Embed Size (px)
Citation preview

Dysproteinemias and Glomerular Disease
Nelson Leung,1,2 Maria E. Drosou,1 and Samih H. Nasr 3
AbstractDysproteinemia is characterized by the overproduction of an Ig by clonal expansion of cells from theB cell lineage.The resultant monoclonal protein can be composed of the entire Ig or its components. Monoclonal proteins areincreasingly recognizedas acontributor to kidneydisease. Theycancause injury in all areasof thekidney, includingthe glomerular, tubular, and vascular compartments. In the glomerulus, the major mechanism of injury isdeposition. Examples of this include Ig amyloidosis, monoclonal Ig deposition disease, immunotactoid glomer-ulopathy, and cryoglobulinemicGN specifically from types 1 and 2 cryoglobulins.Mechanisms that do not involveIg deposition include the activation of the complement system, which causes complement deposition in C3glomerulopathy, and cytokines/growth factors as seen in thrombotic microangiopathy and precipitation, which isinvolved with cryoglobulinemia. It is important to recognize that nephrotoxic monoclonal proteins can beproduced by clones from any of the B cell lineages and that amalignant state is not required for the development ofkidney disease. The nephrotoxic clones that do not meet requirement for a malignant condition are now calledmonoclonal gammopathy of renal significance. Whether it is a malignancy or monoclonal gammopathy of renalsignificance, preservation of renal function requires substantial reduction of the monoclonal protein. Withbetter understanding of the pathogenesis, clone-directed strategies, such as rituximab against CD20 expressingB cell and bortezomib against plasma cell clones, have been used in the treatment of these diseases. Theseclone-directed therapies been found to be more effective than immunosuppressive regimens used in non-monoclonal protein–related kidney diseases.
Clin J Am Soc Nephrol 13: 128–139, 2018. doi: https://doi.org/10.2215/CJN.00560117
IntroductionDysproteinemia results from the overproduction ofa monoclonal Ig (1). All B cell clones can producemonoclonal gammopathy, but the incidence varies. Theyinclude CD201CD382 B cell clones involved in non-Hodgkin lymphoma, CD51CD191 clones in chroniclymphocytic leukemia, CD191CD381 clones in lym-phoplasmacytic lymphoma that produce Waldenströmmacroglobulinemia, and CD192CD381 plasma cellclones in multiple myeloma (2). Monoclonal gammop-athy is nearly universal with the latter two clones butless common in chronic lymphocytic leukemia andCD201CD382 B cell clones. Only one Ig light chaincan be produced by a clone. In rare circumstances, theIg heavy chain is truncated and circulates as a mono-clonal heavy chain.
Because the size of the monoclonal protein oftenreflects the clonal burden, it can be used as a bio-marker for diagnostic and prognostic purposes. Mono-clonal gammopathy of undetermined significance(MGUS) is distinguished from multiple myeloma andWaldenström macroglobulinemia by the size of themonoclonal protein (M protein) or serum free lightchain (sFLC) (2,3). Conversely, response to therapy isdetermined by the reduction of M protein and/or sFLCconcentration (4). The concentration of sFLC at diagnosisis also prognostic in Ig light chain (AL) amyloidosis (5).
SomeM proteins however are toxic and cause organdysfunction. Such is the case in AL amyloidosis. Themisfolded monoclonal free light chain (FLC) forms
amyloid that is deposited in the soft tissues (6). The organis damaged by themonoclonal FLC rather than the clonalmass (7). This explains why most patients have ,10%bone marrow plasma cells. In those with .10% bonemarrow plasma cells, most do not have any myeloma-defining events known as hypercalcemia, AKI as a resultof light-chain cast nephropathy, anemia, or lytic bonelesions (CRAB). In fact, over 80% of the patientswould bebest classified as having monoclonal gammopathy ofrenal significance (MGRS). Animal studies have shownthat only the M protein is necessary for reproduction ofthe renal lesion (8). Unfortunately, efforts to identifyproperties that predict nephrotoxicity have been unsuc-cessful so far. The toxicity is not limited to the kidney,because these proteins can also cause neuropathies,dermopathies, ocular disorders, and other multisystemicdiseases (9).
PathogenesisThe kidney is the organ most affected by M proteins.
A number of factors may contribute to this (10). Thekidneys are exposed to more cardiac output than anyother organ, except the lungs. This increased exposure ispaired with the unique environment, where the pH andelectrolyte concentrations are present in no other tissues.These changes can alter the chemical characteristics ofthe M protein, making it more toxic (11). Finally, recep-tors and proteins are present in the kidney that can in-teract with Igs. The megalin-cubilin receptor actively
Divisions of1Nephrology andHypertension and2Hematology and3Department ofLaboratory Medicineand Pathology, MayoClinic, Rochester,Minnesota
Correspondence:Dr. Nelson Leung, 200First Street SW,Rochester, MN 55905.Email: [email protected]
www.cjasn.org Vol 13 January, 2018128 Copyright © 2018 by the American Society of Nephrology

endocytoses FLC for degradation in proximal tubular cells (12).The inability to degrade certain Ig light chains is the pathogenicbasis in light-chain proximal tubulopathy. On the other hand,formation of obstructive casts by the binding and precipitationof monoclonal FLCs and Tamm Horsfall protein is the char-acteristic of light chain cast nephropathy. (13).Several mechanisms of nephrotoxicity have been de-
scribed, which include deposition, complement activation,cytokine activation, and precipitation (Figure 1) (14,15).Deposition is the most common and seen in Ig-related (AIg)amyloidosis, monoclonal Ig deposition disease, prolifera-tive GN with monoclonal Ig deposits, immunotactoid GN,and fibrillary GN (FGN) (16). The deposits can be orga-nized (as in AL, immunotactoid GN, and FGN) or un-organized (e.g. proliferative GN with monoclonal Igdeposits and monoclonal Ig deposition disease). Extrarenaldeposition can occur especially in AIg amyloidosis andmonoclonal Ig deposition disease (17–21). Extrarenal in-volvement is rare with immunotactoid GN and FGN andhas not been described in proliferative GN with monoclonalIg deposits (22–25).Precipitation is the mechanism of injury in cast nephropa-
thy, cryoglobulinemia, and (cryo)crystalglobulinemia. This oc-curs predominately in the distal tubules in cast nephropathy(10). In contrast, the precipitation occurs intravascularly incryoglobulinemic GN (26). Cryoglobulins are most charac-teristically found in the glomerular capillaries, often result-ing in pseudothrombi formation described as cryoplugs.Complement activation and cytokine activation are also
associated with M proteins. The incidence of monoclonalgammopathy in patients with C3 glomerulopathy has beenfound to far exceed that of the normal population, especially inpeople.50 years of age (27–30). Although C3 nephritic factorand autoantibody against factor H have been reported in somepatients and while others have complement gene polymor-phisms, the mechanism of complement activation remainsundetermined in the majority of patients (27,29–31). Comple-ment is also activated in monoclonal gammopathy–associatedmembranous nephropathy, proliferative GN with monoclonalIg deposits, immunotactoid GN, FGN, and cryoglobulinemicGN. Cytokine activation is involved in the renal lesion ofpatients with polyneuropathy, endocrinopathy, orga-nomegaly, monoclonal gammopathy, and skin changes(POEMS) syndrome (32). The cytokines activation results in
a glomerulopathy that resembles thrombotic microangiopa-thy but without the microangiopathic hemolytic anemia.Interestingly, theM protein is not identifiable in the kidney ineither of these entities, and a direct link to either mechanismhas not been identified.
