Embed Size (px)
Citation preview

BIENVENIDOS A NUESTRA CLASE 7
En esta primera parte de la clase abordaremos las siguientes temáticas:
El Deterioro Cognitivo Leve.
Demencia Tipo Alzheimer.1
Tratamientos farmacológicos.
Teorías y técnicas de rehabilitación y sus debilidades.
La rehabilitación en Demencias. Evaluación, objetivos. Estrategias.
La prevención en las Demencias. Estrategias.
La institucionalización: criterios.
Hacia una intervención integral.
DETERIORO COGNITIVO LEVE
En el deterioro cognitivo leve (MCI), los cambios en la cognición superan los
cambios normales y esperados relacionados con la edad. En una clasificación de
MCI, se distinguen formas amnesicas de la forma no amnésica. La forma
amnésica suele preceder a la enfermedad de Alzheimer. 2
Signos y síntomas
Los síntomas de deterioro cognitivo leve (MCI) a menudo son poco precisos e
incluyen los siguientes:
Pérdida de memoria
Alteración del lenguaje (por ejemplo, dificultad para encontrar palabras)
1 https://emedicine.medscape.com/article/1134817-overview
2 https://emedicine.medscape.com/article/1136393-overview#a1

Déficit de atención (por ejemplo, dificultad para seguir o centrarse en las
conversaciones)
Deterioro de las habilidades visuoespaciales (p. Ej., Desorientación en
entornos familiares en ausencia de condiciones motoras y sensoriales que
podrían explicar la queja)
Ronald C. Petersen postuló que el elemento definitorio de MCI es una esfera única
de deterioro cognitivo lentamente progresivo que no es atribuible a déficits
motores o sensoriales y a la cual se pueden agregar otras áreas de participación,
antes de que el deterioro social u ocupacional sobreviene (porque esto ocurrencia
marca el inicio de la demencia). 3
DIAGNÓSTICO
Aunque no hay una característica única del examen físico general que caracteriza
al MCI, se debe incluir lo siguiente en la evaluación general del paciente:
Evaluación del estado mental
Examen de la presencia de condiciones comórbidas causantes potenciales
Examen de la presencia de deficiencias sensoriales y / o motoras como
causas potenciales o factores exacerbantes.
No existen estudios diagnósticos específicos para el deterioro cognitivo leve. Sin
embargo, la mayoría de los médicos realizan una evaluación básica al mínimo
para excluir posibles causas tratables (p. Ej., Enfermedad de la tiroides, deficiencia
de cobalamina). Se están realizando investigaciones en la búsqueda de
marcadores biológicos que puedan ayudar a diferenciar entre la gran cantidad de
afecciones que pueden progresar desde la ICM hasta la demencia completa.
Entre las imágenes cerebrales, resonancia magnética (RMN O MRI) o tomografía
(TC) a En general, se prefiere la RMN, ya que el volumen total del cerebro y el
3 Petersen RC. Conceptual overview. Petersen RC. Mild Cognitive Impairment: Aging to Alzheimer's
Disease. 1-14. New York, NY: Oxford University Press, Inc; 2003.

hipocampo en la RMN puede predecir la progresión de la MCI a la enfermedad de
Alzheimer (EA).4
Sin embargo, no hay parámetros establecidos para integrar este hallazgo en el
diagnóstico y manejo de rutina de MCI. Además, también hay algunas pruebas
preliminares del uso de la PET-FDG basal del cerebro junto con la evaluación de
la memoria episódica para predecir la conversión a la enfermedad de Alzheimer. 5
No hay pruebas neuropsicológicas estipuladas para pacientes con MCI, ni hay
puntos de corte predeterminados (por ejemplo, 1.0, 1.5 o 2 desviaciones estándar
por debajo de la media). Sin embargo, los clínicos utilizan los resultados de la
memoria estandarizada y las pruebas cognitivas para determinar si estos datos
representan cambios significativos con respecto a la presunta línea de base de un
paciente. En general, se requieren pruebas en serie para establecer si la función
cognitiva del paciente está mejorando, permaneciendo estable o progresando a
una demencia clinica.
TRATAMIENTO:
Aunque no existe un tratamiento establecido para MCI, el donepezilo retrasa la
progresión a la EA en pacientes con depresión con MCI sin afectar sus síntomas
depresivos, y algunas pruebas sugieren que las intervenciones cognitivas pueden
tener un efecto positivo. 6
No se ha encontrado que los inhibidores de la colinesterasa retrasen la aparición
de la EA o la demencia en el MCI. 7
Es esencial identificar y monitorear a los pacientes con MCI, debido a su mayor
riesgo de AD (y, en menor medida, otras condiciones). Además, en la medida de
lo posible, controlar y tratar cualquier manifestación que agrave sus síntomas
cognitivos.
4 Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC; Alzheimer's Disease Neuroimaging
Initiative (ADNI). Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort. Current Alzheimer Research. 2009 Aug. 6(4):347-61. [Medline]. 5 Landau SM, Harvey D, Madison CM, Reiman EM, Foster NL, Aisen PS. Comparing predictors of
conversion and decline in mild cognitive impairment. Neurology. 2010 Jul 20. 75(3):230-8. [Medline]. 6 Simon SS, Yokomizo JE, Bottino CM. Cognitive intervention in amnestic Mild Cognitive Impairment: A
systematic review. Neurosci Biobehav Rev. 2012 Feb 1. 36(4):1163-1178. [Medline]. 7 Panza F, Frisardi V, Capurso C, D'Introno A, Colacicco AM, Chiloiro R, et al. Effect of donepezil on the
continuum of depressive symptoms, mild cognitive impairment, and progression to dementia. Journal of the
American Geriatrics Society. 2010 Feb. 58(2):389-90. [Medline]

La dieta y la actividad cognitiva pueden tener algunos efectos positivos en
pacientes con MCI. El riesgo de desarrollar MCI es menor en las personas que
consumen una dieta mediterránea 8. y las actividades cognitivas que representen
desafíos mentales asi como tambien el ejercicio moderado tienen el potencial
beneficioso en MCI. 9
Se han empleado varios términos para caracterizar el deterioro cognitivo asociado
con el envejecimiento, incluido el olvido senescente benigno, el deterioro de la
memoria asociado con la edad y el deterioro cognitivo asociado con la edad. El
término deterioro cognitivo leve (MCI) pretende representar una etapa intermedia
entre el envejecimiento normal y el desarrollo del envejecimiento patológico y la
demencia.
Otros términos con connotaciones similares a los de MCI incluyen deterioro de
memoria aislado, demencia incipiente y pródromo de demencia. Sin embargo,
estos términos no son tan ampliamente aceptados como MCI y no deben
considerarse sinónimos exactos.
De las funciones de memoria normales, algunas pueden disminuir con la edad, y
otras no.
Las funciones de memoria que permanecen relativamente estables a medida
que aumenta la edad incluyen las siguientes:
Memoria semántica – Significados, entendimiento y conocimiento general del
mundo; aunque esta función generalmente permanece estable con la edad,
especialmente si la información se usa con frecuencia, 10 la recuperación de
información altamente específica (por ejemplo, nombres) generalmente disminuye
8 Roberts RO, Geda YE, Cerhan JR, Knopman DS, Cha RH, Christianson TJ, et al. Vegetables, Unsaturated
Fats, Moderate Alcohol Intake, and Mild Cognitive Impairment. Dementia and Geriatric Cognitive Disorders.
2010 May 22. 29(5):413-423. [Medline] 9 Geda YE, Roberts RO, Knopman DS, Christianson TJ, Pankratz VS, Ivnik RJ, et al. Physical exercise,
aging, and mild cognitive impairment: a population-based study. Archives of Neurology. 2010 Jan. 67(1):80-
6. [Medline]. 10 Luo L, Craik FI. Aging and memory: a cognitive approach. Can J Psychiatry. 2008 Jun. 53(6):346-53.
[Medline]

Memoria procedural o procesal: adquisición y posterior ejecución de habilidades
cognitivas y motoras. 11
Las funciones de la memoria que disminuyen con la edad incluyen las
siguientes:
Memoria de trabajo: retener y manipular información en la mente, como cuando se
reorganiza una lista corta de palabras en orden alfabético. 12 La velocidad de
trabajo verbal y visoespacial, la memoria y el aprendizaje, con la cognición
visuoespacial más afectada por el envejecimiento que la cognición verbal.13
Memoria episódica - Eventos personales y experiencias. 14
Velocidad de procesamiento. 15
Memoria prospectiva: la capacidad de recordar realizar una acción en el futuro
(por ejemplo, recordar cumplir una cita o tomar un medicamento). 16
Capacidad para recordar información de texto nuevo, hacer inferencias sobre la
información de texto nuevo, acceder a conocimientos previos en la memoria a
largo plazo e integrar conocimientos previos con información de texto nuevo. 17
Recuerdo. 18
Para demostrar que la función cognitiva de un paciente es peor de lo que
normalmente se esperaría para su edad, es necesario realizar pruebas
neuropsicológicas para poder comparar el desempeño del paciente con el de un
grupo de control de la misma edad (y, idealmente, de la educación). .
11 Luo L, Craik FI. Aging and memory: a cognitive approach. Can J Psychiatry. 2008 Jun. 53(6):346-53.
[Medline] 12 Luo L, Craik FI. Aging and memory: a cognitive approach. Can J Psychiatry. 2008 Jun. 53(6):346-53.
[Medline]; 13 Jenkins L, Myerson J, Joerding JA, et al. Converging evidence that visuospatial cognition is more age-
sensitive than verbal cognition. Psychol Aging. 2000 Mar. 15(1):157-75. [Medline]. 14 Luo L, Craik FI. Aging and memory: a cognitive approach. Can J Psychiatry. 2008 Jun. 53(6):346-53.
[Medline] 15 Head D, Rodrigue KM, Kennedy KM, et al. Neuroanatomical and cognitive mediators of age-related differences in episodic memory. Neuropsychology. 2008 Jul. 22(4):491-507. [Medline]. 16 Luo L, Craik FI. Aging and memory: a cognitive approach. Can J Psychiatry. 2008 Jun. 53(6):346-53.
[Medline] 17 Hannon B, Daneman M. Age-related changes in reading comprehension: an individual-differences
perspective. Experimental Aging Research. 2009. 35(4):432-56. [Medline]. 18 Parks CM, Decarli C, Jacoby LL, Yonelinas AP. Aging effects on recollection and familiarity: the role of
white matter hyperintensities. Neuropsychology, development, and cognition. Section B, Aging,
neuropsychology and cognition. Feb 2010. 19:1-17. [Medline].

Los grados leves de deterioro cognitivo, en particular cuando los pacientes se
informan a sí mismos, representan un desafío importante para el clínico. El médico
puede estar tratando con un paciente con una condición leve o transitoria, un
efecto adverso inducido por un medicamento o un trastorno depresivo; el paciente
puede estar en las etapas tempranas de una afección que eventualmente llevará a
una demencia; o la queja puede deberse a una condición psicológica más que a
un trastorno cerebral orgánico.
Debido a que una variedad de afecciones puede resultar en una queja de deterioro
cognitivo, se debe buscar un estudio individualizado para tales afecciones y un
consenso sobre un enfoque terapéutico. Hasta la fecha, ningún medicamento ha
sido aprobado por la Administración de Drogas y Alimentos de los Estados Unidos
(FDA) para el tratamiento de MCI.
FISIOPATOLOGÍA
En el deterioro cognitivo leve (MCI), el deterioro cognitivo supera los cambios
normales esperados relacionados con la edad, pero las actividades funcionales se
conservan en gran medida; por lo tanto, MCI no cumple con los criterios para la
demencia. Se reconocen diferentes subtipos de MCI. Una clasificación común
distingue entre las formas amnésicas y no amnésicas de MCI.
El MCI amnésico, en el que predomina el deterioro de la memoria, suele ser un
precursor de la enfermedad de Alzheimer clínica (EA). Las formas no amnésicas
de MCI se caracterizan por una variedad de deficiencias cognitivas, la más común
de las cuales es probablemente la función ejecutiva deteriorada.
La fisiopatología del MCI es multifactorial. La mayoría de los casos de MCI
amnésicos se deben a cambios patológicos de la enfermedad de Alzheimer que
aún no se han vuelto lo suficientemente graves como para causar demencia
clínica. 19
Al menos en las poblaciones de investigación de especialidades, las autopsias
realizadas en pacientes con MCI amnésico han encontrado que la neuropatología
es típica de la enfermedad de Alzheimer. Markesbery WR, Schmitt FA, Kryscio RJ,
19 Morris JC, Storandt M, Miller JP, et al. Mild cognitive impairment represents early-stage Alzheimer
disease. Arch Neurol. 2001 Mar. 58(3):397-405. [Medline].

et al. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006
Jan. 63(1):38-46. [Medline].
El MCI no amnésico puede estar asociado con enfermedad cerebrovascular,
demencias frontotemporales (como precursor) o sin patología específica.
ETIOLOGÍA
El deterioro cognitivo leve (MCI) es heterogéneo tanto en sus manifestaciones
clínicas como en su etiología. Dado que el MCI amnésico a menudo resulta de la
patología de la enfermedad de Alzheimer (EA), no es sorprendente que la mayoría
de los pacientes con MCI amnésica avancen a la EA clínica dentro de los 6 años.
Las formas no amnésicas de MCI pueden deberse a enfermedad cerebrovascular,
demencia con cuerpos de Lewy, enfermedad de Parkinson, demencias
frontotemporales, enfermedad de Alzheimer atípica o ausencia de patología
subyacente específica.
Los trastornos del estado de ánimo, las enfermedades médicas y los
medicamentos pueden afectar la cognición de tal manera que un paciente cumpla
con los criterios de MCI (generalmente MCI no amnésico). Muchos de estos
pacientes tienen resultados de pruebas neuropsicológicas normales cuando se
reevalúan un año después de eliminar cirtos farmacos o restituir enfermedades
clinicas o estados animicos.
EPIDEMIOLOGÍA
Las estimaciones de prevalencia anual para el deterioro cognitivo leve (MCI)
varían del 12% al 18% en personas mayores de 60 años, un hallazgo que se
refleja en varios estudios internacionales. 20
La prevalencia de deterioro cognitivo leve aumenta con edad. La prevalencia es
del 10% en los de 70 a 79 años y del 25% en los de 80 a 89 años. 21
20 Busse A, Hensel A, Gühne U, Angermeyer MC, Riedel-Heller SG. Mild cognitive impairment: long-term course of four clinical subtypes. Neurology. 2006 Dec 26. 67 (12):2176-85. [Medline]. Di Carlo A, Lamassa
M, Baldereschi M, Inzitari M, Scafato E, Farchi G, et al. CIND and MCI in the Italian elderly: frequency,
vascular risk factors, progression to dementia. Neurology. 2007 May 29. 68 (22):1909-16. [Medline]. Ganguli
M, Chang CC, Snitz BE, Saxton JA, Vanderbilt J, Lee CW. Prevalence of mild cognitive impairment by
multiple classifications: The Monongahela-Youghiogheny Healthy Aging Team (MYHAT) project. Am J
Geriatr Psychiatry. 2010 Aug. 18 (8):674-83. [Medline].Larrieu S, Letenneur L, Orgogozo JM, Fabrigoule C,
Amieva H, Le Carret N, et al. Incidence and outcome of mild cognitive impairment in a population-based
prospective cohort. Neurology. 2002 Nov 26. 59 (10):1594-9. [Medline].

Muchos estudios indican que el riesgo de enfermedad de Alzheimer (EA) es
significativamente mayor en las mujeres que en los hombres, y por lo tanto, se
presume que la probabilidad de desarrollar MCI es mayor en las mujeres que en
los hombres. Prácticamente nada se sabe sobre los factores culturales y raciales
que influyen en las manifestaciones clínicas de MCI.
PRESENTACIÓN CLÍNICA
Historia del paciente
Los pacientes con deterioro cognitivo leve (MCI) a menudo presentan síntomas
vagos y subjetivos de disminución del rendimiento cognitivo, lo que puede ser
difícil de distinguir de la disminución típica del rendimiento en personas mayores
sanas. Se dice que el síntoma más común es la pérdida de memoria, de acuerdo
con la tendencia de que el MCI amnésico es el tipo más común. Sin embargo,
algunos estudios demuestran que la forma más común de MCI afecta a múltiples
esferas de la cognición.
Las presentaciones menos comunes de MCI incluyen alteraciones del lenguaje (p.
Ej., dificultad para encontrar palabras), déficit de atención (p. Ej., dificultad para
seguir o centrarse en las conversaciones) y deterioro de las habilidades
visuoespaciales (p. Ej., desorientación en entornos familiares en ausencia de
motricidad y sensibilidad anormales).
La disociación de los síntomas puramente cognitivos de los atribuibles a diversos
grados de privación sensorial (p. Ej., Pérdida de la audición o de la agudeza
visual) que tienden a coexistir en la misma población de pacientes suele ser difícil.
Los médicos y evaluadores deben definir siempre hacer busqueda de procesos
relacionados con la seguridad que sean apropiadas para los pacientes con
demencia, por ejemplo, sobre armas, conducir y posibles incendios en el hogar
que involucren cigarrillos, estufas o chimeneas, a los pacientes con MCI .
EXAMEN FÍSICO
Ninguna característica del examen físico general es característica de MCI. Sin
embargo, se debe realizar un examen físico completo como parte de la evaluación
21 Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling,
participation, baseline measures and sample characteristics. Neuroepidemiology. 2008. 30(1):58-69.
[Medline].

general para determinar si existe alguna afección capaz de causar MCI (por
ejemplo, enfermedad de la tiroides, deficiencia de cobalamina o ETS) y si hay
algún trastorno sensorial y motor. Deficiencias que podrían explicar o agravar los
síntomas. El examen del estado mental también es importante para documentar el
grado de disfunción cognitiva.
DIAGNÓSTICO DIFERENCIAL
El deterioro cognitivo leve (MCI) puede deberse a prácticamente cualquier
trastorno que cause disfunción cerebral. Las causas comunes incluyen lo
siguiente:
- Enfermedad de Alzheimer (AD)
- Enfermedad cerebrovascular
- Parkinson
- Degeneraciones frontotemporales
- Enfermedad de tiroides
- Infeccion por VIH
- Depresión
- Enfermedad metabólica y endocrina
- Efectos adversos sobre el sistema nervioso central de fármacos y tóxicos.
- Infeccion cerebral
- Lesión cerebral traumática
- Efectos adversos cognitivos de los trastornos del sueño.
- Deficiencia de cobalamina
- Estrés psicológico crónico.
Según un análisis de 5150 pacientes de 65 años o más del Estudio de salud
cardiovascular, los pacientes con fibrilación auricular (FA) alcanzan umbrales
clínicos para el deterioro cognitivo y demencia a una edad más temprana que los
pacientes sin FA, incluso en ausencia de accidente cerebrovascular clínico. 22
22 Jeffrey S. AF linked to earlier, more rapid cognitive decline. Medscape Medical News. June 7, 2013. [Full
Text]. Thacker EL, McKnight B, Psaty BM, Longstreth WT Jr, Sitlani CM, Dublin S, et al. Atrial fibrillation
and cognitive decline: A longitudinal cohort study. Neurology. 2013 Jun 5. [Medline].

Los trastornos depresivos son particularmente frecuentes en los adultos mayores
(aproximadamente el 15%), que con frecuencia muestran síntomas somáticos
vagos y ansiedad, y reportan incapacidad para concentrarse y poca memoria.
Estos pacientes suelen negar un estado de ánimo triste, pero a menudo admiten
síntomas de sueño, falta de interés en las cosas que solían disfrutar, pérdida de
apetito y falta de motivación. La depresión puede estar acompañada por una
disfunción cognitiva que disminuye con el tratamiento exitoso de la depresión.
La asociación entre la depresión y la EA y otras demencias probablemente sea
compleja, y la depresión puede diagnosticarse erróneamente en el ámbito de la
demencia. Los datos de Framingham han ayudado a reforzar la asociación
epidemiológica, documentando un aumento del 50% en la EA y la demencia en
aquellos que estaban deprimidos al inicio del estudio. 23Durante un período de
seguimiento de 17 años, un total de 21.6% de los participantes que estaban
deprimidos al inicio del estudio desarrollaron demencia, en comparación con
16.6% de los que no estaban deprimidos.
En otro estudio se observó que la depresión recurrente era particularmente
negativa en este sentido: tener 1 episodio de depresión genero un aumento de 87
a 92% en el riesgo de demencia, mientras que tener 2 o más episodios casi
duplicó el riesgo de demencia (pero no aumentó el riesgo de MCI incidente) ). 24
LABORATORIOS
No se indican estudios específicos de laboratorio para el deterioro cognitivo leve
(MCI). La mayoría de los profesionales realizan al menos un trabajo básico para
descartar afecciones tratables que pueden causar demencia, como la enfermedad
de la tiroides y la deficiencia de cobalamina. Estas evaluaciones no son
obligatorias.
Se está realizando una búsqueda de marcadores biológicos de MCI que puedan
ayudar a distinguir entre las muchas afecciones que conducen de MCI a la
demencia. Hasta ahora, sin embargo, no se ha alcanzado un acuerdo unánime, y
23 Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive symptoms and risk of
dementia: the Framingham Heart Study. Neurology. 2010 Jul 6. 75(1):35-41. [Medline]. 24 Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms and the incidence of dementia
and mild cognitive impairment. Neurology. 2010 Jul 6. 75(1):27-34. [Medline].

marcadores potencialmente útiles, como anomalías funcionales y estructurales
encontradas en estudios de imagen (por ejemplo, atrofia del hipocampo e
hipoperfusión cerebral) y supuestos marcadores bioquímicos (por ejemplo, alelo
de apolipoproteína E epsilon 4), sigue siendo controvertido
PRUEBAS NEUROPSICOLÓGICAS
Se requieren pruebas neuropsicológicas en casos de deterioro cognitivo leve
(MCI) para demostrar que el paciente está por debajo de algún punto de corte en
las pruebas de memoria estandarizadas (así como en otras pruebas cognitivas).
Sin embargo, el punto de corte exacto (ya sea 1.0, 1.5 o 2 desviaciones estándar
por debajo de la media) y las pruebas neuropsicológicas particulares que se
usarán no están totalmente definidos.
Debido a que pocos pacientes con MCI se han sometido a pruebas de referencia
en estas medidas antes del inicio del deterioro, el médico tendrá que determinar si
un puntaje en particular representa un cambio significativo con respecto a la
presunta línea de base de un paciente. Tales determinaciones son inexactas, y
eventualmente se necesitarán pruebas en serie para establecer si la función
cognitiva del paciente está mejorando, permaneciendo estable o progresando a
demencia clínica en toda regla. Un aspecto útil de esta prueba es la capacidad del
neuropsicólogo para establecer un perfil para el paciente según su género, edad y
educación, y luego evaluar si su nivel de función es adecuado para ese perfil.
La Alzheimer's Association publicó pautas, que incluyen un algoritmo, para ayudar
a los médicos en el entorno de atención primaria a detectar el deterioro cognitivo y
determinar si se necesita una referencia o pruebas adicionales.
El algoritmo incluye los siguientes componentes : 25
- Revisión de la información de evaluación de riesgo para la salud del
paciente
- Observación del paciente
- Uso de consultas no estructuradas.
25 Risacher SL, Saykin AJ, West JD, Shen L, Firpi HA, McDonald BC; Alzheimer's Disease Neuroimaging
Initiative (ADNI). Baseline MRI predictors of conversion from MCI to probable AD in the ADNI cohort.
Current Alzheimer Research. 2009 Aug. 6(4):347-61. [Medline]. Landau SM, Harvey D, Madison CM,
Reiman EM, Foster NL, Aisen PS. Comparing predictors of conversion and decline in mild cognitive
impairment. Neurology. 2010 Jul 20. 75(3):230-8. [Medline].

