Embed Size (px)
Citation preview

Division of Pharmacology and Toxicology
Faculty of Pharmacy
University of Helsinki
DIFFERENT RESPONSES OF THE NIGROSTRIATAL AND MESOLIMBIC
DOPAMINERGIC PATHWAYS TO NICOTINIC RECEPTOR AGONISTS
Sanna Janhunen
ACADEMIC DISSERTATION
To be presented, with the permission of the Faculty of Pharmacy of the University of
Helsinki, for public criticism in Auditorium 1041 of Viikki Biocentre, on June 17th,
2005, at 12 o’clock noon.
Helsinki 2005

Supervisors: Professor Liisa Ahtee, M.D. Division of Pharmacology and Toxicology Faculty of Pharmacy University of Helsinki Professor Raimo Tuominen, M.D. Division of Pharmacology and Toxicology Faculty of Pharmacy University of Helsinki Reviewers: Professor Agneta Nordberg, M.D., Ph.D. Division of Molecular Neuropharmacology
Department of Clinical Neuroscience Karolinska Institute, Stockholm
Sweden Docent Pekka Rauhala, M.D. Institute of Biomedicine Faculty of Medicine University of Helsinki Opponent: Professor Ben Westerink, Ph.D.
Department of Biomonitoring and Sensoring University Center for Pharmacy University of Groningen
The Netherlands © Sanna Janhunen 2005 ISBN 952-10-2475-5 (paperback) ISBN 952-10-2476-3 (PDF, http://ethesis.helsinki.fi/) ISSN 1795-7079 Yliopistopaino, University Press Helsinki, Finland 2005

To my parents, Sinikka and Tapio

4
CONTENTS
ABSTRACT ..................................................................................................................................... 6
LIST OF ABBREVIATIONS ........................................................................................................ 7
LIST OF ORIGINAL PUBLICATIONS ..................................................................................... 8
1. INTRODUCTION .................................................................................................................. 9
2. REVIEW OF THE LITERATURE ...................................................................................... 11
2.1. Ascending dopaminergic pathways ......................................................................... 11 2.1.1. Functional implications of nigrostriatal and mesolimbic pathways ............. 13
2.2. Synthesis and release of dopamine .......................................................................... 14
2.3. Metabolism of dopamine............................................................................................ 16
2.4. Dopamine and the neural circuits of basal ganglia ............................................... 17
2.5. Characteristics of Parkinson’s disease ..................................................................... 20
2.6. Neuronal nicotinic acetylcholine receptors (nAChRs) ......................................... 22 2.6.1. Structure and classification of nAChRs ............................................................. 23 2.6.2. Distribution of nAChRs in the brain .................................................................. 25 2.6.3. Activation and desensitization of nAChRs ....................................................... 27 2.6.4. Up-regulation of nAChRs .................................................................................... 28 2.6.5. Influence of subunits on functional properties of nAChRs ............................ 30
2.7. Nicotinic modulation of dopamine release............................................................. 32 2.7.1. Different effects of nicotine on dopamine in the CPu and NAc..................... 34 2.7.2. nAChR-mediated modulation of dopaminergic transmission ....................... 34 2.7.3. Subtypes of presynaptic nAChRs involved in dopamine release .................. 36 2.7.4. Modulation of nicotine-induced dopamine release by other neuro-transmitters, glutamate and GABA .................................................................................... 38
2.8. Nicotine – a substance of abuse................................................................................. 40 2.8.1. Rewarding and reinforcing effects of nicotine in animal models .................. 42 2.8.2. The effects of nicotine on locomotor activity in rats ........................................ 43
2.9. Epibatidine .................................................................................................................... 45
2.10. Nicotinic compounds in Parkinson’s disease ..................................................... 48
2.11. Nicotinic compounds in other CNS disorders.................................................... 51
3. AIMS OF THE STUDY........................................................................................................ 54
4. MATERIALS AND METHODS ........................................................................................ 55
4.1. Animals .......................................................................................................................... 55
4.2. Drugs............................................................................................................................... 55
4.3. Analysis of epibatidine stock solutions................................................................... 56
4.4. In vivo microdialysis (I, II, III) .................................................................................. 57
4.5. Rotational behaviour (I, IV) ....................................................................................... 58

5
4.6. Locomotor activity monitoring (V) ........................................................................... 60
4.7. Place conditioning (V) ................................................................................................. 60
4.8. Statistical analysis ........................................................................................................ 61
5. RESULTS ............................................................................................................................... 62
5.1. Microdialysis studies................................................................................................... 62 5.1.1. Effects of nicotine on the extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the CPu in combination with various dopaminergic drugs (I)......................................................................................................... 62 5.1.2. Effects of nicotine and epibatidine on the extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the CPu and NAc (II) .................................. 64 5.1.3. Effects of nicotine and epibatidine on the extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the CPu and NAc of nomifensine-pretreated rats (III, previously unpublished data) ........................................................... 67
5.2. Behavioural studies...................................................................................................... 69 5.2.1. Effects of acute nicotine on the rotational behaviour of unilaterally 6-OHDA lesioned rats (I, previously unpublished data).................................................... 69 5.2.2. Effects of repeated nicotine and epibatidine on the rotational behaviour of unilaterally 6-OHDA-lesioned rats (IV) ........................................................................ 71 5.2.3. Effects of nicotine and epibatidine on locomotor activity (V, previously unpublished data) ................................................................................................................. 72 5.2.4. Effects of nicotine and epibatidine on conditioned place preference (V) ..... 75
6. DISCUSSION ....................................................................................................................... 77
6.1. Microdialysis studies................................................................................................... 77 6.1.1. Effects of nicotine and dopaminergic drugs on nigrostriatal dopamine (I) .................................................................................................................................. 77 6.1.2. Effects of nicotine and epibatidine on dopaminergic transmission (II) ........ 79 6.1.3. Effects of nicotine and epibatidine on dopamine in combination with nomifensine (III) .................................................................................................................... 83
6.2. Behavioural studies...................................................................................................... 85 6.2.1. Effects of acute nicotine on rotational behaviour in combination with nomifensine or levodopa (I, unpublished results) ........................................................... 85 6.2.2. Effects of repeated nicotine and epibatidine on rotational behaviour (IV) .................................................................................................................................. 86 6.2.2. Effects of nicotine and epibatidine on locomotor activity (V, unpublished results) ............................................................................................................. 88 6.2.3. Effects of nicotine and epibatidine on conditioned place preference (V) ..... 91
7. SUMMARY AND CONCLUSIONS..................................................................................... 93
8. ACKNOWLEDGEMENTS ................................................................................................. 95
9. REFERENCES ....................................................................................................................... 97 APPENDIX: ORIGINAL PUBLICATIONS I-V

6
ABSTRACT
Ascending cerebral dopaminergic pathways, the nigrostriatal and mesolimbic/mesocortical pathways, are involved in both motor control and the rewarding and reinforcing effects of drugs of abuse, such as nicotine. The loss of nigrostriatal dopaminergic neurons is characteristic of Parkinson’s disease. The mesolimbic/mesocortical dopaminergic pathway mediates effects on locomotor activity, but its role is better established in the mediation of reward and reinforcement. Nicotine, the main psychoactive ingredient in tobacco, acts at neuronal nicotinic acetylcholine receptors (nAChRs) to induce release of dopamine in the caudate-putamen and nucleus accumbens, the nerve terminal areas of the nigrostriatal and mesolimbic pathways. Epidemiological studies have shown a reduced incidence of Parkinson’s disease in smokers and even former smokers, compared to those who have never smoked. Once Parkinson’s disease has already developed, there is evidence to suggest that nicotine alleviates both motor and cognitive symptoms of patients. Indeed, nAChRs represent an important modulator of dopaminergic function, both under normal conditions and in pathological states. Discovery of their multiplicity has lead to the search for compounds that selectively bind different neuronal nAChR subtypes and that might be beneficial in the treatment of various diseases. However, the role of nAChRs in the mesolimbic pathway in dependence and reward complicates the development of therapeutic drugs that lack addictive properties. Therefore, this study investigates the differential regulation of nAChRs on the two major dopaminergic pathways, the nigrostriatal and mesolimbic pathways. This is achieved by comparison of two nAChR agonists, nicotine and epibatidine, which differ in their affinity for nAChR subtypes. Our present findings suggest that epibatidine, in contrast to nicotine, stimulates dopamine release preferentially in the nigrostriatal pathway, compared with the mesolimbic pathway. When given repeatedly, epibatidine similarly to nicotine stimulates the nigrostriatal dopamine system sufficiently to activate rotational behaviour. While inhibition of dopamine uptake by nomifensine enhances nicotine-induced dopamine release in both the caudate-putamen and nucleus accumbens, epibatidine-induced dopamine release is enhanced by nomifensine in the caudate-putamen, but inhibited in the nucleus accumbens. The modest effects of epibatidine on mesolimbic dopamine release and on place preference, suggest a reduced abuse potential of epibatidine compared with that of nicotine. Differences between epibatidine and nicotine are probably due to their differing affinities for the nAChR subtypes that mediate their effects. In contrast to dopamine release in the mesolimbic pathway, the dose-response curves of the both nAChR agonists studied on nigrostriatal dopamine release are bell-shaped, suggesting desensitization of the nAChR subtypes that mediate their effects on striatal dopamine. These differences may be due to the variation of nAChR subtypes, as well as distinct localization of subtypes between these two pathways. In conclusion, the present findings suggest that it is possible to develop compounds that selectively affect specific nAChR subtypes located on different dopaminergic pathways. Such compounds alone, or in combination with a dopamine uptake inhibitor, may be beneficial in the treatment of diseases, such as Parkinson’s disease, tobacco abuse or drug addiction.

7
LIST OF ABBREVIATIONS
α-Bgt α-bungarotoxin α-Ctx α-conotoxin ACh acetylcholine ANOVA analysis of variance CNS central nervous system COMT catechol-O-methyl transferase CPu caudate-putamen DAT dopamine transporter DHβE dihydro-β-erythroidine DMPP 1,1-dimethyl-4-phenylpiperazinium DOPA 3,4-dihydroxyphenylalanine DOPAC 3,4-dihydroxyphenylacetic acid GABA γ-aminobutyric acid 5-HIAA 5-hydroxyindoleacetic acid 5-HT 5-hydroxytryptamine, serotonin HPLC high-performance liquid chromatography HVA homovanillic acid i.p. intraperitoneal, intraperitoneally IPN interpeduncular nucleus LDT laterodorsal tegmental nucleus MAO monoamine oxidase MFB medial forebrain bundle MLA methyllycaconitine MPTP 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mRNA messenger ribonucleic acid 3-MT 3-methoxytyramine NAc nucleus accumbens nAChR nicotinic acetylcholine receptor NMDA N-methyl-D-aspartate 6-OHDA 6-hydroxydopamine s.c. subcutaneous, subcutaneously S.E.M. standard error of the mean SN substantia nigra SNc substantia nigra pars compacta SNr substantia nigra pars reticulata TPP tegmental pedunculopontine nucleus TTX tetrodotoxin VTA ventral tegmental area

8
LIST OF ORIGINAL PUBLICATIONS
This dissertation is based on the following publications, herein referred by their Roman
numerals (I-V):
I. Janhunen S, Mielikäinen P, Paldánius P, Tuominen RK, Ahtee L, Kaakkola S
(2005) The effect of nicotine in combination with various dopaminergic drugs on
nigrostriatal dopamine in rats. Naunyn-Schmiedeberg’s Archives of
Pharmacology, in press
II. Janhunen S, Ahtee L (2004) Comparison of the effects of nicotine and epibatidine
on the striatal extracellular dopamine. European Journal of Pharmacology 494 (2-
3), 167-177.
III. Janhunen S, Tuominen RK, Piepponen TP, Ahtee L (2005) Nicotine and
epibatidine alter differently nomifensine-elevated dopamine output in the dorsal
and ventral striatum. European Journal of Pharmacology 511 (2-3), 143-150.
IV. Janhunen S, Tuominen RK, Ahtee L (2005) Comparison of the effects of nicotine
and epibatidine in combination with nomifensine on rotational behaviour.
Neuroscience Letters 381 (3), 314-319.
V. Janhunen S, Linnervuo A, Svensk M, Ahtee L (submitted) The effects of nicotine
and epibatidine on locomotor activity and conditioned place preference in rats.
Reprints were made with permission from the publishers.

9
1. INTRODUCTION
Tobacco smoking is the most prevalent preventable cause of death in developed countries
and is rapidly becoming a significant health problem in developing countries. It is predicted
that every year 3 million people worldwide die from smoking related diseases and that the
rapid increase in smoking in developing countries will increase this number to 10 million by
the year 2025 (Peto et al., 1996; WHO, 2003). Although most smokers are aware of the
harmful effects of smoking, such as increased risks of cardiovascular and pulmonary
diseases and various forms of cancer, many continue to smoke. It has been estimated that
70% of regular smokers want to quit and the majority of them have even tried, but most have
failed. Less than 10% of those who try remain abstinent from smoking after one year
(Cinciripini et al., 1997). As recently as the beginning of the 90’s it was debated whether
smoking should be regarded as a habit or as a true disease (Ochoa, 1994). There is now little
doubt that the majority of tobacco smokers do so in order to experience the
psychopharmacological properties of nicotine that are present in the smoke and to which a
significant proportion of habitual tobacco users become addicted (US Department of Health
and Human Sciences, 1988; Balfour, 1994). In fact, nicotine is now used as a therapeutic
agent in smoking cessation.
The neural mechanisms underlying nicotine dependence are complex and not fully
understood. Similar to other drugs of abuse, such as cocaine and amphetamine, nicotine
stimulates the mesolimbic dopaminergic pathway, which appears to play a critical role in
mediating the reinforcing and rewarding effects of tobacco smoke (Wise and Bozarth, 1987;
Balfour et al., 1998). Smoking (nicotine) improves cognitive function, including learning and
memory, elevates mood, produces a subjective state of well-being, alleviates the stress of
everyday life and prevents the feared withdrawal symptoms caused by the cessation of
smoking (Benowitz, 1996). In animal models of smoking, animals can be trained to self-
administer nicotine and the cessation of regular nicotine can induce physical withdrawal signs
and symptoms (Corrigall, 1999).
The effects of nicotine are mediated by nicotinic acetylcholine receptors (nAChRs), which are
ligand-gated ion channels composed of five subunits. Recent findings on the structural and
functional diversity of nAChR subtypes in the brain have provoked intense research into the

10
role of these receptors in several brain functions and behaviours. The discovery that nAChRs
modulate release of various neurotransmitters, including dopamine, noradrenaline, serotonin,
γ-aminobutyric acid (GABA) and glutamate, has lead to the idea that these receptors could be
therapeutic targets for diverse neurodegenerative and mood disorders, including Parkinson’s
and Alzheimer’s diseases, schizophrenia, depression, anxiety, Tourette’s syndrome and adult
attention deficit hyperactivity disorder (Holladay et al., 1997; Lloyd et al., 1998; Lloyd and
Williams, 2000; Hogg and Bertrand, 2004). In fact, many of these diseases are associated with
a reduction of nAChR number in the brain. Furthermore, discovery of epibatidine, a potent
nAChR agonist with effective analgesic properties (Badio and Daly, 1994; Bannon et al.,
1995), gave rise to the possibility that novel nAChR agonists could also be used to treat pain.
The purpose of present studies was to clarify differences in the nAChR-mediated regulation
of two major ascending dopaminergic pathways, the nigrostriatal and mesolimbic pathways.
The nigrostriatal dopaminergic pathway is involved in motor control and destruction of this
pathway is the main cause of Parkinson’s disease, while the mesolimbic dopaminergic
pathway plays a role in reinforcement and dependence. It is important to clarify the
differences in the nicotinic regulation of these pathways in order to develop novel nicotinic
compounds that elicit therapeutic effects on brain diseases, but lack addictive properties.
Thus, we carried out a series of in vivo brain microdialysis studies in freely moving rats and
studied the effects of two nAChR agonists, nicotine and epibatidine, on dopaminergic
transmission in the nerve terminal regions of the nigrostriatal and mesolimbic pathways. The
affinity of epibatidine at different nAChR subtypes, along with its behavioural properties,
differ from those of nicotine (Sullivan et al., 1994; Reuben et al., 2000). We also studied the
effects of various dopaminergic drugs, including inhibitors of either dopamine reuptake
(nomifensine), dopamine metabolism (tolcapone and selegiline) or presynaptic dopamine
receptors (haloperidol), on striatal dopamine in combination with nicotine. In light of these
results, further studies on the effects of nicotine and epibatidine on dopaminergic transmission
of the nigrostriatal and mesolimbic pathways were carried out in combination with
nomifensine. Furthermore, we studied the effects of nicotine and epibatidine on different
behaviours associated with the two dopaminergic pathways. Rotational behaviour of rats with
unilateral nigrostriatal lesions is thought to mainly reflect an activation of the nigrostriatal
pathway. In contrast, locomotor activity and conditioned place preference preferentially
involve a stimulation of the mesolimbic pathway.

11
2. REVIEW OF THE LITERATURE
2.1. Ascending dopaminergic pathways
Dopamine was thought only to be a precursor for noradrenaline and adrenaline, but not an
independent transmitter in the brain until Carlsson et al. (1958, 1959) found large amounts of
dopamine in the basal ganglia and implicated it in the parkinsonian-like symptoms induced by
reserpine. Findings that dopamine is involved in motor control and that decreased striatal
dopamine is the cause of extrapyramidal symptoms in Parkinson’s disease, resulted in the
clinically most important step in the treatment of Parkinson’s disease so far, the use
dopamine’s prodrug, levodopa.
As shown in Fig. 2-1, dopaminergic neurons form three major ascending pathways, the
nigrostriatal, mesolimbic/mesocortical and tuberohypophyseal pathways (Rang et al., 1999).
Cell bodies of these pathways are located in the midbrain, mainly in the substantia nigra pars
compacta (SNc, A9) and the ventral tegmental area (VTA, A10) (Dahlström and Fuxe, 1964;
Ungerstedt, 1971c). In addition, there are some local dopaminergic interneurons in the
olfactory cortex, medulla and retina (Rang et al., 1999; Cooper et al., 2003).
The nigrostriatal dopaminergic pathway originates from the SNc and terminates in the
dorsal part of the striatum, the caudate-putamen (CPu), also known as the dorsal striatum
(Dahlström and Fuxe, 1964; Ungerstedt, 1971c). A minor part of the nigrostriatal pathway
consists of axons from cell bodies in the A8 area (dopaminergic neurons caudal to A9 and
dorsal to A10) projecting to ventral putamen (Ungerstedt, 1971c). Axons of the nigrostriatal
pathway run alongside fibres containing noradrenaline and 5-hydroxytryptamine (5-HT,
serotonin) through the medial forebrain bundle (MFB) and internal capsule to the striatum.
The nigrostriatal pathway contains about 75% of the dopamine in the brain and it is involved
in the control of posture and motor behaviour, as well as learning of motor programs and
habits.

12
Fig. 2-1. Main dopaminergic pathways in the rat brain. Dopaminergic cell bodies are represented by triangles and axonal fibres by solid lines. (a) Nigrostriatal pathway. (b) Mesolimbic pathway. (c) Mesolimbic/mesocortical and mesothalamic pathways. (d) Tuberohypophyseal pathway and other short dopaminergic pathways. CPu = caudate-putamen, MFB = medial forebrain bundle, NAc = nucleus accumbens, SNc = substantia nigra pars compacta, SNr = substantia nigra pars reticulata. Modified from Feldman et al. (1997), with permission.
The mesolimbic/mesocortical dopaminergic pathway has cell bodies located in the VTA
and its axons ascend medially to the axons of the nigrostriatal pathway through the MFB to
limbic and cortical structures (Dahlström and Fuxe, 1964; Ungerstedt, 1971c). One branch of
these neurons, the mesolimbic pathway, innervates the nucleus accumbens (NAc), amygdala,
hippocampus, septum and olfactory tubercle, while the other, the mesocortical pathway,
innervates the limbic cortex, such as medial prefrontal, cingulate and entorhinal cortices
(Cooper et al., 2003). Some functional properties of the mesolimbic pathway, such as basal
rate of physiological activity and firing pattern differ from those of the mesocortical pathway.
The mesolimbic pathway is involved in the control of motor behaviour as well as motivation,
emotions and reward, while the mesocortical pathway is more involved in the higher
cognitive functions, learning and reward behaviour.

13
The terminal field of the mesolimbic pathway, the NAc, is subdivided into two regions
according to its anatomical projections (Heimer et al., 1991). The NAc shell is part of the
extended amygdala and represents the ventromedial region, while the NAc core represents the
dorsolateral area. Evidence from pharmacological, anatomical and physiological studies
suggest that the shell and core regions are differentially involved in specific behaviours, such
as feeding behaviour and motor activity (Maldonado-Irizarry and Kelley, 1995; Baldo et al.,
2002; Mendoza et al., 2005). The NAc core is linked to the extrapyramidal motor system as it
projects to the dorsolateral ventral pallidum which, in turn, projects to the motor system
through the substantia nigra (SN) and subthalamic nucleus. The pattern of its efferent
connections closely resembles those of the CPu (Heimer et al., 1991) and it is involved in
somatomotor functions. In contrast, the NAc shell is suggested to be more limbic in nature,
because its efferent projections are closely connected to limbic output structures such as the
VTA, amygdala and lateral hypothalamus (Heimer et al., 1991). The shell is involved in
integration and expression of emotions and the projections between the shell and lateral
hypothalamus are relevant for the control of feeding behaviour (Stratford and Kelley, 1997,
1999). Both subdivisions also play a pivotal role in learnt behaviours and Pavlovian
conditioning. The shell is involved in the acquisition of responding for motivationally
significant stimuli, while the core is involved in responding that is elicited, or reinforced, by
conditioned stimuli (Di Chiara, 2002; Ito et al., 2004).
The tuberohypophyseal pathway is a group of short neurons that project from the arcuate
nucleus of the hypothalamus to the median eminence and pituitary gland. This pathway
regulates the function of the pituitary gland. Disruptions in the regulatory control of the
tuberohypophyseal pathway caused by drugs, such as haloperidol that block dopamine D2
receptors, may result in hyperprolactinemia and even lactation.
2.1.1. Functional implications of nigrostriatal and mesolimbic pathways
Both the nigrostriatal and mesolimbic dopaminergic pathways are involved in the control of
movement. Dopamine release in the CPu plays an integral role in the complex circuitry of the
basal ganglia and is crucial for voluntary movement. If this dopaminergic transmission is
disturbed, as occurs in Parkinson’s and Huntington’s diseases, movement generation is

14
damaged. The mesolimbic dopaminergic pathway is involved in locomotor activity, but the
role of the NAc is better established in the mediation of reward and reinforcement.
Recently, in addition to its general role in the initiation and patterning of a variety of
behaviours, the CPu appears to be a brain centre for habit formation as it stores information
about fixed action patterns (reviewed by Gerdeman et al., 2003). While synaptic plasticity in
the NAc appears to play a role in the early stages of drug abuse, the CPu seems to contribute
to the formation of persistent drug-related habits (Berke and Hyman, 2000; Koob and Le
Moal, 2001; Everitt and Wolf, 2002; Gerdeman et al., 2003). In this instance, casual drug-use
progresses towards compulsive, habitual drug-use that exhibits narrowly defined behavioural
repertoires directed mostly at drug-seeking. Recent findings support the idea that participation
of the CPu in the development of addiction is related to mechanisms of long-term synaptic
plasticity, which might occur in combination with similar processes in the NAc (Gerdeman et
al., 2003). Moreover, drug-induced alterations in function of the NAc could subsequently
facilitate synaptic plasticity in the CPu by influencing the flow of information through the
basal ganglia.
2.2. Synthesis and release of dopamine
Dopamine, like other catecholamines, is synthesized from an amino acid precursor, tyrosine
(Cooper et al., 2003). Dietary tyrosine is actively transported across the blood-brain barrier
into dopaminergic neurons and hydroxylated in the cell bodies and nerve terminals by
tyrosine hydroxylase to form 3,4-dihydroxyphenylalanine (DOPA). DOPA is rapidly
converted to dopamine by a cytoplasmic enzyme, aromatic L-amino acid decarboxylase and
newly synthesized dopamine is actively transported into storage vesicles. The conversion of
tyrosine to DOPA is the rate-limiting step of dopamine synthesis and it depends upon the
activity of tyrosine hydroxylase which, in turn, is regulated by several factors (Cooper et al.,
2003). For example, dopamine synthesis is inhibited by dopamine and other catecholamines
that function as inhibitory end-products by competing for a binding site on tyrosine
hydroxylase (Feldman et al., 1997). End-product inhibition is maximal, when neuronal
activity and transmitter release are low and catecholamine concentration in storage vesicles is
high. In addition, the availability of tyrosine may modestly modulate the activity of tyrosine
15
hydroxylase, particularly at high firing rates. Released dopamine activates presynaptic
dopamine receptors on nerve terminals, which may also result in feedback inhibition of
dopamine synthesis (Cooper et al., 2003). The synthesis of dopamine is also dependent on the
rate of impulse flow in dopaminergic neurons so that during increased impulse flow the rate
of tyrosine hydroxylation is increased and results in increased biosynthesis of catecholamines.
Long-term regulation of tyrosine hydroxylase occurs, for example, via increased gene
transcription and synthesis of enzyme molecules (Feldman et al., 1997). In regard to the
nigrostriatal and mesolimbic pathways, the synthesis of dopamine has been shown to be
substantially higher in the limbic areas, compared with the dorsal striatal areas (Andén et al.,
1983).
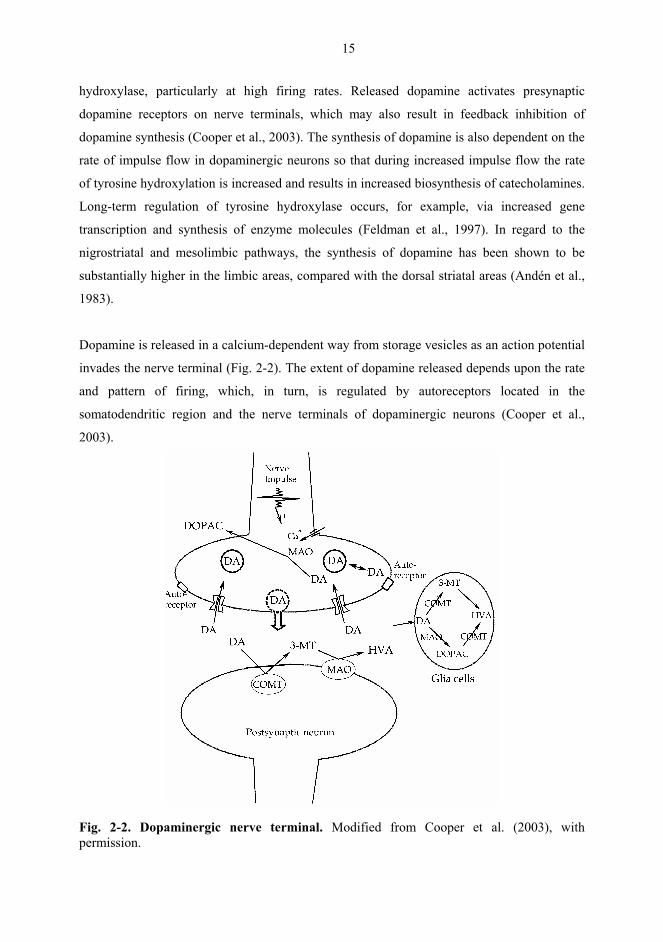
Dopamine is released in a calcium-dependent way from storage vesicles as an action potential
invades the nerve terminal (Fig. 2-2). The extent of dopamine released depends upon the rate
and pattern of firing, which, in turn, is regulated by autoreceptors located in the
somatodendritic region and the nerve terminals of dopaminergic neurons (Cooper et al.,
2003).
Fig. 2-2. Dopaminergic nerve terminal. Modified from Cooper et al. (2003), with permission.

16
Dopamine cells have been observed to fire with two main firing patterns, a slow single
spiking pattern and a burst firing (Grace and Bunney, 1984a, b). The single spike firing
pattern is characterized by trains of spikes that discharge at steady, but irregular, intervals, and
it is the typical firing pattern of the majority of dopamine cells encountered in untreated,
anaesthetised rats. The burst-firing mode consists of consecutive spikes in a burst, displaying
progressively decreasing amplitude and increasing duration. The bursting pattern leads to an
enhanced release of dopamine. Dopamine cells in the VTA have been shown to elicit a higher
level of burst-firing than those in the SN (Grenhoff et al., 1986). Consistently, the release of
dopamine has been found to be more rapid in the NAc than in the CPu (Garris and Wightman,
1994).
The functional activity of released dopamine is primarily terminated through reuptake by a
specific dopamine transporter (DAT) into the nerve terminal. DAT plays a critical role in
maintaining the homeostasis of the dopaminergic systems. For example, mice lacking DAT
protein exhibit hyperdopaminergic state, despite multiple compensatory adaptations
(Gainetdinov et al., 1999). Dopamine reuptake is particularly efficient in the CPu and indeed,
in the CPu there has been found two- to three-fold more DAT binding sites than in the NAc
(Marshall et al., 1990; Garris and Wightman, 1994; Hoffman and Gerhardt, 1998).
2.3. Metabolism of dopamine
As shown in Fig. 2-2, dopamine metabolism can take place in the synaptic cleft, in the
cytoplasm of the nerve terminal and inside glial cells. Dopamine is metabolized by
monoamine oxidase (MAO) and catechol-O-methyl transferase (COMT) to 3,4-
dihydroxyphenylacetic acid (DOPAC), homovanillic acid (HVA) and 3-methoxytyramine (3-
MT). In humans and primates, the major brain metabolite is HVA, while in rat brain it is
DOPAC (Cooper et al., 2003). In rat striatum, the major part of HVA is formed from DOPAC
and the minor part from 3-MT (Westerink and Spaan, 1982a). Accumulation of DOPAC,
HVA and 3-MT in the brain or the cerebrospinal fluid may be used as an index of the
functional activity of dopaminergic neurons (Cooper et al., 2003). Increased dopamine
turnover or electrical stimulation of dopaminergic neurons increases the amount of HVA and

17
3-MT in the brain. It has been suggested that 3-MT is a better indicator of decreased
dopamine release than HVA or DOPAC (Kehr, 1976; Westerink and Spaan, 1982b). There is
also an excellent correlation between changes in the impulse flow of dopaminergic neurons
and changes in the level of DOPAC. Furthermore, DOPAC is thought to reflect the
intraneuronal dopamine metabolism, rather than dopamine release (Roffler-Tarlov et al.,
1971).
COMT is found in soluble and membrane-bound forms in glial cells and postsynaptic neurons
(Rivett et al., 1983; Kaakkola et al., 1987; Männistö and Kaakkola, 1999). Inhibitors of
COMT increase the level of dopamine in the synaptic cleft and thus prolong dopamine
receptor activation. These inhibitors can be coadministered with levodopa to enhance
dopaminergic function in parkinsonian patients. MAO exists in intracellular and extracellular
compartments and inhibition of MAO thus increases presynaptic intracellular concentrations
of dopamine and prolongs the availability of released dopamine. MAO has two forms, MAO-
A and MAO-B, of which MAO-A displays strong affinities for noradrenaline, 5-HT and
dopamine, while MAO-B exhibits the highest affinities for β-phenylethylamine and
dopamine.
2.4. Dopamine and the neural circuits of basal ganglia
The basal ganglia are deep-lying structures of the cerebral hemisphere, including the striatum,
globus pallidus and amygdala, which act in conjunction with allied nuclei, such as the SNc,
substantia nigra pars reticulata (SNr), subthalamic nucleus and endopeduncular nucleus (Fig.
2-3a; Hauber, 1998). The basal ganglia collect inputs from the neocortex, integrate them with
each other and with inputs from the limbic areas and focus the integrated outputs to regions of
the frontal lobes and brainstem involved in motor planning and motor memory (Graybiel,
1990). In fact, abnormalities of the basal ganglia and their allied nuclei can result in disorders
of movement programming, certain forms of drug addiction and some mental disorders, such
as schizophrenia-like states and obsessive-compulsive disorder. The predominant
neurotransmitter in the basal ganglia is GABA and the primary site of functional interactions
in the basal ganglia is the striatum. The GABAergic medium spiny neurons of the striatum
receive dense glutamatergic inputs from the cortex and thalamus, dopaminergic inputs from
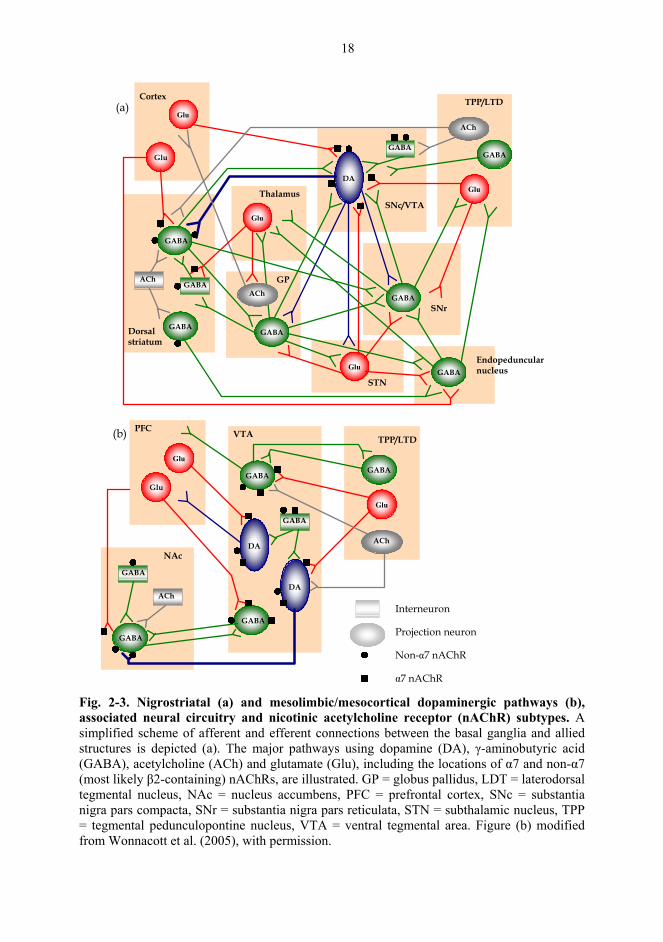
18
(a)
Glu
Glu
Glu
GABA
GABA
GABA
GABA
DA
Dorsal striatum
Cortex
Thalamus SNc/VTA
STN
GP
SNr GABA
GABA
ACh
Glu
Glu
ACh
GABA
GABA
TPP/LTD
Endopeduncular nucleus
ACh
(b)
Glu
Glu
Glu
GABA GABA
GABA
AGABA
GABA
GABA
ACh
ACh
DA
DA
NAc
PFC VTA TPP/LTD
Interneuron
Projection neuron
Non-α7 nAChR
α7 nAChR Fig. 2-3. Nigrostriatal (a) and mesolimbic/mesocortical dopaminergic pathways (b), associated neural circuitry and nicotinic acetylcholine receptor (nAChR) subtypes. A simplified scheme of afferent and efferent connections between the basal ganglia and allied structures is depicted (a). The major pathways using dopamine (DA), γ-aminobutyric acid (GABA), acetylcholine (ACh) and glutamate (Glu), including the locations of α7 and non-α7 (most likely β2-containing) nAChRs, are illustrated. GP = globus pallidus, LDT = laterodorsal tegmental nucleus, NAc = nucleus accumbens, PFC = prefrontal cortex, SNc = substantia nigra pars compacta, SNr = substantia nigra pars reticulata, STN = subthalamic nucleus, TPP = tegmental pedunculopontine nucleus, VTA = ventral tegmental area. Figure (b) modified from Wonnacott et al. (2005), with permission.

19
the midbrain (SNc and VTA) and cholinergic inputs from the tegmental pedunculopontine
nucleus (TPP)(Fig. 2-3a; for reviews e.g. Graybiel, 1990; Parent, 1990; Bolam et al., 2000;
Parent et al., 2000; Smith and Kieval, 2000). The major output from the striatum is provided
by two populations of GABAergic neurons. One projects to the globus pallidus and the other
to the SNr and entopeduncular nucleus and onwards to the thalamus and superior colliculus.
Dopaminergic neurons in the striatum and midbrain interact with neurotransmitter inputs
originating from several brain regions (Fig. 2-3), including glutamatergic projections from the
cortex and subthalamic nucleus, GABA- and peptide-containing inputs from the striatum, a
GABAergic input from the globus pallidus, a serotonergic input from the dorsal raphe
nucleus, a noradrenergic input from the locus coeruleus and a cholinergic input from the
brainstem (Usunoff et al., 1982; Nitsch et al., 1984; Kita and Kitai, 1987; Wirtshafter et al.,
1987; Gariano and Groves, 1988; Smith and Parent, 1988; Gould et al., 1989; Bolam and
Smith, 1990; Smith and Bolam, 1990). In addition, dopaminergic neurons interact with local
GABAergic and cholinergic interneurons (Woolf and Butcher, 1981; Nitsch and Riesenberg,
1988; Koós and Tepper, 1999). Activation of N-methyl-D-aspartate (NMDA)-type glutamate
receptors or excitatory glutamatergic afferents projecting from the frontal cortex, subthalamic
nucleus and TPP, increase the burst firing of dopaminergic neurons and secondarily, the
release of dopamine from nerve terminals (Grenhoff et al., 1988; Svensson and Tung, 1989;
Asencio et al., 1991, Gariano and Groves, 1988; Charléty et al., 1991; Suaud-Chagny et al.,
1992; Westerink et al., 1992b; Chergui et al., 1993; Karreman et al., 1996; Schilström et al.,
1998b). Mesolimbic dopaminergic neurons appear to be more sensitive than nigrostriatal
neurons to the stimulation mediated by NMDA glutamate receptors (Westerink et al., 1992b,
1996).
The TPP ascend excitatory cholinergic and glutamatergic projections to the midbrain (Fig. 2-
3). More specifically, neurons from the TPP project to dopaminergic neurons in the SNc and
run through the VTA. However, the activity of dopaminergic neurons of the VTA is
predominantly regulated by neurons from the laterodorsal tegmental nucleus (LDT)(Satoh and
Fibiger, 1986; Clarke et al., 1987; Bolam et al., 1991; Futami et al., 1995; Blaha et al., 1996).
The cholinergic interneurons in the striatum are tonically active and release acetylcholine
(ACh)(Aosaki et al., 1995; Bennett and Wilson, 1999), which provides an important control
of striatal dopamine release (Zhou et al., 2001). Inhibitory action of a nAChR antagonist

20
mecamylamine, on the activity of dopaminergic neurons suggests that the midbrain
dopaminergic neurons may be physiologically controlled by cholinergic nicotinic mechanisms
(Ahtee and Kaakkola, 1978; Clarke et al., 1985a; Grenhoff et al., 1986; Haikala and Ahtee,
1988). It is suggested that the mesolimbic dopaminergic cells are under a phasic, rather than a
tonic, cholinergic control (Grenhoff and Svensson, 1992).
The burst firing of dopaminergic neurons is tonically inhibited by GABAergic neurons from
the SNr (Fig. 2-3). Inhibition of these inputs by activation of the globus pallidus decreases the
tonic inhibition and facilitates the burst firing of dopaminergic neurons (Tepper et al., 1998).
Pharmacological blocking of GABA receptors in the VTA or SN increases spontaneous
release of dopamine in the nerve terminal areas (NAc or CPu), whereas activation of these
GABA receptors reduces the firing rate and burst firing of dopaminergic neurons (Westerink
et al., 1992a, 1996; Dewey et al., 1999; Cobb and Abercrombie, 2002; Erhardt et al., 2002).
Although the dorsal and ventral striata are very similar with respect to neuronal
cytoarchitecture and neurotransmitter content (i.e. most of the neurons are GABAergic), they
vary with regard to their afferent and efferent circuitry. Dopaminergic and GABAergic
neurons of the CPu are preferentially innervated by neurons from various associative and
sensorimotor cortical areas, while the NAc receives projections from the limbic structures,
prefrontal cortex, amygdala and hippocampus (Parent, 1990). Thus, there are differences in
the interactions of dopaminergic, GABAergic and cholinergic transmission between the
ventral and dorsal striata (Girault et al., 1986). It has been reported that the stimulation of
GABAA receptors in the region of dopaminergic cell bodies stimulates nigrostriatal neurons,
but inhibits mesolimbic neurons, while stimulation of GABAB receptors inhibits both
nigrostriatal and mesolimbic neurons (Santiago and Westerink, 1992; Westerink et al., 1996).
2.5. Characteristics of Parkinson’s disease
Parkinson’s disease was first described in the beginning of the 19th century by James
Parkinson, in whose honour the disease is named (Parkinson, 1817). The major symptoms of
Parkinson’s disease are tremor at rest, rigidity, bradykinesia and even akinesia (reviewed by
Obeso et al., 2000; Olanow, 2004). Postural instability and gait disturbances usually occur as
the disease progresses. The most debilitating of the late-onset symptoms are cognitive defects,

21
such as dementia, general confusion, disorientation and sleepiness. Depression can occur at
any stage of the disease. Pathologically, Parkinson’s disease is characterized by a progressive
preferential degeneration of melanized neurons of the SNc coupled with intracellular protein
aggregates known as Lewy bodies (Jellinger, 1999). Changes in the SNc are most pronounced
in the ventrolateral region. Dopamine deficiency produces dysfunction in the striatum which,
in turn, leads to decreased activity of the direct pathway from striatal GABAergic neurons to
the internal segment of the globus pallidus and the SNr and to increased activity of the
indirect pathway involving the external segment of the globus pallidus and the subthalamic
nucleus (Hamani and Lozano, 2003). As a consequence, the activities in the basal ganglia
output structures are disrupted, which, leads to disruption in the activity of brainstem motor
areas, such as the TPP and thalamocortical motor system. This leads to difficulties in the
initiation of movement and poverty of motion.
The loss of neurons in the SNc and other subcortical nuclei is proportional to striatal
dopamine deficiency and both the duration and clinical severity of the disease. Parkinsonian
motor symptoms appear when dopaminergic neuronal death exceeds a critical threshold: 70-
80% loss of striatal nerve terminals and 50-60% loss of SNc perikarya (Agid, 1991). Tyrosine
hydroxylase activity is decreased parallel to the loss of dopamine (Birkmeyer and Riederer,
1989). Decreased activity of tyrosine hydroxylase is also found in the limbic system and it
may result in psycho-pathological disturbances. In addition, there are deficits in several other
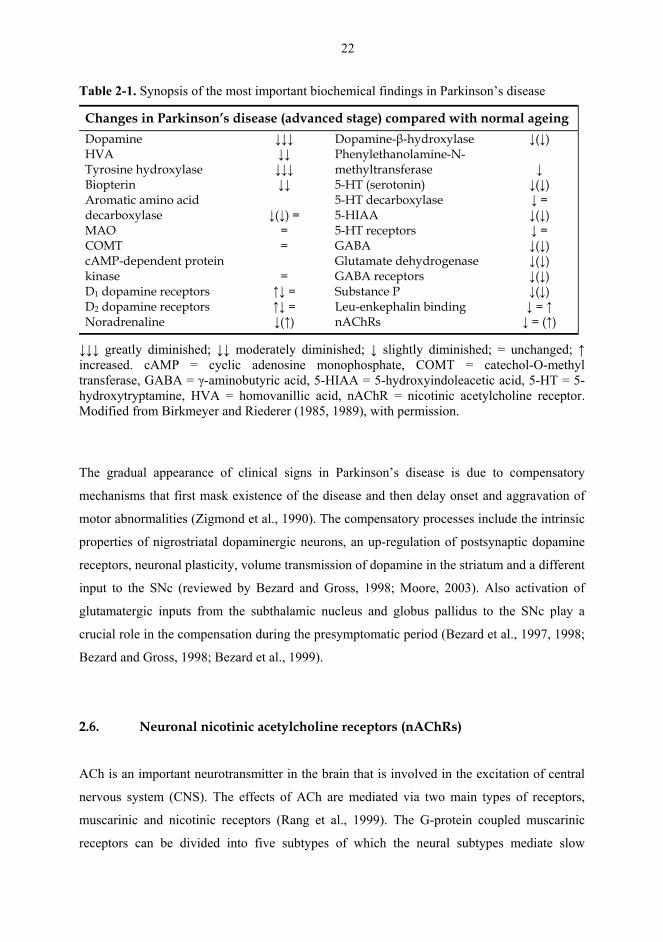
neurotransmitter systems, such as subcortical noradrenergic, serotonergic and cholinergic
ascending pathways (Table 2-1; Nordberg et al., 1985; Jellinger, 1997, 1999). For example,
the loss of two thirds of the cholinergic neurons in the basal ganglia contributes to dementia at
the late stage of Parkinson’s disease. The loss of cortical nAChRs positively correlates with
the degree of cognitive deficits (Paterson and Nordberg, 2000; Quik and Jeyarasasingam,
2000). Many patients have a deficiency of noradrenaline and a reduction of 40-50% of 5-HT
in several brain regions, but these reductions tend not worsen as the disease progresses.
GABA and glutamate decarboxylase in the SN are also found to be altered.
22
Table 2-1. Synopsis of the most important biochemical findings in Parkinson’s disease
Changes in Parkinson’s disease (advanced stage) compared with normal ageing Dopamine HVA Tyrosine hydroxylase Biopterin Aromatic amino acid decarboxylase MAO COMT cAMP-dependent protein kinase D1 dopamine receptors D2 dopamine receptors Noradrenaline
↓↓↓ ↓↓ ↓↓↓ ↓↓
↓(↓) =
= =
= ↑↓ = ↑↓ = ↓(↑)
Dopamine-β-hydroxylase Phenylethanolamine-N-methyltransferase 5-HT (serotonin) 5-HT decarboxylase 5-HIAA 5-HT receptors GABA Glutamate dehydrogenase GABA receptors Substance P Leu-enkephalin binding nAChRs
↓(↓) ↓ ↓(↓) ↓ = ↓(↓) ↓ = ↓(↓) ↓(↓) ↓(↓) ↓(↓) ↓ = ↑ ↓ = (↑)
↓↓↓ greatly diminished; ↓↓ moderately diminished; ↓ slightly diminished; = unchanged; ↑ increased. cAMP = cyclic adenosine monophosphate, COMT = catechol-O-methyl transferase, GABA = γ-aminobutyric acid, 5-HIAA = 5-hydroxyindoleacetic acid, 5-HT = 5-hydroxytryptamine, HVA = homovanillic acid, nAChR = nicotinic acetylcholine receptor. Modified from Birkmeyer and Riederer (1985, 1989), with permission.
The gradual appearance of clinical signs in Parkinson’s disease is due to compensatory
mechanisms that first mask existence of the disease and then delay onset and aggravation of
motor abnormalities (Zigmond et al., 1990). The compensatory processes include the intrinsic
properties of nigrostriatal dopaminergic neurons, an up-regulation of postsynaptic dopamine
receptors, neuronal plasticity, volume transmission of dopamine in the striatum and a different
input to the SNc (reviewed by Bezard and Gross, 1998; Moore, 2003). Also activation of
glutamatergic inputs from the subthalamic nucleus and globus pallidus to the SNc play a
crucial role in the compensation during the presymptomatic period (Bezard et al., 1997, 1998;
Bezard and Gross, 1998; Bezard et al., 1999).
2.6. Neuronal nicotinic acetylcholine receptors (nAChRs)
ACh is an important neurotransmitter in the brain that is involved in the excitation of central
nervous system (CNS). The effects of ACh are mediated via two main types of receptors,
muscarinic and nicotinic receptors (Rang et al., 1999). The G-protein coupled muscarinic
receptors can be divided into five subtypes of which the neural subtypes mediate slow

23
excitatory effects in the CNS that contribute, for instance, to memory. Nicotinic acetylcholine
receptors (nAChRs) are located at the neuromuscular junction, in autonomic ganglia and in
the CNS. Neuronal nAChRs subtypes are involved in fast excitatory transmission in the brain.
2.6.1. Structure and classification of nAChRs
All nAChRs are members of a supergene family of ligand-gated ion channels that also
includes GABAA, glycine and 5-HT3 receptors (reviewed by Karlin and Akabas, 1995;
Lindstrom et al., 1995; McGehee and Role, 1995). They have a pentameric cylindrical
structure, composed of five subunits organised around a central pore that is permeable to Na+,
K+ and Ca2+ ions (Fig. 2-4; Cooper et al., 1991). The muscle-type nAChRs in vertebrate
skeletal muscles and in electric organs of electric eels (Electrophorus electricus) and rays
(Torpedo californica or marmorata) have provided a model for understanding structural,
molecular and biophysical properties of neuronal nAChRs. Muscle nAChRs consist of two
subtypes, either fetal α1β1γδ or adult α1β1εδ subtypes (Karlin, 1993). The first neuronal
nAChR subunit cDNA, ultimately termed as α3, was cloned by Heinemann, Patrick and
colleagues (Boulter et al., 1986). Since this report, eleven homologous genes encoding
neuronal nAChR subunits have been cloned in mammals (α2-α7 and β2-β4 in the brain, α9
and α10 in mechanosensory hair cells)(Elgoyhen et al., 1994; Lukas et al., 1999; Elgoyhen et
al., 2001). In addition, α8 subunit has been identified in avian species (Gotti et al., 1997b).
Neuronal nAChRs are composed of α- and β-subunits (Fig. 2-3d). The α subunits contain two
adjacent cysteines at positions homologues to α1 192-193 in loop C, whereas the β (non-α)
subunits lack these cysteine residues (Le Novere and Changeux, 1995).

