Embed Size (px)
Citation preview

Documentation and Records: Harmonized GMP-PIC/S Requirements
PIC/S PE 009-12 (Introduction Part I, II, ANNEX)
Sirivan Pomchaksin
Expert 10
The Government Pharmaceutical Organization (GPO)
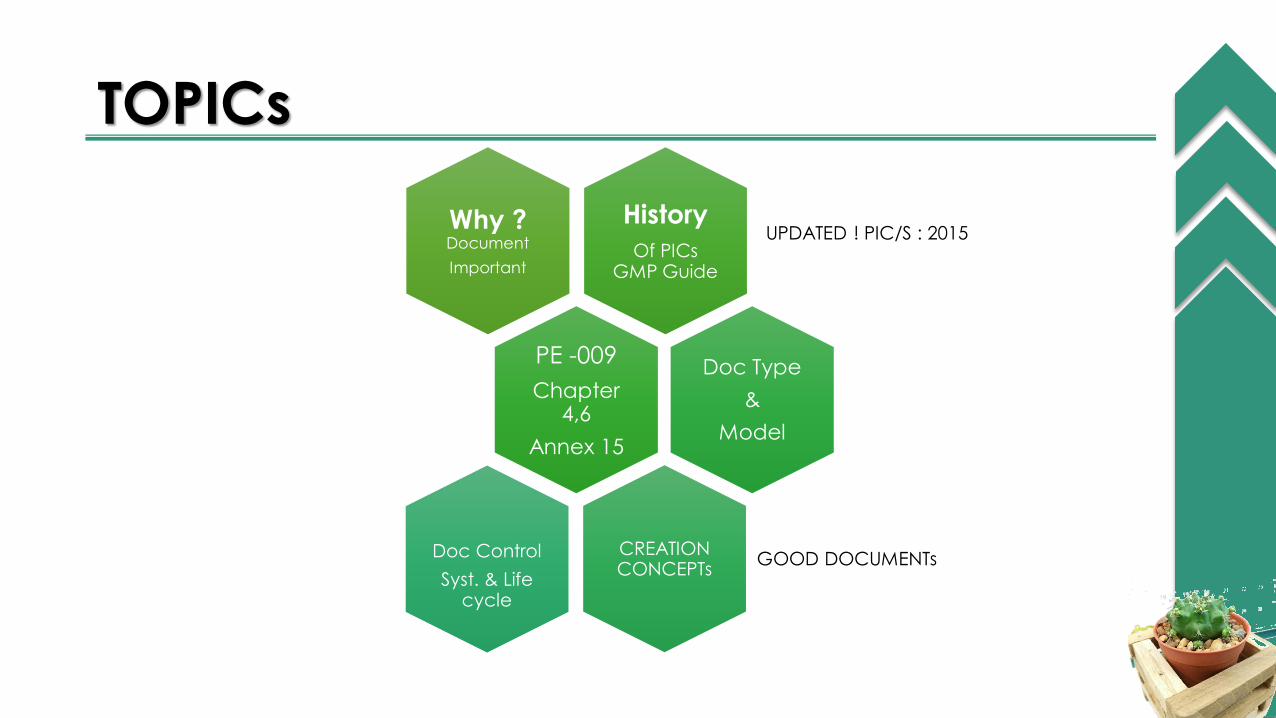
TOPICs
Why ?Document
Important
UPDATED ! PIC/S : 2015History
Of PICs GMP Guide
PE -009
Chapter 4,6
Annex 15
Doc Type
&
Model
CREATION CONCEPTs
GOOD DOCUMENTsDoc Control
Syst. & Life cycle

WHY are documents so important?
Communication
Cost
Audit trail
If it was not written it was not done.

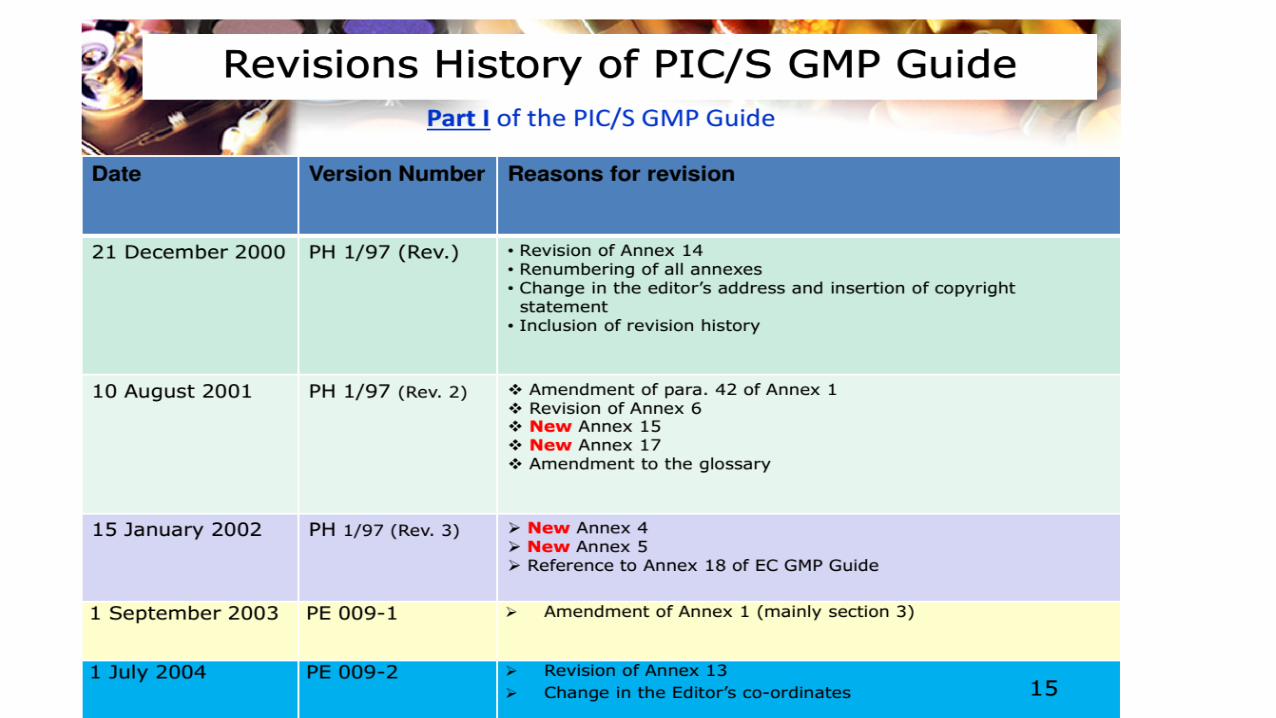

9/19/2016 6
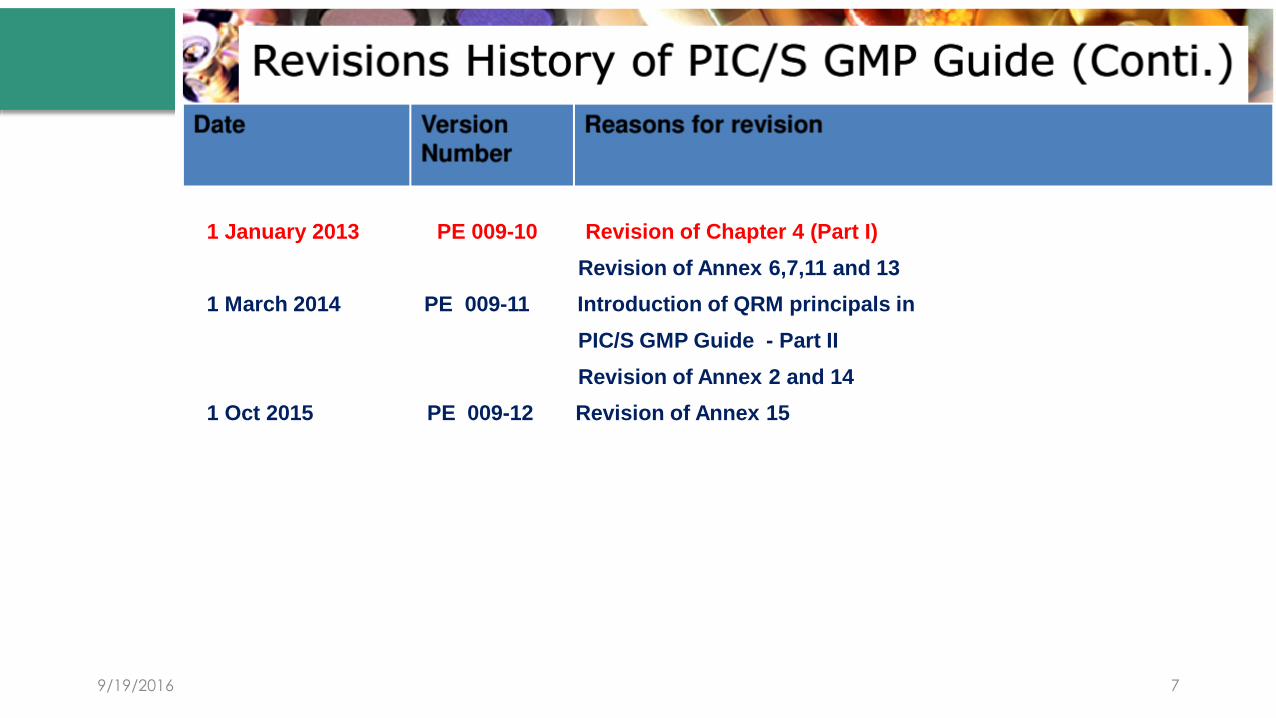
1 January 2013 PE 009-10 Revision of Chapter 4 (Part I)
Revision of Annex 6,7,11 and 13
1 March 2014 PE 009-11 Introduction of QRM principals in
PIC/S GMP Guide - Part II
Revision of Annex 2 and 14
1 Oct 2015 PE 009-12 Revision of Annex 15
9/19/2016 7