Glomerular LesionsAIg AmyloidosisAIg amyloidosis is the most common glomerular lesion
associated with monoclonal gammopathy (33). AL amy-loidosis is the most common subtype, but AH amyloidosisand Ig heavy and light chain (AHL) amyloidosis also exist(Table 1). Clinically, the entire group behaves similarly,with a median age of presentation in the low 60 years ofage and a slight dominance of men (approximately 60%).Patients present with mild renal impairment (medianserum creatinine [Scr] of 1.2 mg/dl) and nephrotic-rangeproteinuria (6 g/d). Nephrotic syndrome is present in morethan two thirds of patients. ESRD predominantly occurs inpatients who present with renal manifestation (34). Recur-rence after kidney transplant has been reported but cantake years to occur (35). AIg amyloid is systemic and can bedeposited in any visceral organ (17). The heart is the mostcommon extrarenal organ involved followed by nerves. Heartinvolvement may be less common in AH and AHL amyloid-osis versus AL amyloidosis (36). Patients with severe heartinvolvement have the poorest prognosis, even with moderntherapies (37). Although 40% of patients have .10% clonalplasma cells on bone marrow biopsy, ,20% actually havesymptomatic multiple myeloma. The lethality of this diseasestems from the progressive organ dysfunction, which is notdependent on clonal expansion as in multiple myeloma.Amyloid deposits can be found in all three compart-
ments of the kidney (33). Amyloid deposits are best seenalong the glomerular basement membranes (GBMs) andmesangium, causing mesangial expansion (Figure 2A). Themesangial deposits can appear nodular. Deposits are typi-cally periodic acid–Schiff (PAS) and silver negative (Figure2A). However, amyloid spicules, which are a parallel align-ment of subepithelial fibrils perpendicular to the GBM,appear silver positive. Amyloid fibrils are congophilic anddisplay an apple green birefringence under a polarized light(Figure 2, B and C). Immunofluorescence studies show a
Figure 1. | Mechanisms of glomerular toxicity bymonoclonal gammopathy. AIg amyloidosis, immunoglobulin-derived amyloidosis.; C3G, C3glomerulopathy; MIDD,monoclonal Ig deposition disease; PGNMID, proliferativeGNwithmonoclonal Ig deposits; POEMS, polyneuropathy,organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes.
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 129

Tab
le1.
Monoclonal
gammopathyofrenal
sign
ificance
glomerularlesions:Clinical
features,pathologicch
arac
teristics,an
doutcomes
Ren
alLesion
Clin
ical
Features
Patholog
icCha
racteristics
Outcomes
andPo
st-Trans
plan
tCou
rse
Ig-related
amyloidosis
(AL,A
HL,A
H)
Proteinu
ria,
NS,
CKD
LM:P
AS(2
),Ag(2
)acellu
lardep
osits
PRDan
dESR
Dcommon
;worse
outcom
esifconc
urren
tcardiacinvo
lvem
ent
HTN
andhe
maturia
uncommon
IF:L
Can
d/or
HCrestricted
Recurrenc
ein
tran
splant
uncommon
EM:ran
dom
,non
bran
chingfibrils
(7–12
nm)
Con
gored(1
)FibrillaryGN
Proteinu
ria,
HTN,h
ematuria,
CKD,N
SLM:M
esGN,m
esan
gial
expa
nsion,
MGN,M
PGN,P
AS(1
),Ag(2
)PR
Dan
dESR
Dcommon
IF:p
olyclona
lIgG
.80
%Recurrenc
ein
tran
splant
common
ifmon
oclona
lgam
mop
athy
ispresen
tEM:ran
dom
fibrils
(9–26
nm)
Con
gored(2
)Im
mun
otactoid
GN/
GOMMID
Proteinu
ria,
NS,
CKD,h
ematuria
LM:M
PGN,M
GN,E
CPGN,P
AS(w
eak),
Ag(2
)PR
common
,PRD,E
SRD
Hyp
ocom
plem
entemia
IF:IgG
,C3,
LCrestricted
Recurrenc
ein
tran
splant
EM:p
arallel,ho
llow
microtubu
les(8–60
nm)
Con
gored(2
),cryo
glob
ulin
(2)
Typ
e1cryo
glob
ulinem
icGN
Proteinu
ria,
hematuria
LM:M
PGN,E
CPG
N,sub
endothe
lial
andintracap
illary,
PAS(1
)CRcommon
,frequ
entrelap
se,
CKD,E
SRD,recurrenc
ein
tran
splant
Nep
hritic/ne
phroticsynd
rome,AKI,CKD,H
TN,
purpura,a
rthralgias,h
ypoc
omplemen
temia
IF:m
onoclona
lIgG
/IgM
andC3,C4,an
dC1q
dep
osits
EM:curvilin
earmicrotubu
les,10
–50
nmMon
oclona
lIgdep
osition
disease
(LCDD,
LHCDD,H
CDD)
Proteinu
ria,
NS,
CKD,A
KI
LM:P
AS(1
),Ag(1
),mesan
gial
expa
nsion,
nodular
GS,
thicke
ned
TBM
PRcommon
,ESR
D
HTN
andhe
maturia
uncommon
IF:linearLC-a
nd/or
HC-restricted
depo
sitsinTB
M,G
BM,and
vascularwall
Recurrenc
ein
tran
splant
common
EM:p
unctate,po
wderyelectron
den
sedep
osits
ProliferativeGN
with
mon
oclona
lIgdep
osits
Proteinu
ria,
hematuria,N
S,CKD,A
KI
LM:M
PGN,E
CPG
N,M
esGN,M
GN,
crescents(occasiona
lly)
PRD,P
R,E
SRD
IF:IgG
(rarelyIgM
andIgA),LCan
dHC
restricted
(mostlyIgG3)
Recurrenc
ein
tran
splant
common
EM:m
esan
gial,sube
ndothe
lial,an
dintram
embran
ousdep
osits
C3glom
erulopathy
Hem
aturia,
proteinu
ria,
CKD
LM:M
PGN,E
CPG
N,M
esGN,M
GN
PRD
andESR
Dcommon
C3GN
Low
C3leve
land
norm
alC4leve
lcom
mon
IF:g
ranu
larC3dep
osits,littleor
noIg
orC1q
dep
osits
C3G
N:recurrenc
ein
tran
splant
most
common
DDD
EM:D
DD,intramem
bran
ous“saus
age-
shap
eddep
osits”;C
3GN,m
esan
gial,
sube
ndothe
lial,an
dsu
bepithelial
dep
osits
DDD:recurrenc
ein
tran
splant
130 Clinical Journal of the American Society of Nephrology

light-chain restriction with AL and AHL (Figure 2F).Monoclonal Ig heavy chain is seen in both AHL andAH, but monoclonal Ig light chain is absent in AH. Amyloiddeposits are typically identified in the vessels (.80%) and theinterstitium (53%–63% of patients). On electron microscopy(EM), amyloid appears as solid randomly arranged fibrilswith a diameter of 7–12 nm irrespective of the precursorprotein (Figure 2, D and E). Typing of some amyloid can bedone by immunohistochemistry/immunofluorescence (38).However, mass spectrometry with proteomic analysis has be-come the gold standard for amyloid typing (39). Most recently,urinary exosomes have been investigated as a potential tool indiagnosis and response assessment in AIg amyloidosis (40).
