otin, C. Nat Rev
Genet 2003, 4, 544-58.
(6) Norman, P. モ Protease Inhibitors: Innovation Drives Drug Pipeline,ヤ Cambridge Healthtech Institute 2009.
(7) Tyndall, J. D.; Nall, T.; Fairlie, D. P. Chem Rev 2005, 105, 973-99. (8) Chatfield, D. C.; Brooks, B. R. J. Am. Chem. Soc. 1995, 117, 5561–
5572. (9) West, M. L.; Fairlie, D. P. Trends Pharmacol Sci 1995, 16, 67-75. (10) Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J. Advanced
Drug Delivery Reviews 1997, 23, 3-25. (11) Veber, D. F.; Johnson, S. R.; Cheng, H. Y.; Smith, B. R.; Ward, K. W.;
Kopple, K. D. J Med Chem 2002, 45, 2615-23. (12) Patrick, L. G. An Introduction to Medicinal Chemistry; Oxford
University Press, 2005; Vol. 3. (13) Alavijeh, M. S.; Chishty, M.; Qaiser, M. Z.; Palmer, A. M. NeuroRx
2005, 2, 554-71. (14) Meredith, J. E., Jr.; Thompson, L. A.; Toyn, J. H.; Marcin, L.; Barten,
D. M.; Marcinkeviciene, J.; Kopcho, L.; Kim, Y.; Lin, A.; Guss, V.; Burton, C.; Iben, L.; Polson, C.; Cantone, J.; Ford, M.; Drexler, D.; Fiedler, T.; Lentz, K. A.; Grace, J. E., Jr.; Kolb, J.; Corsa, J.; Pierdomenico, M.; Jones, K.; Olson, R. E.; Macor, J. E.; Albright, C. F. J Pharmacol Exp Ther 2008, 326, 502-13.

72
(15) Lahiri, D. K.; Farlow, M. R.; Greig, N. H.; Sambamurti, K. Drug Development Research 2002, 56, 267-281.
(16) Citron, M. Trends Pharmacol Sci 2004, 25, 92-7. (17) Hardy, J.; Duff, K.; Hardy, K. G.; Perez-Tur, J.; Hutton, M. Nat
Neurosci 1998, 1, 355-8. (18) Jakob-Roetne, R.; Jacobsen, H. Angew Chem Int Ed Engl 2009, 48,
3030-59. (19) Wolfe, M. S. J Med Chem 2001, 44, 2039-2060. (20) Bertram, L.; Tanzi, R. E. J Clin Invest. 2005, 115, 1449–1457. (21) Zhu, Y.; Xiao, K.; Ma, L.; Xiong, B.; Fu, Y.; Yu, H.; Wang, W.; Wang,
X.; Hu, D.; Peng, H.; Li, J.; Gong, Q.; Chai, Q.; Tang, X.; Zhang, H.; Li, J.; Shen, J. Bioorg Med Chem 2009, 17, 1600-13.
(22) Lleo, A.; Greenberg, S. M.; Growdon, J. H. Annu Rev Med 2006, 57, 513-33.
(23) Reisberg, B.; Doody, R.; Stoffler, A.; Schmitt, F.; Ferris, S.; Mobius, H. J. N Engl J Med 2003, 348, 1333-41.
(24) Selkoe, D. J. Physiol Rev 2001, 81, 741-66. (25) John, V.; Beck, J. P.; Bienkowski, M. J.; Sinha, S.; Heinrikson, R. L. J
Med Chem 2003, 46, 4625-30. (26) Barten, D. M.; Albright, C. F. Mol Neurobiol 2008, 37, 171-86. (27) Brouwers, N.; Sleegers, K.; Van Broeckhoven, C. Ann Med 2008, 40,
562-83. (28) Gandy, S.; Simon, A. J.; Steele, J. W.; Lublin, A. L.; Lah, J. J.; Walker,
L. C.; Levey, A. I.; Krafft, G. A.; Levy, E.; Checler, F.; Glabe, C.; Bilker, W. B.; Abel, T.; Schmeidler, J.; Ehrlich, M. E. Ann Neurol 2010, 68, 220-30.