UPDATED PIC/S : 2015 (PE 009-12)
‘Documentation and Records’GUIDE TO GOOD MANUFACTURING PRACTICE FOR MEDICINAL PRODUCTS
• Introduction
• PART I : CHAPTER 4 DOCUMENTATION
: CHAPTER 6 (6.7-6.10 ) QC DOCUMENTATION
• PART II : SECTION 6 DOCUMENTATION AND RECORDS(API Mfg))
• Annex11 : Data ,Data Storage (ผนวก ๑๐)
• Annex15 : DOCUMENTATION including VMP (ผนวก ๑๔)
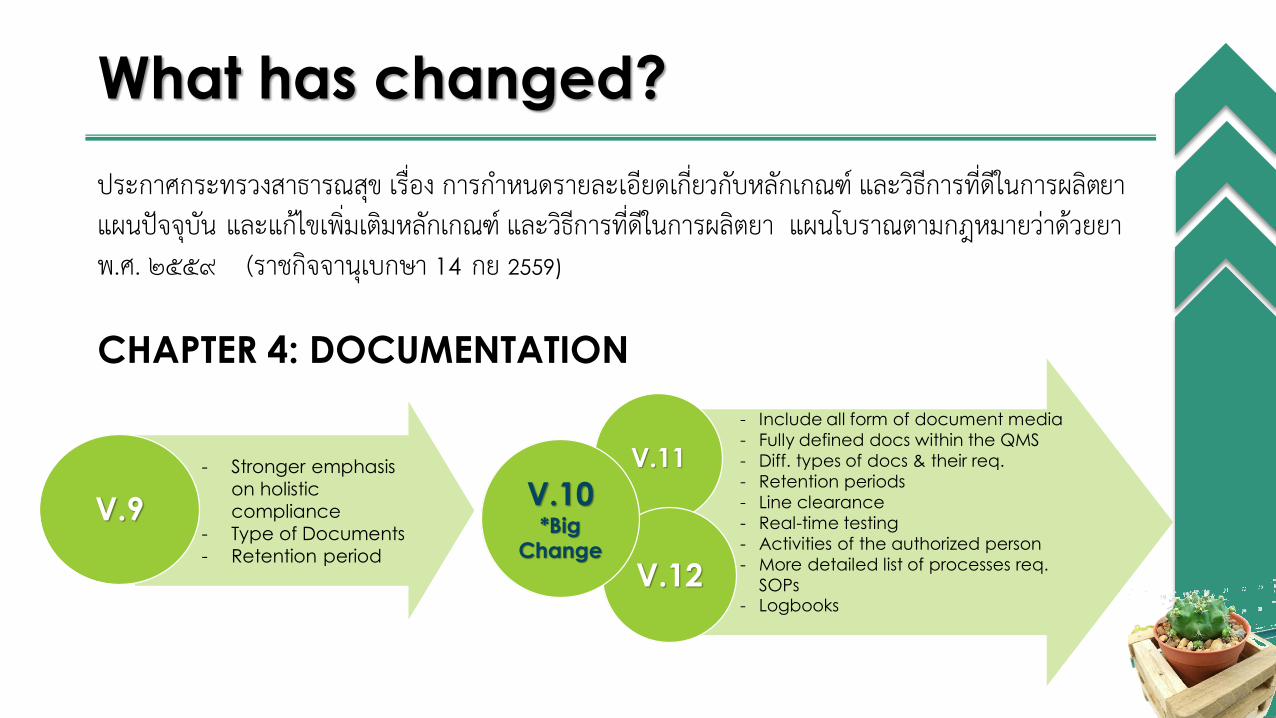
What has changed?
ประกาศกระทรวงสาธารณสขุ เรื่อง การก าหนดรายละเอียดเกี่ยวกับหลักเกณฑ์ และวิธีการที่ดีในการผลิตยา แผนปัจจุบัน และแก้ไขเพิ่มเตมิหลักเกณฑ์ และวิธีการที่ดีในการผลิตยา แผนโบราณตามกฎหมายว่าด้วยยา พ.ศ. ๒๕๕๙ (ราชกิจจานุเบกษา 14 กย 2559)
CHAPTER 4: DOCUMENTATION
- Stronger emphasis
on holistic
compliance
- Type of Documents
- Retention period
V.9
- Include all form of document media
- Fully defined docs within the QMS
- Diff. types of docs & their req.
- Retention periods
- Line clearance
- Real-time testing
- Activities of the authorized person
- More detailed list of processes req.
SOPs- Logbooks
V.11
V.12
V.10*Big
Change

Chapter4 DOCUMENTATION
• Principle
• Required GMP Documentation (by Type)
• Generation and Control Documentation
• Good Documentation Practices
• Retention of Documents
• Specifications
• Manufacturing Formula and Processing /Packaging Instruction• Batch Processing Record• Packaging Record
• Procedures and Records• Receipt • Sampling • Testing• Others

CHAPTER 4 DOCUMENTATION
PRINCIPLE
The main objective of the system of documentation
utilized must be to establish, control, monitor, and record
all activities which directly or indirectly impact on all aspects of the quality of medicinal products.
There are two types of documents used to manage and
record GMP compliance:
Instructions (direction, req.):free from error, available in writing
Records/Reports : be rendered in a human readable form.
To ensure the accuracy, integrity, availability and
legibility of document
CHAPTER 4 DOCUMENTATION
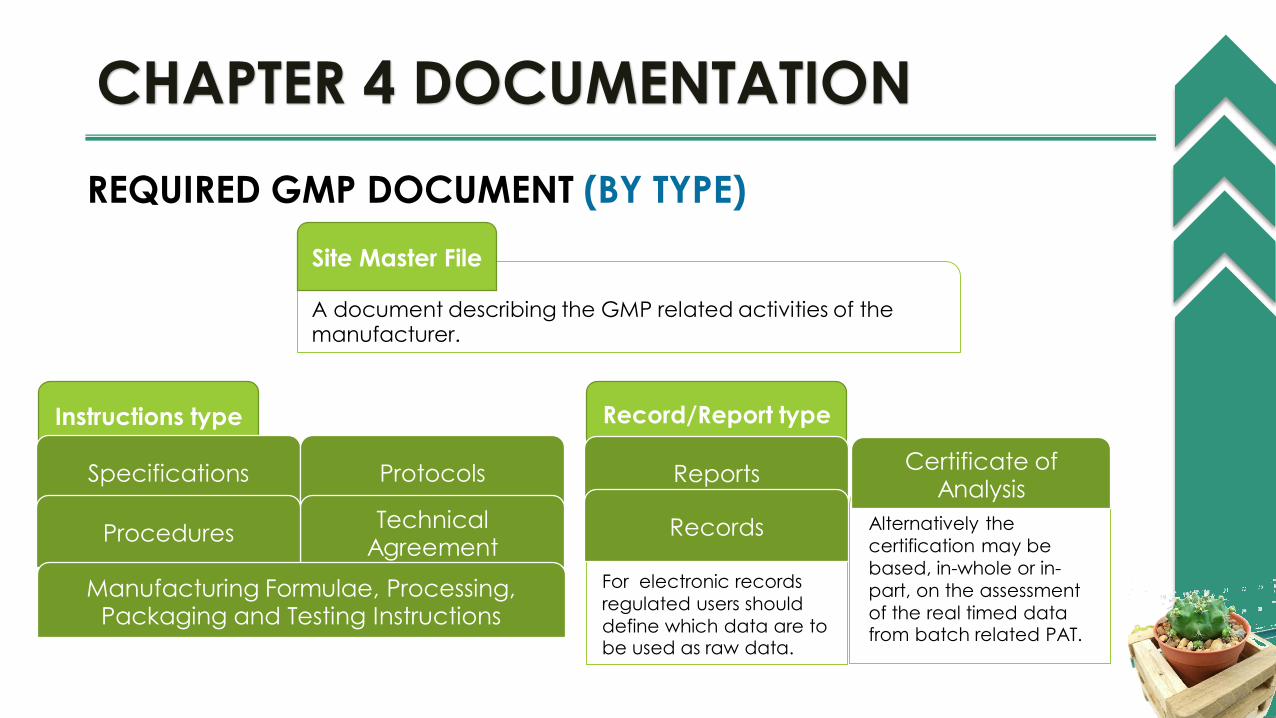
REQUIRED GMP DOCUMENT (BY TYPE)
A document describing the GMP related activities of the manufacturer.
Site Master File
Instructions type
Specifications Protocols
Technical Agreement
Procedures
Manufacturing Formulae, Processing, Packaging and Testing Instructions
Record/Report type
Reports
For electronic records
regulated users should
define which data are to be used as raw data.
Records Alternatively the
certification may be
based, in-whole or in-
part, on the assessment
of the real timed data from batch related PAT.
Certificate of Analysis