Monoclonal Ig Deposition DiseaseMonoclonal Ig deposition disease is also represented by
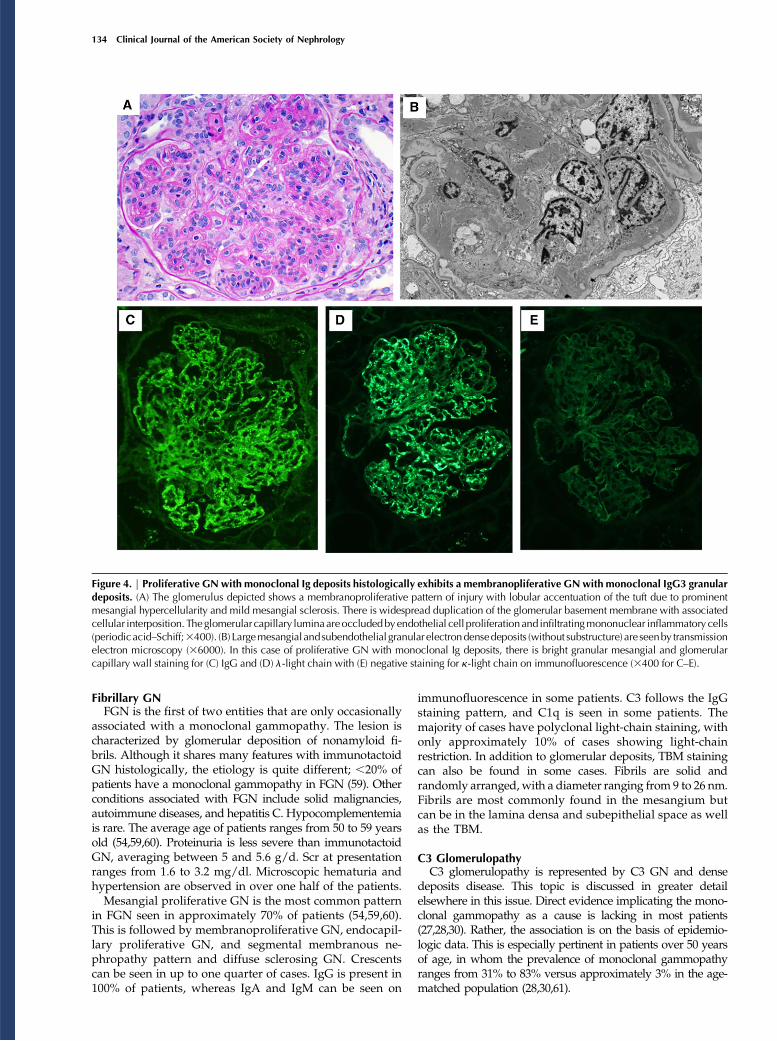
three subtypes (19). Light-chain deposition disease is the mostcommon followed by light- and heavy-chain deposition disease,whereas heavy-chain deposition disease (HCDD) is the rarest.The subtypes are indistinguishable by light microscopy. Theycan only be separated by immunofluorescence. The Ig heavychain in HCDD is typically truncated (41). Patients with HCDDhave heavier proteinuria, but otherwise, all patients with mono-clonal Ig deposition disease behave similarly (19). The medianage of these patients is between 56 and 64 years old. Nearly twothirds are men (19,21,42). Median Scr ranges from 2.2 to 3.8mg/dl. Proteinuria averages between 1.5 and 4.1 g/d.Extrarenal manifestations mainly involve the heart andliver but are much less common than in AL amyloidosis.Most patients with monoclonal Ig deposition disease haveMGRS instead of multiple myeloma. A series from Italyfound multiple myeloma in 65% of patients and chroniclymphocytic leukemia in 3% of patients (20). However,when myeloma was defined by CRAB, the rate dropped to20%–22%, suggesting that most of the patients hadsmoldering multiple myeloma rather than multiple mye-loma (19,43). Mortality of these patients is largely de-pendent on the presence of multiple myeloma. Recurrencein the renal allograft is quite common and rapid (44).The most common renal histologic finding is nodular
mesangial sclerosis (19,42) (Figure 3A). The nodules are PASand silver positive and can mimic those of diabetic nephrop-athy (Figure 3A). Membranoproliferative features, such as GBMduplication and mesangial hypercellularity, may be present.Cellular crescents can be seen focally in patients with rare cases(particularly in aHCDD) (45). Linear staining of the GBM andeven more consistently, the tubular basement membranes(TBMs) by a single Ig light chain is characteristic of light-chaindeposition disease and light- and heavy-chain depositiondisease on immunofluorescence (Figure 3, C and D). Nearly90% of patients have a k-isotype. These deposits are electrondense and have an amorphous or powdery appearance onEM (Figure 3B).
Proliferative GN with Monoclonal Ig DepositsProliferative GN with monoclonal Ig deposits is another
renal disease with nonorganized monoclonal Ig deposits.The deposits are composed of the entire Ig and usually anIgG, although IgA and IgM have rarely been described (46).Patients present with renal impairment (median Scr of 2.8mg/dl), proteinuria (median of 5.7 g/d), and hematuria
Tab
le1.(Continued
)
Ren
alLesion
Clin
ical
Features
Patholog
icCha
racteristics
Outcomes
andPo
st-Trans
plan
tCou
rse
Throm
botic
microan
giop
athy
Proteinu
ria,
hematuria,C
KD
LM:d
oublecontou
rof
GBM,
mesan
giolysis,A
TN,throm
biin
GC
ESR
Dcommon
,PRD
Ane
mia,throm
bocy
topen
ia,sch
istocy
tes
Cou
rsein
tran
splant:ins
ufficien
tev
iden
ceIF:fi
brinog
enmaterialinGClumen
and
wall
EM:d
oublecontou
rof
GBM,
sube
ndothe
lial“
fluff”
Mostcom
mon
symptoms:The
find
ings
aresorted
ontheba
sisof
order
offreq
uen
cy.A
L,Iglig
htch
ainam
yloidosis;A
HL,Ighe
avyan
dlig
htch
ainam
yloidosis;A
H,Ighe
avych
ainam
yloidosis;
NS,
nephroticsynd
rome;
HTN,h
ypertension;
LM,light
microscop
y;PA
S,pe
riod
icacid–Schiff;A
g,silver
stain;
IF,immun
ofluo
rescen
ce;L
C,light
chain;
HC,h
eavy
chain;
EM,electron
microscop
y;PR
D,p
rogressive
rena
ldisease:failure
tomeetcriteriaforeither
completeresp
onse
orpa
rtialrespo
nsebu
tnot
reaching
ESR
D,inc
ludingpa
tien
tswithun
remitting
proteinu
riaor
prog
ressiveCKD;M
esGN,m
esan
gial
proliferativeGN;M
GN,m
embran
ousGN;M
PGN,m
embran
oproliferativeGN;G
OMMID
,GN
withorga
nizedmicrotubu
larIg
dep
osits;ECPGN,
endocap
illaryproliferativeGN;PR,partialresp
onse:red
uctioninproteinu
riaby
atleast50%
andto
,2g/
dwithstab
lerena
lfunc
tion
(#20
%increase
inseru
mcreatinine
);CR,com
pleteremission
:remission
ofproteinu
riato
,50
0mg/
dwithno
rmal
rena
lfunc
tion
;LCDD,light-cha
indep
ositiondisease;L
HCDD,light-a
ndhe
avy-ch
aindep
ositiondisease;H
CDD,h
eavy
-cha
indep
osition
disease;G
S,glom
erulosclerosis;T
BM,tub
ularb
asem
entm
embran
e;GBM,glomerular
basemen
tmem
bran
e;C3G
,C3glom
erulop
athy
;DDD,d
ense
dep
ositsd
isease;A
TN,acu
tetubu
larn
ecrosis;
GC,g
lomerular
capilla
ry.
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 131
(22). Multiple myeloma is rare, with approximately 80% ofpatients having no circulating M protein detectable at thetime of diagnosis. Clonal identification is difficult if an Mprotein is not detected (46).The renal lesion is characterized by proliferative GN (47).