(29) Hardy, J. Trends Neurosci 1997, 20, 154-9. (30) Chen, F.; Hasegawa, H.; Schmitt-Ulms, G.; Kawarai, T.; Bohm, C.;
Katayama, T.; Gu, Y.; Sanjo, N.; Glista, M.; Rogaeva, E.; Wakutani, Y.; Pardossi-Piquard, R.; Ruan, X.; Tandon, A.; Checler, F.; Marambaud, P.; Hansen, K.; Westaway, D.; St George-Hyslop, P.; Fraser, P. Nature 2006, 440, 1208-12.
(31) Citron, M. Nat Rev Neurosci 2004, 5, 677-85. (32) Hardy, J.; Selkoe, D. J. Science 2002, 297, 353-6. (33) Lobello, K.; Ryan, J. M.; Liu, E.; Rippon, G.; Black, R. Int J
Alzheimers Dis 2012, 2012, 628070. (34) Sinha, S.; Anderson, J. P.; Barbour, R.; Basi, G. S.; Caccavello, R.;
Davis, D.; Doan, M.; Dovey, H. F.; Frigon, N.; Hong, J.; Jacobson-Croak, K.; Jewett, N.; Keim, P.; Knops, J.; Lieberburg, I.; Power, M.; Tan, H.; Tatsuno, G.; Tung, J.; Schenk, D.; Seubert, P.; Suomensaari, S. M.; Wang, S.; Walker, D.; Zhao, J.; McConlogue, L.; John, V. Nature 1999, 402, 537-40.
(35) Vassar, R.; Bennett, B. D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E. A.; Denis, P.; Teplow, D. B.; Ross, S.; Amarante, P.; Loeloff, R.; Luo, Y.; Fisher, S.; Fuller, J.; Edenson, S.; Lile, J.; Jarosinski, M. A.; Biere, A.

73
L.; Curran, E.; Burgess, T.; Louis, J. C.; Collins, F.; Treanor, J.; Rogers, G.; Citron, M. Science 1999, 286, 735-41.
(36) Yan, R.; Bienkowski, M. J.; Shuck, M. E.; Miao, H.; Tory, M. C.; Pauley, A. M.; Brashier, J. R.; Stratman, N. C.; Mathews, W. R.; Buhl, A. E.; Carter, D. B.; Tomasselli, A. G.; Parodi, L. A.; Heinrikson, R. L.; Gurney, M. E. Nature 1999, 402, 533-7.
(37) Lin, X.; Koelsch, G.; Wu, S.; Downs, D.; Dashti, A.; Tang, J. Proc Natl Acad Sci U S A 2000, 97, 1456-60.
(38) Hussain, I.; Powell, D.; Howlett, D. R.; Tew, D. G.; Meek, T. D.; Chapman, C.; Gloger, I. S.; Murphy, K. E.; Southan, C. D.; Ryan, D. M.; Smith, T. S.; Simmons, D. L.; Walsh, F. S.; Dingwall, C.; Christie, G. Mol Cell Neurosci 1999, 14, 419-27.
(39) Ghosh, A. K.; Hong, L.; Tang, J. Curr Med Chem 2002, 9, 1135-44. (40) Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A. K.;
Zhang, X. C.; Tang, J. Science 2000, 290, 150-3. (41) Saunders, A. J.; Kim, T.-W.; Tanzi, R. E. Science 1999, 286, 1255. (42) Bennett, B. D.; Babu-Khan, S.; Loeloff, R.; Louis, J. C.; Curran, E.;
Citron, M.; Vassar, R. J Biol Chem 2000, 275, 20647-51. (43) Cummings, J. L. N Engl J Med 2004, 351, 56-67. (44) Salloway, S.; Mintzer, J.; Weiner, M. F.; Cummings, J. L. Alzheimers
Dement 2008, 4, 65-79. (45) De Strooper, B. Neuron 2003, 38, 9-12. (46) Radtke, F.; Wilson, A.; Mancini, S. J.; MacDonald, H. R. Nat Immunol
2004, 5, 247-53. (47) Wu, W. L.; Zhang, L. Drug Dev. Res. 2009, 70, 94-100. (48) Luo, Y.; Bolon, B.; Kahn, S.; Bennett, B. D.; Babu-Khan, S.; Denis, P.;
Fan, W.; Kha, H.; Zhang, J.; Gong, Y.; Martin, L.; Louis, J. C.; Yan, Q.; Richards, W. G.; Citron, M.; Vassar, R. Nat Neurosci 2001, 4, 231-2.