24
Fig 2-4. Structure of nAChRs. (a) Transmembrane topology of a single subunit. (b) Pentameric composition of subunits constituting a nAChR (in this case, α4β2 receptor) and ACh binding site at the extracellular interface between two adjacent subunits. (c) Allosteric transitions of nAChR: resting (B), active (A) and desensitized (I and D). Various ligands bind to different sites as indicated; CB = competitive blocker, NCB = non-competitive blocker. (d) Putative organization of three different types of neuronal nAChRs: α7, α4β2, and α4β2α5. Modified from Lena and Changeux (1997a) and Berkovic et al. (1999), with permission.
Neuronal nAChRs can be divided into subtypes according to their ability to bind the snake
venom α-bungarotoxin (α-Bgt)(Deneris et al., 1991; Le Novere and Changeux, 1995;
McGehee and Role, 1995; Colquhoun and Patrick, 1997). Neuronal nAChRs that do not bind
α-Bgt comprise of both α and β subunits and they can form several different nAChR subtypes
(Boulter et al., 1987; Sargent, 1993). The α2, α3 and α4 subunits can form functional nAChRs
in pairwise combinations with β2 or β4 subunits (Boulter et al., 1987; Luetje and Patrick,
1991; Elliott et al., 1996). While the α5 and β3 subunits do not participate in pairwise
combinations, they may form functional channels when they are coexpressed with other
functional αβ-subunit combinations, such as α4, β2, and β4 as well as α3 and α4 subunits
(Deneris et al., 1989; Ramirez-Latorre et al., 1996; Wang et al., 1996; Gerzanich et al., 1998;

25
Groot-Kormelink et al., 1998). The α6 subunit can form a functional nAChR in combination
with the β4 subunit (Gerzanich et al., 1997), but its expression is more efficient when
coexpressed with at least two other subunits, including β3 (Fucile et al., 1998; Kuryatov et al.,
2000).
Neuronal nAChRs that are inhibited by nanomolar concentrations of α-Bgt consist of α7, α8,
α9 and α10 subunits (Boulter et al., 1987; Sargent, 1993). Only the α7 subunit is widely
distributed in the mammalian CNS. The α7, α8 and α9 subunits are capable of forming robust
functional channels when expressed as homooligomers in Xenopus oocytes (Couturier et al.,
1990b; Elgoyhen et al., 1994; Gerzanich et al., 1994). Some evidence exists that α7 subunits
can also be expressed in heteromeric combinations with α5 and β2 subunits in heterologous
systems (Lukas, 1995; Alkondon et al., 1997; Guo et al., 1998; Yu and Role, 1998; reviewed
by Girod et al., 1999). α7α8 heteromeric nAChRs occur in avian species (Elgoyhen et al.,
1994; Gotti et al., 1994). The α10 subunit does not form functional homomeric channels, but
it can form heteromeric α9α10 nAChRs when coexpressed with α9 subunit (Elgoyhen et al.,
2001; Sgard et al., 2002). The α9 and α10 subunits are expressed in mammals in the cochlea,
pituitary gland, keratinocytes and lymphocytes (Sgard et al., 2002; Peng et al., 2004).
2.6.2. Distribution of nAChRs in the brain
A single neuron may express more than one nAChR subtype, with each subtype localised
separately and serving different functions. This makes predicting the distribution of nAChR
subtypes based purely on subunit gene expression a difficult task. In situ hybridisation and
immunohistochemical studies have shown that α4, β2 and α7 subunit mRNA are the most
abundantly expressed nAChR genes in the mammalian brain (Wada et al., 1989; Marks et al.,
1992; Hill et al., 1993; Seguela et al., 1993). α subunit genes as a group are expressed in most
regions of the brain, but each individual α subunit mRNA is expressed in a unique, although
partly overlapping, set of neuronal structures (Wada et al., 1989). The most predominant α
subunit, α4, is strongly expressed in a large number of brain areas, including the thalamus,
habenula, cerebral cortex and dopaminergic nuclei. The α4 subunit, together with the β2
subunit, accounts for more than 90% of high-affinity nicotine binding sites in the rat brain
(Whiting and Lindstrom, 1986; Wada et al., 1989; Flores et al., 1992). The α7 subunit mRNA

26
accounts for approximately 90% of [125I]α-Bgt binding sites in the brain (Clarke et al., 1985b;
Couturier et al., 1990a; Schoepfer et al., 1990; Seguela et al., 1993). α7 subunits are
distributed throughout the brain with dense localisations in cortical and limbic areas (Orr-
Urtreger et al., 1997). In contrast to α7, the α2, α3 and α5 subunit mRNA are found in limited
areas of the mammalian brain (Wada et al., 1990; Lena and Changeux, 1997a). α2 mRNA is
expressed in the interpeduncular nucleus (IPN), hippocampal formations and olfactory bulb.
α3 and α5 mRNA are expressed in dopaminergic nuclei and cortical, hippocampal and
thalamic areas. Lastly, α6 mRNA, which is often shown to colocalise with β3 mRNA, has
been found in limited regions of the rat brain, including the SN, VTA and locus coeruleus (Le
Novere et al., 1996; Lena and Changeux, 1997a).
The β2 subunit has been shown by both mRNA and protein level, to be widely expressed
throughout the brain, largely, but not exclusively in combination with the α4 subunit (Hill et
al., 1993; Lena and Changeux, 1997a). Their distribution parallels high-affinity nicotine
binding sites in the brain, being strong in the thalamus, intermediate in the striatum and
cortex, and weak in the hypothalamus (Clarke et al., 1985b). β3 and β4 subunit mRNA are
expressed in a limited number of brain areas, such as the habenula, IPN and locus coeruleus
(Lena and Changeux, 1997a). β3 subunits are also expressed in dopaminergic nuclei.
Several radiolabelled nAChR ligands, including agonists such as ACh, nicotine, and more
recently epibatidine and cytisine, as well as antagonists such as α-Bgt, are used to study the
distribution of nAChRs (Clarke et al., 1985b; Marks et al., 1986, 1998). The distribution of
high-affinity binding sites, measured by nAChR agonist binding, differ from that of low-
affinity ([125I]α-Bgt) binding sites, because these ligands apparently bind to different nAChR
subunits (α4β2 vs. α7)(Clarke et al., 1985b; Schoepfer et al., 1990; Flores et al., 1992;
Seguela et al., 1993). Distribution of [3H]nicotine binding is generally consistent with that of
[3H]ACh, [3H]epibatidine or [3H]cytisine, but [3H]epibatidine can also detect nAChRs that
have lower a affinity for nicotine and are not labelled by [3H]nicotine or [3H]ACh (Perry and
Perry, 1995; Marks et al., 1998; Zoli et al., 1998). [3H]nicotine binding sites are distributed
throughout the mammalian brain, with highest levels in the thalamus and nucleus basalis of
Meynert, and lower levels in the IPN, hippocampus, CPu, cortex and superior colliculus
(Clarke et al., 1985b; Cimino et al., 1992; Gotti et al., 1997a; Paterson and Nordberg, 2000;
Han et al., 2003). In comparison with [3H]nicotine binding, [125I]α-Bgt binding is lower in

27
thalamic areas and higher in the hippocampus (Clarke et al., 1985b; Cimino et al., 1992; Han
et al., 2003). [125I]α-Bgt binding is dense in sympathetic ganglia, hypothalamus, cerebellum,
cortex and superior colliculus.
In conclusion, nAChRs seem to be located in brain areas that receive cholinergic innervation,
as measured by immunohistochemistry for ACh-synthesizing enzyme, choline
acetyltransferase (Woolf, 1991). The striatal complex, including the CPu and NAc contains
cholinergic interneurons. Cholinergic neurons of the basal forebrain project to the
hippocampus, limbic cortex, amygdala, olfactory bulbs and neocortex, while those of the
hypothalamic areas and medial habenula project to the IPN. In addition, the thalamus, globus
pallidus, SN, locus coeruleus and raphe nuclei receive cholinergic innervation. The
cholinergic transmission mediated by nAChRs is best characterised in the brain in
dopaminergic nuclei, cerebral cortex, medial habenula and IPN (Clarke, 1993b).
2.6.3. Activation and desensitization of nAChRs
Nicotine and other nAChR agonists can activate and desensitize nAChRs. When a nAChR is
activated upon agonist binding, it rapidly undergoes an allosteric transition from a closed,
resting conformation to an open channel that allows a flow of ions through it and may lead to
depolarisation of the neuron (Fig. 2-3c). Instead of activation, nAChRs can also become
desensitized in response to prolonged exposure to agonists, which has been shown in vitro
(Ogden and Colquhoun, 1985; Bertrand et al., 1990) and in vivo (James et al., 1994). The
desensitization of nAChRs was first described following ACh application in the
neuromuscular junction of the frog by Katz and Thesleff (1957). They introduced a cyclic
model postulating interconvertible high- and low-affinity agonist binding states to explain
desensitization following exposure to non-stimulating and stimulating concentrations of
agonists. Their model consists of two paths, each leading to the nAChR in the desensitized
state. The first path shows receptor activation, but prolonged presence of an agonist induces
ion channel closure and the transition of the nAChR to its desensitized confirmation, which is
refractory to activation, but displays higher affinity for agonists. The second path to
desensitization occurs without significant nAChR activation at low agonist concentrations and
when an agonist binds to an unbound, desensitized nAChR. This is possible because the

28
desensitized form of the nAChR displays higher affinity for agonists than the ground state
form of the receptor and because the unbound ground state and desensitized state are
interconvertible. Therefore, exposure to non-stimulating or stimulating concentrations of
agonists may result in bound desensitized receptors that are refractory to activation.
The cyclic model proposed by Katz and Thesleff was later modified into a more complex
model that includes two ligand bound states (Dilger and Liu, 1992), an inactivated state in
addition to resting, activated and desensitized states (Changeux et al., 1990; Lena and
Changeux, 1993) and multiple desensitized states of nAChRs (Changeux and Edelstein,
1998). Desensitization is characterised by a rapid onset and a quick and fully reversible loss
of nAChR function. As the exposure to an agonist ends, the agonist dissociates from a
receptor and the desensitized form returns to a form that is can be activated (Lena and
Changeux, 1998). The longer-lasting loss of nAChR function (Fig. 2-3c; inactive state) has
been called “specific chronic desensitization” (Ochoa et al., 1990; Collins et al., 1994),
“functional down-regulation” (Marks et al., 1993), “long-lasting inactivation” (Kawai and
Berg, 2001) or “persistent inactivation” (Ke et al., 1998). The desensitized states of nAChRs
are thought to underlie some of the long-term addictive properties of nicotine (Marks et al.,
1983; Schwartz and Kellar, 1985; Kawai and Berg, 2001; Buisson and Bertrand, 2002).
2.6.4. Up-regulation of nAChRs
Generally, chronic agonist administration elicits a decrease in the number of receptors, i.e.
downregulation, accompanied by tolerance to the effects of agonist. On the other hand,
chronic administration of antagonist produces an up-regulation of receptors and enhances
sensitivity to agonistic effects. However, chronic nicotine administration, such as smoking has
been found to increase the number of nAChRs in the human brain post mortem (Benwell et
al., 1988; Breese et al., 1997; Teaktong et al., 2004). Smoking during pregnancy alters the
expression of α4 and α7 subunits in the human fetal brain (Falk et al., 2005). Up-regulation of
nAChRs depends on the number of cigarettes smoked per day (Breese et al., 1997) and a two-
month abstinence from tobacco decreases a number of nAChRs to the levels found in non-
smoking subjects (Breese et al., 1997). Thus, up-regulation of nAChRs seems to be a
consequence of smoking rather than smoking being a consequence of higher number of

29
nAChRs. Smoking up-regulates nAChRs at diverse densities in different brain regions and
up-regulation is highest in the hippocampus, cortical areas and cerebellum, and lowest in the
striatum (Court et al., 1998; Perry et al., 1999). Acutely, tobacco smoke up-regulates α4*
nAChRs (asterisk represents unidentified subunits; Lukas et al., 1999) in axon terminals and
dendrites, as well as α7* nAChRs in perikarya, while chronically it down-regulates α7
expression on astrocytes (Teaktong et al., 2004).
Chronic nicotine administration, either by experimenter or by an animal itself, has been found
to dose-dependently increase the number of high-affinity binding sites (mainly α4β2 nAChRs)
in rodent brain (Marks et al., 1983; Schwartz and Kellar, 1983; Ksir et al., 1985; Schwartz
and Kellar, 1985; Flores et al., 1992, 1997; Pietilä et al., 1998; Nuutinen et al., 2005) and in
transfected cells (Peng et al., 1994a; Bencherif et al., 1995; Zhang et al., 1995). Up-regulation
of nAChRs is consistent with the localization of nAChR subtypes in the brain so that up-
regulation of high-affinity nicotine binding sites is highest in the thalamus, while low-affinity
nicotine binding sites up-regulate mainly in the hippocampus (Schwartz and Kellar, 1985; el-
Bizri and Clarke, 1994b). The functional consequences of nAChR up-regulation are not fully
resolved. In animals, up-regulation is accompanied by tolerance to some of nicotine’s effects,
for instance, on rotarod performance, heart rate and body temperature (Marks et al., 1983).
However, up-regulation is also associated with enhancement of some of the behavioural
effects of nicotine, for example, stimulation of locomotor activity (Ksir et al., 1985) and
responses of frontal cortical neurons to nicotine (Abdulla et al., 1995). Up-regulation is
thought to have implications for nicotine withdrawal, rather than the development of nicotine
addiction (Wonnacott et al., 2005).
Up-regulation has been suggested to result from the conversion of some of the low-affinity
nAChRs into high-affinity nAChRs (Romanelli et al., 1988), but also [125I]α-Bgt binding sites
(mainly α7 nAChRs) are up-regulated, although at higher nicotine concentrations and to a
lesser extent than the high-affinity nicotine binding sites (Marks et al., 1983; Pauly et al.,
1991; el-Bizri and Clarke, 1994b; Nuutinen et al., 2005). Studies in transfected cell lines have
shown that higher nicotine concentrations are needed to up-regulate α3* and α7* than α4β2
nAChRs (Peng et al., 1997; Fenster et al., 1999b). Differences in the regulation of nAChR
subtypes might be due to differences in subtype-specific sensitivities of active and
desensitized receptor states (Fenster et al., 1997; Ke et al., 1998; Wang et al., 1998). Up-

30
regulation of nAChRs following chronic nicotine treatment has been proposed to result from
receptor desensitization, resulting in nicotine as a functional antagonist (Wonnacott, 1990).
However, several studies imply that up-regulation requires neither receptor activation,
desensitization nor an ion flow through the ion channel. Up-regulation requires higher agonist
concentrations than those activating nAChRs and the same or even higher concentrations than
those that desensitize nAChRs (Rowell and Li, 1997; Whiteaker et al., 1998; Fenster et al.,
1999a). After prolonged exposure to nicotine most receptors are permanently unable to open
their ion channels in response to nicotine binding. Although several in vitro studies have
shown that up-regulation is related to loss of nAChR function, there is also evidence that up-
regulated nAChRs, such as α4β2*, α3β2*, and even more α7*, remain functional (Wang et
al., 1998; Buisson and Bertrand, 2001, 2002). Furthermore, an ion channel blocking
antagonist mecamylamine fails to inhibit the nicotine-elicited up-regulation of α3*, α7* and
α4β2* nAChRs in isolated cell lines (Peng et al., 1997) and can even up-regulate [3H]nicotine
binding sites itself in vitro (Peng et al., 1994a) and in vivo (Collins et al., 1994; Abdulla et al.,
1996; Pauly et al., 1996). In addition, other nAChR antagonists, such as the α4β2-selective
dihydro-β-erytroidine (DHβE) and the α7-selective methyllycaconitine (MLA), have been
found to up-regulate α4β2 nAChRs and MLA also up-regulates [125I]α-Bgt binding sites in
transfected cell lines (Molinari et al., 1998; Buisson and Bertrand, 2001). Mechanisms of
nAChR up-regulation do not appear to be related to increased transcription of nAChRs
(Marks et al., 1992; Ke et al., 1998), but rather to a decrease of the turnover of receptor
breakdown (Peng et al., 1994a; Zhang et al., 1995), an increased affinity of the receptors as a
result from their conformational change (Sallette et al., 2004) or an incorporation of
intracellular nAChRs into the cell membrane (Bencherif et al., 1995; Whiteaker et al., 1998).
2.6.5. Influence of subunits on functional properties of nAChRs
Functional properties of nAChRs, primarily permeability for cations and desensitization
properties, vary with nAChR subunit composition. Homomeric nAChRs that consist of either
α7, α8 or α9 subunits exhibit high Ca2+ permeability and they desensitize very rapidly in
response to high concentrations of nAChR agonists (Couturier et al., 1990b; Revah et al.,
1991; Seguela et al., 1993; Elgoyhen et al., 1994; Gerzanich et al., 1994). For example, the
relative permeability value of α7* nAChRs for Ca2+ and Na+ ions is 15 while that of subtypes

31
including α2 to α6 and β2 to β4 subunits ranges between 0.5 and 2.5 (Role, 1992; Role and
Berg, 1996). In heteromeric nAChRs, the α subunit is thought to regulate ligand binding and
determine the apparent agonist affinity of active and desensitized states of a nAChR (Cachelin
and Jaggi, 1991; Fenster et al., 1997). Also the β subunit may modulate the apparent affinity
for some nAChR subtypes, but it is mainly thought to modulate the overall time course of
desensitization of a nAChR and the rate at which agonists and antagonists dissociate from a
receptor, or ion channel opening (Cachelin and Jaggi, 1991; Luetje and Patrick, 1991; Fenster
et al., 1997). The α4* subtypes are more sensitive to desensitizing levels of nicotine and
recover more slowly from desensitization than the α3* subtypes (Fenster et al., 1997). The
β2* subtypes desensitize rapidly, but they also seem to recover faster than β4* subtypes
(Cachelin and Jaggi, 1991; Fenster et al., 1997). Presence of an α5 subunit in α3β2 or α3β4
nAChR complexes alters apparent sensitivity to nAChR agonists and increases Ca2+
permeability as well as the rate and magnitude of desensitization (Wang et al., 1996;
Gerzanich et al., 1998). The α6 subunit alters the properties of α3β2 nAChRs more than those
of α4β2 (Kuryatov et al., 2000).
nAChRs with different subunit compositions show significant pharmacological diversity. This
diversity includes subtype-specific sensitivity to agonists, but also the action elicited by a
ligand may vary, ranging from a full agonist to a partial agonist or even to an antagonist. For
example, nicotine’s affinity to nAChR subtypes varies. Nicotine shows highest affinity for
α4*, moderate for α3* and lowest for α7* subtypes (Table 2-2 on page 46; Fenster et al.,
1997). α7* subtypes are the only nAChRs activated by choline (Alkondon et al., 1997). The
β2 subunit exhibits higher potency for ACh and nicotine than β4 subunits (Gerzanich et al.,
1998) and nicotine is a partial agonist on α3β2 nAChRs, but a full agonist on α3β4 nAChRs
(Wang et al., 1996; Gerzanich et al., 1998). Cytisine is a full agonist on β4* subtypes, but a
potent inhibitor of ACh-induced currents in β2* subtypes (Papke and Heinemann, 1994). The
nAChR antagonist, α-conotoxin-MII (α-CtxMII) competitively inhibits α3β2 nAChRs, but not
α3β4 nAChRs (Cartier et al., 1996). An individual nAChR subtype may also possess different
properties in different brain areas (Wooltorton et al., 2003), which further complicates the
pharmacological characterization of these receptors. Moreover, the pharmacology of nAChRs
also varies between different species. For example, rat α3β4 is more sensitive to ACh than
α3β2 subtype, but the opposite situation exists in corresponding chick nAChR subtypes. 1,1-
dimethyl-4-phenylpiperazinium (DMPP) is a full agonist in rat and human α7 subtypes, but

32
only a partial agonist for chick α7 nAChR (Bertrand et al., 1992; Seguela et al., 1993; Peng et
al., 1994b).
2.7. Nicotinic modulation of dopamine release
Nicotine increases dopamine release in the terminal fields of nigrostriatal, mesolimbic and
mesocortical dopaminergic pathways, to a greater extent in the CPu and NAc and to a lesser
extent in the cerebral cortex and amygdala (Summers and Giacobini, 1995; Marshall et al.,
1997; Rowell, 2002). Nicotine has been reported to stimulate dopamine release in vitro in
rodent striatal slices (Westfall, 1974; Arqueros et al., 1978; Giorguieff-Chesselet et al., 1979;
Sacaan et al., 1995; Teng et al., 1997), accumbal slices (Rowell et al., 1987; Fung, 1989),
hypothalamic slices (Goodman, 1974) and in striatal and frontal cortical synaptosomes
(Sakurai et al., 1982; Rapier et al., 1988; Grady et al., 1992; el-Bizri and Clarke, 1994a;
Rowell, 1995; Whiteaker et al., 1995). In vivo, nicotine-induced dopamine release has been
indirectly shown by increased HVA and DOPAC levels in the dorsal striatum (Nose and
Takemoto, 1974; Lichtensteiger et al., 1976, 1982; Roth et al., 1982; Haikala et al., 1986) and
by increased DOPAC contents in limbic, primarily accumbal, regions (Grenhoff and
Svensson, 1988). In vivo microdialysis studies have shown that acute nicotine increases
extracellular levels of dopamine and its metabolites in the CPu and NAc (Imperato et al.,
1986; Damsma et al., 1988, 1989; Toth et al., 1992; Benwell and Balfour, 1997; Marshall et
al., 1997; Seppä and Ahtee, 2000). Similar to other drugs of abuse, nicotine acutely increases
dopamine overflow in the shell, but not in the core of the NAc (Pontieri et al., 1996; Nisell et
al., 1997; Di Chiara, 2000; Ferrari et al., 2002). Nicotine-induced elevation of dopamine and
its metabolites is inhibited both in vitro and in vivo by a brain penetrating nAChR antagonist
mecamylamine, but not by a peripheral nAChR antagonist hexamethonium (Roth et al., 1982;
Imperato et al., 1986). Nicotine-induced dopamine release is accompanied by stimulation of
dopaminergic cell firing. In electrophysiological studies, nicotine in vivo accelerates the firing
frequency and increases the amount of burst firing in dopaminergic cell bodies of the SN and
VTA (Lichtensteiger et al., 1976, 1982; Clarke et al., 1985a; Grenhoff et al., 1986; Calabresi
et al., 1989; Nisell et al., 1996).

33
Dopamine release induced by chronic nicotine administration is dependent upon the dosing
regimen, brain area studied, species and even conditioning during nicotine administration.
Prolonged exposure to nicotine at low non-activating or higher activating doses attenuates the
nicotine-induced release of dopamine from rat and mouse synaptosomes (Grady et al., 1994;
Rowell and Hillebrand, 1994). Continuous infusion of nicotine at larger doses, that maintain
plasma nicotine concentrations similar to those commonly found in smokers, or repeated
intrastriatal administration of nicotine, have been found to attenuate the acute elevating effect
of nicotine on extracellular levels of dopamine and its metabolites both in the CPu and NAc
(Benwell et al., 1994, 1995; Benwell and Balfour, 1997; Lecca et al., 2000). This tolerance to
nicotine’s acute effects is thought to be due to desensitization of nAChRs mediating effects on
dopamine release. However, repeated nicotine injections to rats have also failed to produce
tolerance to the nicotine-induced increase in the accumbal dopaminergic transmission
(Damsma et al., 1989; Nisell et al., 1996). Indeed, daily nicotine pretreatment has been also
found to enhance nicotine-elevated endogenous dopamine content, dopamine release and
dopamine formation from tyrosine in accumbal slices (Fung, 1989). Repeated, intermittent
administration of nicotine sensitizes the acute elevating effect of nicotine on dopamine release
in vitro in rat striatal slices (Yu and Wecker, 1994) and in vivo in the CPu and to a greater
extent in the NAc in rats (Benwell and Balfour, 1992; Reid et al., 1996; Marshall et al., 1997;
Shim et al., 2001). The subdivisions of the NAc, the shell and core, respond differently to
repeated, discontinuous nicotine exposure. In the core, nicotine-induced increase of dopamine
overflow is thought to become sensitized (Cadoni and Di Chiara, 2000), although another
study failed to show such sensitization (Nisell et al., 1997). In the shell, the dopamine
overflow that nicotine acutely increases is reduced after repeated nicotine administration
(Cadoni and Di Chiara, 2000; Di Chiara, 2000).
The sensitization of dopamine release evoked by daily nicotine injections has been suggested
to relate to a regionally selective downregulation of the control of mesoaccumbens
dopaminergic neurons by inhibitory autoreceptors and to depend upon co-stimulation of
NMDA-type glutamate receptors (Shoaib et al., 1994; Balfour et al., 1998; Ferrari et al.,
2002). Also up-regulation of nAChRs requires stimulation of NMDA receptors, suggesting
that it may relate to enhanced responsiveness to repeated nicotine (Shoaib et al., 1997). The
sensitized responses of the mesolimbic dopaminergic pathway elicited by nicotine as well as

34
other drugs of abuse are suggested to be critical for the mechanisms underlying development
of addiction to these drugs (Robinson and Berridge, 1993).
2.7.1. Different effects of nicotine on dopamine in the CPu and NAc
Nicotine stimulates the firing rate of dopaminergic neurons more effectively in the VTA than
in the SNc in locally anaesthesized rats (Mereu et al., 1987; Westfall et al., 1989), whereas in
generally anaesthesized rats the response to nicotine does not differ between these two brain
areas (Grenhoff et al., 1986; Mereu et al., 1987; Westfall et al., 1989). Nicotine-evoked
currents in dopaminergic neurons have been found to be of a larger amplitude in the VTA
than in the SN (Klink et al., 2001). Nicotine preferentially increases dopamine metabolism
and synthesis in limbic areas, compared with striatal areas (Clarke et al., 1988; Grenhoff and
Svensson, 1988). The nicotine-induced increase of dopamine overflow is comparatively
greater and occurs at lower doses in the NAc than in the CPu (Imperato et al., 1986; Benwell
and Balfour, 1997). Furthermore, the dose-response curves of dopamine overflow in response
to nicotine differ between the CPu and NAc. In the CPu, the dose-response curve for nicotine
is bell-shaped so that maximal elevation of dopamine overflow occurs at the 0.4 mg/kg dose
and increase in the dose attenuates the elevation of dopamine. In the NAc however, the
dopamine overflow is nearly maximally elevated already at the 0.1 mg/kg dose and increase
in the nicotine dose prolongs the elevation of dopamine (Benwell and Balfour, 1997). Also,
when nicotine is given repeatedly, desensitization of dopamine overflow in the CPu requires
higher doses of nicotine than is needed in the NAc (Benwell and Balfour, 1997). Thus, there
appears to be physiological and functional differences between the nigrostriatal and
mesolimbic dopaminergic pathways and the mechanisms mediating nicotine’s effects on the
nigrostriatal pathway differ from those of the mesolimbic pathway.
2.7.2. nAChR-mediated modulation of dopaminergic transmission
Despite the wide distribution of nAChRs, these receptors do not play a major role in direct
mediation of central synaptic transmission, but rather modulate release of several
neurotransmitters, including dopamine, GABA, glutamate, noradrenaline and ACh (Role and

35
Berg, 1996). nAChRs can be divided into somatodendritic, preterminal and presynaptic
receptors on the basis of their location in the neuron (Wonnacott, 1997). Stimulation of
somatodendritic or preterminal nAChRs results in Na+ influx and local depolarisation of a
neuron, sufficient to activate voltage-sensitive Ca2+ channels that, in turn, provide calcium for
exocytosis of transmitter, such as dopamine, into the synaptic cleft (Harsing et al., 1992;
Prince et al., 1996; Soliakov and Wonnacott, 1996). Somatodendritic nAChRs have little
influence in the modulation of transmitter release once it has been initiated, while presynaptic
nAChRs appear to function as synaptic or non-synaptic receptors and effectively modulate
chemical transmission at the release site (Vizi and Lendvai, 1999). Stimulation of presynaptic
nAChRs can either modulate transmitter release initiated by axonal firing or itself induce a
sufficient Na+ and Ca2+ influx to depolarise the neuron and release transmitter. Transmitter
release mediated by either somatodendritic or presynaptic nAChRs is Ca2+-dependent, but
while the modulation by somatodendritic nAChRs is sensitive to tetrodotoxin (TTX), the
modulation by presynaptic nAChRs is mostly insensitive to TTX.
On the nigrostriatal and mesolimbic dopaminergic neurons, nAChRs are located at cell bodies
in the SN and VTA, as well as on nerve terminals in the CPu and NAc (Clarke and Pert, 1985;
Clarke et al., 1985b). Local administration of nicotine into the SN increases the firing of
nigrostriatal dopaminergic neurons and concentrations of dopamine metabolites in the dorsal
striatum (Lichtensteiger et al., 1976, 1982). Nicotine administered into the VTA increases
extracellular level of dopamine in the NAc (Yoshida et al., 1993), and this increase in the
accumbal dopamine overflow is significantly greater when nicotine is given into the VTA
rather than into the NAc (Nisell et al., 1994a; Ferrari et al., 2002). Indeed, nicotine-evoked
accumbal dopamine release is thought to be predominantly mediated by somatodendritic
nAChRs, because when the nAChR antagonist mecamylamine, is infused into the VTA,
dopamine release is abolished, but this does not occur when mecamylamine is infused into the
NAc (Nisell et al., 1994b). In addition, dopamine release is inhibited by TTX in the VTA
(Benwell et al., 1993; Marshall et al., 1996). However, nicotine infused directly into the
terminal field (CPu or NAc) does elevate dopamine release from the nerve terminals
(Marshall et al., 1997) and this effect is blocked by prior administration of the α7-selective
antagonist, MLA (Alkondon et al., 1992; Lecca et al., 2000). Thus, different types of nAChRs
located in the SN/VTA and CPu/NAc appear to be required for the full range of nicotine’s
effects on the dopaminergic pathways.

36
2.7.3. Subtypes of presynaptic nAChRs involved in dopamine release
Although there is strong evidence for the nAChR-mediated modulation of transmitter release,
the lack of knowledge about subunit compositions of native nAChRs and the limited number
of subtype-selective tools has restricted our understanding of nAChR subtypes involved in
this modulation. Moreover, precise localization of nAChRs in relation to synapses remains to
be elucidated. The array of subunit mRNA expressed by neurons in the SN and VTA has been
shown to be α3, α4, α5, α6, α7, β2, β3 and β4 (Wada et al., 1990; Göldner et al., 1997; Sgard
et al., 1999). In detail, 80-100% of midbrain dopaminergic neurons appear to express α4, α5,
α6, β2, and β3 subunits, 40-60% express α3 and α7 subunits, and a few neurons express β4
subunits (Le Novere et al., 1996; Klink et al., 2001; Azam et al., 2002). Characterization of
presynaptic nAChRs on striatal dopaminergic terminals has been significantly aided by the
discovery of the α-conotoxins, particularly α-CtxMII, the selective α3β2* and α6* receptor
antagonist (Cartier et al., 1996; Mogg et al., 2002). Presynaptic nAChRs of the striatum can
be divided into α-CtxMII-sensitive and α-CtxMII-resistant nAChRs (Kulak et al., 1997;
Kaiser et al., 1998). Immunoprecipitation and 6-OHDA lesioning studies identified the α-
CtxMII-sensitive nAChRs as α6β2(β3) and α6α4β2(β3) subtypes and the α-CtxMII-
insensitive nAChRs as α4β2 and α4α5β2 subtypes (Fig. 2-5; Zoli et al., 2002).
Fig 2-5. The subunit composition of striatal presynaptic nAChRs that mediate dopamine release. The α-CtxMII-sensitive receptors have one (α4α6β2β3) or two (α6β2β3) α-CtxMII-sensitive ACh-binding sites. The α-CtxMII-resistant receptors (α4β2, α4α5β2) each have two α-CtxMII-resistant ACh-binding sites. Modified from Luetje (2004), with permission.

37
In regard to the function of presynaptic nAChRs, it is suggested that striatal dopamine release
is mediated to a great extent by α4* nAChRs and to a lesser extent by non-α4* nAChRs
(Grady et al., 1992; Kulak et al., 1997; Kaiser et al., 1998; Sharples et al., 2000). Agonist-
stimulated dopamine release is almost completely removed both in vivo and in vitro in knock-
out mice lacking either α4 or α4α6 subunits (Champtiaux et al., 2003; Marubio et al., 2003).
Furthermore, nAChRs mediating striatal dopamine release seem to contain β2 subunit,
because nicotine-stimulated dopamine release is suppressed in mice lacking this subunit in
vivo (Picciotto et al., 1998) and in vitro (Grady et al., 2001; Zhou et al., 2001; Champtiaux et
al., 2003). The non-α4 subtype was first identified as the α3β2 subtype when it was observed
that the α3β2 antagonists, α-CtxMII (Cartier et al., 1996) and neuronal bungarotoxin (Luetje
et al., 1990), substantially inhibited dopamine release from striatal synaptosomes and slices
(Grady et al., 1992; Kulak et al., 1997; Kaiser et al., 1998). Shortly after this discovery α-
CtxMII, neuronal bungarotoxin and α-CtxPnIa, the toxins previously characterized as α3β2-
specific antagonists, were found to also block α6* nAChRs expressed in oocytes (Kuryatov et
al., 2000; Dowell et al., 2003; Everhart et al., 2003). Furthermore, both α-CtxMII binding in
dopaminergic nuclei and α-CtxMII-sensitive nicotine-stimulated [3H]dopamine release, were
abolished by deletion of the α6 (Whiteaker et al., 2002) or β3 subunit (Cui et al., 2003), but
not by deletion of the α3 subunit in knock-out mice (Champtiaux et al., 2002). The α6 and β3
subunits are often coexpressed with β2 in the SN and VTA (Le Novere et al., 1996), which
further supports the idea that the α-CtxMII-sensitive component of nicotine-induced
dopamine release is controlled by these subunits. Consistent with the localization of nAChR
subtypes in striatal dopaminergic terminals (Fig. 2-5; Zoli et al., 2002), the α-CtxMII-
sensitive dopamine release was recently suggested to be mediated by α6β2β3 and α4α6β2β3
subtypes and the α-CtxMII-resistant dopamine release by α4β2 and α4α5β2 subtypes
(Salminen et al., 2004). Thus, β2 is present in all these receptors, α4 in all resistant and some
sensitive receptors, and β3 in most or all sensitive, but no resistant receptors.
The α7 and β4 subunits are not significantly involved in the function of nAChRs on the
presynaptic terminals of dopaminergic projections to the striatum. Neither deletion of the β4
subunit in knock-out mice (Salminen et al., 2004) nor use of the α3β4 selective blocker, α-
CtxAuIB (Luo et al., 1998), altered agonist-stimulated dopamine release from synaptosomes
(Grady et al., 2001). Furthermore, β4 subunits are only expressed at low levels in the SN
(Klink et al., 2001). Both deletion of the α7 subunit (Salminen et al., 2004) and use of the α7
selective blockers, α-Bgt and α-CtxImI (Johnson et al., 1995; Pereira et al., 1996), failed to

38
suppress agonist-stimulated dopamine release in synaptosomal preparations (Rapier et al.,
1990; Kulak et al., 1997). However, synaptosomes represent isolated nerve terminals and
modulation of transmitter release reflects only direct effects via presynaptic receptors. On the
other hand, slice preparations allow the cross talk between nerve terminals that is present in
vivo to be observed. In striatal slices an additional component of dopamine release has been
observed. Dopamine release in striatal slices can be blocked by the α7-selective antagonists,
α-Bgt, α-CtxImI and MLA and also by glutamate receptor antagonists (Kaiser and Wonnacott,
2000; Wonnacott et al., 2000). Furthermore, in vivo infusion of MLA or NMDA-type
glutamate receptor antagonists into the VTA abolishes nicotine-induced accumbal dopamine
overflow, implying that this dopamine release is also indirectly controlled by presynaptic α7*
nAChRs, as well as by NMDA receptors located in dopaminergic nuclei (Schilström et al.,
1998a, b). While activation of nAChRs located in the VTA is required for the accumbal
dopamine release induced by systemic nicotine, nAChRs located in the NAc modulate this
response (Fu et al., 2000a). This fine tuning of the mesolimbic dopaminergic pathway through
α7* nAChRs in the NAc may amplify the release of dopamine, allowing a subthreshold brain
concentration of nicotine to become an effective stimulus for dopamine release. Furthermore,
α7 nAChRs are preferentially expressed in the VTA, compared with the SN (Wooltorton et
al., 2003), suggesting that these nAChRs might preferentially modulate dopaminergic
transmission in the mesolimbic pathway.
2.7.4. Modulation of nicotine-induced dopamine release by other neuro-transmitters, glutamate and GABA
The effects of nicotine on dopaminergic transmission are modulated by several other
neurotransmitters, including glutamate and GABA. Intra-striatally administered nicotine
elevates extracellular levels of both dopamine and glutamate and blocking glutamate receptors
diminishes nicotine-induced dopamine overflow in the striatum (Toth et al., 1992, 1993).
Both glutamate and α7* nAChRs seem to be involved in nicotine-induced dopamine release
in the NAc, because the release of dopamine is attenuated by intrategmental administration of
NMDA or α7* nAChR antagonists (Schilström et al., 1998a, b; Svensson et al., 1998; Fu et
al., 2000a, b; Grillner and Svensson, 2000). Indeed, glutamatergic neurotransmission and α7*
nAChRs are thought to amplify nicotine’s effects on dopamine secretion particularly in the
reward-mediating mesolimbic pathway. The α7* receptors can be localised to glutamatergic

39
nerve terminals (Fig. 2-3b on page 18; Marchi et al., 2002) and are thought to evoke
glutamate release, which, in turn, acts on glutamate receptors located on dopaminergic
terminals to stimulate dopamine release (Kaiser and Wonnacott, 2000; Nomikos et al., 2000).
However, in vivo glutamate release may be dependent on the nicotine dose. At low nicotine
doses, tonic activation of NMDA receptors in the VTA may be necessary for dopamine
release, while at higher doses the phasic release of glutamate in the VTA is involved in the
accumbal dopamine response (Fu et al., 2000b). There may also be somatodendritic or
postsynaptic α7* nAChRs that modulate glutamatergic transmission (Alkondon et al., 1996b;
Fisher and Dani, 2000).
Pharmacological stimulation of GABA receptors inhibits nicotine-induced dopamine release
in the NAc (Dewey et al., 1999; Schiffer et al., 2000), more precisely, in the NAc shell (Fadda
et al., 2003). In vitro, nicotine concentration-dependently stimulates GABA release in
synaptosomes, particularly at concentrations also found in smokers (Wonnacott et al., 1989;
Lu et al., 1998) and in slices prepared from rabbit caudate nucleus (Limberger et al., 1986),
rat IPN (Lena et al., 1993), rat hippocampus (Alkondon et al., 1997; Chiodini et al., 1999), rat
SN and globus pallidus (Kayadjanian et al., 1994) and mouse thalamus (Lena and Changeux,
1997b). It is thought that higher concentrations of nicotine fail to release GABA due to
desensitization of nAChRs (Chiodini et al., 1999). Nicotine also promotes the release of
GABA in vivo (Yang et al., 1996; Bertolino et al., 1997). It is proposed that nicotine directly
stimulates release of GABA via activation of nAChRs located on GABAergic neurons (Fig.
2-3 on page 18; Wonnacott et al., 1989; Lena et al., 1993; Alkondon et al., 1996a; Lena and
Changeux, 1997b). However, nicotine may also release other neurotransmitters, such as
dopamine and 5-HT, which indirectly modulate nicotine-stimulated GABA release in vivo
(Kayadjanian et al., 1994; Bianchi et al., 1995; Neal et al., 2001). The nAChRs that mediate
nicotine’s effects on GABA release are thought to be located at a preterminal site on
GABAergic axons (Lena et al., 1993). Expression of α2, α4, β2 or β3 subunits are not affected
by 6-OHDA lesioning of dopaminergic neurons, which suggests that these subunits may
preferentially be located on non-dopaminergic neurons including GABAergic neurons or
interneurons in the SN and VTA (Fig. 2-3; Göldner et al., 1997; Charpantier et al., 1998;
Kaiser et al., 1998; Sgard et al., 1999; Zoli et al., 2002). Recently, α7 and β2 nAChR mRNAs
have been found in the majority of GABAergic neurons (Fig. 2-3; Azam et al., 2003). Indeed,
the effects of nicotine on GABA release appear to be mediated via both α7* and α4β2*

40
receptors (Alkondon et al., 1997; Lena and Changeux, 1997b; Whiteaker et al., 2000;
Alkondon and Albuquerque, 2001). Consistent with this, deletion of the β2 subunit in knock-
out mice eliminates nicotine-induced GABA release (Lu et al., 1998).
Glutamate and GABA might offer an explanation for the controversial finding that a single
nicotine exposure is sufficient to increase dopamine level in the mesolimbic reward system
for hours (Nisell et al., 1994a; Ferrari et al., 2002), although nicotine concentrations
experienced by smokers desensitize nAChRs on dopaminergic neurons in seconds, or perhaps
minutes (Wonnacott et al., 1990; Alkondon et al., 2000; Mansvelder et al., 2002). Nicotine at
low concentrations increases the firing rate of dopaminergic neurons and transiently enhances
GABAergic transmission (Fisher et al., 1998) before these inhibitory GABAergic inputs are
persistently inhibited by nAChR desensitization (Mansvelder et al., 2002). The non-
dopaminergic, probably GABAergic neurons, in the VTA have been found to respond
robustly to nicotine at non-activating concentrations, but to desensitize (Lu et al., 1999; Yin
and French, 2000; Pidoplichko et al., 2004). Simultaneously, nicotine enhances glutamatergic
transmission via nAChRs that have a slower desensitization rate than than those mediating
GABA release. The released glutamate activates NMDA receptors and induces long-term
potentiation of excitatory inputs in the reward-related mesolimbic pathway (Mansvelder and
McGehee, 2000). Hence, spatial and temporal differences in nAChR activity on both
excitatory and inhibitory neurons in reward areas might coordinate to reinforce nicotine
reward and self-administration (Mansvelder et al., 2003).
2.8. Nicotine – a substance of abuse
The terms drug addiction or drug dependence are usually described as a compulsion to take a
drug with a loss of control over apparently voluntary acts of drug seeking and drug taking
(Wise and Bozarth, 1987). Addiction is a chronic disorder since even after treatment and
extended periods of drug abstinence, the risk of relapse to active drug use remains high.
Craving, intensely wanting drugs, is fundamental to addiction (Robinson and Berridge, 1993).
A drug that is able to support drug-seeking behaviour is a reinforcer (Stolerman and Shoaib,
1991). Phenomena that are pivotal to dependence-producing drugs are tolerance and
sensitization. Tolerance (attenuation of acute effects, such as aversive effects of a drug) and

41
sensitization (progressive increase in an acute effect with repeated treatment) may be
important for the dependence-producing properties. Withdrawal signs and symptoms result
from physiological adaptations induced by chronic drug exposure and are relieved when the
drug is used again.
Nicotine is known to play an essential role in maintaining a tobacco smoking habit and many
habitual smokers become dependent upon the drug (US Department of Health and Human
Sciences, 1988; Stolerman and Shoaib, 1991; Benowitz, 1996). Addiction to nicotine depends
on the rate and route of nicotine intake. Inhalation of nicotine via cigarette smoke appears to
be the most addictive method of intake (Benowitz, 1996). After inhalation, nicotine passes
rapidly to the arterial bloodstream and into the brain within 10-19 s (Russell et al., 1978). This
exposure results in relatively intense CNS effects that occur in close temporal proximity to
smoking, an ideal situation for behavioural reinforcement (Benowitz, 1996). Smoking is
characterized by intermittent peak concentrations of nicotine with each cigarette smoked,
followed by a low, steady-state baseline level until the next cigarette is smoked (Benowitz et
al., 1990; Clarke, 1993a). The level of nicotine in venous blood after smoking a single
cigarette ranges from 5 to 30 ng/ml depending upon how the cigarette is smoked (Herning et
al., 1983). During the day, the baseline levels of nicotine rise so that the influence of peak
levels becomes less important (Benowitz et al., 1990). In the afternoon, blood or plasma
concentrations of smokers generally range from 10 to 50 ng/ml and if a smoker smokes until
bedtime, measurable nicotine levels persist all night. The prolonged exposure to nicotine leads
to desensitization of nAChRs, which is though to explain tachyphylaxis (short-term tolerance
of rapid onset) and perhaps tolerance to nicotine associated with up-regulation of nAChRs in
the brain of a smoker (Benwell et al., 1988). Decline in the brain nicotine level between
cigarettes and especially overnight, provide an opportunity for nAChRs to return from a
desensitized state to a resting state, so positive reinforcement can, to some extent, occur with
successive cigarettes, despite the development of tolerance (Clarke, 1993a; Benowitz, 1996).
In smokers, nicotine self-administration appears to be motivated by both positive and negative
reinforcement (Benowitz, 1996; Koob, 1996). Positive reinforcing effects include relaxation,
reduced stress, enhanced vigilance, improved cognitive function, mood modulation and lower
body weight. Negative reinforcement refers to the relief of nicotine withdrawal symptoms,
such as nervousness, restlessness, irritability, anxiety, impaired concentration and cognitive