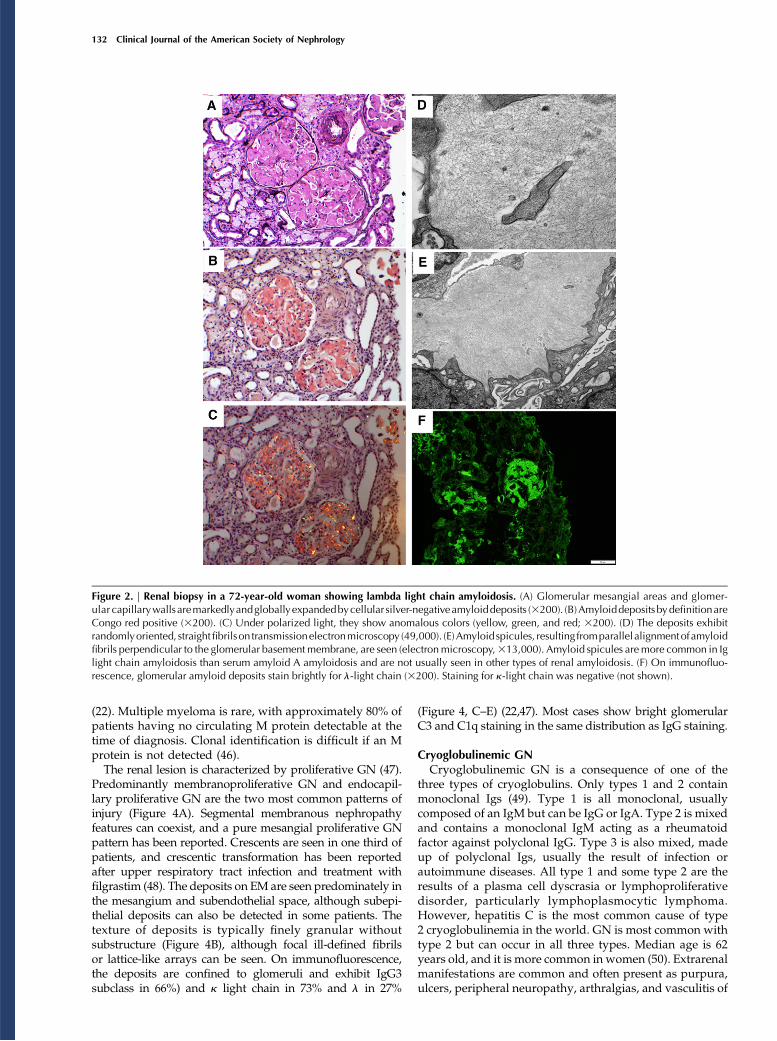
Predominantly membranoproliferative GN and endocapil-lary proliferative GN are the two most common patterns ofinjury (Figure 4A). Segmental membranous nephropathyfeatures can coexist, and a pure mesangial proliferative GNpattern has been reported. Crescents are seen in one third ofpatients, and crescentic transformation has been reportedafter upper respiratory tract infection and treatment withfilgrastim (48). The deposits on EM are seen predominately inthe mesangium and subendothelial space, although subepi-thelial deposits can also be detected in some patients. Thetexture of deposits is typically finely granular withoutsubstructure (Figure 4B), although focal ill-defined fibrilsor lattice-like arrays can be seen. On immunofluorescence,the deposits are confined to glomeruli and exhibit IgG3subclass in 66%) and k light chain in 73% and l in 27%
(Figure 4, C–E) (22,47). Most cases show bright glomerularC3 and C1q staining in the same distribution as IgG staining.
Cryoglobulinemic GNCryoglobulinemic GN is a consequence of one of the
three types of cryoglobulins. Only types 1 and 2 containmonoclonal Igs (49). Type 1 is all monoclonal, usuallycomposed of an IgM but can be IgG or IgA. Type 2 is mixedand contains a monoclonal IgM acting as a rheumatoidfactor against polyclonal IgG. Type 3 is also mixed, madeup of polyclonal Igs, usually the result of infection orautoimmune diseases. All type 1 and some type 2 are theresults of a plasma cell dyscrasia or lymphoproliferativedisorder, particularly lymphoplasmocytic lymphoma.However, hepatitis C is the most common cause of type2 cryoglobulinemia in the world. GN is most common withtype 2 but can occur in all three types. Median age is 62years old, and it is more common in women (50). Extrarenalmanifestations are common and often present as purpura,ulcers, peripheral neuropathy, arthralgias, and vasculitis of
Figure 2. | Renal biopsy in a 72-year-old woman showing lambda light chain amyloidosis. (A) Glomerular mesangial areas and glomer-ular capillarywallsaremarkedlyandgloballyexpandedbycellular silver-negativeamyloiddeposits (3200). (B)AmyloiddepositsbydefinitionareCongo red positive (3200). (C) Under polarized light, they show anomalous colors (yellow, green, and red; 3200). (D) The deposits exhibitrandomlyoriented, straightfibrilson transmissionelectronmicroscopy (49,000). (E)Amyloidspicules, resulting fromparallelalignmentofamyloidfibrils perpendicular to the glomerular basementmembrane, are seen (electronmicroscopy,313,000). Amyloid spicules aremore common in Iglight chain amyloidosis than serum amyloid A amyloidosis and are not usually seen in other types of renal amyloidosis. (F) On immunofluo-rescence, glomerular amyloid deposits stain brightly for l-light chain (3200). Staining for k-light chain was negative (not shown).
132 Clinical Journal of the American Society of Nephrology

other organs (49). Severe hypertension is also commonlyobserved.Cryoglobulinemic GN presents mainly in a membrano-
proliferative pattern (Figure 5A) (51). Eosinophilic cryo-globulins are commonly observed in the glomerular capillarylumina. These are strongly PAS positive and trichrome red(Figure 5A). Leukocytoclastic vasculitis is common withneutrophils in the acute phase, whereas macrophages andmonocytes are in the acute and chronic phases. Crescents arepresent in some cases. The Ig deposits are dependent on themakeup of the cryoglobulin; however, C3 is often prominent.In type 2, both IgM and IgG will be present, but light-chainrestriction may not be shown, because the IgG is polyclonal.Light-chain restriction is usually present in type 1. In somecases, the deposits do not stain on routine immunofluores-cence on frozen tissue and require paraffin immunofluores-cence with protease digestion for detection (52). Intraluminal,mesangial, and subendothelial deposits are seen on EMalong with double contouring of the GBM. The deposits inmost cases are focally organized, showing curved or straightmicrotubules (Figure 5B).
Immunotactoid GNImmunotactoid GN is another glomerular disorder
resulting from deposition of microtubules (53). By defini-tion, cryoglobulinemic GN must be ruled out, because theyare difficult to differentiate histopathologically. Other thanthe absence of cryoglobulins, extrarenal manifestations areuncommon in immunotactoid GN. Rarely, vasculitic skinlesions, immunotactoid corneal and liver deposits, andmononeuritis multiplex have been reported (54–56). Me-dian age ranges from 57 to 61 years old, with a slightincreased prevalence in men (53,54). Most patients presentwith nephrotic-range proteinuria, with a range of 6–11 g/d.
Median creatinine at presentation is 1.5 mg/dl. Micro-scopic hematuria can be seen in some patients on urinal-ysis. A monoclonal gammopathy is identified in 80%–90%of cases. Hypocomplementemia is not uncommon. Oneparticular characteristic of immunotactoid GN is its associationwith chronic lymphocytic leukemia. Approximately 50% of thepatients had chronic lymphocytic leukemia or monoclonalB cell lymphocytosis (MBL; a chronic lymphocytic leukemiaclonal disorder) (54,57).The most common pattern in immunotactoid GN is
membranoproliferative with mesangial expansion, hyper-cellularity, and double contouring (53). Large subendothelialdeposits mimickingwire loop–type deposits of lupus nephritiscan be seen in some cases (Figure 6A). Immunotactoid GNcould also present with membranous, like features, withthickening basement membranes and spikes either globally orsegmentally. The least common pattern is endocapillary pro-liferative GN characterized by leukocyte infiltration, resultingin luminal occlusion but without GBM duplication. Focalcrescents and fibrinoid necrosis are seen in a small number ofpatients (53,58). Patients who have underlying chronic lym-phocytic leukemia or lymphoplasmocytic lymphoma oftenhave neoplastic lymphocytic infiltration in the kidney. Onimmunofluorescence, nearly 90% have IgG staining of 2–31intensity. The rest stain for IgA and IgM. C3 staining followsthe same pattern as the IgG. C1q deposits are seen in over onehalf of cases. Light-chain restriction can be shown in 69%–93%of cases. IgG1 represents about one half of the IgG subtypesin immunotactoid GN. The deposits are uniformly composedof microtubules with hollow centers that have an averagediameter of 31 nm but can range from 9 to 52 nm (53,54). Theyare typically arranged in parallel arrays (Figure 6B). Depositscan be found in the mesangium and the subendothelial andsubepithelial spaces.
Figure 3. | Light-chain deposition disease on renal biopsy exhibits nodularmesangial sclerosis andpunctate linearmonoclonal deposits alongrenal basement membranes. (A) Glomeruli exhibit marked nodular mesangial expansion by periodic acid–Schiff-positive material, mimickingdiabetic glomerulosclerosis (3200). (B) On electron microscopy, punctate powdery electron dense deposits are seen along tubular basementmembranes (315,000). Similar deposits are frequently seen along glomerular basement membranes and in the mesangium (not shown). Onimmunofluorescence in this case of l–light-chain deposition disease, there is diffuse linear glomerular and tubular basement membrane stainingfor (C) l (3100) with (D) negative k (3100).