- Uso de herramientas estructuradas de evaluación cognitiva para pacientes
e informantes.
Los siguientes 3 herramientas de evaluación cognitiva se recomiendan para uso
de rutina por los médicos de atención primaria: 26Evaluación general de la
cognición (GPCOG)
- Mini-Cog
- Pantalla de deterioro de la memoria (MIS)
Además, la Asociación de Alzheimer recomienda las siguientes 3 herramientas de
evaluación cognitiva para usar con el cónyuge, la familia o los amigos del paciente
[8, 9]:
- Evaluación de la cognición del médico general informante (informante
GPCOG)
- Pantalla de 8 preguntas AD (AD8)
- Cuestionario breve para informantes sobre el deterioro cognitivo en los
ancianos (IQCODE breve)
TRATAMIENTO Y MANEJO
Atención médica
En la actualidad, no existe un tratamiento establecido para el deterioro cognitivo
leve (MCI). No se ha encontrado que los inhibidores de la colinesterasa retrasen la
aparición de la enfermedad de Alzheimer (EA) o la demencia en individuos con
MCI; sin embargo, se ha encontrado que el donepezilo retrasa la progresión a la
EA en pacientes con MCI con depresión sin afectar sus síntomas de depresión.
Hay algunas pruebas que sugieren que las intervenciones cognitivas pueden tener
un efecto positivo.
Una recomendación de parámetros de práctica de la Academia Americana de
Neurología establece que los pacientes con MCI deben ser identificados y
monitoreados debido a su mayor riesgo de AD y, en menor medida, a otras
condiciones de demencia. Obviamente, corregir (en la medida de lo posible)
26 KRAL VA. Senescent forgetfulness: benign and malignant. Can Med Assoc J. 1962 Feb 10. 86:257-60.
[Medline].

cualquier manifestación motora y sensorial que agrave los síntomas cognitivos es
importante para minimizar su impacto en la ICM.
Se debe prestar especial atención a la necesidad de hacer una declaración legal
sobre la competencia de los pacientes para manejar sus propios asuntos. Debido
a que los pacientes con MCI, por definición, no tienen demencia, generalmente no
necesitan asignar un poder legal a nadie más, a diferencia de los pacientes con
AD, quienes eventualmente necesitarán esa ayuda.
Dieta
Roberts et al descubrieron que el riesgo de desarrollar MCI es menor en las
personas que consumen una dieta mediterránea, que es alta en verduras y grasas
no saturadas. 27
Un ensayo aleatorizado, doble ciego, controlado con placebo que incluyó a 25
sujetos ancianos con LMI determinó que la suplementación dietética con una
emulsión oleosa de ácido docosahexaenoico (DHA) -fosfolípidos que contienen
melatonina y triptófano produjo mejoras significativas en varias medidas de la
función cognitiva en comparación con la suplementación con el placebo. 28
De acuerdo con un estudio de 663 adultos en Singapur de 60 años de edad y
mayores, el consumo de más de dos porciones estándar de hongos por semana
puede reducir las probabilidades de desarrollar MCI hasta en un 50%. 29
Actividad general
Debido a que la actividad física, social y mental a menudo se recomiendan para
pacientes con AD y porque MCI a menudo anuncia AD, muchos expertos han
sugerido que las actividades desafiantes mentales (por ejemplo, crucigramas y
rompecabezas) pueden ser útiles para los pacientes con MCI. Aunque no hay
pruebas definitivas de que estos ejercicios sean eficaces, se recomienda
recomendarlos a pacientes con MCI.
27 Roberts RO, Geda YE, Cerhan JR, Knopman DS, Cha RH, Christianson TJ, et al. Vegetables, Unsaturated Fats, Moderate Alcohol Intake, and Mild Cognitive Impairment. Dementia and Geriatric Cognitive Disorders.
2010 May 22. 29(5):413-423. [Medline]. 28 Mariangela R, Annalisa O, Milena F, Marco M, Neldo A, Roberta C, et al. Effects of a diet integration with
an oily emulsion of DHA-phospholipids containing melatonin and tryptophan in elderly patients suffering
from mild cognitive impairment. Nutr Neurosci. 2011 Dec 20. [Medline]. 29 Feng L, Cheah IK, Ng MM, Li J, Chan SM, Lim SL, et al. The Association between Mushroom
Consumption and Mild Cognitive Impairment: A Community-Based Cross-Sectional Study in Singapore. J
Alzheimers Dis. 2019. 68 (1):197-203. [Medline].

Tales ejercicios deben mantenerse a un nivel de dificultad que sea razonable para
el paciente. Idealmente, deberían ser interactivos en lugar de pasivos, y deberían
administrarse de una manera que no cause frustración excesiva. Si una actividad
no es agradable o estimulante para el paciente, es poco probable que ofrezca
muchos beneficios cognitivos. En tales casos, la búsqueda de otras actividades
cognitivas similares puede ser beneficiosa.
El aislamiento social se puede minimizar mediante la derivación a centros
comunitarios para personas mayores o un programa de tratamiento diurno. Las
estrategias cognitivas de rehabilitación y rehabilitación ofrecen una promesa
considerable en MCI 30y, por lo tanto, se están explorando.
Un creciente cuerpo de evidencia sugiere que la actividad física y el ejercicio son
beneficiosos para la salud del cerebro. Un estudio prospectivo sugirió que la
participación en el ejercicio moderado de cualquier frecuencia en la mediana edad
o en la vida tardía se asociaba con la reducción de las probabilidades de tener
MCI. 31
Según un estudio, el ejercicio aeróbico se asoció con una leve mejoría en la
cognición. 32La actualización de 2017 de la guía de la Academia Americana de
Neurología sobre el deterioro cognitivo leve (MCI), que cuenta con el respaldo de
la Alzheimer's Association, recomienda que los pacientes con MCI hagan ejercicio
regularmente como parte de un enfoque general para controlar sus síntomas. 33
Sin embargo, un estudio de 2018 publicado en BMJ sugiere que el ejercicio
riguroso no es eficaz contra la demencia. Los investigadores asignaron a 329
participantes a un programa de ejercicios aeróbicos y de fuerza y 165 a la atención
habitual (n = 494). El resultado primario fue la puntuación en la subescala
30 Galante E, Venturini G, Fiaccadori C. Computer-based cognitive intervention for dementia: preliminary
results of a randomized clinical trial. G Ital Med Lav Ergon. 2007 Jul-Sep. 29(3 Suppl B):B26-32. [Medline] 31 Geda YE, Roberts RO, Knopman DS, Christianson TJ, Pankratz VS, Ivnik RJ, et al. Physical exercise,
aging, and mild cognitive impairment: a population-based study. Archives of Neurology. 2010 Jan. 67(1):80-6. [Medline]. 32 Lautenschlager NT, Cox KL, Flicker L, Foster JK, van Bockxmeer FM, Xiao J, et al. Effect of physical
activity on cognitive function in older adults at risk for Alzheimer disease: a randomized trial. JAMA. 2008
Sep 3. 300 (9):1027-37. [Medline]. 33 Petersen RC, Lopez O, Armstrong MJ, Getchius TSD, Ganguli M, Gloss D, et al. Practice guideline update
summary: Mild cognitive impairment: Report of the Guideline Development, Dissemination, and
Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018 Jan 16. 90 (3):126-
135. [Medline].

cognitiva de escala de evaluación de la enfermedad de Alzheimer (ADAS-cog) a
los 12 meses.
Los resultados secundarios incluyeron actividades de la vida diaria, síntomas
neuropsiquiátricos, calidad de vida relacionada con la salud y calidad de vida del
cuidador y carga. Los resultados muestran que la puntuación media de ADAS-cog
había aumentado a 25.2 (SD 12.3) en el brazo de ejercicio y a 23.8 (SD 10.4) en el
brazo de atención habitual, lo que indica un mayor deterioro cognitivo en el grupo
de ejercicio, pero la importancia clínica de este hallazgo es incierta . En general, el
estudio encontró que un programa de ejercicios aeróbicos y de fuerza de
intensidad moderada a alta no retarda el deterioro cognitivo en personas con
demencia leve a moderada. 34Otro estudio mostró ciertas actividades para reducir
el riesgo de MCI en individuos cognitivamente normales mayores de 70 años.
Estos incluían juegos, leyendo revistas, participando en manualidades, uso de
computadoras y actividades sociales. Entre estos, se demostró que ser social y
usar computadoras reduce el riesgo de MCI en personas que también eran
portadoras de APOE4. 35
Pronóstico
Muchos pacientes con deterioro cognitivo leve (MCI) eventualmente experimentan
un deterioro progresivo en sus habilidades para realizar actividades de la vida
diaria, la cognición y el comportamiento.
Los subtipos de MCI progresan a la enfermedad de Alzheimer (EA) a diferentes
tasas. Un estudio realizado por Rountree et al mostró que la tasa de conversión a
AD fue del 56% para el MCI amnésico, del 50% para el MCI por debajo del umbral
amnésico y del 52% para el MCI no amnésico. 36Para todos los subtipos de MCI,
34 Lamb SE, Sheehan B, Atherton N, et al. Dementia And Physical Activity (DAPA) trial of moderate to high
intensity exercise training for people with dementia: randomised controlled trial. BMJ. May 16, 2018. 361:[Full Text]. 35 Krell-Roesch, J., Roberts, R., Pink, A., et al. Mentally Stimulating Activities in Late-Life and the Risk of
Incident Mild Cognitive Impairment: A Prospective Cohort Study. Neurology. 2016. 86:S35-007.
36 Rountree SD, Waring SC, Chan WC, et al. Importance of subtle amnestic and nonamnestic deficits in mild
cognitive impairment: prognosis and conversion to dementia. Dement Geriatr Cogn Disord. 2007. 24(6):476-
82. [Medline].

la tasa de conversión de 4 años a la demencia fue del 56% (14% anual), y la de la
enfermedad de Alzheimer fue del 46% (11% anual). En comparación, las personas
de edad avanzada sanas desarrollan EA a una tasa de 1-2% por año.
Boyle et al informaron que los pacientes con MCI tienen casi 7 veces más
probabilidades de desarrollar AD que los individuos de mayor edad sin deterioro
cognitivo. 37De los pacientes con MCI, se dice que el 80% progresa a demencia
después de aproximadamente 6 años.
Al menos un estudio bien diseñado ha demostrado que el MCI, según se identifica
en el Cuestionario de estado mental portátil corto, es un predictor independiente
de mortalidad. 38
La gravedad del deterioro de la memoria es predictiva de la progresión a la
enfermedad de Alzheimer: los pacientes con un deterioro de la memoria más
grave tienen más probabilidades de progresar. Hay ciertas características
neurorradiológicas que predicen la progresión de MCI. Estos incluyen los
hallazgos de atrofia y pérdida de volumen en el lóbulo temporal mediano, así como
un patrón hipometabólico en la exploración FDG-PET.39 Además, los portadores
del genotipo APOE4 tienen un mayor riesgo de progresión, pero la prueba APoE4
no se recomienda para uso de rutina. [46] Una nueva modalidad que podría
resultar útil para predecir y monitorear la progresión de MCI es un nuevo marcador
PET que se centra en el papel de tau. Los primeros datos sugieren que la
diseminación de tau lateralmente, fuera del lóbulo temporal medial, puede predecir
un pronóstico desfavorable y una progresión más rápida.40
Guias de diagnostico:
Asociación de Alzheimer
37 . Boyle PA, Wilson RS, Aggarwal NT, et al. Mild cognitive impairment: risk of Alzheimer disease and rate
of cognitive decline. Neurology. 2006 Aug 8. 67(3):441-5. [Medline]. 38 Sachs GA, Carter R, Holtz LR, Smith F, Stump TE, Tu W, et al. Cognitive impairment: an independent predictor of excess mortality: a cohort study. Ann Intern Med. 2011 Sep 6. 155(5):300-8. [Medline]. 39 Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, et al. Impact of the Alzheimer's
Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement. 2015 Jul. 11 (7):865-84. [Medline].
Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, et al. Increased hippocampal
activation in mild cognitive impairment compared to normal aging and AD. Neurology. 2005 Aug 9. 65
(3):404-11. [Medline] 40 Johnson KA, Schultz A, Betensky RA, Becker JA, Sepulcre J, et al. Tau positron emission tomographic
imaging in aging and early Alzheimer disease. Ann Neurol. 2016 Jan. 79 (1):110-9. [Medline].

Las pautas de la Asociación de Alzheimer, que incluyen un algoritmo, ayudan a los
médicos en el ámbito de la atención primaria a detectar el deterioro cognitivo y
determinar si se necesita una referencia o pruebas adicionales. Crook T, Bartus
RT, Ferris SH. Age-associated memory impairment: proposed diagnostic criteria
and measures of clinical change. Dev Neuropsychol. 1986. 2:261-276
KRAL VA. Senescent forgetfulness: benign and malignant. Can Med Assoc J. 1962
Feb 10. 86:257-60. [Medline].El algoritmo incluye los siguientes componentes:
Revisión de la información de evaluación de riesgo para la salud del
paciente (HRA)
Observación del paciente
Uso de consultas no estructuradas.
Uso de herramientas estructuradas de evaluación cognitiva para pacientes
e informantes.
Las siguientes 3 herramientas de evaluación cognitiva se recomiendan para uso
de rutina por los médicos de atención primaria: 41Evaluación general de la
cognición (GPCOG)
Mini-Cog
Pantalla de deterioro de la memoria (MIS)
Además, la Asociación de Alzheimer recomienda las siguientes 3 herramientas de
evaluación cognitiva para usar con el cónyuge, la familia o los amigos del
paciente: 42
Evaluación de la cognición del médico general informante (informante
GPCOG)
Pantalla de 8 preguntas AD (AD8)
41 jeffrey S. New guidelines on screening for cognitive impairment. Medscape Medical News. December 21,
2012. January 8, 2013. Cordell CB, Borson S, Boustani M, Chodosh J, Reuben D, Verghese J, et al.
Alzheimer's Association recommendations for operationalizing the detection of cognitive impairment during
the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 2012 Dec 19. [Medline]. 42 Jeffrey S. New guidelines on screening for cognitive impairment. Medscape Medical News. December 21,
2012. January 8, 2013. Cordell CB, Borson S, Boustani M, Chodosh J, Reuben D, Verghese J, et al.
Alzheimer's Association recommendations for operationalizing the detection of cognitive impairment during
the Medicare Annual Wellness Visit in a primary care setting. Alzheimers Dement. 2012 Dec 19. [Medline].

Cuestionario breve para informantes sobre el deterioro cognitivo en los
ancianos (IQCODE breve)
Academia Americana de Neurología
La actualización de 2017 de la guía de la Academia Americana de Neurología
sobre el deterioro cognitivo leve (MCI), que cuenta con el respaldo de la
Alzheimer's Association, recomienda que los pacientes con MCI hagan ejercicio
regularmente como parte de un enfoque general para controlar sus síntomas. 43
La guía también aconseja a los clínicos hacer lo siguiente:
Evaluar para MCI usando herramientas validadas en escenarios apropiados
(nivel B);
evalúe a los pacientes con MCI para determinar factores de riesgo
modificables, evalúe el deterioro funcional y evalúe y trate los síntomas de
comportamiento / neuropsiquiátricos (nivel B);
monitorear el estado cognitivo de los pacientes con MCI a lo largo del
tiempo (nivel B);
cuando sea posible, deje de tomar medicamentos que afecten la capacidad
cognitiva y trate los síntomas conductuales (nivel B);
considere no ofrecer inhibidores de la colinesterasa (nivel B), y si ofrece,
primero discuta la falta de evidencia (nivel A);
recomendar el ejercicio regular (nivel B);
Considera recomendar entrenamiento cognitivo (nivel C);
discutir el diagnóstico, el pronóstico, la planificación a largo plazo y la falta
de opciones farmacológicas efectivas (nivel B); y
Considere discutir la investigación de biomarcadores con pacientes con
MCI y familias (nivel C).
ENFERMEDAD DE ALZHEIMER
43 Petersen RC, Lopez O, Armstrong MJ, Getchius TSD, Ganguli M, Gloss D, et al. Practice guideline update
summary: Mild cognitive impairment: Report of the Guideline Development, Dissemination, and
Implementation Subcommittee of the American Academy of Neurology. Neurology. 2018 Jan 16. 90 (3):126-
135. [Medline].
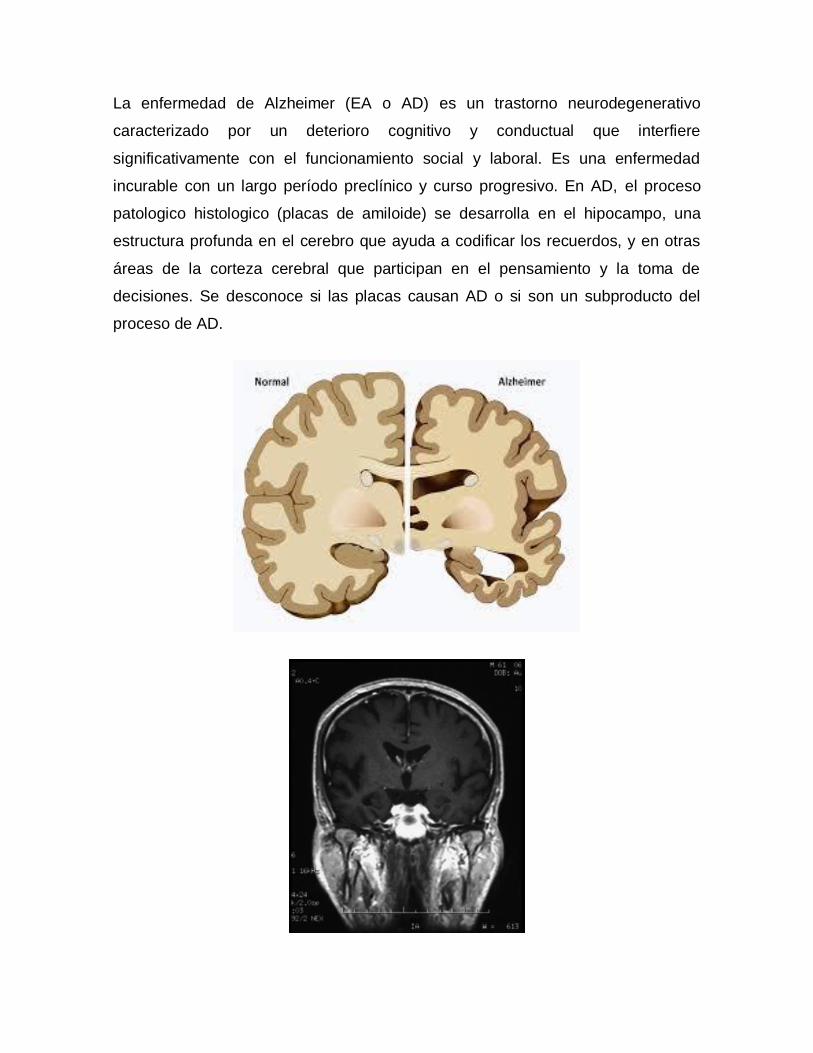
La enfermedad de Alzheimer (EA o AD) es un trastorno neurodegenerativo
caracterizado por un deterioro cognitivo y conductual que interfiere
significativamente con el funcionamiento social y laboral. Es una enfermedad
incurable con un largo período preclínico y curso progresivo. En AD, el proceso
patologico histologico (placas de amiloide) se desarrolla en el hipocampo, una
estructura profunda en el cerebro que ayuda a codificar los recuerdos, y en otras
áreas de la corteza cerebral que participan en el pensamiento y la toma de
decisiones. Se desconoce si las placas causan AD o si son un subproducto del
proceso de AD.

Resonancia magnética ponderada en T1 coronal (RM)
Imagen coronal de resonancia magnética ponderada en T1 (MRI) en un paciente
con enfermedad de Alzheimer moderada. La imagen del cerebro revela atrofia del
hipocampo, especialmente en el lado derecho.
SIGNOS Y SINTOMAS
Enfermedad de Alzheimer preclínica
Un paciente con AD preclínica puede parecer completamente normal en el
examen físico y en las pruebas de estado mental. Es probable que las regiones
específicas del cerebro (p. Ej., La corteza entorrinal, el hipocampo) se vean
afectadas décadas antes de que aparezcan signos o síntomas.
Enfermedad de Alzheimer leve
Los signos de EA leve pueden incluir los siguientes:
- Pérdida de memoria
- Confusión sobre la ubicación de lugares familiares.
- Tomando más tiempo para realizar las tareas normales, diarias
- Problemas para manejar el dinero y pagar las cuentas.
- Juicio comprometido, que a menudo lleva a malas decisiones.
- Pérdida de espontaneidad y sentido de iniciativa.
- Cambios de humor y personalidad; mayor ansiedad
Enfermedad de Alzheimer moderada
Los síntomas de esta etapa pueden incluir los siguientes:
- Aumento de la pérdida de memoria y confusión.
- Capacidad de atención reducida
- Problemas para reconocer amigos y familiares.

- Dificultad con el lenguaje; Problemas con la lectura, la escritura, el trabajo
con números.
- Dificultad para organizar los pensamientos y pensar de forma lógica.
- Incapacidad para aprender cosas nuevas o para hacer frente a situaciones
nuevas o inesperadas
- Inquietud, agitación, ansiedad, lágrimas, vagar, especialmente al final de la
tarde o en la noche.
- Acciones o movimientos repetitivos; contracciones musculares ocasionales
tipo mioclonias.
- Alucinaciones, delirios, desconfianza o paranoia, irritabilidad.
- Pérdida de control de impulsos: se muestra a través de comportamientos
como desvestirse en momentos o lugares inapropiados o lenguaje vulgar
- Problemas apraxicos: como problemas para levantarse de una silla o poner
la mesa
Enfermedad de Alzheimer grave
Los pacientes con EA grave no pueden reconocer a sus familiares o seres
queridos y no pueden comunicarse de manera efectiva. Son completamente
dependientes de los demás para su cuidado, y todo sentido de sí mismo parece
desaparecer.
- Otros síntomas de la EA severa pueden incluir los siguientes:
- Pérdida de peso
- Convulsiones, infecciones de la piel, dificultad para tragar.
- Gemidos o gruñidos
- Dormir más
- Falta de control de la vejiga y del intestino.
En la etapa final de la EA, los pacientes pueden estar en cama mucho o todo el
tiempo. La muerte es a menudo el resultado de otras enfermedades,
frecuentemente neumonía por aspiración.
Diagnóstico
Los medios para diagnosticar la EA incluyen los siguientes:

Examen clínico: el diagnóstico clínico de la enfermedad de Alzheimer
generalmente se realiza durante la etapa leve de la enfermedad, utilizando los
signos enumerados anteriormente.
Punción lumbar: los niveles de tau y tau fosforilada en el líquido cefalorraquídeo a
menudo se elevan en la EA, mientras que los niveles de amiloide suelen ser bajos;
en la actualidad, sin embargo, no se recomienda la medición rutinaria de tau y
amiloide en LCR, excepto en entornos de investigación
Estudios por imágenes: los estudios por imágenes son particularmente
importantes para descartar causas potencialmente tratables de deterioro cognitivo
progresivo, como el hematoma subdural crónico o la hidrocefalia de presión
normal. [1] Además, se han empleado estudios volumétricos de hipocampo y
tomografía por emisión de positrones de fluoro-2-desoxi-D-glucosa (FDG-PET)
con o sin imágenes amiloides para la detección temprana y la diferenciación de las
etiologías de demencia. [2]
MEDICACION
Todos los medicamentos aprobados por FDA para el tratamiento de la enfermedad
de Alzheimer son terapias sintomáticas que modulan los neurotransmisores, ya
sea acetilcolina o glutamato. El tratamiento médico estándar para la EA incluye
inhibidores de la colinesterasa (ChEI) y un antagonista parcial de N-metil-D-
aspartato (NMDA). [3, 4] No tratan la causa subyacente de la EA ni detienen la
tasa de disminución.
Las siguientes clases de medicamentos psicotrópicos se han utilizado para tratar
los síntomas secundarios de la enfermedad de Alzheimer, como depresión,
agitación, agresión, alucinaciones, delirios y trastornos del sueño [5]:
- Antidepresivos
- Ansiolíticos
- Agentes antiparkinsonianos
- Bloqueadores beta
- Medicamentos antiepilépticos
- Neurolépticos