CHAPTER 4 DOCUMENTATION
GENERATION AND CONTROL OF DOCUMENTATION
4.1
Many documents (instructions and/or records) may exist in hybrid
forms, i.e. some elements as electronic and others as paper based.
Relationships and control measures for master documents, official
copies, data handling and records need to be stated for both
hybrid and homogenous systems.
Controls for electronic documentsTo ensure the integrity of the record throughout the retention period

CHAPTER 4 DOCUMENTATION
GENERATION AND CONTROL OF DOCUMENTATION
4.2
Document should be designed, prepared, reviewed, and
distributed with care.
They should comply with the relevant parts of Product specification
files, Manufacturing and marketing authorization dossiers.
The reproduction of working documents from master documents
should not allow any error to be introduced through the
reproduction process.

CHAPTER 4 DOCUMENTATION
GENERATION AND CONTROL OF DOCUMENTATION
4.3
Documents containing instructions should be approved, signed and date by
appropriate and authorized persons.(Qualified :EU )
Documents should have unambiguous contents and be uniquely identifiable. The
effective should be defined.
4.4
Document containing instructions should be laid out in an orderly fashion and be
easy to check
4.5
Documents within the QMS should be regularly review and kept up-to-date.
When a document has been revised, systems should be operated to prevent inadvertent use of superseded documents.
4.6 Documents should not be hand written, ….sufficient space
should be provided for such entries.

CHAPTER 4 DOCUMENTATION
GOOD DOCUMENTATION PRACTICE
4.7 Handwritten : clear, legible, indelible way
4.8 Records : at the time each action, traceable
4.9 Alteration : signed and dated
permit the reading of the original information
the reason for the alteration should be recorded

CHAPTER 4 DOCUMENTATION
RETENTION OF DOCUMENT
4.10
Records is located, ensure the integrity, validated
4.11
Specific requirements apply to batch documentation which must be kept for one
year after expiry of the batch to which it relates or at least five years after certification of the batch by the Authorized Person, whichever is longer.
4.12
For other types of documentation, the retention period will depend on the
business activity which the documentation supports. Critical documentation,
including raw data (for example relating to validation or stability), which support
information in the Market Authorization should be retained whilst the
authorization remains in force. It may be considered acceptable to retire certain documentation where the data has been superseded by a full set of new data.

CHAPTER 4 DOCUMENTATION
SPECIFICATIONS 4.13-16
Starting and Packaging Materials
Finished ProductsIntermediate and
Bulk Products
Starting and packaging material:
a)Name and internal code reference
Reference
approved suppliers ; original producer
specimen of printed material
b) Direction for sampling and testing
c) Requirement and acceptance limits
d) Storage conditions and precautions
e) The maximum period of storage before re examination

CHAPTER 4 DOCUMENTATION
SPECIFICATIONS
Intermediate and Bulk Products:
Should be available for critical steps or if these are purchased or
dispatched. The specifications should be similar to specifications for
starting materials or for finished products, as appropriate.
Finished Products:
as starting
+ The Formula
+ Storage conditions and special handling precautions, where
application
+ The shelf life

CHAPTER 4 DOCUMENTATION
MANUFACTURING FORMULA AND PROCESSING INSTRUCTIONS 4.17-4.21
Approved, written Manufacturing Formula and Processing Instructions should exist for each product and batch size to be manufactured.
Should include :
a) The name of the product, with a product reference code relating to its specification
b) A description of the pharmaceutical form, strength of the product and batch size
c) A list of all starting materials to be used, with the amount of each, described; mention
should be made of any substance that may disappear in the course of processing;
d) A statement of the expected final yield with the acceptable limits, and of relevant
intermediate yields, where applicable
Manufacturing Formula

CHAPTER 4 DOCUMENTATION
Processing Instructions
work station are clear of previous products
equipment is clean and suitable for use.
Line Clearance
Reconciliation (Batch Packaging Record):
The quantities and reference number or identification of all printed
packaging materials and bulk products issued, used, destroyed or
returned to stock and the quantities of obtained product, in order to provide for an adequate reconciliation.
Packaging Instructions
Batch Processing Record
Batch Packaging Record

CHAPTER 4 DOCUMENTATION
Name batch no. /dates /times Significant step, who check ,sign/date Actually weighed including batch no, IPC /Environment Major equipment Product yield /Reconciliation Deviation from Formula, processing operation
Approval for processing operation (validated)
++ Formula + Instruction
Batch Processing Record

Chapter 4 : Procedure and Records
Receipt (4.22-4.24)
• each delivery of starting material, Intermediate, primary, secondary
and printed PM
• Record + COA , supplier, manufacturer
Sampling (4.25) + Chapter 6 QC documentation (6.11)
+ Annex 8 sampling of starting and packaging material
• SOP : Methods, equipment, quantity
• Precaution avoid contamination, any deterioration
Testing (4.26) + Chapter 6 QC testing
• SOP at different stage ;approved method, equipment
• Record (6.17)

Procedure and Records
SAMPLING chapter66.11. The sample taking should be done in accordance with approved
written procedures that describe:
the method of sampling;
the equipment to be used;
the amount of the sample to be taken;
instructions for any required sub-division of the sample;
the type and condition of the sample container to be used;
the identification of containers sampled;
any special precautions to be observed, especially with regard to the
sampling of sterile or noxious materials;
the storage conditions;
instructions for the cleaning and storage of sampling equipment

Procedure and Records
TESTING chapte6
6.17. The tests performed should be recorded and the records should include
at least the following data:
a) name of the material or product and, where applicable, dosage form;
b) batch number and, where appropriate, the manufacturer and/or supplier;
c) references to the relevant specifications and testing procedures;
d) test results, including observations and calculations, and reference to any
certificates of analysis;
e) dates of testing;
f) initials of the persons who performed the testing;
g) initials of the persons who verified the testing and the calculations, where
appropriate;
h) a clear statement of release or rejection (or other status decision) and the
dated signature of the designated responsible person.

Procedure and Records (cont.)
4.27 Batch release ; authorized person(s) ,COA
4.28 Records; to facilitate recall
4.29 There should be written policies, procedures, protocols, reports, and the
associated records of actions taken or conclusions reached, where appropriate, for the following examples;
Validation and Qualification of
processes, equipment and systems Equipment assembly and calibration
Technology Transfer Maintenance, cleaning and sanitation
Personnel matter including signature
lists, training in GMP and technical
matters, clothing and hygiene and
verification of the effectiveness training Environment monitoring
Pest control
Complaints
Returns
Recalls
Change Control
Investigation into deviations and non-
conformances
Internal quality / GMP compliance
audits
Summaries of records where
appropriate (e.g. product quality
review)
Supplier audit

Procedure and Records (cont.)
Others SOP (4.27-4.32)
4.30 Major/critical manufacturing and testing equipment
4.31 Log books; chronological order , dates, identity of people who carried
4.32 An inventory of documents within the QMS should be maintained

Part I CHAPTER 6 : QUALITY CONTROL
6.7 LABORATORY DOCUMENTATION; Should be readily available to the Quality Control Department
Specifications Sampling procedures +Annex8
Testing Procedures and Records Analytical Reports and/or certificates Data from environmental monitoring, where required Validation records of test methods, where applicable Procedures for and records of the calibration of
instruments and maintenance of equipment.