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 133
Fibrillary GNFGN is the first of two entities that are only occasionally
associated with a monoclonal gammopathy. The lesion ischaracterized by glomerular deposition of nonamyloid fi-brils. Although it shares many features with immunotactoidGN histologically, the etiology is quite different; ,20% ofpatients have a monoclonal gammopathy in FGN (59). Otherconditions associated with FGN include solid malignancies,autoimmune diseases, and hepatitis C. Hypocomplementemiais rare. The average age of patients ranges from 50 to 59 yearsold (54,59,60). Proteinuria is less severe than immunotactoidGN, averaging between 5 and 5.6 g/d. Scr at presentationranges from 1.6 to 3.2 mg/dl. Microscopic hematuria andhypertension are observed in over one half of the patients.Mesangial proliferative GN is the most common pattern
in FGN seen in approximately 70% of patients (54,59,60).This is followed by membranoproliferative GN, endocapil-lary proliferative GN, and segmental membranous ne-phropathy pattern and diffuse sclerosing GN. Crescentscan be seen in up to one quarter of cases. IgG is present in100% of patients, whereas IgA and IgM can be seen on
immunofluorescence in some patients. C3 follows the IgGstaining pattern, and C1q is seen in some patients. Themajority of cases have polyclonal light-chain staining, withonly approximately 10% of cases showing light-chainrestriction. In addition to glomerular deposits, TBM stainingcan also be found in some cases. Fibrils are solid andrandomly arranged,with a diameter ranging from 9 to 26 nm.Fibrils are most commonly found in the mesangium butcan be in the lamina densa and subepithelial space as wellas the TBM.
C3 GlomerulopathyC3 glomerulopathy is represented by C3 GN and dense
deposits disease. This topic is discussed in greater detailelsewhere in this issue. Direct evidence implicating the mono-clonal gammopathy as a cause is lacking in most patients(27,28,30). Rather, the association is on the basis of epidemio-logic data. This is especially pertinent in patients over 50 yearsof age, in whom the prevalence of monoclonal gammopathyranges from 31% to 83% versus approximately 3% in the age-matched population (28,30,61).
Figure 4. | Proliferative GN with monoclonal Ig deposits histologically exhibits a membranopliferative GNwith monoclonal IgG3 granulardeposits. (A) The glomerulus depicted shows a membranoproliferative pattern of injury with lobular accentuation of the tuft due to prominentmesangial hypercellularity and mild mesangial sclerosis. There is widespread duplication of the glomerular basement membrane with associatedcellular interposition. Theglomerularcapillary luminaareoccludedbyendothelial cell proliferationand infiltratingmononuclear inflammatory cells(periodicacid–Schiff;3400). (B) Largemesangialandsubendothelial granular electrondensedeposits (without substructure) areseenby transmissionelectron microscopy (36000). In this case of proliferative GN with monoclonal Ig deposits, there is bright granular mesangial and glomerularcapillary wall staining for (C) IgG and (D) l-light chain with (E) negative staining for k-light chain on immunofluorescence (3400 for C–E).
134 Clinical Journal of the American Society of Nephrology
Hematologic ConditionsMonoclonal gammopathy is a sequela of lymphoproli-
ferative disorders or plasma cell dyscrasia. These disordersare classified as an MGUS or MBL if they do not meetcriteria for symptomatic lymphoma or multiple myeloma.However, if the clone produces a nephrotoxic M protein, itwould be renamed as MGRS. Each hematologic disorderhas its own preferential kidney diseases, although no renallesion is exclusive to any hematologic disorder.
Multiple MyelomaMultiple myeloma is a malignant condition of the plasma
cells. Symptomatic multiple myeloma is diagnosed either bythe presence of a CRAB lesion or when the patient is at high
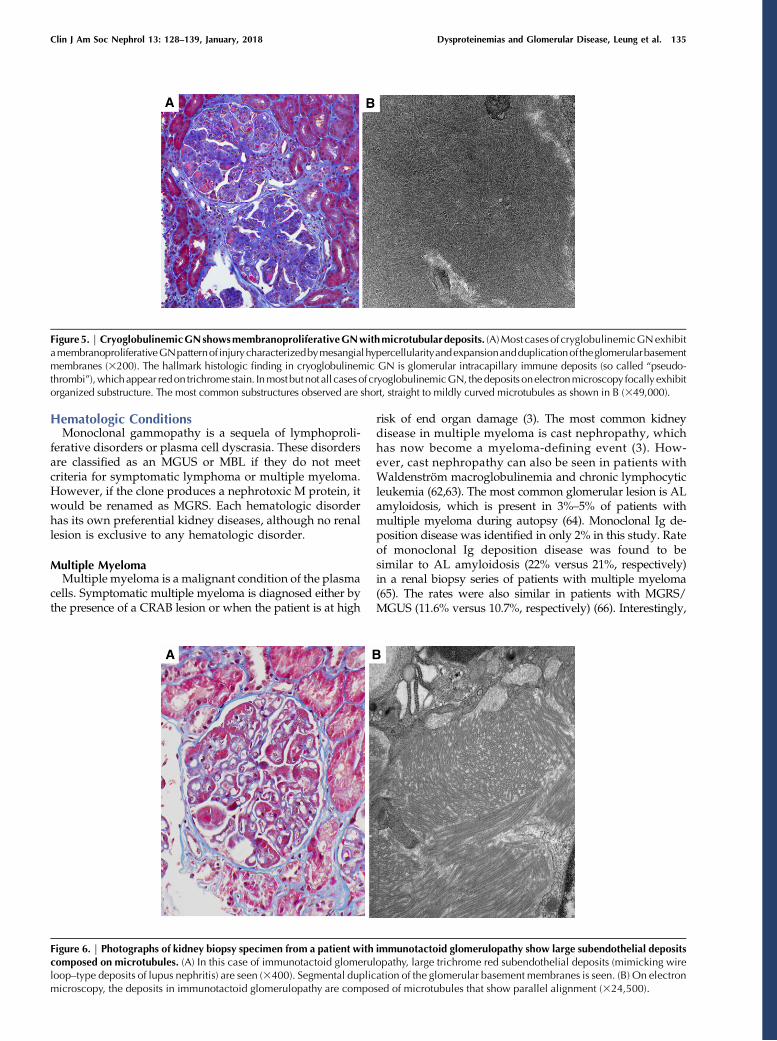
risk of end organ damage (3). The most common kidneydisease in multiple myeloma is cast nephropathy, whichhas now become a myeloma-defining event (3). How-ever, cast nephropathy can also be seen in patients withWaldenström macroglobulinemia and chronic lymphocyticleukemia (62,63). The most common glomerular lesion is ALamyloidosis, which is present in 3%–5% of patients withmultiple myeloma during autopsy (64). Monoclonal Ig de-position disease was identified in only 2% in this study. Rateof monoclonal Ig deposition disease was found to besimilar to AL amyloidosis (22% versus 21%, respectively)in a renal biopsy series of patients with multiple myeloma(65). The rates were also similar in patients with MGRS/MGUS (11.6% versus 10.7%, respectively) (66). Interestingly,
Figure5. | CryoglobulinemicGNshowsmembranoproliferativeGNwithmicrotubulardeposits. (A)Most casesof cryglobulinemicGNexhibitamembranoproliferativeGNpatternof injurycharacterizedbymesangialhypercellularityandexpansionandduplicationof theglomerularbasementmembranes (3200). The hallmark histologic finding in cryoglobulinemic GN is glomerular intracapillary immune deposits (so called “pseudo-thrombi”),whichappear redon trichromestain. Inmostbutnot all casesof cryoglobulinemicGN, thedeposits onelectronmicroscopy focallyexhibitorganized substructure. The most common substructures observed are short, straight to mildly curved microtubules as shown in B (349,000).
Figure 6. | Photographs of kidney biopsy specimen from a patient with immunotactoid glomerulopathy show large subendothelial depositscomposed on microtubules. (A) In this case of immunotactoid glomerulopathy, large trichrome red subendothelial deposits (mimicking wireloop–type deposits of lupus nephritis) are seen (3400). Segmental duplication of the glomerular basement membranes is seen. (B) On electronmicroscopy, the deposits in immunotactoid glomerulopathy are composed of microtubules that show parallel alignment (324,500).