Prevención
No existen modalidades comprobadas para prevenir la EA [3], pero la evidencia,
en gran parte epidemiológica, sugiere que los estilos de vida saludables pueden
reducir el riesgo de desarrollar la enfermedad; lo siguiente puede ser protector [6,
7]:
- Actividad física
- Ejercicio
- Fitness cardiorrespiratorio
- Dieta: aunque no se pueden hacer recomendaciones dietéticas definitivas,
en general, los patrones nutricionales que parecen beneficiosos para la
prevención de la EA se ajustan a la dieta mediterránea
La enfermedad de Alzheimer (EA) es la forma más común de demencia.[8]
Económicamente, la enfermedad de Alzheimer es un importante problema de
salud pública [9] [10]
Actualmente, una autopsia o una biopsia cerebral es la única forma de hacer un
diagnóstico definitivo de la enfermedad de Alzheimer. En la práctica clínica, el
diagnóstico generalmente se realiza sobre la base de la historia y los hallazgos del
examen de estado mental .
Las terapias sintomáticas son los únicos tratamientos disponibles para la EA. Los
tratamientos médicos estándar incluyen inhibidores de la colinesterasa y un
antagonista parcial de N -metil-D-aspartato (NMDA). Los medicamentos
psicotrópicos a menudo se usan para tratar los síntomas secundarios de la
enfermedad de Alzheimer, como la depresión, la agitación y los trastornos del
sueño. (Ver Tratamiento).
Antecedentes históricos
En 1901, un psiquiatra alemán llamado Alois Alzheimer observó a una paciente en
el Asilo de Frankfurt llamada Sra. Auguste D. Esta mujer de 51 años sufrió una
pérdida de memoria a corto plazo, entre otros síntomas de comportamiento que
desconcertaron al Dr. Alzheimer. Cinco años después, en abril de 1906, el
paciente falleció y el Dr. Alzheimer envió su cerebro y sus registros médicos a
Munich, donde trabajaba en el laboratorio del Dr. Emil Kraeplin. Al teñir secciones

de su cerebro en el laboratorio, pudo identificar las placas amiloides y los ovillos
neurofibrilares. [11]
Un discurso pronunciado por el Dr. Alzheimer el 3 de noviembre de 1906 fue la
primera vez que se presentaron juntos la patología y los síntomas clínicos del
trastorno, que en ese momento se denominaba demencia presenil. Alzheimer
publicó sus hallazgos en 1907. [12]
En los últimos 15 a 20 años, se ha logrado un progreso espectacular en la
comprensión de la neurogenética y la fisiopatología de la EA (consulte
Fisiopatología y etiología). Cuatro genes diferentes se han asociado
definitivamente con la EA, y se han identificado otros que tienen un papel
probable. Se están dilucidando los mecanismos por los cuales el metabolismo
alterado de la proteína amiloide y tau, la inflamación, el estrés oxidativo y los
cambios hormonales pueden producir degeneración neuronal en la EA, y se están
desarrollando intervenciones farmacológicas racionales basadas en estos
descubrimientos.
Anatomía Patológica
Las neuronas sanas tienen una estructura de soporte interno que se compone en
parte de estructuras llamadas microtúbulos. Estos microtúbulos actúan como
pistas, guiando nutrientes y moléculas desde el cuerpo de la célula hasta los
extremos del axón. Un tipo especial de proteína, el tau, se une a los microtúbulos
y los estabiliza.
En la EA, la proteina tau se modifica químicamente. Comienza a emparejarse con
otros hilos de tau, que se enredan entre sí. Cuando esto sucede, los microtúbulos
se desintegran y colapsan el sistema de transporte de la neurona. La formación de
estos ovillos neurofibrilares (NFT, por sus siglas en inglés) puede provocar primero
un mal funcionamiento de la comunicación entre las neuronas y, posteriormente, la
muerte de las células.
Además de las NFT, la patología anatómica de la EA incluye placas seniles (SP),
también conocidas como placas beta-amiloides, a nivel microscópico y atrofia
cerebral cortical a nivel macroscópico. El hipocampo y el lóbulo temporal medial

son los sitios iniciales de depositos de ovillos y atrofia. [13] Esto se puede ver en
las imágenes de resonancia magnética cerebral en una etapa temprana de la
enfermedad de Alzheimer y ayuda a apoyar un diagnóstico clínico.
Alois Alzheimer describió la anatomia patologica en su informe original sobre el
trastorno en 1907. [12] Ahora son aceptadas universalmente como el sello
patológico de la enfermedad.
Fisiopatologia
Existe un continuo entre la fisiopatología del envejecimiento normal y la de la
enfermedad de Alzheimer. [14] Se han identificado características patológicas de
AD en el cerebro de personas cognitivamente intactas. Por ejemplo, en un estudio
en el que los neuropatólogos estaban cegados a los datos clínicos, identificaron
que el 76% de los cerebros de pacientes ancianos cognitivamente intactos
demostraron fisiopatologia de EA. [15]
La EA afecta los 3 procesos que mantienen a las neuronas saludables:
comunicación, metabolismo y reparación. Ciertas células nerviosas en el cerebro
dejan de funcionar, pierden conexiones con otras células nerviosas y finalmente
mueren. La destrucción y la muerte de estas células nerviosas causan falla de la
memoria, cambios de personalidad, problemas para llevar a cabo las actividades
diarias y otras características de la enfermedad.
La acumulación de placas seniles precede principalmente al inicio clínico de la EA.
Los NFT, la pérdida de neuronas y la pérdida de sinapsis acompañan la
progresión del deterioro cognitivo. [14]
Se ha dedicado mucha atención a dilucidar la composición de SP y NFT para
encontrar pistas sobre la patogénesis molecular y la bioquímica de la EA. El
principal constituyente de las NFT es la proteína tau asociada a los microtúbulos.
Desde la época de Alois Alzheimer, se sabe que las placas seniles SP incluyen
una sustancia (amiloide), generalmente en el centro de estas lesiones. La
sustancia amiloide está rodeada por un halo o capa de neuritas degenerativas
(distróficas) y glía reactiva (tanto astrocitos como microglia).

Uno de los avances más importantes en las últimas décadas ha sido la
caracterización química de esta proteína amiloide, la secuenciación de su cadena
de aminoácidos y la clonación del gen que codifica su proteína precursora (en el
cromosoma 21). Estos avances han proporcionado una gran cantidad de
información sobre los mecanismos que subyacen a los depositos de amiloide en el
cerebro, incluida la información sobre las formas familiares de la enfermedad de
Alzheimer.
Los biomarcadores de la enfermedad de Alzheimer pueden seguir un patrón
secuencial en el cerebro, según un estudio de trayectorias de biomarcadores de
EA. El estudio incluyó portadores sintomáticos y asintomáticos de mutaciones
genéticas autosómicas dominantes vinculadas a AD, incluidas APP, PSEN1 y
PSEN2. Los investigadores no abordaron la tauopatía. Los resultados muestran
que los depositos de amiloide en el cerebro ocurre primero, seguida por una
disminución en el metabolismo de la glucosa y luego atrofia cerebral estructural.
La tasa de acumulación de Ab fue significativamente mayor en los portadores de
mutación en comparación con los no portadores, y se descubrió que comenzó más
de 2 décadas antes del inicio esperado de demencia. En los portadores, el
metabolismo comenzó a disminuir a una media de 14.1 años antes de la aparición
de los síntomas esperados, y los cambios estructurales en el cerebro comenzaron
4.7 años antes de la aparición de los síntomas esperados. Es importante tener en
cuenta que solo alrededor del 1% de los pacientes con AD tienen una mutación
autosómica dominante, por lo que los resultados pueden no ser generalizables a la
AD esporádica. [16]
Aunque la hipótesis de la cascada de amiloide ha reunido la mayor parte del
financiamiento de la investigación, se han propuesto otras hipótesis interesantes.
Entre ellas se encuentran la hipótesis de la cascada mitocondrial. [17]
Además de las NFT y SP, se han reconocido muchas otras lesiones de la
enfermedad de Alzheimer desde que se publicaron los artículos originales de
Alzheimer. Estos incluyen la degeneración granulovacuolar de Shimkowicz; los
hilos de Braak et al [17]; y la pérdida neuronal y la degeneración sináptica, que se

cree que median en última instancia las manifestaciones cognitivas y conductuales
del trastorno.
Ovillos neurofibrilares y placas seniles.
Las placas son densas, en su mayoría depósitos insolubles de proteína y material
celular fuera y alrededor de las neuronas. Las placas están hechas de beta-
amiloide (Ab), un fragmento de proteína cortado de una proteína más grande
llamada proteína precursora de amiloide (APP). Estos fragmentos se agrupan y se
mezclan con otras moléculas, neuronas y células no nerviosas.
En el AD, las placas se desarrollan en el hipocampo, una estructura profunda en el
cerebro que ayuda a codificar los recuerdos, y en otras áreas de la corteza
cerebral que se usan para pensar y tomar decisiones. Las placas pueden
comenzar a desarrollarse tan pronto como la quinta década de la vida. [18] Aún no
se sabe si las placas Ab causan EA o si son un subproducto del proceso.
Los ovillos son fibras retorcidas insolubles que se acumulan dentro de la célula
nerviosa. Aunque muchas personas mayores desarrollan algunas placas y ovillos,
el cerebro de las personas con EA las tiene en mayor medida, especialmente en
ciertas regiones del cerebro que son importantes en la memoria. Es probable que
haya diferencias significativas relacionadas con la edad en la medida en que la
presencia de placas y ovillos son indicativos de la presencia de demencia.
Las NFT se distribuyen inicialmente y de forma más densa en region medial y en
el polo del lóbulo temporal; afectan más severamente a la corteza entorrinal y al
hipocampo (sin embargo, Braak et al descubrieron que en la EA esporádica, la
tauopatía puede aparecer primero en el tronco cerebral inferior en lugar de en la
región transentorinal [18]). A medida que avanza la EA, las NFT se acumulan en
muchas otras regiones corticales, comenzando en regiones de asociación de alto
orden y menos frecuentemente en las regiones motoras y sensoriales primarias.
Los SP también se acumulan principalmente en cortezas de asociación y en el
hipocampo. Las placas y los ovillos tienen patrones relativamente discretos y
estereotipados de distribución laminar en la corteza cerebral, lo que indica una
participación predominante de las conexiones corticocorticales.

Aunque las NFT y las SP son características de la AD, no son patognomónicas.
Las NFT se encuentran en varios otros trastornos neurodegenerativos, incluida la
parálisis supranuclear progresiva y la demencia pugilística (encefalopatía
traumática crónica). SPs pueden ocurrir en el envejecimiento normal.
Por lo tanto, la mera presencia de estas lesiones no es suficiente para apoyar el
diagnóstico de EA. Estas lesiones deben estar presentes en cantidades suficientes
y en una distribución topográfica característica para cumplir con los criterios
histopatológicos actuales para la EA. Existe consenso en que la presencia de
incluso números bajos de NFT en el neocórtex cerebral con SP concomitantes es
característica de la EA.
Algunas autoridades creían que los NFT, cuando estaban presentes en
densidades bajas y se limitaban esencialmente al hipocampo, formaban parte del
envejecimiento normal. Sin embargo, las etapas histológicas para AD que Braak et
al formuló incluyen una etapa temprana en la que las NFT están presentes en una
densidad baja en las cortezas entorrinal y perirhinal (es decir, transentorhinal). [19]
Por lo tanto, incluso pequeñas cantidades de NFT en estas áreas del lóbulo
temporal medio pueden ser anormales.
Hipótesis amiloide versus hipótesis tau.
Un tema central pero controvertido en la patogenia de la EA es la relación entre
los depositos de amiloide y la formación de NFT. La evidencia muestra que el
metabolismo anormal del amiloide desempeña un papel patógeno clave. En
concentraciones altas, se ha demostrado que la forma fibrilar de Ab es neurotóxica
para las neuronas cultivadas.
Las neuronas corticales y del hipocampo cultivadas tratadas con proteína Ab
presentan cambios característicos de la apoptosis (destrucción celular
autorregulada), incluida la condensación de la cromatina nuclear, la formación de
vacuolas en la membrana plasmática y la fragmentación del ADN
internucleosomal. También se ha demostrado que la forma fibrilar de Ab altera el
estado de fosforilación de la proteína tau.
El alelo de la apolipoproteína E (APOE) E4, que se ha relacionado con un riesgo
significativamente mayor de desarrollar EA, puede estar vinculada con

incapacidad de suprimir la producción de amiloide, aumento de la producción de
amiloide y la eliminacion reducida del amiloide.
Las autopsias han demostrado que los pacientes con 1 o 2 copias del alelo APOE
E4 tienden a tener más amiloide. Pruebas adicionales provienen de datos
experimentales recientes que apoyan el papel de las presenilinas en el
metabolismo del Ab, así como los hallazgos de producción anormal de la proteína
Ab en la enfermedad de Alzheimer familiar con mutación de presenilina.
Aunque es muy popular, la hipótesis amiloide no se acepta de manera uniforme.
En el análisis post-mortem, las placas de amiloide pueden ser indetectables en los
cerebros de pacientes con AD grave, pero pueden estar presentes en los cerebros
de pacientes ancianos que no tenían demencia. [20]
La severidad de la demencia se correlaciona mejor con el número de NFT
neocorticales que con los SP. La proteína tau estabiliza los microtúbulos
neuronales. Se especula que la desestabilización del sistema microtubular
interrumpe el aparato de Golgi, lo que a su vez induce un procesamiento anormal
de proteínas y aumenta la producción de Ab. Además, esta desestabilización
puede disminuir el flujo axoplásmico, generando neuritas distróficas y
contribuyendo a la pérdida sináptica.
Degeneración granulovacuolar y hilos de neuropilo
La degeneración granulovacuolar ocurre casi exclusivamente en el hipocampo.
Los hilos de neuropilo son una serie de neuritas distróficas distribuidas de manera
difusa en el neuropilo cortical, más o menos independientemente de las placas y
los ovillos. Esta lesión sugiere alteraciones más allá de aquellas meramente
debidas a NFT y SP e indica un mayor compromiso y más generalizado a los
circuitos corticales que el que se visualiza al estudiar solo placas y ovillos.
Neurotransmisión colinérgica y enfermedad de Alzheimer.
El sistema colinérgico está involucrado en la función de la memoria, y la
deficiencia colinérgica se ha implicado en el deterioro cognitivo y los cambios de
comportamiento de la EA. La actividad de la enzima sintética colina
acetiltransferasa (CAT) y la enzima catabólica acetilcolinesterasa se reducen

significativamente en la corteza cerebral, el hipocampo y la amígdala en pacientes
con EA.
El núcleo basal de Meynert y la banda diagonal de Broca proporcionan el aporte
colinérgico principal para el hipocampo, la amígdala y el neocórtex, que se pierden
en pacientes con EA. Se ha encontrado que la pérdida de CAT cortical y la
disminución en la síntesis de acetilcolina en muestras de biopsia se correlacionan
con el deterioro cognitivo y el rendimiento en el tiempo de reacción. Debido a que
la disfunción colinérgica puede contribuir a los síntomas de los pacientes con AD,
el aumento de la neurotransmisión colinérgica constituye una base racional para el
tratamiento sintomático.
Estrés oxidativo y daños.
Los estudios han demostrado que se produce un aumento en el daño oxidativo en
las regiones del cerebro involucradas en la regulación del rendimiento cognitivo.
[21]
Se cree que el estrés oxidativo es un factor crítico en el envejecimiento normal y
en enfermedades neurodegenerativas como la enfermedad de Parkinson, la
esclerosis lateral amiotrófica y la EA. Las placas y los ovillos muestran
inmunorreactividad a las enzimas antioxidantes.
Existen múltiples mecanismos por los cuales las alteraciones celulares pueden ser
inducidas por el estrés oxidativo, incluida la producción de especies reactivas de
oxígeno (ROS) en la membrana celular (peroxidación de lípidos). Esto, a su vez,
perjudica las diversas proteínas de la membrana implicadas en la homeostasis
iónica, como los canales receptores de N -metil-D-aspartato o las adenosina
trifosfatasas motivadas por iones.
El aumento subsiguiente del calcio intracelular, junto con la acumulación de ROS,
daña varios componentes celulares, como las proteínas, el ADN y los lípidos, y
puede provocar la muerte celular apoptótica. El aumento del calcio intracelular
también puede alterar la actividad de la enzima dependiente del calcio, como la
implicación de la proteína quinasa C en el metabolismo de la proteína amiloide y la
fosforilación de tau.

La participación del calcio en la AD ha sugerido que el bloqueo del aumento del
calcio intracelular libre puede disminuir la lesión neuronal. Sin embargo, los
ensayos clínicos de nimodipina, un bloqueador de los canales de calcio lipófilos
que está mediado a través de la inactivación de los canales de calcio de tipo L (de
larga duración) dependientes del voltaje, no han dado resultados favorables en
pacientes con AD.
Reacciones inflamatorias
Los mecanismos inflamatorios e inmunitarios pueden jugar un papel en el proceso
degenerativo en la EA. En las placas neuriticas se encuentran celular reactivas de
la microglia. Se observan mayores niveles de citoquinas en el suero, las placas
corticales y las neuronas de los pacientes con AD, en comparación con los
pacientes de control emparejados de edad.
Los fragmentos de la vía del complemento clásico también se encuentran en los
cerebros de los pacientes con EA, y el amiloide puede activar directamente la vía
del complemento clásico de una manera independiente del anticuerpo.
Aún no está claro si los marcadores de los procesos inmunes e inflamatorios
participan activamente en el proceso neurodegenerativo o si representan un
epifenómeno. Las muestras de cerebro de pacientes ancianos con artritis tratadas
con medicamentos antiinflamatorios no esteroides (AINE) tienen un número similar
de placas seniles que los cerebros de control.
Sin embargo, se observa menos activación microglial en los cerebros de los
pacientes con artritis. Esto sugiere que si bien los AINE no pueden impedir la
formación de placa senil, pueden retrasar o prevenir los síntomas clínicos al limitar
la inflamación asociada.
Clusterin
La clusterina, una proteína plasmática, desempeña un papel importante en la
patogénesis de la EA. En un estudio, la clusterina se asoció con atrofia de la
corteza entorrinal, gravedad de la enfermedad basal y progresión clínica rápida en
la AD. [23] Un estudio realizado por Schrijvers et al señala que aunque los niveles
de clusterina plasmática están significativamente asociados con la prevalencia y la

gravedad de la línea de base de la AD, no están relacionados con el riesgo de la
AD. [24]
Presenilinas
Una proporción significativa de los casos de AD autosómica dominante de inicio
temprano se ha relacionado con un gen en el cromosoma 14 (14q24.3) llamado
presenilina-1 (PS1) y un gen en el cromosoma 1 llamado presenilina-2 (PS2). Los
2 productos de estos genes, PS1 y PS2, comparten aminoácidos importantes y
similitudes estructurales, lo que sugiere que pueden estar relacionados
funcionalmente. Además, los patrones de expresión de PS1 y PS2 en el cerebro
son similares, si no idénticos.
Tanto el ARN mensajero (ARNm) de PS1 como el ARNm de PS2 son detectables
solo dentro de poblaciones neuronales. Los análisis inmunoquímicos indican que
la PS1 se localiza en compartimentos intracelulares, como el retículo
endoplásmico y el complejo de Golgi, que participan en funciones similares. La
evidencia apoya el papel de las presenilinas en el metabolismo del Ab.
Tanto los sujetos asintomáticos como los pacientes con EA portadores de la
mutación PS1 han aumentado la producción de la isoforma amiloidogénica Ab
42/43 en fibroblastos de la piel y plasma. Los depositos de Ab 42/43 se
encuentran en muchas regiones del cerebro de pacientes con mutaciones de PS1.
Estos hallazgos ofrecen nuevas vías terapéuticas.
Reduccion de estrógeno:
Las mujeres posmenopáusicas tienen un riesgo más alto que los hombres de AD.
Algunos estudios han demostrado que la disminucion de estrógeno puede
provocar deterioro cognitivo y degeneración neuronal, y también disminuye la
expresión del factor de crecimiento nervioso y del ARNm del factor neurotrófico
derivado del cerebro.
También se ha demostrado que el estrógeno ejerce efectos citoprotectores y
previene la toxicidad amiloide en cultivos de células de neuroblastoma humano.
Sin embargo, un ensayo clínico aleatorizado de estrógeno en mujeres
cognitivamente normales de 65 años de edad y mayores con un familiar de primer

grado con AD mostró que la terapia con estrógenos en realidad podría aumentar el
riesgo de accidente cerebrovascular y demencia. [25]
Etiología
La causa de la AD es desconocida. Varios investigadores ahora creen que los
factores de riesgo genéticos y ambientales convergentes desencadenan una
cascada fisiopatológica que, durante décadas, conduce a la patología del
Alzheimer y la demencia. [26]
Se identificaron los siguientes factores de riesgo para la demencia de tipo
Alzheimer: [27, 28, 29, 30]
- Edad avanzada
- Historia familiar
- Genotipo APOE 4
- Obesidad
- Resistencia a la insulina
- Factores vasculares
- Dislipidemia
- Hipertensión
- Marcadores inflamatorios
- Síndrome de Down
- Lesión cerebral traumática
La hipertensión de la mediana edad es un factor de riesgo establecido para la
demencia tardía, de la cual la EA es el tipo más común. Un estudio de autopsia
cerebral que evaluó el vínculo entre la hipertensión y la enfermedad de Alzheimer
encontró que los pacientes que usan bloqueadores beta para controlar la presión
arterial tenían menos lesiones cerebrales de tipo Alzheimer en la autopsia en
comparación con los pacientes que no reciben terapia con medicamentos o los
que toman otros medicamentos. [31]
Además, los estudios epidemiológicos han sugerido algunos posibles factores de
riesgo como el aluminio [32, 33] y la depresión previa [34]. Otros estudios han

sugerido factores de protección (por ejemplo, educación [35, 36], uso a largo plazo
de medicamentos antiinflamatorios no esteroides [37]).
Causas genéticas
Aunque la mayoría de los casos de AD son esporádicos (es decir, no se heredan),
existen formas familiares de AD. La EA autosómica dominante, que representa
menos del 5% de los casos, es casi exclusivamente una EA de inicio temprano;
los casos ocurren en al menos 3 individuos en 2 o más generaciones, y 2 de ellos
son familiares de primer grado. [38]
El agrupamiento familiar representa aproximadamente el 15–25% de los casos de
AD de inicio tardío. En la agrupación familiar, al menos 2 de los individuos
afectados son parientes de tercer grado o más cercanos. [38]
Las mutaciones en los siguientes genes causan EA autosómica dominante de
inicio temprano:
- El gen de la proteína precursora de amiloide (APP) en el cromosoma 21
- El gen presenilin-1 (PS1) en el cromosoma 14
- El gen presenilin-2 (PS2) en el cromosoma 1
- Estos 3 genes conducen a un exceso en la producción del péptido Ab.
Se postula que este péptido beta tiene propiedades neurotóxicas y conduce a una
cascada de eventos (que aún no se han comprendido completamente) que
resultan en muerte neuronal, pérdida de sinapsis y la formación de NFT y SP,
entre otras lesiones. No obstante, las mutaciones que se han encontrado hasta la
fecha representan menos de la mitad de todos los casos de EA de inicio temprano.
Aparte del genotipo de la apolipoproteína E épsilon 4 (APOE E4), no se ha
encontrado que los polimorfismos en otros genes estén asociados con la EA de
inicio tardío. Sin embargo, los estudios de asociación en todo el genoma han
identificado los siguientes loci de susceptibilidad adicionales [39]:
- Gen Clusterin (CLU)
- Gen de la proteína de ensamblaje de la clatrina de unión a fosfatidilinositol
(PICALM)
- Complemento del gen receptor 1 (CR1)