Part I CHAPTER 6 : QUALITY CONTROL (cont.)
6.8
Any Quality Control documentation relating to a batch record
should be retained for one year after the expiry date of the batch.
6.9
For some kinds of data (e.g. analytical test results, yields,
environmental controls,…) it is recommended that records in a
manner permitting trend evaluation be kept.
6.10
In addition to the information which is part of the batch record, other
original data such as laboratory notebooks and/or records should
be retained and readily available.

Part II Chapter 6 DOCUMENTATION AND RECORDS (API)
6.1 Documentation and Specification
6.2 Equipment Cleaning and Use Record
6.3 Records of RM, Intermediate, API Labelling and PM
6.4 Master Production Instructions and Control Records
6.5 BPR
6.6 Laboratory Control Records
6.7 BPR Review
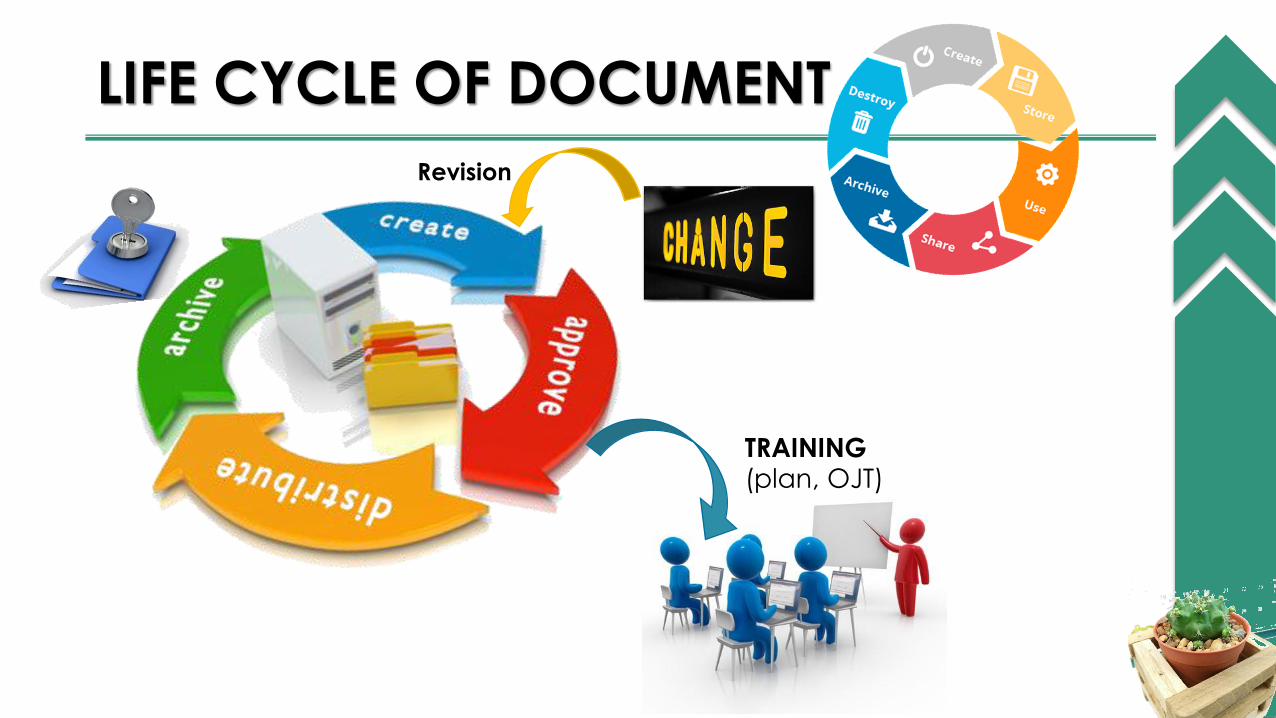
LIFE CYCLE OF DOCUMENT
TRAINING (plan, OJT)
Revision
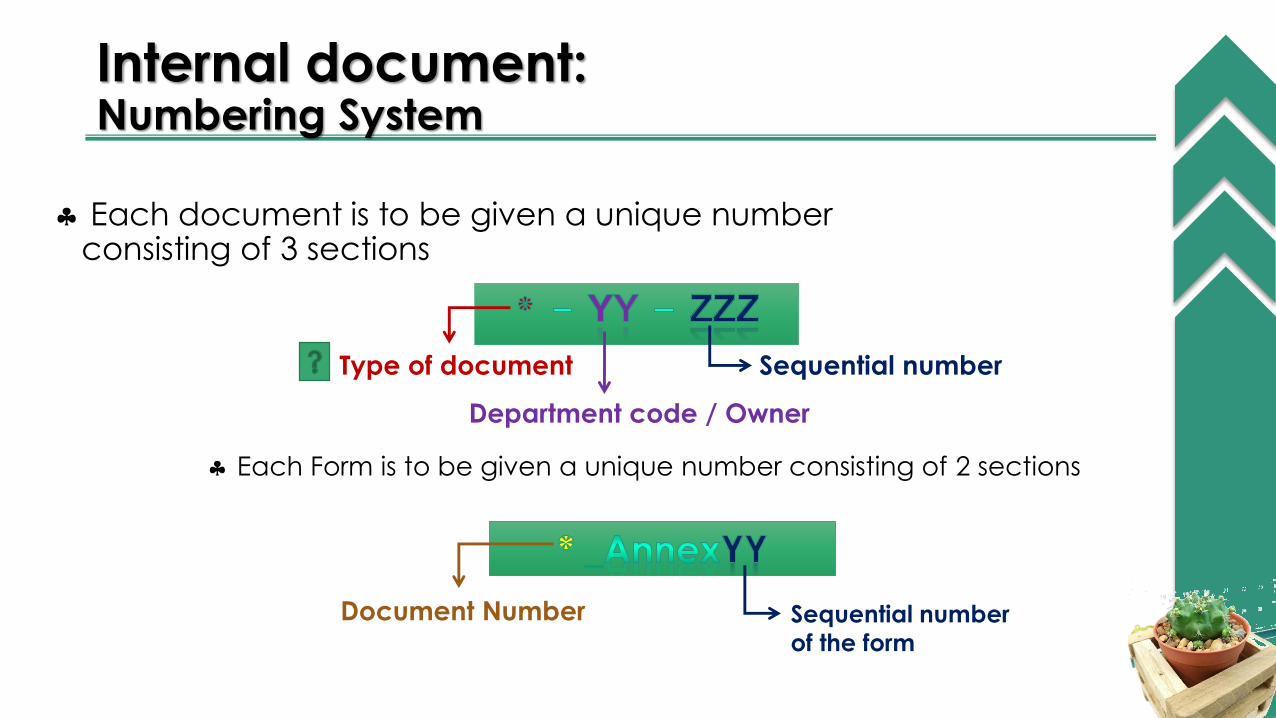
Internal document:Numbering System
Each document is to be given a unique number consisting of 3 sections
Type of document
Department code / Owner
Sequential number
Each Form is to be given a unique number consisting of 2 sections
Document Number Sequential number
of the form

The DCC personnel issues hard copies of approved documents
on an as-requested basis.
COPY No.
Controlled copy will be stamped a unique,
controlled copy number with blue ink on
every page.
Uncontrolled copy will be stamped a unique,
controlled copy number with red ink
on every page. UNCONTROLLED
sign & symbol

The Obsolete or Superseded copies will be shredded to ensure they
are not used to perform work. Only the master document will be kept
as reference in Document Control Center after each of the pages has
been stamped in red with
S U P E R S E D E D
The cancellation of controlled copies and documents will be
shredded to ensure they are not used to perform work. Only the
master document will be kept as reference in Document Control
Center after each of the pages has been stamped in red with
C A N C E L L E D
sign & symbol

• All new or revised documents must pass through the document approval process by using the Document Action Request
• Document Preparer or Designee requests a controlled number from Document Control Center (DCC) for a new document.
• The DCC Personnel notify the Document Preparer at least 30-day before the periodic review date is due by using the Periodic Review Notice form
DOCUMENT CREATION/REVISION

DOCUMENT REQUEST & DISTRIBUTION
• The hard copies both Controlled and Uncontrolled can be requested by using Document Request Form.
• The DCC personnel will generate and send a Controlled Document Receipt Acknowledgement Form, included with the controlled hard copies to all users in the distribution list or the requestors while the users shall acknowledge the receipt of the document and must be returned the
superseded version to the DCC personnel.

THE USE OF ANNEX/FORM
• The use of annex (form for recording), the DCC personnel shall distribute the controlled copy to each unit operation as defined in distribution list.
• The form can be copied without the cover page for routine operation and each operation shall control them to stay in the current status.