42
function. In addition to its reinforcing properties, nicotine is particularly effective in
establishing or magnifying the incentive properties and reinforcing the effects of associated,
non-pharmacological stimuli, such as the predictive visual and auditory cues typically found
in animal self-administration paradigms, or the taste and smell of tobacco smoke for smokers
(Rose and Corrigall, 1997; Balfour et al., 2000; Perkins, 2002).
2.8.1. Rewarding and reinforcing effects of nicotine in animal models
In habitual smokers, nicotine acts as a reinforcer and it is self-administered by volunteers
deprived of cigarettes (Henningfield and Goldberg, 1983). In rats, nicotine can act as a
discriminative stimulus (cue) so that rats can differentiate the effects of nicotine from those of
saline in order to obtain a food reinforcement (Rosecrans and Meltzer, 1981). Nicotine has
been found to effectively reinforce operant responding at doses comparable to those taken in
by human smokers in a self-administration model in several species, such as squirrel monkeys
(Goldberg et al., 1981), mice (Picciotto et al., 1998; Epping-Jordan et al., 1999) and rats
(Corrigall and Coen, 1989; Corrigall et al., 1992; Donny et al., 1995, 1998; Corrigall, 1999;
Donny et al., 1999). In self-administration studies in rats, the dose-response curve for nicotine
is bell-shaped, with the maximal response occurring at the dose range 30-60 µg/injection
(Corrigall and Coen, 1989). The lack of self-administration at higher nicotine doses may be
due to either aversive effects produced by these doses or desensitization of nAChRs
mediating the rewarding effects of nicotine (Benwell et al., 1995).
In the place conditioning paradigm, the primary motivational properties of a drug or non-drug
treatment serve as an unconditioned stimulus that is repeatedly paired with neutral
environmental stimuli (Tzschentke, 1998). During the conditioning the neutral stimuli
acquires motivational properties and may thus act as conditioned stimuli and elicit approach
(or withdrawal, if the primary motivational properties of the treatment were aversive) when
the animal is subsequently exposed to these stimuli. Nicotine has also been reported to
produce conditioned place preference in rats (Fudala et al., 1985; Fudala and Iwamoto, 1986;
Acquas et al., 1989; Carboni et al., 1989a; Calcagnetti and Schechter, 1994; Vastola et al.,
2002; Le Foll and Goldberg, 2005). However, the opposite results have also been obtained,
suggesting that nicotine-induced conditioned place preference is effected by the experimental

43
paradigm used (Clarke and Fibiger, 1987; Fudala and Iwamoto, 1987; Jorenby et al., 1990;
Calcagnetti and Schechter, 1994; Le Foll and Goldberg, 2005).
Similar to other drugs of abuse, the reinforcing and rewarding effects of nicotine appear to
depend upon direct and/or indirect stimulation of the mesolimbic dopaminergic pathway,
because lesions or pharmacological blockade of these neurons attenuate self-administration
(Singer et al., 1982; Corrigall and Coen, 1991; Corrigall et al., 1992). The pattern of increased
immediate-early gene expression exhibited in the terminal fields of the mesolimbic
dopaminergic neurons is similar for cocaine and nicotine self-administration in rats (Pagliusi
et al., 1996; Pich et al., 1997). Nicotine’s reinforcing effects seem to be mediated by nAChRs
located on dopaminergic neurons in the VTA, because they are diminished by intra-
ventricular administration of a nAChR antagonist chlorisondamine (Corrigall et al., 1992) or
by intrategmental administration of the nAChR antagonist DHβE (Corrigall et al., 1994).
These nAChR subtypes are likely to contain β2 subunit, because mice lacking this subunit
will not lever-press for nicotine, but they do lever-press for cocaine and food (Picciotto et al.,
1998; Epping-Jordan et al., 1999).
Additional neuronal connections to the VTA are thought to be important in drug reward.
Nicotine injected into the TPP supports place preference conditioning (Iwamoto, 1990) and
cholinergic lesions of the TPP or DHβE infused into this nucleus attenuate nicotine self-
administration (Lança et al., 2000b). Also GABA receptor agonists administered into the
VTA or TPP attenuate nicotine self-administration in rats (Corrigall et al., 2000, 2001; Fattore
et al., 2002; Markou et al., 2004; reviewed by Cousins et al., 2002). Furthermore, systemic
nicotine has been found to increase expression of immediate-early genes in the TPP and LDT
and this effect is possibly mediated via nAChRs located in non-cholinergic, presumably
GABAergic neurons that modulate the activity of cholinergic cells in the TPP, which, in turn,
regulates dopamine release in the mesolimbic system (Lança et al., 2000a).
2.8.2. The effects of nicotine on locomotor activity in rats
In man, nicotine is considered to have both depressant and stimulant actions (Gilbert, 1979).
In rats also, acute nicotine can induce either depressant or stimulant effects on locomotor

44
activity. The depressant effect occurs particularly in experimentally naïve rats at high nicotine
doses (Stolerman et al., 1973; Clarke and Kumar, 1983b). Depression of activity is readily
followed by stimulation as rats habituate to the test environment (Morrison and Stephenson,
1972; Clarke and Kumar, 1983a, b) and furthermore, if the rats have been previously
habituated to test apparatus, nicotine preferentially stimulates activity (O'Neill et al., 1991;
Benwell and Balfour, 1992). Repeated, intermittent administration of nicotine rapidly
produces tolerance to depressant effects and dose-dependently enhances stimulant effects, i.e.
produces behavioural sensitization (Morrison and Stephenson, 1972; Stolerman et al., 1973;
Bättig et al., 1976; Clarke and Kumar, 1983b; Ksir et al., 1985; Benwell and Balfour, 1992;
Whiteaker et al., 1995; Nisell et al., 1996). The sensitized locomotor response has been found
to be accompanied by the up-regulation of brain nAChRs (Ksir et al., 1985; Marks et al.,
1985). However, when nicotine is infused continuously, the stimulant effect is attenuated
(Benwell et al., 1994, 1995), which is suggested to be a consequence of down-regulation of
nAChR function (Marks et al., 1993).
The mesolimbic dopaminergic pathway is thought to play a pivotal role in mediating the
stimulant effects of nicotine, because lesions or pharmacological blockade of these neurons
attenuates the stimulation of locomotor activity (Clarke et al., 1988; Corrigall and Coen,
1991; Louis and Clarke, 1998). Stimulation of GABA receptors in the NAc reduces activity,
possibly by inhibiting accumbal dopamine release (Wachtel and Andén, 1978), and inhibits
hyperactivity induced by intra-accumbal injection of dopamine or amphetamine (Pycock and
Horton, 1979). Systemic nicotine elevates accumbal dopamine release at doses that stimulate
locomotion (Imperato et al., 1986; Clarke et al., 1988; Di Chiara and Imperato, 1988).
Repeated, intermittent administration of nicotine that results in the sensitized locomotor
stimulant response, also causes sensitization of its effects on dopamine overflow in the NAc
(Benwell and Balfour, 1992; Reid et al., 1996) and the prefrontal cortex (Nisell et al., 1996)
and possibly also in the CPu (Shim et al., 2001). Behavioural sensitization to nicotine is
associated with a reduction in dopamine overflow in the NAc shell and with an increase in
dopamine overflow in the core (Cadoni and Di Chiara, 2000).
Like nicotine’s actions on accumbal dopamine release (Nisell et al., 1994a; Ferrari et al.,
2002), the locomotor stimulant effect of nicotine appears to be primarily mediated by its
binding to nAChRs in the somatodendritic region (VTA), rather than in the terminal area

45
(NAc)(Museo and Wise, 1990; Reavill and Stolerman, 1990; Leikola-Pelho and Jackson,
1992; Panagis et al., 1996; Ferrari et al., 2002). However, nicotine’s actions on the
mesolimbic dopaminergic terminal fields in the NAc probably modulate its effects on
locomotor activity (Welzl et al., 1990). The α6* nAChR appears to mediate nicotine’s
stimulant effects on locomotor activity, because an infusion of α6 antisense oligonucleotides
reduces the effect (Le Novere et al., 1999). DHβE inhibits the locomotor stimulant effect of
nicotine at doses that do not antagonize its locomotor depressant effects, which suggests that
these two effects are mediated via different mechanisms (Stolerman et al., 1997).
Furthermore, deletion of the β2 subunit reduces the locomotor activity of habituated mice, but
not the exploratory activity of naïve mice (Picciotto et al., 1998).
2.9. Epibatidine
Epibatidine is an alkaloid originally purified from the skin of the Ecuadorian, poisonous frog
Epipedobates tricolor (Spande et al., 1992). It is a potent nAChR agonist that has proved to
be 80 to 2000 times more potent in preventing nociception than morphine (Badio and Daly,
1994; Sullivan et al., 1994). Epibatidine-induced analgesia is not blocked by the opioid
receptor antagonist naloxone, but it is abolished by mecamylamine and slightly reduced by
DHβE, which suggests that the effect is mediated by nAChRs (Badio and Daly, 1994; Damaj
et al., 1994; Bannon et al., 1995), particularly α4β2 nAChRs (Marubio et al., 1999). Indeed,
epibatidine displaces [3H]cytisine and [3H]nicotine binding in rat brain preparations, but fails
to displace ligands for several other CNS receptors, including opioid, muscarinic, adrenergic,
dopamine, 5-HT and GABA receptors (Qian et al., 1993; Badio and Daly, 1994). It displays
higher affinity at heteromeric nAChRs than at homomeric nAChRs (Table 2-2; Alkondon and
Albuquerque, 1995; Gerzanich et al., 1995; Bertrand et al., 1999). In contrast to nicotine,
epibatidine desensitizes α4β2 nAChRs at concentrations that do not activate the receptor and
up-regulates α4β2 nAChRs to a lesser extent than nicotine (Whiteaker et al., 1998; Buisson et
al., 2000). It also attenuates nicotine-induced up-regulation of α4β2 nAChRs, suggesting that
it might function as a partial up-regulatory agonist (Whiteaker et al., 1998).
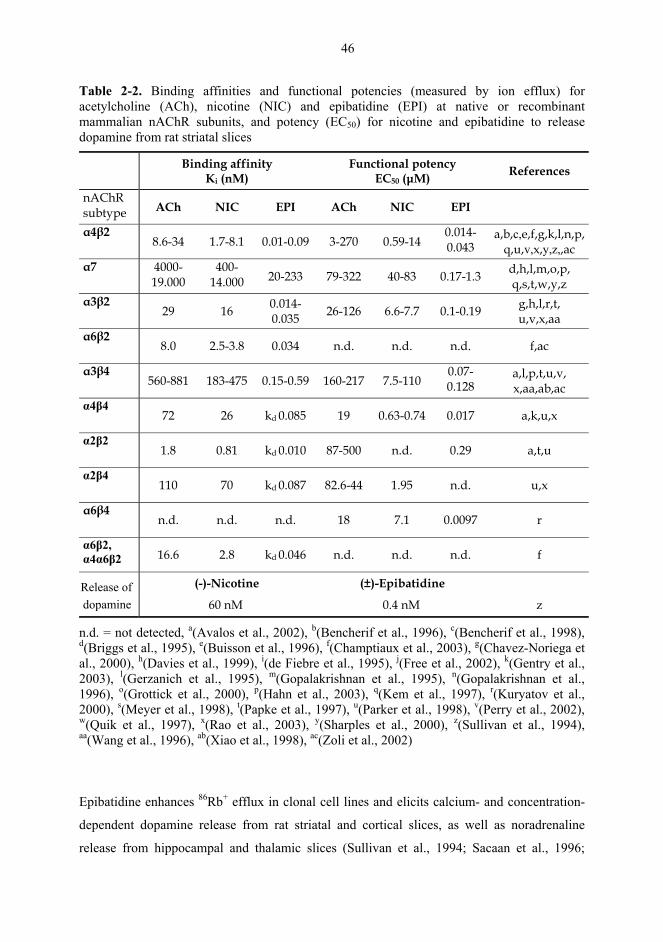
46
Table 2-2. Binding affinities and functional potencies (measured by ion efflux) for acetylcholine (ACh), nicotine (NIC) and epibatidine (EPI) at native or recombinant mammalian nAChR subunits, and potency (EC50) for nicotine and epibatidine to release dopamine from rat striatal slices
Binding affinity Ki (nM)
Functional potency EC50 (µM) References
nAChR subtype ACh NIC EPI ACh NIC EPI
α4β2 8.6-34 1.7-8.1 0.01-0.09 3-270 0.59-14
0.014-0.043
a,b,c,e,f,g,k,l,n,p, q,u,v,x,y,z,,ac
α7 4000-19.000
400-14.000
20-233 79-322 40-83 0.17-1.3 d,h,l,m,o,p, q,s,t,w,y,z
α3β2 29 16 0.014-
0.035 26-126 6.6-7.7 0.1-0.19 g,h,l,r,t, u,v,x,aa
α6β2 8.0 2.5-3.8 0.034 n.d. n.d. n.d. f,ac
α3β4 560-881 183-475 0.15-0.59 160-217 7.5-110
0.07-0.128
a,l,p,t,u,v, x,aa,ab,ac
α4β4 72 26 kd 0.085 19 0.63-0.74 0.017 a,k,u,x
α2β2 1.8 0.81 kd 0.010 87-500 n.d. 0.29 a,t,u
α2β4 110 70 kd 0.087 82.6-44 1.95 n.d. u,x
α6β4 n.d. n.d. n.d. 18 7.1 0.0097 r
α6β2, α4α6β2 16.6 2.8 kd 0.046 n.d. n.d. n.d. f
Release of (-)-Nicotine (±)-Epibatidine
dopamine 60 nM 0.4 nM z
n.d. = not detected, a(Avalos et al., 2002), b(Bencherif et al., 1996), c(Bencherif et al., 1998), d(Briggs et al., 1995), e(Buisson et al., 1996), f(Champtiaux et al., 2003), g(Chavez-Noriega et al., 2000), h(Davies et al., 1999), i(de Fiebre et al., 1995), j(Free et al., 2002), k(Gentry et al., 2003), l(Gerzanich et al., 1995), m(Gopalakrishnan et al., 1995), n(Gopalakrishnan et al., 1996), o(Grottick et al., 2000), p(Hahn et al., 2003), q(Kem et al., 1997), r(Kuryatov et al., 2000), s(Meyer et al., 1998), t(Papke et al., 1997), u(Parker et al., 1998), v(Perry et al., 2002), w(Quik et al., 1997), x(Rao et al., 2003), y(Sharples et al., 2000), z(Sullivan et al., 1994), aa(Wang et al., 1996), ab(Xiao et al., 1998), ac(Zoli et al., 2002)
Epibatidine enhances 86Rb+ efflux in clonal cell lines and elicits calcium- and concentration-
dependent dopamine release from rat striatal and cortical slices, as well as noradrenaline
release from hippocampal and thalamic slices (Sullivan et al., 1994; Sacaan et al., 1996;

47
Jacobs et al., 2002). Epibatidine-evoked currents are characterized by late onset and offset,
compared with those evoked by ACh (Gerzanich et al., 1995; Bertrand et al., 1999; Buisson et
al., 2000). Epibatidine is also more efficacious in enhancing 86Rb+ efflux and dopamine
release from striatal synaptosomes than nicotine (Sullivan et al., 1994; Reuben et al., 2000),
but appears to be less efficacious in enhancing dopamine release from dendrosomal
preparations of the SN/VTA (Reuben et al., 2000). In slice preparations, it has been found to
evoke striatal dopamine release 150-240-fold more potently and cortical dopamine release
400-fold more potently than nicotine (Table 2-2; Sullivan et al., 1994; Puttfarcken et al.,
2000). It is also 180-fold more potent and more efficacious in releasing GABA from mouse
synaptosomes (Lu et al., 1998). Unlike nicotine, in vivo epibatidine preferentially increases
the extracellular dopamine level in the nigrostriatal pathway, compared with the mesolimbic
pathway (Seppä and Ahtee, 2000). In fact, it may even decrease dopamine overflow in the
NAc and frontal cortex (Bednar et al., 2004). However, epibatidine increases dopamine
metabolism in these brain areas (Seppä and Ahtee, 2000; Bednar et al., 2004). Prior treatment
with repeated nicotine attenuates the epibatidine-induced decrease in dopamine overflow, but
not the increase in dopamine metabolism in the NAc (Bednar et al., 2004).
Epibatidine provokes nicotine-like responding in the discrimination test in rats with 100-fold
greater potency than nicotine (Damaj et al., 1994). In a similar way to nicotine, it improves
attentional performance, and response rate and speed in rats (Hahn et al., 2003). In contrast to
nicotine, epibatidine lacks reinforcing effects in a self-administration test in mice (Rasmussen
and Swedberg, 1998) and anxiolytic activity on a plusmaze test (Sullivan et al., 1994). It
decreases locomotor activity and body temperature in mice, and prevents nociception 300 to
1000 times more potently and somewhat more efficaciously than nicotine (Damaj et al., 1994;
Sullivan et al., 1994; Bannon et al., 1995; Bonhaus et al., 1995). Like nicotine, acute
epibatidine decreases locomotor activity in naïve, unhabituated rats, but increases the activity
after a delay in habituated rats (Sacaan et al., 1996; Menzaghi et al., 1997b; Reuben et al.,
2000). Epibatidine increases locomotor activity to a lesser extent than nicotine and the
maximal increase by it occurs at the 3.0 µg/kg dose and that by nicotine at the 0.4 mg/kg dose
(Clarke and Kumar, 1983a; Menzaghi et al., 1997b; Reuben et al., 2000). In addition, prior
repeated treatment with nicotine enhances the acute locomotor stimulant effect of nicotine,
but attenuates that of epibatidine (Reuben et al., 2000).

48
Apart from ACh and nicotine, no other agent has had such an impact on nAChR research as
has epibatidine had (Dukat and Glennon, 2003). Its picomolar affinity for α4β2 nAChR
subtype and potent antinociceptive properties have stimulated interest in developing novel
nicotinic agents and nonopioid analgesics. Nowadays, several nicotinic compounds inspired
by epibatidine and nicotine are under evaluation in preclinical and clinical trials as possible
new treatments for neurodegenerative and mood disorders (Lloyd and Williams, 2000; Dukat
and Glennon, 2003; Hogg and Bertrand, 2004).
2.10. Nicotinic compounds in Parkinson’s disease
Although it is well-documented that nicotine (the main component in tobacco) has many
harmful effects, it has also been shown to have a number of positive effects in relation to
Parkinson’s disease. Several epidemiological studies have shown that people who smoke or
who have smoked during their lifetime, are 50% less likely to develop Parkinson’s disease
than those who have never smoked (Baron, 1986; Morens et al., 1995; Gorell et al., 1999;
Fratiglioni and Wang, 2000; Allam et al., 2004; Scott et al., 2005; Wirdefeldt et al., 2005).
This negative association between smoking and Parkinson’s disease is reproducible, dose-
related and not a result of selective mortality. However, the risk of Parkinson’s disease among
smokers is increased by other factors, such as old age and family history of Parkinson’s
disease (Elbaz et al., 2000). The mechanisms responsible for this decline in the incidence of
Parkinson’s disease are not known and several compounds in tobacco are able to alter
biological processes. One explanation for this may be the alteration of metabolic enzyme
activity. For example, activities of MAO-A and B are decreased in the brains of smokers,
which may attenuate the enzymatic oxidation of dopamine and thereby reduce oxidative stress
in dopaminergic neurons, or block MAO-mediated toxic xenobiotics (Yong and Perry, 1986;
Fowler et al., 1996, 2003). Another explanation may be nicotine’s interaction with
dopaminergic systems. Indeed, dopaminergic activity in the caudate and putamen is elevated
in smokers, compared with non-smokers (Salokangas et al., 2000).
Nicotine has been shown to have neuroprotective effects in several in vitro primary culture
models, for example, in striatal, cortical, cerebellar and mesencephalic neurons against several
toxic insults, including NMDA (Akaike et al., 1994; Marin et al., 1994; Sullivan et al., 1997;

49
Meyer et al., 1998; Minana et al., 1998) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
(MPTP), a toxin that selectively destroys dopaminergic neurons (Quik and Jeyarasasingam,
2000; Jeyarasasingam et al., 2002). In vivo, chronic nicotine administration to rats protects
dopaminergic cell bodies in the SN and nerve terminals in the striatum from mechanical
lesioning (Janson et al., 1988; Fuxe et al., 1990; Janson et al., 1991; Janson and Moller,
1993), restores glucose utilization in lesioned areas (Owman et al., 1989), counteracts lesion-
induced dopamine receptor up-regulation (Janson et al., 1994) and increases the rate of
dopamine turnover (Fuxe et al., 1990). In addition, smoking and acute and chronic nicotine
treatment have been shown to protect the nigrostriatal dopaminergic neurons from MPTP or
6-hydroxydopamine (6-OHDA)(Carr and Rowell, 1990; Janson et al., 1992; Maggio et al.,
1998; Costa et al., 2001; Ryan et al., 2001; Parain et al., 2003). However, at high
desensitizing doses nicotine fails to protect neurons and may even increase MPTP-mediated
toxicity (Behmand and Harik, 1992; Janson et al., 1992; Hadjiconstantinou et al., 1994).
Studies into the mechanisms of the neuroprotective actions of nicotine (reviewed by Picciotto
and Zoli, 2002) suggest that it may exert its effects by inducing expression of neurotrophic
factors that are known to promote survival of dopaminergic neurons (Maggio et al., 1998;
Roceri et al., 2001). Epibatidine is also shown to up-regulate the expression of neurotrophic
factors (Belluardo et al., 2000). The α7* nAChR appears to be involved in cell survival and
apoptosis (Shimohama et al., 1996; Kaneko et al., 1997; Li et al., 1999). Consistent with this,
α7-selective nAChR agonists (Briggs et al., 1997) have been found to elicit neuroprotective
effects in cultured cells (Buccafusco et al., 2004; Marrero et al., 2004).
Nicotine may also play a role in alleviating the symptoms of Parkinson’s disease once it has
developed. Smoking/nicotine has been found to ameliorate parkinsonian motor symptoms,
including tremor, bradykinesia, rigidity and lack of energy (Moll, 1926; Marshall and
Schnieden, 1966; Ishikawa and Miyatake, 1993; Fagerström et al., 1994; Kelton et al., 2000).
Motor symptoms are reduced shortly after smoking a cigarette for 10-30 min and this
reduction is more prominent after smoking, rather than chewing nicotine gum (Ishikawa and
Miyatake, 1993; Fagerström et al., 1994). One of the least understood areas of Parkinson’s
disease are the cognitive deficits, such as problems of attention, planning and cognitive
flexibility, and these symptoms also respond least to current pharmacological treatments.
Levodopa has been shown to alleviate (Pillon et al., 1989), fail to effect (Schneider et al.,
1999) or even exacerbate some cognitive deficits (Gotham et al., 1988; Lange et al., 1995).

50
Furthermore, levodopa has been reported to reduce several nAChR subtypes in monkey
striatum at doses ameliorating parkinsonian symptoms in both humans and monkeys (Quik et
al., 2003). Nicotine is known to improve attention and cognitive and memory performances
both in healthy subjects (Warburton et al., 1992; Levin et al., 1998; Levin and Rezvani, 2000,
2002) and in young and old animals (Brioni and Arneric, 1993; Levin and Torry, 1996;
Zarrindast et al., 1996; Grilly et al., 2000; reviewed by Decker et al., 1995; Levin and Simon,
1998). Furthermore, nicotine improves cognitive performance in parkinsonian patients
(Fagerström et al., 1994; Kelton et al., 2000), and it is the hippocampal α4β2 nAChRs that
have been proposed to mediate nicotine’s effects on memory functioning (Bancroft and
Levin, 2000). Consistent with this, nicotine and several novel β2-selective nAChR agonists,
such as altinicline and its racemates (Cosford et al., 2000), have been found to improve
MPTP-induced cognitive deficits (Schneider et al., 1999) and reverse cognitive and motor
deficits in combination with the parkinsonian drugs, levodopa and benserazide, in reserpine-
treated rats (Menzaghi et al., 1997a), as well as in MPTP-treated monkeys (Schneider et al.,
1998a, b; Domino et al., 1999). These nAChR agonists also stimulate locomotor activity and
dopamine turnover in rats (Menzaghi et al., 1997b; Sacaan et al., 1997). Potentiation of
levodopa’s effects by nAChR agonists may allow patients to reduce their levodopa dose and
eliminate complications of long-term levodopa treatment (Schneider et al., 1998b).
Another important aspect of Parkinson’s disease with respect to nAChRs, is the loss of high-
affinity nicotine binding sites in the caudate, putamen and SN, as well as in the cortex and
hippocampus (Aubert et al., 1992; Lange et al., 1993; Perry et al., 1995; Court et al., 2000b;
Paterson and Nordberg, 2000; Guan et al., 2002). The loss of nAChRs in the parkinsonian
brain is accompanied by a loss of choline acetyltransferase, indicating reduced cholinergic
transmission preferentially in cortical areas as compared with striatal areas (Nordberg et al.,
1985; Rinne et al., 1991; Aubert et al., 1992; Lange et al., 1993; Perry et al., 1993).
Furthermore, the loss of nAChRs parallels a decline in dopaminergic neurons (Piggott et al.,
1999; Quik et al., 2004) and is not associated with an overall loss of synaptic density, as
measured with synaptophysin immunoreactivity (Martin-Ruiz et al., 2000), but a specific loss
of nigrostriatal dopaminergic neurons (Court et al., 2000b). To date, the exact nAChR
subtypes lost in the human brain in Parkinson’s disease patients are not fully resolved. Low-
and high-affinity α-CtxMII binding sites are known to be present in the striatum and they are
both decreased in Parkinson’s disease (Quik et al., 2004). Furthermore, expression of α4, α7

51
and β2 subunits in putamen appear unaltered, both in parkinsonian subjects (Martin-Ruiz et
al., 2000) and in monkeys with dopaminergic lesions (Quik et al., 2000, 2005), suggesting
that these subunits are preferentially located on non-dopaminergic neurons. However,
expression of α3* subtypes has been found to be reduced in the caudate and temporal cortex
in parkinsonian patients (Guan et al., 2002). There appear also to be reduced expression of
β2* subtypes in the hippocampus and cortex, but an increased expression of α7 subtypes in
the cortex (Guan et al., 2002).
In rodent and monkey brain, destruction of the SN neurons by 6-OHDA or MPTP leads to
reduction in number of high-affinity nicotine binding sites in the SN and striatum (Schwartz
et al., 1984; Clarke and Pert, 1985), particularly those including α3, α5, α6, β3 and β4
subunits (Charpantier et al., 1998; Elliott et al., 1998; Quik et al., 2001, 2002). Consistent
with this, these nAChR subunits are reported to be located on dopaminergic neurons in
dopaminergic nuclei (Le Novere et al., 1996; Klink et al., 2001; Azam et al., 2002; Zoli et al.,
2002). The α6 subunit also appears to play a key role in the mediation of locomotor activity
(Le Novere et al., 1999), which further supports the importance of this nAChR subunit in
Parkinson’s disease. The α7 subunit may also be important in Parkinson’s disease (Quik and
Kulak, 2002), because it is implicated in neuromodulation, synaptic plasticity and
development (Role and Berg, 1996; Wonnacott, 1997; Dani, 2001), as well as in
neuroprotection (Shimohama et al., 1996; Kaneko et al., 1997; Li et al., 1999). The α7 subunit
has also been suggested to be involved in the cognition enhancing activity of nicotine via
modulation of glutamate release in the cerebral cortex (Levin and Simon, 1998).
2.11. Nicotinic compounds in other CNS disorders
nAChRs in the brain are suggested to play a role in attention, memory, sensation and
perception. If not causal, the involvement of nAChR function is likely to be important in
several neurodegenerative and mood disorders. Different nAChR subtypes seem to be
selectively implicated in different brain diseases in which nAChRs are shown to be modified.
For example, autosomal nocturnal frontal lobe epilepsy is related to a mutation in the α4
subunit, which reduces α4β2 subtype function and may cause nAChR-mediated seizures via
GABAergic systems (Steinlein et al., 1995; Weiland et al., 1996; Kuryatov et al., 1997;

52
Bertrand et al., 1998; Dobelis et al., 2003; Fonck et al., 2003). Abnormalities in the size and
number of nAChRs have been found in the cortex and cerebellum of autistic patients (Perry et
al., 2001; Lee et al., 2002; Martin-Ruiz et al., 2004) and in the hippocampus of older
individuals with Down’s syndrome (Court et al., 2000a). The status of nAChRs in Tourette’s
syndrome is not clear, but nicotine delivered by gum or transdermal patch can provide short-
term relief and may prevent symptoms of this syndrome from being exacerbated. However,
the use of nicotine is limited by frequent toxicity, primarily nausea (Dursun and Kutcher,
1999). Nicotine also improves attention in attention deficit and hyperactivity disorder in
adults (Levin et al., 2001).
Even though schizophrenic subjects are consistently found to smoke tobacco, which is likely
to up-regulate nAChRs, they were actually found to have reduced levels of both high- (α4β2)
and low-affinity (α7) nAChRs in several brain regions, including the striatum, hippocampus
and frontal cortex (Freedman et al., 1995; Guan et al., 1999; Breese et al., 2000; Durany et al.,
2000). These decreases are unlikely to result from neuroleptic medication, but they seem to be
associated with a high incidence of smoking among schizophrenics (Glassman, 1993; Dalack
et al., 1998). In fact, smoking is suggested to be a form of self-medication and nicotine
appears to normalize the sensory gating deficit associated with schizophrenia (Adler et al.,
1998). This deficit in schizophrenia is linked to a mutation in the α7 subunit (Stevens et al.,
1996; Leonard et al., 2002) and thus α7 nAChR agonists may be reasonable candidates for the
treatment of cognitive and perceptual disturbances in schizophrenia (Martin et al., 2004).
Indeed, in a similar way to nicotine, a novel partial α7-selective nAChR agonist has been
found to normalize an auditory evoked-potential deficit in mice expressing decreased levels of
α7 subunits (Stevens et al., 1998). In addition to schizophrenia, the incidence of smoking is
higher in people with bipolar disorder (manic depression) or with depression, and smoking
cessation therapies are less successful among these patients (Glassman, 1993; McEvoy and
Allen, 2002). Smoking may be a form of self-medication in subgroups of depressed
individuals that show a genetic predisposition (Lerman et al., 1998) and nicotine is thought to
modulate stress-induced responses that occur when a subject is not smoking (Balfour and
Ridley, 2000).
The loss of cholinergic neurons appears to result in learning and memory deficits associated,
for example, with Alzheimer’s disease. Drugs that enhance cholinergic neurotransmission,

53
such as cholinesterase inhibitors are used to compensate these deficits. The number of both
low- and high-affinity nAChRs decline with age (Nordberg, 1994; Court et al., 1997; Marutle
et al., 1998), but in Alzheimer’s disease and in dementia with Lewy bodies, there are
selective, region specific decreases in high-affinity nicotine binding sites post mortem
(Nordberg et al., 1992; Warpman and Nordberg, 1995; Nordberg et al., 1997; Court et al.,
2000b; Rusted et al., 2000). For example, in Alzheimer’s disease expression of α4 and
possibly α3 subunits is reduced in the neocortex (Hellström-Lindahl et al., 1999; Martin-Ruiz
et al., 1999; Guan et al., 2000), while expression of α7 subunits may increase (Hellström-
Lindahl et al., 1999) or decrease in the hippocampus (Guan et al., 2000), but appear to remain
unaltered in the cerebral cortex (Sugaya et al., 1990). Changes in expression of nAChRs are
shown to differ at various stages of Alzheimer’s disease and this may lead to inconsistencies
between reports (reviewed by Court et al., 2000a).
In addition to a loss of cortical innervation and basal forebrain neurons (Cummings and
Benson, 1987), the pathology of Alzheimer's disease is characterised by β-amyloid plaques
and neurofibrillary tangles, and inhibition of β-amyloid accumulation has been proposed to be
essential for effective therapy of this disease (Picciotto and Zoli, 2002). Nicotine has been
found to reduce β-amyloid aggregation, possibly via α7 nAChRs in the mouse brain
(Nordberg et al., 2002; Hellström-Lindahl et al., 2004b) and to antagonize β-amyloid-induced
memory deficits in mice (Maurice et al., 1996). Recently, smoking was reported to decrease
the level of β-amyloid in patients with Alzheimer’s disease (Hellström-Lindahl et al., 2004a).
However, the results of epidemiological studies into the protective effects of smoking in
Alzheimer’s disease are less consistent than those in Parkinson’s disease (Fratiglioni and
Wang, 2000; Sabbagh et al., 2002). Nicotine, as well as nAChR agonists showing selectivity
for nAChRs in the CNS, more precisely those consisting of α4β2 or α7 subunits, have been
found to improve overall cognitive performance, including memory in rats (Lippiello et al.,
1996; Meyer et al., 1997; Bencherif et al., 2000; Hahn et al., 2003), in monkeys (Bontempi et
al., 2003; Schneider et al., 2003) and in humans (Jones et al., 1992; Levin and Simon, 1998;
Gatto et al., 2004; Newhouse et al., 2004). The novel nAChR agonists appear to have less
harmful effects than nicotine which indicates a potential use for these compounds as treatment
for Alzheimer's disease (Lloyd et al., 1998; Meyer et al., 1998; Vernier et al., 1998; Cosford
et al., 2000; Gatto et al., 2004).

54
3. AIMS OF THE STUDY
The discovery of the multiplicity of nAChRs has stimulated interest in developing compounds
that selectively affect different nAChR subtypes in the brain and that might be therapeutically
beneficial in diseases, such as Parkinson’s and Alzheimer’s diseases, Tourette’s syndrome,
depression, schizophrenia, anxiety and pain. However, nAChRs also mediate effects on the
dopaminergic pathways involved in dependence and reward, which complicates the
development of compounds that affect nAChRs, but lack addictive properties. Thus, we
clarified differences in the nicotinic regulation of the nigrostriatal and mesolimbic
dopaminergic pathways by comparing the effects of two different nAChR agonists, nicotine
and epibatidine, on dopaminergic transmission in the CPu and NAc, and on dopamine-
mediated behaviours.
The detailed aims of the study were:
To clarify the mode of action of nicotine on dopaminergic transmission. Nicotine’s effects on
extracellular levels of dopamine and its metabolites in the CPu were studied in combination
with several drugs affecting dopaminergic transmission. These drugs affected endogenous
synaptic dopamine via inhibition of either dopamine reuptake (nomifensine), dopamine
metabolism (tolcapone, selegiline) or presynaptic dopamine receptors (haloperidol). (I)
To compare the effects of epibatidine and nicotine on dopamine overflow in the CPu and
NAc. The two nAChR agonists were given to rats acutely either alone or after pretreatment
with saline (II) or the dopamine uptake inhibitor nomifensine (III).
To explore if changes in striatal dopaminergic transmission result in altered behaviour of rats.
The effects of nicotine and epibatidine were studied in the well-known parkinsonian animal
model, rotational behaviour of unilaterally 6-OHDA-lesioned rats. Nicotine and epibatidine
were given acutely and repeatedly either alone or after pretreatment with saline or
nomifensine. (I, IV)
To explore if the effects of nicotine and epibatidine on dopaminergic transmission lead to
changes in locomotor activity and conditioned place preference. (V) Additionally, nicotine’s
effects on locomotor activity were studied at an elevated ambient temperature (+30-33ºC).

55
4. MATERIALS AND METHODS
4.1. Animals
Naïve male Wistar rats were used in all experiments. The rats were bred at the Laboratory
Animal Center, University of Helsinki (I-IV) or purchased from the Harlan Netherlands BV
(II, V). The rats used in the microdialysis and rotational behaviour studies weighed 250-380 g
and those used in the locomotion and place preference studies weighed 230-300 g. The animal
room was kept at an ambient temperature of 20-23°C and a 12-h light-dark cycle, with lights
on from 6:00 to 18:00. Standard rat chow and tap water were available ad libitum. The rats
were housed in groups of four, except after the stereotaxic surgeries. In the microdialysis
studies, the rats were housed one rat per cage during the recovery and experiment. In the
rotational behaviour studies, the rats lesioned with 6-OHDA were allowed to recover for 1-2
days in single cages and thereafter housed in the same group of four rats as before the surgery.
The experimental designs were approved by the Committee for Animal Experiments of the
University of Helsinki (I-V) and the chief veterinarian of the county administrative board (I-
IV). All experiments were conducted according to the European Convention for the
Protection of Vertebrate Animals used for Experimental and other Scientific Purposes.
4.2. Drugs
nAChR agonists, (-)-nicotine base (Fluka Chemie, Buchs, Switzerland) and (±)-epibatidine
hydrochloride (Sigma, MO, U.S.A.) were diluted with saline (0.9% NaCl solution). The pH of
the nicotine solution was adjusted to 7.0-7.4 with 0.05M HCl. nAChR antagonists,
hexamethonium chloride (Sigma, MO, U.S.A.) and mecamylamine hydrochloride (Merck, NJ,
U.S.A.) were dissolved in saline. Haloperidol (Orion-Pharma, Finland), selegiline (a gift from
Orion-Pharma, Finland) and tramadol hydrochloride (Orion-Pharma, Finland) were dissolved
in saline. The pH of the haloperidol solution was adjusted to 7.2 with 1M NaOH.
Nomifensine maleate (RBI, MA, U.S.A. or Sigma, MO, U.S.A.) and desmethylimipramine
hydrochloride (Sigma, MO, U.S.A.) were dissolved in sterile water. Tolcapone (a gift from
Orion-Pharma, Finland) was suspended in phosphate buffer (pH 7.4) containing a drop of
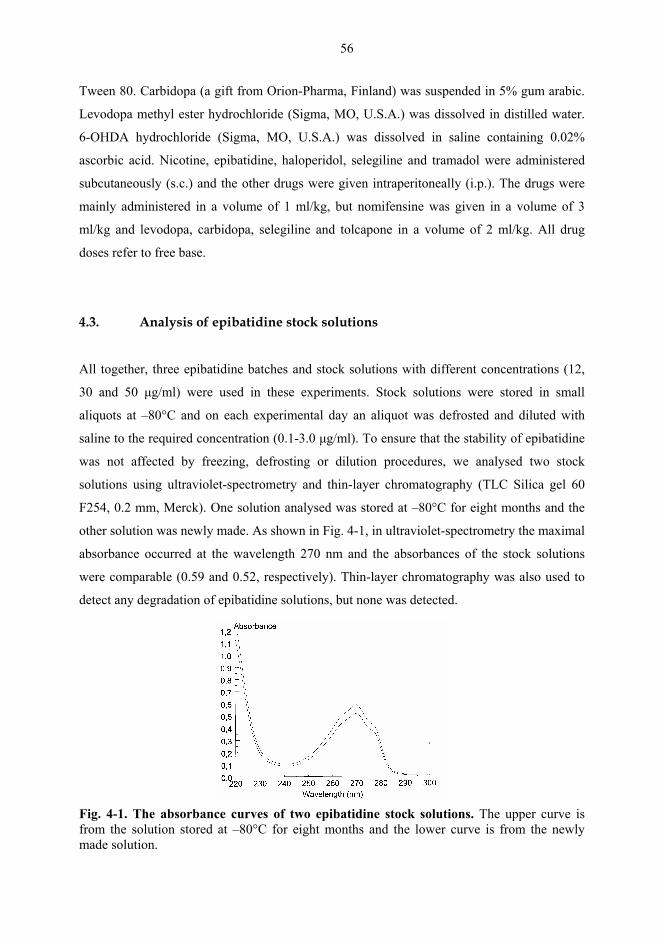
56
Tween 80. Carbidopa (a gift from Orion-Pharma, Finland) was suspended in 5% gum arabic.
Levodopa methyl ester hydrochloride (Sigma, MO, U.S.A.) was dissolved in distilled water.
6-OHDA hydrochloride (Sigma, MO, U.S.A.) was dissolved in saline containing 0.02%
ascorbic acid. Nicotine, epibatidine, haloperidol, selegiline and tramadol were administered
subcutaneously (s.c.) and the other drugs were given intraperitoneally (i.p.). The drugs were
mainly administered in a volume of 1 ml/kg, but nomifensine was given in a volume of 3
ml/kg and levodopa, carbidopa, selegiline and tolcapone in a volume of 2 ml/kg. All drug
doses refer to free base.
4.3. Analysis of epibatidine stock solutions
All together, three epibatidine batches and stock solutions with different concentrations (12,
30 and 50 µg/ml) were used in these experiments. Stock solutions were stored in small
aliquots at –80°C and on each experimental day an aliquot was defrosted and diluted with
saline to the required concentration (0.1-3.0 µg/ml). To ensure that the stability of epibatidine
was not affected by freezing, defrosting or dilution procedures, we analysed two stock
solutions using ultraviolet-spectrometry and thin-layer chromatography (TLC Silica gel 60
F254, 0.2 mm, Merck). One solution analysed was stored at –80°C for eight months and the
other solution was newly made. As shown in Fig. 4-1, in ultraviolet-spectrometry the maximal
absorbance occurred at the wavelength 270 nm and the absorbances of the stock solutions
were comparable (0.59 and 0.52, respectively). Thin-layer chromatography was also used to
detect any degradation of epibatidine solutions, but none was detected.
Fig. 4-1. The absorbance curves of two epibatidine stock solutions. The upper curve is from the solution stored at –80°C for eight months and the lower curve is from the newly made solution.

57
4.4. In vivo microdialysis (I, II, III)
The rats were under general halothane (3.5% induction, 2% maintenance) and local lidocaine
anaesthesia as microdialysis guides were implanted using stereotaxic guidance. In the CPu,
we used microdialysis probes with 2.00 mm (II) or 4.00 mm (I-III) membrane (BAS MD-
2200, 0.32 O.D., Bioanalytical Systems, IN, U.S.A.) and therefore, coordinates relative to
bregma and dura in the insertion of the BAS MD-2250 guide cannula (Bioanalytical Systems,
IN, U.S.A.) were A/P +1.0, L/M ±2.7, and D/V -2.0 or -4.0 (Paxinos and Watson, 1986).
Thus, final location of the tip of the microdialysis probe was D/V -6.0 for the CPu in all
experiments. In the NAc, coordinates of the guides were A/P +1.7, L/M ±1.4 and D/V -6.3
and final location of the tip of the probe was D/V -8.3. After the surgery, the rats were given
tramadol 1.0 mg/kg for postoperative pain, after which each rat was placed in its own test
cage and allowed to recover for five to eight days before conducting experiments.
The microdialysis experiments were performed in freely moving animals. In experiments
described in (I), we used a dummy probe to shorten the equilibration period on the
experimental day. The dummy probe was inserted through the guide cannula one day prior to
the dialysis experiment and it was replaced by an identical experimental probe the next
morning. In experiments (II, III), no dummy probe was used, but the experimental probe was
inserted between 7:00 and 8:00 on the experimental day. Input of the probe was connected to
a microperfusion pump and artificial cerebrospinal fluid (Ringer solution, pH 7.4 containing
147 mM NaCl, 1.2 mM CaCl2, 2.7 mM KCl, 1.0 mM MgCl2 and 0.04 mM ascorbic acid) was
infused through the probe at a flow rate of 2 µl/min. Ringer solution was infused for 2 h in
experiment (I) and for 3-3.5 hours in experiments (II, III) before collecting baseline samples.
When a stable outflow was shown by four consecutive samples of dopamine, the rats were
given drugs and microdialysate samples (30 µl) were collected in 15 min intervals for 2.5-4 h
after the last drug administration. When the nAChR agonists were studied in combination
with other drugs, the drugs were administered in 30 min intervals in all microdialysis
experiments, except for those studying selegiline. Microdialysate levels of dopamine,
DOPAC, HVA and 5-hydroxyindoleacetic acid (5-HIAA) were analysed immediately with
high-performance liquid chromatography (HPLC) with electrochemical detection.

58
The HPLC system consisted of a Coulochem II detector (ESA Inc., MA, U.S.A.) equipped
with a model 5014B microdialysis cell, a model 2248 HPLC pump (Pharmacia LKB,
Sweden) and a model LP-21 pulse damper (Scientific Systems Inc., PA, U.S.A.). The column
(Spherisorb ODS2, 3 µm, 4.6x100 mm, Waters, U.S.A.) was kept at 40°C with a CROCO-
CIL column heater (Cluzeau Info-Labo, France). The mobile phase was 0.1 M NaH2PO4
buffer, pH 4.0 (adjusted with 1.0 mM citric acid), 0.6-0.95 mM octane sulphonic acid, 10-
15% (v/v) methanol and 1.2 mM EDTA. A flow rate of the mobile phase was 1.0 ml/min. A
CMA/200 autoinjector (CMA/Microdialysis, Stockholm, Sweden) was utilized for injecting
20 µl of the microdialysate sample into the HPLC system. Dopamine was reduced with an
amperometric detector (potential –100 mV) after having been oxidized with a coulometric
detector (+300 mV). Chromatograms were processed with a C-R5A chromatointegrator
(Shimadzu Co., Kyoto, Japan). Values were not corrected for in vitro probe recovery
efficiency, which was approximately 25% for dopamine and 9-12% for DOPAC, HVA and 5-
HIAA. The average concentration of the first four stable dialysis samples (< 20% variation)
was determined as baseline (= 100%) and data are expressed as percentage change of the
baseline values.
At the end of the experiments, the rats were anaesthetised with CO2, decapitated and brains
were rapidly removed and frozen on dry ice (-80ºC). Positions of the probes in the CPu and
NAc were examined visually or microscopically from 100-µm coronal sections stained with
thionine.
4.5. Rotational behaviour (I, IV)
The rats were under general halothane (3.5% induction, 2% maintenance) and local lidocaine
anaesthesia as 6-OHDA was injected using stereotaxic guidance. The rats were pretreated
with a noradrenaline uptake inhibitor, desmethylimipramine (15 mg/kg i.p.) 30 min before
injecting 6-OHDA in order to protect noradrenergic nerves from 6-OHDA. The 6-OHDA
solution (2 µg/µl) was prepared just before the surgery. It was kept on ice throughout the
surgery and if the solution developed a reddish colour, a sign of oxidation of 6-OHDA, a new
solution was prepared. 6-OHDA solution was injected with a self-made 30 gauge stainless
steel cannula attached by polyethylene tubing to a 10 µl Hamilton syringe driven by a syringe

59
pump. The injection cannula filled with 6-OHDA solution was carefully inserted through a 1
mm hole in the skull into the MFB using coordinates relative to bregma and dura (A/P –4.2,
L/M ±1.4, D/V –8.2) according to the rat brain atlas (Paxinos and Watson, 1986). A total of 8
µg of 6-OHDA base was infused slowly at a flow rate of 1 µl/min during 4 min and the
injection cannula was kept in place for an additional minute to prevent the backflow of
solution. After the surgery, the rats were allowed to recover in individual cages for 1-2 days
and thereafter in groups of four up to a total of 14 days before conducting experiments.
The rotational experiments were conducted for each group of rats every day at the same time
(±1 h). Rotational behaviour of the rat was measured in a circular metal bowl (35 cm
diameter, 15 cm high) surrounded by a transparent 40 cm high cylinder. The rat was attached
to a rotation sensor with a spring steel tether connected to a plastic collar around the neck of
the rat. The rotation sensor detected full (360°) clockwise (in this case, ipsilateral) and counter
clockwise (contralateral) turns, and the rotations were counted in 15 min intervals for 3-4 h.
When nAChR agonists were studied in combination with other drugs, the drugs were
administered in 15 min intervals in all rotational experiments. In experiments (IV) the drugs
were administered repeatedly on five consecutive days and once more on the day 8. The
rotations were measured on test days 1, 3 and 5 and challenge day 8.
In order to verify depletion of dopamine in the CPu, the rats were decapitated two days after
the rotational experiments. The brains were removed from the skull and placed into the rat
brain mould. A 3x3 mm sized sample was punched out of the dorsal striatum and the
dissected samples were immediately frozen on dry ice and stored at –80 °C until the assay
was carried out. The samples were homogenized and purified as described earlier (Haikala,
1987) and thereafter assayed for dopamine concentration with the HPLC system using a C-18
reverse-phase column (Spherisorb ODS2, 3 µm, 4.6x250 mm, Waters, U.S.A.) and
electrochemical detection (+780 mV, Waters Model 464 detector, Millipore, MA, U.S.A.).
Only rats with dopamine depletion of more than 95% in the lesioned right dorsal striatum,
compared with the intact left dorsal striatum, were included in the final data analysis. With
this criterion, about 90% of the rats were qualified and the mean dopamine depletion of these
rats was 100%. Except for the experiments with levodopa, all the qualified rats rotated
predominently in the ipsilateral direction and thus the few contralateral rotations seen were
excluded from the data.