- Casco de unión a ATP subfamilia A miembro 7 gen (ABCA7)
- Grupo de genes que abarcan la membrana (MS4A6A / MS4A4E)
- Receptor de efrina A1 (EPHA1)
- CD33
- CD2AP
Mutaciones de APP
La observación de que los pacientes con síndrome de Down (trisomía 21)
desarrollan un deterioro cognitivo y las características patológicas típicas de la EA
en la mediana edad llevó al descubrimiento del gen APP en el cromosoma 21.
Simultáneamente, un locus se segrega con una minoría de familias con AD de
inicio temprano. se asignó a este cromosoma, en la misma región que el gen APP.
Posteriormente, se identificaron varias mutaciones sen el gen APP que dieron
lugar a sustituciones de aminoácidos en APP en estas familias con EA.
Los fibroblastos de la piel de individuos portadores de mutaciones de APP
producen un aumento de Ab 42/43. El aumento de la concentración plasmática de
Ab 42/43 también se observa en estos pacientes, independientemente de la edad,
el sexo o el estado clínico. Curiosamente, algunos pacientes con EA esporádica
pueden exhibir elevaciones similares de plasma Ab 42/43.
Mutaciones PS1 y PS2
Aproximadamente el 50-70% de los casos de AD autosómica dominante de inicio
temprano parecen estar asociados con un locus (AD3) mapeado por enlace
genético al brazo largo del cromosoma 14 (14q24.3). Se han identificado
numerosas mutaciones sin sentido en un gen candidato fuerte, llamado PS1.
Al mismo tiempo, otro locus autosómico dominante responsable de la AD de inicio
temprano se localizó en el cromosoma 1. Se identificaron dos mutaciones en el
gen candidato, denominado PS2. El papel fisiológico de las presenilinas y los
efectos patógenos de sus mutaciones aún no se conocen bien.
APOE
El gen que codifica la apolipoproteína E (APOE) en el cromosoma 19 se ha
relacionado con un mayor riesgo de AD, principalmente de inicio tardío, pero
también en algunos casos de inicio temprano. El gen se hereda como un rasgo

autosómico codominante con 3 alelos. El alelo APOE E2, el menos prevalente de
los 3 alelos APOE comunes, está asociado con el riesgo más bajo de desarrollar
AD, [40] con una tasa más baja de atrofia anual del hipocampo y mayor Aβ de
líquido cefalorraquídeo y fosfotau inferior, lo que sugiere una patología de AD
menor. [41]
El alelo E3 confiere un riesgo intermedio de desarrollar AD, con menos riesgo que
el alelo E4. El alelo E3, que es más común que el alelo E2, puede proteger a tau
de la hiperfosforilación, y el efecto del alelo E2 en la fosforilación de tau es
complejo.
La "cantidad" del gen APOE E4 se correlaciona con un mayor riesgo y un inicio
más temprano de la EA. [42] Se recomienda a las personas genéticamente
predispuestas a la EA que controlen de cerca su presión arterial. Se ha
demostrado que la hipertensión interactúa con el genotipo APOE E4 para
aumentar los depositos de amiloide en adultos de mediana edad y adultos
cognitivamente sanos; el control de la hipertensión puede disminuir
significativamente el riesgo de desarrollar depósitos de amiloide, incluso en
aquellos con riesgo genético. [43, 44]
Las personas con 2 copias del alelo APOE E4 (genotipo 4/4) tienen un riesgo
significativamente mayor de desarrollar AD que las personas con otros subtipos de
APOE. La edad media de inicio es significativamente menor en presencia de 2
copias APOE E4. Un estudio colaborativo ha sugerido que APOE E4 ejerce su
efecto máximo antes de los 70 años.
Muchos portadores de APOE E4 no desarrollan AD, y muchos pacientes con AD
no tienen este alelo. Por lo tanto, la presencia de un alelo APOE E4 no asegura el
diagnóstico de EA, sino que, por el contrario, el alelo APOE E4 actúa como un
factor de riesgo biológico para la enfermedad, especialmente en los menores de
70 años.
Resistencia a la insulina
Un pequeño estudio realizado por Baker et al. Implica que la resistencia a la
insulina, como lo demuestra la disminución de la tasa metabólica de la glucosa

cerebral medida por un tipo específico de tomografía por emisión de positrones
(TEP), puede ser útil como un marcador temprano del riesgo de AD, incluso antes
del inicio de MCI. [46] La exploración PET reveló un patrón de activación
cualitativamente diferente en pacientes con Diabetes o con resistencia a la
insulina, durante una tarea de codificación de memoria, en comparación con
individuos sanos que no eran resistentes a la insulina.
Aunque el estudio de Baker et al tuvo muy pocos sujetos (n = 23) para que los
resultados alcanzaran significación estadística, un estudio de Schrijvers et al en
una población mucho más grande (3,139 sujetos) encontró una asociación similar
entre la resistencia a la insulina y la EA más de 3 Años, que luego desaparecieron
después de ese tiempo. [47] Estos investigadores utilizaron una medida diferente
de la resistencia a la insulina, la evaluación del modelo de homeostasis. Las
alteraciones en el metabolismo de la insulina pueden no causar cambios
neurológicos, pero pueden influir y acelerar estos cambios, lo que lleva a un inicio
más temprano de la enfermedad de Alzheimer.
Infecciones
Un campo de investigación emergente sugiere una asociación significativa entre la
enfermedad de Alzheimer y la infección crónica con varias especies de
espiroquetas, incluyendo el patógeno periodontal Treponemas y Borrelia
burgdorferi, así como patógenos como el virus del herpes simple tipo 1. [48] Los
estudios in vitro y en animales apoyan El concepto de infección que resulta en
inflamación crónica y destrucción neuronal. Se ha demostrado que el Ab es un
péptido antimicrobiano, por lo que su acumulación podría representar una
respuesta a la infección.
Depresión
La depresión se ha identificado como un factor de riesgo para la EA y otras
demencias. Los datos recientes de Framingham han ayudado a reforzar la
asociación epidemiológica. El estudio mostró un aumento del 50% en la EA y la
demencia en aquellos que estaban deprimidos al inicio del estudio. [49] Durante
un período de seguimiento de 17 años, un total de 21.6% de los participantes que

estaban deprimidos al inicio del estudio desarrollaron demencia, en comparación
con 16.6% de los que no estaban deprimidos.
En otro estudio relacionado, se observó que la depresión recurrente era
particularmente riesgosa. Un episodio de depresión confirió un aumento de 87 a
92% en el riesgo de demencia, mientras que tener 2 o más episodios casi duplicó
el riesgo. [50]
De acuerdo con los resultados de un metanálisis de 23 estudios prospectivos de
cohorte basados en la población, la depresión tardía se asocia con un mayor
riesgo de demencia por todas las causas, demencia vascular y EA. [51, 52] El
riesgo de demencia vascular parecía ser significativamente mayor que el riesgo de
AD. El análisis incluyó datos de pacientes de 50 años o más que estaban libres de
demencia al inicio del estudio. La muestra total incluida en el análisis agrupado
para la demencia por todas las causas fue 49,612 participantes, 5116 de los
cuales tenían depresión tardía.
Alternativamente, un gran estudio longitudinal encontró que la depresión que
comienza temprano en la vida aumenta el riesgo de AD. Los investigadores
utilizaron datos del Estudio de población prospectiva de mujeres en Gotemburgo,
Suecia, que comenzó en 1968. La muestra del estudio incluyó 800 mujeres (edad
media, 46 años), nacidas entre 1914 y 1930, que fueron seguidas en 1974, 1980,
1992 , 2000, 2009 y 2012. Los datos muestran que las mujeres que
experimentaron el inicio de la depresión antes de los 20 años de edad tenían tres
veces más probabilidades de desarrollar EA (HR ajustada, 3,41; IC del 95%, 1,78 -
6,54). [34]
Trauma cefalico
El traumatismo craneal moderado a severo se ha documentado como un factor de
riesgo para el desarrollo de la EA así como para otras formas de demencia en el
futuro. [53] Chen et al. Han propuesto que la lesión cerebral traumática conduce a
la acumulación de proteína precursora de amiloide con sus enzimas proteolíticas
en los sitios de lesión axonal, aumento de la producción intracelular de Ab,
liberación de Ab de los axones lesionados al espacio extracelular y deposición de
Ab En placas extracelulares. [54]

Un estudio que siguió a más de 7,000 veteranos de la Segunda Guerra Mundial en
EE. UU. mostró que aquellos que sufrieron lesiones en la cabeza tenían el doble
de riesgo de desarrollar demencia más adelante en la vida, y los veteranos que
sufrieron un traumatismo craneal más grave tenían un riesgo aún mayor. El
estudio también encontró que la presencia del gen APOE y el traumatismo craneal
sostenido parecían actuar de manera aditiva para aumentar el riesgo de
desarrollar AD, aunque no hubo una correlación directa. [55]
Epigenética
La epigenética es un cambio en la expresión génica que resulta de las
interacciones gen-ambiente. Esto está mediado por la metilación del ADN, la
edición del ARN y la interferencia del ARN sin cambios en la secuencia del ADN.
Los elementos epigenéticos en la EA se sugieren por hechos que la mayoría de
los casos de EA son esporádicos, ocurren en pacientes sin antecedentes
familiares de la enfermedad y se presentan más tarde en la vida.
Un factor ambiental que ha demostrado daños en animales de laboratorio
compatibles con AD humana es el plomo. La exposición temprana al plomo en
investigacion dio lugar a la formación de placas. [56] Un aspecto de la exposición
temprana al plomo parece ser el aumento del estrés oxidativo en las células
cerebrales. El estrés oxidativo es la acumulación de radicales libres en exceso que
alteran los patrones de metilación en las células.
El estrés oxidativo temprano, se ha postulado como una causa de EA esporádica.
[57]
Dado que la exposición al plomo en animales en la vida temprana no produce
manifestaciones hasta más tarde en la vida, el estrés ambiental continuo puede
contribuir a la expresión. En consecuencia, es posible que el uso de suplementos
antioxidantes a partir de la infancia pueda disminuir el estrés oxidativo a largo
plazo y disminuir la incidencia de la EA. El trabajo de Harman indica que los
antioxidantes pueden disminuir el daño celular y el envejecimiento al disminuir el
exceso de estrés oxidativo. [58]
El único estudio importante de un antioxidante, la vitamina E, arrojó resultados
decepcionantes. Sin embargo, el estudio implicó un uso del tiempo muy limitado.

En la actualidad, no se han estudiado otros cambios en la exposición ambiental
para la prevención de la EA, pero esta área será crítica en futuros estudios a largo
plazo.
Distribución por edad de la enfermedad de Alzheimer
La prevalencia de la EA aumenta con la edad. La EA es más frecuente en
individuos mayores de 60 años. Algunas formas de EA familiar de aparición
temprana pueden aparecer tan temprano como en la tercera década, pero los
casos familiares constituyen menos del 10% de la EA general.
Más del 90% de los casos de AD son esporádicos y ocurren en personas mayores
de 60 años. De interés, sin embargo, los resultados de algunos estudios de
nonagenarios y centenarios sugieren que el riesgo puede disminuir en personas
mayores de 90 años. Si es así, la edad no seria un factor de riesgo para la
enfermedad, pero se necesitan más estudios.
Savva et al encontraron que en la población anciana, la asociación entre la
demencia y las características patológicas de la EA (p. Ej., Placas neuríticas,
placas difusas, ovillos) es más fuerte en personas de 75 años que en personas de
95 años. Estos resultados se lograron mediante la evaluación de 456 cerebros
donados al Estudio de Función Cognitiva y Envejecimiento del Consejo de
Investigación Médica basado en la población de personas de 69 a 103 años de
edad. [64]
Diferencias sexuales en la incidencia.
Algunos estudios han reportado un mayor riesgo de AD en mujeres que en
hombres [10]; sin embargo, otros estudios, incluidos el estudio de envejecimiento,
demografía y memoria, no encontraron diferencias en el riesgo entre hombres y
mujeres. [65] En general, entre los pacientes con EA, cualquier disparidad sexual
puede reflejar simplemente la mayor esperanza de vida de las mujeres. Sin
embargo, entre aquellos que son heterocigotos para el alelo APOE E4, Payami et
al encontraron un doble aumento en el riesgo en las mujeres. [66]
Diferencias de incidencia relacionadas con la raza.

La enfermedad de Alzheimer y otras demencias son más comunes en los
afroamericanos que en los blancos. Según la Asociación de Alzheimer, en la
población de 71 años y más, los afroamericanos tienen casi el doble de
probabilidades de tener EA y otras demencias que los blancos (21.3% de los
afroamericanos frente al 11.2% de los blancos). El número de pacientes hispanos
estudiados en este grupo de edad era demasiado pequeño para determinar la
prevalencia de demencia en esta población.
En un estudio de 1255 personas, incluidos participantes afroamericanos (n = 173)
y caucásicos, los investigadores encontraron que el líquido cefalorraquídeo de los
afroamericanos tenia niveles más bajos de proteína cerebral tau, un biomarcador
asociado con la enfermedad de Alzheimer. Sin embargo, esto no pareció
protegerlos de la enfermedad; eran tan propensos como los caucásicos a tener
problemas cognitivos. Estos resultados sugieren que el análisis de los
biomarcadores para la EA debe ajustarse según la raza. [67]
Pronóstico
La AD se asocia inicialmente con un deterioro de la memoria que empeora
progresivamente. Con el tiempo, los pacientes con AD también pueden mostrar
ansiedad, depresión, insomnio, agitación y paranoia. A medida que su enfermedad
progresa, los pacientes con EA llegan a requerir asistencia con las actividades
básicas de la vida diaria, como vestirse, bañarse e ir al baño. Eventualmente, se
pueden desarrollar dificultades para caminar y tragar. La alimentación en estados
avanzados puede requerir alimentacion enteral. La dificultad para tragar puede
llevar a una neumonía por aspiración.
El tiempo desde el diagnóstico hasta la muerte varía desde tan poco como 3 años
hasta 10 o más años. Los pacientes con AD de inicio temprano tienden a tener un
curso más agresivo y rápido que aquellos con AD de inicio tardío. La causa
principal de muerte es la enfermedad intercurrente, como la neumonía.
Educación del paciente
Al asesorar a los pacientes después de un diagnóstico de EA, es esencial
involucrar a la familia del paciente y otras personas que desempeñarán un papel
de apoyo en la discusión. Es importante enfatizar que no solo el paciente, sino

también aquellos que lo apoyan, probablemente experimentarán reacciones de
tristeza y enojo y que estas son reacciones normales ante tal diagnóstico.
A medida que los síntomas del paciente se vuelven más pronunciados, se debe
abrir un diálogo sobre los deseos de atención del paciente cuando ya no pueda
tomar las decisiones necesarias. El poder notarial duradero debe ser discutido,
con especial atención a quién tomará las decisiones tanto por cuestiones médicas
como financieras. Se deben tener en cuenta las directivas médicas anticipadas
mientras el paciente aún pueda participar en el proceso de toma de decisiones.
A lo largo del curso de la enfermedad, los miembros de la familia deben tener
cuidado de seleccionar a individuos calificados y confiables para que participen en
el manejo diario del paciente. Los cuidadores deben equilibrar la atención a las
necesidades físicas del paciente y mantener el respeto por el paciente como
adulto competente, en la medida en que lo permita la progresión de la
enfermedad. Cualquier sospecha de maltrato a personas mayores debe abordarse
de inmediato.
Por encima de todo, la asesoría a pacientes y familias con EA debe enfatizar que
los pacientes deben continuar participando en actividades que disfrutan. Mantener
una calidad de vida óptima es clave.
Examen físico
En el momento del diagnóstico inicial, se debe realizar un examen físico completo,
incluido un examen neurológico detallado y un examen del estado mental, para
evaluar el estadio de la enfermedad y descartar condiciones comórbidas. La
prueba de estado mental inicial debe incluir la evaluación de lo siguiente:
- Atención y concentración.
- Memoria reciente y remota
- Idioma
- Praxis (es decir, capacidad para realizar tareas motorizadas especializadas
sin indicaciones no verbales)
- Función ejecutiva
- Función visuoespacial

Las características cognitivas de la EA temprana incluyen pérdida de memoria,
afasia anómica leve y disfunción visuoespacial. En todas las visitas de
seguimiento posteriores, se debe realizar un examen completo del estado mental
para evaluar la progresión de la enfermedad e identificar el desarrollo de cualquier
nuevo síntoma neuropsiquiátrico.
Los exámenes breves y estandarizados, como el Mini-Mental Status Examination
(MMSE), son menos sensibles y específicos que las baterías más largas que
están específicamente diseñadas para pacientes individuales. Otros ejemplos
incluyen el examen de evaluación cognitiva de Montreal (MoCA) y el estado
mental de la Universidad de Saint Louis (SLUMS). No obstante, los exámenes de
detección tienen un papel, especialmente como una línea de base. Para obtener
más información, consulte el artículo de Evaluación de Medscape sobre el
deterioro cognitivo.
Se realiza un examen neurológico completo para buscar signos de otras
enfermedades que podrían causar demencia, como la enfermedad de Parkinson o
varios accidentes cerebrovasculares. En los pacientes con AD, el examen
neurológico es generalmente normal pero puede revelar anomalías menores,
como hiposmia o anosmia.
pidemiología
Etapas de la enfermedad de Alzheimer
AD se puede clasificar en las siguientes etapas:
Preclínica
Leve
Moderado
Grave
Enfermedad de Alzheimer preclínica
Los cambios patológicos asociados con la AD comienzan en la corteza entorrinal,
que está cerca del hipocampo y está directamente conectada a él. Con el paso del
tiempo afecta al hipocampo, que es la estructura que es esencial para la formación
de memorias a corto y largo plazo. Las regiones afectadas comienzan a atrofiarse.
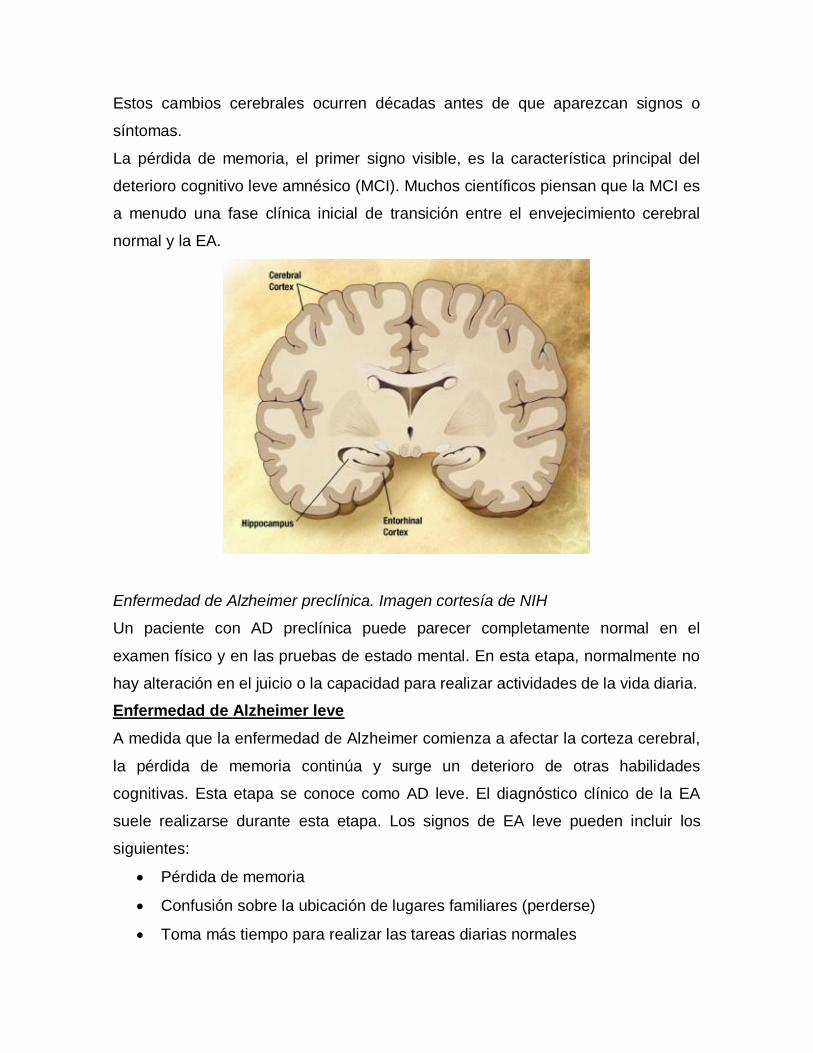
Estos cambios cerebrales ocurren décadas antes de que aparezcan signos o
síntomas.
La pérdida de memoria, el primer signo visible, es la característica principal del
deterioro cognitivo leve amnésico (MCI). Muchos científicos piensan que la MCI es
a menudo una fase clínica inicial de transición entre el envejecimiento cerebral
normal y la EA.
Enfermedad de Alzheimer preclínica. Imagen cortesía de NIH
Un paciente con AD preclínica puede parecer completamente normal en el
examen físico y en las pruebas de estado mental. En esta etapa, normalmente no
hay alteración en el juicio o la capacidad para realizar actividades de la vida diaria.
Enfermedad de Alzheimer leve
A medida que la enfermedad de Alzheimer comienza a afectar la corteza cerebral,
la pérdida de memoria continúa y surge un deterioro de otras habilidades
cognitivas. Esta etapa se conoce como AD leve. El diagnóstico clínico de la EA
suele realizarse durante esta etapa. Los signos de EA leve pueden incluir los
siguientes:
Pérdida de memoria
Confusión sobre la ubicación de lugares familiares (perderse)
Toma más tiempo para realizar las tareas diarias normales

Problemas para manejar el dinero y pagar las cuentas.
Juicio comprometido que a menudo conduce a malas decisiones
Pérdida de espontaneidad y sentido de iniciativa.
Cambios de humor y personalidad; mayor ansiedad
El número creciente de placas y ovillos primero dañan áreas del cerebro que
controlan la memoria, el lenguaje y el razonamiento
Más tarde en la enfermedad, las habilidades físicas disminuyen. La persona
parece estar sana pero en realidad tiene cada vez más problemas para vincularse
con el entorno. La comprensión de que algo está mal a menudo se produce
gradualmente porque los primeros signos pueden confundirse con los cambios
que pueden ocurrir normalmente con el envejecimiento.
Enfermedad de Alzheimer leve. Imagen cortesía de NIH
Reconocer estos signos de EA y decidir hacer pruebas de diagnóstico es lo mas
relevante en esta etapa. En muchos casos, la familia tiene más dificultades para
manejar el diagnóstico y reconocer la enfermedad que el paciente, probablemente
debido a la anosognosia del AD. Después del diagnóstico inicial, los pacientes
deben ser monitoreados cuidadosamente para detectar tambien alteraciones del
estado de animo, como depression.
Enfermedad de Alzheimer moderada
En el momento en que AD alcanza el estado moderado, el daño se ha extendido

más allá de las áreas de la corteza cerebral que controlan el lenguaje, el
razonamiento, el procesamiento sensorial y el pensamiento consciente. Las
regiones afectadas se atrofian mas, y los signos y síntomas de la enfermedad se
vuelven más pronunciados y generalizados. Pueden ocurrir problemas de
comportamiento, tales como el deambular y la agitación. La supervisión y el
cuidado más intensivos se vuelven necesarios, y esto puede ser difícil para
muchos cónyuges y familias.
Los síntomas de esta etapa pueden incluir los siguientes:
Aumento de la pérdida de memoria y confusión.
Capacidad de atención reducida
Problemas para reconocer amigos y familiares.
Dificultad con el lenguaje; Problemas con la lectura, la escritura, el trabajo
con números.
Dificultad para organizar los pensamientos y pensar de forma lógica.
Incapacidad para aprender cosas nuevas o para hacer frente a situaciones
nuevas o inesperadas
Inquietud, agitación, ansiedad, lágrimas, vagar, especialmente al final de la
tarde o en la noche.
Declaraciones o movimientos repetitivos; contracciones musculares
ocasionales
Alucinaciones, delirios, desconfianza o paranoia, irritabilidad.
Pérdida del control de los impulsos (que se muestra a través de
comportamientos como desnudarse en momentos o lugares inapropiados o
lenguaje vulgar)
Problemas de percepción perceptiva (como problemas para levantarse de
una silla o poner la mesa)
El comportamiento es el resultado de procesos cerebrales complejos, todos los
cuales tienen lugar en una fracción de segundo en el cerebro sano. En AD,
muchos de estos procesos están perturbados, y esta es la base de muchos
comportamientos angustiosos o inapropiados. Por ejemplo, los pacientes pueden