DOCUMENT CANCELLATION
• The Document Preparer or Designee must be completed a DAR Form when he/she wants to cancel a controlled document. Approval signatures must include the following at minimum: • Document Preparer • Responsible Manager
• The DCC Personnel shall dispose of cancellation of controlled copies and documents by shredding to ensure they are not used to perform work.
• Only the master document will be kept as reference in Document Control Center after each of the pages has been stamped in red with “CANCELLED”.
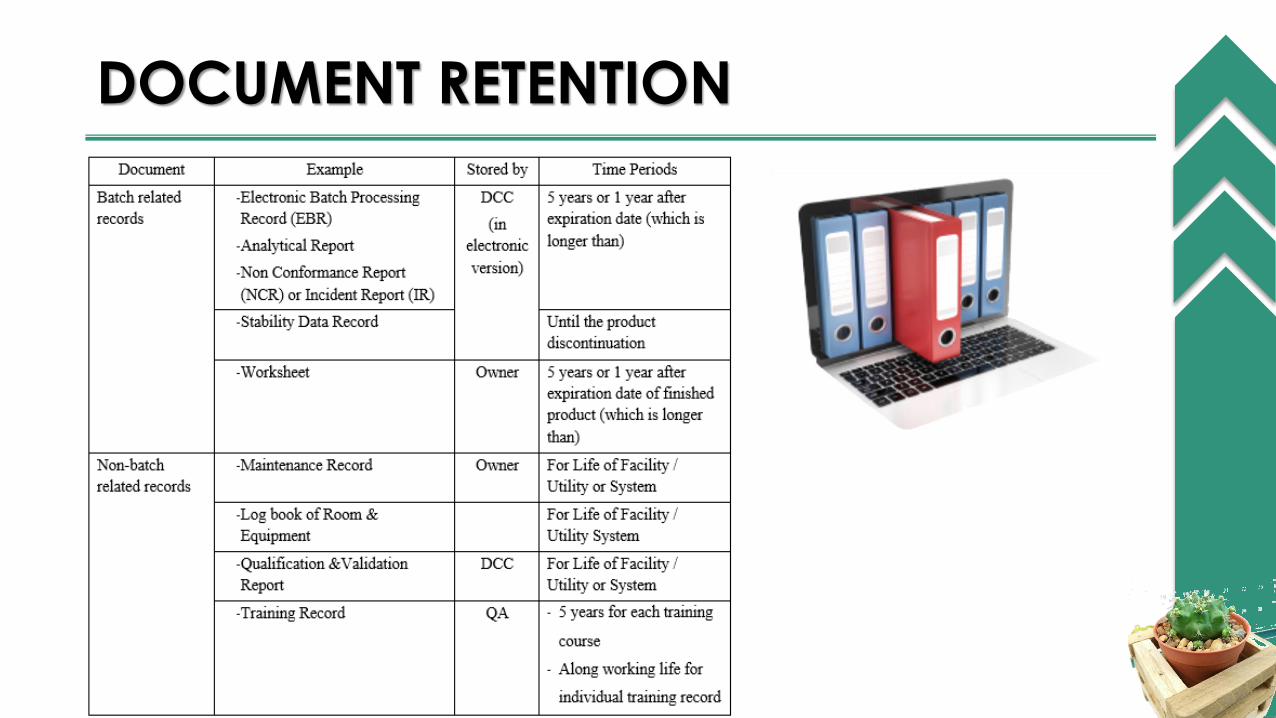
DOCUMENT RETENTION

PINK DOCUMENT
• In training and trial period, the DCC will generate the documents for training purposes ::: Signed & Copied on Pink paper.
• Purposes of pink paper are also warning the operator who read them, these instructions have some changes. They will stay in pink paper for a month.
• When the Pink document is ready for removal from the training /warning status, the DCC should generate the master of White document with original approval same revision but set the new effective date .
• DCC distribute the white hard copy version to each operation units and get the pink document return to DCC.

DOCUMENT FORMAT : Header
THE GOVERNMENT PHARMACEUTICAL ORGANIZATION
RANGSIT PHARMACEUTICAL PRODUCTION PLANT
Document No.
SOP-QA-001
Rev. 02
Eff. Date:
4 Jul 2014TITLE DOCUMENT CONTROL SYSTEM
DEPARTMENT QUALITY ASSURANCE PAGE 12 of 24
THE GOVERNMENT PHARMACEUTICAL ORGANIZATION
RANGSIT PHARMACEUTICAL PRODUCTION PLANT
Document No.
SOP-QA-001_Annex01
Rev.02
Eff. Date :
4 Jul 2014TITLE DOCUMENT ACTION REQUEST (DAR) FORM
DEPARTMENT QUALITY ASSURANCE PAGE 4 of 5
THE GOVERNMENT PHARMACEUTICAL ORGANIZATION
RANGSIT PHARMACEUTICAL PRODUCTION PLANTDocument No.
SOP-EN-001
Rev.01
Eff. Date :
20 Oct 2013TITLE PREVENTIVE MAINTENANCE PLAN
DEPARTMENT ENGINEERING DIVISION PAGE 1 of 10

DOCUMENT FORMAT : Distribution List
Copy No. Area / Position
Master Document Control Center - QA
1 QA Manager
2 Manager of Rangsit Pharmaceutical Production Plant 1
3 Director of Validation Division
4 Director of Compliance and Quality System Division
5 Director of Quality Control Division
6 Director of Administration Division
7 Director of Production Division
8 Director of Warehouse Division
9 Director of Engineering Division

DOCUMENT FORMAT : REVISION HISTORYRev. No.
Change & Reason For changesEffective
Date
01 Initial Release 19 Sep 2011
02
02
03
Revised:
- Header: Change the plant name from
“Rangsit Manufacturing Plant” to “Rangsit
Pharmaceutical Production Plant”.
- Rearrange the sequence of topics.
- Distribution List: Rename and add the newly
divisions and sections as per the approved
organization chart. (p. 2/23)
- Internal Document Control System: remove
the section of Numbering System. (p. 10/23)
- Internal Document Control System: change
the process and lifecycle of pink document.
(p. 10/23)
- Attachment: remove template of VMP, QM,
BPR, SOP
Release in white paper
Revised:
- Updated the format of document header for
using in eQMS.
- Add definition of term that used in eQMS.
- Add detail for document control procedure in
eQMS
- Cancel the use of pink document
- Cancel the use of SOP-QA-001_Annex04
4 Jul 2014
4 Aug 2014
As per an
effective
date in
electronic
system