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 135

cryoglobulinemic GN was seen in 16.5% of patients withMGRS/MGUS but has not been reported in patients withmultiple myeloma. FGN, immunotactoid GN, and light-chain proximal tubulopathy are rarely reportedwithmultiplemyeloma.
Lymphoplasmocytic Lymphoma/WaldenstromMacroglobulinemiaLymphoplasmocytic lymphoma is a lymphoproliferative
disorder that possesses both B cell (CD19) and plasma cell(CD38) characteristics. Waldenström macroglobulinemia isthe condition characterized by a serum monoclonal IgMproduced by the lymphoplasmocytic lymphoma. Morethan one set of diagnostic criteria exist for Waldenströmmacroglobulinemia. One requires .10% involvement oflymphoplasmocytic lymphoma in the bone marrow. Pa-tients with less lymphoplasmocytic lymphoma involvementare considered to have IgM MGUS, and those with .10%but no end organ damage are classified as smolderingWaldenström macroglobulinemia (67). In the past, denseeosinophilic occlusive capillary thrombi were commonlyfound (68). All patients with these endocapillary thrombihad either cryoglobulins or high concentrations of mono-clonal IgM. EM was not performed in this study. Two morerecent series, however, found that AIg amyloidosis wasthe most common renal lesion in Waldenström macro-globulinemia. Cryoglobulinemic GN had dropped to sec-ond (43,63). Reasons for this are unclear, but kidney biopsymay have been skipped if cryoglobulins were detected inthe blood. Cast nephropathy and monoclonal Ig depositiondisease were the next most common in one series, whereascast nephropathy and MPGN were the next most commonin another series. Intracapillary IgM deposits were muchless commonly found. Other lesions seen were thromboticmicroangiopathy and light-chain proximal tubulopathywith Fanconi syndrome. Lymphoplasmocytic lymphomainfiltration was common.
Chronic lymphocytic leukemiaChronic lymphocytic leukemia is the blood component
of the same disease represented by small lymphocyticlymphoma in the lymphoid tissue. MBL is the MGUSequivalent in chronic lymphocytic leukemia. Kidney dis-ease is less common in chronic lymphocytic leukemiathan in Waldenström macroglobulinemia or multiplemyeloma. A large study of 1043 patients newly diagnosedwith chronic lymphocytic leukemia/small lymphocyticlymphoma found that 10% of patients had renal impair-ment, whereas another 14.5% developed renal impairmentduring the course of their disease (69). Another study fromthe same institution found that only 1.2% of all patients withchronic lymphocytic leukemia or MBL (n54024)underwent a kidney biopsy (62). Despite the lower prev-alence, the variety of kidney diseases was similar to that ofmultiple myeloma and Waldenström macroglobulinemia.chronic lymphocytic leukemia infiltration was the mostcommon histopathologic finding, accounting for 73% ofpatients (70). The chronic lymphocytic leukemia infiltrationoften coexisted with other lesions, and granulomatousreaction was noted in some (62,71). CryoglobulinemiaGN (20%) was the most common glomerular lesion fol-lowed by minimal change disease (13.3%). Immunotactoid
GN, FGN, AHL amyloidosis, and MPGN each accountedfor 6.6%. In another study, MPGN was the most commonglomerular lesion (20%) followed by minimal changedisease (10%). Amyloidosis, membranous nephropathy,and mesangial proliferative GN were also reported alongwith TMA (12%) and light-chain cast nephropathy (7%).
Monoclonal Gammopathy of Renal SignificanceMonoclonal gammopathy of renal significance is a term
used to describe hematologic conditions that produce anephrotoxic M protein but do not meet criteria for symp-tomatic multiple myeloma or lymphoma (72). It differsfrom MGUS, which cannot have any end organ damage, bythe presence of the kidney disease. Like MGUS, however,monoclonal gammopathy of renal significance could arisefrom any clone from the B cell lineage. However, bydefinition, cast nephropathy could not be the result ofmonoclonal gammopathy of renal significance, because itis a myeloma-defining event (3). It is now recognized thatmost glomerular lesions associated with an M protein aredue to MGRS rather than its malignant counterpart (16). InAL amyloidosis and monoclonal Ig deposition disease,evidence suggests that patients with coexistence of multiplemyeloma have aworse prognosis comparedwith those withMGRS (73,74). This suggests that patients with kidneydiseases associated with MGRS may require differentmanagement from those secondary to multiple myeloma/Waldenström macroglobulinemia/chronic lymphocyticleukemia.
ManagementThe management of glomerular disease secondary to
monoclonal gammopathy has changed significantly sincethe introduction of the concept of MGRS (72,75). Treatmentis no longer decided by the renal histology but rather, isdecided by the nature of the clone producing the M protein.Initiation of therapy also no longer relies on presence ofmalignant characteristics. Traditional immunosuppressivedrugs and renin-angiotensin system blockade are generallyineffective at preventing ESRD (76). Clone-directed therapyis the key. Thus, clonal identification is extremely impor-tant in choosing the right treatment. The renal histology,however, remains useful for predicting the natural history,clinical features, and recurrence after kidney transplanta-tion. One should not forget about extrarenal involvement inpatients with AL amyloidosis and monoclonal Ig deposi-tion disease (17,21,42). Patients with severe heart involve-ment may need to have modified chemotherapy,aggressive fluid management, and care for CKD (77–79).The speed of recurrence after kidney transplant should betaken into account when deciding on transplantation(35,44,80). A good understanding of the differences canhelp select the best options for these patients.The most important drug in the treatment of glomerular
diseases associated with monoclonal gammopathy isbortezomib, a proteosome inhibitor. Other proteosomeinhibitors are now available, but bortezomib has themost robust data in the treatment of these diseases (81).Bortezomib represents the most important advance in thetreatment of AIg amyloidosis since the introductionof high-dose melphalan followed by autologous stem cell
136 Clinical Journal of the American Society of Nephrology

transplant, melphalan, and dexamethasone (82,83). In com-bination with cyclophosphamide and dexamethasone,bortezomib produced response rates as high as 94%, with66%–71% having a very good partial response (VGPR)(84,85). Achievement of VGPR, which is measured byeither a difference of the involved minus the uninvolvedsFLC of ,40 mg/L or .90% reduction of difference of theinvolved minus the uninvolved sFLC, has been found to beassociated with significant improvement in overall andrenal survival (86,87). Patients with complete response areless likely to have recurrence after kidney transplant (88).Bortezomib seems to be the most effective agent in mono-clonal Ig deposition disease (21,42,74,89). In one study,immunomodulatory drugs represented by thalidomide andlenalidomide were found to be less effective in achievingVGPR or better responses (21). Fortunately, the hematologicresponse can be improved by the addition of autologousstem cell transplant. Achievement of VGPR in monoclonal Igdeposition disease similarly has been found to improve renaland patient survival (21,42,74,89). Kidney transplant has beensuccessfully performed in patients with hematologic com-plete response without recurrence. A complete response isdefined by negative serum and urine immunofixation andnormal sFLC ratio. In multiple myeloma, it is confirmed by anegative bone marrow biopsy. More recently, achievement ofVGPR or better was found to be effective at preventing thedevelopment of ESRD in patients with C3 glomerulopathywith monoclonal gammopathy (76). In this study, 76% of thepatients used a bortezomib-based regimen, whereas 7%used a rituximab-based chemotherapy. Rituximab shouldbe the first choice in clones expressing strong CD20. Thecurrent evidence strongly supports the strategy of clone-directed therapy, and the goal of therapy should be ahematologic response of VGPR or better.Monoclonal gammopathy is a well recognized cause of
kidney disease. The monoclonal gammopathy is producedby a number of clones from the B cell lineage. Symptomaticmultiple myeloma or lymphoma is no longer required forthe development of kidney disease. However, because ofthe clonal nature of these diseases, clone-directed ther-apy is required for disease control. Multiple studies havefound that a VGPR is needed for preservation of kidneyfunction and improvement in patient survival. Achieve-ment of a complete hematologic response has been foundto reduce the risk of recurrence after kidney transplanta-tion. Given the complexity of these diseases, a multidis-ciplinary approach is preferred for the best outcome ofthese patients.