(49) Roberds, S. L.; Anderson, J.; Basi, G.; Bienkowski, M. J.; Branstetter, D. G.; Chen, K. S.; Freedman, S. B.; Frigon, N. L.; Games, D.; Hu, K.; Johnson-Wood, K.; Kappenman, K. E.; Kawabe, T. T.; Kola, I.; Kuehn, R.; Lee, M.; Liu, W.; Motter, R.; Nichols, N. F.; Power, M.; Robertson, D. W.; Schenk, D.; Schoor, M.; Shopp, G. M.; Shuck, M. E.; Sinha, S.; Svensson, K. A.; Tatsuno, G.; Tintrup, H.; Wijsman, J.; Wright, S.; McConlogue, L. Hum Mol Genet 2001, 10, 1317-24.
(50) Dominguez, D.; Tournoy, J.; Hartmann, D.; Huth, T.; Cryns, K.; Deforce, S.; Serneels, L.; Camacho, I. E.; Marjaux, E.; Craessaerts, K.; Roebroek, A. J.; Schwake, M.; D'Hooge, R.; Bach, P.; Kalinke, U.; Moechars, D.; Alzheimer, C.; Reiss, K.; Saftig, P.; De Strooper, B. J Biol Chem 2005, 280, 30797-806.
(51) Bursavich, M. G.; Rich, D. H. J Med Chem 2002, 45, 541-58. (52) John, V. Curr Top Med Chem 2006, 6, 569-78. (53) Stachel, S. J.; Coburn, C. A.; Steele, T. G.; Jones, K. G.; Loutzenhiser,
E. F.; Gregro, A. R.; Rajapakse, H. A.; Lai, M. T.; Crouthamel, M. C.; Xu, M.; Tugusheva, K.; Lineberger, J. E.; Pietrak, B. L.; Espeseth, A.

74
S.; Shi, X. P.; Chen-Dodson, E.; Holloway, M. K.; Munshi, S.; Simon, A. J.; Kuo, L.; Vacca, J. P. J Med Chem 2004, 47, 6447-50.
(54) Ghosh, A. K.; Kumaragurubaran, N.; Hong, L.; Kulkarni, S.; Xu, X.; Miller, H. B.; Reddy, D. S.; Weerasena, V.; Turner, R.; Chang, W.; Koelsch, G.; Tang, J. Bioorg Med Chem Lett 2008, 18, 1031-6.
(55) Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. Nat Genet 1992, 1, 345-7.
(56) Tung, J. S.; Davis, D. L.; Anderson, J. P.; Walker, D. E.; Mamo, S.; Jewett, N.; Hom, R. K.; Sinha, S.; Thorsett, E. D.; John, V. J Med Chem 2002, 45, 259-62.
(57) Ghosh, A. K.; Bilcer, G.; Harwood, C.; Kawahama, R.; Shin, D.; Hussain, K. A.; Hong, L.; Loy, J. A.; Nguyen, C.; Koelsch, G.; Ermolieff, J.; Tang, J. J Med Chem 2001, 44, 2865-8.
(58) Kimura, T.; Shuto, D.; Hamada, Y.; Igawa, N.; Kasai, S.; Liu, P.; Hidaka, K.; Hamada, T.; Hayashi, Y.; Kiso, Y. Bioorg Med Chem Lett 2005, 15, 211-5.
(59) Asai, M.; Hattori, C.; Iwata, N.; Saido, T. C.; Sasagawa, N.; Szabo, B.; Hashimoto, Y.; Maruyama, K.; Tanuma, S.; Kiso, Y.; Ishiura, S. J Neurochem 2006, 96, 533-40.
(60) Beswick, P.; Charrier, N.; Clarke, B.; Demont, E.; Dingwall, C.; Dunsdon, R.; Faller, A.; Gleave, R.; Hawkins, J.; Hussain, I.; Johnson, C. N.; MacPherson, D.; Maile, G.; Matico, R.; Milner, P.; Mosley, J.; Naylor, A.; O'Brien, A.; Redshaw, S.; Riddell, D.; Rowland, P.; Skidmore, J.; Soleil, V.; Smith, K. J.; Stanway, S.; Stemp, G.; Stuart, A.; Sweitzer, S.; Theobald, P.; Vesey, D.; Walter, D. S.; Ward, J.; Wayne, G. Bioorg Med Chem Lett 2008, 18, 1022-6.