60
4.6. Locomotor activity monitoring (V)
Drugs were administrated and the locomotor activity of each group of rats was monitored
every day at the same time (±1 h) between 8:00-13:00. The rats were individually placed in
43x43x30 cm3 Plexiglas boxes (MED Associates ENV-515, Vermont, GA, U.S.A.). The
computer registered and analysed infrared photo beam interruptions. The rats were first
habituated to the activity monitoring boxes on two consecutive days for 2 h each. On the first
habituation day, the rats were placed in the boxes and their activity was measured, while on
the second day the rats were given s.c. saline before placing them in the boxes and measuring
the activity. On the following day, test day 1, the rats were given drugs and the locomotor
activity was measured in 5 min intervals for 2 h. The drugs were given repeatedly on five
consecutive days, and the locomotor activity was measured on test days 1, 3 and 5. On days 2
and 4 the drugs were given in the home cages. The rats repeatedly given epibatidine (or saline
as control) were given an additional challenge dose of epibatidine (or saline) on test day 7 and
the locomotor activity was monitored similarly to previous test days. The locomotion
experiments were normally carried out at an ambient temperature of 20-23°C, but some
experiments were conducted at the higher ambient temperature of 30-33°C and for those
experiments, the rats were habituated to 30-33°C for two days before conducting the
experiments.
4.7. Place conditioning (V)
The place conditioning apparatus consisted of identical Plexiglas chambers divided into two
equally sized compartments (41x41x28 cm3) by a separating black wall with guillotine door
(MED Associates, Vermont, GA, U.S.A.). In one compartment there was a grid rod floor and
the walls were covered outside with a white layer (“white” compartment), whereas in the
other compartment there was a wire mesh floor and the walls were covered with a black layer
(“black” compartment). The computer registered interruptions of infrared photo beams, which
were used both to measure the locomotor activity and to determine the rat’s position in the
compartments. The rats were accustomed to experimenter by handling and weighing them for
5 min on 2-3 days before experiments. On test days, the experiments were conducted each
day at the same time (±1 h) during 8:00-14:00 and the rats were habituated to the experiment

61
room for 30 min before the experiments. White noise was used to cover background noise.
Each experiment was performed over six consecutive days and consisted of three phases:
habituation (days 1 and 2, one session per day), conditioning (days 3, 4 and 5, two sessions
per day), and testing (day 6, one session).
Habituation. The rats were in the test chambers with free access to both compartments (door
open). We used a biased conditioning design, in which rats initially prefer one compartment,
and thus, only the rats that initially spent more time in the black compartment with a wire
mesh floor were included in the data. The time that the rats spent in the non-preferred (white)
compartment during the first 15 min on the second habituation day was used as an initial
preference level for each rat (preconditioning time).
Conditioning. In the morning, the rats were given saline and immediately exposed to the
preferred black compartment (door closed). Three to four hours later, the rats were given
drugs or saline (controls) and immediately exposed to the non-preferred white (drug-paired)
compartment (door closed). The conditioning time was either 20 min (nicotine) or 60 min
(epibatidine).
Testing. On day 6, the preference for compartments was tested without any drug
administration similarly to the testing on the second habituation day. The rats had free access
to both compartments (door open) and the time in the drug-paired (white) compartment
(postconditioning time) was measured. The differences in the postconditioning and
preconditioning times spent in the drug-paired compartment were calculated. When the
change was positive, a drug was considered to induce place preference.
4.8. Statistical analysis
All data are expressed as means ± standard errors of the mean (S.E.M.). The microdialysis,
rotational behaviour and locomotor activity data (I-V) were analysed with one-, two- or three-
way analysis of variance (ANOVA) for repeated measures (Statview 5.0, SAS Institute Inc.,
U.S.A. or GraphPad Prism 3.0 for Windows, GraphPad Software, San Diego, CA, U.S.A.).
When appropriate (P < 0.05), multiple comparisons were conducted by using Student
Newman Keuls (I, IV, V) post hoc tests or contrast analysis with Bonferroni levels (II, III).
The basal concentrations in the microdialysis samples and part of the rotational data were
analysed with Student’s t-test (I-IV).
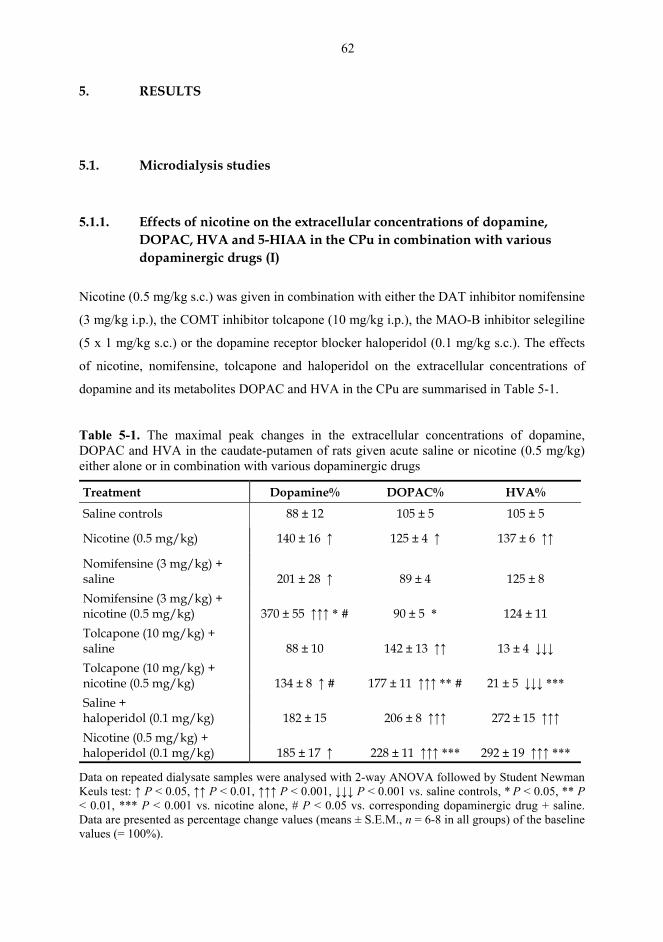
62
5. RESULTS
5.1. Microdialysis studies
5.1.1. Effects of nicotine on the extracellular concentrations of dopamine,
DOPAC, HVA and 5-HIAA in the CPu in combination with various dopaminergic drugs (I)
Nicotine (0.5 mg/kg s.c.) was given in combination with either the DAT inhibitor nomifensine
(3 mg/kg i.p.), the COMT inhibitor tolcapone (10 mg/kg i.p.), the MAO-B inhibitor selegiline
(5 x 1 mg/kg s.c.) or the dopamine receptor blocker haloperidol (0.1 mg/kg s.c.). The effects
of nicotine, nomifensine, tolcapone and haloperidol on the extracellular concentrations of
dopamine and its metabolites DOPAC and HVA in the CPu are summarised in Table 5-1.
Table 5-1. The maximal peak changes in the extracellular concentrations of dopamine, DOPAC and HVA in the caudate-putamen of rats given acute saline or nicotine (0.5 mg/kg) either alone or in combination with various dopaminergic drugs
Treatment Dopamine% DOPAC% HVA%
Saline controls 88 ± 12 105 ± 5 105 ± 5
Nicotine (0.5 mg/kg) 140 ± 16 ↑ 125 ± 4 ↑ 137 ± 6 ↑↑
Nomifensine (3 mg/kg) + saline
201 ± 28 ↑
89 ± 4
125 ± 8
Nomifensine (3 mg/kg) + nicotine (0.5 mg/kg)
370 ± 55 ↑↑↑ * #
90 ± 5 *
124 ± 11
Tolcapone (10 mg/kg) + saline
88 ± 10
142 ± 13 ↑↑
13 ± 4 ↓↓↓
Tolcapone (10 mg/kg) + nicotine (0.5 mg/kg)
134 ± 8 ↑ #
177 ± 11 ↑↑↑ ** #
21 ± 5 ↓↓↓ ***
Saline + haloperidol (0.1 mg/kg)
182 ± 15
206 ± 8 ↑↑↑
272 ± 15 ↑↑↑
Nicotine (0.5 mg/kg) + haloperidol (0.1 mg/kg)
185 ± 17 ↑
228 ± 11 ↑↑↑ ***
292 ± 19 ↑↑↑ ***
Data on repeated dialysate samples were analysed with 2-way ANOVA followed by Student Newman Keuls test: ↑ P < 0.05, ↑↑ P < 0.01, ↑↑↑ P < 0.001, ↓↓↓ P < 0.001 vs. saline controls, * P < 0.05, ** P < 0.01, *** P < 0.001 vs. nicotine alone, # P < 0.05 vs. corresponding dopaminergic drug + saline. Data are presented as percentage change values (means ± S.E.M., n = 6-8 in all groups) of the baseline values (= 100%).

63
Nicotine (0.5 mg/kg) significantly elevated the extracellular levels of dopamine, DOPAC and
HVA in the CPu (P < 0.05). The elevations were inhibited by the brain penetrating nAChR
antagonist mecamylamine (5 mg/kg i.p., P < 0.05), but not by the peripheral nAChR
antagonist hexamethonium (5 mg/kg i.p.). Nomifensine (3 mg/kg) significantly increased the
extracellular level of dopamine (P < 0.05), but did not alter dopamine metabolites in the CPu.
When nicotine was given after nomifensine, the dopamine level was further elevated,
compared with the elevation induced by either nomifensine or nicotine alone (P < 0.05).
Tolcapone (10 mg/kg) did not alter the dopamine level, but increased the level of DOPAC (P
< 0.01) and decreased that of HVA (P < 0.001). When nicotine was given after tolcapone, the
dopamine level was elevated to the same degree as when nicotine was given alone. However,
the DOPAC level was further elevated, compared with the elevation induced by either
tolcapone or nicotine alone (P < 0.05). The level of HVA was decreased to the same extent as
after tolcapone alone. Although haloperidol (0.1 mg/kg) did not significantly elevate the
dopamine level, it significantly elevated the levels of DOPAC and HVA (P < 0.001). When
nicotine was given before haloperidol, the elevation in the levels of dopamine, DOPAC and
HVA was the same as after haloperidol alone. None of the drugs studied altered the
extracellular 5-HIAA concentration in the CPu (data not shown). Furthermore, when the
nAChR antagonist mecamylamine (5 mg/kg i.p.) was given in combination with the
dopaminergic drugs, it did not alter the effects of nomifensine, tolcapone or haloperidol on the
levels of dopamine, DOPAC, HVA or 5-HIAA (unpublished data; data not shown).
Repeated daily pretreatment with selegiline (5 x 1 mg/kg s.c.) elevated the basal extracellular
dopamine concentration and decreased the basal level of dopamine metabolites, particularly
that of DOPAC, in the CPu (Table 5-2; P < 0.05). Selegiline did not alter the basal
extracellular 5-HIAA concentration in the CPu. Nicotine did not significantly alter the
extracellular levels of dopamine, DOPAC, HVA or 5-HIAA when given acutely 3 h after the
fifth selegiline administration (Table 5-2).
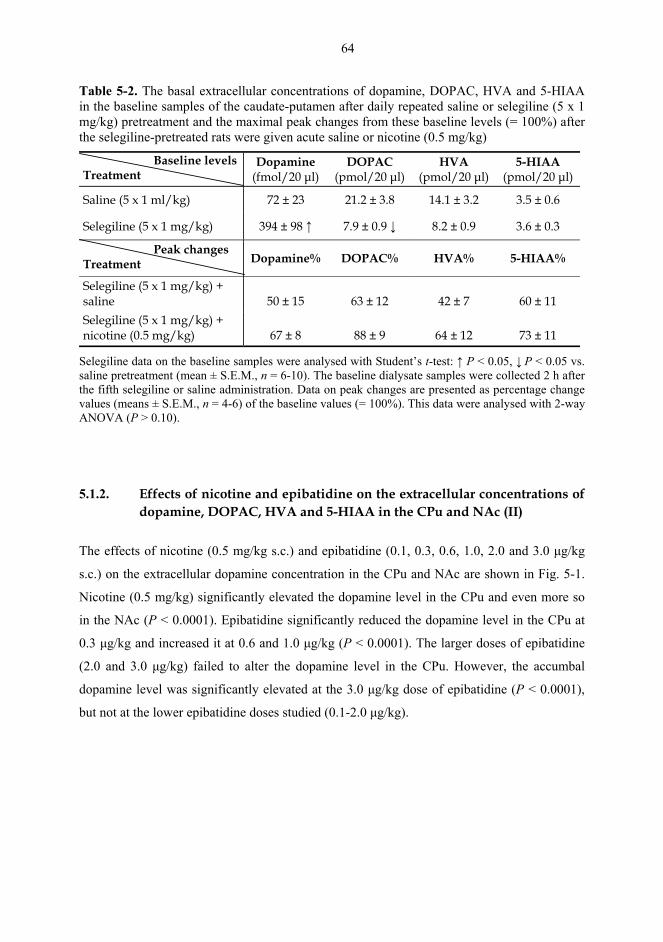
64
Table 5-2. The basal extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the baseline samples of the caudate-putamen after daily repeated saline or selegiline (5 x 1 mg/kg) pretreatment and the maximal peak changes from these baseline levels (= 100%) after the selegiline-pretreated rats were given acute saline or nicotine (0.5 mg/kg)
Baseline levels Treatment
Dopamine (fmol/20 µl)
DOPAC (pmol/20 µl)
HVA (pmol/20 µl)
5-HIAA (pmol/20 µl)
Saline (5 x 1 ml/kg) 72 ± 23 21.2 ± 3.8 14.1 ± 3.2 3.5 ± 0.6
Selegiline (5 x 1 mg/kg) 394 ± 98 ↑ 7.9 ± 0.9 ↓ 8.2 ± 0.9 3.6 ± 0.3
Peak changes Treatment Dopamine% DOPAC% HVA% 5-HIAA%
Selegiline (5 x 1 mg/kg) + saline
50 ± 15
63 ± 12
42 ± 7
60 ± 11
Selegiline (5 x 1 mg/kg) + nicotine (0.5 mg/kg)
67 ± 8
88 ± 9
64 ± 12
73 ± 11
Selegiline data on the baseline samples were analysed with Student’s t-test: ↑ P < 0.05, ↓ P < 0.05 vs. saline pretreatment (mean ± S.E.M., n = 6-10). The baseline dialysate samples were collected 2 h after the fifth selegiline or saline administration. Data on peak changes are presented as percentage change values (means ± S.E.M., n = 4-6) of the baseline values (= 100%). This data were analysed with 2-way ANOVA (P > 0.10).
5.1.2. Effects of nicotine and epibatidine on the extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the CPu and NAc (II)
The effects of nicotine (0.5 mg/kg s.c.) and epibatidine (0.1, 0.3, 0.6, 1.0, 2.0 and 3.0 µg/kg
s.c.) on the extracellular dopamine concentration in the CPu and NAc are shown in Fig. 5-1.
Nicotine (0.5 mg/kg) significantly elevated the dopamine level in the CPu and even more so
in the NAc (P < 0.0001). Epibatidine significantly reduced the dopamine level in the CPu at
0.3 µg/kg and increased it at 0.6 and 1.0 µg/kg (P < 0.0001). The larger doses of epibatidine
(2.0 and 3.0 µg/kg) failed to alter the dopamine level in the CPu. However, the accumbal
dopamine level was significantly elevated at the 3.0 µg/kg dose of epibatidine (P < 0.0001),
but not at the lower epibatidine doses studied (0.1-2.0 µg/kg).
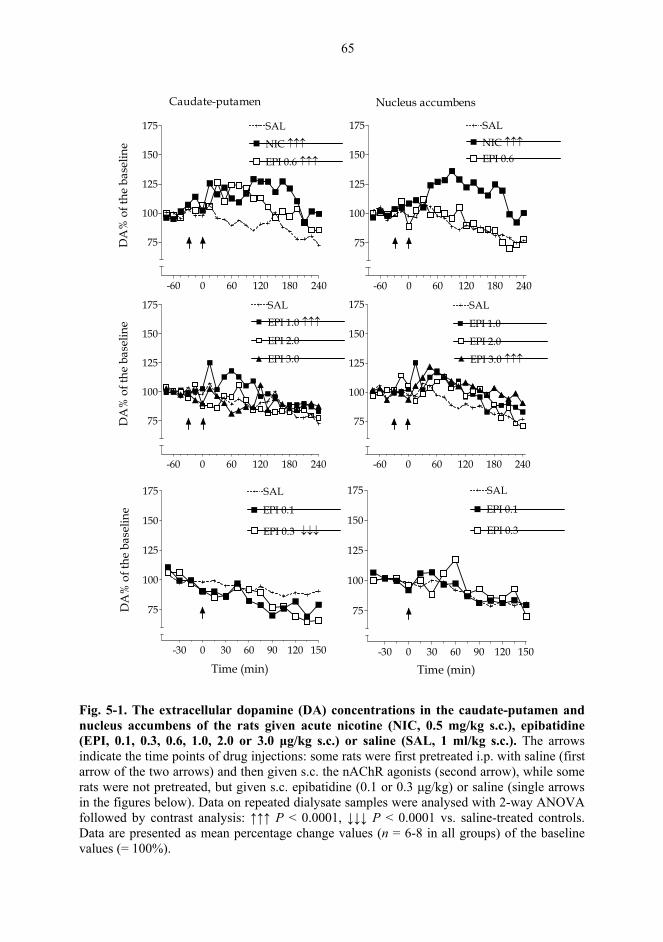
65
-60 0 60 120 180 240
NIC ↑↑↑
SAL
EPI 0.6 ↑↑↑
75
100
125
150
175D
A%
of t
he b
asel
ine
-60 0 60 120 180 240
SALNIC ↑↑↑
EPI 0.6
75
100
125
150
175
-60 0 60 120 180 240
SAL
EPI 3.0
EPI 2.0
EPI 1.0 ↑↑↑
75
100
125
150
175
DA
% o
f the
bas
elin
e
-60 0 60 120 180 240
SAL
EPI 3.0 ↑↑↑
EPI 2.0
EPI 1.0
75
100
125
150
175
-30 0 30 60 90 120 150
EPI 0.1
75
100
125
150
175
EPI 0.3 ↓↓↓
SAL
Time (min)
DA
% o
f the
bas
elin
e
-30 0 30 60 90 120 150
EPI 0.1
75
100
125
150
175
EPI 0.3
SAL
Time (min)
Caudate-putamen Nucleus accumbens
Fig. 5-1. The extracellular dopamine (DA) concentrations in the caudate-putamen and nucleus accumbens of the rats given acute nicotine (NIC, 0.5 mg/kg s.c.), epibatidine (EPI, 0.1, 0.3, 0.6, 1.0, 2.0 or 3.0 µg/kg s.c.) or saline (SAL, 1 ml/kg s.c.). The arrows indicate the time points of drug injections: some rats were first pretreated i.p. with saline (first arrow of the two arrows) and then given s.c. the nAChR agonists (second arrow), while some rats were not pretreated, but given s.c. epibatidine (0.1 or 0.3 µg/kg) or saline (single arrows in the figures below). Data on repeated dialysate samples were analysed with 2-way ANOVA followed by contrast analysis: ↑↑↑ P < 0.0001, ↓↓↓ P < 0.0001 vs. saline-treated controls. Data are presented as mean percentage change values (n = 6-8 in all groups) of the baseline values (= 100%).
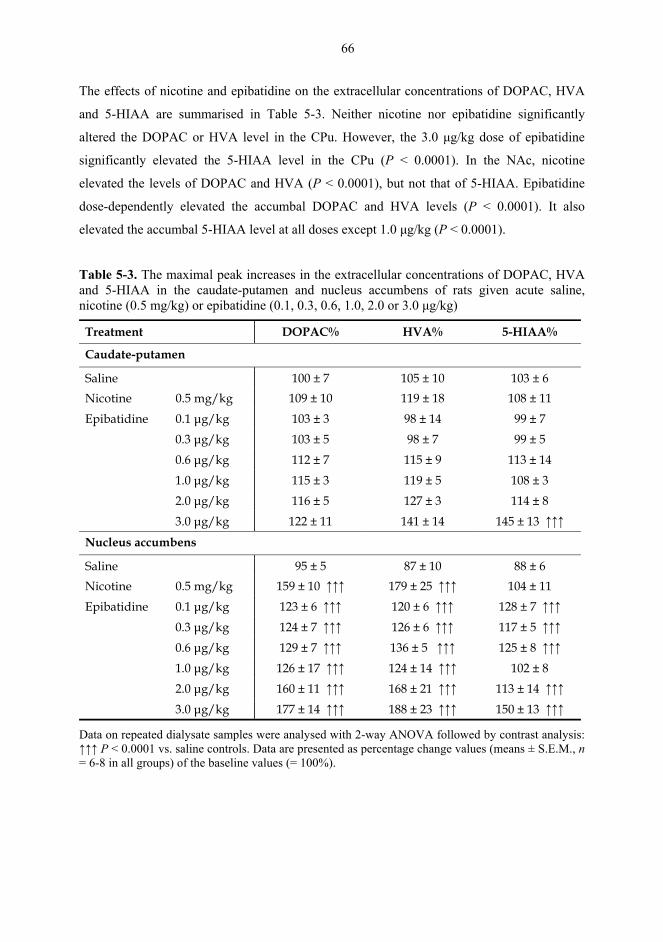
66
The effects of nicotine and epibatidine on the extracellular concentrations of DOPAC, HVA
and 5-HIAA are summarised in Table 5-3. Neither nicotine nor epibatidine significantly
altered the DOPAC or HVA level in the CPu. However, the 3.0 µg/kg dose of epibatidine
significantly elevated the 5-HIAA level in the CPu (P < 0.0001). In the NAc, nicotine
elevated the levels of DOPAC and HVA (P < 0.0001), but not that of 5-HIAA. Epibatidine
dose-dependently elevated the accumbal DOPAC and HVA levels (P < 0.0001). It also
elevated the accumbal 5-HIAA level at all doses except 1.0 µg/kg (P < 0.0001).
Table 5-3. The maximal peak increases in the extracellular concentrations of DOPAC, HVA and 5-HIAA in the caudate-putamen and nucleus accumbens of rats given acute saline, nicotine (0.5 mg/kg) or epibatidine (0.1, 0.3, 0.6, 1.0, 2.0 or 3.0 µg/kg)
Treatment DOPAC% HVA% 5-HIAA%
Caudate-putamen
Saline 100 ± 7 105 ± 10 103 ± 6 Nicotine 0.5 mg/kg 109 ± 10 119 ± 18 108 ± 11 Epibatidine 0.1 µg/kg 103 ± 3 98 ± 14 99 ± 7 0.3 µg/kg 103 ± 5 98 ± 7 99 ± 5 0.6 µg/kg 112 ± 7 115 ± 9 113 ± 14 1.0 µg/kg 115 ± 3 119 ± 5 108 ± 3 2.0 µg/kg 116 ± 5 127 ± 3 114 ± 8 3.0 µg/kg 122 ± 11 141 ± 14 145 ± 13 ↑↑↑
Nucleus accumbens
Saline 95 ± 5 87 ± 10 88 ± 6 Nicotine 0.5 mg/kg 159 ± 10 ↑↑↑ 179 ± 25 ↑↑↑ 104 ± 11 Epibatidine 0.1 µg/kg 123 ± 6 ↑↑↑ 120 ± 6 ↑↑↑ 128 ± 7 ↑↑↑ 0.3 µg/kg 124 ± 7 ↑↑↑ 126 ± 6 ↑↑↑ 117 ± 5 ↑↑↑ 0.6 µg/kg 129 ± 7 ↑↑↑ 136 ± 5 ↑↑↑ 125 ± 8 ↑↑↑ 1.0 µg/kg 126 ± 17 ↑↑↑ 124 ± 14 ↑↑↑ 102 ± 8 2.0 µg/kg 160 ± 11 ↑↑↑ 168 ± 21 ↑↑↑ 113 ± 14 ↑↑↑ 3.0 µg/kg 177 ± 14 ↑↑↑ 188 ± 23 ↑↑↑ 150 ± 13 ↑↑↑
Data on repeated dialysate samples were analysed with 2-way ANOVA followed by contrast analysis: ↑↑↑ P < 0.0001 vs. saline controls. Data are presented as percentage change values (means ± S.E.M., n = 6-8 in all groups) of the baseline values (= 100%).
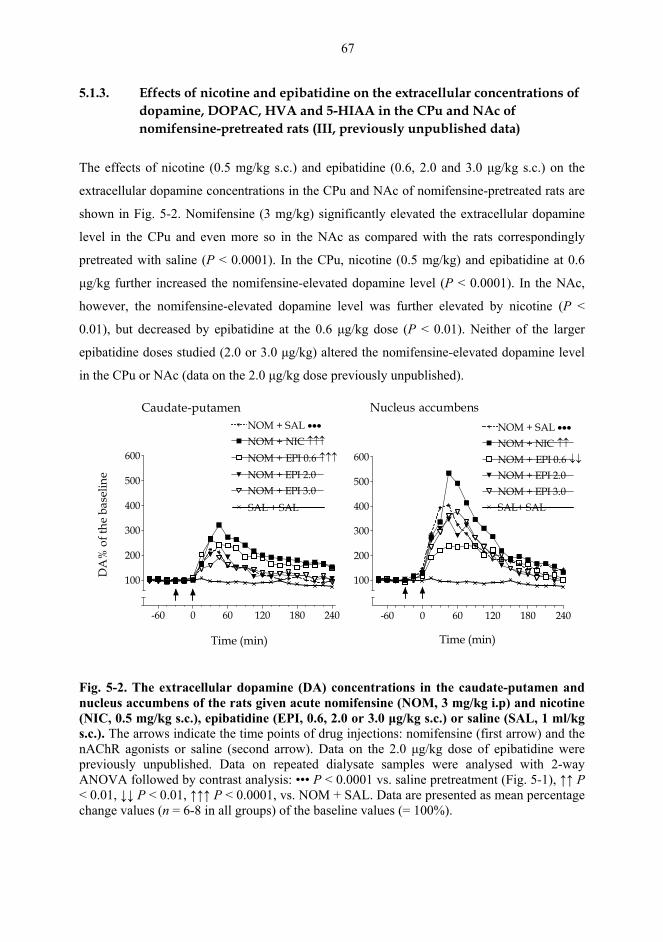
67
5.1.3. Effects of nicotine and epibatidine on the extracellular concentrations of dopamine, DOPAC, HVA and 5-HIAA in the CPu and NAc of nomifensine-pretreated rats (III, previously unpublished data)
The effects of nicotine (0.5 mg/kg s.c.) and epibatidine (0.6, 2.0 and 3.0 µg/kg s.c.) on the
extracellular dopamine concentrations in the CPu and NAc of nomifensine-pretreated rats are
shown in Fig. 5-2. Nomifensine (3 mg/kg) significantly elevated the extracellular dopamine
level in the CPu and even more so in the NAc as compared with the rats correspondingly
pretreated with saline (P < 0.0001). In the CPu, nicotine (0.5 mg/kg) and epibatidine at 0.6
µg/kg further increased the nomifensine-elevated dopamine level (P < 0.0001). In the NAc,
however, the nomifensine-elevated dopamine level was further elevated by nicotine (P <
0.01), but decreased by epibatidine at the 0.6 µg/kg dose (P < 0.01). Neither of the larger
epibatidine doses studied (2.0 or 3.0 µg/kg) altered the nomifensine-elevated dopamine level
in the CPu or NAc (data on the 2.0 µg/kg dose previously unpublished).
-60 0 60 120 180 240
NOM + SAL •••
NOM + NIC ↑↑↑
NOM + EPI 0.6 ↑↑↑
100
200
300
400
500
600
NOM + EPI 3.0NOM + EPI 2.0
SAL + SAL
Time (min)
DA
% o
f the
bas
elin
e
-60 0 60 120 180 240
NOM + NIC ↑↑
NOM + SAL •••
NOM + EPI 0.6 ↓↓
100
200
300
400
500
600
NOM + EPI 3.0NOM + EPI 2.0
SAL+ SAL
Time (min)
Caudate-putamen Nucleus accumbens
Fig. 5-2. The extracellular dopamine (DA) concentrations in the caudate-putamen and nucleus accumbens of the rats given acute nomifensine (NOM, 3 mg/kg i.p) and nicotine (NIC, 0.5 mg/kg s.c.), epibatidine (EPI, 0.6, 2.0 or 3.0 µg/kg s.c.) or saline (SAL, 1 ml/kg s.c.). The arrows indicate the time points of drug injections: nomifensine (first arrow) and the nAChR agonists or saline (second arrow). Data on the 2.0 µg/kg dose of epibatidine were previously unpublished. Data on repeated dialysate samples were analysed with 2-way ANOVA followed by contrast analysis: ••• P < 0.0001 vs. saline pretreatment (Fig. 5-1), ↑↑ P < 0.01, ↓↓ P < 0.01, ↑↑↑ P < 0.0001, vs. NOM + SAL. Data are presented as mean percentage change values (n = 6-8 in all groups) of the baseline values (= 100%).
68
The effects of nicotine and epibatidine on the extracellular concentrations of DOPAC, HVA
and 5-HIAA in the CPu and NAc of nomifensine-pretreated rats are summarized in Table 5-4.
Nomifensine (3 mg/kg) significantly decreased the extracellular DOPAC level in the CPu (P
< 0.01) and to an even greater extent in the NAc (P < 0.0001). Nicotine or epibatidine did not
alter the nomifensine-decreased DOPAC level in the CPu, but significantly elevated that in
the NAc (P < 0.0001). Nomifensine alone or in combination with the nAChR agonists did not
alter the HVA levels in either brain area studied. Furthermore, nomifensine significantly
increased the 5-HIAA levels in both brain areas and two-way ANOVA for repeated measures
revealed a significant pretreatment-effect for nomifensine [previously unpublished data; F(1,21)
= 11.8, P = 0.0025].
Table 5-4. The maximal peak changes in the extracellular concentrations of DOPAC, HVA and 5-HIAA in the caudate-putamen and nucleus accumbens of the rats given acute saline, nicotine (0.5 mg/kg) or epibatidine (0.6, 2.0 or 3.0 µg/kg) after nomifensine (3 mg/kg)
Treatment DOPAC% HVA% 5-HIAA%
Caudate-putamen
Saline + saline controls 100 ± 7 105 ± 10 103 ± 6 Nomifensine + saline 81 ± 4 •• 104 ± 7 129 ± 12 ↑↑↑ Nomifensine + nicotine 0.5 mg/kg 102 ± 5 117 ± 6 129 ± 5 ↑↑↑ Nomifensine + epibatidine 0.6 µg/kg 96 ± 6 109 ± 9 117 ± 11
2.0 µg/kg 97 ± 4 112 ± 5 136 ± 7 ↑↑↑ 3.0 µg/kg 104 ± 7 121 ± 8 143 ± 8 ↑↑↑
Nucleus accumbens
Saline + saline controls 95 ± 5 87 ± 10 88 ± 6 Nomifensine + saline 69 ± 3 ••• 92 ± 7 142 ± 12 ↑↑↑ Nomifensine + nicotine 0.5 mg/kg 118 ± 8 ↑↑↑ 114 ± 12 118 ± 15 ↑↑↑ Nomifensine + epibatidine 0.6 µg/kg 99 ± 10 ↑↑↑ 109 ± 10 150 ± 16 ↑↑↑
2.0 µg/kg 110 ± 8 ↑↑↑ 114 ± 13 121 ± 21 ↑↑↑ 3.0 µg/kg 117 ± 10 ↑↑↑ 120 ± 2 126 ± 8 ↑↑↑
Data on repeated dialysate samples were analysed with 2-way ANOVA followed by contrast analysis: •• P < 0.01, ••• P < 0.0001 vs. saline + saline controls, ↑↑↑ P < 0.0001 vs. nomifensine + saline. Data on the 2.0 µg/kg dose of epibatidine and the 5-HIAA levels were not previously unpublished. Data are presented as percentage change values (means ± S.E.M., n = 6-8 in all groups) of the baseline values (= 100%).
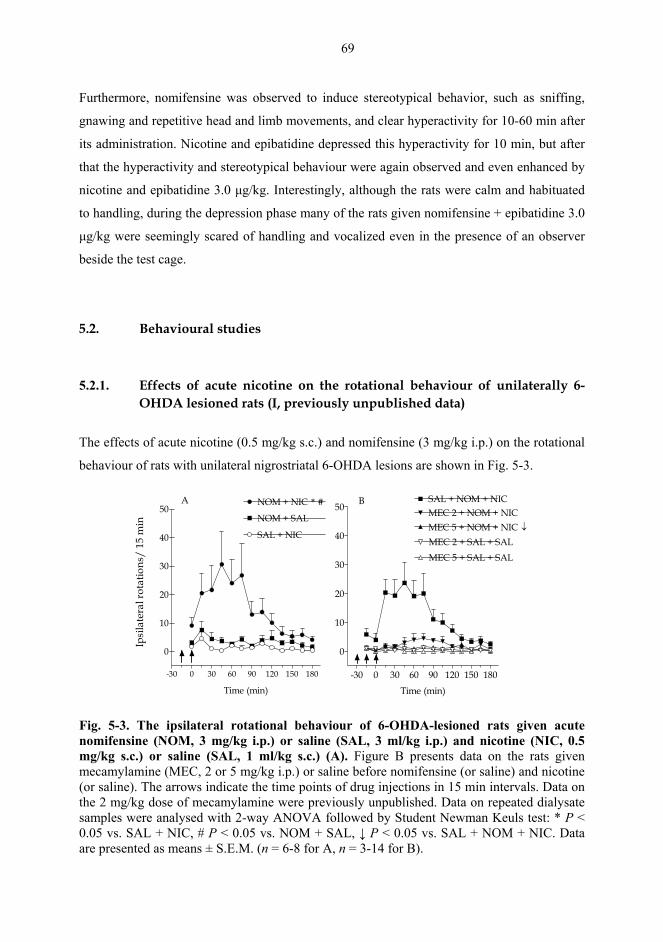
69
Furthermore, nomifensine was observed to induce stereotypical behavior, such as sniffing,
gnawing and repetitive head and limb movements, and clear hyperactivity for 10-60 min after
its administration. Nicotine and epibatidine depressed this hyperactivity for 10 min, but after
that the hyperactivity and stereotypical behaviour were again observed and even enhanced by
nicotine and epibatidine 3.0 µg/kg. Interestingly, although the rats were calm and habituated
to handling, during the depression phase many of the rats given nomifensine + epibatidine 3.0
µg/kg were seemingly scared of handling and vocalized even in the presence of an observer
beside the test cage.
5.2. Behavioural studies
5.2.1. Effects of acute nicotine on the rotational behaviour of unilaterally 6-
OHDA lesioned rats (I, previously unpublished data)
The effects of acute nicotine (0.5 mg/kg s.c.) and nomifensine (3 mg/kg i.p.) on the rotational
behaviour of rats with unilateral nigrostriatal 6-OHDA lesions are shown in Fig. 5-3.
-30 0 30 60 90 120 150 180
0
10
20
30
40
50
SAL + NIC
NOM + SAL
NOM + NIC * #
Time (min)
Ipsi
late
ral r
otat
ions
/ 15
min
-30 0 30 60 90 120 150 180
0
10
20
30
40
50
MEC 2 + SAL + SAL
MEC 2 + NOM + NIC
MEC 5 + SAL + SAL
MEC 5 + NOM + NIC ↓
SAL + NOM + NIC
Time (min)
A B
Fig. 5-3. The ipsilateral rotational behaviour of 6-OHDA-lesioned rats given acute nomifensine (NOM, 3 mg/kg i.p.) or saline (SAL, 3 ml/kg i.p.) and nicotine (NIC, 0.5 mg/kg s.c.) or saline (SAL, 1 ml/kg s.c.) (A). Figure B presents data on the rats given mecamylamine (MEC, 2 or 5 mg/kg i.p.) or saline before nomifensine (or saline) and nicotine (or saline). The arrows indicate the time points of drug injections in 15 min intervals. Data on the 2 mg/kg dose of mecamylamine were previously unpublished. Data on repeated dialysate samples were analysed with 2-way ANOVA followed by Student Newman Keuls test: * P < 0.05 vs. SAL + NIC, # P < 0.05 vs. NOM + SAL, ↓ P < 0.05 vs. SAL + NOM + NIC. Data are presented as means ± S.E.M. (n = 6-8 for A, n = 3-14 for B).

70
The effect of nomifensine or nicotine alone on ipsilateral rotations was modest, whereas the
combination of these two drugs produced a strong ipsilateral rotational behaviour. Indeed, the
rats given both nomifensine and nicotine rotated ipsilaterally significantly more than those
given either nomifensine or nicotine alone (P < 0.05). Some of the rats were pretreated with
the nAChR antagonist mecamylamine (2 or 5 mg/kg i.p; data on the 2 mg/kg dose previously
unpublished). Mecamylamine did not induce rotational behaviour. However, mecamylamine
dose-dependently inhibited the rotational behaviour induced by the combination of
nomifensine and nicotine and this effect reached significance at the 5 mg/kg dose (P < 0.05).
As shown in Table 5-5, the effect of nicotine (0.5 mg/kg s.c.) on the rotational behaviour of 6-
OHDA-lesioned rats was also studied in combination with parkinsonian drugs (levodopa, 10
mg/kg i.p., the DOPA decarboxylase inhibitor carbidopa, 20 mg/kg i.p., and the COMT-
inhibitor tolcapone, 10 mg/kg i.p.; previously unpublished data). The rats given levodopa in
combination with carbidopa and tolcapone rotated mainly contralaterally. Nicotine (0.5
mg/kg) tended to enhance the rotational behaviour as compared with the rats correspondingly
given saline (Student’s t-test, P = 0.097).
Table 5-5. The cumulative contralateral and ipsilateral rotational behaviour of 6-OHDA-lesioned rats given saline or nicotine (0.5 mg/kg) in combination with levodopa treatment
Rotational behaviour/ 4 h Treatment
Contralateral Ipsilateral
Levodopa (10 mg/kg) + carbidopa (20 mg/kg) + tolcapone (10 mg/kg) +
Saline 203 ± 98 14 ± 6 Nicotine 0.5 mg/kg 710 ± 264 29 ± 13
The following drugs were given: carbidopa (10 mg/kg i.p.), followed 15 min later by tolcapone (10 mg/kg i.p.) and nicotine or saline, and another 15 min later by carbidopa (10 mg/kg i.p.) and levodopa (10 mg/kg i.p.), after which the rotations were counted in 15 min intervals up to 4 h. Data are presented as means ± S.E.M., n = 11-12.
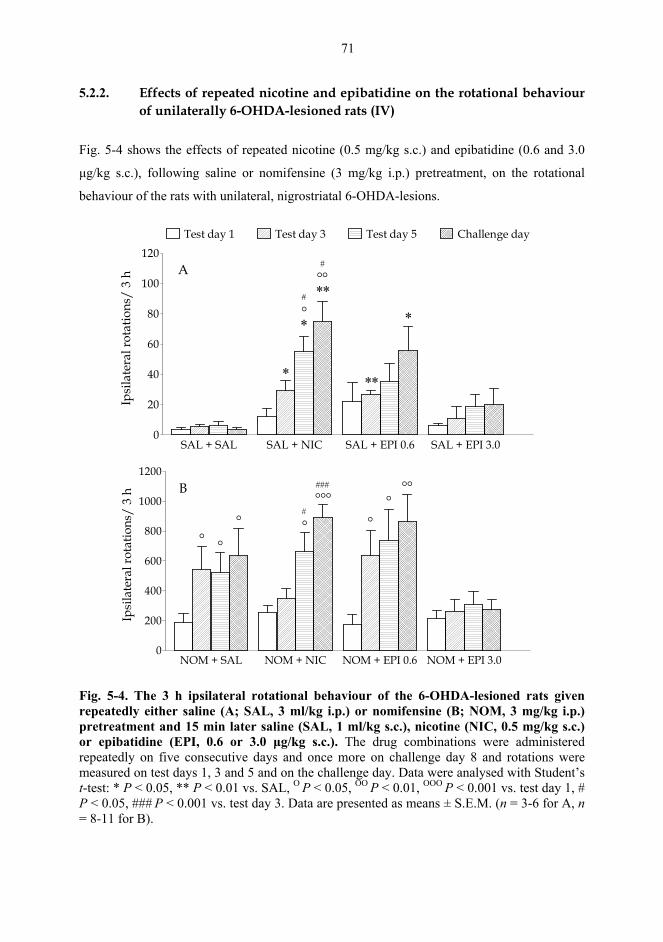
71
5.2.2. Effects of repeated nicotine and epibatidine on the rotational behaviour of unilaterally 6-OHDA-lesioned rats (IV)
Fig. 5-4 shows the effects of repeated nicotine (0.5 mg/kg s.c.) and epibatidine (0.6 and 3.0
µg/kg s.c.), following saline or nomifensine (3 mg/kg i.p.) pretreatment, on the rotational
behaviour of the rats with unilateral, nigrostriatal 6-OHDA-lesions.
SAL + SAL SAL + NIC SAL + EPI 0.6 SAL + EPI 3.00
20
40
60
80
100
120
Test day 1 Test day 5 Test day 3 Challenge day
***
*°
**
*
°°#
#
Ipsi
late
ral r
otat
ions
/ 3
h
NOM + SAL NOM + NIC NOM + EPI 0.6 NOM + EPI 3.00
200
400
600
800
1000
1200
°°°#
###
Ipsi
late
ral r
otat
ions
/ 3
h
°
°°
° °°
°°
A
B
Fig. 5-4. The 3 h ipsilateral rotational behaviour of the 6-OHDA-lesioned rats given repeatedly either saline (A; SAL, 3 ml/kg i.p.) or nomifensine (B; NOM, 3 mg/kg i.p.) pretreatment and 15 min later saline (SAL, 1 ml/kg s.c.), nicotine (NIC, 0.5 mg/kg s.c.) or epibatidine (EPI, 0.6 or 3.0 µg/kg s.c.). The drug combinations were administered repeatedly on five consecutive days and once more on challenge day 8 and rotations were measured on test days 1, 3 and 5 and on the challenge day. Data were analysed with Student’s t-test: * P < 0.05, ** P < 0.01 vs. SAL, O P < 0.05, OO P < 0.01, OOO P < 0.001 vs. test day 1, # P < 0.05, ### P < 0.001 vs. test day 3. Data are presented as means ± S.E.M. (n = 3-6 for A, n = 8-11 for B).

72
In the saline-pretreated rats, acute nicotine and epibatidine produced only few ipsilateral
rotations. The effect of nicotine on rotational behaviour was gradually enhanced by repeated
administration and the rats rotated significantly more on the challenge day than on test day 1
(P < 0.01). The rotational effect of epibatidine at 0.6 µg/kg, but not at 3.0 µg/kg, was also
enhanced by repeated administration, although to a lesser extent than that of nicotine.
Nomifensine’s effect on the rotational behaviour was significantly enhanced by repeated
administration even by test day 3, compared with its acute effect on test day 1 (P < 0.05).
Nicotine and epibatidine at 0.6 µg/kg tended to enhance the effect of repeated nomifensine on
the rotational behaviour, but the higher dose of epibatidine (3.0 µg/kg) tended to prevent the
nomifensine-induced sensitization of rotational behaviour.
5.2.3. Effects of nicotine and epibatidine on locomotor activity (V, previously
unpublished data)
As shown in Fig. 5-5, the effects of repeated nicotine (0.5 and 0.8 mg/kg s.c.) on locomotor
activity were studied at a normal ambient temperature of 20-23°C, as well as at an elevated
ambient temperature of 30-33°C (data on the effects at 30-33°C previously unpublished).
Analysis of basal locomotor activity on the second habituation day revealed that the rats were
significantly more active at the ambient temperature of 30-33°C than at 20-23°C (Fig. 5-5
Insert; Student’s t-test, P = 0.0082). When the activity on test day 1 was analysed, two-way
ANOVA revealed that during 0-60 min there was a significant temperature(20-23 vs. 30-
33°C)-effect [F(1,90) = 5.6, P = 0.0198] and during 0-120 min a significant treatment-effect
was seen [0-60 min: F(2,90) = 6.6, P = 0.0021; 61-120 min: F(2,90) = 8.4, P = 0.0004], but there
were no significant treatment × temperature-interactions (P > 0.08). Thus, acute nicotine
increased the activity at 0.5 mg/kg during both test hours and at 0.8 mg/kg during the second
test hour at both ambient temperatures (Student Newman Keuls P < 0.05: 0-60 min: NIC 0.5
vs. SAL/NIC 0.8, 61-120 min: NIC 0.5/0.8 vs. SAL).
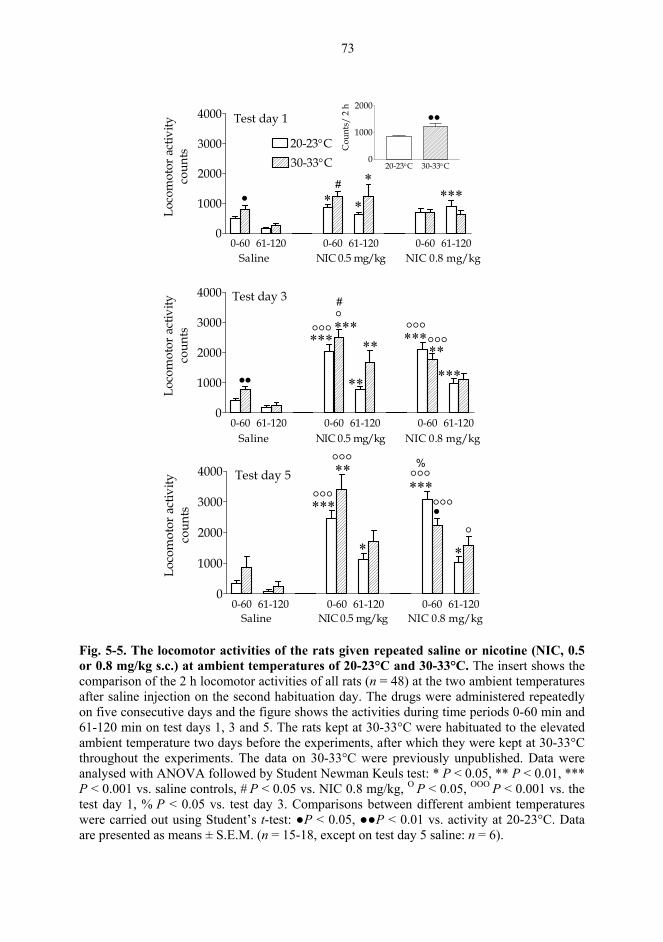
73
0-60 61-120 0-60 61-120 0-60 61-1200
1000
2000
3000
4000
20-23°C30-33°C
Test day 1
• * ****
*#Lo
com
otor
act
ivity
coun
ts20-23°C 30-33°C
0
1000
2000••
Cou
nts/
2 h
0-60 61-120 0-60 61-120 0-60 61-1200
1000
2000
3000
4000 Test day 3
***°°°
••
***°°°°***
**
** *****
°°°
#
Loco
mot
or a
ctiv
ityco
unts
0-60 61-120 0-60 61-120 0-60 61-1200
1000
2000
3000
4000 Test day 5
******°°°°°°
•
**°°°
* *
°°°
°
%
Loco
mot
or a
ctiv
ityco
unts
Saline NIC 0.5 mg/kg NIC 0.8 mg/kg
Saline NIC 0.5 mg/kg NIC 0.8 mg/kg
Saline NIC 0.5 mg/kg NIC 0.8 mg/kg
Fig. 5-5. The locomotor activities of the rats given repeated saline or nicotine (NIC, 0.5 or 0.8 mg/kg s.c.) at ambient temperatures of 20-23°C and 30-33°C. The insert shows the comparison of the 2 h locomotor activities of all rats (n = 48) at the two ambient temperatures after saline injection on the second habituation day. The drugs were administered repeatedly on five consecutive days and the figure shows the activities during time periods 0-60 min and 61-120 min on test days 1, 3 and 5. The rats kept at 30-33°C were habituated to the elevated ambient temperature two days before the experiments, after which they were kept at 30-33°C throughout the experiments. The data on 30-33°C were previously unpublished. Data were analysed with ANOVA followed by Student Newman Keuls test: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. saline controls, # P < 0.05 vs. NIC 0.8 mg/kg, O P < 0.05, OOO P < 0.001 vs. the test day 1, % P < 0.05 vs. test day 3. Comparisons between different ambient temperatures were carried out using Student’s t-test: ●P < 0.05, ●●P < 0.01 vs. activity at 20-23°C. Data are presented as means ± S.E.M. (n = 15-18, except on test day 5 saline: n = 6).