DisclosuresNone.
References1. Kyle RA,Durie BG, Rajkumar SV, LandgrenO, Blade J, Merlini G,
Kroger N, Einsele H, Vesole DH, Dimopoulos M, San Miguel J,Avet-Loiseau H, Hajek R, Chen WM, Anderson KC, Ludwig H,Sonneveld P, Pavlovsky S, PalumboA, Richardson PG, Barlogie B,Greipp P, Vescio R, Turesson I, Westin J, Boccadoro M; In-ternational MyelomaWorking Group: Monoclonal gammopathyof undetermined significance (MGUS) and smoldering (asymp-tomatic) multiple myeloma: IMWG consensus perspectives riskfactors for progression and guidelines for monitoring and man-agement. Leukemia 24: 1121–1127, 2010
2. Rajkumar SV, Dispenzieri A, Kyle RA: Monoclonal gammopathyofundetermined significance,Waldenstrommacroglobulinemia,AL amyloidosis, and related plasma cell disorders: Diagnosis andtreatment. Mayo Clin Proc 81: 693–703, 2006
3. Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G,Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P,LandgrenO,PaivaB,DispenzieriA,Weiss B, LeLeuX, ZweegmanS, Lonial S, Rosinol L, Zamagni E, Jagannath S, Sezer O,Kristinsson SY, Caers J, Usmani SZ, Lahuerta JJ, Johnsen HE,Beksac M, CavoM, Goldschmidt H, Terpos E, Kyle RA, AndersonKC, Durie BG,Miguel JF: International MyelomaWorking Groupupdated criteria for the diagnosis of multiple myeloma. LancetOncol 15: e538–e548, 2014
4. Kumar S, Paiva B, Anderson KC, Durie B, Landgren O, Moreau P,Munshi N, Lonial S, Blade J, MateosMV, DimopoulosM, KastritisE, Boccadoro M, Orlowski R, Goldschmidt H, Spencer A, Hou J,ChngWJ,UsmaniSZ,ZamagniE, ShimizuK, JagannathS, JohnsenHE, Terpos E, Reiman A, Kyle RA, Sonneveld P, Richardson PG,McCarthyP, LudwigH,ChenW,CavoM,Harousseau JL, LentzschS,Hillengass J, PalumboA,OrfaoA,Rajkumar SV,Miguel JS,Avet-Loiseau H: International Myeloma Working Group consensuscriteria for response and minimal residual disease assessment inmultiple myeloma. Lancet Oncol 17: e328–e346, 2016
5. Kumar S, Dispenzieri A, Lacy MQ, Hayman SR, Buadi FK, ColbyC, Laumann K, Zeldenrust SR, LeungN,Dingli D,Greipp PR, LustJA, Russell SJ, Kyle RA, Rajkumar SV, Gertz MA: Revised prog-nostic staging system for light chain amyloidosis incorporatingcardiac biomarkers and serum free light chain measurements.J Clin Oncol 30: 989–995, 2012
6. Merlini G, Bellotti V: Molecular mechanisms of amyloidosis. NEngl J Med 349: 583–596, 2003
7. Merlini G, Stone MJ: Dangerous small B-cell clones. Blood 108:2520–2530, 2006
8. Solomon A, Weiss DT, Kattine AA: Nephrotoxic potential ofBence Jones proteins. N Engl J Med 324: 1845–1851, 1991
9. GlaveySV,LeungN:Monoclonalgammopathy:Thegood, thebadand the ugly. Blood Rev 30: 223–231, 2016
10. Sanders PW, Booker BB: Pathobiology of cast nephropathy fromhuman Bence Jones proteins. J Clin Invest 89: 630–639, 1992
11. Sanders PW, Booker BB, Bishop JB, Cheung HC: Mechanisms ofintranephronal proteinaceous cast formation by low molecularweight proteins. J Clin Invest 85: 570–576, 1990
12. Klassen RB, Allen PL, Batuman V, Crenshaw K, Hammond TG:Light chains are a ligand for megalin. J Appl Physiol (1985) 98:257–263, 2005
13. HuangZQ,SandersPW:Biochemical interactionbetweenTamm-Horsfall glycoprotein and Ig light chains in the pathogenesis ofcast nephropathy. Lab Invest 73: 810–817, 1995
14. NakamotoY, ImaiH, Yasuda T,WakuiH,MiuraAB: A spectrumofclinicopathological features of nephropathy associated withPOEMS syndrome. Nephrol Dial Transplant 14: 2370–2378,1999
15. Sanders PW: Mechanisms of light chain injury along the tubularnephron. J Am Soc Nephrol 23: 1777–1781, 2012
16. Bridoux F, LeungN,Hutchison CA, TouchardG, Sethi S, FermandJP, Picken MM, Herrera GA, Kastritis E, Merlini G, Roussel M,Fervenza FC, Dispenzieri A, Kyle RA, Nasr SH; InternationalKidney and Monoclonal Gammopathy Research Group: Di-agnosis ofmonoclonal gammopathyof renal significance.KidneyInt 87: 698–711, 2015
17. Kyle RA, Gertz MA: Primary systemic amyloidosis: Clinical andlaboratory features in 474 cases. Semin Hematol 32: 45–59,1995
18. Buxbaum JN, Chuba JV, Hellman GC, Solomon A, Gallo GR:Monoclonal immunoglobulindepositiondisease: Light chainandlight and heavy chain deposition diseases and their relation tolight chainamyloidosis.Clinical features, immunopathology, andmolecular analysis. Ann Intern Med 112: 455–464, 1990
19. Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, D’Agati VD,LeungN:Renalmonoclonal immunoglobulin depositiondisease:A report of 64 patients from a single institution. Clin J Am SocNephrol 7: 231–239, 2012
20. PozziC,D’AmicoM, FogazziGB,Curioni S, Ferrario F, Pasquali S,Quattrocchio G, Rollino C, Segagni S, Locatelli F: Light chaindeposition disease with renal involvement: Clinical
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 137

characteristics and prognostic factors.Am J KidneyDis 42: 1154–1163, 2003
21. Sayed RH, Wechalekar AD, Gilbertson JA, Bass P, Mahmood S,SachchithananthamS, FontanaM, Patel K,WhelanCJ, LachmannHJ, Hawkins PN, Gillmore JD: Natural history and outcome oflight chain deposition disease. Blood 126: 2805–2810, 2015
22. Nasr SH, Satoskar A, Markowitz GS, Valeri AM, Appel GB, StokesMB, Nadasdy T, D’Agati VD: Proliferative glomerulonephritis withmonoclonal IgG deposits. J Am Soc Nephrol 20: 2055–2064, 2009
23. Rovin BH, Bou-Khalil P, Sedmak D: Pulmonary-renal syndromein a patient with fibrillary glomerulonephritis. Am J Kidney Dis22: 713–716, 1993
24. JenKY, FixOK, Foster EN, Laszik ZG, Ferrell LD:Monoclonal lightchain depositswithin the stomachmanifesting as immunotactoidgastropathy. Ultrastruct Pathol 39: 62–68, 2015
25. Wallner M, Prischl FC, Hobling W, Haidenthaler A, Regele H,Ulrich W, Kramar R: Immunotactoid glomerulopathy with ex-trarenal deposits in the bone, and chronic cholestatic liver dis-ease. Nephrol Dial Transplant 11: 1619–1624, 1996
26. Karras A, Noel LH, Droz D, Delansorne D, Saint-Andre JP,Aucouturier P, Alyanakian MA, Grunfeld JP, Lesavre P: Renalinvolvement inmonoclonal (type I) cryoglobulinemia: Two casesassociated with IgG3 kappa cryoglobulin. Am J Kidney Dis 40:1091–1096, 2002
27. BridouxF,Desport E, Fremeaux-BacchiV,ChongCF,Gombert JM,Lacombe C, Quellard N, Touchard G: Glomerulonephritis withisolated C3 deposits and monoclonal gammopathy: A fortuitousassociation? Clin J Am Soc Nephrol 6: 2165–2174, 2011