(61) Hussain, I.; Hawkins, J.; Harrison, D.; Hille, C.; Wayne, G.; Cutler, L.; Buck, T.; Walter, D.; Demont, E.; Howes, C.; Naylor, A.; Jeffrey, P.; Gonzalez, M. I.; Dingwall, C.; Michel, A.; Redshaw, S.; Davis, J. B. J Neurochem 2007, 100, 802-9.
(62) Ghosh, A. K.; Kumaragurubaran, N.; Hong, L.; Kulkarni, S. S.; Xu, X.; Chang, W.; Weerasena, V.; Turner, R.; Koelsch, G.; Bilcer, G.; Tang, J. J Med Chem 2007, 50, 2399-407.
(63) Stachel, S. J.; Coburn, C. A.; Sankaranarayanan, S.; Price, E. A.; Wu, G.; Crouthamel, M.; Pietrak, B. L.; Huang, Q.; Lineberger, J.; Espeseth, A. S.; Jin, L.; Ellis, J.; Holloway, M. K.; Munshi, S.; Allison, T.; Hazuda, D.; Simon, A. J.; Graham, S. L.; Vacca, J. P. J Med Chem 2006, 49, 6147-50.
(64) Luo, X.; Yan, R. Int J Clin Exp Pathol 2010, 3, 618-28. (65) Björklund, C.; Oscarson, S.; Benkestock, K.; Borkakoti, N.; Jansson,
K.; Lindberg, J.; Vrang, L.; Hallberg, A.; Rosenquist, Å.; Samuelsson, B. J Med Chem 2010, 53, 1458-64.
(66) Silvestri, R. Med Res Rev 2009, 29, 295-338. (67) Stachel, S. J. Drug Dev. Res. 2009, 70, 101-110.

75
(68) Johansson, P. O.; Bäck, M.; Kvarnström, I.; Jansson, K.; Vrang, L.; Hamelink, E.; Hallberg, A.; Rosenquist, Å.; Samuelsson, B. Bioorg Med Chem 2006, 14, 5136-51.
(69) Bäck, M.; Nyhlén, J.; Kvarnström, I.; Appelgren, S.; Borkakoti, N.; Jansson, K.; Lindberg, J.; Nyström, S.; Hallberg, A.; Rosenquist, Å.; Samuelsson, B. Bioorg Med Chem 2008, 16, 9471-86.
(70) Dhaon, M. K.; Olsen, R. K.; Ramasamy, K. J. Org. Chem. 1982, 47, 1962-1965.
(71) Thierry, J.; Yue, C.; Potier, P. Tetrahedron Lett. 1998, 39, 1557-1560. (72) Elliott, R. P.; Hui, A.; Fairbanks, A. J.; Nash, R. J.; Winchester, B. G.;
Way, G.; Smith, C.; Lamont, R. B.; Storer, R.; Fleet, G. W. J. Tetrahedron Lett. 1993, 34, 7949-7952.
(73) Zuccarello, G.; Bouzide, A.; Kvarnström, I.; Niklasson, G.; Svensson, S. C. T.; Brisander, M.; Danielsson, H.; Nillroth, U.; Karlén, A.; Hallberg, A.; Classon, B.; Samuelsson, B. J.Org. Chem. 1998, 63, 4898–4906.
(74) Rosenquist, Å.; Kvarnström, I.; Svensson, S. C. T.; Classon, B.; Samuelsson, B. Acta Chem. Scand. 1992, 46, 1127-1129.
(75) Bartlett, P. A.; Green III, F. R. J. Am. Chem. Soc. 1978, 100, 4858–4865.
(76) Masafumi, A.; Kunitomo, A.; Yukishige, I.; Hiroaki, S.; Masaji, O. J. Am. Chem. Soc. 1983, 105, 4049–4055.
(77) Mehta, A.; Jaouhari, R.; Benson, T. J.; Douglas, K. T. Tetrahedron Lett. 1992, 33, 5441-5444.