74
When locomotor activities at the elevated ambient temperature of 30-33°C on test days 1, 3
and 5 were analysed, the two-way ANOVA for repeated measures revealed that during 0-120
min there were significant treatment-effects [Fig. 5-5; 0-60 min: F(2,66) = 21.4, P < 0.0001; 61-
120 min: F(2,66) = 6.8, P = 0.0021] and during 0-60 min a significant day(1, 3 and 5)-effect
[F(2,132) = 51.5, P < 0.0001] and treatment × day-interaction [F(4,132) = 8.1, P < 0.0001]. Thus,
when given repeatedly, nicotine increased the activity throughout the 2 h test at 0.5 and 0.8
mg/kg (Student Newman Keuls P < 0.05: NIC 0.5/0.8 vs. SAL) and the stimulant effects of
nicotine were significantly enhanced by repeated treatment as compared with the saline
controls. However, the elevated ambient temperature of 30-33°C did not significantly alter the
acute or sensitized effects of nicotine as compared with the effects at 20-23°C.
The rats given either saline or nicotine 0.5 mg/kg tended to be more active at 30-33°C than at
20-23°C, while the rats given nicotine 0.8 mg/kg tended to be less active at 30-33°C than at
20-23°C. Compared with the corresponding group at 20-23°C, the rats given nicotine 0.8
mg/kg at 30-33°C were less active during the second test hour on test day 1, but when
nicotine was given repeatedly the rats were less active also during the first test hour on test
days 3 and 5. In fact, on test day 5 the rats given 0.8 mg/kg at 30-33°C were significantly less
active during 0-60 min than the corresponding rats at 20-23°C (Student’s t-test, P = 0.0337).
The effects of repeated epibatidine (0.6 and 3.0 µg/kg s.c.) on locomotor activity are
presented in Fig. 5-6. Both acute doses of epibatidine significantly increased locomotor
activity during the period of 31-60 min on test day 1 (P < 0.05). The locomotor stimulant
effects of epibatidine were enhanced after the repeated administration, at the 3.0 µg/kg dose
more markedly than at the 0.6 µg/kg dose. The rats given epibatidine 0.6 µg/kg were more
active on test day 5 than on test days 1 and 3 (P < 0.05) and the rats given epibatidine 3.0
µg/kg were more active on test days 3 and 5 than on test day 1 (P < 0.05). On the challenge
day, the activity of the rats repeatedly treated with epibatidine 3.0 µg/kg was significantly
enhanced from that on the test day 1, compared with the corresponding saline controls.
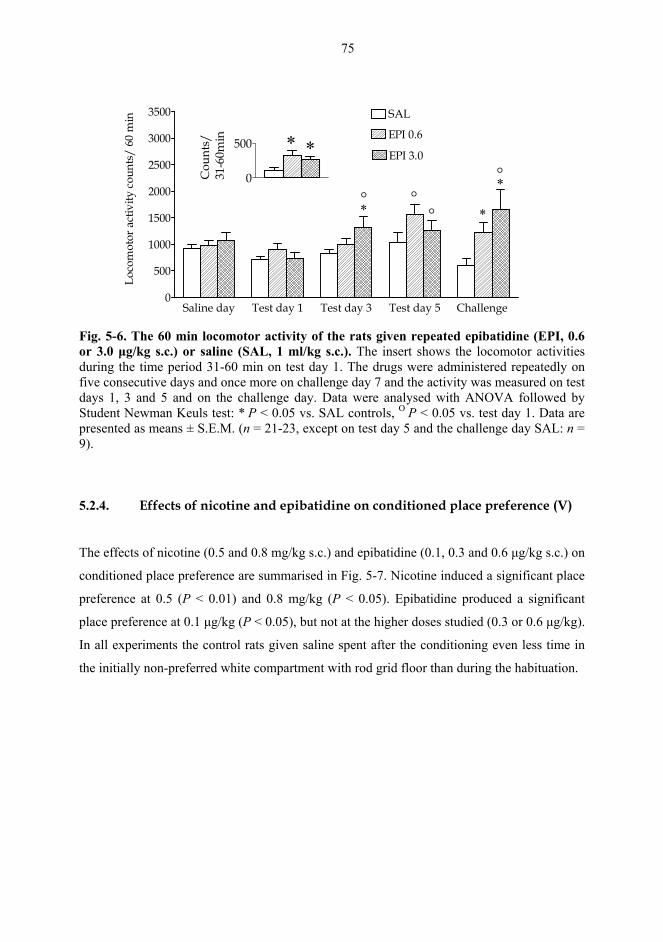
75
Saline day Test day 1 Test day 3 Test day 5 Challenge 0
500
1000
1500
2000
2500
3000
3500
EPI 3.0
EPI 0.6
SAL
* *
*
Loco
mot
or a
ctiv
ity c
ount
s/ 6
0 m
in
° °°
°0
500 * *
Cou
nts/
31-6
0min
Fig. 5-6. The 60 min locomotor activity of the rats given repeated epibatidine (EPI, 0.6 or 3.0 µg/kg s.c.) or saline (SAL, 1 ml/kg s.c.). The insert shows the locomotor activities during the time period 31-60 min on test day 1. The drugs were administered repeatedly on five consecutive days and once more on challenge day 7 and the activity was measured on test days 1, 3 and 5 and on the challenge day. Data were analysed with ANOVA followed by Student Newman Keuls test: * P < 0.05 vs. SAL controls, O P < 0.05 vs. test day 1. Data are presented as means ± S.E.M. (n = 21-23, except on test day 5 and the challenge day SAL: n = 9).
5.2.4. Effects of nicotine and epibatidine on conditioned place preference (V)
The effects of nicotine (0.5 and 0.8 mg/kg s.c.) and epibatidine (0.1, 0.3 and 0.6 µg/kg s.c.) on
conditioned place preference are summarised in Fig. 5-7. Nicotine induced a significant place
preference at 0.5 (P < 0.01) and 0.8 mg/kg (P < 0.05). Epibatidine produced a significant
place preference at 0.1 µg/kg (P < 0.05), but not at the higher doses studied (0.3 or 0.6 µg/kg).
In all experiments the control rats given saline spent after the conditioning even less time in
the initially non-preferred white compartment with rod grid floor than during the habituation.
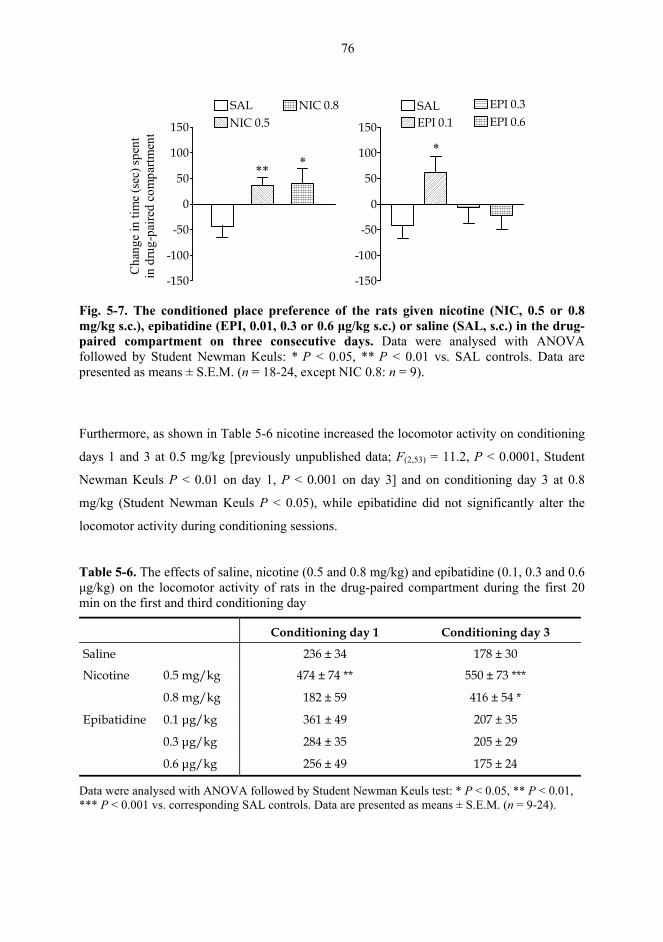
76
-150
-100
-50
0
50
100
150
SALNIC 0.5
NIC 0.8
** *
Cha
nge
in ti
me
(sec
) spe
ntin
dru
g-pa
ired
com
partm
ent
-150
-100
-50
0
50
100
150
SALEPI 0.1
EPI 0.3EPI 0.6
*
Fig. 5-7. The conditioned place preference of the rats given nicotine (NIC, 0.5 or 0.8 mg/kg s.c.), epibatidine (EPI, 0.01, 0.3 or 0.6 µg/kg s.c.) or saline (SAL, s.c.) in the drug-paired compartment on three consecutive days. Data were analysed with ANOVA followed by Student Newman Keuls: * P < 0.05, ** P < 0.01 vs. SAL controls. Data are presented as means ± S.E.M. (n = 18-24, except NIC 0.8: n = 9).
Furthermore, as shown in Table 5-6 nicotine increased the locomotor activity on conditioning
days 1 and 3 at 0.5 mg/kg [previously unpublished data; F(2,53) = 11.2, P < 0.0001, Student
Newman Keuls P < 0.01 on day 1, P < 0.001 on day 3] and on conditioning day 3 at 0.8
mg/kg (Student Newman Keuls P < 0.05), while epibatidine did not significantly alter the
locomotor activity during conditioning sessions.
Table 5-6. The effects of saline, nicotine (0.5 and 0.8 mg/kg) and epibatidine (0.1, 0.3 and 0.6 µg/kg) on the locomotor activity of rats in the drug-paired compartment during the first 20 min on the first and third conditioning day
Conditioning day 1 Conditioning day 3
Saline 236 ± 34 178 ± 30
Nicotine 0.5 mg/kg 474 ± 74 ** 550 ± 73 ***
E i
0.8 mg/kg 182 ± 59 416 ± 54 *
Epibatidine 0.1 µg/kg 361 ± 49 207 ± 35
0.3 µg/kg 284 ± 35 205 ± 29
0.6 µg/kg 256 ± 49 175 ± 24
Data were analysed with ANOVA followed by Student Newman Keuls test: * P < 0.05, ** P < 0.01, *** P < 0.001 vs. corresponding SAL controls. Data are presented as means ± S.E.M. (n = 9-24).

77
6. DISCUSSION
6.1. Microdialysis studies
6.1.1. Effects of nicotine and dopaminergic drugs on nigrostriatal dopamine (I)
In parkinsonian patients, smoking/nicotine is known to alleviate motor symptoms (Moll,
1926; Marshall and Schnieden, 1966; Ishikawa and Miyatake, 1993; Fagerström et al., 1994;
Kelton et al., 2000), suggesting that it stimulates dopaminergic systems in the brain. Indeed,
acute nicotine increases in vivo extracellular dopamine concentration in the CPu of rats when
given systemically (Damsma et al., 1988) or locally (Toth et al., 1992). When given
intranigrally, acute nicotine also produces contralateral rotations, a behaviour associated with
increased release of dopamine in the dorsal striatum (Kaakkola, 1980). In parkinsonian
monkeys, nicotine and some novel nAChR agonists have been found to enhance the effects of
levodopa (Schneider et al., 1998b; Domino et al., 1999). However, although acute nicotine
activates the nigrostriatal pathway, it fails to induce ipsilateral rotations in unilaterally 6-
OHDA lesioned rats when given systemically (Fuxe et al., 1977; Kaakkola, 1981). This might
be due to the particularly effective neuronal reuptake of dopamine in the CPu (Marshall et al.,
1990; Garris and Wightman, 1994; Hoffman and Gerhardt, 1998), which terminates the effect
of dopamine in the synapse. Benwell and Balfour (1992, 1997) have shown that nicotine
increases dopamine release in the dorsal striatum in the presence of the DAT inhibitor
nomifensine.
The present study is the first to collectively examine how the inhibition of either DAT,
COMT, MAO-B or presynaptic dopamine receptors, affects nicotine-induced dopamine
release in the nigrostriatal pathway of rats. Moreover, the effects of COMT inhibition on
nicotine-induced synaptic dopamine release have not previously been studied. When nicotine
was given acutely (s.c.) in the present study, it increased the extracellular level of dopamine in
the CPu, which corresponds with earlier reports (Imperato et al., 1986; Damsma et al., 1988).
The DAT inhibitor nomifensine, elevated the extracellular dopamine level to a greater extent
than nicotine alone, but in combination these two drugs produced an additive effect on the
elevation of the dopamine level. As discussed in detail below, this elevation was also reflected

78
by the ipsilateral rotational behaviour of 6-OHDA-lesioned rats, because nomifensine and
nicotine in combination induced significantly more ipsilateral rotations than either drug alone.
However, similar additive or synergistic effects on the extracellular dopamine level in the
CPu were not found when nicotine was administered in combination with the other drugs
studied. The COMT inhibitor tolcapone, or the dopamine receptor blocker haloperidol, did
not significantly increase the extracellular dopamine level or the nicotine-elevated dopamine
level in the CPu. Pretreatment with the MAO-B inhibitor selegiline, significantly increased
the basal extracellular dopamine concentration, but did not further enhance the nicotine-
elevated dopamine level. Furthermore, mecamylamine did not alter the effects of
nomifensine, tolcapone or haloperidol on the extracellular levels of dopamine, its metabolites
or 5-HIAA in the CPu (unpublished results). This differs from previous studies in which
mecamylamine inhibited the activity of dopaminergic neurons, for example, by reducing the
haloperidol-induced elevation of striatal HVA concentrations (Ahtee and Kaakkola, 1978;
Clarke et al., 1985a; Grenhoff et al., 1986; Haikala and Ahtee, 1988). To summarize, these
findings support the important role of dopamine reuptake in the termination of dopamine’s
effect in the synapse, while the role of COMT or MAO-B seems to be minimal under normal
metabolic conditions in the rat striatum (Huotari et al., 2002).
Nicotine significantly elevated the extracellular levels of DOPAC and HVA in the present
study. Previously, nicotine has been shown to slightly increase (Benwell and Balfour, 1997)
or fail to alter the levels of dopamine metabolites in the dorsal striatum (Imperato et al., 1986;
Damsma et al., 1988). Nomifensine did not significantly alter the levels of dopamine
metabolites, but it significantly attenuated the nicotine-induced increase in the DOPAC level.
Changes in the level of DOPAC are thought to reflect changes in the intracellular dopamine
metabolism, rather than changes in dopamine release (Roffler-Tarlov et al., 1971). Thus, it
seems that nomifensine, by inhibiting the reuptake of dopamine into the nerve terminal,
attenuates the nicotine-induced elevation in intracellular dopamine metabolism. Consistent
with its mechanism of action, inhibition of COMT, tolcapone increased the level of DOPAC
and decreased that of HVA in the CPu. This is in agreement with earlier reports (Kaakkola
and Wurtman, 1992). Tolcapone had an additive effect on the nicotine-induced increase in the
DOPAC level when compared with the elevation induced by either tolcapone or nicotine
alone. Thus, nicotine’s effect on dopamine metabolism is enhanced by inhibition of COMT.
However, neither inhibition of MAO-B (selegiline) nor blockade of presynaptic dopamine

79
receptors (haloperidol), had such additive effects on the DOPAC level in combination with
nicotine. Thus, the nicotine-induced increase in dopamine metabolism is attenuated by
inhibition of DAT and enhanced by inhibition of COMT.
6.1.2. Effects of nicotine and epibatidine on dopaminergic transmission (II)
The aim of Study II was to clarify the effects of the nAChR agonist epibatidine, on
dopaminergic transmission in the CPu and NAc, and to compare those effects with that of
nicotine. (±)-Epibatidine has higher relative affinities at α4β2, α3β2, α3β4 and α7 nAChR
subtypes than nicotine (Table 2-2; Gerzanich et al., 1995; Xiao et al., 1998; Hahn et al.,
2003). Epibatidine also desensitizes α4β2 nAChRs at concentrations that do not activate the
receptor (Buisson et al., 2000). The α4β2 subtype is the most abundant nAChR subtype in the
brain (Whiting and Lindstrom, 1986; Wada et al., 1989; Seguela et al., 1993) and has an
important role in the regulation of dopamine release (Kulak et al., 1997; Kaiser et al., 1998;
Picciotto et al., 1998; Salminen et al., 2004). Epibatidine is considerably more potent in the
stimulation of dopamine release from rat striatal slices than (-)-nicotine (Sullivan et al., 1994).
In addition, epibatidine’s effects on dopamine overflow in the dorsal and ventral striatum
seem to differ from those of nicotine (Seppä and Ahtee, 2000). In the present study,
epibatidine was studied at a range of doses from 0.1 to 3.0 µg/kg and nicotine was studied at
the dose 0.5 mg/kg, at which it has been found to increase striatal dopamine overflow without
causing toxic side effects (Imperato et al., 1986; Benwell and Balfour, 1997; Ferger and
Kuschinsky, 1997; Seppä and Ahtee, 2000).
The dose-response curve of epibatidine-induced dopamine release in the CPu was bell-
shaped, because epibatidine increased dopamine overflow in the CPu at moderate doses (0.6
and 1.0 µg/kg), but not at higher doses (2.0 or 3.0 µg/kg). The low dose of epibatidine (0.3
µg/kg) modestly, but significantly decreased dopamine overflow in the CPu. A similar dose-
response curve has previously been reported for nicotine, with the maximal increase in
dopamine overflow in the CPu at the 0.4 mg/kg dose (Benwell and Balfour, 1997). The bell-
shaped dose-response curves of dopamine release in the CPu may result from dose-dependent
desensitization of nAChR subtypes, which mediate dopamine release (Haikala et al., 1986;
Haikala and Ahtee, 1988; Benwell et al., 1995). Moreover, these findings might further

80
explain why nicotine at large doses fails to sufficiently stimulate the nigrostriatal pathway to
produce stereotypical behavioural responses, but instead produces catalepsy, a behaviour
associated with reduced activity of the nigrostriatal pathway (Zetler, 1968, 1971; Costall and
Naylor, 1973). The finding that very low or high doses of nAChR agonists do not stimulate
and probably desensitize nAChRs may explain the reportedly diverse effects of nicotine on
parkinsonian symptoms. The repeated effects of nicotine on these symptoms are
contradictory, ranging from a failure to improve motor deficits in MPTP-treated animals
(Fung et al., 1991) or even to worsen parkinsonian motor symptoms (Ebersbach et al., 1999),
to an improvement of motor symptoms in animals (Sershen et al., 1987) and parkinsonian
patients (Moll, 1926; Marshall and Schnieden, 1966; Ishikawa and Miyatake, 1993;
Fagerström et al., 1994; Kelton et al., 2000). The diversity of findings from epidemiological
studies that have investigated the beneficial effects of smoking on the occurrence of
Parkinson’s disease (reviewed by Allam et al., 2004), may also be explained by the bell-
shaped dose-response curve for nicotinic compounds on nigrostriatal dopamine release.
Epibatidine-induced dopamine overflow in the NAc differed from that in the CPu. Epibatidine
increased dopamine overflow in the NAc at the highest dose tested (3.0 µg/kg), but not at the
lower doses (0.1-2.0 µg/kg). This is consistent with Bednar et al. (2004), who recently
reported that epibatidine failed to increase accumbal dopamine overflow at the 2.5 µg/kg
dose. Thus, it seems that three to five times higher dose of epibatidine is required to elevate
dopamine release in the NAc, compared with the CPu. This might be related to structural
and/or physiological differences in the two brain nuclei. Furthermore, the mesolimbic
dopaminergic pathway may contain a different set of nAChR subtypes compared with the
nigrostriatal pathway. Indeed, α7* nAChRs are expressed at higher levels in the VTA than in
the SN (Wooltorton et al., 2003) and nicotine-induced dopamine release in the NAc can be
inhibited by MLA (a selective α7 nAChR antagonist) into the VTA (Schilström et al., 1998a).
This suggests a role for α7* receptors in the VTA, in the mediation of this effect. Although it
is probable that the dopamine release is regulated by more than one nAChR subtype, α7*
nAChRs are suggested to be preferentially involved in accumbal dopamine release, whereas
nAChRs containing α3/α6 subunit mediate effects on dopamine release in the CPu (Kulak et
al., 1997; Kaiser et al., 1998; Schilström et al., 1998a). According to Mansvelder et al. (2002),
long-lasting dopamine release in the mesolimbic system after acute nicotine administration
may be the result of differential sensitivity of nAChRs to desensitization. It is suggested that

81
nAChRs mediating GABA release may be more prone to desensitization than those involved
in glutamate release. α4β2* and α7* nAChR subtypes are thought to mediate GABA release
(Wonnacott et al., 1989; Alkondon et al., 1997; Alkondon and Albuquerque, 2001), while
glutamate release is mediated via α7* nAChRs (Kaiser and Wonnacott, 2000). Epibatidine has
a higher relative affinity at α7 nAChRs than nicotine (Table 2-2; Gerzanich et al., 1995; Xiao
et al., 1998) and the effects of epibatidine on α4β2 nAChRs are characterized by slow offset,
possibly a consequence of longer-lasting binding of epibatidine to the receptor (Buisson et al.,
2000). The pronounced activation of nAChRs that mediate inhibitory GABAergic
transmission, may explain the present finding that the lower doses of epibatidine failed to
increase accumbal dopamine release. Increasing the dose of epibatidine to 3.0 µg/kg may
preferentially desensitize/inhibit these receptors and thus inhibit GABA release, while the
excitatory effects of glutamate on dopamine release remain.
Table 6-1 summarizes the effects of nicotine 0.5 mg/kg and epibatidine 0.6 µg/kg on
dopamine overflow in the CPu and NAc. These doses induce about similar effects on striatal
dopamine in rats without producing toxic side effects that higher doses, such as the 0.8 mg/kg
dose of nicotine and the 3.0 µg/kg dose of epibatidine produce. The present finding that
epibatidine enhances dopamine overflow in the CPu at lower doses than that in the NAc is the
reverse of the effects of nicotine, because nicotine enhances dopamine overflow from the
NAc at lower doses than from the CPu (Benwell and Balfour, 1997). Moreover, in the present
study nicotine increased dopamine overflow in both brain nuclei to a higher level than
epibatidine. These findings are probably due to the different affinities of epibatidine and
nicotine at various nAChR subtypes that are involved in the regulation of dopamine release,
as well as regional variations in the expression of these receptors. Indeed, as a result of its
higher affinity for nAChRs, epibatidine may desensitize/inactivate the nAChRs in the
mesolimbic dopaminergic pathway more easily than nicotine, which could result in a failure
by epibatidine to increase dopamine overflow in the NAc (Table 6-1). However, when
nicotine is applied, the nAChRs mediating the stimulatory effects on dopamine release may
remain functional. The modest stimulatory effect of epibatidine on the mesolimbic
dopaminergic pathway, compared to nicotine, may also explain why mice self-administer
nicotine, but do not self-administer epibatidine (Rasmussen and Swedberg, 1998).
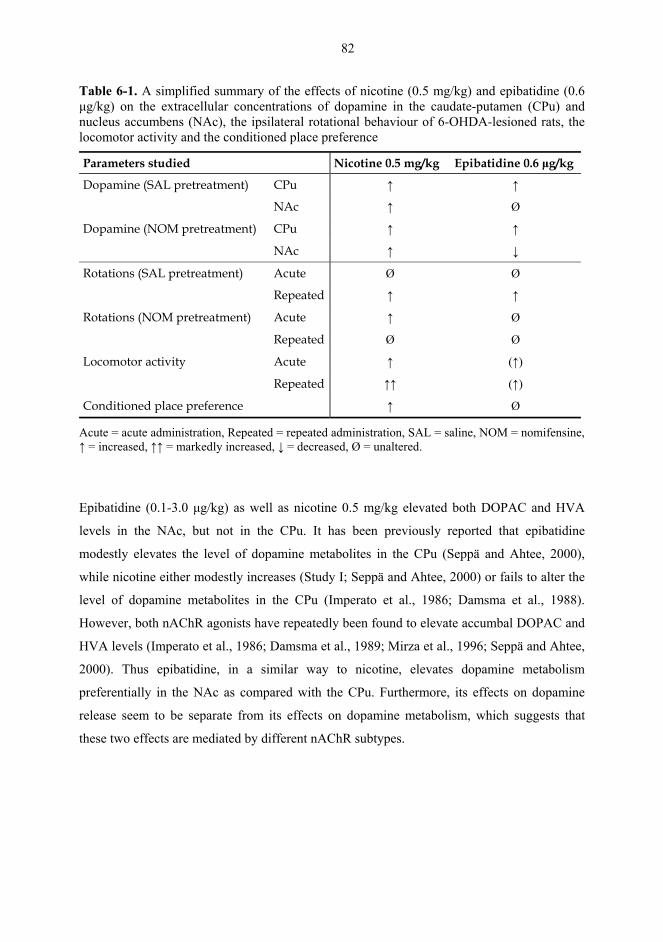
82
Table 6-1. A simplified summary of the effects of nicotine (0.5 mg/kg) and epibatidine (0.6 µg/kg) on the extracellular concentrations of dopamine in the caudate-putamen (CPu) and nucleus accumbens (NAc), the ipsilateral rotational behaviour of 6-OHDA-lesioned rats, the locomotor activity and the conditioned place preference
Parameters studied Nicotine 0.5 mg/kg Epibatidine 0.6 µg/kg
Dopamine (SAL pretreatment) CPu ↑ ↑
NAc ↑ Ø
Dopamine (NOM pretreatment) CPu ↑ ↑
NAc ↑ ↓
Rotations (SAL pretreatment) Acute Ø Ø
Repeated ↑ ↑
Rotations (NOM pretreatment) Acute ↑ Ø
Repeated Ø Ø
Locomotor activity Acute ↑ (↑)
Repeated ↑↑ (↑)
Conditioned place preference ↑ Ø
Acute = acute administration, Repeated = repeated administration, SAL = saline, NOM = nomifensine, ↑ = increased, ↑↑ = markedly increased, ↓ = decreased, Ø = unaltered.
Epibatidine (0.1-3.0 µg/kg) as well as nicotine 0.5 mg/kg elevated both DOPAC and HVA
levels in the NAc, but not in the CPu. It has been previously reported that epibatidine
modestly elevates the level of dopamine metabolites in the CPu (Seppä and Ahtee, 2000),
while nicotine either modestly increases (Study I; Seppä and Ahtee, 2000) or fails to alter the
level of dopamine metabolites in the CPu (Imperato et al., 1986; Damsma et al., 1988).
However, both nAChR agonists have repeatedly been found to elevate accumbal DOPAC and
HVA levels (Imperato et al., 1986; Damsma et al., 1989; Mirza et al., 1996; Seppä and Ahtee,
2000). Thus epibatidine, in a similar way to nicotine, elevates dopamine metabolism
preferentially in the NAc as compared with the CPu. Furthermore, its effects on dopamine
release seem to be separate from its effects on dopamine metabolism, which suggests that
these two effects are mediated by different nAChR subtypes.

83
6.1.3. Effects of nicotine and epibatidine on dopamine in combination with nomifensine (III)
When the DAT inhibitor nomifensine is administered locally, it has been found to intensify
the effects of nicotine on striatal dopamine overflow (Benwell and Balfour, 1992, 1997). In
Study I, pretreatment with systemic nomifensine was found to enhance the effects of nicotine
on the extracellular dopamine level in the CPu. In the present study (III), nomifensine
increased the extracellular dopamine concentration preferentially in the NAc, which agrees
with previous findings (Carboni et al., 1989b; Cass et al., 1993). Nomifensine also decreased
the extracellular level of DOPAC in the CPu and to an even greater extent in the NAc. Indeed,
as nomifensine prevents the interaction of dopamine with intraneuronal MAO, the increased
level of extracellular dopamine and decreased level of DOPAC are expected. Both nicotine
and epibatidine elevated accumbal DOPAC level that was reduced by nomifensine, which
corresponds with their effects on accumbal dopamine metabolites shown in Studies I and II.
In the present study, nomifensine also increased 5-HIAA levels in the CPu and NAc, but this
effect was not altered by nicotine or epibatidine. It can be hypothesed that serotonergic
transmission might be altered as a result of increased extracellular dopamine level.
Consistent with the bell-shaped dose-response curve of dopamine overflow in the CPu found
in Study II, epibatidine at a moderate dose (0.6 µg/kg), but not at a higher dose (3.0 µg/kg)
elevated nomifensine-induced increase in extracellular dopamine in the CPu. Nicotine 0.5
mg/kg enhanced this nomifensine-elevated extracellular dopamine in the CPu. Thus, the
effects of nAChR agonists on dopamine overflow in the CPu appear to be similar in the
presence and absence of dopamine uptake inhibition (Table 6-1). Consistent with Study II and
with nicotine’s effects in the CPu, nicotine enhanced the increase in extracellular dopamine in
the NAc of nomifensine-pretreated rats. This finding agrees with studies by Benwell and
Balfour (1992, 1997) in which local nomifensine intensified nicotine’s effects on accumbal
dopaminergic transmission. It also agrees with previous reports that nicotine enhances the
elevation of accumbal dopamine induced by other DAT inhibitors (cocaine, methylphenidate)
(Zernig et al., 1997; Gerasimov et al., 2000). However, in contrast to nicotine, epibatidine at a
moderate dose (0.6 µg/kg) decreased (Table 6-1) and at a higher dose (3.0 µg/kg) did not
alter, the nomifensine-increased extracellular dopamine level. Thus, the different affinities of
epibatidine and nicotine for different nAChR subtypes (Table 2-2 on page 46), as well as

84
regional variations in the expression of these receptors, probably explain our findings that
epibatidine’s effects on accumbal dopamine overflow differ from those of nicotine.
As discussed in the previous chapter, spatial and temporal differences in nAChR activity on
both excitatory and inhibitory neurons in the mesolimbic pathways might result in the
elevated release of mesolimbic dopamine after systemic administration of nicotine
(Mansvelder et al., 2002). Interestingly, smoking tobacco (10-50 ng/ml; Benowitz et al.,
1990) or subcutaneous administration of the 0.5 mg/kg dose of nicotine to rats (80 ng/ml;
Salminen et al., 1999), leads to plasma nicotine concentrations at which non-α7 nAChRs (e.g.
β2-subunit) are desensitized, while those containing the α7-subunit remain functional and are
likely to mediate the effects on accumbal dopamine release (Wooltorton et al., 2003).
However epibatidine, when compared to nicotine, has a higher relative affinity at α7 nAChRs
(Table 2-2; Gerzanich et al., 1995; Xiao et al., 1998) and at low concentration it desensitizes
nAChRs (e.g. α4β2*) more markedly than nicotine (Buisson et al., 2000). Thus, similar to the
findings in the saline-pretreated rats in Study II, the inhibitory effect of epibatidine at the
lower dose (0.6 µg/kg) on accumbal dopamine overflow in nomifensine-pretreated rats, could
also be due to desensitization of nAChR subtypes that mediate the effects on inhibitory
GABAergic transmission, at low epibatidine concentrations. In contrast, nicotine enhances
accumbal dopamine overflow in nomifensine-pretreated rats and in previous studies nicotine
has been found to elevate dopamine overflow at even lower doses (e.g. 0.1 mg/kg) when the
DAT is inhibited by intra-accumbal nomifensine (Benwell and Balfour, 1997).
The inhibitory effect of epibatidine in nomifensine-pretreated rats also differ from its effects
in saline-pretreated rats (Study II; Table 6-1). In saline-pretreated rats epibatidine did not alter
accumbal dopamine overflow at 0.6 µg/kg, but elevated it at 3.0 µg/kg. Thus, the effects of
the potent nAChR agonist epibatidine, on dopamine overflow in the NAc are different in the
presence of DAT inhibition by nomifensine, compared with epibatidine’s effects under
normal physiological conditions. Elevation of extracellular dopamine by nomifensine causes a
hyperpolarization of dopaminergic neurons and reduces their firing rate (Mercuri et al., 1992).
The present study has shown that the NAc is more sensitive to nomifensine than the CPu.
Recently, the effects of nicotine on dopamine release were shown to be dependent on the
firing rate of dopaminergic neurons. Desensitization of nAChRs inhibits dopamine release at
low firing rates, but accelerating the firing rate attenuates this effect (Rice and Cragg, 2004;

85
Zhang and Sulzer, 2004). Thus, hyperpolarization induced by nomifensine might be expected
to shift the effects of nAChR agonists to low frequency effects, i.e. to a depression of
dopamine release. This might explain why the low dose of epibatidine, which probably
desensitized nicotinic receptors (Buisson et al., 2000), was found to decrease dopamine
overflow in the presence of nomifensine. Consistent with this, epibatidine was recently
reported to decrease accumbal dopamine overflow in Sprague-Dawley rats (Bednar et al.,
2004). The rats in the study by Bednar et al. recovered for 17-22 h after the surgery, while our
rats were tested 5-7 days after the implantation of the guide cannula. Hence, it is possible that
the extracellular dopamine level was elevated in the same manner as in our nomifensine-
pretreated rats, which elevation, in turn, could emphasize epibatidine’s effects on mechanisms
inhibiting the accumbal dopamine release.
6.2. Behavioural studies
6.2.1. Effects of acute nicotine on rotational behaviour in combination with
nomifensine or levodopa (I, unpublished results)
The rotational behaviour experiments were carried out to see if changes in the striatal
extracellular dopamine level correspond with rotational behaviour in unilaterally 6-OHDA-
lesioned rats. These rats represent a parkinsonian animal model, and their ipsilateral rotational
behaviour is thought to reflect an increase in dopamine release in the intact nigrostriatal
pathway (Ungerstedt, 1968; Ungerstedt and Arbuthnott, 1970; Ungerstedt, 1971a, b). In the
present study we investigated the effects of nicotine in combination with nomifensine,
because these two drugs produced the most substantial increase in extracellular dopamine
level in Study I. The modest increase in dopamine overflow induced by nicotine was
accompanied by few ipsilateral rotations, while the greater increase in dopamine induced by
nomifensine was accompanied by a greater number of ipsilateral rotations. However, the
combination of both nicotine and nomifensine had an additive effect on the number of
ipsilateral rotations, and that correlated with the elevated dopamine level in the CPu. The
combined effect of these drugs on rotations was inhibited by mecamylamine, suggesting that
it is mediated by neuronal nAChRs. Thus, even when acute nicotine releases dopamine in the
CPu, the released dopamine is inadequate to induce rotational behaviour, because the DAT

86
termination of dopamine’s action in the synaptic cleft is so effective. Nicotine, or other
nAChR agonists, in combination with a DAT inhibitor could thus alleviate the motor
symptoms of the early stage of Parkinson’s disease when functional dopaminergic neurons
remain.
In the advanced stage of Parkinson’s disease, the most effective drug treatment is levodopa in
combination with peripheral inhibitors of dopamine metabolism (Olanow, 2004). Thus, we
carried out rotational experiments to see whether nicotine enhances the effects of levodopa on
contralateral rotational behaviour. Indeed, nicotine tended to potentiate the rotational
behaviour, although the response failed to reach significance, thought to be due to the
variability between animals. Differences are likely to have arisen in the concentrations of
carbidopa and tolcapone administered, because these drugs had to be given as suspension, due
to their poor solubility. Thus, the dose of levodopa crossing the blood-brain barrier probably
varied between animals. Although more experiments are needed to confirm these findings, the
data agrees with previous studies in which levodopa produced greater contraversive circling
in MPTP-treated monkeys in combination with nicotine than with saline (Domino et al.,
1999). In addition, novel nAChR agonists have been reported to potentiate levodopa’s effects
on motor symptoms in MPTP-treated monkeys (Schneider et al., 1998a, b), as well as to
reverse both cognitive and motor deficits in combination with levodopa and benserazide, in
reserpine-treated rats (Menzaghi et al., 1997a). Thus, the use of subtype-selective nAChR
agonist in combination with levodopa may allow a reduction in levodopa dosage in some
parkinsonian patients and thus reduce complications associated with long-term levodopa use.
6.2.2. Effects of repeated nicotine and epibatidine on rotational behaviour (IV)
The aim of Study IV was to investigate the acute and repeated effects of epibatidine and
nicotine on the rotational behaviour of 6-OHDA-lesioned rats. Although epibatidine
preferentially stimulates nigrostriatal dopaminergic transmission (Study II; Seppä and Ahtee,
2000) and therefore is likely to affect rotational behaviour, this has not previously been
investigated. However, the effect of systemic nicotine on rotational behaviour has previously
been studied following acute and chronic administration (Fuxe et al., 1977; Kaakkola, 1981;
Lapin et al., 1987; Domino et al., 1999), but a profound investigation on the development of

87
sensitization of rotational behaviour after repeated nicotine administration has not. Acute
nomifensine administered intrastriatally, has been reported to produce contralateral rotations
in mice (Worms et al., 1986a) and when given systemically to produce ipsilateral rotations in
rodents with unilateral nigrostriatal lesions (Costall et al., 1975; Worms et al., 1986b; Nakachi
et al., 1995). To our knowledge, the effects of repeated nomifensine administration have not
previously been studied.
In the present study, acute epibatidine and nicotine produced only few ipsilateral rotations in
rats and the effect of nicotine was similar to that found in Study I (Table 6-1). Hence, it seems
that acute epibatidine, in a similar way to acute nicotine (Fuxe et al., 1977; Kaakkola, 1981),
fails to elevate synaptic dopamine concentration in the CPu sufficiently to produce rotational
behaviour. When the nAChR agonists were given repeatedly, the effect of nicotine on the
ipsilateral rotational behaviour was the most enhanced, that of epibatidine 0.6 µg/kg was
somewhat less enhanced (Table 6-1), and the effect of epibatidine 3.0 µg/kg was not
significantly enhanced. These findings are consistent with those from the microdialysis
studies (Study II). Study II showed that nicotine and epibatidine at 0.6 µg/kg, but not at 3.0
µg/kg, elevated extracellular dopamine in the CPu, which implies that the effects of nicotine
and epibatidine (0.6 µg/kg) on dopaminergic transmission in the nigrostriatal pathway were
sensitized by repeated administration of nAChR agonists.
In light of the results from Study I that nAChR agonists do not induce rotational behaviour,
due to the effective dopamine reuptake in the CPu, we studied the effects of either acute or
repeated epibatidine and nicotine in combination with nomifensine. Nomifensine alone
induced ipsilateral rotational behaviour and its effects were sensitized when administered
repeatedly. Indeed, acute systemic nomifensine has been reported to elevate extracellular
striatal dopamine level in rats (Carboni et al., 1989b). Acute nicotine enhanced the
nomifensine-induced rotational behaviour during the first 45 min, while acute epibatidine did
not alter this rotational behaviour (Table 6-1). In Study III, acute nicotine enhanced the
nomifensine-elevated dopamine level both in the CPu and NAc, while epibatidine enhanced
that in the CPu, but inhibited that in the NAc (Table 6-1). Hence, the more pronounced effect
of nicotine on rotational behaviour, compared with epibatidine, may be due to its elevating
effects in both dopaminergic nerve terminals. Furthermore, injection of 6-OHDA into the
MFB leads to an almost complete dopamine depletion in the nigrostriatal pathway, but also

88
causes up to 90% depletion of the mesolimbic dopamine (Mikkola et al., 2002). These
findings thus suggest that the rotational behaviour is mainly mediated by the nigrostriatal
dopaminergic system, but the mesolimbic system probably modulates it (Pycock and
Marsden, 1978).
Repeated administration of nicotine and epibatidine (0.6 µg/kg) slightly enhanced the effect
of repeated nomifensine on rotational behaviour, but this effect was not significant (Table 6-
1). This might be due to a ceiling effect in the rotational behaviour induced by the elevated
endogenous dopamine in the synaptic cleft. In contrast to nicotine and the lower dose of
epibatidine (0.6 µg/kg), the 3.0 µg/kg dose of epibatidine tended to inhibit the sensitized
effect of repeated nomifensine on rotational behaviour, which may be due to desensitization
of nAChRs. Indeed, psychostimulant-induced sensitization evoked by amphetamine or
cocaine is associated with enhanced endogenous activation of nAChRs and blocking nAChRs
with antagonists inhibits the induction of behavioural sensitization (Schoffelmeer et al., 2002;
de Rover et al., 2004). Thus, it seems possible that a high dose of nAChR agonist that is likely
to desensitize at least some nAChR subtypes, could inhibit the development of sensitization in
the nomifensine-induced rotational behaviour. However, there may also be other indirect
mechanisms involved, such as release of neurotransmitters like GABA and glutamate that
modulate dopamine release. In fact, an irreversible GABA transaminase inhibitor has been
found to inhibit the sensitization of stereotypical behaviour and locomotor activity associated
with cocaine (Gardner et al., 2002). The sensitization of rotational behaviour of 6-OHDA-
lesioned rats has been associated with the potential of dopaminergic drugs to induce motor
complications, such as dyskinesia (Carey, 1991; Henry et al., 1998). Thus, the finding that the
3.0 µg/kg dose of epibatidine inhibited nomifensine-induced sensitization might be of
therapeutic interest.
6.2.2. Effects of nicotine and epibatidine on locomotor activity (V, unpublished results)
Nicotine is known to stimulate locomotor activity in habituated rats, an effect that becomes
sensitized after repeated nicotine administration (Morrison and Stephenson, 1972; Clarke and
Kumar, 1983a; Ksir et al., 1985; O'Neill et al., 1991; Benwell and Balfour, 1992). This
enhanced behavioural response to nicotine and other addictive drugs is thought to relate to

89
neuronal adaptations, which lead to compulsive drug use. Drugs of abuse, such as nicotine are
known to stimulate cerebral dopaminergic transmission and both stimulation and sensitization
of the mesolimbic/ mesocortical dopaminergic pathway in particular is associated with the
locomotor stimulant actions (Clarke et al., 1988; Corrigall and Coen, 1991; Leikola-Pelho and
Jackson, 1992; Louis and Clarke, 1998). The effects of epibatidine on locomotor activity have
previously been studied after acute administration (Sacaan et al., 1996; Menzaghi et al.,
1997b; Reuben et al., 2000) and only one study has compared the acute effects epibatidine
with those of nicotine (Reuben et al., 2000). Epibatidine’s effects on dopamine release in the
dorsal and ventral striatum have been found to differ from those of nicotine in microdialysis
studies (Study II; Seppä and Ahtee, 2000), suggesting that also its effects on locomotor
activity might differ from those of nicotine.
At the ambient temperature 20-23°C, acute nicotine at 0.5 mg/kg significantly increased
locomotor activity of rats throughout the 2 h experiment. At the higher dose (0.8 mg/kg),
nicotine did not alter the locomotor activity during the first 30 min from the control level, but
increased thereafter the activity up to 2 h. When nicotine was given repeatedly, the acute
locomotor stimulant effects were enhanced at both doses (0.5 and 0.8 mg/kg). These findings
agree with several earlier studies in which repeated administration of nicotine enhanced its
stimulant effects (Ksir et al., 1985; O'Neill et al., 1991; Benwell and Balfour, 1992). These
sensitized effects of nicotine on locomotor activity are probably due to enhanced mesolimbic
dopaminergic transmission (Benwell and Balfour, 1992; Balfour et al., 1998).
The stimulatory effects of nicotine on striatal dopamine release and metabolism have
previously been found to be enhanced by an elevated ambient temperature of 30-33°C
(Haikala, 1986; Leikola-Pelho et al., 1990; Seppä et al., 2000). Therefore, we investigated
whether this enhancement of dopamine release is accompanied by enhanced locomotor
activity. At the ambient temperature of 30-33°C, the basal activity of the rats was significantly
higher than at 20-23°C and repeated exposure to the test boxes and saline injections did not
alter this effect. In earlier studies, the elevation of ambient temperature alone was not found to
alter basal extracellular concentrations of dopamine or its metabolites in the CPu or NAc
(Seppä et al., 2000; Seppä, 2001), suggesting that other mechanisms and/or neurotransmitters
may underlie this phenomenon. Furthermore, rats given either acute or repeated nicotine at
0.5 mg/kg were somewhat more active, while rats given nicotine at 0.8 mg/kg were somewhat

90
less active, particularly on test day 5, at the elevated ambient temperature of 30-33°C,
compared to 20-23°C. However, the responses to acute or repeated nicotine were not
significantly different at 30-33°C from those at 20-23°C. Acute nicotine increased activity of
rats at 0.5 mg/kg immediately and at 0.8 mg/kg after a delay, and the stimulant response was
sensitized after repeated administration at both doses. This is in line with the findings that a
higher ambient temperature enhances nicotine-induced dopamine release only in the CPu, but
not in the NAc (Seppä et al., 2000; Seppä, 2001).
Acute epibatidine depressed locomotor activity at the highest dose (3.0 µg/kg) during the first
10 min, but activity was increased during 31-60 min at both 0.6 and 3.0 µg/kg. These findings
are consistent with earlier studies in which acute epibatidine at 3.0 µg/kg depressed the
activity of habituated rats for 10-15 min, but increased it between 30-40 min after
administration (Sacaan et al., 1996; Menzaghi et al., 1997b; Reuben et al., 2000). After
repeated administration of epibatidine, its locomotor depressant effects were attenuated and
its stimulant effects were enhanced, particularly at the higher dose (3.0 µg/kg). Sensitization
of locomotor activity was more pronounced at 3.0 µg/kg than at 0.6 µg/kg and this is
consistent with the findings in Study II that epibatidine elevates accumbal dopamine release at
3.0 µg/kg, but not at 0.6 µg/kg. In contrast, the 0.6 µg/kg dose of epibatidine, but not 3.0
µg/kg, elevates dopamine release in the CPu. Thus, stimulation of locomotor activity and its
sensitization appear to relate to increased dopaminergic transmission in the mesolimbic
terminal regions as suggested previously (Clarke et al., 1988; Corrigall and Coen, 1991;
Benwell and Balfour, 1992; Leikola-Pelho and Jackson, 1992; Louis and Clarke, 1998).
Comparatively, stimulation of locomotor activity by nicotine is greater than by epibatidine
(Table 6-1), which is consistent with a previous study (Reuben et al., 2000). Furthermore,
repeated epibatidine did not produce such a clear and rapid sensitization as nicotine (Table 6-
1). When compared with nicotine, the more modest stimulatory effect of epibatidine on
locomotor activity, is in line with its modest stimulatory effect on mesolimbic dopaminergic
transmission (Study II; Seppä and Ahtee, 2000). However, dopamine is unlikely to be the
only neurotransmitter mediating the locomotor effects studied here.