negarse a tomar un baño o vestirse porque no entienden lo que el cuidador les ha
pedido que hagan. Si lo comprenden, es posible que no recuerden cómo hacer lo
que se les pidió.
Esta alteracion de conducta es una máscara para la confusión y la ansiedad
subyacentes. En consecuencia, el riesgo de comportamientos agresivos es mayor
en esta etapa de progresión de la enfermedad. Los pacientes deben ser
monitoreados cuidadosamente para detectar cualquier comportamiento que pueda
comprometer la seguridad de quienes los rodean.
Para una persona que no puede recordar el pasado o anticipar el futuro, el mundo
que lo rodea puede ser extraño y aterrador. Mantenerse cerca de un cuidador
familiar y de confianza puede ser lo único que tiene sentido y proporciona
seguridad. Una persona con AD puede seguir constantemente a su cuidador y
preocuparse cuando la persona está fuera de la vista.
El juicio y el control de los impulsos continúan disminuyendo en esta etapa. Por
ejemplo, quitarse la ropa puede parecer razonable para una persona con AD que
se siente acalorada y no entiende o recuerda que no es aceptable desnudarse en
público.
Enfermedad de Alzheimer grave
En la última etapa, la enfermedad de Alzheimer severa, las placas y los ovillos se
extienden por todo el cerebro, y algunas áreas del cerebro se han atrofiado aún
más
Los pacientes no pueden reconocer a familiares y seres queridos ni comunicarse
de ninguna manera. Son completamente dependientes de otros para el cuidado.
Todo sentido del yo parece desvanecerse.
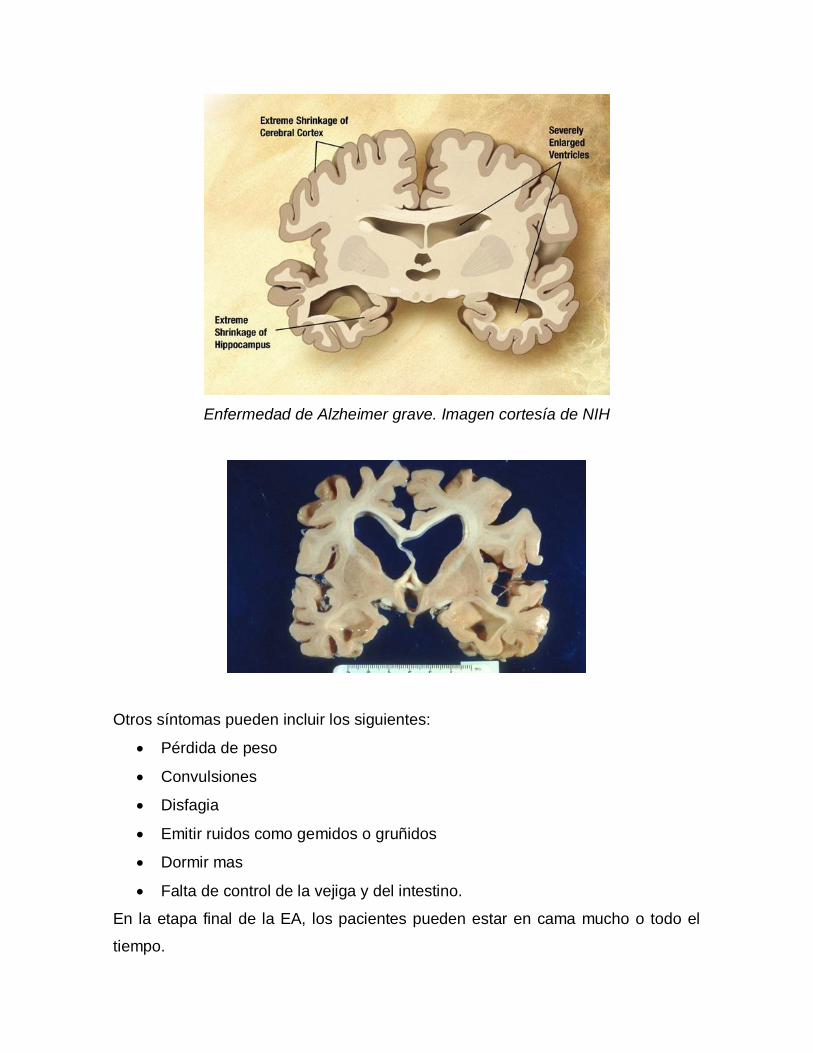
Enfermedad de Alzheimer grave. Imagen cortesía de NIH
Otros síntomas pueden incluir los siguientes:
Pérdida de peso
Convulsiones
Disfagia
Emitir ruidos como gemidos o gruñidos
Dormir mas
Falta de control de la vejiga y del intestino.
En la etapa final de la EA, los pacientes pueden estar en cama mucho o todo el
tiempo.
Generalmente el paciente puede fallecer por
causas diferentes a la enfermedad en si,
como infecciones, neumonia por aspiracion,
lesions en la piel por decubito y posterior
sobreinfeccion y otras tantas mas.
PROGRESION POR LOCALIZACION
DEL COMPROMISO PATOGENICO:
Pautas clínicas para el diagnostico
(GUIAS DE DIAGNOSTICO)
Las directrices clínicas para el
diagnóstico de la enfermedad de Alzheimer han sido formuladas por los Institutos
Nacionales de la Salud, Asociación de Enfermedades de Alzheimer y Trastornos
Relacionados (NIH-ADRDA); la Asociación Americana de Psiquiatría, en el Manual
Diagnóstico y Estadístico de los Trastornos Mentales, Quinta Edición (DSM5); y el
Consorcio para establecer un registro de la enfermedad de Alzheimer (CERAD).
En 2011, el grupo de trabajo del Instituto Nacional sobre el Envejecimiento (NIA) y
la Asociación de Alzheimer (AA) publicó nuevas investigaciones y criterios de
diagnóstico clínico para la enfermedad de Alzheimer.44
Los criterios de NIH-ADRDA para el diagnóstico de EA requieren el hallazgo de
una pérdida de memoria lentamente progresiva de inicio insidioso en un paciente
completamente consciente. La AD no se puede diagnosticar en pacientes con
conciencia nublada o delirio. Las condiciones metabólicas tóxicas y las neoplasias
cerebrales también deben excluirse como posibles causas de la demencia del
paciente.
El enfoque de los criterios NIA-AA de 2011
44 Jack CR Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, et al. Introduction to the
recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic
guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May. 7(3):257-62. [Medline]

es la necesidad de crear un diagnóstico más preciso de la enfermedad preclínica
para que el tratamiento pueda comenzar antes de que las neuronas se dañen
significativamente, mientras que es más probable que respondan. Por lo tanto, el
informe incluye criterios para el diagnóstico de lo siguiente:
EA asintomática, preclínica (para fines de investigación, no para diagnóstico
clínico) 45
Deterioro cognitivo leve (MCI), una fase temprana sintomática pero de
predementia de AD 46
Demencia AD 47
El Manual estadístico y de diagnóstico de trastornos mentales, Quinta edición,
publicado en 2013, reemplaza el término demencia por trastorno neurocognitivo
mayor y trastorno neurocognitivo leve. Los nuevos términos se centran en una
disminución, en lugar de un déficit, en la función.
Para cumplir con los criterios del DSM5 para AD, el individuo debe cumplir con los
criterios para un trastorno neurocognitivo mayor o menor y debe haber un inicio
insidioso y una progresión gradual de deterioro en uno o más dominios cognitivos
(para el trastorno neurocognitivo mayor, al menos dos dominios deben estar
impedido). El individuo también debe cumplir con los criterios de AD probable o
posible como se describe en el DSM5
DIAGNOSTICOS DIFERENCIALES
Consideraciones diagnósticas
Depresión
La depresión es una consideración importante en el diagnóstico diferencial de la
enfermedad de Alzheimer (EA). Las manifestaciones clínicas de la depresión se
45 • Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the
preclinical stages of Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's
Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May.
7(3):280-92. [Medline]. 46 • Albert MS, Dekosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild
cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging-
Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement.
2011 May. 7(3):270-9. [Medline]. 47 • McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis
of dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's
Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May.
7(3):263-9. [Medline].

superponen con las de la enfermedad de Alzheimer. El término pseudodemencia
se refiere a la aparición de disfunción cognitiva (demencia) debido a la depresión.
Además, se estima que entre el 30 y el 50% de los pacientes con AD tienen
depresión comórbida. 48
La depresión en pacientes con AD parece diferir de la depresión en pacientes
ancianos cognitivamente sanos. La depresión en la EA con mayor frecuencia
presenta trastornos motivacionales (por ejemplo, fatiga, desaceleración
psicomotora, apatía), mientras que la depresión en pacientes geriátricos sin
deterioro cognitivo tiende a presentar síntomas del estado de ánimo (por ejemplo,
estado de ánimo depresivo, ansiedad, suicidio, trastornos del sueño y apetito). 49
Los instrumentos comúnmente utilizados para evaluar la depresión (p. Ej., La
Escala de Hamilton para la Depresión, el Inventario de Depresión de Beck, la
Escala de Depresión Geriátrica) se diseñaron para su uso en otras poblaciones de
pacientes y pueden ser menos confiables en pacientes con EA. En consecuencia,
el Instituto Nacional de Salud Mental ha desarrollado criterios diagnósticos
provisionales para la depresión en la EA. Teng E, Ringman JM, Ross LK, et al.
Diagnosing depression in Alzheimer disease with the national institute of mental
health provisional criteria. Am J Geriatr Psychiatry. 2008 Jun. 16(6):469-
77. [Medline].
Encefalopatía traumática crónica.
El traumatismo craneal repetitivo ha sido reconocido durante mucho tiempo como
una causa de degeneración cerebral en los boxeadores (es decir, la demencia
pugilística). Más recientemente, la enfermedad cerebral degenerativa progresiva
(encefalopatía traumática crónica [ETC]) se ha reconocido en atletas con
antecedentes de múltiples conmociones cerebrales, así como golpes más leves en
la cabeza que no causan conmoción cerebral. 50
ETC confirmado neuropatológicamente ha sido reportado en jugadores de fútbol y
48 Teng E, Ringman JM, Ross LK, et al. Diagnosing depression in Alzheimer disease with the national
institute of mental health provisional criteria. Am J Geriatr Psychiatry. 2008 Jun. 16(6):469-77. [Medline]. 49 Teng E, Ringman JM, Ross LK, et al. Diagnosing depression in Alzheimer disease with the national
institute of mental health provisional criteria. Am J Geriatr Psychiatry. 2008 Jun. 16(6):469-77. [Medline]. 50 Lakhan SE, Kirchgessner A. Chronic traumatic encephalopathy: the dangers of getting "dinged".
Springerplus. 2012. 1:2. [Medline].

hockey profesionales retirados y otros atletas con un historial de repetidas lesiones
en la cabeza.
Las características patológicas de la CTE, que pueden no aparecer hasta mucho
después del final de la participación atlética activa, incluyen lo siguiente51Ovillos
neurofibrilares de proteina Tau positivos (NFT) en el neocórtex, concentrado
alrededor de los vasos del parénquima penetrante
Placas de amiloide difusas neocorticales, con o sin placas neuríticas
Sin compromiso hipocampo
La distribución de NFT en ETC difiere notablemente de la del envejecimiento
normal y la EA, en la que hay una afectación temprana de la corteza entorrinal y el
hipocampo con afectación posterior de la neocorteza en estadios avanzados.
Los síntomas de CTE pueden incluir los siguientes:
Dolores de cabeza recurrentes
Mareo
Trastornos del estado de ánimo
Agresión
Juicio deteriorado y control de impulsos.
Trastornos del movimiento con indicadores de parkinsonismo
Demencia progresiva
Otros trastornos
Otros trastornos a considerar en el diagnóstico diferencial de AD incluyen los
siguientes:
- Deterioro de la memoria asociado a la edad
- Abuso de alcohol o drogas
- Depresión
- Deficiencia de vitamina B12
- Enfermedad cerebrovascular (y demencia vascular)
- Discapacidad auditiva o visual.
- Hipernatremia 51 : Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH. Chronic traumatic
encephalopathy in a National Football League player. Neurosurgery. 2005 Jul. 57(1):128-34; discussion 128-
34. [Medline].

- Hipoglucemia
- Hipotiroidismo o hipertiroidismo.
- Demencia por cuerpos de Lewy
- Hidrocefalia de presión normal o normotensiva
- Enfermedad de Parkinson con demencia
- Polifarmacia
- Síndrome de Wernicke-Korsakoff
Diagnósticos diferenciales
- Afasia
- Estenosis de la arteria carótida
- Leucemia mielógena crónica (LMC)
- Degeneración Gangliónica Basal Cortical
- Demencia en la enfermedad de la neurona motora o motoneurona
- Demencia Con Cuerpos De Lewy
- Depresión
- Demencia frontotemporal
- Demencia de la enfermedad de Huntington
- Hidrocefalia normotensiva
- Parkinson
- Síndromes de Parkinson Plus
- Enfermedades provocadas por priones
- Demencia vascular
- Enfermedad de Wilson
APROXIMACION DIAGNOSTICA:
La enfermedad de Alzheimer (EA) es una patologia de diagnóstico clínico. Sin
embargo, se pueden usar estudios de imágenes auxiliares (p. Ej., Tomografía
computarizada [TC]; imagen de resonancia magnética [RMN]; TC de emisión de
fotón único [SPECT]; o tomografía de emisión de positrones [PET]) y pruebas de
laboratorio. Estas pruebas ayudan a excluir otras posibles causas de demencia (p.
Ej., Enfermedad cerebrovascular, deficiencia de cobalamina [vitamina B12], sífilis,

enfermedades de la tiroides).
Los criterios de diagnóstico establecidos por el Instituto Nacional sobre el
Envejecimiento (NIA) y la Asociación de Alzheimer tienen como objetivo principal
facilitar la investigación. Sin embargo, la NIA-AA también propuso "criterios
clínicos centrales" para el diagnóstico de deterioro cognitivo leve (MCI) por parte
de los proveedores de atención médica sin acceso a pruebas de líquido
cefalorraquídeo (LCR) o imágenes avanzadas. 52
Los criterios NIA-AA para el diagnóstico de la demencia debida a la enfermedad
de Alzheimer son clínicos, con biomarcadores como un papel auxiliar, no
esencial.53
Estudios de sangre
Se pueden realizar pruebas de laboratorio para descartar otras afecciones que
pueden causar deterioro cognitivo. Las recomendaciones actuales de la Academia
Americana de Neurología (AAN) incluyen la medición del nivel de cobalamina
(vitamina B12) y una prueba de detección de la función tiroidea. Las
investigaciones adicionales se dejan al médico, para que se adapte a las
necesidades particulares de cada paciente. Los resultados de la prueba inicial que
requieren mayor investigación incluyen lo siguiente:
Las anomalías en el recuento completo de células sanguíneas y los niveles de
cobalamina (vitamina B12) requieren un estudio adicional para descartar una
enfermedad hematológica
Las anomalías encontradas en la detección de los niveles de enzimas hepáticas
requieren un estudio adicional para descartar una enfermedad hepática.
Las anomalías en los niveles de hormona estimulante de la tiroides (TSH)
requieren un estudio adicional para descartar la enfermedad tiroidea.
Las anomalías en el reactivo rápido de plasma (RPR) requieren un estudio
52 Albert MS, Dekosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild
cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging-
Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement.
2011 May. 7(3):270-9. [Medline]. 53 McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, et al. The diagnosis of
dementia due to Alzheimer's disease: Recommendations from the National Institute on Aging-Alzheimer's
Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May.
7(3):263-9. [Medline].

adicional para descartar la sífilis.
Las anomalías en la serología y / o PCR del VIH requieren un estudio adicional
para descartar el VIH / SIDA.
Las anomalías en los anticuerpos paraneoplásicos requieren un estudio adicional
para descartar una encefalitis autoinmune.
Las anomalías en las proteínas del LCR tau, P-tau y 14-3-3 requieren un estudio
adicional para descartar la enfermedad de Creutzfeldt-Jakob.
Existe un posible vínculo entre la deficiencia de vitamina D y el deterioro cognitivo.
54
Sin embargo, la deficiencia de vitamina D no se ha identificado como una causa
reversible de demencia.
RM cerebral o tomografía computarizada
Las recomendaciones de la Academia Americana de Neurología (AAN) indican
que la neuroimagen estructural con una tomografía computarizada (TC) sin
contraste o una imagen de resonancia magnética (RMN) es apropiada en la
evaluación inicial de pacientes con demencia, para detectar lesiones que pueden
resultar en cognición deterioro (p. ej., ACV, enfermedad de pequeños vasos,
tumor).
Los estudios de imagen son particularmente importantes para descartar causas
potencialmente tratables de deterioro cognitivo progresivo, como el hematoma
subdural crónico o la hidrocefalia de presión normal.55
En pacientes con EA, las RMN cerebral o la TC pueden mostrar atrofia cortical y /
o cerebral difusa, pero estos hallazgos no son diagnósticos de EA.
En estudios de investigación clínica, la atrofia del hipocampo (estructuras
importantes en la mediación de los procesos de memoria) en la RMN coronal se
considera un biomarcador válido de la neuropatología de la EA. No obstante, la
54 Annweiler C, Schott AM, Allali G, Bridenbaugh SA, Kressig RW, Allain P, et al. Association of vitamin D deficiency with cognitive impairment in older women: cross-sectional study. Neurology. 2010 Jan 5.
74(1):27-32. [Medline].
Buell JS, Dawson-Hughes B, Scott TM, Weiner DE, Dallal GE, Qui WQ, et al. 25-Hydroxyvitamin D,
dementia, and cerebrovascular pathology in elders receiving home services. Neurology. 2010 Jan 5. 74(1):18-
26. [Medline]. [Full Text]. 55 [Guideline] Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J, Relkin N, et al. Practice
parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee
of the American Academy of Neurology. Neurology. 2001 May 8. 56(9):1143-53. [Medline]

medición del volumen del hipocampo no se utiliza en la atención clínica de rutina
en el diagnóstico de la enfermedad de Alzheimer.
Un estudio realizado por Chen y cols. Sugiere que la RMN funcional en estado de
reposo puede ayudar a clasificar a los pacientes con EA, los pacientes con
deterioro cognitivo leve amnésico (MCI) y los pacientes cognitivamente sanos.56
Un estudio realizado por McMillan et al sugiere que la RMN puede proporcionar un
sustituto no invasivo razonablemente preciso para los biomarcadores de líquido
cefalorraquídeo (LCR), lo que reduce la necesidad de punción lumbar en la AD
discriminatoria de la degeneración lobar frontotemporal (FTLD).57 Los
investigadores detectaron un patrón cerebral estructural de RMN que predice la
proporción de tau total y β-amiloide en el LCR, para discriminar la AD del FTLD.
De esta manera, pudieron diferenciar entre los 2 tipos de demencia en un 75% de
precision.58
Otros investigadores han sugerido que los biomarcadores de RMN pueden ser
clave para diagnosticar la AD temprana. 59
Además, se encontró que la corteza cingulada posterior y el lóbulo temporal
medial, 2 regiones que con frecuencia se ven afectadas por AD, tienen las
disminuciones más prominentes en la conectividad funcional.
SPECT y PET
No se recomienda la exploración cerebral con SPECT o PET para el diagnostico
de rutina de los pacientes con presentaciones típicas de EA. Estas modalidades
pueden ser útiles en casos atípicos o cuando un diagnóstico más probable es una
56 Chen G, Ward BD, Xie C, Li W, Wu Z, Jones JL, et al. Classification of Alzheimer Disease, Mild
Cognitive Impairment, and Normal Cognitive Status with Large-Scale Network Analysis Based on Resting-
State Functional MR Imaging. Radiology. 2011 Apr. 259(1):213-21. [Medline] 57 Brooks M. MRI may offer noninvasive option for Alzheimer’s Diagnosis. Medscape Medical News. Dec
27 2012. Available at http://www.medscape.com/viewarticle/776766. Accessed: Jan 8 2012. McMillan CT,
Avants B, Irwin DJ, Toledo JB, et al. Can MRI screen for CSF biomarkers in neurodegenerative disease?. Neurology. 2012 Dec 26. [Medline]. 58 Brooks M. MRI may offer noninvasive option for Alzheimer’s Diagnosis. Medscape Medical News. Dec
27 2012. Available at http://www.medscape.com/viewarticle/776766. Accessed: Jan 8 2012.
McMillan CT, Avants B, Irwin DJ, Toledo JB, et al. Can MRI screen for CSF biomarkers in
neurodegenerative disease?. Neurology. 2012 Dec 26. [Medline]. 59 Brooks M. Functional Brain Imaging May Spot Alzheimer's Early. Medscape Medical News. Aug 21 2013.
[Full Text]. Wang L, Brier MR, Snyder AZ, et al. Cerebrospinal fluid Aß42, phosphorylated tau181, and
resting-state functional connectivity. JAMA Neurol. 2013 Aug 19. [Medline].
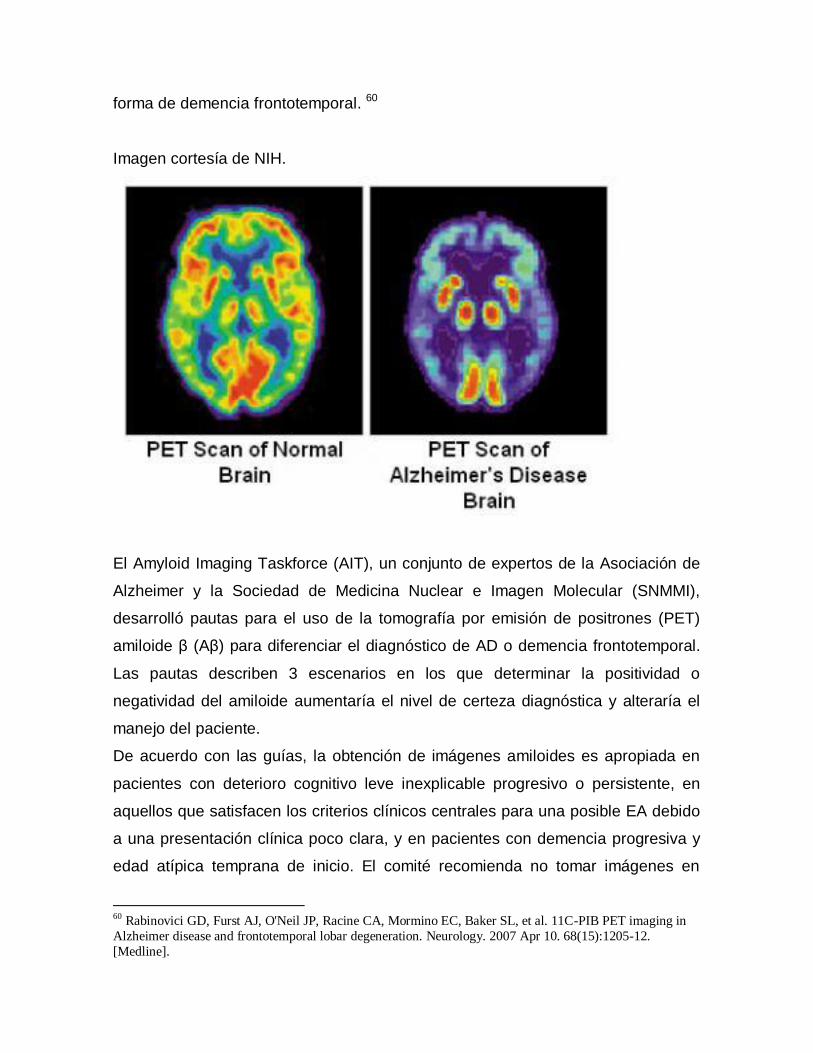
forma de demencia frontotemporal. 60
Imagen cortesía de NIH.
El Amyloid Imaging Taskforce (AIT), un conjunto de expertos de la Asociación de
Alzheimer y la Sociedad de Medicina Nuclear e Imagen Molecular (SNMMI),
desarrolló pautas para el uso de la tomografía por emisión de positrones (PET)
amiloide β (Aβ) para diferenciar el diagnóstico de AD o demencia frontotemporal.
Las pautas describen 3 escenarios en los que determinar la positividad o
negatividad del amiloide aumentaría el nivel de certeza diagnóstica y alteraría el
manejo del paciente.
De acuerdo con las guías, la obtención de imágenes amiloides es apropiada en
pacientes con deterioro cognitivo leve inexplicable progresivo o persistente, en
aquellos que satisfacen los criterios clínicos centrales para una posible EA debido
a una presentación clínica poco clara, y en pacientes con demencia progresiva y
edad atípica temprana de inicio. El comité recomienda no tomar imágenes en
60 Rabinovici GD, Furst AJ, O'Neil JP, Racine CA, Mormino EC, Baker SL, et al. 11C-PIB PET imaging in
Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007 Apr 10. 68(15):1205-12.
[Medline].

individuos asintomáticos y pacientes con un diagnóstico claro de EA con una edad
típica de inicio. La exploración no se puede usar para clasificar la demencia o
determinar su gravedad, y no se debe usar en lugar de la genotipificación de los
portadores sospechosos de mutación autosómica. 61
Florbetapir F 18 (AMYViD) fue aprobado por la FDA en abril de 2012 como agente
de diagnóstico por imagen. Está indicado para la PET de imágenes de cerebro de
plagas neuríticas beta-amiloides en adultos que se están evaluando para la
enfermedad de Alzheimer u otro deterioro cognitivo. 62
La aprobación de florbetapir F 18 se basó en 3 estudios clínicos que examinaron
imágenes de pacientes adultos sanos así como pacientes con una variedad de
trastornos cognitivos, incluidos algunos pacientes con enfermedades terminales
que aceptaron participar en un programa de donación de cerebro postmortem. Las
mediciones de la carga amiloide cortical postmortem se correlacionaron con las
puntuaciones medias de florbetapir F 18. 63
En un estudio realizado por Clark y cols., la presencia y la densidad de beta
amiloide se correlacionaron estrechamente en individuos con imágenes de
florbetapir-PET dentro de los 99 días anteriores a la muerte y luego de la
autopsia.64
Los pacientes con enfermedad de Alzheimer probable, deterioro cognitivo leve o
controles sanos mayores mostraron relaciones de valor de captación de florbetapir
61 Anderson, P. New Guidelines for Amyloid PET Imaging. Available at
http://www.medscape.com/viewarticle/778274. Accessed: February 5, 2013. 62 Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al. Correlation of amyloid PET
ligand florbetapir F 18 binding with Aß aggregation and neuritic plaque deposition in postmortem brain
tissue. Alzheimer Dis Assoc Disord. 2012 Jan. 26(1):8-16. [Medline]. Clark CM, Schneider JA, Bedell
BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for imaging beta-amyloid pathology.
JAMA. 2011 Jan 19. 305(3):275-83. [Medline]. Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, et al. Using positron emission tomography and florbetapir F18 to image cortical amyloid in
patients with mild cognitive impairment or dementia due to Alzheimer disease. Arch Neurol. 2011 Nov.
68(11):1404-11. [Medline]. 63 Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al. Correlation of amyloid PET
ligand florbetapir F 18 binding with Aß aggregation and neuritic plaque deposition in postmortem brain
tissue. Alzheimer Dis Assoc Disord. 2012 Jan. 26(1):8-16. [Medline]. [Full Text]. 64 Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al. Use of florbetapir-PET for
imaging beta-amyloid pathology. JAMA. 2011 Jan 19. 305(3):275-83. [Medline].