DOCUMENT FORMAT : Table of Contents
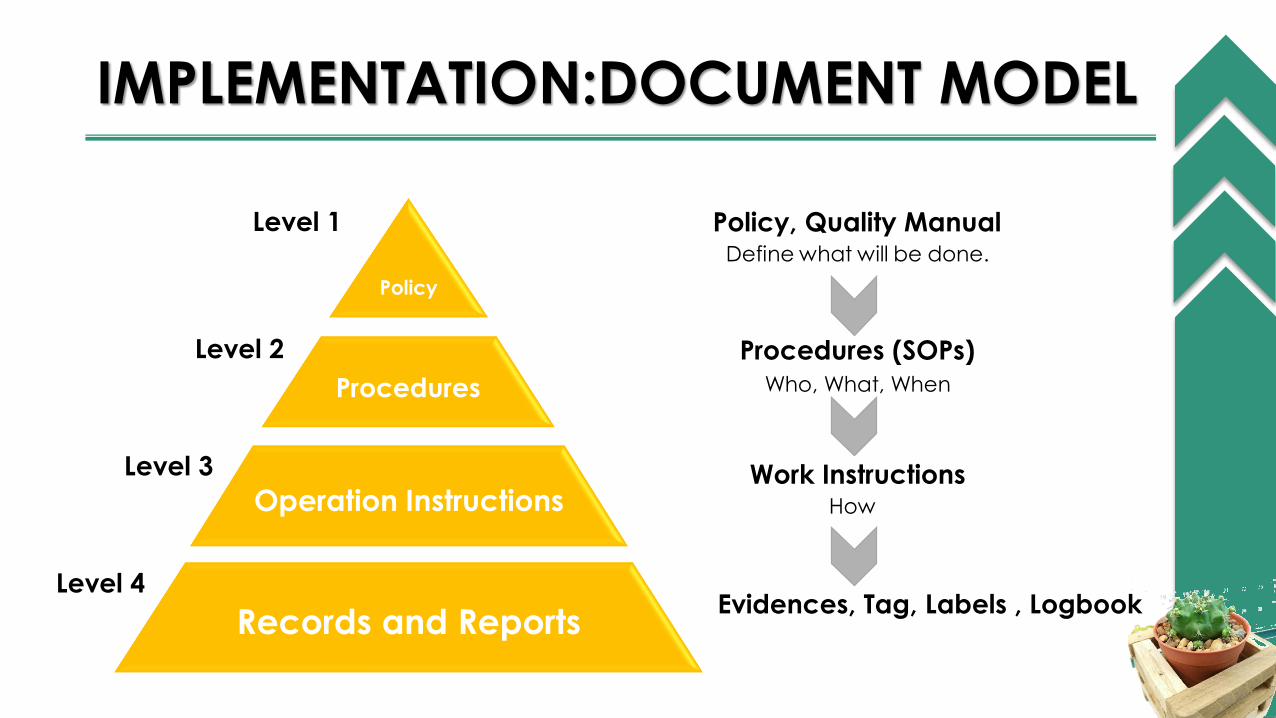
IMPLEMENTATION:DOCUMENT MODEL
Procedures
Operation Instructions
Records and Reports
Policy
Level 1
Level 2
Level 3
Level 4
Policy, Quality ManualDefine what will be done.
Procedures (SOPs)
Who, What, When
Work InstructionsHow
Evidences, Tag, Labels , Logbook
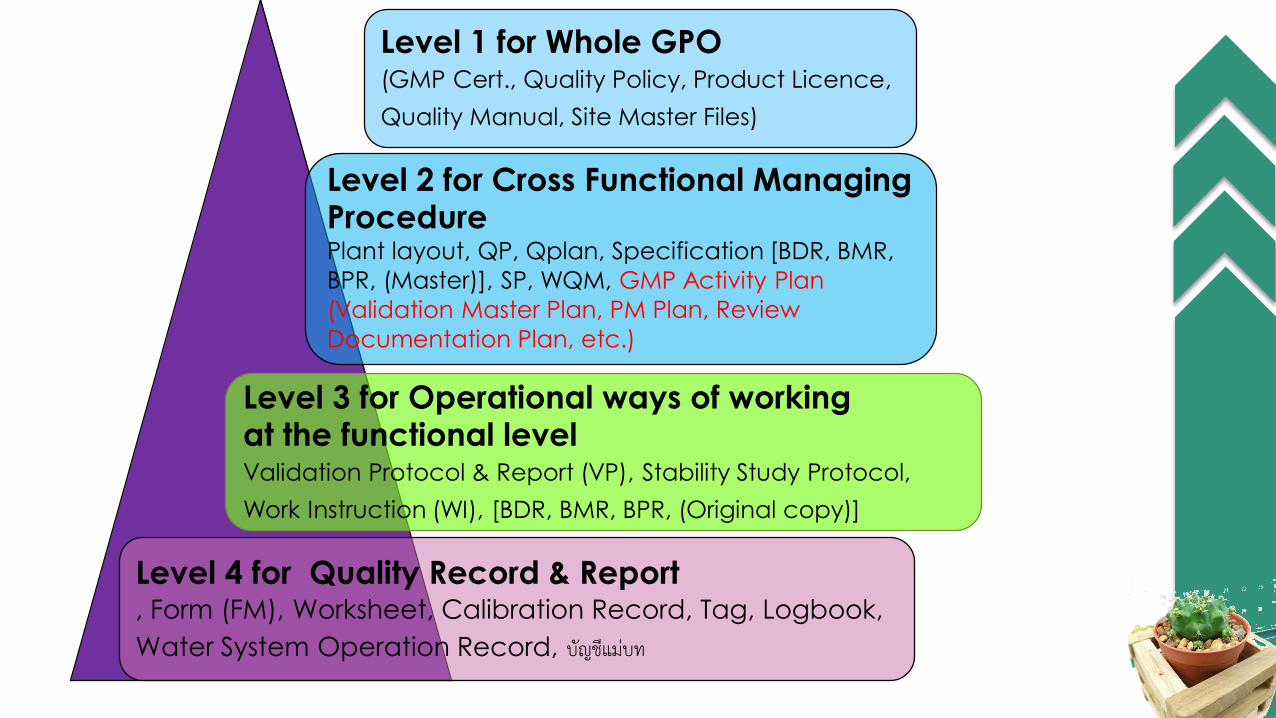
Level 1 for Whole GPO(GMP Cert., Quality Policy, Product Licence,
Quality Manual, Site Master Files)
Level 2 for Cross Functional Managing ProcedurePlant layout, QP, Qplan, Specification [BDR, BMR,
BPR, (Master)], SP, WQM, GMP Activity Plan
(Validation Master Plan, PM Plan, Review
Documentation Plan, etc.)
Level 4 for Quality Record & Report, Form (FM), Worksheet, Calibration Record, Tag, Logbook,
Water System Operation Record, บัญชีแม่บท
Level 3 for Operational ways of working at the functional levelValidation Protocol & Report (VP), Stability Study Protocol,
Work Instruction (WI), [BDR, BMR, BPR, (Original copy)]
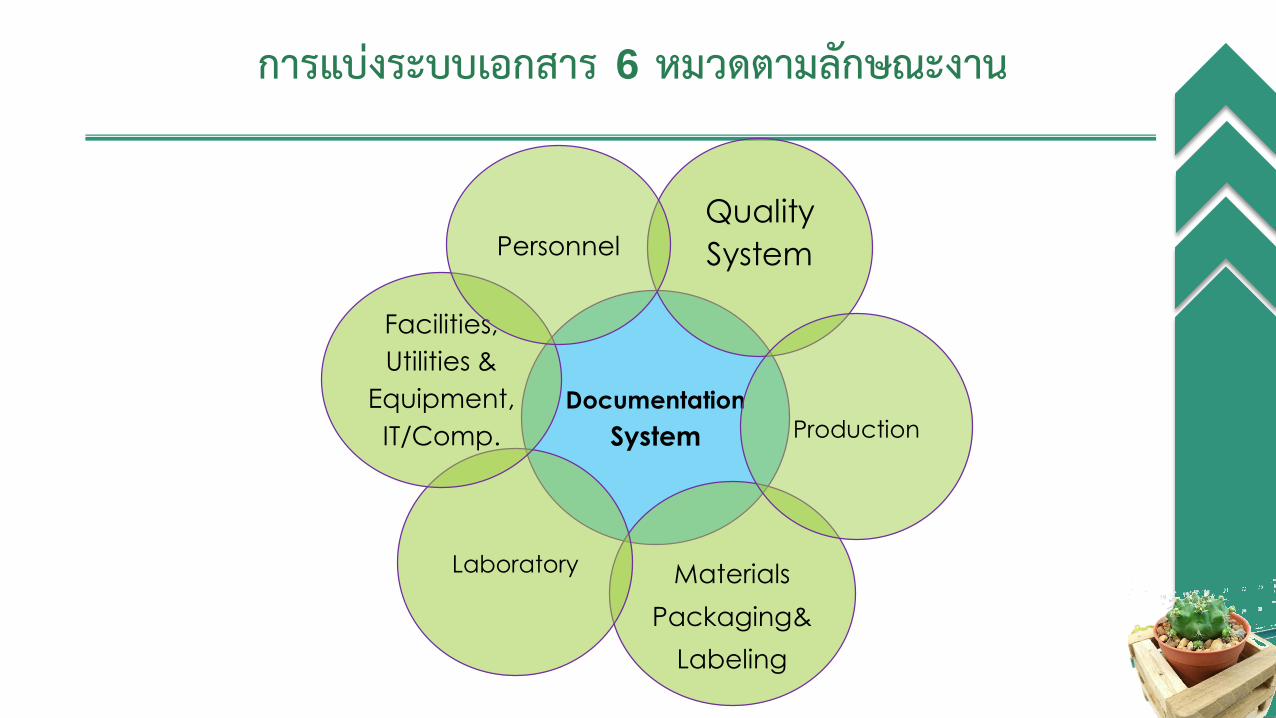
Documentation
System
Quality
System
Materials
Packaging&
Labeling
Laboratory
Facilities,
Utilities &
Equipment,
IT/Comp.
Personnel
Production
การแบ่งระบบเอกสาร 6 หมวดตามลักษณะงาน

Document Control Instruction
• Document Numbering System
• Operation for Procedure and Instruction
• Specification and
• Operation of Record
• Document Distribution
• Generation of Validation Documents
• Generation of BDR/BMR/BPR

The first documents auditor asks ??
• SMF PE 008
• QM
• VMP Annex 15
• SOP list
• SOP PLANT GOWNING

SMF and Quality Manual
• Site Master Files
• What does the document contain?
• A SMF contains information about the GMP activities occurring specifically at a site – quality management, production and/or QC operations, or any closely integrated operations at nearby buildings. It doesn’t contain information about GMP operations completed elsewhere.