(78) Garber, S. B.; Kingsbury, J. S.; Gray, B. L.; Hoveyda, A. H. J. Am. Chem. Soc. 2000, 122, 8168–8179.
(79) Coburn, C. A.; Stachel, S. J.; Li, Y. M.; Rush, D. M.; Steele, T. G.; Chen-Dodson, E.; Holloway, M. K.; Xu, M.; Huang, Q.; Lai, M. T.; DiMuzio, J.; Crouthamel, M. C.; Shi, X. P.; Sardana, V.; Chen, Z.; Munshi, S.; Kuo, L.; Makara, G. M.; Annis, D. A.; Tadikonda, P. K.; Nash, H. M.; Vacca, J. P.; Wang, T. J Med Chem 2004, 47, 6117-9.
(80) Johansson, P. O.; Lindberg, J.; Blackman, M. J.; Kvarnström, I.; Vrang, L.; Hamelink, E.; Hallberg, A.; Rosenquist, Å.; Samuelsson, B. J Med Chem 2005, 48, 4400-9.
(81) Rahuel, J.; Rasetti, V.; Maibaum, J.; Rueger, H.; Goschke, R.; Cohen, N. C.; Stutz, S.; Cumin, F.; Fuhrer, W.; Wood, J. M.; Grutter, M. G. Chem Biol 2000, 7, 493-504.
(82) Barber, C. G.; Bunnage, M. E.; Harvey, J. W.; Mathias, J. P. U.S. Patent 2005/0020611, 2005.
(83) Simas, A. B. C.; Coelho, A. L.; Costa, P. R. R. Synthesis-Stuttgart 1999, 1017-1021.
(84) Arslan, T.; Abraham, A. T.; Hecht, S. M. Nucleosides Nucleotides 1998, 17, 515-30.
(85) Wångsell, F.; Gustafsson, K.; Kvarnström, I.; Borkakoti, N.; Edlund, M.; Jansson, K.; Lindberg, J.; Hallberg, A.; Rosenquist, Å.; Samuelsson, B. Eur J Med Chem 2010, 45, 870-82.

76
(86) Rasmussen, J. R.; Slinger, C. J.; Kordish, R. J.; Newman-Evans, D. D. J. Org. Chem. 1981, 46, 4843-4846.
(87) Rondot, B.; Durand, T.; Girard, J. P.; Rossi, J. C.; Schio, L.; Khanapure, S. P.; Rokach, J. Tetrahedron Letters 1993, 34, 8245-8248.
(88) Müller, M.; Huchel, U.; Geyer, A.; Schmidt, R. R. J Org Chem 1999, 64, 6190-6201.
(89) Crich, D.; Vinogradova, O. J Org Chem 2007, 72, 3581-3584. (90) Beher, D.; Graham, S. L. Expert Opin Investig Drugs 2005, 14, 1385-
409. (91) Loughlin, W. A.; Tyndall, J. D.; Glenn, M. P.; Fairlie, D. P. Chem Rev
2004, 104, 6085-117. (92) Lerchner, A.; Machauer, R.; Betschart, C.; Veenstra, S.; Rueeger, H.;
McCarthy, C.; Tintelnot-Blomley, M.; Jaton, A. L.; Rabe, S.; Desrayaud, S.; Enz, A.; Staufenbiel, M.; Paganetti, P.; Rondeau, J. M.; Neumann, U. Bioorg Med Chem Lett 2010, 20, 603-7.
(93) Ghosh, A. K.; Devasamudram, T.; Hong, L.; DeZutter, C.; Xu, X.; Weerasena, V.; Koelsch, G.; Bilcer, G.; Tang, J. Bioorg Med Chem Lett 2005, 15, 15-20.
(94) Fürstner, A.; Davies, P. W. Chem Commun (Camb) 2005, 2307-20. (95) Esch, P. M.; Hiemstra, H.; de Boer, R. F.; Speckamp, W. N.