cortical medias significativamente diferentes en un estudio de Fleisher et al. 65
En octubre de 2013, la FDA aprobó un segundo derivado del compuesto B de
Pittsburgh (PIB) marcado con 18F, inyección de flutemetamol F18 (Vizamyl), para
uso con imágenes de PET en adultos que se someten a una evaluación de la
enfermedad de Alzheimer y la demencia. Al igual que el florbetapir F18, el
flutemetamol F18 se adhiere al beta-amiloide en el cerebro y produce una imagen
PET que se puede usar para evaluar su presencia. Una exploración positiva indica
que es probable que haya una cantidad moderada o mayor de amiloide en el
cerebro, pero no establece un diagnóstico de enfermedad de Alzheimer u otra
demencia. La efectividad del flutemetamol F18 se estableció en 2 estudios clínicos
con 384 participantes que tenían un rango de función cognitiva. 66
Un tercer agente, florbetaben F18 (Neuraceq), fue aprobado por la FDA en marzo
de 2014. Las imágenes se pueden obtener entre 45 y 130 minutos después de la
dosis inyectada. La aprobación de la FDA se basó en los datos de seguridad de
872 pacientes que participaron en ensayos clínicos globales, así como en 3
estudios que examinaron imágenes de adultos con una variedad de funciones
cognitivas, incluidos 205 pacientes que habían aceptado participar en un estudio
posterior de donación de cerebro mortem. Se analizaron las imágenes de 82
sujetos con confirmación post mortem de la presencia o ausencia de placas
neuríticas de beta-amiloide. 67
Un estudio de 129 adultos cognitivamente normales de 65 a 87 años (media, 73,7
años) indicó que una combinación de pruebas de memoria e imágenes cerebrales
puede ayudar a identificar las etapas más tempranas de la EA, antes de que
65 Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, et al. Using positron emission
tomography and florbetapir F18 to image cortical amyloid in patients with mild cognitive impairment or
dementia due to Alzheimer disease. Arch Neurol. 2011 Nov. 68(11):1404-11. [Medline]. 66 US Food and Drug Administration (FDA). FDA approves second brain imaging drug to help evaluate
patients for Alzheimer’s disease, dementia [press release]. October 25, 2013. Available at
http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm. Accessed: October 25,
2013. 67 Neuraceq (florbetaben F 18) prescribing information. [package insert]. Matran Switzerland: Piramal
Imaging, S.A. 2014.

aparezcan los síntomas68 Los investigadores encontraron que el compromiso de la
memoria episódica en el contexto de disfunción sináptica y los niveles elevados de
amiloide identificaron a los sujetos que tenían un alto riesgo de progresión a
demencia de la EA.
Los sujetos en este estudio se sometieron a pruebas de memoria y función
ejecutiva junto con la tomografía por emisión de positrones de fluorodeoxiglucosa
con flúor-18 (FDG-PET) y la deposición de amiloide con C 11 Pittsburgh
Compound B (PiB PET). 69 Los investigadores encontraron que la carga de
amiloide y el metabolismo de FDG más bajo (disfunción sináptica) predecían de
forma independiente el rendimiento de la memoria episódica. Los sujetos con peor
rendimiento de la memoria tuvieron mayor PiB y menor metabolismo de FDG en
regiones del cerebro comúnmente afectadas en la EA.
Electroencefalografía:
La electroencefalografía (EEG) es valiosa cuando la enfermedad de Creutzfeldt-
Jakob u otra enfermedad relacionada con los priones es un diagnóstico probable
(ver EEG en Demencia y Encefalopatía). En la mayoría de los casos de la
enfermedad de Creutzfeldt-Jakob, eventualmente se pueden detectar ondas
agudas periódicas.
El EEG también es útil en casos de pseudodemencia, cuando existe un EEG
normal en un paciente que parece profundamente demente. La demencia
raramente presenta convulsiones en la enfermedad de Alzheimer, pero de
presentarlas, un EEG sería valioso para evaluar.
Punción lumbar
La punción lumbar puede ser util en casos seleccionados, para descartar
afecciones como hidrocefalia, infecciónes del sistema nervioso central (p. Ej.,
Neurosífilis, neuroborreliosis, criptococosis), enfermedad por priones.
Los niveles de tau y tau fosforilado a menudo se elevan en la AD, mientras que los
niveles de amiloide suelen ser bajos. La razón de esto no se conoce, pero quizás
68 . Brooks M. Memory tests plus brain scans detect earliest stages of AD. Medscape Medical News. July 15,
2013. 69 Brooks M. Memory tests plus brain scans detect earliest stages of AD. Medscape Medical News. July 15,
2013.

los niveles de amiloide son bajos porque el amiloide se deposita en el cerebro en
lugar de en el LCR. Al medir ambas proteínas, se puede lograr una sensibilidad y
una especificidad de al menos el 80%, y más a menudo el 90%.
En la actualidad, sin embargo, no se recomienda la medición rutinaria de tau y
amiloide en LCR, excepto en entornos de investigación. La punción lumbar para la
medición de tau y amiloide puede formar parte del trabajo de diagnóstico cuando
se desarrollan terapias efectivas que disminuyen la tasa de progresión de la EA,
especialmente si las terapias son específicas para la EA y tienen una morbilidad
significativa.
Genodiagnostico
La genotipificación de los alelos de apolipoproteína E (APOE) es una herramienta
de investigación que ha sido útil para determinar el riesgo de AD en las
poblaciones, pero hasta hace poco tenía poco o ningún valor en hacer un
diagnóstico clínico y desarrollar un plan de manejo en pacientes individuales .
Numerosas declaraciones de consenso han recomendado no utilizar el genotipado
APOE para predecir el riesgo de EA.70
Investigadores del Estudio de población general de Copenhague informaron que
los niveles plasmáticos de APOE épsilon 4 (APOE ε4) están asociados con el
riesgo de demencia, independientemente del genotipo APOE.71 El riesgo de
enfermedad de Alzheimer aumentó con niveles decrecientes de niveles de APOE,
con un riesgo altamente significativo de 3 veces mayor para el tercil más bajo de
los niveles de APOE en relación con el tercil más alto, una asociación que se
mantuvo incluso después de ajustar la APOE genotipo. Los genotipos APOE con
mayor riesgo de enfermedad de Alzheimer fueron ε43 y ε44, mientras que aquellos
con los menores riesgos fueron ε22, ε32, ε42 y ε33. 72
Previamente, se ha expresado la preocupación de que, en individuos
70 Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical
Genetics and the National Society of Genetic Counselors. Genet Med. 2011 Jun. 13(6):597-605. [Medline]. 71 Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R. Apolipoprotein E plasma level
and genotype -- risk of dementia in 76,000 individuals from the general population. Presented at: XXI World
Congress of Neurology (WNC), 2013; September 24, 2013; Vienna, Austria. Keller DM. APOE plasma
levels predict dementia independent of genotype. Medscape Medical News. October 4, 2013. 72 Keller DM. APOE plasma levels predict dementia independent of genotype. Medscape Medical News.
October 4, 2013.

asintomáticos, las pruebas genéticas que identifican un mayor riesgo pueden
desencadenar una respuesta psicológica desfavorable. Sin embargo, en un
ensayo sobre el efecto de la divulgación de los resultados de genotipificación de
APOE en 162 adultos asintomáticos que tuvieron un padre con AD, Green et al
encontraron que las pruebas de seguimiento durante el transcurso de un año no
mostraron diferencias significativas con la divulgación en comparación con la no
divulgación a tiempo medidas promediadas de ansiedad, depresión o angustia
relacionada con la prueba73 La angustia relacionada con la prueba se redujo en
aquellos que se enteraron de que no tenían el alelo APOE E4). Las personas que
tenían altos niveles de estrés emocional antes de someterse a pruebas genéticas
tenían más probabilidades de tener dificultades emocionales después de la
divulgación. Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE
genotype for risk of Alzheimer's disease. N Engl J Med. 2009 Jul 16. 361(3):245-
54. [Medline].
Pruebas genéticas para APP y mutaciones de presenilina
De acuerdo con las pautas del Colegio Americano de Genética Médica y la
Sociedad Nacional de Consejeros Genéticos, las pruebas para los genes de APP
y presenilina asociados con el AD autosómico dominante de inicio temprano
deben ofrecerse en las siguientes situaciones: 74En pacientes sintomáticos con EA
de inicio temprano que tienen antecedentes familiares de demencia o un historial
familiar desconocido (por ejemplo, debido a la adopción)
En personas con antecedentes familiares de demencia autosómica
dominante con uno o más casos de AD de inicio temprano
En familiares con una mutación compatible con AD de inicio temprano (es
decir, PS-1, PS-2, APP)
Antes de someterse a las pruebas, los pacientes deben recibir asesoramiento
adecuado sobre el valor predictivo de las pruebas genéticas para la EA. Después,
los pacientes deben recibir asesoramiento sobre las implicaciones de los
73 . Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE genotype for risk of Alzheimer's disease. N
Engl J Med. 2009 Jul 16. 361(3):245-54. [Medline]. 74 Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic
counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical
Genetics and the National Society of Genetic Counselors. Genet Med. 2011 Jun. 13(6):597-605. [Medline].

resultados. Las recomendaciones sobre el asesoramiento previo y posterior a la
prueba se detallan en las directrices del Colegio Americano de Genética Médica y
la Sociedad Nacional de Consejeros Genéticos. 75
TRATAMIENTO:
Consideraciones de enfoque
Hasta la fecha, solo se dispone de terapias sintomáticas para la enfermedad de
Alzheimer (EA) y, por lo tanto, no actúan sobre la evolución de la enfermedad. El
tratamiento médico estándar para la AD incluye los inhibidores de la colinesterasa
(ChEI) y un antagonista parcial de N -metil-D-aspartato (NMDA). [3, 4]
Los síntomas secundarios de la EA (p. Ej., Depresión, agitación, agresión,
alucinaciones, delirios, trastornos del sueño) pueden ser problemáticos. Los
síntomas conductuales en particular son comunes y pueden exacerbar el deterioro
cognitivo y funcional. Las siguientes clases de medicamentos psicotrópicos se han
utilizado para tratar estos síntomas secundarios [5]:
- Antidepresivos
- Ansiolíticos
- Agentes antiparkinsonianos
- Bloqueadores beta
- Fármacos antiepilépticos (por sus efectos sobre el comportamiento).
- Neurolépticos
La mayoría de los estudios de fármacos psicotrópicos para la EA han demostrado
una eficacia limitada o nula. Sin embargo, muchos problemas dificultan la
interpretación de los datos de estos estudios.
La investigación farmacológica actual en la EA se centra principalmente en el
desarrollo de fármacos modificadores de la enfermedad que pueden retardar o
revertir la progresión de la EA. Los objetivos de estos agentes en investigación
han incluido la producción, la agregación y la eliminacion de beta-amiloide, así
como la fosforilación y el ensamblaje de tau. Hasta la fecha, ninguno de estos
fármacos ha demostrado eficacia en los ensayos de fase III. [98] Sin embargo, un
75 Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB, Rumbaugh M, et al. Genetic
counseling and testing for Alzheimer disease: joint practice guidelines of the American College of Medical
Genetics and the National Society of Genetic Counselors. Genet Med. 2011 Jun. 13(6):597-605. [Medline].

estudio de 2018 fase II mostró resultados prometedores para un agente
antiamiloide en pacientes con AD en estadio temprano. El estudio incluyó a 856
pacientes con EA temprana (deterioro cognitivo leve debido a EA o demencia leve
a EA) y patología amiloide confirmada por tomografía por emisión de positrones
(PET) o trazador del líquido cefalorraquídeo (LCR). Se encontró que el agente,
BAN2401, reducía significativamente el amiloide cerebral en dosis altas. El estudio
también mostró un beneficio de la función cognitiva , más significativa en función
de la dosis. [99]
Los posibles tratamientos quirúrgicos en el futuro pueden incluir el uso de
dispositivos para infundir factores neurotróficos, como los factores de crecimiento.
Se debe considerar la hospitalización para cualquier condición médica inestable
que pueda complicar el tratamiento del paciente. Si el paciente se convierte en un
peligro para sí mismo o para otras personas, la hospitalización a corto plazo puede
estar indicada para facilitar la eliminación de procesos infecciosos y metabólicos y
el ajuste de los medicamentos psicotrópicos. La razón más común para la
admisión a un centro de atención a largo plazo es la necesidad de supervisión las
24 horas que no se puede administrar en el hogar y / o el estrés / agotamiento del
cuidador.
Tratamiento de la enfermedad leve a moderada
El diagnóstico y tratamiento tempranos permiten a los pacientes con EA mantener
los niveles más altos de capacidad cognitiva y funcional posible. Los inhibidores
de la colinesterasa (ChEI) y la rehabilitacion cognitiva se usan para intentar
prevenir o retrasar el deterioro de la cognición en pacientes con AD.
Inhibidores de la colinesterasa (ChEI)
Numerosas líneas de evidencia sugieren que los sistemas colinérgicos que
modulan el procesamiento de la información en el hipocampo y el neocórtex se
deterioran en una etapa temprana del curso de la EA. Estas observaciones han
sugerido que algunas de las manifestaciones clínicas de la EA se deben a la
pérdida de la inervación colinérgica de la corteza cerebral.
Los ChEI de acción central previenen la descomposición de la acetilcolina. Tres de
estos agentes han sido aprobados por la FDA para el tratamiento de la EA, de la

siguiente manera:
- Donepecilo
- Rivastigmina
- Galantamina
Todos los ChEI han demostrado un beneficio modesto en las medidas de la
función cognitiva y las actividades de la vida diaria. Los pacientes tratados con
ChEI han mostrado disminuciones más lentas en las medidas cognitivas y
funcionales que los pacientes que reciben placebo. Sin embargo, los ChEI no
abordan la causa subyacente de la degeneración de las neuronas colinérgicas,
que continúa durante la enfermedad. Los ChEI también pueden aliviar las
manifestaciones no cognitivas de la EA, como la agitación, el deambular y el
comportamiento socialmente inapropiado. [100]
Aunque originalmente se esperaba que la utilidad de los ChEI se limitara a las
etapas iniciales e intermedias de la EA (debido a que el déficit colinérgico se
agrava más tarde en la enfermedad y porque hay menos sinapsis colinérgicas
intactas), también son útiles en la enfermedad avanzada. [101] Los ChEI también
son útiles en pacientes con EA con infartos concomitantes y en pacientes con
demencia con cuerpos de Lewy.
Los ChEI comparten un perfil común de efectos adversos, los más frecuentes son
náuseas, vómitos, diarrea y mareos. Estos están típicamente relacionados con la
dosis y se pueden mitigar con una titulación lenta hacia la dosis de mantenimiento
deseada. Además, los efectos secundarios gastrointestinales pueden reducirse
utilizando el parche transdérmico en lugar de la forma oral del medicamento.
Como los medicamentos antimucarínicos se usan para el tratamiento de la
incontinencia, lógicamente, los ChEI podrían exacerbar la incontinencia. [102]
Los ChEI prescritos para tratar la demencia pueden provocar bradicardia
sintomática y síncope y precipitar lesiones relacionadas con caídas, incluida la
fractura de cadera. En un estudio de adultos mayores con demencia que tomaban
inhibidores de la colinesterasa, se observó que las visitas al hospital para el
síncope eran más frecuentes en los pacientes que recibían ChEI que en los
pacientes de control (31.5 versus 18.6 eventos por 1000 personas-año). [103]

También se observó que otros eventos relacionados con el síncope, como visitas
al hospital por bradicardia, inserción de marcapasos permanente y fractura de
cadera, son más comunes en pacientes que reciben inhibidores de la
colinesterasa. El uso de ChEI en adultos mayores con demencia se asocia con un
mayor riesgo de eventos relacionados con el síncope; estos riesgos deben
sopesarse frente a los beneficios de tomar ChEIs. [103]
Discontinuar o cambiar de medicacion:
Existen informes de deterioro cognitivo y conductual agudo asociado con la
terminación abrupta de los ChEI. En varios de estos casos, reiniciar el ChEI no dio
lugar a una mejora sustancial. Estos informes tienen implicaciones con respecto a
las mejores prácticas cuando se cambia de un paciente de un ChEI a otro en esta
clase. Las razones para el cambio pueden incluir efectos secundarios indeseables
o una aparente falta de eficacia. No obstante, no hay datos publicados disponibles
para ayudar a los clínicos a saber cuándo sería útil cambiar a otro ChEI.
No se debe seguir la práctica común de reducir gradualmente a un paciente de un
medicamento activo para el SNC antes de comenzar con uno nuevo al cambiar los
ChEI. Por ejemplo, un paciente que estaba tomando 10 mg de donepezilo debe
iniciarse al día siguiente con galantamina en una dosis de al menos 8 mg / día y
posiblemente 16 mg / día. Ninguna evidencia actual apoya el uso de más de 1
ChEI a la vez. Se deben evitar los medicamentos anticolinérgicos de acción
central.
No es infrecuente que los pacientes reciban ChEI y agentes anticolinérgicos, que
se contrarrestan entre sí. Los medicamentos con efectos anticolinérgicos, como la
difenhidramina, los antidepresivos tricíclicos (p. Ej., Amitriptilina, nortriptilina) y la
oxibutinina (comúnmente utilizada para la espasticidad de la vejiga), pueden
causar disfunción cognitiva. Por lo tanto, una lista cuidadosa de los medicamentos
del paciente es importante para que el médico pueda reducir las dosis de, o
idealmente eliminar, todos los agentes anticolinérgicos de acción central.
Actividad mental para apoyar la cognición.
A muchos pacientes con cognición normal o aquellos con deterioro leve les
preocupa desarrollar AD. Muchos expertos creen que las actividades mentalmente

desafiantes, como hacer crucigramas y acertijos, pueden reducir el riesgo en tales
pacientes. Se desconoce si dichas actividades pueden reducir la tasa de
progresión de la enfermedad en pacientes que ya tienen AD. Se están realizando
ensayos clínicos para determinar el efecto que tienen estas actividades cognitivas
en la progresión de la EA.
Las actividades mentales deben mantenerse dentro de un nivel razonable de
dificultad. Las actividades deberían ser preferiblemente interactivas, y deberían
estar diseñadas para permitir que el paciente reconozca y corrija los errores. Lo
más importante es que estas actividades se deben administrar de una manera que
no cause frustración excesiva y que, idealmente, motive al paciente a participar en
ellas con frecuencia.
Tratamiento de la enfermedad moderada a severa
Se cree que el antagonista parcial de N-metil-D-aspartato (NMDA) memantina
(Namenda, Namenda XR) funciona mejorando la relación señal-ruido de la
transmisión glutamatérgica en el receptor NMDA. Se cree que el bloqueo de los
receptores de NMDA por la memantina retarda la acumulación de calcio
intracelular y por lo tanto ayuda a prevenir un mayor daño a los nervios. Este
agente está aprobado por la FDA para el tratamiento de la EA moderada y grave.
Varios estudios han demostrado que la memantina se puede usar de manera
segura en combinación con los ChEI. Se ha demostrado que la combinación de
memantina con un ChEI retrasa significativamente la institucionalización en
pacientes con AD. [104] Los estudios sugieren que el uso de memantina con
donepezilo afecta la cognición en la AD moderada a grave [105] pero no en la EA
leve a moderada. [106, 107] En 2014, la FDA aprobó una combinación de dosis
fijas de memantina (ER) y donepezil (Namzaric) una vez al día. [108] El mareo, el
dolor de cabeza y la confusión son algunos de los más comunes Efectos
secundarios de la memantina.
En junio de 2013, la FDA aprobó la rivastigmina transdérmica para la EA grave.
[109] La aprobación se basó en el estudio ACCIÓN (Actividades de la vida diaria y
cognición en pacientes con demencia grave del tipo de Alzheimer), en el que una
dosis más alta del fármaco (13,3 mg / 24 hr) demostró una mejora

estadísticamente significativa en la cognición general y la función en comparación
con una dosis más baja (4,6 mg / 24 h). [110
Tratamiento de los síntomas secundarios
Una variedad de intervenciones conductuales y farmacológicas pueden aliviar las
manifestaciones clínicas de la EA, como ansiedad, agitación, depresión,
comportamiento psicótico y problemas para dormir. La efectividad de tales
intervenciones varía de moderada y temporal a excelente y prolongada. No se
acepta por unanimidad ningún agente o dosis específica de agentes individuales
para la amplia gama de manifestaciones clínicas. En la actualidad, la FDA no ha
aprobado ningún agente psicotrópico para el tratamiento de la EA.
Intervenciones conductuales
Las intervenciones conductuales van desde los enfoques centrados en el paciente
hasta la capacitación de cuidadores para ayudar a controlar las manifestaciones
cognitivas y conductuales de la EA. Estas intervenciones a menudo se combinan
con las intervenciones farmacológicas más utilizadas, como ansiolíticos para la
ansiedad y la agitación, neurolépticos para delirios o alucinaciones, y
antidepresivos o estabilizadores del estado de ánimo para los trastornos del
estado de ánimo y manifestaciones específicas (por ejemplo, episodios de ira).
Un estudio francés descubrió que un programa psicoeducativo para cuidadores
primarios de pacientes con EA les proporcionó a los cuidadores una comprensión
más efectiva de la enfermedad y una mejor capacidad de afrontamiento. Sin
embargo, el programa no tuvo efecto en las actividades de la vida diaria de los
pacientes. [111]
Agentes neurolepticos
En 2005, la FDA agregó una advertencia sobre el uso de neurolépticos atípicos en
el tratamiento de síntomas secundarios de la EA, como la agitación. [112] Los
análisis sugirieron que los pacientes con neurolépticos atípicos tenían un mayor
riesgo de muerte o accidente cerebrovascular en comparación con los pacientes
con placebo. En 2008, se incluyó una advertencia sobre haloperidol,
proclorperazina, tioridazina y clorpromazina por la misma razón.
Un estudio piloto en pacientes con EA con psicosis o agitación que respondieron

al tratamiento con haloperidol encontró que la suspensión del fármaco después de
6 meses se asociaba con un mayor riesgo de recaída. Los investigadores
aconsejaron que en los pacientes que responden a este medicamento
antipsicótico, el mayor riesgo de recaída después de la interrupción debe
sopesarse frente a los efectos secundarios asociados con la continuación de la
medicación. [113]
Otra preocupación es el riesgo de que estos agentes puedan contribuir al deterioro
cognitivo. Los estudios clínicos antipsicóticos de la efectividad de la intervención,
el estudio de la enfermedad de Alzheimer (CATIE-AD) encontraron que los
antipsicóticos atípicos olanzapina, quetiapina y risperidona se asociaron con un
empeoramiento de la función cognitiva en una magnitud consistente con un
deterioro de 1 año. [114]
La recomendación general es usar dichos agentes con la menor frecuencia posible
y en las dosis más bajas posibles para minimizar los efectos adversos,
especialmente en pacientes ancianos frágiles. Se ha expresado especial
preocupación sobre el potencial de los agentes que agotan la dopamina para
agravar las manifestaciones motoras de la demencia con cuerpos de Lewy (DLB),
porque los pacientes con DLB pueden ser extremadamente sensibles a estos
agentes.
Antidepresivos y moduladores del estado de ánimo.
Los antidepresivos tienen un papel importante en el tratamiento de los trastornos
del estado de ánimo en pacientes con EA. La depresión se observa en más del
30% de los pacientes con EA, y con frecuencia comienza antes de que se
diagnostique clínicamente la EA. Por lo tanto, la paliación de esta condición
comórbida frecuente puede mejorar el rendimiento cognitivo y no cognitivo.
Nyth encontró que el citalopram es beneficioso en el estado de ánimo y otros
síntomas neuropsiquiátricos en pacientes en la etapa moderada de la EA. [115]
Debido a que citalopram puede causar aumentos dependientes de la dosis en el
intervalo QT, la FDA recomienda usar un máximo de 40 mg al día y considerar 20
mg al día en los ancianos. [116]
En un estudio aleatorizado, controlado con placebo en 186 pacientes con EA

probable y agitación clínicamente significativa, el tratamiento con 30 mg de
citalopram redujo la agitación diaria y la angustia del cuidador. Sin embargo,
citalopram también aumentó el riesgo de eventos cardíacos adversos y redujo
ligeramente la cognición, lo que los autores del estudio señalan que puede limitar
su aplicación práctica. Además, mientras se realizaba el estudio, la FDA emitió
una advertencia de que el citalopram puede prolongar el intervalo QT y que los
pacientes mayores no deben usar dosis superiores a 20 mg. [117, 118, 119]
Weintraub et al [120] y Petracca et al [121] encontraron que la sertralina y la
fluoxetina no tienen beneficios a corto o largo plazo en el estado de ánimo sobre el
placebo. De manera similar, Banerjee et al encontraron que el tratamiento de la
depresión con sertralina o mirtazapina no proporcionó beneficios en comparación
con el placebo y aumentó el riesgo de eventos adversos. [122]
Los resultados de varios estudios indican que los anticonvulsivos (por ejemplo,
gabapentina, ácido valproico) pueden tener un papel en el tratamiento de los
problemas de conducta en pacientes con enfermedad de Alzheimer. Sin embargo,
un ensayo de 313 pacientes con EA moderada encontró que 24 meses de
tratamiento con valproato no retrasó la aparición de agitación o psicosis, no
disminuyó el deterioro cognitivo o funcional y se asoció con efectos tóxicos
significativos. [125]
Otros agentes
Un metaanálisis de siete estudios realizado en 2016 demostró un tiempo total de
sueño prolongado durante la noche con melatonina, sin mejoría en las
capacidades cognitivas evaluadas mediante el examen del estado mini-mental y la
subescala cognitiva de evaluación de la enfermedad de Alzheimer. [126]
La investigación sobre los efectos de la prescripción de hipnóticos como zolpidem,
zopiclon o zaleplon para tratar los problemas del sueño en pacientes con
demencia encontró que estos llamados medicamentos Z aumentan
significativamente el riesgo de fractura. En el estudio, a los dos años de
seguimiento, el riesgo general de fractura aumentó en un 40% y el riesgo de
fractura de cadera en un 59%. [127]
Antinflamatorios