SMF PE008-4
EXPLANATORY NOTES FOR PHARMACEUTICAL MANYFACTURERS ON THE PREPARATION OF A SITE MASTERFILE
1. GENERAL INFORMATION ON THE MANUFACTURER
2. QUALITY MANAGEMENT SYSTEM OF THE MANUFACTURER
3. PERSONNEL
4. PREMISES AND EQUIPMENT
5. DOCUMENTATION
6. PRODUCTION
7. QUALITY CONTROL (QC)
8. DISTRIBUTION, COMPLAINTS, PRODUCT DEFECTS AND RECALLS
9. SELF INSPECTIONS

SMF PE008-4
EXPLANATORY NOTES FOR PHARMACEUTICAL MANYFACTURERS ON THE PREPARATION OF A SITE MASTERFILE
Contents 5. Documentation
• Description of documentation system (i.e. electronic ,manual)
• When documents and records are stored or archived off-site (including pharmacovigilance data when applicable)
• List of types of documents/records
• Name and address of storage site and estimate of time required retrieving documents from the off-site archive

SMF PE008
• Appendix 1 Copy of valid manufacturing authorisation
• Appendix 2 List of dosage forms manufactured including the INN-names or common name (as available) of active pharmaceutical ingredients (API) used
• Appendix 3 Copy of valid GMP Certificate
• Appendix 4 List of contract manufacturers and laboratories including the addresses and contact information, and flow-charts of the supplychains for these outsourced activities
• Appendix 5 Organisation charts
• Appendix 6 Lay outs of production areas including material and personnel flows, general flow charts of manufacturing processes of each product type (dosage form)
• Appendix 7 Schematic drawings of water systems
• Appendix 8 List of major production and laboratory equipment

QM
• Quality Manuals
• What does the document contain?
• The Quality Manual is the overarching document of the QMS used to describe:• the quality policy of the business entity
• the boundaries, operations and process improvement of the QMS throughout the product lifecycle
• management responsibilities
• the road map of the key processes of the QMS and their relationship to each other.
• The Quality Manual may encompass multiple sites or a business entity operating within a larger site. Larger companies may not have a single document, or even call it a Quality Manual, but implement the quality policy using a series of individual documents – this is perfectly acceptable. Smaller companies may use the Quality Manual alone to describe their QMS (particularly in ISO 9001, though not so much in pharmaceutical companies).
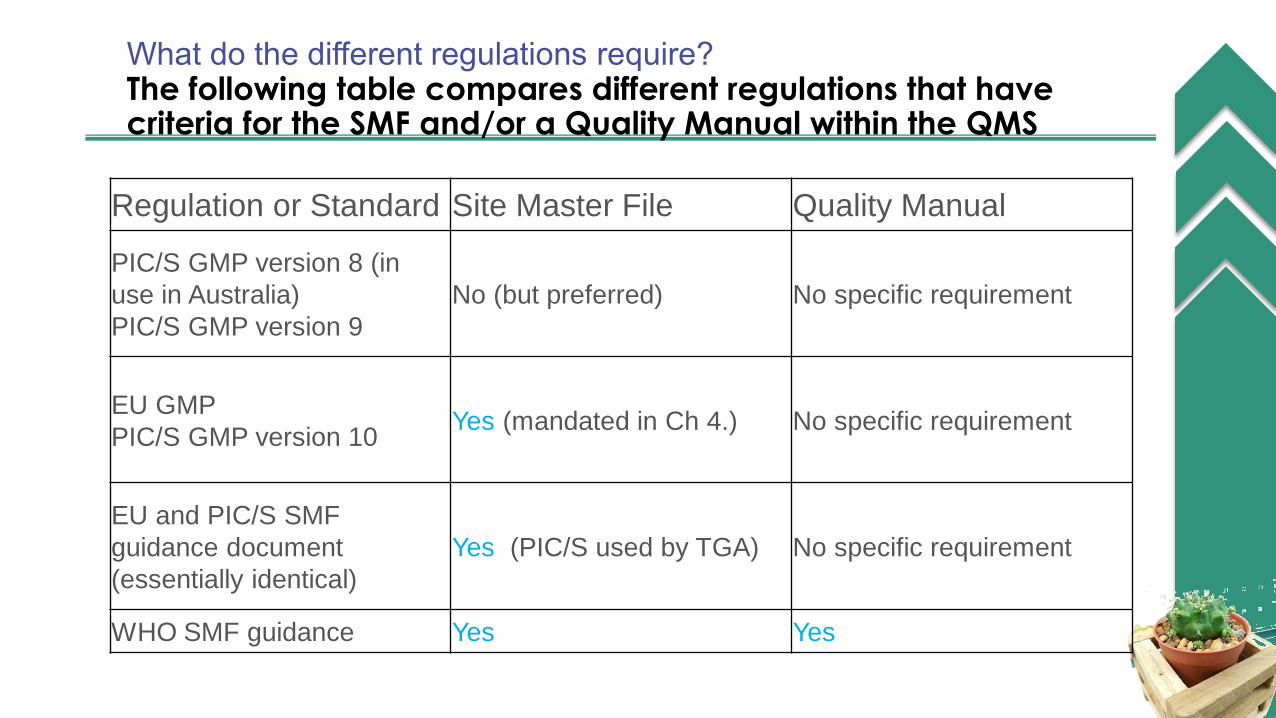
What do the different regulations require?ollowinable comparesThe following table compares different regulations that have criteria for the SMF and/or a Quality Manual within the QMSegulations that have criteria for the SMF and/or a Quality Manual within the QMS.Regulation or Standard Site Master File Quality Manual
PIC/S GMP version 8 (in
use in Australia)
PIC/S GMP version 9
No (but preferred) No specific requirement
EU GMP
PIC/S GMP version 10Yes (mandated in Ch 4.) No specific requirement
EU and PIC/S SMF
guidance document
(essentially identical)
Yes (PIC/S used by TGA) No specific requirement
WHO SMF guidance Yes Yes

SMF Checklist

ANNEX15 VALIDATION
The key elements of the site qualification and validation program should be clearly defined and documented in a
validation master plan (VMP) or equivalent document.

ANNEX15 VALIDATION
I. Qualification and Validation policy;
II. The organizational structure including
roles and responsibilities for
qualification and validation activities
III. Summary of the facilities, equipment,
systems, processes on site and the
qualification and validation status
IV. Change control and deviation management for qualification and validation
V. Guidance on developing acceptance criteria
VI. References to existing documents
VII. The qualification and validation strategy, including requalification, where applicable
The VMP or equivalent document should define the
qualification/validation system and include or reference
information on at least the following:

ANNEX15 VALIDATION
DOCUMENTATION, INCLUDING VMP (2.1-2.10)
2.1 To support Km throughout the product lifecycle
2.2 All documents ;approved and authorized by appropriate personnel as defined in the pharmaceutical quality system.
2.3 Inter-relationship between document ; clearly defined
2.4Validation protocols should be prepared which defines the
critical systems, attributes and parameters and the associated
acceptance criteria.
2.5 Qualification documents may be combined

ANNEX15 VALIDATION
2.6
Where validation protocols and other documentation are
supplied by a third party providing validation services,
appropriate personnel at the manufacturing site should
confirm suitability and compliance with internal procedures
before approval.
Vendor protocols may be supplemented by additional
documentation/test protocols before use.