Tetrahedron 1992, 48, 4659–4676. (96) He, L.; Byun, H.; Bittman, R. Tetrahedron Letters 1998, 39, 2071-2074. (97) Calderón, F.; Doyagüez, E. G.; Fernández-Mayoralas, A. J Org Chem
2006, 71, 6258-61. (98) Mori, K.; Kinsho, T. Liebigs Annalen der Chemie 1991, 1991, 1309–
1315. (99) Bayley, H.; Standring, D. N.; Knowles, J. R. Tetrahedron Letters 1978,
19, 3633–3634. (100) Listemann, M. L.; Schrock, R. R. Organometallics 1985, 4, 74–83. (101) Vintonyak, V. V.; Maier, M. E. Org Lett 2007, 9, 655-8. (102) Fürstner, A.; Rumbo, A. J Org Chem 2000, 65, 2608-11. (103) Rojo, I.; Martin, J. A.; Broughton, H.; Timm, D.; Erickson, J.; Yang, H.
C.; McCarthy, J. R. Bioorg Med Chem Lett 2006, 16, 191-5. (104) Machauer, R.; Laumen, K.; Veenstra, S.; Rondeau, J. M.; Tintelnot-
Blomley, M.; Betschart, C.; Jaton, A. L.; Desrayaud, S.; Staufenbiel, M.; Rabe, S.; Paganetti, P.; Neumann, U. Bioorg Med Chem Lett 2009, 19, 1366-70.
(105) Huang, Y.; Strobel, E. D.; Ho, C. Y.; Reynolds, C. H.; Conway, K. A.; Piesvaux, J. A.; Brenneman, D. E.; Yohrling, G. J.; Moore Arnold, H.; Rosenthal, D.; Alexander, R. S.; Tounge, B. A.; Mercken, M.; Vandermeeren, M.; Parker, M. H.; Reitz, A. B.; Baxter, E. W. Bioorg Med Chem Lett 2010, 20, 3158-60.
(106) Lindsley, S. R.; Moore, K. P.; Rajapakse, H. A.; Selnick, H. G.; Young, M. B.; Zhu, H.; Munshi, S.; Kuo, L.; McGaughey, G. B.; Colussi, D.; Crouthamel, M. C.; Lai, M. T.; Pietrak, B.; Price, E. A.; Sankaranarayanan, S.; Simon, A. J.; Seabrook, G. R.; Hazuda, D. J.;

77
Pudvah, N. T.; Hochman, J. H.; Graham, S. L.; Vacca, J. P.; Nantermet, P. G. Bioorg Med Chem Lett 2007, 17, 4057-61.
(107) Moore, K. P.; Zhu, H.; Rajapakse, H. A.; McGaughey, G. B.; Colussi, D.; Price, E. A.; Sankaranarayanan, S.; Simon, A. J.; Pudvah, N. T.; Hochman, J. H.; Allison, T.; Munshi, S. K.; Graham, S. L.; Vacca, J. P.; Nantermet, P. G. Bioorg Med Chem Lett 2007, 17, 5831-5.
(108) Maillard, M. C.; Hom, R. K.; Benson, T. E.; Moon, J. B.; Mamo, S.; Bienkowski, M.; Tomasselli, A. G.; Woods, D. D.; Prince, D. B.; Paddock, D. J.; Emmons, T. L.; Tucker, J. A.; Dappen, M. S.; Brogley, L.; Thorsett, E. D.; Jewett, N.; Sinha, S.; John, V. J Med Chem 2007, 50, 776-81.
(109) Calderon, N.; Chen, H. Y.; Scott, K. W. Tetrahedron Letters 1967, 8, 3327–3329.
(110) Astruc, D. New J. Chem. 2005, 29, 42-56. (111) Trnka, T. M.; Grubbs, R. H. Acc Chem Res 2001, 34, 18-29. (112) Hérisson, J.-L.; Chauvin, Y. Makromol. Chem. 1971, 141, 161–176. (113) Bazan, G. C.; Oskam, J. H.; Cho, H. N.; Park, L. Y.; Schrock, R. R. J.
Am. Chem. Soc. 1991, 113, 6899–6907. (114) Schwab, P.; France, M. B.; Ziller, J. W.; Grubbs, R. H. Angew. Chem.
In. Ed. Engl. 1995, 34, 2039–2041. (115) Scholl, M.; Ding, S.; Lee, C. W.; Grubbs, R. H. Org Lett 1999, 1, 953-6. (116) Zhan, Z.-Y. J. WO Patent 2007003135, 2007. (117) Paterson, I.; Muhlthau, F. A.; Cordier, C. J.; Housden, M. P.; Burton, P.