Muchos estudios han sugerido que se produce una inflamación en el cerebro de
pacientes con AD. Los estudios epidemiológicos sugieren que algunos pacientes
con terapia antiinflamatoria a largo plazo tienen un riesgo menor de desarrollar EA.
No obstante, ningún ensayo clínico aleatorizado de más de 6 meses ha
demostrado la eficacia de los fármacos antiinflamatorios para reducir la tasa de
progresión de la EA.
Si bien los informes anteriores reflejan el inicio tardío de la EA en individuos que
usaron antiinflamatorios no esteroides (AINE), un estudio de Breitner et al
demostró que los AINE no protegen contra la EA, al menos en personas muy
mayores. Al basarse en los registros de dispensación de farmacias
computarizados y en el cribado bienal de la demencia, estos investigadores
encontraron que la incidencia de AD se incrementó en los usuarios de AINE
pesados. Estos hallazgos pueden representar el aplazamiento de los síntomas de
la EA desde una edad más temprana hasta una edad avanzada. [128]
Terapias Experimentales
Se han propuesto una variedad de terapias experimentales para la EA. Estos
incluyen la terapia antiamiloide, la reversión del exceso de fosforilación de tau, la
terapia con estrógenos, la terapia con vitamina E y la terapia del eliminador de
radicales libres. Los estudios de estas terapias han dado resultados en su mayoría
decepcionantes.
En los últimos 10 años, se han realizado numerosos estudios de terapia
antiamiloide para disminuir los fragmentos amiloides tóxicos en el cerebro,
incluidos los estudios de los siguientes:
- Vacunación con especies amiloides.
- Administración de anticuerpos antiamiloides monoclonales.
- Administración de inmunoglobulina intravenosa que puede contener
anticuerpos de unión a amiloide
- Agentes selectivos para la disminución de amiloide
- Agentes quelantes para prevenir la polimerización amiloide.
- Inhibidores de la beta-secretasa para prevenir la generación del fragmento
amiloide A-beta

Hasta la fecha, ningún estudio de fase III de terapias antiamiloides ha mostrado
una combinación de eficacia aceptable y efectos secundarios.
La creciente conciencia de que tau es un fenomeno central en la patogénesis de la
EA ha sugerido que esta proteína puede ofrecer una vía para la intervención
terapéutica. Se están realizando estudios con agentes que pueden prevenir o
revertir el exceso de fosforilación de tau y, por lo tanto, disminuir la formación de
ovillos neurofibrilares. [129]
La terapia antagonista de radicales libres también ha atraído la atención, porque
los niveles excesivos de radicales libres en el cerebro son neurotóxicos. Sin
embargo, ningún estudio ha demostrado la eficacia de los antagonistas de
radicales libres en el tratamiento de los síntomas cognitivos de la enfermedad de
Alzheimer.
Varios estudios indican que el estrés oxidativo puede ser parte de la patogénesis
de la AD. En el Estudio cooperativo de la enfermedad de Alzheimer, las dosis altas
de vitamina E (2000 unidades por día de alfa-tocoferol) durante 2 años redujeron
la progresión de la enfermedad en pacientes con EA moderada. [130] Este
beneficio probablemente se debió a los efectos antioxidantes de la vitamina E.
De manera similar, un ensayo en 613 pacientes con EA leve a moderada encontró
que una dosis diaria de 2000 UI de vitamina E ralentizaba el deterioro funcional.
En el estudio, los pacientes fueron asignados al azar al tratamiento con vitamina E
sola, una combinación de vitamina E y memantina, o placebo. Durante el
seguimiento medio de 2,27 años, los pacientes tratados con vitamina E sola
demostraron un retraso en la progresión clínica del 19% por año en comparación
con el placebo. [131, 132]
Además, los aumentos en el tiempo del cuidador, medidos en la Escala de
actividad del cuidador, fueron más bajos en el grupo de vitamina E en
comparación con los otros grupos. No se observó beneficio para la memantina o el
tratamiento combinado con memantina y vitamina E. [131, 132]
Sin embargo, otros estudios han sugerido que la suplementación con vitamina E
puede aumentar el riesgo de resultados cardiovasculares adversos. Por lo tanto, el
uso de vitamina E no se recomienda actualmente.

A pesar de la evidencia in vitro de un efecto protector del estrógeno, no hay datos
que muestren que las mujeres con EA que reciben tratamiento con estrógenos
(ET) tengan menos síntomas o progresen más lentamente que las mujeres que
recibieron placebo. Además, un ensayo clínico aleatorizado de estrógeno en
mujeres cognitivamente normales de 65 años de edad y mayores con un familiar
de primer grado con EA mostró que la ET podría aumentar el riesgo de accidente
cerebrovascular y demencia. [25] No se sabe si la ET podría disminuir el riesgo si
se inicia mucho antes de los 65 años.
Los niveles elevados de colesterol son un factor de riesgo para la EA, y los datos
epidemiológicos sugieren que el uso de estatinas puede reducir este riesgo. Sin
embargo, un ensayo de simvastatina en pacientes con EA de leve a moderada y
niveles normales de lípidos encontró que aunque el tratamiento con estatinas
disminuyó significativamente los niveles de colesterol, no disminuyó la progresión
de los síntomas. [133]
La estimulación magnética transcraneal (TMS) se ha utilizado para identificar
objetivos terapéuticos en la EA y para monitorear los efectos de los agentes
farmacológicos, y se está explorando la estimulación de la corriente directa tanto
de la EMT como de la transcraneal para un posible papel terapéutico en la EA. Sin
embargo, la evidencia del beneficio terapéutico de estas modalidades es
altamente preliminar. [134]
Terapia presintomática modificadora de la enfermedad.
Las terapias que modifican la enfermedad retrasarían la aparición de la EA y / o
disminuirían la velocidad de progresión.
Dado que los cambios cerebrales asociados con la AD probablemente comiencen
décadas antes de que la demencia se vuelva clínicamente evidente, muchos
investigadores creen que las terapias que modifican la enfermedad tienen muchas
más probabilidades de ser eficaces si se inician en una etapa presintomática.
Los métodos neuropsicológicos, de neuroimagen y genéticos están identificando a
los pacientes con mayor riesgo. Aunque los ensayos de fase III para varias
terapias potenciales que modifican la enfermedad se han completado, ninguno de
estos agentes ha demostrado tener una eficacia clara y, por lo tanto, aún no ha

sido aprobado por la FDA.
Actividad física
La actividad física y el ejercicio de rutina pueden tener un impacto en la progresión
de la EA y quizás tengan un efecto protector en la salud del cerebro. [6] El
aumento de los niveles de aptitud cardiorrespiratoria se asocia con mayores
volúmenes de hipocampo en pacientes con EA leve, lo que sugiere que la aptitud
cardiorrespiratoria puede modificar la atrofia cerebral relacionada con la AD. [7]
La actividad de cada paciente debe ser individualizada. El entorno del paciente
debe ser seguro y familiar. Demasiada actividad puede causar agitación, pero muy
poca puede hacer que el paciente se retire y quizás se deprima. Mantener rutinas
estructuradas puede ser útil para disminuir el estrés del paciente con respecto a
las comidas, los medicamentos y otras actividades terapéuticas dirigidas a
mantener el funcionamiento cognitivo.
El paciente necesita contacto con el entorno exterior. El médico debe alentar la
participación en actividades que interesen al paciente y resulten en estimulación
cognitiva, pero que no estresen al paciente. El rango de posibilidades es amplio y
puede incluir visitas a museos, parques o restaurantes.
Guias de la Asociación de Alzheimer
En 2018, la Asociación de Alzheimer publicó las primeras guías de práctica clínica
para la evaluación del deterioro cognitivo que se sospecha es el resultado de la
enfermedad de Alzheimer y las demencias relacionadas, tanto en la atención
primaria como en el entorno de atención especializada. Las pautas contienen 20
recomendaciones, 16 de las cuales están clasificadas como recomendaciones "A".
La recomendación principal es que todas las personas de mediana edad o
mayores que se autoinformen o para quienes su compañero de atención informa
sobre cambios cognitivos, de comportamiento o funcionales, se sometan a una
evaluación oportuna. Otras recomendaciones enfatizan la importancia de obtener
una historia del paciente y de alguien que conozca bien al paciente para
establecer la presencia y las características de cualquier cambio sustancial para
categorizar el síndrome de comportamiento cognitivo, investigar las posibles
causas y los factores que contribuyen para llegar a un diagnóstico / diagnóstico. y

educar adecuadamente, comunicar los hallazgos y el diagnóstico, y garantizar la
administración, atención y apoyo continuos. [145]
Resumen de la medicación
El pilar de la terapia para pacientes con enfermedad de Alzheimer (EA) es el uso
de inhibidores de la colinesterasa de acción central para intentar compensar el
agotamiento de la acetilcolina (ACh) en la corteza cerebral y el hipocampo. Un
antagonista parcial de N-metil-D-aspartato (NMDA) está aprobado para el
tratamiento de la EA moderada y grave. Se usan varios medicamentos para el
tratamiento de los síntomas secundarios de la EA, incluidos los antidepresivos, los
agentes contra la ansiedad y los agentes antipsicóticos.
Inhibidores de la colinesterasa
Los inhibidores de la colinesterasa (ChEI) se usan para paliar la deficiencia
colinérgica. Los 4 ChEI aprobados actualmente (es decir, donepezilo, rivastigmina,
galantamina) inhiben la acetilcolinesterasa (AChE) en la sinapsis (colinesterasa
específica).
La rivastigmina también inhibe la butirilcolinesterasa (BuChE). Aunque los niveles
de BuChE pueden incrementarse en la EA, no está claro que la rivastigmina tenga
mayor eficacia clínica que el donepezilo y la galantamina.
Galantamine tiene un segundo mecanismo de acción diferente; También es un
modulador nicotínico presináptico. No existen datos que indiquen que este
segundo mecanismo es de importancia clínica.
Donepecilo
Donepecilo está indicado para el tratamiento de la demencia de tipo Alzheimer.
Donepecilo ha demostrado eficacia en pacientes con EA leve a moderada, así
como moderada a grave. Inhibe selectivamente la acetilcolinesterasa, la enzima
responsable de la destrucción de la acetilcolina, y mejora la disponibilidad de la
acetilcolina. La larga vida media de Donepezil proporciona una disponibilidad de
medicamentos de larga duración para la unión en los sitios receptores. No hay
evidencia que sugiera que el proceso de la enfermedad subyacente de la
demencia se vea afectado por la administración de donepezil.
Las recomendaciones de dosificación para la EA leve a moderada son de 5 a 10

mg administrados una vez al día. Los pacientes con EA moderada a grave pueden
recibir 10 o 23 mg una vez al día.
Rivastigmina
La rivastigmina PO está indicada para el tratamiento de la demencia leve a
moderada del tipo Alzheimer. Las recomendaciones iniciales de dosificación son
1,5 mg PO BID, con una dosis máxima de 12 mg / día PO. La rivastigmina es un
potente inhibidor selectivo de la AChE y la BChE del cerebro. La rivastigmina se
considera un inhibidor pseudo irreversible de la AChE.
Si bien se desconoce el mecanismo preciso de la acción de rivastigmina, se
postula que ejerce su efecto terapéutico al mejorar la función colinérgica. Esto se
logra aumentando la concentración de acetilcolina a través de la inhibición
reversible de su hidrólisis por colinesterasa.
El parche transdérmico de 13,3 mg / 24 h está aprobado para todas las etapas de
la enfermedad de Alzheimer, incluidas las graves. La titulación de dosis es
necesaria al iniciar.
Galantamine
Galantamine está indicado para el tratamiento de la demencia leve a moderada
del tipo Alzheimer. Mejora la función colinérgica central y probablemente inhibe la
AChE. No hay evidencia de que galantamina altere el curso del proceso de
demencia subyacente. La recomendación de dosificación para la formulación de
liberación inmediata es 4 mg dos veces al día. La formulación de liberación
prolongada se administra en una dosis de 8 mg una vez al día. La dosis de
mantenimiento después de la titulación de la dosis es de 16-24 mg / día.
Antagonistas de N-metil-D-aspartato
El único medicamento en la clase de antagonistas de N -metil-D-aspartato (NMDA)
que está aprobado por la Administración de Drogas y Alimentos de EE. UU. Es la
memantina. Este agente se puede usar solo o en combinación con inhibidores de
AChE.
Memantina (Namenda, Namenda XR)
Namenda está aprobado para el tratamiento de la demencia moderada a grave en

pacientes con AD. La dosis inicial para la formulación de liberación inmediata es
de 5 mg una vez al día, y se puede ajustar a una dosis máxima de 20 mg / día. La
dosis inicial para la formulación de liberación prolongada es de 7 mg una vez al
día, y se puede ajustar a una dosis máxima de 28 mg / día
Drogas combinadas
Los productos combinados pueden ayudar en la facilidad de administración
(disminución de la carga de la píldora) y mejorar el cumplimiento.
Memantina / donepezil (Namzaric)
Cápsula de combinación de dosis fija que contiene memantina de liberación
prolongada y donepecilo para pacientes con enfermedad de Alzheimer de
moderada a grave actualmente estabilizados con donepecilo 10 mg una vez al día.
Administrar una vez al día por la noche. La memantina es un antagonista del
receptor de NMDA y el donepecilo es un inhibidor de la acetilcolinesterasa.
Eliana Roldán Gerschcovich
Valeria Casal Passion.
BIBLIOGRAFÍA CONSULTADA.
1. Guideline] Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J,
Relkin N, et al. Practice parameter: diagnosis of dementia (an evidence-based
review). Report of the Quality Standards Subcommittee of the American Academy of
Neurology. Neurology. 2001 May 8. 56(9):1143-53. [Medline]. [Full Text].
2. Mosconi L, Berti V, Glodzik L, Pupi A, De Santi S, de Leon MJ. Pre-clinical
detection of Alzheimer's disease using FDG-PET, with or without amyloid imaging. J
Alzheimers Dis. 2010. 20 (3):843-54. [Medline].
3. Winslow BT, Onysko MK, Stob CM, Hazlewood KA. Treatment of Alzheimer
disease. Am Fam Physician. 2011 Jun 15. 83(12):1403-12. [Medline].
4. Massoud F, Léger GC. Pharmacological treatment of Alzheimer disease. Can
J Psychiatry. 2011 Oct. 56(10):579-88. [Medline].
5. Madhusoodanan S, Shah P, Brenner R, Gupta S. Pharmacological treatment
of the psychosis of Alzheimer's disease: what is the best approach?. CNS Drugs.
2007. 21(2):101-15. [Medline].

6. Rolland Y, Abellan van Kan G, Vellas B. Healthy brain aging: role of exercise
and physical activity. Clin Geriatr Med. 2010 Feb. 26(1):75-87. [Medline].
7. Honea RA, Thomas GP, Harsha A, Anderson HS, Donnelly JE, Brooks WM, et
al. Cardiorespiratory fitness and preserved medial temporal lobe volume in
Alzheimer disease. Alzheimer Dis Assoc Disord. 2009 Jul-Sep. 23(3):188-97.
[Medline]. [Full Text].
8. Brookmeyer R, Abdalla N, Kawas CH, Corrada MM. Forecasting the
prevalence of preclinical and clinical Alzheimer's disease in the United States.
Alzheimers Dement. 2017 Nov 29. [Medline].
9. Taylor CA, Greenlund SF, McGuire LC, Lu H, Croft JB. Deaths from
Alzheimer's Disease - United States, 1999-2014. MMWR Morb Mortal Wkly Rep. 2017
May 26. 66 (20):521-526. [Medline].
10. Alzheimer's Association. 2017 Alzheimer's disease facts and figures.
Alzheimers Dement. 2017. 13:325-373.
11. Maurer K, Maurer U. Alzheimer: The Life of a Physician and Career of a
Disease. New York: Columbia University Press; 2003.
12. Alzheimer A. Uber eine eigenartige Erkangkung der Hirnrinde. Allgemeine
Zeitschrift fur Psychiatrie und Psychisch-Gerichtliche Medizin. 1907. 64: 146-148.
13. Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes.
Acta Neuropathol. 1991. 82(4):239-59. [Medline].
14. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological
alterations in Alzheimer disease. Cold Spring Harb Perspect Biol. 2011 Sep.
3(9):a006189. [Medline].
15. Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of
dementia in over-80-year-old brain donors from the population-based Cambridge
city over-75s cohort (CC75C) study. J Alzheimers Dis. 2009. 18(3):645-58. [Medline].
16. Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, et al. Spatial patterns of
neuroimaging biomarker change in individuals from families with autosomal
dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018 Mar. 17
(3):241-250. [Medline].
17. Swerdlow RH, Khan SM. The Alzheimer's disease mitochondrial cascade
hypothesis: an update. Exp Neurol. 2009 Aug. 218(2):308-15. [Medline]. [Full Text].

18. Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic
process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol
Exp Neurol. 2011 Nov. 70(11):960-9. [Medline].
19. Braak H, Braak E, Grundke-Iqbal I, Iqbal K. Occurrence of neuropil threads in
the senile human brain and in Alzheimer's disease: a third location of paired helical
filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci Lett. 1986
Apr 24. 65(3):351-5. [Medline].
20. Davinelli S, Intrieri M, Russo C, Di Costanzo A, Zella D, Bosco P, et al. The
"Alzheimer's disease signature": potential perspectives for novel biomarkers.
Immun Ageing. 2011 Sep 20. 8:7. [Medline]. [Full Text].
21. Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, Nagley P. Oxidative
stress: emerging mitochondrial and cellular themes and variations in neuronal
injury. J Alzheimers Dis. 2010. 20 Suppl 2:S453-73. [Medline].
22. Ding Q, Dimayuga E, Keller JN. Oxidative damage, protein synthesis, and
protein degradation in Alzheimer's disease. Curr Alzheimer Res. 2007 Feb. 4(1):73-9.
[Medline].
23. Thambisetty M, Simmons A, Velayudhan L, Hye A, Campbell J, Zhang Y, et al.
Association of plasma clusterin concentration with severity, pathology, and
progression in Alzheimer disease. Arch Gen Psychiatry. 2010 Jul. 67(7):739-48.
[Medline].
24. Schrijvers EM, Koudstaal PJ, Hofman A, Breteler MM. Plasma clusterin and
the risk of Alzheimer disease. JAMA. 2011 Apr 6. 305(13):1322-6. [Medline].
25. Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, et al.
Estrogen plus progestin and the incidence of dementia and mild cognitive
impairment in postmenopausal women: the Women's Health Initiative Memory
Study: a randomized controlled trial. JAMA. 2003 May 28. 289(20):2651-62. [Medline].
26. Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, et al. Meta-analysis of
modifiable risk factors for Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2015
Dec. 86 (12):1299-306. [Medline].
27. Rocchi A, Orsucci D, Tognoni G, Ceravolo R, Siciliano G. The role of vascular
factors in late-onset sporadic Alzheimer's disease. Genetic and molecular aspects.
Curr Alzheimer Res. 2009 Jun. 6(3):224-37. [Medline].

28. S Roriz-Filho J, Sá-Roriz TM, Rosset I, Camozzato AL, Santos AC, Chaves ML,
et al. (Pre)diabetes, brain aging, and cognition. Biochim Biophys Acta. 2009 May.
1792(5):432-43. [Medline].
29. Naderali EK, Ratcliffe SH, Dale MC. Obesity and Alzheimer's disease: a link
between body weight and cognitive function in old age. Am J Alzheimers Dis Other
Demen. 2009 Dec-2010 Jan. 24(6):445-9. [Medline].
30. de la Monte SM. Insulin resistance and Alzheimer's disease. BMB Rep. 2009
Aug 31. 42(8):475-81. [Medline].
31. Hughes S. Beta-Blockers Linked to Fewer Alzheimer's Lesions. Available at
http://www.medscape.com/viewarticle/777239. Accessed: January 15, 2013.
32. Perl DP. Relationship of aluminum to Alzheimer's disease. Environ Health
Perspect. 1985 Nov. 63:149-53. [Medline]. [Full Text].
33. Perl DP, Moalem S. Aluminum and Alzheimer's disease, a personal
perspective after 25 years. J Alzheimers Dis. 2006. 9(3 Suppl):291-300. [Medline].
34. Anderson, P. Early-Life Depression Boosts Alzheimer's Risk. Medscape
Medical News. Available at http://www.medscape.com/viewarticle/883327. July 24,
2017; Accessed: July 26, 2017.
35. Goldbourt U, Schnaider-Beeri M, Davidson M. Socioeconomic status in
relationship to death of vascular disease and late-life dementia. J Neurol Sci. 2007
Jun 15. 257(1-2):177-81. [Medline].
36. McDowell I, Xi G, Lindsay J, Tierney M. Mapping the connections between
education and dementia. J Clin Exp Neuropsychol. 2007 Feb. 29(2):127-41.
[Medline].
37. Szekely CA, Zandi PP. Non-steroidal anti-inflammatory drugs and Alzheimer's
disease: the epidemiological evidence. CNS Neurol Disord Drug Targets. 2010 Apr.
9(2):132-9. [Medline].
38. Goldman JS, Hahn SE, Catania JW, LaRusse-Eckert S, Butson MB,
Rumbaugh M, et al. Genetic counseling and testing for Alzheimer disease: joint
practice guidelines of the American College of Medical Genetics and the National
Society of Genetic Counselors. Genet Med. 2011 Jun. 13(6):597-605. [Medline]. [Full
Text].
39. Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7,
MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's
disease. Nat Genet. 2011 May. 43(5):429-35. [Medline]. [Full Text].

40. Caselli RJ, Dueck AC. APOE varepsilon2 and presymptomatic stage
Alzheimer disease: how much is not enough?. Neurology. 2010 Nov 30. 75(22):1952-
3. [Medline].
41. Chiang GC, Insel PS, Tosun D, et al. Hippocampal atrophy rates and CSF
biomarkers in elderly APOE2 normal subjects. Neurology. 2010 Nov 30. 75(22):1976-
81. [Medline].
42. Finch CE, Morgan TE. Systemic inflammation, infection, ApoE alleles, and
Alzheimer disease: a position paper. Curr Alzheimer Res. 2007 Apr. 4(2):185-9.
[Medline].
43. Anderson P. Hypertension Interacts With APOE Epsilon 4 to Increase
Amyloid Load. Available at http://www.medscape.com/viewarticle/781430.
Accessed: April 8, 2013.
44. Rodrigue KM, Rieck JR, Kennedy KM, Devous MD, Diaz-Arrastia R, Park DC.
Risk Factors for ß-Amyloid Deposition in Healthy Aging: Vascular and Genetic
Effects. JAMA Neurol. 2013 Mar 18. 1-7. [Medline].
45. Cacciottolo M, Wang X, Driscoll I, Woodward N, Saffari A, Reyes J, et al.
Particulate air pollutants, APOE alleles and their contributions to cognitive
impairment in older women and to amyloidogenesis in experimental models. Transl
Psychiatry. 2017 Jan 31. 7 (1):e1022. [Medline].
46. Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin
resistance and Alzheimer-like reductions in regional cerebral glucose metabolism
for cognitively normal adults with prediabetes or early type 2 diabetes. Arch Neurol.
2011 Jan. 68(1):51-7. [Medline].
47. Schrijvers JCM, Witteman EJG, Sijbrands, et al. Insulin metabolism and the
risk of Alzheimer disease: The Rotterdam Study. Neurology. 2010;75:1982-1987.
48. Miklossy J. Emerging roles of pathogens in Alzheimer disease. Expert Rev
Mol Med. 2011 Sep 20. 13:e30. [Medline].
49. Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA, Au R. Depressive
symptoms and risk of dementia: the Framingham Heart Study. Neurology. 2010 Jul
6. 75(1):35-41. [Medline]. [Full Text].
50. Dotson VM, Beydoun MA, Zonderman AB. Recurrent depressive symptoms
and the incidence of dementia and mild cognitive impairment. Neurology. Jul 6
2010. 75(1):27-34.