ANNEX15 VALIDATION
2.7
Any significant changes to the approved protocol during
execution, e.g. acceptance criteria, operating parameters
etc., should be documented as a deviation and be
scientifically justified.
2.8
Results which fail to meet the pre-defined acceptance criteria
should be recorded as a deviation, and be fully investigated
according to local procedures. Any implications for the
validation should be discussed in the report.
2.9
The review and conclusions ; against criteria/justification
change/recommendation

ANNEX15 VALIDATION
2.10A formal release for the next stage in the qualification and validation process should be authorized by the relevant responsible personnel either as part of the validation
report approval or as a separate summary document.
Conditional approval to proceed to the next qualification stage can be given where certain acceptance criteria or deviations have not been fully addressed and there is a
documented assessment that there is no significant impact on the next activity

LIST OF PROCEDURES
QMS Laboratory • Document Control System• การทบทวนของฝ่ายบรหิาร• การบรหิารความเสีย่งดา้นคณุภาพ Quality Risk Management• PERMIT TO OPERATE• ระบบการแกไ้ข / ป้องกัน (CAPA SYSTEM)• การควบคมุการเปลีย่นแปลง (Change Control System)• Product Quality Review• การตรวจสอบตนเอง (GMP Self-Inspection/Internal audit)• การสบืหาสาเหตขุองผลติภัณฑท์ีไ่มเ่ป็นไปตามขอ้ก าหนด
(Failure Investigation) • การอนุมัตปิลอ่ยผา่นยาส าเร็จรปู (Batch Release)• Incident Report• Investigating Out of Specification /Out of Trend Test
Results and control of Non-Conformance• Supplier Approval and Evaluation System• การรับคนืยา (Returned and Salvaged Drug Products)• การเรยีกเก็บยาคนื (Recall Procedure)• วธิกีารจัดการกับค ารอ้งเรยีนจากลกูคา้ Customer Complaint
Record (CCR)• การก าหนดรหัสสนิคา้ (Item Number)• การจา้งผลติยา เครือ่งส าอาง และการจา้งตรวจวเิคราะห์• การรับรองและทดลองผลติยาใหม ่
• การตรวจคณุภาพวตัถดุบิ• การจัดการวตัถดุบิและผลติภัณฑร์ายการใหมใ่นชว่งทดลอง• การตรวจคณุภาพบรรจภัุณฑแ์ละสิง่พมิพ์• Packaging and Printing Material Supplier • Approval and Evaluation• การตรวจและทดสอบยาส าเร็จรปู • การจัดท าขอ้ก าหนดมาตรฐานของยา (Product Specification) • การแสดงสถานะ การตรวจและทดสอบ • Analytical Results and Trends• การทดสอบความคงสภาพของยา• Microbiological Laboratory Management and Safety
Procedure• Storage and Handling of Reference Working Standards• Microbiological Laboratory Instrument Management• Microorganism and Media Management• Microbiological Testing Procedure• การส ารองขอ้มลูส าหรับคอมพวิเตอร ์(Backup of Electronic Data)

LIST OF PROCEDURES
Production Material
ตรวจสอบเฝ้าระวงัการปนเป้ือนของยากลุม่ Penicillinขัน้ตอนการผลติและควบคมุคณุภาพระบบการผลติการวางแผนการผลติและการควบคมุการผลติการจัดท าเอกสารประกอบการผลติและการก าหนด Lot No.การตรวจการปนเป้ือนของเชือ้จลุนิทรยีใ์นอากาศการวางแผนการผลติและการควบคมุการผลติการชีบ้ง่และสอบกลบัไดข้องผลติภัณฑ ์การเคลือ่นยา้ย จัดบรรจ ุจัดเก็บของหน่วยงานการผลติ การควบคมุสภาวะแวดลอ้ม การผลติ และการจัดเก็บ
การควบคมุกระบวนการผลติสิง่พมิพ์การจัดซือ้วตัถดุบิและบรรจภัุณฑ์การรับคนื / แลกเปลีย่นยาและเวชภัณฑ์การท าลายทรัพยส์นิทีเ่ป็นยาและเวชภัณฑ ์การจัดสง่สนิคา้แกล่กูคา้การรับ การเคลือ่นยา้ย การเก็บรักษา การจา่ยสนิคา้ส าเร็จรปูการสง่มอบสนิคา้ควบคมุอณุหภมูดิว้ยระบบ VMIการวางแผนความตอ้งการใชพั้สดขุองคลังวตัถดุบิ/บรรจภุัณฑ์การเคลือ่นยา้ย การจัดเก็บและการเบกิจา่ยของคลังวตัถดุบิ /บรรจภุัณฑ ์

LIST OF PROCEDURES
Facility Utility and Equipment Personnel
• การซอ่มบ ารงุ• การบ ารงุรักษาเชงิป้องกัน• การควบคมุการส ารองอะไหลแ่ละวสัดชุา่ง • การ Shut Down ระบบโรงงาน• การสอบเทยีบเครือ่งตรวจ เครือ่งวดั และเครือ่งทดสอบ • การสรา้ง การตดิตัง้ การควบคมุ และการรับรองงานทาง
วศิวกรรม• การออกแบบและการออกขอ้ก าหนดทางวศิวกรรม• การผลติและควบคมุคณุภาพระบบการผลติน ้าบรสิทุธิ ์
(Purified Water) ทีใ่ชใ้นการผลติยา• การก าหนดรหัสเอกสารแบบกอ่สรา้งงานสถาปัตยกรรม และ
วศิวกรรมระบบ
• Process Validation Procedure• Method Validation Procedure• Cleaning Validation Procedure• Validation and Monitoring of Microbiological
Laboratory Cleanroom and Aseptic Process• Excel Spreadsheet Validation
• การบรหิารทรัพยากรบคุคล• การฝึกอบรมพนักงาน• การควบคมุการเขา้พืน้ที่
Computer System นโยบายการตรวจสอบความถกูตอ้งของระบบคอมพวิเตอร์การขึน้ทะเบยีนระบบเพือ่ตรวจสอบความถกูตอ้งการบรหิารการเปลีย่นแปลงระบบเทคโนโลยสีารสนเทศการเก็บขอ้มลู GMP และทีเ่ก็บขอ้มลูการบรหิารการส ารองและกูค้นืขอ้มลูการบรหิารการเขา้ถงึของผูใ้ชง้านการบรหิารจัดการสนิทรัพยเ์ทคโนโลยสีารสนเทศ
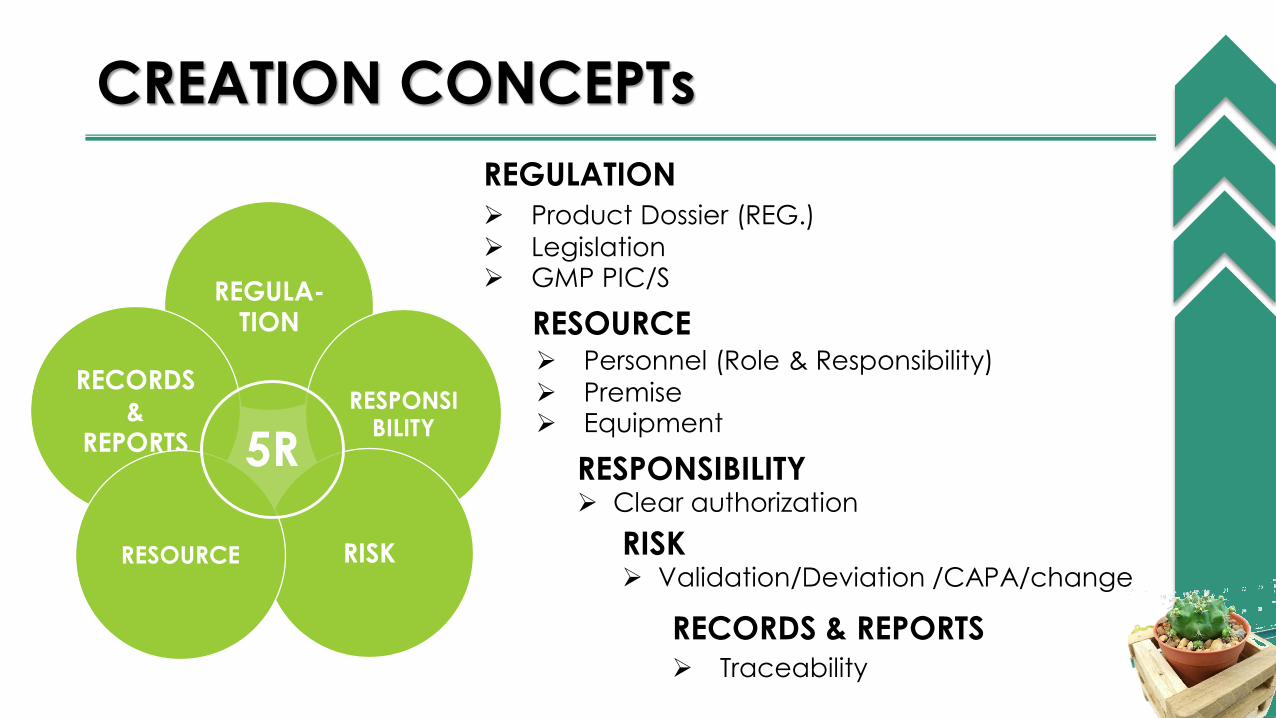
CREATION CONCEPTs
REGULATION
Product Dossier (REG.)
Legislation GMP PIC/S
RESOURCE Personnel (Role & Responsibility)
Premise Equipment
RECORDS & REPORTS Traceability
REGULA-TION
RECORDS
& REPORTS
RESPONSIBILITY
RISKRESOURCE
5R RESPONSIBILITY Clear authorization
RISK Validation/Deviation /CAPA/change





