91
6.2.3. Effects of nicotine and epibatidine on conditioned place preference (V)
Similarly to other drugs of abuse, nicotine has been found to effectively support drug-seeking
behaviour in rats, as demonstrated by the self-administration model (Corrigall and Coen,
1989; Corrigall et al., 1992; Donny et al., 1995, 1998; Corrigall, 1999; Donny et al., 1999)
and the conditioned place preference paradigm (Fudala et al., 1985; Fudala and Iwamoto,
1986; Acquas et al., 1989; Carboni et al., 1989a; Calcagnetti and Schechter, 1994; Vastola et
al., 2002; Le Foll and Goldberg, 2005). For both nicotine and other drugs of abuse, the
reinforcing and rewarding effects appear to depend upon the direct and/or indirect stimulation
of mesolimbic dopaminergic projections, because lesioning or pharmacological blockade of
these neurons attenuates self-administration (Singer et al., 1982; Corrigall and Coen, 1991;
Corrigall et al., 1992). In addition, the pedunculopontine nucleus that projects into the VTA
seems to be involved in the mediation of the rewarding effects of nicotine (Lança et al.,
2000b; Cousins et al., 2002). The modest effects of epibatidine on the mesolimbic
dopaminergic pathways, compared to those of nicotine (Study II; Seppä and Ahtee, 2000),
suggest that epibatidine might possess a low abuse potential. Indeed, mice did not self-
administer epibatidine (Rasmussen and Swedberg, 1998). Therefore, in the present study we
wanted to compare the effects of nicotine and epibatidine on the conditioned place preference.
Nicotine produced place preference in rats at 0.5 and 0.8 mg/kg and also increased locomotor
activity during the conditioning days. Correspondingly, nicotine has previously been reported
to elicit place preference in rats at 0.1-1.4 mg/kg under similar biased conditions (Fudala et
al., 1985; Fudala and Iwamoto, 1986; Acquas et al., 1989; Carboni et al., 1989a; Calcagnetti
and Schechter, 1994; Le Foll and Goldberg, 2005). In studies by Vastola et al. (2002) nicotine
induced place preference at 0.6 mg/kg in adolescent Sprague-Dawley rats (4-6 weeks old), but
not in adult rats (8-10 weeks). Epibatidine’s effects have not previously been studied using
the conditioned place preference, but Rasmussen et al. (1998) reported that mice did not self-
administer epibatidine infused i.v. at 0.25-1.25 µg/kg, although they did self-administer
nicotine (at doses up to 0.075 mg/kg per i.v. infusion). In the present study, epibatidine
induced place preference in rats at the lowest dose, 0.1 µg/kg, but not at 0.3 or 0.6 µg/kg.
When compared to nicotine, epibatidine’s effect on place preference was modest (Table 6-1),
although at 0.1 µg/kg it did elicit place preference. However, in contrast to nicotine,
epibatidine failed to significantly alter locomotor activity during the conditioning days when

92
given acutely or repeatedly. Thus, the present findings imply that the actions of epibatidine on
the nAChR subtypes that regulate neurotransmitters involved in the mediation of locomotor
activity and motivation, differ from those of nicotine.
In Study II, epibatidine at the doses studied here (0.1, 0.3 or 0.6 µg/kg) did not alter accumbal
dopamine release, although it slightly, but significantly, elevated accumbal extracellular
concentration of dopamine metabolites and the serotonin metabolite 5-HIAA. The mesolimbic
dopaminergic pathway has been suggested to directly and/or indirectly mediate the
motivational effects of nicotine (Corrigall et al., 1992). However, other neurotransmitters,
such as endogenous opioids, may play a role in the acquisition of the reinforcing properties of
nAChR agonists (Pomerleau, 1998) and in the conditioning effects of 0.1 µg/kg of
epibatidine. The nAChR subtypes that mediate effects on these neurotransmitters may
desensitize at higher doses of epibatidine, which fail to elicit place preference. In conclusion,
epibatidine’s rewarding and reinforcing effects seem to occur transiently only at very low
doses. If the epibatidine dose is increased to levels found to alter striatal dopamine release
(Study II) and to stimulate locomotor activity (Study V) this effect is attenuated. Thus, these
findings support the idea that it is possible to develop subtype-selective nAChR agonists that
lack dependence-producing properties.

93
7. SUMMARY AND CONCLUSIONS
In contrast to nicotine, which preferentially stimulated the mesolimbic dopaminergic pathway,
epibatidine increased dopamine overflow at lower doses in the nigrostriatal pathway than in
the mesolimbic pathway. This is probably due to the different affinities of nicotine and
epibatidine at various nAChR subtypes, as well as the fact that there are different nAChR
subtypes located on neurons in the nigrostriatal pathway compared to the mesolimbic
pathway. Indeed, α3/α6* nAChRs appear to mediate effects on dopamine release in the CPu,
while α7* nAChRs are preferentially involved in accumbal dopamine release.
Similar to nicotine, the dose-response curve of epibatidine-induced dopamine release in the
CPu was bell-shaped. Thus, high doses of nAChR agonists probably desensitize nAChR
subunits that mediate effects on dopamine release in the nigrostriatal terminal areas, while
those of the mesolimbic pathway remain functional. These findings may explain the
conflicting results regarding nicotine’s therapeutic effects in animals, as well as in
parkinsonian patients. The pronounced effects of nicotine on mesolimbic dopamine are likely
to be related to its ability to cause addiction.
Nicotine-induced dopamine release was enhanced by inhibition of dopamine reuptake by
nomifensine, but not by inhibition of COMT (tolcapone), MAO-B (selegiline) or presynaptic
dopamine receptors (haloperidol). Hence, effective dopamine reuptake mechanisms in the
CPu appear to terminate the actions of synaptic dopamine release stimulated by nicotine. This
may also explain why dopamine release induced by systemic nicotine fails to induce
ipsilateral rotations in unilaterally 6-OHDA lesioned rats. These findings imply that a DAT
inhibitor, such as nomifensine, could be used to potentiate the effects nicotine or other
nAChR agonists on nigrostriatal dopaminergic transmission.
When given in combination with nomifensine, both nicotine and epibatidine at a low dose
enhanced the nomifensine-induced increase in extracellular dopamine level in the CPu. In the
NAc, however, nicotine enhanced, while epibatidine inhibited the nomifensine-induced
elevation in dopamine level. Thus, nomifensine revealed the inhibitory effect of epibatidine
on accumbal dopamine release. This effect was characteristic for the NAc and was not found
in the CPu. This supports the idea that there are differences in nicotinic regulation of the

94
mesolimbic and nigrostriatal dopaminergic pathways. In addition, drugs that substantially
elevate synaptic dopamine and thus induce hyperpolarization of dopaminergic neurons, may
modulate the net effects of nAChR agonists in the direction of inhibition.
The effects of nicotine and epibatidine on dopaminergic transmission of the nigrostriatal and
mesolimbic pathways corresponded with the rotational behaviour results. Both nicotine and
epibatidine induced ipsilateral rotational behaviour after repeated administration, but not
acutely, which suggests sensitization of dopaminergic transmission, particularly in the
nigrostriatal pathway.
Consistent with its modest effects on mesolimbic dopamine release, acute epibatidine
stimulated locomotor activity less than nicotine. When given repeatedly, epibatidine induced
sensitization of locomotor activity to a lesser extent than nicotine. Furthermore, contrary to
nicotine, epibatidine elicited transient conditioned place preference at a very low dose, but
failed to alter the preference of rats at doses that increase striatal dopamine release and
stimulate locomotor activity. These findings imply that epibatidine does not produce
dependence to the same degree as nicotine.
In conclusion, the present study shows that the effects of epibatidine on the nigrostriatal
dopaminergic pathway and the behaviours preferentially mediated by nigrostriatal dopamine,
resemble those of nicotine, while the effects on the mesolimbic dopaminergic pathways and
the behaviours mediated by mesolimbic dopamine, differ from those of nicotine. Thus, it
seems possible to stimulate the nigrostriatal dopaminergic pathway with nAChR agonists
without significantly affecting the mesolimbic dopaminergic pathway. This supports the
possibility that novel subtype-selective nAChR agonists that enhance dopaminergic
transmission, but lack addictive properties, could be developed. Such compounds may be
clinically useful for controlling tobacco and drug addiction, and improving brain function in
neurodegenerative brain diseases, such as Parkinson’s disease.

95
8. ACKNOWLEDGEMENTS
This study was carried out in the Division of Pharmacology and Toxicology, Department/
Faculty of Pharmacy, University of Helsinki, during the years 2000-2005.
I wish to express my sincere gratitude to the following persons:
Professor Liisa Ahtee, M.D., my teacher and supervisor, for welcoming me to her lab and for
introducing the fascinating field of neuropharmacology to me. Her profound knowledge and
experience, as well as her endless encouragement, optimism and support have enabled me to
successfully complete this project. I will always appreciate her hospitality and kindness
during the preparation of the papers of this thesis.
Professor Raimo Tuominen, M.D., for his support and confidence in me during this thesis
project. I also wish to thank docent Seppo Kaakkola, M.D., for his contribution to the first
study and for valuable discussions during the preparation of its manuscript.
Professor Agneta Nordberg, M.D., Ph.D., and docent Pekka Rauhala, M.D., the reviewers of
this thesis, for devoting their time to review the manuscript, and for their valuable and
encouraging comments for its improvement. I am grateful to Miss Kate Hanrott, B.Sc.(Hons),
University of Bath, United Kingdom, for her skillful revision of the English language of the
manuscript.
All the co-authors of these studies. I am grateful to Anna Linnervuo, M.Sc.(Pharm), Paula
Mielikäinen, M.Sc.(Pharm), Päivi Paldánius, Lic.Med., and Minna Svensk, B.Sc.(Pharm) for
their excellent collaboration and contribution to the studies. I wish to thank Dr. Petteri
Piepponen for his support with the microdialysis studies. I am furthermore thankful to Marjo
Vaha and Kati Valkonen for their skillful technical assistance and friendship throughout this
project.
All my colleagues at the Division of Pharmacology and Toxicology for creating a pleasant
atmosphere to work in, and for all their help, support and both scientific and non-scientific
discussions during these years. Special thanks to the people in the nicotine group. I am

96
particularly grateful to Saara Nuutinen, M.Sc.(Pharm) for numerous inspiring and helpful
scientific discussions, for her constant encouragement and optimism on the days of both
success and problems, and for her valuable friendship, in and outside the lab. I wish to thank
Dr. Elina Ekokoski for all those scientific and non-scientific lunches between the
experiments, her strong knowledge and professionality is really something to look up to. I am
grateful to Anne Tammimäki, M.Sc.(Pharm), Mikko Airavaara, M.Sc.(Pharm), Jelena
Mijatovic, M.Sc.(Pharm), and Johanna Salimäki, Lic.Pharm., for their help, support and
friendship during these studies. I also wish to thank Dr. Ulla-Mari Parkkisenniemi-Kinnunen,
Dr. Helena Raattamaa and Dr. Tiina Seppä for their guidance, support and helpful advises
concerning both teaching and research.
All my dear friends. I particularly wish to thank Hanna Heikkilä, M.Sc.(Pharm), Elina Harju,
M.Sc.(Pharm), Sanna Heikkilä, M.Sc.(Pharm) and Laura Kasslin, M.Sc.(Pharm) for their
interest, encouragement and friendship, not to mention their endless confidence that I will pull
it through.
My warmest gratitude belongs to my family. I wish to thank my parents Sinikka and Tapio
Janhunen and my brother Olli Janhunen for their patience, support and love during my
studies. I am also deeply grateful to my grandparents, especially my grandmother Aune
Talvio, for her effortless support and encouragement throughout all my years at school, and
for teaching me the importance of education. Finally, I wish to thank Fernand for his love,
encouragement and positive thinking during my long period of writing, for all the happiness
that he has brought into my life, and for making me a cup of coffee when I needed it the most.
The financial support from the Academy of Finland, the Sigrid Jusélius Foundation, the
University of Helsinki’s Fund, the Graduate School in Pharmaceutical Research, the Finnish
Cultural Foundation (Elli Turunen Foundation), the Finnish Pharmaceutical Society, the
Finnish Parkinson Foundation and the Paulo Foundation is gratefully acknowledged.
Helsinki, May 2005
Sanna Janhunen

97
9. REFERENCES Abdulla FA, Calaminici M, Wonnacott S, Gray JA, Sinden JD, Stephenson JD (1995) Sensitivity of rat frontal cortical
neurones to nicotine is increased by chronic administration of nicotine and by lesions of the nucleus basalis magnocellularis: comparison with numbers of 3H nicotine binding sites. Synapse 21:281-288.
Abdulla FA, Bradbury E, Calaminici MR, Lippiello PM, Wonnacott S, Gray JA, Sinden JD (1996) Relationship between up-regulation of nicotine binding sites in rat brain and delayed cognitive enhancement observed after chronic or acute nicotinic receptor stimulation. Psychopharmacology 124:323-331.
Acquas E, Carboni E, Leone P, Di Chiara G (1989) SCH 23390 blocks drug-conditioned place-preference and place-aversion: anhedonia (lack of reward) or apathy (lack of motivation) after dopamine-receptor blockade? Psychopharmacology 99:151-155.
Adler LE, Olincy A, Waldo M, Harris JG, Griffith J, Stevens K, Flach K, Nagamoto H, Bickford P, Leonard S, Freedman R (1998) Schizophrenia, sensory gating, and nicotinic receptors. Schizophrenia Bull 24:189-202.
Agid Y (1991) Parkinson's disease: pathophysiology. Lancet 337:1321-1324. Ahtee L, Kaakkola S (1978) Effect of mecamylamine on the fate of dopamine in striatal and mesolimbic areas of rat brain;
interaction with morphine and haloperidol. Br J Pharmacol 62:213-218. Akaike A, Tamura Y, Yokota T, Shimohama S, Kimura J (1994) Nicotine-induced protection of cultured cortical neurons
against N-methyl-D-aspartate receptor-mediated glutamate cytotoxicity. Brain Res 644:181-187. Alkondon M, Albuquerque EX (1995) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. III. Agonist
actions of the novel alkaloid epibatidine and analysis of type II current. J Pharmacol Exp Ther 274:771-782. Alkondon M, Albuquerque EX (2001) Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially
control GABAergic input to CA1 neurons in rat hippocampus. J Neurophysiol 86:3043-3055. Alkondon M, Pereira EF, Albuquerque EX (1996a) Mapping the location of functional nicotinic and gamma-aminobutyric
acidA receptors on hippocampal neurons. J Pharmacol Exp Ther 279:1491-1506. Alkondon M, Pereira EF, Wonnacott S, Albuquerque EX (1992) Blockade of nicotinic currents in hippocampal neurons
defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol 41:802-808. Alkondon M, Rocha ES, Maelicke A, Albuquerque EX (1996b) Diversity of nicotinic acetylcholine receptors in rat brain. V.
Alpha-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther 278:1460-1471.
Alkondon M, Pereira EF, Barbosa CT, Albuquerque EX (1997) Neuronal nicotinic acetylcholine receptor activation modulates gamma-aminobutyric acid release from CA1 neurons of rat hippocampal slices. J Pharmacol Exp Ther 283:1396-1411.
Alkondon M, Pereira EF, Almeida LE, Randall WR, Albuquerque EX (2000) Nicotine at concentrations found in cigarette smokers activates and desensitizes nicotinic acetylcholine receptors in CA1 interneurons of rat hippocampus. Neuropharmacology 39:2726-2739.
Allam MF, Campbell MJ, Hofman A, Del Castillo AS, Fernandez-Crehuet Navajas R (2004) Smoking and Parkinson's disease: systematic review of prospective studies. Mov Disord 19:614-621.
Andén NE, Grabowska-Andén M, Lindgren S, Thornström U (1983) Synthesis rate of dopamine: difference between corpus striatum and limbic system as a possible explanation of variations in reactions to drugs. Naunyn-Schmiedebergs Arch Pharmacol 323:193-198.
Aosaki T, Kimura M, Graybiel AM (1995) Temporal and spatial characteristics of tonically active neurons of the primate's striatum. J Neurophysiol 73:1234-1252.
Arqueros L, Naquira D, Zunino E (1978) Nicotine-induced release of catecholamines from rat hippocampus and striatum. Biochem Pharmacol 27:2667-2674.
Asencio H, Bustos G, Gysling K, Labarca R (1991) N-Methyl-D-aspartate receptors and release of newly-synthesized 3H dopamine in nucleus accumbens slices and its relationship with neocortical afferences. Prog Neuro-Psychopharmacology Biol Psychiatry 15:663-676.
Aubert I, Araujo DM, Cecyre D, Robitaille Y, Gauthier S, Quirion R (1992) Comparative alterations of nicotinic and muscarinic binding sites in Alzheimer's and Parkinson's diseases. J Neurochem 58:529-541.
Avalos M, Parker MJ, Maddox FN, Carroll FI, Luetje CW (2002) Effects of pyridine ring substitutions on affinity, efficacy, and subtype selectivity of neuronal nicotinic receptor agonist epibatidine. J Pharmacol Exp Ther 302:1246-1252.
Azam L, Winzer-Serhan U, Leslie FM (2003) Co-expression of alpha7 and beta2 nicotinic acetylcholine receptor subunit mRNAs within rat brain cholinergic neurons. Neuroscience 119:965-977.
Azam L, Winzer-Serhan UH, Chen Y, Leslie FM (2002) Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol 444:260-274.
Badio B, Daly JW (1994) Epibatidine, a potent analgetic and nicotinic agonist. Mol Pharmacol 45:563-569. Baldo BA, Sadeghian K, Basso AM, Kelley AE (2002) Effects of selective dopamine D1 or D2 receptor blockade within
nucleus accumbens subregions on ingestive behavior and associated motor activity. Behav Brain Res 137:165-177. Balfour DJ (1994) Neural mechanisms underlying nicotine dependence. Addiction 89:1419-1423. Balfour DJ, Ridley DL (2000) The effects of nicotine on neural pathways implicated in depression: a factor in nicotine
addiction? Pharmacol Biochem Behav 66:79-85. Balfour DJ, Wright AE, Benwell ME, Birrell CE (2000) The putative role of extra-synaptic mesolimbic dopamine in the
neurobiology of nicotine dependence. Behav Brain Res 113:73-83. Balfour DJ, Benwell ME, Birrell CE, Kelly RJ, Al-Aloul M (1998) Sensitization of the mesoaccumbens dopamine response
to nicotine. Pharmacol Biochem Behav 59:1021-1030. Bancroft A, Levin ED (2000) Ventral hippocampal alpha4beta2 nicotinic receptors and chronic nicotine effects on memory.
Neuropharmacology 39:2770-2778.

98
Bannon AW, Gunther KL, Decker MW (1995) Is epibatidine really analgesic? Dissociation of the activity, temperature, and analgesic effects of (+/-)-epibatidine. Pharmacol Biochem Behav 51:693-698.
Baron JA (1986) Cigarette smoking and Parkinson's disease. Neurology 36:1490-1496. Bednar I, Friberg L, Nordberg A (2004) Modulation of dopamine release by the nicotinic agonist epibatidine in the frontal
cortex and the nucleus accumbens of naive and chronic nicotine treated rats. Neurochem Int 45:1049-1055. Behmand RA, Harik SI (1992) Nicotine enhances 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. J Neurochem
58:776-779. Belluardo N, Mudo G, Blum M, Fuxe K (2000) Central nicotinic receptors, neurotrophic factors and neuroprotection. Behav
Brain Res 113:21-34. Bencherif M, Fowler K, Lukas RJ, Lippiello PM (1995) Mechanisms of up-regulation of neuronal nicotinic acetylcholine
receptors in clonal cell lines and primary cultures of fetal rat brain. J Pharmacol Exp Ther 275:987-994. Bencherif M, Bane AJ, Miller CH, Dull GM, Gatto GJ (2000) TC-2559: a novel orally active ligand selective at neuronal
acetylcholine receptors. Eur J Pharmacol 409:45-55. Bencherif M, Lovette ME, Fowler KW, Arrington S, Reeves L, Caldwell WS, Lippiello PM (1996) RJR-2403: a nicotinic
agonist with CNS selectivity I. In vitro characterization. J Pharmacol Exp Ther 279:1413-1421. Bencherif M, Schmitt JD, Bhatti BS, Crooks P, Caldwell WS, Lovette ME, Fowler K, Reeves L, Lippiello PM (1998) The
heterocyclic substituted pyridine derivative (+/-)-2-(-3-pyridinyl)-1-azabicyclo2.2.2 octane (RJR-2429): a selective ligand at nicotinic acetylcholine receptors. J Pharmacol Exp Ther 284:886-894.
Bennett BD, Wilson CJ (1999) Spontaneous activity of neostriatal cholinergic interneurons in vitro. J Neurosci 19:5586-5596.
Benowitz NL (1996) Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol 36:597-613. Benowitz NL, Porchet H, Jacob PI (1990) Pharmacokinetics, metabolism, and pharmacodynamics of nicotine. In: Nicotine
psychopharmacology: Molecular, cellular and behavioural aspects (Wonnacott S, Russell MAH, Stolerman IP, eds), pp 112-157. Oxford: Oxford University Press.
Benwell ME, Balfour DJ (1992) The effects of acute and repeated nicotine treatment on nucleus accumbens dopamine and locomotor activity. Br J Pharmacol 105:849-856.
Benwell ME, Balfour DJ (1997) Regional variation in the effects of nicotine on catecholamine overflow in rat brain. Eur J Pharmacol 325:13-20.
Benwell ME, Balfour DJ, Anderson JM (1988) Evidence that tobacco smoking increases the density of (-)3H nicotine binding sites in human brain. J Neurochem 50:1243-1247.
Benwell ME, Balfour DJ, Lucchi HM (1993) Influence of tetrodotoxin and calcium on changes in extracellular dopamine levels evoked by systemic nicotine. Psychopharmacology 112:467-474.
Benwell ME, Balfour DJ, Khadra LF (1994) Studies on the influence of nicotine infusions on mesolimbic dopamine and locomotor responses to nicotine. Clin Investigator 72:233-239.
Benwell ME, Balfour DJ, Birrell CE (1995) Desensitization of the nicotine-induced mesolimbic dopamine responses during constant infusion with nicotine. Br J Pharmacol 114:454-460.
Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25:515-532. Berkovic S, Curtis L, Bertrand D (1999) Neuronal nicotinic receptor channel dysfunction in a human epilepsy syndrome. In:
Neuronal nicotinic receptors. Pharmacology and therapeutic opportunities (Arneric SP, Brioni JD, eds), 1st Edition, pp 287-306. New York: Wiley-Liss, Inc.
Bertolino M, Kellar KJ, Vicini S, Gillis RA (1997) Nicotinic receptor mediates spontaneous GABA release in the rat dorsal motor nucleus of the vagus. Neuroscience 79:671-681.
Bertrand D, Ballivet M, Rungger D (1990) Activation and blocking of neuronal nicotinic acetylcholine receptor reconstituted in Xenopus oocytes. Proc Natl Acad Sci U S A 87:1993-1997.
Bertrand D, Bertrand S, Ballivet M (1992) Pharmacological properties of the homomeric alpha7 receptor. Neurosci Lett 146:87-90.
Bertrand S, Weiland S, Berkovic SF, Steinlein OK, Bertrand D (1998) Properties of neuronal nicotinic acetylcholine receptor mutants from humans suffering from autosomal dominant nocturnal frontal lobe epilepsy. Br J Pharmacol 125:751-760.
Bertrand S, Patt JT, Spang JE, Westera G, Schubiger PA, Bertrand D (1999) Neuronal nAChR stereoselectivity to non-natural epibatidine derivatives. FEBS Lett 450:273-279.
Bezard E, Gross CE (1998) Compensatory mechanisms in experimental and human parkinsonism: towards a dynamic approach. Prog Neurobiol 55:93-116.
Bezard E, Bioulac B, Gross CE (1998) Glutamatergic compensatory mechanisms in experimental parkinsonism. Prog Neuro-Psychopharmacology Biol Psychiatry 22:609-623.
Bezard E, Boraud T, Bioulac B, Gross CE (1997) Compensatory effects of glutamatergic inputs to the substantia nigra pars compacta in experimental parkinsonism. Neuroscience 81:399-404.
Bezard E, Boraud T, Bioulac B, Gross CE (1999) Involvement of the subthalamic nucleus in glutamatergic compensatory mechanisms. Eur J Neurosci 11:2167-2170.
Bianchi C, Ferraro L, Tanganelli S, Morari M, Spalluto G, Simonato M, Beani L (1995) 5-Hydroxytryptamine-mediated effects of nicotine on endogenous GABA efflux from guinea-pig cortical slices. Br J Pharmacol 116:2724-2728.
Birkmeyer W, Riederer P (1985) Die Parkinson-Krankheit, 2nd Edition. Wien: Springer-Verlag. Birkmeyer W, Riederer P (1989) Understanding the Neurotransmitters: Key to the workings of the brain, 1st Edition. Wien:
Springer-Verlag. Blaha CD, Allen LF, Das S, Inglis WL, Latimer MP, Vincent SR, Winn P (1996) Modulation of dopamine efflux in the
nucleus accumbens after cholinergic stimulation of the ventral tegmental area in intact, pedunculopontine tegmental nucleus-lesioned, and laterodorsal tegmental nucleus-lesioned rats. J Neurosci 16:714-722.

99
Bolam JP, Smith Y (1990) The GABA and substance P input to dopaminergic neurones in the substantia nigra of the rat. Brain Res 529:57-78.
Bolam JP, Francis CM, Henderson Z (1991) Cholinergic input to dopaminergic neurons in the substantia nigra: a double immunocytochemical study. Neuroscience 41:483-494.
Bolam JP, Hanley JJ, Booth PA, Bevan MD (2000) Synaptic organisation of the basal ganglia. J Anat 196:527-542. Bonhaus DW, Bley KR, Broka CA, Fontana DJ, Leung E, Lewis R, Shieh A, Wong EH (1995) Characterization of the
electrophysiological, biochemical and behavioral actions of epibatidine. J Pharmacol Exp Ther 272:1199-1203. Bontempi B, Whelan KT, Risbrough VB, Lloyd GK, Menzaghi F (2003) Cognitive enhancing properties and tolerability of
cholinergic agents in mice: a comparative study of nicotine, donepezil, and SIB-1553A, a subtype-selective ligand for nicotinic acetylcholine receptors. Neuropsychopharmacology 28:1235-1246.
Boulter J, Connolly J, Deneris E, Goldman D, Heinemann S, Patrick J (1987) Functional expression of two neuronal nicotinic acetylcholine receptors from cDNA clones identifies a gene family. Proc Natl Acad Sci U S A 84:7763-7767.
Boulter J, Evans K, Goldman D, Martin G, Treco D, Heinemann S, Patrick J (1986) Isolation of a cDNA clone coding for a possible neural nicotinic acetylcholine receptor alpha-subunit. Nature 319:368-374.
Breese CR, Marks MJ, Logel J, Adams CE, Sullivan B, Collins AC, Leonard S (1997) Effect of smoking history on 3H nicotine binding in human postmortem brain. J Pharmacol Exp Ther 282:7-13.
Breese CR, Lee MJ, Adams CE, Sullivan B, Logel J, Gillen KM, Marks MJ, Collins AC, Leonard S (2000) Abnormal regulation of high affinity nicotinic receptors in subjects with schizophrenia. Neuropsychopharmacology 23:351-364.
Briggs CA, McKenna DG, Piattoni-Kaplan M (1995) Human alpha7 nicotinic acetylcholine receptor responses to novel ligands. Neuropharmacology 34:583-590.
Briggs CA, Anderson DJ, Brioni JD, Buccafusco JJ, Buckley MJ, Campbell JE, Decker MW, Donnelly-Roberts D, Elliott RL, Gopalakrishnan M, Holladay MW, Hui YH, Jackson WJ, Kim DJ, Marsh KC, O'Neill A, Prendergast MA, Ryther KB, Sullivan JP, Arneric SP (1997) Functional characterization of the novel neuronal nicotinic acetylcholine receptor ligand GTS-21 in vitro and in vivo. Pharmacol Biochem Behav 57:231-241.
Brioni JD, Arneric SP (1993) Nicotinic receptor agonists facilitate retention of avoidance training: participation of dopaminergic mechanisms. Behav Neural Biol 59:57-62.
Buccafusco JJ, Beach JW, Terry AV, Jr., Doad GS, Sood A, Arias E, Misawa H, Masai M, Fujii T, Kawashima K (2004) Novel analogs of choline as potential neuroprotective agents. J Alzheimers Dis 6(Suppl):S85-92.
Buisson B, Bertrand D (2001) Chronic exposure to nicotine upregulates the human alpha4beta2 nicotinic acetylcholine receptor function. J Neurosci 21:1819-1829.
Buisson B, Bertrand D (2002) Nicotine addiction: the possible role of functional upregulation. Trends Pharmacol Sci 23:130-136.
Buisson B, Vallejo YF, Green WN, Bertrand D (2000) The unusual nature of epibatidine responses at the alpha4beta2 nicotinic acetylcholine receptor. Neuropharmacology 39:2561-2569.
Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D (1996) Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: A patch-clamp study. J Neurosci 16:7880-7891.
Bättig K, Driscoll P, Schlatter J, Uster HJ (1976) Effects of nicotine on the exploratory locomotion patterns of female Roman high- and low-avoidance rats. Pharmacol Biochem Behav 4:435-439.
Cachelin AB, Jaggi R (1991) Beta subunits determine the time course of desensitization in rat alpha3 neuronal nicotinic acetylcholine receptors. Pflügers Arch Eur J Physiol 419:579-582.
Cadoni C, Di Chiara G (2000) Differential changes in accumbens shell and core dopamine in behavioral sensitization to nicotine. Eur J Pharmacol 387:R23-25.
Calabresi P, Lacey MG, North RA (1989) Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. Br J Pharmacol 98:135-140.
Calcagnetti DJ, Schechter MD (1994) Nicotine place preference using the biased method of conditioning. Prog Neuro-Psychopharmacology Biol Psychiatry 18:925-933.
Carboni E, Acquas E, Leone P, Di Chiara G (1989a) 5HT3 receptor antagonists block morphine- and nicotine- but not amphetamine-induced reward. Psychopharmacologia 97:175-178.
Carboni E, Imperato A, Perezzani L, Di Chiara G (1989b) Amphetamine, cocaine, phencyclidine and nomifensine increase extracellular dopamine concentrations preferentially in the nucleus accumbens of freely moving rats. Neuroscience 28:653-661.
Carey RJ (1991) Chronic L-dopa treatment in the unilateral 6-OHDA rat: evidence for behavioral sensitization and biochemical tolerance. Brain Res 568:205-214.
Carlsson A (1959) The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev 11:490-493.
Carlsson A, Lindqvist M, Magnusson T, Waldeck B (1958) On the presence of 3-hydroxytyramine in brain. Science 127:471. Carr LA, Rowell PP (1990) Attenuation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by tobacco
smoke. Neuropharmacology 29:311-314. Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM (1996) A new alpha-conotoxin which targets
alpha3beta2 nicotinic acetylcholine receptors. J Biol Chem 271:7522-7528. Cass WA, Zahniser NR, Flach KA, Gerhardt GA (1993) Clearance of exogenous dopamine in rat dorsal striatum and nucleus
accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem 61:2269-2278. Champtiaux N, Han ZY, Bessis A, Rossi FM, Zoli M, Marubio L, McIntosh JM, Changeux JP (2002) Distribution and
pharmacology of alpha6-containing nicotinic acetylcholine receptors analyzed with mutant mice. J Neurosci 22:1208-1217.

100
Champtiaux N, Gotti C, Cordero-Erausquin M, David DJ, Przybylski C, Lena C, Clementi F, Moretti M, Rossi FM, Le Novere N, McIntosh JM, Gardier AM, Changeux JP (2003) Subunit composition of functional nicotinic receptors in dopaminergic neurons investigated with knock-out mice. J Neurosci 23:7820-7829.
Changeux JP, Edelstein SJ (1998) Allosteric receptors after 30 years. Neuron 21:959-980. Changeux JP, Benoit P, Bessis A, Cartaud J, Devillers-Thiery A, Fontaine B, Galzi JL, Klarsfeld A, Laufer R, Mulle C
(1990) The acetylcholine receptor: functional architecture and regulation. Adv Second Messenger Phosphoprotein Res 24:15-19.
Charléty PJ, Grenhoff J, Chergui K, De la Chapelle B, Buda M, Svensson TH, Chouvet G (1991) Burst firing of mesencephalic dopamine neurons is inhibited by somatodendritic application of kynurenate. Acta Physiol Scand 142:105-112.
Charpantier E, Barneoud P, Moser P, Besnard F, Sgard F (1998) Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport 9:3097-3101.
Chavez-Noriega LE, Gillespie A, Stauderman KA, Crona JH, Claeps BO, Elliott KJ, Reid RT, Rao TS, Velicelebi G, Harpold MM, Johnson EC, Corey-Naeve J (2000) Characterization of the recombinant human neuronal nicotinic acetylcholine receptors alpha3beta2 and alpha4beta2 stably expressed in HEK293 cells. Neuropharmacology 39:2543-2560.
Chergui K, Charléty PJ, Akaoka H, Saunier CF, Brunet JL, Buda M, Svensson TH, Chouvet G (1993) Tonic activation of NMDA receptors causes spontaneous burst discharge of rat midbrain dopamine neurons in vivo. Eur J Neurosci 5:137-144.
Chiodini FC, Tassonyi E, Hulo S, Bertrand D, Muller D (1999) Modulation of synaptic transmission by nicotine and nicotinic antagonists in hippocampus. Brain Res Bull 48:623-628.
Cimino M, Marini P, Fornasari D, Cattabeni F, Clementi F (1992) Distribution of nicotinic receptors in cynomolgus monkey brain and ganglia: localization of alpha3 subunit mRNA, alpha-bungarotoxin and nicotine binding sites. Neuroscience 51:77-86.
Cinciripini PM, Hecht SS, Henningfield JE, Manley MW, Kramer BS (1997) Tobacco addiction: implications for treatment and cancer prevention. J Natl Cancer Inst 89:1852-1867.
Clarke PB (1993a) Nicotine dependence - mechanisms and therapeutic strategies. Biochem Soc Symp 59:83-95. Clarke PB (1993b) Nicotinic receptors in mammalian brain: localization and relation to cholinergic innervation. Prog Brain
Res 98:77-83. Clarke PB, Kumar R (1983a) Characterization of the locomotor stimulant action of nicotine in tolerant rats. Br J Pharmacol
80:587-594. Clarke PB, Kumar R (1983b) The effects of nicotine on locomotor activity in non-tolerant and tolerant rats. Br J Pharmacol
78:329-337. Clarke PB, Pert A (1985) Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic
neurons. Brain Res 348:355-358. Clarke PB, Fibiger HC (1987) Apparent absence of nicotine-induced conditioned place preference in rats.
Psychopharmacology 92:84-88. Clarke PB, Hommer DW, Pert A, Skirboll LR (1985a) Electrophysiological actions of nicotine on substantia nigra single
units. Br J Pharmacol 85:827-835. Clarke PB, Hommer DW, Pert A, Skirboll LR (1987) Innervation of substantia nigra neurons by cholinergic afferents from
pedunculopontine nucleus in the rat: neuroanatomical and electrophysiological evidence. Neuroscience 23:1011-1019. Clarke PB, Fu DS, Jakubovic A, Fibiger HC (1988) Evidence that mesolimbic dopaminergic activation underlies the
locomotor stimulant action of nicotine in rats. J Pharmacol Exp Ther 246:701-708. Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A (1985b) Nicotinic binding in rat brain: autoradiographic comparison of
3Hacetylcholine, 3Hnicotine, and 125Ialpha-bungarotoxin. J Neurosci 5:1307-1315. Cobb WS, Abercrombie ED (2002) Distinct roles for nigral GABA and glutamate receptors in the regulation of dendritic
dopamine release under normal conditions and in response to systemic haloperidol. J Neurosci 22:1407-1413. Collins AC, Luo Y, Selvaag S, Marks MJ (1994) Sensitivity to nicotine and brain nicotinic receptors are altered by chronic
nicotine and mecamylamine infusion. J Pharmacol Exp Ther 271:125-133. Colquhoun LM, Patrick JW (1997) Pharmacology of neuronal nicotinic acetylcholine receptor subtypes. Adv Pharmacol
39:191-220. Cooper E, Couturier S, Ballivet M (1991) Pentameric structure and subunit stoichiometry of a neuronal nicotinic
acetylcholine receptor. Nature 350:235-238. Cooper JR, Bloom FE, Roth RH (2003) The biochemical basis of neuropharmacology, 8th Edition. New York: Oxford
University Press. Corrigall WA (1999) Nicotine self-administration in animals as a dependence model. Nicotine Tobacco Res 1:11-20. Corrigall WA, Coen KM (1989) Nicotine maintains robust self-administration in rats on a limited-access schedule.
Psychopharmacology 99:473-478. Corrigall WA, Coen KM (1991) Selective dopamine antagonists reduce nicotine self-administration. Psychopharmacology
104:171-176. Corrigall WA, Coen KM, Adamson KL (1994) Self-administered nicotine activates the mesolimbic dopamine system through
the ventral tegmental area. Brain Res 653:278-284. Corrigall WA, Franklin KB, Coen KM, Clarke PB (1992) The mesolimbic dopaminergic system is implicated in the
reinforcing effects of nicotine. Psychopharmacology 107:285-289. Corrigall WA, Coen KM, Zhang J, Adamson KL (2001) GABA mechanisms in the pedunculopontine tegmental nucleus
influence particular aspects of nicotine self-administration selectively in the rat. Psychopharmacology 158:190-197.

101
Corrigall WA, Coen KM, Adamson KL, Chow BL, Zhang J (2000) Response of nicotine self-administration in the rat to manipulations of mu-opioid and gamma-aminobutyric acid receptors in the ventral tegmental area. Psychopharmacologia 149:107-114.
Cosford ND, Bleicher L, Vernier JM, Chavez-Noriega L, Rao TS, Siegel RS, Suto C, Washburn M, Lloyd GK, McDonald IA (2000) Recombinant human receptors and functional assays in the discovery of altinicline (SIB-1508Y), a novel acetylcholine-gated ion channel (nAChR) agonist. Pharm Acta Helv 74:125-130.
Costa G, Abin-Carriquiry JA, Dajas F (2001) Nicotine prevents striatal dopamine loss produced by 6-hydroxydopamine lesion in the substantia nigra. Brain Res 888:336-342.
Costall B, Naylor RJ (1973) Neuroleptic and non-neuroleptic catalepsy. Arzneimittelforschung 23:674-683. Costall B, Kelly DM, Naylor RJ (1975) Nomifensine: a potent dopaminergic agonist of antiparkinson potential.
Psychopharmacologia 41:153-164. Court JA, Martin-Ruiz C, Graham A, Perry E (2000a) Nicotinic receptors in human brain: topography and pathology. J Chem
Neuroanat 20:281-298. Court JA, Piggott MA, Lloyd S, Cookson N, Ballard CG, McKeith IG, Perry RH, Perry EK (2000b) Nicotine binding in
human striatum: elevation in schizophrenia and reductions in dementia with Lewy bodies, Parkinson's disease and Alzheimer's disease and in relation to neuroleptic medication. Neuroscience 98:79-87.
Court JA, Lloyd S, Johnson M, Griffiths M, Birdsall NJ, Piggott MA, Oakley AE, Ince PG, Perry EK, Perry RH (1997) Nicotinic and muscarinic cholinergic receptor binding in the human hippocampal formation during development and aging. Brain Res Dev Brain Res 101:93-105.
Court JA, Lloyd S, Thomas N, Piggott MA, Marshall EF, Morris CM, Lamb H, Perry RH, Johnson M, Perry EK (1998) Dopamine and nicotinic receptor binding and the levels of dopamine and homovanillic acid in human brain related to tobacco use. Neuroscience 87:63-78.
Cousins MS, Roberts DC, de Wit H (2002) GABA(B) receptor agonists for the treatment of drug addiction: a review of recent findings. Drug Alcohol Depend 65:209-220.
Couturier S, Erkman L, Valera S, Rungger D, Bertrand S, Boulter J, Ballivet M, Bertrand D (1990a) Alpha5, alpha3, and non-alpha3. Three clustered avian genes encoding neuronal nicotinic acetylcholine receptor-related subunits. J Biol Chem 265:17560-17567.
Couturier S, Bertrand D, Matter JM, Hernandez MC, Bertrand S, Millar N, Valera S, Barkas T, Ballivet M (1990b) A neuronal nicotinic acetylcholine receptor subunit alpha7 is developmentally regulated and forms a homo-oligomeric channel blocked by alpha-BTX. Neuron 5:847-856.
Cui C, Booker TK, Allen RS, Grady SR, Whiteaker P, Marks MJ, Salminen O, Tritto T, Butt CM, Allen WR, Stitzel JA, McIntosh JM, Boulter J, Collins AC, Heinemann SF (2003) The beta3 nicotinic receptor subunit: a component of alpha-conotoxin MII-binding nicotinic acetylcholine receptors that modulate dopamine release and related behaviors. J Neurosci 23:11045-11053.
Cummings JL, Benson DF (1987) The role of the nucleus basalis of Meynert in dementia: review and reconsideration. Alzheimer Dis Assoc Dis 1:128-155.
Dahlström A, Fuxe K (1964) Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand 62:1-55.
Dalack GW, Healy DJ, Meador-Woodruff JH (1998) Nicotine dependence in schizophrenia: clinical phenomena and laboratory findings. Am J Psychiatry 155:1490-1501.
Damaj MI, Creasy KR, Grove AD, Rosecrans JA, Martin BR (1994) Pharmacological effects of epibatidine optical enantiomers. Brain Res 664:34-40.
Damsma G, Day J, Fibiger HC (1989) Lack of tolerance to nicotine-induced dopamine release in the nucleus accumbens. Eur J Pharmacol 168:363-368.
Damsma G, Westerink BH, de Vries JB, Horn AS (1988) The effect of systemically applied cholinergic drugs on the striatal release of dopamine and its metabolites, as determined by automated brain dialysis in conscious rats. Neurosci Lett 89:349-354.
Dani JA (2001) Nicotinic receptor activity alters synaptic plasticity. ScientificWorldJournal 1:393-395. Davies AR, Hardick DJ, Blagbrough IS, Potter BV, Wolstenholme AJ, Wonnacott S (1999) Characterisation of the binding
of 3H methyllycaconitine: a new radioligand for labelling alpha7-type neuronal nicotinic acetylcholine receptors. Neuropharmacology 38:679-690.
de Fiebre CM, Meyer EM, Henry JC, Muraskin SI, Kem WR, Papke RL (1995) Characterization of a series of anabaseine-derived compounds reveals that the 3-(4)-dimethylaminocinnamylidine derivative is a selective agonist at neuronal nicotinic alpha7/125I-alpha-bungarotoxin receptor subtypes. Mol Pharmacol 47:164-171.
de Rover M, Mansvelder HD, Lodder JC, Wardeh G, Schoffelmeer AN, Brussaard AB (2004) Long-lasting nicotinic modulation of GABAergic synaptic transmission in the rat nucleus accumbens associated with behavioural sensitization to amphetamine. Eur J Neurosci 19:2859-2870.
Decker MW, Brioni JD, Bannon AW, Arneric SP (1995) Diversity of neuronal nicotinic acetylcholine receptors: lessons from behavior and implications for CNS therapeutics. Life Sci 56:545-570.
Deneris ES, Connolly J, Rogers SW, Duvoisin R (1991) Pharmacological and functional diversity of neuronal nicotinic acetylcholine receptors. Trends Pharmacol Sci 12:34-40.
Deneris ES, Boulter J, Swanson LW, Patrick J, Heinemann S (1989) Beta 3: a new member of nicotinic acetylcholine receptor gene family is expressed in brain. J Biol Chem 264:6268-6272.
Dewey SL, Brodie JD, Gerasimov M, Horan B, Gardner EL, Ashby CR, Jr. (1999) A pharmacologic strategy for the treatment of nicotine addiction. Synapse 31:76-86.
Di Chiara G (2000) Role of dopamine in the behavioural actions of nicotine related to addiction. Eur J Pharmacol 393:295-314.

102
Di Chiara G (2002) Nucleus accumbens shell and core dopamine: differential role in behavior and addiction. Behav Brain Res 137:75-114.
Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci U S A 85:5274-5278.
Dilger JP, Liu Y (1992) Desensitization of acetylcholine receptors in BC3H-1 cells. Pflügers Arch Eur J Physiol 420:479-485.
Dobelis P, Hutton S, Lu Y, Collins AC (2003) GABAergic systems modulate nicotinic receptor-mediated seizures in mice. J Pharmacol Exp Ther 306:1159-1166.
Domino EF, Ni L, Zhang H (1999) Nicotine alone and in combination with L-DOPA methyl ester or the D(2) agonist N-0923 in MPTP-induced chronic hemiparkinsonian monkeys. Exp Neurol 158:414-421.
Donny EC, Caggiula AR, Knopf S, Brown C (1995) Nicotine self-administration in rats. Psychopharmacology 122:390-394. Donny EC, Caggiula AR, Mielke MM, Jacobs KS, Rose C, Sved AF (1998) Acquisition of nicotine self-administration in
rats: the effects of dose, feeding schedule, and drug contingency. Psychopharmacology 136:83-90. Donny EC, Caggiula AR, Mielke MM, Booth S, Gharib MA, Hoffman A, Maldovan V, Shupenko C, McCallum SE (1999)
Nicotine self-administration in rats on a progressive ratio schedule of reinforcement. Psychopharmacology 147:135-142. Dowell C, Olivera BM, Garrett JE, Staheli ST, Watkins M, Kuryatov A, Yoshikami D, Lindstrom JM, McIntosh JM (2003)
Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J Neurosci 23:8445-8452.
Dukat M, Glennon RA (2003) Epibatidine: impact on nicotinic receptor research. Cell Mol Neurobiol 23:365-378. Durany N, Zochling R, Boissl KW, Paulus W, Ransmayr G, Tatschner T, Danielczyk W, Jellinger K, Deckert J, Riederer P
(2000) Human post-mortem striatal alpha4beta2 nicotinic acetylcholine receptor density in schizophrenia and Parkinson's syndrome. Neurosci Lett 287:109-112.
Dursun SM, Kutcher S (1999) Smoking, nicotine and psychiatric disorders: evidence for therapeutic role, controversies and implications for future research. Med Hypotheses 52:101-109.
Ebersbach G, Stock M, Muller J, Wenning G, Wissel J, Poewe W (1999) Worsening of motor performance in patients with Parkinson's disease following transdermal nicotine administration. Mov Disord 14:1011-1013.
Elbaz A, Manubens-Bertran JM, Baldereschi M, Breteler MM, Grigoletto F, Lopez-Pousa S, Dartigues JF, Alperovitch A, Rocca WA, Tzourio C (2000) Parkinson's disease, smoking, and family history. EUROPARKINSON Study Group. J Neurol 247:793-798.
el-Bizri H, Clarke PB (1994a) Blockade of nicotinic receptor-mediated release of dopamine from striatal synaptosomes by chlorisondamine and other nicotinic antagonists administered in vitro. Br J Pharmacol 111:406-413.
el-Bizri H, Clarke PB (1994b) Regulation of nicotinic receptors in rat brain following quasi-irreversible nicotinic blockade by chlorisondamine and chronic treatment with nicotine. Br J Pharmacol 113:917-925.
Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S (1994) Alpha9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79:705-715.
Elgoyhen AB, Vetter DE, Katz E, Rothlin CV, Heinemann SF, Boulter J (2001) Alpha10: a determinant of nicotinic cholinergic receptor function in mammalian vestibular and cochlear mechanosensory hair cells. Proc Natl Acad Sci U S A 98:3501-3506.
Elliott KJ, Jones JM, Sacaan AI, Lloyd GK, Corey-Naeve J (1998) 6-Hydroxydopamine lesion of rat nigrostriatal dopaminergic neurons differentially affects nicotinic acetylcholine receptor subunit mRNA expression. J Mol Neurosci 10:251-260.
Elliott KJ, Ellis SB, Berckhan KJ, Urrutia A, Chavez-Noriega LE, Johnson EC, Velicelebi G, Harpold MM (1996) Comparative structure of human neuronal alpha2-alpha7 and beta2-beta4 nicotinic acetylcholine receptor subunits and functional expression of the alpha2, alpha3, alpha4, alpha7, beta2, and beta4 subunits. J Mol Neurosci 7:217-228.
Epping-Jordan MP, Picciotto MR, Changeux JP, Pich EM (1999) Assessment of nicotinic acetylcholine receptor subunit contributions to nicotine self-administration in mutant mice. Psychopharmacologia 147:25-26.
Erhardt S, Mathe JM, Chergui K, Engberg G, Svensson TH (2002) GABA(B) receptor-mediated modulation of the firing pattern of ventral tegmental area dopamine neurons in vivo. Naunyn-Schmiedebergs Arch Pharmacol 365:173-180.
Everhart D, Reiller E, Mirzoian A, McIntosh JM, Malhotra A, Luetje CW (2003) Identification of residues that confer alpha-conotoxin-PnIA sensitivity on the alpha 3 subunit of neuronal nicotinic acetylcholine receptors. J Pharmacol Exp Ther 306:664-670.
Everitt BJ, Wolf ME (2002) Psychomotor stimulant addiction: a neural systems perspective. J Neurosci 22:3312-3320. Fadda P, Scherma M, Fresu A, Collu M, Fratta W (2003) Baclofen antagonizes nicotine-, cocaine-, and morphine-induced
dopamine release in the nucleus accumbens of rat. Synapse 50:1-6. Fagerström KO, Pomerleau O, Giordani B, Stelson F (1994) Nicotine may relieve symptoms of Parkinson's disease.
Psychopharmacology 116:117-119. Falk L, Nordberg A, Seiger A, Kjaeldgaard A, Hellström-Lindahl E (2005) Smoking during early pregnancy affects the
expression pattern of both nicotinic and muscarinic acetylcholine receptors in human first trimester brainstem and cerebellum. Neuroscience 132:389-397.
Fattore L, Cossu G, Martellotta MC, Fratta W (2002) Baclofen antagonizes intravenous self-administration of nicotine in mice and rats. Alcohol Alcoholism 37:495-498.
Feldman RS, Meyer JS, Quenzer LF (1997) Principles in Neuropsychopharmacology. Massachusetts: Sinauer Associates, Inc.
Fenster CP, Rains MF, Noerager B, Quick MW, Lester RA (1997) Influence of subunit composition on desensitization of neuronal acetylcholine receptors at low concentrations of nicotine. J Neurosci 17:5747-5759.
Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RA (1999a) Upregulation of surface alpha4beta2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci 19:4804-4814.