51. Lowry F. Late-life depression linked to increased risk for dementia. Medscape
Medical News. May 7, 2013. [Full Text].
52. Diniz BS, Butters MA, Albert SM, Dew MA, Reynolds CF 3rd. Late-life
depression and risk of vascular dementia and Alzheimer's disease: systematic
review and meta-analysis of community-based cohort studies. Br J Psychiatry. 2013
May. 202:329-35. [Medline]. [Full Text].
53. Magnoni S, Brody DL. New perspectives on amyloid-beta dynamics after
acute brain injury: moving between experimental approaches and studies in the
human brain. Arch Neurol. 2010 Sep. 67(9):1068-73. [Medline]. [Full Text].
54. Chen XH, Siman R, Iwata A, Meaney DF, Trojanowski JQ, Smith DH. Long-
term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in
damaged axons following brain trauma. Am J Pathol. 2004 Aug. 165(2):357-71.
[Medline]. [Full Text].
55. Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, et
al. Documented head injury in early adulthood and risk of Alzheimer's disease and
other dementias. Neurology. 2000 Oct 24. 55(8):1158-66. [Medline].
56. Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, et al. The fetal
basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid
precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005 Jan 26.
25(4):823-9. [Medline].
57. Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced
damage to DNA: mechanisms and measurement. Free Radic Biol Med. 2002 Jun 1.
32(11):1102-15. [Medline].
58. Harman, D. (1956). Aging: a theory based on free radical and radiation
chemistry. J Gerontol. 11, 298-300.
59. Genin E, Hannequin D, Wallon D, et al. APOE and Alzheimer disease: a major
gene with semi-dominant inheritance. Mol Psychiatry. 2011 Sep. 16(9):903-7.
[Medline]. [Full Text].
60. Kochanek KD, Murphy SL, Xu J, Tejada-Vera B. Deaths: Final data for 2014.
National Vital Statistics Reports. June 30, 2016. Available at
https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_04.pdf.
61. Heron M. Deaths: Leading Causes for 2014. National Vital Statistics Reports.
June 30, 2016. Available at
https://www.cdc.gov/nchs/data/nvsr/nvsr65/nvsr65_05.pdf.

62. Ives DG, Samuel P, Psaty BM, Kuller LH. Agreement between nosologist and
Cardiovascular Health Study review of deaths: Implications of coding differences.
Journal of the American Geriatrics Society. 2009. 57:133-139.
63. Mathers C., Leonardi M. Global burden of dementia in the year 2000:summary
of methods and data sources. [Full Text].
64. Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age,
neuropathology, and dementia. N Engl J Med. 2009 May 28. 360(22):2302-9.
[Medline]. [Full Text].
65. Plassman BL, Langa KM, Fisher GG, et al. Prevalence of dementia in the
United States: the aging, demographics, and memory study. Neuroepidemiology.
2007. 29(1-2):125-32.
66. Payami H, Zareparsi S, Montee KR, et al. Gender difference in apolipoprotein
E-associated risk for familial Alzheimer disease: a possible clue to the higher
incidence of Alzheimer disease in women. Am J Hum Genet. 1996 Apr. 58(4):803-11.
[Medline]. [Full Text].
67. Morris J, Schindler S, McCue L, et al. Assessment of Racial Disparities in
Biomarkers for Alzheimer Disease. Jama Neurol. 2019 Jan 7.
68. Jack CR Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC,
et al. Introduction to the recommendations from the National Institute on Aging-
Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's
disease. Alzheimers Dement. 2011 May. 7(3):257-62. [Medline]. [Full Text].
69. Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al.
Toward defining the preclinical stages of Alzheimer's disease: Recommendations
from the National Institute on Aging-Alzheimer's Association workgroups on
diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May.
7(3):280-92. [Medline].
70. Albert MS, Dekosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The
diagnosis of mild cognitive impairment due to Alzheimer's disease:
Recommendations from the National Institute on Aging-Alzheimer's Association
workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement.
2011 May. 7(3):270-9. [Medline].
71. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH,
et al. The diagnosis of dementia due to Alzheimer's disease: Recommendations
from the National Institute on Aging-Alzheimer's Association workgroups on

diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011 May.
7(3):263-9. [Medline].
72. American Psychiatric Association. Neurocognitive Disorders. Diagnostic and
Statistical Manual of Mental Disorders, Fifth Edition. Washington, DC: American
Psychiatric Association; 2013. 611-614.
73. Teng E, Ringman JM, Ross LK, et al. Diagnosing depression in Alzheimer
disease with the national institute of mental health provisional criteria. Am J Geriatr
Psychiatry. 2008 Jun. 16(6):469-77. [Medline]. [Full Text].
74. Lakhan SE, Kirchgessner A. Chronic traumatic encephalopathy: the dangers
of getting "dinged". Springerplus. 2012. 1:2. [Medline].
75. Omalu BI, DeKosky ST, Minster RL, Kamboh MI, Hamilton RL, Wecht CH.
Chronic traumatic encephalopathy in a National Football League player.
Neurosurgery. 2005 Jul. 57(1):128-34; discussion 128-34. [Medline].
76. Annweiler C, Schott AM, Allali G, Bridenbaugh SA, Kressig RW, Allain P, et al.
Association of vitamin D deficiency with cognitive impairment in older women:
cross-sectional study. Neurology. 2010 Jan 5. 74(1):27-32. [Medline].
77. Buell JS, Dawson-Hughes B, Scott TM, Weiner DE, Dallal GE, Qui WQ, et al.
25-Hydroxyvitamin D, dementia, and cerebrovascular pathology in elders receiving
home services. Neurology. 2010 Jan 5. 74(1):18-26. [Medline]. [Full Text].
78. Chen G, Ward BD, Xie C, Li W, Wu Z, Jones JL, et al. Classification of
Alzheimer Disease, Mild Cognitive Impairment, and Normal Cognitive Status with
Large-Scale Network Analysis Based on Resting-State Functional MR Imaging.
Radiology. 2011 Apr. 259(1):213-21. [Medline]. [Full Text].
79. Petrella JR, Sheldon FC, Prince SE, Calhoun VD, Doraiswamy PM. Default
mode network connectivity in stable vs progressive mild cognitive impairment.
Neurology. 2011 Feb 8. 76(6):511-7. [Medline]. [Full Text].
80. Brooks M. MRI may offer noninvasive option for Alzheimer’s Diagnosis.
Medscape Medical News. Dec 27 2012. Available at
http://www.medscape.com/viewarticle/776766. Accessed: Jan 8 2012.
81. McMillan CT, Avants B, Irwin DJ, Toledo JB, et al. Can MRI screen for CSF
biomarkers in neurodegenerative disease?. Neurology. 2012 Dec 26. [Medline].
82. Brooks M. Functional Brain Imaging May Spot Alzheimer's Early. Medscape
Medical News. Aug 21 2013. [Full Text].

83. Wang L, Brier MR, Snyder AZ, et al. Cerebrospinal fluid Aß42, phosphorylated
tau181, and resting-state functional connectivity. JAMA Neurol. 2013 Aug 19.
[Medline].
84. Rabinovici GD, Furst AJ, O'Neil JP, Racine CA, Mormino EC, Baker SL, et al.
11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration.
Neurology. 2007 Apr 10. 68(15):1205-12. [Medline].
85. Anderson, P. New Guidelines for Amyloid PET Imaging. Available at
http://www.medscape.com/viewarticle/778274. Accessed: February 5, 2013.
86. Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al.
Correlation of amyloid PET ligand florbetapir F 18 binding with Aß aggregation and
neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord.
2012 Jan. 26(1):8-16. [Medline]. [Full Text].
87. Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al.
Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011 Jan 19.
305(3):275-83. [Medline].
88. Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, et al.
Using positron emission tomography and florbetapir F18 to image cortical amyloid
in patients with mild cognitive impairment or dementia due to Alzheimer disease.
Arch Neurol. 2011 Nov. 68(11):1404-11. [Medline].
89. Choi SR, Schneider JA, Bennett DA, Beach TG, Bedell BJ, Zehntner SP, et al.
Correlation of amyloid PET ligand florbetapir F 18 binding with Aß aggregation and
neuritic plaque deposition in postmortem brain tissue. Alzheimer Dis Assoc Disord.
2012 Jan. 26(1):8-16. [Medline]. [Full Text].
90. Clark CM, Schneider JA, Bedell BJ, Beach TG, Bilker WB, Mintun MA, et al.
Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA. 2011 Jan 19.
305(3):275-83. [Medline].
91. Fleisher AS, Chen K, Liu X, Roontiva A, Thiyyagura P, Ayutyanont N, et al.
Using positron emission tomography and florbetapir F18 to image cortical amyloid
in patients with mild cognitive impairment or dementia due to Alzheimer disease.
Arch Neurol. 2011 Nov. 68(11):1404-11. [Medline].
92. US Food and Drug Administration (FDA). FDA approves second brain
imaging drug to help evaluate patients for Alzheimer’s disease, dementia [press
release]. October 25, 2013. Available at

http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm372261.htm.
Accessed: October 25, 2013.
93. Neuraceq (florbetaben F 18) prescribing information. [package insert]. Matran
Switzerland: Piramal Imaging, S.A. 2014.
94. Brooks M. Memory tests plus brain scans detect earliest stages of AD.
Medscape Medical News. July 15, 2013. [Full Text].
95. Rasmussen KL, Tybjaerg-Hansen A, Nordestgaard BG, Frikke-Schmidt R.
Apolipoprotein E plasma level and genotype -- risk of dementia in 76,000 individuals
from the general population. Presented at: XXI World Congress of Neurology (WNC),
2013; September 24, 2013; Vienna, Austria. [Full Text].
96. Keller DM. APOE plasma levels predict dementia independent of genotype.
Medscape Medical News. October 4, 2013. [Full Text].
97. Green RC, Roberts JS, Cupples LA, et al. Disclosure of APOE genotype for
risk of Alzheimer's disease. N Engl J Med. 2009 Jul 16. 361(3):245-54. [Medline]. [Full
Text].
98. Salomone S, Caraci F, Leggio GM, Fedotova J, Drago F. New pharmacological
strategies for treatment of Alzheimer's disease: focus on disease-modifying drugs.
Br J Clin Pharmacol. 2011 Oct 28. [Medline].
99. Anderson P. Finally, a Winner for Alzheimer's? Antiamyloid Agent Shows
Promise. Medscape Medical News. Available at
https://www.medscape.com/viewarticle/899841. July 26, 2018; Accessed: July 26,
2018.
100. Kavanagh S, Gaudig M, Van Baelen B, et al. Galantamine and behavior in
Alzheimer disease: analysis of four trials. Acta Neurol Scand. 2011 Nov. 124(5):302-
8. [Medline].
101. Farlow M, Veloso F, Moline M, et al. Safety and tolerability of donepezil 23 mg
in moderate to severe Alzheimer's disease. BMC Neurol. 2011 May 25. 11:57.
[Medline]. [Full Text].
102. Starr JM. Cholinesterase inhibitor treatment and urinary incontinence in
Alzheimer's disease. J Am Geriatr Soc. 2007 May. 55(5):800-1. [Medline].
103. Gill SS, Anderson GM, Fischer HD, Bell CM, Li P, Normand SL, et al. Syncope
and its consequences in patients with dementia receiving cholinesterase inhibitors:
a population-based cohort study. Arch Intern Med. 2009 May 11. 169(9):867-73.
[Medline].

104. Lachaine J, Beauchemin C, Legault M, Bineau S. Economic evaluation of the
impact of memantine on time to nursing home admission in the treatment of
Alzheimer disease. Can J Psychiatry. 2011 Oct. 56(10):596-604. [Medline].
105. Schmitt FA, van Dyck CH, Wichems CH, Olin JT. Cognitive response to
memantine in moderate to severe Alzheimer disease patients already receiving
donepezil: an exploratory reanalysis. Alzheimer Dis Assoc Disord. 2006 Oct-Dec.
20(4):255-62. [Medline].
106. Porsteinsson AP, Grossberg GT, Mintzer J, Olin JT. Memantine treatment in
patients with mild to moderate Alzheimer's disease already receiving a
cholinesterase inhibitor: a randomized, double-blind, placebo-controlled trial. Curr
Alzheimer Res. 2008 Feb. 5(1):83-9. [Medline].
107. Schneider LS, Dagerman KS, Higgins JP, McShane R. Lack of evidence for
the efficacy of memantine in mild Alzheimer disease. Arch Neurol. 2011 Aug.
68(8):991-8. [Medline].
108. Greig SL. Memantine ER/Donepezil: A Review in Alzheimer's Disease. CNS
Drugs. 2015 Nov. 29 (11):963-70. [Medline].
109. Jeffrey S. FDA Approves Exelon Patch for Severe Alzheimer's. Medscape
[serial online]. Available at http://www.medscape.com/viewarticle/807062. Accessed:
July 8, 2013.
110. Farlow MR, Grossberg G, Gauthier S, Meng X, Olin JT. The ACTION study:
methodology of a trial to evaluate safety and efficacy of a higher dose rivastigmine
transdermal patch in severe Alzheimer's disease. Curr Med Res Opin. 2010 Oct.
26(10):2441-7. [Medline].
111. de Rotrou J, Cantegreil I, Faucounau V, et al. Do patients diagnosed with
Alzheimer's disease benefit from a psycho-educational programme for family
caregivers? A randomised controlled study. Int J Geriatr Psychiatry. 2011 Aug.
26(8):833-42. [Medline].
112. Deaths with Antipsychotics in Elderly Patients with Behavioral Disturbances.
US Food and Drug Administration Website. Available at
http://www.fda.gov/Drugs/DrugSafety/PublicHealthAdvisories/ucm053171.htm.
Accessed: August 11, 2009.
113. Devanand DP, Pelton GH, Cunqueiro K, Sackeim HA, Marder K. A 6-month,
randomized, double-blind, placebo-controlled pilot discontinuation trial following

response to haloperidol treatment of psychosis and agitation in Alzheimer's
disease. Int J Geriatr Psychiatry. 2011 Sep. 26(9):937-43. [Medline].
114. Vigen CL, Mack WJ, Keefe RS, et al. Cognitive effects of atypical
antipsychotic medications in patients with Alzheimer's disease: outcomes from
CATIE-AD. Am J Psychiatry. 2011 Aug. 168(8):831-9. [Medline].
115. Nyth AL, Gottfries CG. The clinical efficacy of citalopram in treatment of
emotional disturbances in dementia disorders. A Nordic multicentre study. Br J
Psychiatry. 1990 Dec. 157:894-901. [Medline].
116. US Food and Drug Administration. August 24, 2011. FDA Drug Safety
Communication: Abnormal heart rhythms associated with high doses of Celexa
(citalopram hydrobromide). Available at
http://www.fda.gov/Drugs/DrugSafety/ucm269086.htm. Accessed: December 27,
2011.
117. Porsteinsson AP, Drye LT, Pollock BG, Devanand DP, Frangakis C, Ismail Z,
et al. Effect of citalopram on agitation in Alzheimer disease: the CitAD randomized
clinical trial. JAMA. 2014 Feb 19. 311(7):682-91. [Medline].
118. Small GW. Treating dementia and agitation. JAMA. 2014 Feb 19. 311(7):677-8.
[Medline].
119. Anderson P. Citalopram Reduces Agitation in Patients With Alzheimer's.
Medscape Medical News. Available at http://www.medscape.com/viewarticle/820741.
Accessed: February 24, 2014.
120. Weintraub D, Rosenberg PB, Drye LT, et al. Sertraline for the treatment of
depression in Alzheimer disease: week-24 outcomes. Am J Geriatr Psychiatry. 2010
Apr. 18(4):332-40. [Medline]. [Full Text].
121. Petracca GM, Chemerinski E, Starkstein SE. A double-blind, placebo-
controlled study of fluoxetine in depressed patients with Alzheimer's disease. Int
Psychogeriatr. 2001 Jun. 13(2):233-40. [Medline].
122. Banerjee S, Hellier J, Dewey M, et al. Sertraline or mirtazapine for depression
in dementia (HTA-SADD): a randomised, multicentre, double-blind, placebo-
controlled trial. Lancet. 2011 Jul 30. 378(9789):403-11. [Medline].
123. Boggs W. Trazodone Treats Sleep Disturbance in Alzheimer's Disease.
Medscape Medical News. Available at http://www.medscape.com/viewarticle/819383.
Accessed: January 26, 2014.

124. Camargos E, Louzada L, Quintas J, Naves J, Louzada F, Nóbrega O.
Trazodone improves sleep parameters in Alzheimer's disease patients: a
randomized, double-blind and placebo-controlled study. Am J Geriatr Psychiatry.
2014 Jan 4. [Epub ahead of print].
125. Tariot PN, Schneider LS, Cummings J, et al. Chronic divalproex sodium to
attenuate agitation and clinical progression of Alzheimer disease. Arch Gen
Psychiatry. 2011 Aug. 68(8):853-61. [Medline].
126. Wang YY, Zheng W, Ng CH, Ungvari GS, Wei W, Xiang YT. Meta-analysis of
randomized, double-blind, placebo-controlled trials of melatonin in Alzheimer's
disease. Int J Geriatr Psychiatry. 2016 Sep 19. [Medline].
127. Anderson P. 'Z' Drugs Significantly Boost Fracture Risk in Dementia Patients.
Medscape Medical News. Available at
https://www.medscape.com/viewarticle/899792. July 25, 2018; Accessed: July 30,
2018.
128. Breitner JC, Haneuse S, Walker R, Dublin S, Crane PK, Gray SL, et al. Risk of
dementia and AD with prior exposure to NSAIDs in an elderly community-based
cohort. Neurology. Jun 2 2009. 72(22):1899-1905.
129. Pritchard SM, Dolan PJ, Vitkus A, Johnson GV. The toxicity of tau in
Alzheimer disease: turnover, targets and potential therapeutics. J Cell Mol Med.
2011 Aug. 15(8):1621-35. [Medline].
130. Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, alpha-
tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease
Cooperative Study. N Engl J Med. 1997 Apr 24. 336(17):1216-22. [Medline].
131. Brooks M. Vitamin E May Slow Functional Decline in Mild Alzheimer's.
Medscape Medical News. Available at http://www.medscape.com/viewarticle/818533.
Accessed: January 8, 2014.
132. Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, et al.
Effect of vitamin E and memantine on functional decline in Alzheimer disease: the
TEAM-AD VA cooperative randomized trial. JAMA. 2014 Jan 1. 311(1):33-44.
[Medline].
133. Sano M, Bell KL, Galasko D, Galvin JE, Thomas RG, van Dyck CH, et al. A
randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer
disease. Neurology. 2011 Aug 9. 77(6):556-63. [Medline]. [Full Text].

134. Freitas C, Mondragón-Llorca H, Pascual-Leone A. Noninvasive brain
stimulation in Alzheimer's disease: systematic review and perspectives for the
future. Exp Gerontol. 2011 Aug. 46(8):611-27. [Medline].
135. Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of
the ketogenic agent AC-1202 in mild to moderate Alzheimer's disease: a
randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond).
2009 Aug 10. 6:31. [Medline]. [Full Text].
136. Harrison P. Novel Intervention May Reverse Alzheimer's Memory Loss.
Medscape Medical News. Available at http://www.medscape.com/viewarticle/832752.
Accessed: 10/8/14.
137. Bredesen D. Reversal of cognitive decline: A novel therapeutic program.
Aging. September 2014. [Full Text].
138. Hörder H, Johansson L, Guo X, Grimby G, Kern S, Östling S, et al. Midlife
cardiovascular fitness and dementia: A 44-year longitudinal population study in
women. Neurology. 2018 Mar 14. [Medline].
139. Barberger-Gateau P, Raffaitin C, Letenneur L, Berr C, Tzourio C, Dartigues JF,
et al. Dietary patterns and risk of dementia: the Three-City cohort study. Neurology.
2007 Nov 13. 69(20):1921-30. [Medline].
140. Solfrizzi V, Panza F, Frisardi V, Seripa D, Logroscino G, Imbimbo BP, et al.
Diet and Alzheimer's disease risk factors or prevention: the current evidence. Expert
Rev Neurother. 2011 May. 11(5):677-708. [Medline].
141. Witte AV, Fobker M, Gellner R, Knecht S, Flöel A. Caloric restriction improves
memory in elderly humans. Proc Natl Acad Sci U S A. 2009 Jan 27. 106(4):1255-60.
[Medline]. [Full Text].
142. Anstey KJ, Mack HA, Cherbuin N. Alcohol consumption as a risk factor for
dementia and cognitive decline: meta-analysis of prospective studies. Am J Geriatr
Psychiatry. 2009 Jul. 17(7):542-55. [Medline].
143. Virtaa JJ, Järvenpää T, Heikkilä K, Perola M, Koskenvuo M, Räihä I, et al.
Midlife alcohol consumption and later risk of cognitive impairment: a twin follow-up
study. J Alzheimers Dis. 2010. 22(3):939-48. [Medline].
144. Schwarzinger M, Pollock BG, Hasan OSM, Dufouil C, Rehm J, QalyDays Study
Group. Contribution of alcohol use disorders to the burden of dementia in France
2008-13: a nationwide retrospective cohort study. Lancet Public Health. 2018 Mar. 3
(3):e124-e132. [Medline].

145. Brooks M. First Alzheimer's Guidelines for Clinical Practice Released.
Medscape Medical News. Available at
https://www.medscape.com/viewarticle/899674. July 23, 2018; Accessed: July 30,
2018.
146. Brooks M. Brain Insulin Resistance Marker May Diagnose Alzheimer's.
Medscape Medical News. Available at
http://www.medscape.com/viewarticle/835489.. Accessed: November 27, 2014.
147. Billioti de Gage S, Moride Y, Ducruet T, Kurth T, Verdoux H, Tournier M, et al.
Benzodiazepine use and risk of Alzheimer's disease: case-control study. BMJ. 2014
Sep 9. 349:g5205. [Medline]. [Full Text].
148. Brauser D. Benzodiazepines Linked to Increased Alzheimer's Risk. Medscape
Medical News. Available at http://www.medscape.com/viewarticle/831403. Accessed:
September 14, 2014.
149. Cassels C. Simple Eye Tests to Detect Alzheimer's Disease in the Works.
Medscape Medical News. Jul 16 2014. [Full Text].
150. Davenport L. Herpes simplex may double Alzheimer's risk. Medscape Medical
New. October 27, 2014. [Full Text].
151. Dysken MW, Sano M, Asthana S, Vertrees JE, Pallaki M, Llorente M, et al.
Effect of vitamin E and memantine on functional decline in Alzheimer disease: the
TEAM-AD VA cooperative randomized trial. JAMA. 2014 Jan 1. 311(1):33-44.
[Medline].
152. Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease
in the US population: prevalence estimates using the 2000 census. Arch Neurol.
2003 Aug. 60(8):1119-22. [Medline].
153. Jeffrey S. FDA Approves Third Amyloid PET Tracer for Alzheimer's.
Medscape Medical News. Available at http://www.medscape.com/viewarticle/822370.
Accessed: March 31, 2014.
154. Lovheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes
simplex infection increases the risk of Alzheimer's disease. Alzheimers Dement.
2014 Jul 17. [Medline].
155. Lovheim H, Gilthorpe J, Johansson A, Eriksson S, Hallmans G, Elgh F.
Herpes simplex infection and the risk of Alzheimer's disease-A nested case-control
study. Alzheimers Dement. 2014 Oct 7. [Medline].

156. Yaffe K, Boustani M. Benzodiazepines and risk of Alzheimer's disease. BMJ.
2014 Sep 9. 349:g5312. [Medline].
IMPORTANTE: El contenido de este Seminario está protegido por Derechos de
Autor ®. Podrá ser utilizado para estudio en el curso de este Seminario. Está
prohibida su reproducción total o parcial por cualquier medio de impresión o digital,
en forma idéntica, extractada o modificada, en castellano o en cualquier otro
idioma, sin autorización expresa del autor.
Copyright 2019 Red de Salud enLazos – Todos los derechos reservados





