M.; Loiseleur, O. Org. Lett. 2009, 11, 353–356. (118) Betschart, C.; Lerchner, A.; Machauer, R.; Rueger, H.; Tintelnot-
Blomley, M.; Veenstra, S. J. U.S. Patent 2008/132477, 2008. (119) Caddick, S.; Haynes, A. K. K.; Judd, D. B.; Williams, M. R. V.
Tetrahedron Letters 2000, 41, 3513–3516. (120) Martinelli, M. J.; Nayyar, N. K.; Moher, E. D.; Dhokte, U. P.; Pawlak,
J. M.; Vaidyanathan, R. Org. Lett. 1999, 1, 447–450. (121) Sealy, J. M.; Truong, A. P.; Tso, L.; Probst, G. D.; Aquino, J.; Hom, R.
K.; Jagodzinska, B. M.; Dressen, D.; Wone, D. W.; Brogley, L.; John, V.; Tung, J. S.; Pleiss, M. A.; Tucker, J. A.; Konradi, A. W.; Dappen, M. S.; Toth, G.; Pan, H.; Ruslim, L.; Miller, J.; Bova, M. P.; Sinha, S.; Quinn, K. P.; Sauer, J. M. Bioorg Med Chem Lett 2009, 19, 6386-91.
(122) Probst, G. D.; Bowers, S.; Sealy, J. M.; Stupi, B.; Dressen, D.; Jagodzinska, B. M.; Aquino, J.; Gailunas, A.; Truong, A. P.; Tso, L.; Xu, Y. Z.; Hom, R. K.; John, V.; Tung, J. S.; Pleiss, M. A.; Tucker, J. A.; Konradi, A. W.; Sham, H. L.; Jagodzinski, J.; Toth, G.; Brecht, E.; Yao, N.; Pan, H.; Lin, M.; Artis, D. R.; Ruslim, L.; Bova, M. P.; Sinha, S.; Yednock, T. A.; Gauby, S.; Zmolek, W.; Quinn, K. P.; Sauer, J. M. Bioorg Med Chem Lett 2010, 20, 6034-9.
(123) Clarke, B.; Cutler, L.; Demont, E.; Dingwall, C.; Dunsdon, R.; Hawkins, J.; Howes, C.; Hussain, I.; Maile, G.; Matico, R.; Mosley, J.; Naylor, A.; O'Brien, A.; Redshaw, S.; Rowland, P.; Soleil, V.; Smith, K.

78
J.; Sweitzer, S.; Theobald, P.; Vesey, D.; Walter, D. S.; Wayne, G. Bioorg Med Chem Lett 2010, 20, 4639-44.
(124) Stachel, S. J.; Coburn, C. A.; Steele, T. G.; Crouthamel, M. C.; Pietrak, B. L.; Lai, M. T.; Holloway, M. K.; Munshi, S. K.; Graham, S. L.; Vacca, J. P. Bioorg Med Chem Lett 2006, 16, 641-4.
(125) Pardridge, W. M. J Neurochem 1998, 70, 1781-92. (126) Schmid, C. R.; Bryant, J. D.; Dowlatzedah, M.; Phillips, J. L.; Prather,
D. E.; Schantz, R. D.; Sear, N. L.; Vianco, C. S. J. Org. Chem. 1991, 56, 4056–4058.
(127) Parikh, J. R.; Doering, W. v. E. J. Am. Chem. Soc. 1967, 89, 5505–5507.
(128) Dess, D. B.; Martin, J. C. J. Org. Chem. 1983, 48, 4155–4156.














![RESEARCHARTICLE OpAccess Transformer-C: nif A terpretation · 2020. 3. 18. · BCF Bioconcentrationfactor[47] 378 BACE Human-secretase1(BACE-1)inhibitors[46] 1513 FreeSolv Freesolvationenergy[46]](https://img.pdfslide.net/doc/110x75/6148dbf92918e2056c22f67d/researcharticle-opaccess-transformer-c-nif-a-terpretation-2020-3-18-bcf-bioconcentrationfactor47.jpg)