103
Fenster CP, Hicks JH, Beckman ML, Covernton PJ, Quick MW, Lester RA (1999b) Desensitization of nicotinic receptors in the central nervous system. Ann NY Acad Sci 868:620-623.
Ferger B, Kuschinsky K (1997) Biochemical studies support the assumption that dopamine plays a minor role in the EEG effects of nicotine. Psychopharmacology 129:192-196.
Ferrari R, Le Novere N, Picciotto MR, Changeux JP, Zoli M (2002) Acute and long-term changes in the mesolimbic dopamine pathway after systemic or local single nicotine injections. Eur J Neurosci 15:1810-1818.
Fisher JL, Dani JA (2000) Nicotinic receptors on hippocampal cultures can increase synaptic glutamate currents while decreasing the NMDA-receptor component. Neuropharmacology 39:2756-2769.
Fisher JL, Pidoplichko VI, Dani JA (1998) Nicotine modifies the activity of ventral tegmental area dopaminergic neurons and hippocampal GABAergic neurons. J Physiol (Paris) 92:209-213.
Flores CM, Davila-Garcia MI, Ulrich YM, Kellar KJ (1997) Differential regulation of neuronal nicotinic receptor binding sites following chronic nicotine administration. J Neurochem 69:2216-2219.
Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ (1992) A subtype of nicotinic cholinergic receptor in rat brain is composed of alpha4 and beta2 subunits and is up-regulated by chronic nicotine treatment. Mol Pharmacol 41:31-37.
Fonck C, Nashmi R, Deshpande P, Damaj MI, Marks MJ, Riedel A, Schwarz J, Collins AC, Labarca C, Lester HA (2003) Increased sensitivity to agonist-induced seizures, straub tail, and hippocampal theta rhythm in knock-in mice carrying hypersensitive alpha4 nicotinic receptors. J Neurosci 23:2582-2590.
Fowler JS, Logan J, Wang GJ, Volkow ND (2003) Monoamine oxidase and cigarette smoking. Neurotoxicology 24:75-82. Fowler JS, Volkow ND, Wang GJ, Pappas N, Logan J, MacGregor R, Alexoff D, Shea C, Schlyer D, Wolf AP, Warner D,
Zezulkova I, Cilento R (1996) Inhibition of monoamine oxidase B in the brains of smokers. Nature 379:733-736. Fratiglioni L, Wang HX (2000) Smoking and Parkinson's and Alzheimer's disease: review of the epidemiological studies.
Behav Brain Res 113:117-120. Free RB, Bryant DL, McKay SB, Kaser DJ, McKay DB (2002) 3H Epibatidine binding to bovine adrenal medulla: evidence
for alpha3beta4* nicotinic receptors. Neurosci Lett 318:98-102. Freedman R, Hall M, Adler LE, Leonard S (1995) Evidence in postmortem brain tissue for decreased numbers of
hippocampal nicotinic receptors in schizophrenia. Biol Psychiatry 38:22-33. Fu Y, Matta SG, Gao W, Sharp BM (2000a) Local alpha-bungarotoxin-sensitive nicotinic receptors in the nucleus accumbens
modulate nicotine-stimulated dopamine secretion in vivo. Neuroscience 101:369-375. Fu Y, Matta SG, Gao W, Brower VG, Sharp BM (2000b) Systemic nicotine stimulates dopamine release in nucleus
accumbens: re-evaluation of the role of N-methyl-D-aspartate receptors in the ventral tegmental area. J Pharmacol Exp Ther 294:458-465.
Fucile S, Matter JM, Erkman L, Ragozzino D, Barabino B, Grassi F, Alema S, Ballivet M, Eusebi F (1998) The neuronal alpha6 subunit forms functional heteromeric acetylcholine receptors in human transfected cells. Eur J Neurosci 10:172-178.
Fudala PJ, Iwamoto ET (1986) Further studies on nicotine-induced conditioned place preference in the rat. Pharmacol Biochem Behav 25:1041-1049.
Fudala PJ, Iwamoto ET (1987) Conditioned aversion after delay place conditioning with nicotine. Psychopharmacology 92:376-381.
Fudala PJ, Teoh KW, Iwamoto ET (1985) Pharmacologic characterization of nicotine-induced conditioned place preference. Pharmacol Biochem Behav 22:237-241.
Fung YK (1989) Effects of chronic nicotine pretreatment on (+)-amphetamine and nicotine-induced synthesis and release of 3H dopamine from 3H tyrosine in rat nucleus accumbens. J Pharm Pharmacol 41:66-68.
Fung YK, Fiske LA, Lau YS (1991) Chronic administration of nicotine fails to alter the MPTP-induced neurotoxicity in mice. Gen Pharmacol 22:669-672.
Futami T, Takakusaki K, Kitai ST (1995) Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci Res 21:331-342.
Fuxe K, Janson AM, Jansson A, Andersson K, Eneroth P, Agnati LF (1990) Chronic nicotine treatment increases dopamine levels and reduces dopamine utilization in substantia nigra and in surviving forebrain dopamine nerve terminal systems after a partial di-mesencephalic hemitransection. Naunyn-Schmiedebergs Arch Pharmacol 341:171-181.
Fuxe K, Agnati L, Eneroth P, Gustafsson JA, Hökfelt T, Lofström A, Skett B, Skett P (1977) The effect of nicotine on central catecholamine neurons and gonadotropin secretion. I. Studies in the male rat. Med Biol 55:148-157.
Gainetdinov RR, Jones SR, Caron MG (1999) Functional hyperdopaminergia in dopamine transporter knock-out mice. Biol Psychiatry 46:303-311.
Gardner EL, Schiffer WK, Horan BA, Highfield D, Dewey SL, Brodie JD, Ashby CR, Jr. (2002) Gamma-vinyl GABA, an irreversible inhibitor of GABA transaminase, alters the acquisition and expression of cocaine-induced sensitization in male rats. Synapse 46:240-250.
Gariano RF, Groves PM (1988) Burst firing induced in midbrain dopamine neurons by stimulation of the medial prefrontal and anterior cingulate cortices. Brain Res 462:194-198.
Garris PA, Wightman RM (1994) Different kinetics govern dopaminergic transmission in the amygdala, prefrontal cortex, and striatum: an in vivo voltammetric study. J Neurosci 14:442-450.
Gatto GJ, Bohme GA, Caldwell WS, Letchworth SR, Traina VM, Obinu MC, Laville M, Reibaud M, Pradier L, Dunbar G, Bencherif M (2004) TC-1734: an orally active neuronal nicotinic acetylcholine receptor modulator with antidepressant, neuroprotective and long-lasting cognitive effects. CNS Drug Rev 10:147-166.
Gentry CL, Wilkins LH, Jr., Lukas RJ (2003) Effects of prolonged nicotinic ligand exposure on function of heterologously expressed, human alpha4beta2- and alpha4beta4-nicotinic acetylcholine receptors. J Pharmacol Exp Ther 304:206-216.
Gerasimov MR, Franceschi M, Volkow ND, Rice O, Schiffer WK, Dewey SL (2000) Synergistic interactions between nicotine and cocaine or methylphenidate depend on the dose of dopamine transporter inhibitor. Synapse 38:432-437.

104
Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM (2003) It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26:184-192.
Gerzanich V, Anand R, Lindstrom J (1994) Homomers of alpha8 and alpha7 subunits of nicotinic receptors exhibit similar channel but contrasting binding site properties. Mol Pharmacol 45:212-220.
Gerzanich V, Kuryatov A, Anand R, Lindstrom J (1997) "Orphan" alpha6 nicotinic AChR subunit can form a functional heteromeric acetylcholine receptor. Mol Pharmacol 51:320-327.
Gerzanich V, Wang F, Kuryatov A, Lindstrom J (1998) Alpha5 subunit alters desensitization, pharmacology, Ca++ permeability and Ca++ modulation of human neuronal alpha3 nicotinic receptors. J Pharmacol Exp Ther 286:311-320.
Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J (1995) Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Mol Pharmacol 48:774-782.
Gilbert DG (1979) Paradoxical tranquilizing and emotion-reducing effects of nicotine. Psychol Bull 86:643-661. Giorguieff-Chesselet MF, Kemel ML, Wandscheer D, Glowinski J (1979) Regulation of dopamine release by presynaptic
nicotinic receptors in rat striatal slices: effect of nicotine in a low concentration. Life Sci 25:1257-1262. Girault JA, Spampinato U, Savaki HE, Glowinski J, Besson MJ (1986) In vivo release of 3H gamma-aminobutyric acid in the
rat neostriatum I. Characterization and topographical heterogeneity of the effects of dopaminergic and cholinergic agents. Neuroscience 19:1101-1108.
Girod R, Crabtree G, Ernstrom G, Ramirez-Latorre J, McGehee D, Turner J, Role L (1999) Heteromeric complexes of alpha5 and/or alpha7 subunits. Effects of calcium and potential role in nicotine-induced presynaptic facilitation. Ann NY Acad Sci 868:578-590.
Glassman AH (1993) Cigarette smoking: implications for psychiatric illness. Am J Psychiatry 150:546-553. Goldberg SR, Spealman RD, Goldberg DM (1981) Persistent behavior at high rates maintained by intravenous self-
administration of nicotine. Science 214:573-575. Goodman FR (1974) Effects of nicotine on distribution and release of 14C-norepinephrine and 14C-dopamine in rat brain
striatum and hypothalamus slices. Neuropharmacology 13:1025-1032. Gopalakrishnan M, Monteggia LM, Anderson DJ, Molinari EJ, Piattoni-Kaplan M, Donnelly-Roberts D, Arneric SP,
Sullivan JP (1996) Stable expression, pharmacologic properties and regulation of the human neuronal nicotinic acetylcholine alpha4beta2 receptor. J Pharmacol Exp Ther 276:289-297.
Gopalakrishnan M, Buisson B, Touma E, Giordano T, Campbell JE, Hu IC, Donnelly-Roberts D, Arneric SP, Bertrand D, Sullivan JP (1995) Stable expression and pharmacological properties of the human alpha7 nicotinic acetylcholine receptor. Eur J Pharmacol 290:237-246.
Gorell JM, Rybicki BA, Johnson CC, Peterson EL (1999) Smoking and Parkinson's disease: a dose-response relationship. Neurology 52:115-119.
Gotham AM, Brown RG, Marsden CD (1988) 'Frontal' cognitive function in patients with Parkinson's disease 'on' and 'off' levodopa. Brain 111:299-321.
Gotti C, Fornasari D, Clementi F (1997a) Human neuronal nicotinic receptors. Prog Neurobiol 53:199-237. Gotti C, Hanke W, Maury K, Moretti M, Ballivet M, Clementi F, Bertrand D (1994) Pharmacology and biophysical
properties of alpha7 and alpha7-alpha8 alpha-bungarotoxin receptor subtypes immunopurified from the chick optic lobe. Eur J Neurosci 6:1281-1291.
Gotti C, Moretti M, Maggi R, Longhi R, Hanke W, Klinke N, Clementi F (1997b) Alpha7 and alpha8 nicotinic receptor subtypes immunopurified from chick retina have different immunological, pharmacological and functional properties. Eur J Neurosci 9:1201-1211.
Gould E, Woolf NJ, Butcher LL (1989) Cholinergic projections to the substantia nigra from the pedunculopontine and laterodorsal tegmental nuclei. Neuroscience 28:611-623.
Grace AA, Bunney BS (1984a) The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci 4:2877-2890.
Grace AA, Bunney BS (1984b) The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci 4:2866-2876.
Grady S, Marks MJ, Wonnacott S, Collins AC (1992) Characterization of nicotinic receptor-mediated 3H dopamine release from synaptosomes prepared from mouse striatum. J Neurochem 59:848-856.
Grady SR, Marks MJ, Collins AC (1994) Desensitization of nicotine-stimulated 3H dopamine release from mouse striatal synaptosomes. J Neurochem 62:1390-1398.
Grady SR, Meinerz NM, Cao J, Reynolds AM, Picciotto MR, Changeux JP, McIntosh JM, Marks MJ, Collins AC (2001) Nicotinic agonists stimulate acetylcholine release from mouse interpeduncular nucleus: a function mediated by a different nAChR than dopamine release from striatum. J Neurochem 76:258-268.
Graybiel AM (1990) Neurotransmitters and neuromodulators in the basal ganglia. Trends Neurosci 13:244-254. Grenhoff J, Svensson TH (1988) Selective stimulation of limbic dopamine activity by nicotine. Acta Physiol Scand 133:595-
596. Grenhoff J, Svensson TH (1992) Nicotinic and muscarinic components of rat brain dopamine synthesis stimulation induced
by physostigmine. Naunyn-Schmiedebergs Arch Pharmacol 346:395-398. Grenhoff J, Aston-Jones G, Svensson TH (1986) Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta
Physiol Scand 128:351-358. Grenhoff J, Tung CS, Svensson TH (1988) The excitatory amino acid antagonist kynurenate induces pacemaker-like firing of
dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand 134:567-568. Grillner P, Svensson TH (2000) Nicotine-induced excitation of midbrain dopamine neurons in vitro involves ionotropic
glutamate receptor activation. Synapse 38:1-9. Grilly DM, Simon BB, Levin ED (2000) Nicotine enhances stimulus detection performance of middle- and old-aged rats: a
longitudinal study. Pharmacol Biochem Behav 65:665-670.

105
Groot-Kormelink PJ, Luyten WH, Colquhoun D, Sivilotti LG (1998) A reporter mutation approach shows incorporation of the "orphan" subunit beta3 into a functional nicotinic receptor. J Biol Chem 273:15317-15320.
Grottick AJ, Trube G, Corrigall WA, Huwyler J, Malherbe P, Wyler R, Higgins GA (2000) Evidence that nicotinic alpha7 receptors are not involved in the hyperlocomotor and rewarding effects of nicotine. J Pharmacol Exp Ther 294:1112-1119.
Guan ZZ, Zhang X, Blennow K, Nordberg A (1999) Decreased protein level of nicotinic receptor alpha7 subunit in the frontal cortex from schizophrenic brain. Neuroreport 10:1779-1782.
Guan ZZ, Zhang X, Ravid R, Nordberg A (2000) Decreased protein levels of nicotinic receptor subunits in the hippocampus and temporal cortex of patients with Alzheimer's disease. J Neurochem 74:237-243.
Guan ZZ, Nordberg A, Mousavi M, Rinne JO, Hellström-Lindahl E (2002) Selective changes in the levels of nicotinic acetylcholine receptor protein and of corresponding mRNA species in the brains of patients with Parkinson's disease. Brain Res 956:358-366.
Guo JZ, Tredway TL, Chiappinelli VA (1998) Glutamate and GABA release are enhanced by different subtypes of presynaptic nicotinic receptors in the lateral geniculate nucleus. J Neurosci 18:1963-1969.
Göldner FM, Dineley KT, Patrick JW (1997) Immunohistochemical localization of the nicotinic acetylcholine receptor subunit alpha6 to dopaminergic neurons in the substantia nigra and ventral tegmental area. Neuroreport 8:2739-2742.
Hadjiconstantinou M, Hubble JP, Wemlinger TA, Neff NH (1994) Enhanced MPTP neurotoxicity after treatment with isoflurophate or cholinergic agonists. J Pharmacol Exp Ther 270:639-644.
Hahn B, Sharples CG, Wonnacott S, Shoaib M, Stolerman IP (2003) Attentional effects of nicotinic agonists in rats. Neuropharmacology 44:1054-1067.
Haikala H (1986) Different changes in striatal dopamine metabolism induced by nicotine in mice kept at different ambient temperatures. Evidence for partly separate metabolic routes of dopamine derived from separate compartmentations. Naunyn-Schmiedebergs Arch Pharmacol 334:373-376.
Haikala H (1987) Use of a novel type of rotating disc electrode and a flow cell with laminar flow pattern for the electrochemical detection of biogenic monoamines and their metabolites after Sephadex gel chromatographic purification and high-performance liquid chromatographic isolation from rat brain. J Neurochem 49:1033-1041.
Haikala H, Ahtee L (1988) Antagonism of the nicotine-induced changes of the striatal dopamine metabolism in mice by mecamylamine and pempidine. Naunyn-Schmiedebergs Arch Pharmacol 338:169-173.
Haikala H, Karmalahti T, Ahtee L (1986) The nicotine-induced changes in striatal dopamine metabolism of mice depend on body temperature. Brain Res 375:313-319.
Hamani C, Lozano AM (2003) Physiology and pathophysiology of Parkinson's disease. Ann NY Acad Sci 991:15-21. Han Z-Y, Zoli M, Cardona A, Bourgeois J-P, Changeux J-P, Le Novere N (2003) Localization of 3H nicotine, 3H cytisine,
3H epibatidine, and 125I bungarotoxin binding sites in the brain of macaca mulatta. J Comp Neurol 461:49-60. Harsing LG, Sershen H, Vizi SE, Lajtha A (1992) N-type calcium channels are involved in the dopamine releasing effect of
nicotine. Neurochem Res 17:729-734. Hauber W (1998) Involvement of basal ganglia transmitter systems in movement initiation. Prog Neurobiol 56:507-540. Heimer L, Zahm DS, Churchill L, Kalivas PW, Wohltmann C (1991) Specificity in the projection patterns of accumbal core
and shell in the rat. Neuroscience 41:89-125. Hellström-Lindahl E, Mousavi M, Ravid R, Nordberg A (2004a) Reduced levels of Abeta 40 and Abeta 42 in brains of
smoking controls and Alzheimer's patients. Neurobiol Dis 15:351-360. Hellström-Lindahl E, Mousavi M, Zhang X, Ravid R, Nordberg A (1999) Regional distribution of nicotinic receptor subunit
mRNAs in human brain: comparison between Alzheimer and normal brain. Brain Res Mol Brain Res 66:94-103. Hellström-Lindahl E, Court J, Keverne J, Svedberg M, Lee M, Marutle A, Thomas A, Perry E, Bednar I, Nordberg A (2004b)
Nicotine reduces Abeta in the brain and cerebral vessels of APPsw mice. Eur J Neurosci 19:2703-2710. Henningfield JE, Goldberg SR (1983) Control of behavior by intravenous nicotine injections in human subjects. Pharmacol
Biochem Behav 19:1021-1026. Henry B, Crossman AR, Brotchie JM (1998) Characterization of enhanced behavioral responses to L-DOPA following
repeated administration in the 6-hydroxydopamine-lesioned rat model of Parkinson's disease. Exp Neurol 151:334-342. Herning RI, Jones RT, Benowitz NL, Mines AH (1983) How a cigarette is smoked determines blood nicotine levels. Clin
Pharmacol Ther 33:84-90. Hill JA, Zoli M, Bourgeois JP, Changeux JP (1993) Immunocytochemical localization of a neuronal nicotinic receptor: the
beta2-subunit. J Neurosci 13:1551-1568. Hoffman AF, Gerhardt GA (1998) In vivo electrochemical studies of dopamine clearance in the rat substantia nigra: effects
of locally applied uptake inhibitors and unilateral 6-hydroxydopamine lesions. J Neurochem 70:179-189. Hogg RC, Bertrand D (2004) Nicotinic acetylcholine receptors as drug targets. Curr Drug Target CNS Neurol Disord 3:123-
130. Holladay MW, Dart MJ, Lynch JK (1997) Neuronal nicotinic acetylcholine receptors as targets for drug discovery. J Med
Chem 40:4169-4194. Huotari M, Santha M, Lucas LR, Karayiorgou M, Gogos JA, Mannisto PT (2002) Effect of dopamine uptake inhibition on
brain catecholamine levels and locomotion in catechol-O-methyltransferase-disrupted mice. J Pharmacol Exp Ther 303:1309-1316.
Imperato A, Mulas A, Di Chiara G (1986) Nicotine preferentially stimulates dopamine release in the limbic system of freely moving rats. Eur J Pharmacol 132:337-338.
Ishikawa A, Miyatake T (1993) Effects of smoking in patients with early-onset Parkinson's disease. J Neurol Sci 117:28-32. Ito R, Robbins TW, Everitt BJ (2004) Differential control over cocaine-seeking behavior by nucleus accumbens core and
shell. Nat Neuroscience 7:389-397.

106
Iwamoto ET (1990) Nicotine conditions place preferences after intracerebral administration in rats. Psychopharmacology 100:251-257.
Jacobs I, Anderson DJ, Surowy CS, Puttfarcken PS (2002) Differential regulation of nicotinic receptor-mediated neurotransmitter release following chronic (-)-nicotine administration. Neuropharmacology 43:847-856.
James JR, Villanueva HF, Johnson JH, Arezo S, Rosecrans JA (1994) Evidence that nicotine can acutely desensitize central nicotinic acetylcholinergic receptors. Psychopharmacology 114:456-462.
Janson AM, Moller A (1993) Chronic nicotine treatment counteracts nigral cell loss induced by a partial mesodiencephalic hemitransection: an analysis of the total number and mean volume of neurons and glia in substantia nigra of the male rat. Neuroscience 57:931-941.
Janson AM, Fuxe K, Goldstein M (1992) Differential effects of acute and chronic nicotine treatment on MPTP-(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) induced degeneration of nigrostriatal dopamine neurons in the black mouse. Clin Investigator 70:232-238.
Janson AM, Meana JJ, Goiny M, Herrera-Marschitz M (1991) Chronic nicotine treatment counteracts the decrease in extracellular neostriatal dopamine induced by a unilateral transection at the mesodiencephalic junction in rats: a microdialysis study. Neurosci Lett 134:88-92.
Janson AM, Hedlund PB, Fuxe K, von Euler G (1994) Chronic nicotine treatment counteracts dopamine D2 receptor upregulation induced by a partial meso-diencephalic hemitransection in the rat. Brain Res 655:25-32.
Janson AM, Fuxe K, Agnati LF, Kitayama I, Harfstrand A, Andersson K, Goldstein M (1988) Chronic nicotine treatment counteracts the disappearance of tyrosine-hydroxylase-immunoreactive nerve cell bodies, dendrites and terminals in the mesostriatal dopamine system of the male rat after partial hemitransection. Brain Res 455:332-345.
Jellinger KA (1997) Morphological substrates of dementia in parkinsonism. A critical update. J Neural Transm Suppl 51:57-82.
Jellinger KA (1999) Post mortem studies in Parkinson's disease--is it possible to detect brain areas for specific symptoms? J Neural Transm Suppl 56:1-29.
Jeyarasasingam G, Tompkins L, Quik M (2002) Stimulation of non-alpha7 nicotinic receptors partially protects dopaminergic neurons from 1-methyl-4-phenylpyridinium-induced toxicity in culture. Neuroscience 109:275-285.
Johnson DS, Martinez J, Elgoyhen AB, Heinemann SF, McIntosh JM (1995) Alpha-conotoxin ImI exhibits subtype-specific nicotinic acetylcholine receptor blockade: preferential inhibition of homomeric alpha7 and alpha9 receptors. Mol Pharmacol 48:194-199.
Jones GM, Sahakian BJ, Levy R, Warburton DM, Gray JA (1992) Effects of acute subcutaneous nicotine on attention, information processing and short-term memory in Alzheimer's disease. Psychopharmacology 108:485-494.
Jorenby DE, Steinpreis RE, Sherman JE, Baker TB (1990) Aversion instead of preference learning indicated by nicotine place conditioning in rats. Psychopharmacology 101:533-538.
Kaakkola S (1980) Contralateral circling behaviour induced by intranigral injection of morphine and enkephalin analogue FK 33-824 in rats. Acta Pharmacol Toxicol 47:385-393.
Kaakkola S (1981) Effect of nicotinic and muscarinic drugs on amphetamine- and apomorphine-induced circling behaviour in rats. Acta Pharmacol Toxicol 48:162-167.
Kaakkola S, Wurtman RJ (1992) Effects of COMT inhibitors on striatal dopamine metabolism: a microdialysis study. Brain Res 587:241-249.
Kaakkola S, Männistö PT, Nissinen E (1987) Striatal membrane-bound and soluble catechol-O-methyl-transferase after selective neuronal lesions in the rat. J Neural Transm Gen Sect 69:221-228.
Kaiser S, Wonnacott S (2000) Alpha-bungarotoxin-sensitive nicotinic receptors indirectly modulate 3H dopamine release in rat striatal slices via glutamate release. Mol Pharmacol 58:312-318.
Kaiser SA, Soliakov L, Harvey SC, Luetje CW, Wonnacott S (1998) Differential inhibition by alpha-conotoxin-MII of the nicotinic stimulation of 3H-dopamine release from rat striatal synaptosomes and slices. J Neurochem 70:1069-1076.
Kaneko S, Maeda T, Kume T, Kochiyama H, Akaike A, Shimohama S, Kimura J (1997) Nicotine protects cultured cortical neurons against glutamate-induced cytotoxicity via alpha7-neuronal receptors and neuronal CNS receptors. Brain Res 765:135-140.
Karlin A (1993) Structure of nicotinic acetylcholine receptors. Curr Opin Neurobiol 3:299-309. Karlin A, Akabas MH (1995) Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins.
Neuron 15:1231-1244. Karreman M, Westerink BH, Moghaddam B (1996) Excitatory amino acid receptors in the ventral tegmental area regulate
dopamine release in the ventral striatum. J Neurochem 67:601-607. Katz B, Thessleff S (1957) A study of desensitization produced by acetylcholine at the motor end-plate. J Physiol (Lond)
138:63-80. Kawai H, Berg DK (2001) Nicotinic acetylcholine receptors containing alpha7 subunits on rat cortical neurons do not
undergo long-lasting inactivation even when up-regulated by chronic nicotine exposure. J Neurochem 78:1367-1378. Kayadjanian N, Retaux S, Menetrey A, Besson MJ (1994) Stimulation by nicotine of the spontaneous release of 3H gamma-
aminobutyric acid in the substantia nigra and in the globus pallidus of the rat. Brain Res 649:129-135. Ke L, Eisenhour CM, Bencherif M, Lukas RJ (1998) Effects of chronic nicotine treatment on expression of diverse nicotinic
acetylcholine receptor subtypes. I. Dose- and time-dependent effects of nicotine treatment. J Pharmacol Exp Ther 286:825-840.
Kehr W (1976) 3-Methoxytyramine as an indicator of impulse-induced dopamine release in rat brain in vivo. Naunyn-Schmiedebergs Arch Pharmacol 293:209-215.
Kelton MC, Kahn HJ, Conrath CL, Newhouse PA (2000) The effects of nicotine on Parkinson's disease. Brain Cogn 43:274-282.

107
Kem WR, Mahnir VM, Papke RL, Lingle CJ (1997) Anabaseine is a potent agonist on muscle and neuronal alpha-bungarotoxin-sensitive nicotinic receptors. J Pharmacol Exp Ther 283:979-992.
Kita H, Kitai ST (1987) Efferent projections of the subthalamic nucleus in the rat: light and electron microscopic analysis with the PHA-L method. J Comp Neurol 260:435-452.
Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP (2001) Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21:1452-1463.
Koob GF (1996) Drug addiction: the yin and yang of hedonic homeostasis. Neuron 16:893-896. Koob GF, Le Moal M (2001) Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24:97-129. Koós T, Tepper JM (1999) Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat
Neuroscience 2:467-472. Ksir C, Hakan R, Hall DP, Jr., Kellar KJ (1985) Exposure to nicotine enhances the behavioral stimulant effect of nicotine and
increases binding of 3H acetylcholine to nicotinic receptors. Neuropharmacology 24:527-531. Kulak JM, Nguyen TA, Olivera BM, McIntosh JM (1997) Alpha-conotoxin MII blocks nicotine-stimulated dopamine release
in rat striatal synaptosomes. J Neurosci 17:5263-5270. Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J (1997) Mutation causing autosomal dominant nocturnal frontal
lobe epilepsy alters Ca2+ permeability, conductance, and gating of human alpha4beta2 nicotinic acetylcholine receptors. J Neurosci 17:9035-9047.
Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J (2000) Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology 39:2570-2590.
Lança AJ, Adamson KL, Coen KM, Chow BL, Corrigall WA (2000a) The pedunculopontine tegmental nucleus and the role of cholinergic neurons in nicotine self-administration in the rat: a correlative neuroanatomical and behavioral study. Neuroscience 96:735-742.
Lança JA, Sanelli TR, Corrigall WA (2000b) Nicotine-induced fos expression in the pedunculopontine mesencephalic tegmentum in the rat. Neuropharmacology 39:2808-2817.
Lange KW, Wells FR, Jenner P, Marsden CD (1993) Altered muscarinic and nicotinic receptor densities in cortical and subcortical brain regions in Parkinson's disease. J Neurochem 60:197-203.
Lange KW, Paul GM, Naumann M, Gsell W (1995) Dopaminergic effects on cognitive performance in patients with Parkinson's disease. J Neural Transm Suppl 46:423-432.
Lapin EP, Maker HS, Sershen H, Hurd Y, Lajtha A (1987) Dopamine-like action of nicotine: lack of tolerance and reverse tolerance. Brain Res 407:351-363.
Le Foll B, Goldberg SR (2005) Nicotine induces conditioned place preferences over a large range of doses in rats. Psychopharmacology 178:481-492.
Le Novere N, Changeux JP (1995) Molecular evolution of the nicotinic acetylcholine receptor: an example of multigene family in excitable cells. J Mol Evol 40:155-172.
Le Novere N, Zoli M, Changeux JP (1996) Neuronal nicotinic receptor alpha6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci 8:2428-2439.
Le Novere N, Zoli M, Lena C, Ferrari R, Picciotto MR, Merlo-Pich E, Changeux JP (1999) Involvement of alpha6 nicotinic receptor subunit in nicotine-elicited locomotion, demonstrated by in vivo antisense oligonucleotide infusion. Neuroreport 10:2497-2501.
Lecca D, Shim I, Costa E, Javaid JI (2000) Striatal application of nicotine, but not of lobeline, attenuates dopamine release in freely moving rats. Neuropharmacology 39:88-98.
Lee M, Martin-Ruiz C, Graham A, Court J, Jaros E, Perry R, Iversen P, Bauman M, Perry E (2002) Nicotinic receptor abnormalities in the cerebellar cortex in autism. Brain 125:1483-1495.
Leikola-Pelho T, Jackson DM (1992) Preferential stimulation of locomotor activity by ventral tegmental microinjections of (-)-nicotine. Pharmacol Toxicol 70:50-52.
Leikola-Pelho T, Heinämäki J, Laakso I, Ahtee L (1990) Chronic nicotine treatment changes differentially the effects of acute nicotine on the three main dopamine metabolites in mouse striatum. Naunyn-Schmiedebergs Arch Pharmacol 342:400-406.
Lena C, Changeux JP (1993) Allosteric modulations of the nicotinic acetylcholine receptor. Trends Neurosci 16:181-186. Lena C, Changeux JP (1997a) Pathological mutations of nicotinic receptors and nicotine-based therapies for brain disorders.
Curr Opin Neurobiol 7:674-682. Lena C, Changeux JP (1997b) Role of Ca2+ ions in nicotinic facilitation of GABA release in mouse thalamus. J Neurosci
17:576-585. Lena C, Changeux JP (1998) Allosteric nicotinic receptors, human pathologies. J Physiol (Paris) 92:63-74. Lena C, Changeux JP, Mulle C (1993) Evidence for "preterminal" nicotinic receptors on GABAergic axons in the rat
interpeduncular nucleus. J Neurosci 13:2680-2688. Leonard S, Gault J, Hopkins J, Logel J, Vianzon R, Short M, Drebing C, Berger R, Venn D, Sirota P, Zerbe G, Olincy A,
Ross RG, Adler LE, Freedman R (2002) Association of promoter variants in the alpha7 nicotinic acetylcholine receptor subunit gene with an inhibitory deficit found in schizophrenia. Arch Gen Psychiatry 59:1085-1096.
Lerman C, Caporaso N, Main D, Audrain J, Boyd NR, Bowman ED, Shields PG (1998) Depression and self-medication with nicotine: the modifying influence of the dopamine D4 receptor gene. Health Psychol 17:56-62.
Levin ED, Torry D (1996) Acute and chronic nicotine effects on working memory in aged rats. Psychopharmacology 123:88-97.
Levin ED, Simon BB (1998) Nicotinic acetylcholine involvement in cognitive function in animals. Psychopharmacologia 138:217-230.
Levin ED, Rezvani AH (2000) Development of nicotinic drug therapy for cognitive disorders. Eur J Pharmacol 393:141-146.

108
Levin ED, Rezvani AH (2002) Nicotinic treatment for cognitive dysfunction. Curr Drug Target CNS Neurol Disord 1:423-431.
Levin ED, Conners CK, Silva D, Canu W, March J (2001) Effects of chronic nicotine and methylphenidate in adults with attention deficit/hyperactivity disorder. Exp Clin Psychopharmacol 9:83-90.
Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, Rose JE (1998) Transdermal nicotine effects on attention. Psychopharmacology 140:135-141.
Li Y, Papke RL, He YJ, Millard WJ, Meyer EM (1999) Characterization of the neuroprotective and toxic effects of alpha7 nicotinic receptor activation in PC12 cells. Brain Res 830:218-225.
Lichtensteiger W, Felix D, Lienhart R, Hefti F (1976) A quantitative correlation between single unit activity and fluorescence intensity of dopamine neurones in zona compacta of substantia nigra, as demonstrated under the influence of nicotine and physostigmine. Brain Res 117:85-103.
Lichtensteiger W, Hefti F, Felix D, Huwyler T, Melamed E, Schlumpf M (1982) Stimulation of nigrostriatal dopamine neurones by nicotine. Neuropharmacology 21:963-968.
Limberger N, Spath L, Starke K (1986) A search for receptors modulating the release of gamma3H aminobutyric acid in rabbit caudate nucleus slices. J Neurochem 46:1109-1117.
Lindstrom J, Anand R, Peng X, Gerzanich V, Wang F, Li Y (1995) Neuronal nicotinic receptor subtypes. Ann NY Acad Sci 757:100-116.
Lippiello PM, Bencherif M, Gray JA, Peters S, Grigoryan G, Hodges H, Collins AC (1996) RJR-2403: a nicotinic agonist with CNS selectivity II. In vivo characterization. J Pharmacol Exp Ther 279:1422-1429.
Lloyd GK, Williams M (2000) Neuronal nicotinic acetylcholine receptors as novel drug targets. J Pharmacol Exp Ther 292:461-467.
Lloyd GK, Menzaghi F, Bontempi B, Suto C, Siegel R, Akong M, Stauderman K, Velicelebi G, Johnson E, Harpold MM, Rao TS, Sacaan AI, Chavez-Noriega LE, Washburn MS, Vernier JM, Cosford ND, McDonald LA (1998) The potential of subtype-selective neuronal nicotinic acetylcholine receptor agonists as therapeutic agents. Life Sci 62:1601-1606.
Louis M, Clarke PB (1998) Effect of ventral tegmental 6-hydroxydopamine lesions on the locomotor stimulant action of nicotine in rats. Neuropharmacology 37:1503-1513.
Lu Y, Marks MJ, Collins AC (1999) Desensitization of nicotinic agonist-induced 3H gamma-aminobutyric acid release from mouse brain synaptosomes is produced by subactivating concentrations of agonists. J Pharmacol Exp Ther 291:1127-1134.
Lu Y, Grady S, Marks MJ, Picciotto M, Changeux JP, Collins AC (1998) Pharmacological characterization of nicotinic receptor-stimulated GABA release from mouse brain synaptosomes. J Pharmacol Exp Ther 287:648-657.
Luetje CW (2004) Getting past the asterisk: the subunit composition of presynaptic nicotinic receptors that modulate striatal dopamine release. Mol Pharmacol 65:1333-1335.
Luetje CW, Patrick J (1991) Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci 11:837-845.
Luetje CW, Wada K, Rogers S, Abramson SN, Tsuji K, Heinemann S, Patrick J (1990) Neurotoxins distinguish between different neuronal nicotinic acetylcholine receptor subunit combinations. J Neurochem 55:632-640.
Lukas RJ (1995) Diversity and patterns of regulation of nicotinic receptor subtypes. Ann NY Acad Sci 757:153-168. Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, Bertrand D, Chiappinelli VA, Clarke PB,
Collins AC, Dani JA, Grady SR, Kellar KJ, Lindstrom JM, Marks MJ, Quik M, Taylor PW, Wonnacott S (1999) International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev 51:397-401.
Luo S, Kulak JM, Cartier GE, Jacobsen RB, Yoshikami D, Olivera BM, McIntosh JM (1998) Alpha-conotoxin AuIB selectively blocks alpha3 beta4 nicotinic acetylcholine receptors and nicotine-evoked norepinephrine release. J Neurosci 18:8571-8579.
Maggio R, Riva M, Vaglini F, Fornai F, Molteni R, Armogida M, Racagni G, Corsini GU (1998) Nicotine prevents experimental parkinsonism in rodents and induces striatal increase of neurotrophic factors. J Neurochem 71:2439-2446.
Maldonado-Irizarry CS, Kelley AE (1995) Excitotoxic lesions of the core and shell subregions of the nucleus accumbens differentially disrupt body weight regulation and motor activity in rat. Brain Res Bull 38:551-559.
Mansvelder HD, McGehee DS (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27:349-357.
Mansvelder HD, Keath JR, McGehee DS (2002) Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron 33:905-919.
Mansvelder HD, De Rover M, McGehee DS, Brussaard AB (2003) Cholinergic modulation of dopaminergic reward areas: upstream and downstream targets of nicotine addiction. Eur J Pharmacol 480:117-123.
Marchi M, Risso F, Viola C, Cavazzani P, Raiteri M (2002) Direct evidence that release-stimulating alpha7* nicotinic cholinergic receptors are localized on human and rat brain glutamatergic axon terminals. J Neurochem 80:1071-1078.
Marin P, Maus M, Desagher S, Glowinski J, Premont J (1994) Nicotine protects cultured striatal neurones against N-methyl-D-aspartate receptor-mediated neurotoxicity. Neuroreport 5:1977-1980.
Markou A, Paterson NE, Semenova S (2004) Role of gamma-aminobutyric acid (GABA) and metabotropic glutamate receptors in nicotine reinforcement: potential pharmacotherapies for smoking cessation. Ann NY Acad Sci 1025:491-503.
Marks MJ, Burch JB, Collins AC (1983) Effects of chronic nicotine infusion on tolerance development and nicotinic receptors. J Pharmacol Exp Ther 226:817-825.
Marks MJ, Stitzel JA, Collins AC (1985) Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther 235:619-628.

109
Marks MJ, Grady SR, Collins AC (1993) Downregulation of nicotinic receptor function after chronic nicotine infusion. J Pharmacol Exp Ther 266:1268-1276.
Marks MJ, Smith KW, Collins AC (1998) Differential agonist inhibition identifies multiple epibatidine binding sites in mouse brain. J Pharmacol Exp Ther 285:377-386.
Marks MJ, Stitzel JA, Romm E, Wehner JM, Collins AC (1986) Nicotinic binding sites in rat and mouse brain: comparison of acetylcholine, nicotine, and alpha-bungarotoxin. Mol Pharmacol 30:427-436.
Marks MJ, Pauly JR, Gross SD, Deneris ES, Hermans-Borgmeyer I, Heinemann SF, Collins AC (1992) Nicotine binding and nicotinic receptor subunit RNA after chronic nicotine treatment. J Neurosci 12:2765-2784.
Marrero MB, Papke RL, Bhatti BS, Shaw S, Bencherif M (2004) The neuroprotective effect of 2-(3-pyridyl)-1-azabicyclo3.2.2 nonane (TC-1698), a novel alpha7 ligand, is prevented through angiotensin II activation of a tyrosine phosphatase. J Pharmacol Exp Ther 309:16-27.
Marshall D, Soliakov L, Redfern P, Wonnacott S (1996) Tetrodotoxin-sensitivity of nicotine-evoked dopamine release from rat striatum. Neuropharmacology 35:1531-1536.
Marshall DL, Redfern PH, Wonnacott S (1997) Presynaptic nicotinic modulation of dopamine release in the three ascending pathways studied by in vivo microdialysis: comparison of naive and chronic nicotine-treated rats. J Neurochem 68:1511-1519.
Marshall J, Schnieden H (1966) Effect of adrenaline, noradrenaline, atropine and nicotine on some types of human tremor. J Neurol Neurosurg Psychiatry 29.
Marshall JF, O'Dell SJ, Navarrete R, Rosenstein AJ (1990) Dopamine high-affinity transport site topography in rat brain: major differences between dorsal and ventral striatum. Neuroscience 37:11-21.
Martin LF, Kem WR, Freedman R (2004) Alpha-7 nicotinic receptor agonists: potential new candidates for the treatment of schizophrenia. Psychopharmacology 174:54-64.
Martin-Ruiz CM, Lee M, Perry RH, Baumann M, Court JA, Perry EK (2004) Molecular analysis of nicotinic receptor expression in autism. Brain Res Mol Brain Res 123:81-90.
Martin-Ruiz CM, Piggott M, Gotti C, Lindstrom J, Mendelow AD, Siddique MS, Perry RH, Perry EK, Court JA (2000) Alpha and beta nicotinic acetylcholine receptors subunits and synaptophysin in putamen from Parkinson's disease. Neuropharmacology 39:2830-2839.
Martin-Ruiz CM, Court JA, Molnar E, Lee M, Gotti C, Mamalaki A, Tsouloufis T, Tzartos S, Ballard C, Perry RH, Perry EK (1999) Alpha4 but not alpha3 and alpha7 nicotinic acetylcholine receptor subunits are lost from the temporal cortex in Alzheimer's disease. J Neurochem 73:1635-1640.
Marubio LM, del Mar Arroyo-Jimenez M, Cordero-Erausquin M, Lena C, Le Novere N, de Kerchove d'Exaerde A, Huchet M, Damaj MI, Changeux JP (1999) Reduced antinociception in mice lacking neuronal nicotinic receptor subunits. Nature 398:805-810.
Marubio LM, Gardier AM, Durier S, David D, Klink R, Arroyo-Jimenez MM, McIntosh JM, Rossi F, Champtiaux N, Zoli M, Changeux JP (2003) Effects of nicotine in the dopaminergic system of mice lacking the alpha4 subunit of neuronal nicotinic acetylcholine receptors. Eur J Neurosci 17:1329-1337.
Marutle A, Warpman U, Bogdanovic N, Nordberg A (1998) Regional distribution of subtypes of nicotinic receptors in human brain and effect of aging studied by (+/-) 3H epibatidine. Brain Res 801:143-149.
Maurice T, Lockhart BP, Privat A (1996) Amnesia induced in mice by centrally administered beta-amyloid peptides involves cholinergic dysfunction. Brain Res 706:181-193.
McEvoy JP, Allen TB (2002) The importance of nicotinic acetylcholine receptors in schizophrenia, bipolar disorder and Tourette's syndrome. Curr Drug Target CNS Neurol Disord 1:433-442.
McGehee DS, Role LW (1995) Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol 57:521-546.
Mendoza J, Angeles-Castellanos M, Escobar C (2005) Differential role of the accumbens shell and core subterritories in food-entrained rhythms of rats. Behav Brain Res 158:133-142.
Menzaghi F, Whelan KT, Risbrough VB, Rao TS, Lloyd GK (1997a) Interactions between a novel cholinergic ion channel agonist, SIB-1765F and L-DOPA in the reserpine model of Parkinson's disease in rats. J Pharmacol Exp Ther 280:393-401.
Menzaghi F, Whelan KT, Risbrough VB, Rao TS, Lloyd GK (1997b) Effects of a novel cholinergic ion channel agonist SIB-1765F on locomotor activity in rats. J Pharmacol Exp Ther 280:384-392.
Mercuri NB, Calabresi P, Bernardi G (1992) The electrophysiological actions of dopamine and dopaminergic drugs on neurons of the substantia nigra pars compacta and ventral tegmental area. Life Sci 51:711-718.
Mereu G, Yoon KW, Boi V, Gessa GL, Naes L, Westfall TC (1987) Preferential stimulation of ventral tegmental area dopaminergic neurons by nicotine. Eur J Pharmacol 141:395-399.
Meyer EM, Kuryatov A, Gerzanich V, Lindstrom J, Papke RL (1998) Analysis of 3-(4-hydroxy, 2-methoxybenzylidene)anabaseine selectivity and activity at human and rat alpha-7 nicotinic receptors. J Pharmacol Exp Ther 287:918-925.
Meyer EM, Tay ET, Papke RL, Meyers C, Huang GL, de Fiebre CM (1997) 32,4-Dimethoxybenzylidene anabaseine (DMXB) selectively activates rat alpha7 receptors and improves memory-related behaviors in a mecamylamine-sensitive manner. Brain Res 768:49-56.
Mihailescu S, Drucker-Colin R (2000) Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res 31:131-144.
Mikkola JA, Janhunen S, Hyytiä P, Kiianmaa K, Ahtee L (2002) Rotational behaviour in AA and ANA rats after repeated administration of morphine and cocaine. Pharmacol Biochem Behav 73:697-702.

110
Minana MD, Montoliu C, Llansola M, Grisolia S, Felipo V (1998) Nicotine prevents glutamate-induced proteolysis of the microtubule-associated protein MAP-2 and glutamate neurotoxicity in primary cultures of cerebellar neurons. Neuropharmacology 37:847-857.
Mirza NR, Pei Q, Stolerman IP, Zetterström TS (1996) The nicotinic receptor agonists (-)-nicotine and isoarecolone differ in their effects on dopamine release in the nucleus accumbens. Eur J Pharmacol 295:207-210.
Mogg AJ, Whiteaker P, McIntosh JM, Marks M, Collins AC, Wonnacott S (2002) Methyllycaconitine is a potent antagonist of alpha-conotoxin-MII-sensitive presynaptic nicotinic acetylcholine receptors in rat striatum. J Pharmacol Exp Ther 302:197-204.
Molinari EJ, Delbono O, Messi ML, Renganathan M, Arneric SP, Sullivan JP, Gopalakrishnan M (1998) Up-regulation of human alpha7 nicotinic receptors by chronic treatment with activator and antagonist ligands. Eur J Pharmacol 347:131-139.
Moll H (1926) The treatment of post-encephalitic parkinsonism by nicotine. Br Med J 1:1079-1081. Moore RY (2003) Organization of midbrain dopamine systems and the pathophysiology of Parkinson's disease. Parkinsonism
Rel Disord 9 Suppl 2:S65-71. Morens DM, Grandinetti A, Reed D, White LR, Ross GW (1995) Cigarette smoking and protection from Parkinson's disease:
false association or etiologic clue? Neurology 45:1041-1051. Morrison CF, Stephenson JA (1972) The occurrence of tolerance to a central depressant effect of nicotine. Br J Pharmacol
46:151-156. Museo E, Wise RA (1990) Microinjections of a nicotinic agonist into dopamine terminal fields: effects on locomotion.
Pharmacol Biochem Behav 37:113-116. Männistö PT, Kaakkola S (1999) Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology,
and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev 51:593-628. Nakachi N, Kiuchi Y, Inagaki M, Inazu M, Yamazaki Y, Oguchi K (1995) Effects of various dopamine uptake inhibitors on
striatal extracellular dopamine levels and behaviours in rats. Eur J Pharmacol 281:195-203. Neal MJ, Cunningham JR, Matthews KL (2001) Activation of nicotinic receptors on GABAergic amacrine cells in the rabbit
retina indirectly stimulates dopamine release. Vis Neurosci 18:55-64. Newhouse PA, Potter A, Singh A (2004) Effects of nicotinic stimulation on cognitive performance. Curr Opin Pharmacol
4:36-46. Nisell M, Nomikos GG, Svensson TH (1994a) Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of
the rat differentially affects accumbal dopamine release. Pharmacol Toxicol 75:348-352. Nisell M, Nomikos GG, Svensson TH (1994b) Systemic nicotine-induced dopamine release in the rat nucleus accumbens is
regulated by nicotinic receptors in the ventral tegmental area. Synapse 16:36-44. Nisell M, Marcus M, Nomikos GG, Svensson TH (1997) Differential effects of acute and chronic nicotine on dopamine
output in the core and shell of the rat nucleus accumbens. J Neural Transm (Budapest) 104:1-10. Nisell M, Nomikos GG, Hertel P, Panagis G, Svensson TH (1996) Condition-independent sensitization of locomotor
stimulation and mesocortical dopamine release following chronic nicotine treatment in the rat. Synapse 22:369-381. Nitsch C, Riesenberg R (1988) Immunocytochemical demonstration of GABAergic synaptic connections in rat substantia
nigra after different lesions of the striatonigral projection. Brain Res 461:127-142. Nitsch C, Mews K, Wagner A, Hassler R (1984) Effects of frontal motor cortex ablation on the ultrastructure of cat
substantia nigra. Acta Anat 119:193-202. Nomikos GG, Schilström B, Hildebrand BE, Panagis G, Grenhoff J, Svensson TH (2000) Role of alpha7 nicotinic receptors
in nicotine dependence and implications for psychiatric illness. Behav Brain Res 113:97-103. Nordberg A (1994) Human nicotinic receptors - their role in aging and dementia. Neurochem Int 25:93-97. Nordberg A, Nyberg P, Windblad B (1985) Topographic distribution of choline acetyltransferase activity and muscarinic and
nicotinic receptors in Parkinson brains. Neurochem Pathol 3:223-236. Nordberg A, Lundqvist H, Hartvig P, Andersson J, Johansson M, Hellström-Lindahi E, Langström B (1997) Imaging of
nicotinic and muscarinic receptors in Alzheimer's disease: effect of tacrine treatment. Dement Geriatr Cogn Disord 8:78-84.
Nordberg A, Hellström-Lindahl E, Lee M, Johnson M, Mousavi M, Hall R, Perry E, Bednar I, Court J (2002) Chronic nicotine treatment reduces beta-amyloidosis in the brain of a mouse model of Alzheimer's disease (APPsw). J Neurochem 81:655-658.
Nordberg A, Lilja A, Lundqvist H, Hartvig P, Amberla K, Viitanen M, Warpman U, Johansson M, Hellström-Lindahl E, Bjurling P, et al. (1992) Tacrine restores cholinergic nicotinic receptors and glucose metabolism in Alzheimer patients as visualized by positron emission tomography. Neurobiol Aging 13:747-758.
Nose T, Takemoto H (1974) Effect of oxotremorine on homovanillic acid concentration in the striatum of the rat. Eur J Pharmacol 25:51-55.
Nuutinen S, Ahtee L, Tuominen RK (2005) Time and brain region specific up-regulation of low affinity neuronal nicotinic receptors during chronic nicotine administration in mice. Eur J Pharmacol, in press
Obeso JA, Olanow CW, Nutt JG (2000) Levodopa motor complications in Parkinson's disease. Trends Neurosci 23:S2-7. Ochoa EL (1994) Nicotine-related brain disorders: the neurobiological basis of nicotine dependence. Cell Mol Neurobiol
14:195-225. Ochoa EL, Li L, McNamee MG (1990) Desensitization of central cholinergic mechanisms and neuroadaptation to nicotine.
Mol Neurobiol 4:251-287. Ogden DC, Colquhoun D (1985) Ion channel block by acetylcholine, carbachol and suberyldicholine at the frog
neuromuscular junction. Proc R Soc Lond B Biol Sci 225:329-355. Olanow CW (2004) The scientific basis for the current treatment of Parkinson's disease. Ann Rev Med 55:41-60.

111
O'Neill MF, Dourish CT, Iversen SD (1991) Evidence for an involvement of D1 and D2 dopamine receptors in mediating nicotine-induced hyperactivity in rats. Psychopharmacology 104:343-350.
Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL (1997) Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci 17:9165-9171.
Owman C, Fuxe K, Janson AM, Kahrstrom J (1989) Studies of protective actions of nicotine on neuronal and vascular functions in the brain of rats: comparison between sympathetic noradrenergic and mesostriatal dopaminergic fiber systems, and the effect of a dopamine agonist. Prog Brain Res 79:267-276.
Pagliusi SR, Tessari M, DeVevey S, Chiamulera C, Pich EM (1996) The reinforcing properties of nicotine are associated with a specific patterning of c-fos expression in the rat brain. Eur J Neurosci 8:2247-2256.
Panagis G, Nisell M, Nomikos GG, Chergui K, Svensson TH (1996) Nicotine injections into the ventral tegmental area increase locomotion and Fos-like immunoreactivity in the nucleus accumbens of the rat. Brain Res 730:133-142.
Papke RL, Heinemann SF (1994) Partial agonist properties of cytisine on neuronal nicotinic receptors containing the beta2 subunit. Mol Pharmacol 45:142-149.
Papke RL, Thinschmidt JS, Moulton BA, Meyer EM, Poirier A (1997) Activation and inhibition of rat neuronal nicotinic receptors by ABT-418. Br J Pharmacol 120:429-438.
Parain K, Hapdey C, Rousselet E, Marchand V, Dumery B, Hirsch EC (2003) Cigarette smoke and nicotine protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine Parkinsonian toxin. Brain Res 984:224-232.
Parent A (1990) Extrinsic connections of the basal ganglia. Trends Neurosci 13:254-258. Parent A, Sato F, Wu Y, Gauthier J, Levesque M, Parent M (2000) Organization of the basal ganglia: the importance of
axonal collateralization. Trends Neurosci 23:S20-27. Parker MJ, Beck A, Luetje CW (1998) Neuronal nicotinic receptor beta2 and beta4 subunits confer large differences in
agonist binding affinity. Mol Pharmacol 54:1132-1139. Parkinson J (1817) An Essay on the Shaking Palsy. London: Sherwood. Paterson D, Nordberg A (2000) Neuronal nicotinic receptors in the human brain. Prog Neurobiol 61:75-111. Pauly JR, Marks MJ, Gross SD, Collins AC (1991) An autoradiographic analysis of cholinergic receptors in mouse brain
after chronic nicotine treatment. J Pharmacol Exp Ther 258:1127-1136. Pauly JR, Marks MJ, Robinson SF, van de Kamp JL, Collins AC (1996) Chronic nicotine and mecamylamine treatment
increase brain nicotinic receptor binding without changing alpha4 or beta2 mRNA levels. J Pharmacol Exp Ther 278:361-369.
Paxinos G, Watson C (1986) The Rat Brain in Stereotaxic Coordinates, 2nd Edition. San Diego, CA: Academic Press. Peng H, Ferris RL, Matthews T, Hiel H, Lopez-Albaitero A, Lustig LR (2004) Characterization of the human nicotinic
acetylcholine receptor subunit alpha9 (CHRNA9) and alpha10 (CHRNA10) in lymphocytes. Life Sci 76:263-280. Peng X, Gerzanich V, Anand R, Whiting PJ, Lindstrom J (1994a) Nicotine-induced increase in neuronal nicotinic receptors
results from a decrease in the rate of receptor turnover. Mol Pharmacol 46:523-530. Peng X, Katz M, Gerzanich V, Anand R, Lindstrom J (1994b) Human alpha7 acetylcholine receptor: cloning of the alpha7
subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha7 homomers expressed in Xenopus oocytes. Mol Pharmacol 45:546-554.
Peng X, Gerzanich V, Anand R, Wang F, Lindstrom J (1997) Chronic nicotine treatment up-regulates alpha3 and alpha7 acetylcholine receptor subtypes expressed by the human neuroblastoma cell line SH-SY5Y. Mol Pharmacol 51:776-784.
Pereira EF, Alkondon M, McIntosh JM, Albuquerque EX (1996) Alpha-conotoxin-ImI: a competitive antagonist at alpha-bungarotoxin-sensitive neuronal nicotinic receptors in hippocampal neurons. J Pharmacol Exp Ther 278:1472-1483.
Perkins KA (2002) Chronic tolerance to nicotine in humans and its relationship to tobacco dependence. Nicotine Tobacco Res 4:405-422.
Perry DC, Davila-Garcia MI, Stockmeier CA, Kellar KJ (1999) Increased nicotinic receptors in brains from smokers: membrane binding and autoradiography studies. J Pharmacol Exp Ther 289:1545-1552.
Perry DC, Xiao Y, Nguyen HN, Musachio JL, Davila-Garcia MI, Kellar KJ (2002) Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem 82:468-481.
Perry EK, Perry RH (1995) Acetylcholine and hallucinations: disease-related compared to drug-induced alterations in human consciousness. Brain Cogn 28:240-258.
Perry EK, Morris CM, Court JA, Cheng A, Fairbairn AF, McKeith IG, Irving D, Brown A, Perry RH (1995) Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience 64:385-395.
Perry EK, Irving D, Kerwin JM, McKeith IG, Thompson P, Collerton D, Fairbairn AF, Ince PG, Morris CM, Cheng AV, et al. (1993) Cholinergic transmitter and neurotrophic activities in Lewy body dementia: similarity to Parkinson's and distinction from Alzheimer disease. Alzheimer Dis Assoc Disord 7:69-79.
Perry EK, Lee ML, Martin-Ruiz CM, Court JA, Volsen SG, Merrit J, Folly E, Iversen PE, Bauman ML, Perry RH, Wenk GL (2001) Cholinergic activity in autism: abnormalities in the cerebral cortex and basal forebrain. Am J Psychiatry 158:1058-1066.
Peto R, Lopez AD, Boreham J, Thun M, Heath C, Jr., Doll R (1996) Mortality from smoking worldwide. Br Med Bull 52:12-21.
Picciotto MR, Zoli M (2002) Nicotinic receptors in aging and dementia. J Neurobiol 53:641-655. Picciotto MR, Zoli M, Rimondini R, Lena C, Marubio LM, Pich EM, Fuxe K, Changeux JP (1998) Acetylcholine receptors
containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature 391:173-177.

112
Pich EM, Pagliusi SR, Tessari M, Talabot-Ayer D, Hooft van Huijsduijnen R, Chiamulera C (1997) Common neural substrates for the addictive properties of nicotine and cocaine. Science 275:83-86.
Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA (2004) Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem 11:60-69.
Pietilä K, Lähde T, Attila M, Ahtee L, Nordberg A (1998) Regulation of nicotinic receptors in the brain of mice withdrawn from chronic oral nicotine treatment. Naunyn-Schmiedebergs Arch Pharmacol 357:176-182.
Piggott MA, Marshall EF, Thomas N, Lloyd S, Court JA, Jaros E, Burn D, Johnson M, Perry RH, McKeith IG, Ballard C, Perry EK (1999) Striatal dopaminergic markers in dementia with Lewy bodies, Alzheimer's and Parkinson's diseases: rostrocaudal distribution. Brain 122:1449-1468.
Pillon B, Dubois B, Bonnet AM, Esteguy M, Guimaraes J, Vigouret JM, Lhermitte F, Agid Y (1989) Cognitive slowing in Parkinson's disease fails to respond to levodopa treatment: the 15-objects test. Neurology 39:762-768.
Pomerleau OF (1998) Endogenous opioids and smoking: a review of progress and problems. Psychoneuroendocrinology 23:115-130.
Pontieri FE, Tanda G, Orzi F, Di Chiara G (1996) Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature 382:255-257.
Prince RJ, Fernandes KG, Gregory JC, Martyn ID, Lippiello PM (1996) Modulation of nicotine-evoked 3H dopamine release from rat striatal synaptosomes by voltage-sensitive calcium channel ligands. Biochem Pharmacol 52:613-618.
Puttfarcken PS, Jacobs I, Faltynek CR (2000) Characterization of nicotinic acetylcholine receptor-mediated 3H-dopamine release from rat cortex and striatum. Neuropharmacology 39:2673-2680.
Pycock CJ, Marsden CD (1978) The rotating rodent: a two component system? Eur J Pharmacol 47:167-175. Pycock CJ, Horton RW (1979) Dopamine-dependent hyperactivity in the rat following manipulation of GABA mechanisms
in the region of the nucleus accumbens. J Neural Transm Gen Sect 45:17-33. Qian C, Li T, Shen TY, Libertine-Garahan L, Eckman J, Biftu T, Ip S (1993) Epibatidine is a nicotinic analgesic. Eur J
Pharmacol 250:R13-14. Quik M, Jeyarasasingam G (2000) Nicotinic receptors and Parkinson's disease. Eur J Pharmacol 393:223-230. Quik M, Kulak JM (2002) Nicotine and nicotinic receptors; relevance to Parkinson's disease. Neurotoxicology 23:581-594. Quik M, Philie J, Choremis J (1997) Modulation of alpha7 nicotinic receptor-mediated calcium influx by nicotinic agonists.
Mol Pharmacol 51:499-506. Quik M, Polonskaya Y, Kulak JM, McIntosh JM (2001) Vulnerability of 125I-alpha-conotoxin MII binding sites to
nigrostriatal damage in monkey. J Neurosci 21:5494-5500. Quik M, Polonskaya Y, McIntosh JM, Kulak JM (2002) Differential nicotinic receptor expression in monkey basal ganglia:
effects of nigrostriatal damage. Neuroscience 112:619-630. Quik M, Bordia T, Forno L, McIntosh JM (2004) Loss of alpha-conotoxinMII- and A85380-sensitive nicotinic receptors in
Parkinson's disease striatum. J Neurochem 88:668-679. Quik M, Polonskaya Y, Gillespie A, G KL, Langston JW (2000) Differential alterations in nicotinic receptor alpha6 and
beta3 subunit messenger RNAs in monkey substantia nigra after nigrostriatal degeneration. Neuroscience 100:63-72. Quik M, Bordia T, Okihara M, Fan H, Marks MJ, McIntosh JM, Whiteaker P (2003) L-DOPA treatment modulates nicotinic
receptors in monkey striatum. Mol Pharmacol 64:619-628. Quik M, Vailati S, Bordia T, Kulak JM, Fan H, McIntosh JM, Clementi F, Gotti C (2005) Subunit composition of nicotinic
receptors in monkey striatum: effect of treatments with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine or L-DOPA. Mol Pharmacol 67:32-41.
Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L (1996) Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature 380:347-351.
Rang HP, Dale MM, Ritter JM (1999) Pharmacology, 4th Edition. Edinburgh: Churchill Livingstone. Rao TS, Adams PB, Correa LD, Santori EM, Sacaan AI, Reid RT, Suto CM, Vernier JM (2003) In vitro pharmacological
characterization of (+/-)-42-(1-methyl-2-pyrrolidinyl)ethyl thio]phenol hydrochloride (SIB-1553A), a nicotinic acetylcholine receptor ligand. Brain Res 981:85-98.
Rapier C, Lunt GG, Wonnacott S (1988) Stereoselective nicotine-induced release of dopamine from striatal synaptosomes: concentration dependence and repetitive stimulation. J Neurochem 50:1123-1130.
Rapier C, Lunt GG, Wonnacott S (1990) Nicotinic modulation of 3H dopamine release from striatal synaptosomes: pharmacological characterisation. J Neurochem 54:937-945.
Rasmussen T, Swedberg MD (1998) Reinforcing effects of nicotinic compounds: intravenous self-administration in drug-naive mice. Pharmacol Biochem Behav 60:567-573.
Reavill C, Stolerman IP (1990) Locomotor activity in rats after administration of nicotinic agonists intracerebrally. Br J Pharmacol 99:273-278.
Reid MS, Ho LB, Berger SP (1996) Effects of environmental conditioning on the development of nicotine sensitization: behavioral and neurochemical analysis. Psychopharmacology 126:301-310.
Reuben M, Boye S, Clarke PB (2000) Nicotinic receptors modulating somatodendritic and terminal dopamine release differ pharmacologically. Eur J Pharmacol 393:39-49.
Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP (1991) Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature 353:846-849.
Rice ME, Cragg SJ (2004) Nicotine amplifies reward-related dopamine signals in striatum. Nat Neuroscience 7:583-584. Rinne JO, Myllykylä T, Lonnberg P, Marjamäki P (1991) A postmortem study of brain nicotinic receptors in Parkinson's and
Alzheimer's disease. Brain Res 547:167-170. Rivett AJ, Francis A, Roth JA (1983) Localization of membrane-bound catechol-O-methyltransferase. J Neurochem 40:1494-
1496.

113
Robinson TE, Berridge KC (1993) The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18:247-291.
Roceri M, Molteni R, Fumagalli F, Racagni G, Gennarelli M, Corsini G, Maggio R, Riva M (2001) Stimulatory role of dopamine on fibroblast growth factor-2 expression in rat striatum. J Neurochem 76:990-997.
Roffler-Tarlov S, Sharman DF, Tegerdine P (1971) 3,4-Dihydroxyphenylacetic acid and 4-hydroxy-3-methoxyphenylacetic acid in the mouse striatum: a reflection of intra- and extra-neuronal metabolism of dopamine? Br J Pharmacol 42:343-351.
Role LW (1992) Diversity in primary structure and function of neuronal nicotinic acetylcholine receptor channels. Curr Opin Neurobiol 2:254-262.
Role LW, Berg DK (1996) Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16:1077-1085. Romanelli L, Ohman B, Adem A, Nordberg A (1988) Subchronic treatment of rats with nicotine: interconversion of nicotinic
receptor subtypes in brain. Eur J Pharmacol 148:289-291. Rose JE, Corrigall WA (1997) Nicotine self-administration in animals and humans: similarities and differences.
Psychopharmacology 130:28-40. Rosecrans JA, Meltzer LT (1981) Central sites and mechanisms of action of nicotine. Neurosci Biobehav Rev 5:497-501. Roth KA, McIntire SL, Barchas JD (1982) Nicotinic-catecholaminergic interactions in rat brain: evidence for cholinergic
nicotinic and muscarinic interactions with hypothalamic epinephrine. J Pharmacol Exp Ther 221:416-420. Rowell PP (1995) Nanomolar concentrations of nicotine increase the release of 3H dopamine from rat striatal synaptosomes.
Neurosci Lett 189:171-175. Rowell PP (2002) Effects of nicotine on dopaminergic neurotransmission. In: Nicotinic receptors in the nervous system
(Levin ED, ed), pp 51-80. New York: CRC Press. Rowell PP, Hillebrand JA (1994) Characterization of nicotine-induced desensitization of evoked dopamine release from rat
striatal synaptosomes. J Neurochem 63:561-569. Rowell PP, Li M (1997) Dose-response relationship for nicotine-induced up-regulation of rat brain nicotinic receptors. J
Neurochem 68:1982-1989. Rowell PP, Carr LA, Garner AC (1987) Stimulation of 3H dopamine release by nicotine in rat nucleus accumbens. J
Neurochem 49:1449-1454. Russell MA, Raw M, Taylor C, Feyerabend C, Saloojee Y (1978) Blood nicotine and carboxyhemoglobin levels after rapid-
smoking aversion therapy. J Consult Clin Psychol 46:1423-1431. Rusted JM, Newhouse PA, Levin ED (2000) Nicotinic treatment for degenerative neuropsychiatric disorders such as
Alzheimer's disease and Parkinson's disease. Behav Brain Res 113:121-129. Ryan RE, Ross SA, Drago J, Loiacono RE (2001) Dose-related neuroprotective effects of chronic nicotine in 6-
hydroxydopamine treated rats, and loss of neuroprotection in alpha4 nicotinic receptor subunit knockout mice. Br J Pharmacol 132:1650-1656.
Sabbagh MN, Lukas RJ, Sparks DL, Reid RT (2002) The nicotinic acetylcholine receptor, smoking, and Alzheimer's disease. J Alzheimers Dis 4:317-325.
Sacaan AI, Dunlop JL, Lloyd GK (1995) Pharmacological characterization of neuronal acetylcholine gated ion channel receptor-mediated hippocampal norepinephrine and striatal dopamine release from rat brain slices. J Pharmacol Exp Ther 274:224-230.
Sacaan AI, Menzaghi F, Dunlop JL, Correa LD, Whelan KT, Lloyd GK (1996) Epibatidine: a nicotinic acetylcholine receptor agonist releases monoaminergic neurotransmitters: in vitro and in vivo evidence in rats. J Pharmacol Exp Ther 276:509-515.
Sacaan AI, Reid RT, Santori EM, Adams P, Correa LD, Mahaffy LS, Bleicher L, Cosford ND, Stauderman KA, McDonald IA, Rao TS, Lloyd GK (1997) Pharmacological characterization of SIB-1765F: a novel cholinergic ion channel agonist. J Pharmacol Exp Ther 280:373-383.
Sakurai Y, Takano Y, Kohjimoto Y, Honda K, Kamiya HO (1982) Enhancement of 3H dopamine release and its 3H metabolites in rat striatum by nicotinic drugs. Brain Res 242:99-106.
Sallette J, Bohler S, Benoit P, Soudant M, Pons S, Le Novere N, Changeux JP, Corringer PJ (2004) An extracellular protein microdomain controls up-regulation of neuronal nicotinic acetylcholine receptors by nicotine. J Biol Chem 279:18767-18775.
Salminen O, Seppä T, Gäddnäs H, Ahtee L (1999) The effects of acute nicotine on the metabolism of dopamine and the expression of Fos protein in striatal and limbic brain areas of rats during chronic nicotine infusion and its withdrawal. J Neurosci 19:8145-8151.
Salminen O, Murphy KL, McIntosh JM, Drago J, Marks MJ, Collins AC, Grady SR (2004) Subunit composition and pharmacology of two classes of striatal presynaptic nicotinic acetylcholine receptors mediating dopamine release in mice. Mol Pharmacol 65:1526-1535.
Salokangas RK, Vilkman H, Ilonen T, Taiminen T, Bergman J, Haaparanta M, Solin O, Alanen A, Syvälahti E, Hietala J (2000) High levels of dopamine activity in the basal ganglia of cigarette smokers. Am J Psychiatry 157:632-634.
Santiago M, Westerink BH (1992) The role of GABA receptors in the control of nigrostriatal dopaminergic neurons: dual-probe microdialysis study in awake rats. Eur J Pharmacol 219:175-181.
Sargent PB (1993) The diversity of neuronal nicotinic acetylcholine receptors. Ann Rev Neurosci 16:403-443. Satoh K, Fibiger HC (1986) Cholinergic neurons of the laterodorsal tegmental nucleus: efferent and afferent connections. J
Comp Neurol 253:277-302. Schiffer WK, Gerasimov MR, Bermel RA, Brodie JD, Dewey SL (2000) Stereoselective inhibition of dopaminergic activity
by gamma vinyl-GABA following a nicotine or cocaine challenge: a PET/microdialysis study. Life Sci 66:L169-173.

114
Schilström B, Svensson HM, Svensson TH, Nomikos GG (1998a) Nicotine and food induced dopamine release in the nucleus accumbens of the rat: putative role of alpha7 nicotinic receptors in the ventral tegmental area. Neuroscience 85:1005-1009.
Schilström B, Nomikos GG, Nisell M, Hertel P, Svensson TH (1998b) N-methyl-D-aspartate receptor antagonism in the ventral tegmental area diminishes the systemic nicotine-induced dopamine release in the nucleus accumbens. Neuroscience 82:781-789.
Schneider JS, Van Velson M, Menzaghi F, Lloyd GK (1998a) Effects of the nicotinic acetylcholine receptor agonist SIB-1508Y on object retrieval performance in MPTP-treated monkeys: comparison with levodopa treatment. Ann Neurol 43:311-317.
Schneider JS, Tinker JP, Menzaghi F, Lloyd GK (2003) The subtype-selective nicotinic acetylcholine receptor agonist SIB-1553A improves both attention and memory components of a spatial working memory task in chronic low dose 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated monkeys. J Pharmacol Exp Ther 306:401-406.
Schneider JS, Pope-Coleman A, Van Velson M, Menzaghi F, Lloyd GK (1998b) Effects of SIB-1508Y, a novel neuronal nicotinic acetylcholine receptor agonist, on motor behavior in parkinsonian monkeys. Mov Disord 13:637-642.
Schneider JS, Tinker JP, Van Velson M, Menzaghi F, Lloyd GK (1999) Nicotinic acetylcholine receptor agonist SIB-1508Y improves cognitive functioning in chronic low-dose MPTP-treated monkeys. J Pharmacol Exp Ther 290:731-739.
Schoepfer R, Conroy WG, Whiting P, Gore M, Lindstrom J (1990) Brain alpha-bungarotoxin binding protein cDNAs and MAbs reveal subtypes of this branch of the ligand-gated ion channel gene superfamily. Neuron 5:35-48.
Schoffelmeer AN, De Vries TJ, Wardeh G, van de Ven HW, Vanderschuren LJ (2002) Psychostimulant-induced behavioral sensitization depends on nicotinic receptor activation. J Neurosci 22:3269-3276.
Schwartz RD, Kellar KJ (1983) Nicotinic cholinergic receptor binding sites in the brain: regulation in vivo. Science 220:214-216.
Schwartz RD, Kellar KJ (1985) In vivo regulation of 3H acetylcholine recognition sites in brain by nicotinic cholinergic drugs. J Neurochem 45:427-433.
Schwartz RD, Lehmann J, Kellar KJ (1984) Presynaptic nicotinic cholinergic receptors labeled by 3H acetylcholine on catecholamine and serotonin axons in brain. J Neurochem 42:1495-1498.
Scott WK, Zhang F, Stajich JM, Scott BL, Stacy MA, Vance JM (2005) Family-based case-control study of cigarette smoking and Parkinson disease. Neurology 64:442-447.
Seguela P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW (1993) Molecular cloning, functional properties, and distribution of rat brain alpha7: a nicotinic cation channel highly permeable to calcium. J Neurosci 13:596-604.
Seppä T (2001) Role of nicotinic acetylcholine receptors in crérebral dopaminergic transmission and expression of Fos protein. Helsinki: University Press.
Seppä T, Ahtee L (2000) Comparison of the effects of epibatidine and nicotine on the output of dopamine in the dorsal and ventral striatum of freely-moving rats. Naunyn-Schmiedebergs Arch Pharmacol 362:444-447.
Seppä T, Ruotsalainen M, Laakso I, Tuominen R, Ahtee L (2000) Effect of acute nicotine administration on striatal dopamine output and metabolism in rats kept at different ambient temperatures. Br J Pharmacol 130:1147-1155.
Sershen H, Hashim A, Lajtha A (1987) Behavioral and biochemical effects of nicotine in an MPTP-induced mouse model of Parkinson's disease. Pharmacol Biochem Behav 28:299-303.
Sgard F, Charpantier E, Barneoud P, Besnard F (1999) Nicotinic receptor subunit mRNA expression in dopaminergic neurons of the rat brain. Ann NY Acad Sci 868:633-635.
Sgard F, Charpantier E, Bertrand S, Walker N, Caput D, Graham D, Bertrand D, Besnard F (2002) A novel human nicotinic receptor subunit, alpha10, that confers functionality to the alpha9-subunit. Mol Pharmacol 61:150-159.
Sharples CG, Kaiser S, Soliakov L, Marks MJ, Collins AC, Washburn M, Wright E, Spencer JA, Gallagher T, Whiteaker P, Wonnacott S (2000) UB-165: a novel nicotinic agonist with subtype selectivity implicates the alpha4beta2* subtype in the modulation of dopamine release from rat striatal synaptosomes. J Neurosci 20:2783-2791.
Shim I, Javaid JI, Wirtshafter D, Jang SY, Shin KH, Lee HJ, Chung YC, Chun BG (2001) Nicotine-induced behavioral sensitization is associated with extracellular dopamine release and expression of c-Fos in the striatum and nucleus accumbens of the rat. Behav Brain Res 121:137-147.
Shimohama S, Akaike A, Kimura J (1996) Nicotine-induced protection against glutamate cytotoxicity. Nicotinic cholinergic receptor-mediated inhibition of nitric oxide formation. Ann NY Acad Sci 777:356-361.
Shoaib M, Schindler CW, Goldberg SR, Pauly JR (1997) Behavioural and biochemical adaptations to nicotine in rats: influence of MK801, an NMDA receptor antagonist. Psychopharmacology 134:121-130.
Shoaib M, Benwell ME, Akbar MT, Stolerman IP, Balfour DJ (1994) Behavioural and neurochemical adaptations to nicotine in rats: influence of NMDA antagonists. Br J Pharmacol 111:1073-1080.
Singer G, Wallace M, Hall R (1982) Effects of dopaminergic nucleus accumbens lesions on the acquisition of schedule induced self injection of nicotine in the rat. Pharmacol Biochem Behav 17:579-581.
Smith Y, Parent A (1988) Neurons of the subthalamic nucleus in primates display glutamate but not GABA immunoreactivity. Brain Res 453:353-356.
Smith Y, Bolam JP (1990) The output neurones and the dopaminergic neurones of the substantia nigra receive a GABA-containing input from the globus pallidus in the rat. J Comp Neurol 296:47-64.
Smith Y, Kieval JZ (2000) Anatomy of the dopamine system in the basal ganglia. Trends Neurosci 23:S28-33. Soliakov L, Wonnacott S (1996) Voltage-sensitive Ca2+ channels involved in nicotinic receptor-mediated 3H dopamine
release from rat striatal synaptosomes. J Neurochem 67:163-170. Spande TF, Garraffo HM, Edwards MW, Yeh HJC, Pannell L, Daly JW (1992) Epibatidine: A novel
(Chloropyridyl)azabicycloheptane with potent analgesic activity from an Equadoran poison frog. J Am Chem Soc 114:3475-3478.

115
Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF (1995) A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genetics 11:201-203.
Stevens KE, Kem WR, Mahnir VM, Freedman R (1998) Selective alpha7-nicotinic agonists normalize inhibition of auditory response in DBA mice. Psychopharmacology 136:320-327.
Stevens KE, Freedman R, Collins AC, Hall M, Leonard S, Marks MJ, Rose GM (1996) Genetic correlation of inhibitory gating of hippocampal auditory evoked response and alpha-bungarotoxin-binding nicotinic cholinergic receptors in inbred mouse strains. Neuropsychopharmacology 15:152-162.
Stolerman IP, Shoaib M (1991) The neurobiology of tobacco addiction. Trends Pharmacol Sci 12:467-473. Stolerman IP, Fink R, Jarvik ME (1973) Acute and chronic tolerance to nicotine measured by activity in rats.
Psychopharmacologia 30:329-342. Stolerman IP, Chandler CJ, Garcha HS, Newton JM (1997) Selective antagonism of behavioural effects of nicotine by
dihydro-beta-erythroidine in rats. Psychopharmacology 129:390-397. Stratford TR, Kelley AE (1997) GABA in the nucleus accumbens shell participates in the central regulation of feeding
behavior. J Neurosci 17:4434-4440. Stratford TR, Kelley AE (1999) Evidence of a functional relationship between the nucleus accumbens shell and lateral
hypothalamus subserving the control of feeding behavior. J Neurosci 19:11040-11048. Suaud-Chagny MF, Chérgui K, Chouvet G, Gonon F (1992) Relationship between dopamine release in the rat nucleus
accumbens and the discharge activity of dopaminergic neurons during local in vivo application of amino acids in the ventral tegmental area. Neuroscience 49:63-72.
Sugaya K, Giacobini E, Chiappinelli VA (1990) Nicotinic acetylcholine receptor subtypes in human frontal cortex: changes in Alzheimer's disease. J Neurosci Res 27:349-359.
Sullivan JP, Decker MW, Brioni JD, Donnelly-Roberts D, Anderson DJ, Bannon AW, Kang CH, Adams P, Piattoni-Kaplan M, Buckley MJ, et al. (1994) (+/-)-Epibatidine elicits a diversity of in vitro and in vivo effects mediated by nicotinic acetylcholine receptors. J Pharmacol Exp Ther 271:624-631.
Sullivan JP, Donnelly-Roberts D, Briggs CA, Anderson DJ, Gopalakrishnan M, Xue IC, Piattoni-Kaplan M, Molinari E, Campbell JE, McKenna DG, Gunn DE, Lin NH, Ryther KB, He Y, Holladay MW, Wonnacott S, Williams M, Arneric SP (1997) ABT-089 2-methyl-3-(2-(S)-pyrrolidinylmethoxy)pyridine: I. A potent and selective cholinergic channel modulator with neuroprotective properties. J Pharmacol Exp Ther 283:235-246.
Summers KL, Giacobini E (1995) Effects of local and repeated systemic administration of (-)nicotine on extracellular levels of acetylcholine, norepinephrine, dopamine, and serotonin in rat cortex. Neurochem Res 20:753-759.
Svensson TH, Tung CS (1989) Local cooling of pre-frontal cortex induces pacemaker-like firing of dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand 136:135-136.
Svensson TH, Mathe JM, Nomikos GG, Schilström B (1998) Role of excitatory amino acids in the ventral tegmental area for central actions of non-competitive NMDA-receptor antagonists and nicotine. Amino Acids 14:51-56.
Teaktong T, Graham AJ, Johnson M, Court JA, Perry EK (2004) Selective changes in nicotinic acetylcholine receptor subtypes related to tobacco smoking: an immunohistochemical study. Neuropathol Appl Neurobiol 30:243-254.
Teng L, Crooks PA, Sonsalla PK, Dwoskin LP (1997) Lobeline and nicotine evoke 3H overflow from rat striatal slices preloaded with 3H dopamine: differential inhibition of synaptosomal and vesicular 3H dopamine uptake. J Pharmacol Exp Ther 280:1432-1444.
Tepper JM, Paladini CA, Celada P (1998) GABAergic control of the firing pattern of substantia nigra dopaminergic neurons. Adv Pharmacol 42:694-699.
Toth E, Vizi ES, Lajtha A (1993) Effect of nicotine on levels of extracellular amino acids in regions of the rat brain in vivo. Neuropharmacology 32:827-832.
Toth E, Sershen H, Hashim A, Vizi ES, Lajtha A (1992) Effect of nicotine on extracellular levels of neurotransmitters assessed by microdialysis in various brain regions: role of glutamic acid. Neurochem Res 17:265-271.
Tzschentke TM (1998) Measuring reward with the conditioned place preference paradigm: a comprehensive review of drug effects, recent progress and new issues. Prog Neurobiol 56:613-672.
Ungerstedt U (1968) 6-Hydroxydopamine induced degeneration of central monoamine neurons. Eur J Pharmacol 5:107-110. Ungerstedt U (1971a) Postsynaptic supersensitivity after 6-hydroxydopamine induced degeneration of the nigrostriatal
dopamine system. Acta Physiol Scand Suppl 367:69-93. Ungerstedt U (1971b) Striatal dopamine release after amphetamine or nerve degeneration revealed by rotational behaviour.
Acta Physiol Scand Suppl 367:49-68. Ungerstedt U (1971c) Stereotaxic mapping of the monoamine pathways in the rat brain. Acta Physiol Scand Suppl 367:1-48. Ungerstedt U, Arbuthnott GW (1970) Quantitative recording of rotational behavior in rats after 6-hydroxydopamine lesions
of the nigrostriatal dopamine system. Brain Res 24:485-493. US Department of Health and Human Sciences (1988) The health consequences of smoking: nicotine addiction. A report of
the Surgeon General Maryland: Office on Smoking and health. Usunoff KG, Romansky KV, Malinov GB, Ivanov DP, Blagov ZA, Galabov GP (1982) Electron microscopic evidence for
the existence of a corticonigral tract in the cat. J Hirnforschung 23:23-29. Vastola BJ, Douglas LA, Varlinskaya EI, Spear LP (2002) Nicotine-induced conditioned place preference in adolescent and
adult rats. Physiol Behav 77:107-114. Vernier JM, Holsenback H, Cosford ND, Whitten JP, Menzaghi F, Reid R, Rao TS, Sacaan AI, Lloyd GK, Suto CM,
Chavez-Noriega LE, Washburn MS, Urrutia A, McDonald IA (1998) Conformationally restricted analogues of nicotine and anabasine. Bioorganic Med Chem Lett 8:2173-2178.
Vizi ES, Lendvai B (1999) Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res Brain Res Rev 30:219-235.

116
Wachtel H, Andén NE (1978) Motor activity of rats following intracerebral injections of drugs influencing GABA mechanisms. Naunyn-Schmiedebergs Arch Pharmacol 302:133-139.
Wada E, McKinnon D, Heinemann S, Patrick J, Swanson LW (1990) The distribution of mRNA encoded by a new member of the neuronal nicotinic acetylcholine receptor gene family (alpha5) in the rat central nervous system. Brain Res 526:45-53.
Wada E, Wada K, Boulter J, Deneris E, Heinemann S, Patrick J, Swanson LW (1989) Distribution of alpha2, alpha3, alpha4, and beta2 neuronal nicotinic receptor subunit mRNAs in the central nervous system: a hybridization histochemical study in the rat. J Comp Neurol 284:314-335.
Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J (1996) Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem 271:17656-17665.
Wang F, Nelson ME, Kuryatov A, Olale F, Cooper J, Keyser K, Lindstrom J (1998) Chronic nicotine treatment up-regulates human alpha3 beta2 but not alpha3 beta4 acetylcholine receptors stably transfected in human embryonic kidney cells. J Biol Chem 273:28721-28732.
Warburton DD, Rusted JJ, Muller CC (1992) Patterns of facilitation of memory by nicotine. Behav Pharmacol 3:375-378. Warpman U, Nordberg A (1995) Epibatidine and ABT 418 reveal selective losses of alpha4 beta2 nicotinic receptors in
Alzheimer brains. Neuroreport 6:2419-2423. Weiland S, Witzemann V, Villarroel A, Propping P, Steinlein O (1996) An amino acid exchange in the second
transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Lett 398:91-96.
Welzl H, Bättig K, Berz S (1990) Acute effects of nicotine injection into the nucleus accumbens on locomotor activity in nicotine-naive and nicotine-tolerant rats. Pharmacol Biochem Behav 37:743-746.
Westerink BH, Spaan SJ (1982a) Estimation of the turnover of 3-methoxytyramine in the rat striatum by HPLC with electrochemical detection: implications for the sequence in the cerebral metabolism of dopamine. J Neurochem 38:342-347.
Westerink BH, Spaan SJ (1982b) On the significance of endogenous 3-methoxytyramine for the effects of centrally acting drugs on dopamine release in the rat brain. J Neurochem 38:680-686.
Westerink BH, Santiago M, De Vries JB (1992a) In vivo evidence for a concordant response of terminal and dendritic dopamine release during intranigral infusion of drugs. Naunyn-Schmiedebergs Arch Pharmacol 346:637-643.
Westerink BH, Santiago M, De Vries JB (1992b) The release of dopamine from nerve terminals and dendrites of nigrostriatal neurons induced by excitatory amino acids in the conscious rat. Naunyn-Schmiedebergs Arch Pharmacol 345:523-529.
Westerink BH, Kwint HF, deVries JB (1996) The pharmacology of mesolimbic dopamine neurons: a dual-probe microdialysis study in the ventral tegmental area and nucleus accumbens of the rat brain. J Neurosci 16:2605-2611.
Westfall TC (1974) Effect of nicotine and other drugs on the release of 3H-norepinephrine and 3H-dopamine from rat brain slices. Neuropharmacology 13:693-700.
Westfall TC, Mereu G, Vickery L, Perry H, Naes L, Yoon KW (1989) Regulation by nicotine of midbrain dopamine neurons. Prog Brain Res 79:173-185.
Whiteaker P, Sharples CG, Wonnacott S (1998) Agonist-induced up-regulation of alpha4beta2 nicotinic acetylcholine receptors in M10 cells: pharmacological and spatial definition. Mol Pharmacol 53:950-962.
Whiteaker P, Garcha HS, Wonnacott S, Stolerman IP (1995) Locomotor activation and dopamine release produced by nicotine and isoarecolone in rats. Br J Pharmacol 116:2097-2105.
Whiteaker P, Marks MJ, Grady SR, Lu Y, Picciotto MR, Changeux JP, Collins AC (2000) Pharmacological and null mutation approaches reveal nicotinic receptor diversity. Eur J Pharmacol 393:123-135.
Whiteaker P, Peterson CG, Xu W, McIntosh JM, Paylor R, Beaudet AL, Collins AC, Marks MJ (2002) Involvement of the alpha3 subunit in central nicotinic binding populations. J Neurosci 22:2522-2529.
Whiting P, Lindstrom J (1986) Pharmacological properties of immuno-isolated neuronal nicotinic receptors. J Neurosci 6:3061-3069.
WHO (2003) World Health Report 2003: Shaping the future. Geneva: World Health Organization. Wirdefeldt K, Gatz M, Pawitan Y, Pedersen NL (2005) Risk and protective factors for Parkinson's disease: a study in
Swedish twins. Ann Neurol 57:27-33. Wirtshafter D, Stratford TR, Asin KE (1987) Evidence that serotonergic projections to the substantia nigra in the rat arise in
the dorsal, but not the median, raphe nucleus. Neurosci Lett 77:261-266. Wise RA, Bozarth MA (1987) A psychomotor stimulant theory of addiction. Psychol Rev 94:469-492. Wonnacott S (1990) The paradox of nicotinic acetylcholine receptor upregulation by nicotine. Trends Pharmacol Sci 11:216-
219. Wonnacott S (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20:92-98. Wonnacott S, Sidhpura N, Balfour DJ (2005) Nicotine: from molecular mechanisms to behaviour. Curr Opin Pharmacol
5:53-59. Wonnacott S, Drasdo A, Sanderson E, Rowell P (1990) Presynaptic nicotinic receptors and the modulation of transmitter
release. Ciba Found Symp 152:87-101; discussion 102-105. Wonnacott S, Irons J, Rapier C, Thorne B, Lunt GG (1989) Presynaptic modulation of transmitter release by nicotinic
receptors. Prog Brain Res 79:157-163. Wonnacott S, Kaiser S, Mogg A, Soliakov L, Jones IW (2000) Presynaptic nicotinic receptors modulating dopamine release
in the rat striatum. Eur J Pharmacol 393:51-58. Woolf NJ (1991) Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol 37:475-524. Woolf NJ, Butcher LL (1981) Cholinergic neurons in the caudate-putamen complex proper are intrinsically organized: a
combined Evans blue and acetylcholinesterase analysis. Brain Res Bull 7:487-507.

117
Wooltorton JR, Pidoplichko VI, Broide RS, Dani JA (2003) Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J Neurosci 23:3176-3185.
Worms P, Gueudet C, Biziere K (1986a) Induction of turning by direct intrastriatal injection of dopaminomimetic drugs in mice: pharmacological analysis of a simple screening model. Life Sci 39:2199-2208.
Worms P, Kan JP, Wermuth CG, Biziere K (1986b) Dopamine-like activities of an aminopyridazine derivative, CM 30366: a behavioural study. Naunyn-Schmiedebergs Arch Pharmacol 334:246-252.
Xiao Y, Meyer EL, Thompson JM, Surin A, Wroblewski J, Kellar KJ (1998) Rat alpha3/beta4 subtype of neuronal nicotinic acetylcholine receptor stably expressed in a transfected cell line: pharmacology of ligand binding and function. Mol Pharmacol 54:322-333.
Yang X, Criswell HE, Breese GR (1996) Nicotine-induced inhibition in medial septum involves activation of presynaptic nicotinic cholinergic receptors on gamma-aminobutyric acid-containing neurons. J Pharmacol Exp Ther 276:482-489.
Yin R, French ED (2000) A comparison of the effects of nicotine on dopamine and non-dopamine neurons in the rat ventral tegmental area: an in vitro electrophysiological study. Brain Res Bull 51:507-514.
Yong VW, Perry TL (1986) Monoamine oxidase B, smoking, and Parkinson's disease. J Neurol Sci 72:265-272. Yoshida M, Yokoo H, Tanaka T, Mizoguchi K, Emoto H, Ishii H, Tanaka M (1993) Facilitatory modulation of mesolimbic
dopamine neuronal activity by a mu-opioid agonist and nicotine as examined with in vivo microdialysis. Brain Res 624:277-280.
Yu CR, Role LW (1998) Functional contribution of the alpha7 subunit to multiple subtypes of nicotinic receptors in embryonic chick sympathetic neurones. J Physiol 509:651-665.
Yu ZJ, Wecker L (1994) Chronic nicotine administration differentially affects neurotransmitter release from rat striatal slices. J Neurochem 63:186-194.
Zarrindast MR, Sadegh M, Shafaghi B (1996) Effects of nicotine on memory retrieval in mice. Eur J Pharmacol 295:1-6. Zernig G, O'Laughlin IA, Fibiger HC (1997) Nicotine and heroin augment cocaine-induced dopamine overflow in nucleus
accumbens. Eur J Pharmacol 337:1-10. Zetler G (1968) Cataleptic state and hypothermia in mice, caused by central cholinergic stimulation and antagonized by
anticholinergic and antidepressant drugs. Intl J Neuropharmacol 7:325-335. Zetler G (1971) Pharmacological differentiation of "nicotinic" and "muscarinic" catalepsy. Neuropharmacology 10:289-296. Zhang H, Sulzer D (2004) Frequency-dependent modulation of dopamine release by nicotine. Nat Neuroscience 7:581-582. Zhang X, Gong ZH, Hellström-Lindahl E, Nordberg A (1995) Regulation of alpha4 beta2 nicotinic acetylcholine receptors in
M10 cells following treatment with nicotinic agents. Neuroreport 6:313-317. Zhou FM, Liang Y, Dani JA (2001) Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat
Neuroscience 4:1224-1229. Zigmond MJ, Abercrombie ED, Berger TW, Grace AA, Stricker EM (1990) Compensations after lesions of central
dopaminergic neurons: some clinical and basic implications. Trends Neurosci 13:290-296. Zoli M, Lena C, Picciotto MR, Changeux JP (1998) Identification of four classes of brain nicotinic receptors using beta2
mutant mice. J Neurosci 18:4461-4472. Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C (2002) Identification of the nicotinic receptor subtypes
expressed on dopaminergic terminals in the rat striatum. J Neurosci 22:8785-8789.





