28. Lloyd IE,GallanA,HustonHK,RaphaelKL,MillerDV,ReveloMP,Khalighi MA: C3 glomerulopathy in adults: A distinct patientsubset showing frequent association with monoclonal gamm-opathy and poor renal outcome. Clin Kidney J 9: 794–799, 2016
29. Sethi S, Sukov WR, Zhang Y, Fervenza FC, Lager DJ, Miller DV,Cornell LD, Krishnan SG, Smith RJ: Dense deposit disease asso-ciated with monoclonal gammopathy of undetermined signifi-cance. Am J Kidney Dis 56: 977–982, 2010
30. Zand L, KattahA, Fervenza FC, Smith RJ, Nasr SH, Zhang Y, VranaJA, Leung N, Cornell LD, Sethi S: C3 glomerulonephritis associ-ated with monoclonal gammopathy: A case series. Am J KidneyDis 62: 506–514, 2013
31. Zipfel PF, Skerka C, Chen Q, Wiech T, Goodship T, Johnson S,Fremeaux-Bacchi V,Nester C, de Cordoba SR,NorisM, PickeringM, Smith R: The role of complement in C3 glomerulopathy. MolImmunol 67: 21–30, 2015
32. Soubrier M, Sauron C, Souweine B, Larroche C, Wechsler B,Guillevin L, Piette JC, Rousset H, Deteix P: Growth factors andproinflammatory cytokines in the renal involvement of POEMSsyndrome. Am J Kidney Dis 34: 633–638, 1999
33. Said SM, Sethi S, Valeri AM, Leung N, Cornell LD, Fidler ME,Herrera Hernandez L, Vrana JA, Theis JD, Quint PS, Dogan A, NasrSH: Renal amyloidosis: Origin and clinicopathologic correlationsof 474 recent cases. Clin J Am Soc Nephrol 8: 1515–1523, 2013
34. Gertz MA, LacyMQ, Dispenzieri A: Immunoglobulin light chainamyloidosis and the kidney. Kidney Int 61: 1–9, 2002
35. Pinney JH, Lachmann HJ, Sattianayagam PT, Gibbs SD,Wechalekar AD, Venner CP, Whelan CJ, Gilbertson JA,RowczenioD,HawkinsPN,Gillmore JD:Renal transplantation insystemic amyloidosis-importance of amyloid fibril type andprecursorproteinabundance.AmJTransplant13:433–441,2013
36. Nasr SH,SaidSM,Valeri AM, Sethi S, FidlerME,Cornell LD,GertzMA,Dispenzieri A, Buadi FK, Vrana JA, Theis JD,DoganA, LeungN: The diagnosis and characteristics of renal heavy-chain andheavy/light-chain amyloidosis and their comparison with renallight-chain amyloidosis. Kidney Int 83: 463–470, 2013
37. LeungN:Definingultrahigh-riskALamyloidosiswithVWF.Blood128: 320–322, 2016
38. Leung N, Nasr SH, Sethi S: How I treat amyloidosis: The impor-tance of accurate diagnosis and amyloid typing. Blood 120:3206–3213, 2012
39. Vrana JA,Gamez JD,MaddenBJ, Theis JD, Bergen 3rdHR,DoganA:Classificationofamyloidosisby lasermicrodissectionandmassspectrometry-based proteomic analysis in clinical biopsy speci-mens. Blood 114: 4957–4959, 2009
40. Ramirez-Alvarado M, Barnidge DR, Murray DL, Dispenzieri A,Marin-ArganyM,DickCJ,Cooper SA,Nasr SH,WardCJ,Dasari S,
Jimenez-ZepedaVH, LeungN:Assessment of renal responsewithurinary exosomes in patients with AL amyloidosis: A proof ofconcept. Am J Hematol 92: 536–541, 2017
41. Bridoux F, Javaugue V, Bender S, Leroy F, Aucouturier P,Debiais-Delpech C, Goujon JM, Quellard N, Bonaud A, ClavelM, Trouillas P, Di Meo F, Gombert JM, Fermand JP, Jaccard A,Cogne M, Touchard G, Sirac C: Unravelling the immuno-pathological mechanisms of heavy chain deposition diseasewith implications for clinicalmanagement.Kidney Int 91: 423–434, 2017
42. Cohen C, Royer B, Javaugue V, Szalat R, El Karoui K, Caulier A,Knebelmann B, Jaccard A, Chevret S, Touchard G, Fermand JP,Arnulf B, Bridoux F: Bortezomib produces high hematologicalresponse rates with prolonged renal survival in monoclonal im-munoglobulin deposition disease. Kidney Int 88: 1135–1143,2015
43. Chauvet S, Bridoux F, Ecotiere L, Javaugue V, Sirac C, Arnulf B,Thierry A, Quellard N, Milin S, Bender S, Goujon JM, Jaccard A,Fermand JP, Touchard G: Kidney diseases associated withmonoclonal immunoglobulin M-secreting B-cell lymphoproli-ferativedisorders:A case series of 35patients.AmJKidneyDis66:756–767, 2015
44. Leung N, Lager DJ, Gertz MA, Wilson K, Kanakiriya S, FervenzaFC: Long-term outcome of renal transplantation in light-chaindeposition disease. Am J Kidney Dis 43: 147–153, 2004
45. Alexander MP, Nasr SH, Watson DC, Mendez GP, Rennke HG:Renalcrescenticalphaheavychaindepositiondisease:A reportof3 cases and reviewof the literature.Am JKidneyDis58: 621–625,2011
46. Bhutani G, Nasr SH, Said SM, Sethi S, Fervenza FC, Morice WG,Kurtin PJ, Buadi FK,DingliD,Dispenzieri A,GertzMA, LacyMQ,Kapoor P, Kumar S, Kyle RA, Rajkumar SV, LeungN:Hematologiccharacteristics of proliferative glomerulonephritides with non-organized monoclonal immunoglobulin deposits. Mayo ClinProc 90: 587–596, 2015
47. Nasr SH, Markowitz GS, Stokes MB, Seshan SV, Valderrama E,Appel GB, Aucouturier P, D’Agati VD: Proliferative glomerulo-nephritis with monoclonal IgG deposits: A distinct entity mim-icking immune-complex glomerulonephritis. Kidney Int 65:85–96, 2004
48. Batal I, Markowitz GS, Wong W, Avasare R, Mapara MY, AppelGB, D’Agati VD: Filgrastim-induced crescentic transformation ofrecurrent IgG2l GN. J Am Soc Nephrol 27: 1911–1915, 2016
49. Ramos-Casals M, Stone JH, Cid MC, Bosch X: The cry-oglobulinaemias. Lancet 379: 348–360, 2012
50. Zaidan M, Plasse F, Rabant M, Javaugue V, Knebelmann B,AlyanakianMA, Joly D, Nochy D, Bridoux F: [Renal involvementduring type1cryoglobulinemia].Nephrol Ther12[Suppl 1]: S71–S81, 2016
51. Fogo AB, Lusco MA, Najafian B, Alpers CE: AJKD atlas of renalpathology: Cryoglobulinemic glomerulonephritis. Am J KidneyDis 67: e5–e7, 2016
52. Larsen CP, Messias NC, Walker PD, Fidler ME, Cornell LD,Hernandez LH, Alexander MP, Sethi S, Nasr SH: Mem-branoproliferative glomerulonephritis with masked monotypicimmunoglobulin deposits. Kidney Int 88: 867–873, 2015
53. Nasr SH, Fidler ME, Cornell LD, Leung N, Cosio FG, Sheikh SS,Amir AA, Vrana JA, Theis JD, Dogan A, Sethi S: Immunotactoidglomerulopathy: Clinicopathologic and proteomic study.Nephrol Dial Transplant 27: 4137–4146, 2012
54. BridouxF,HugueV,ColdefyO,Goujon JM,BauwensM,SechetA,Preud’Homme JL, Touchard G: Fibrillary glomerulonephritis andimmunotactoid (microtubular) glomerulopathy are associatedwith distinct immunologic features. Kidney Int 62: 1764–1775,2002
55. Singh K: Immunotactoid microtubular corneal deposits in bi-lateral paraprotein crystalline keratopathy. Cornea 28: 829–831,2009
56. Strøm EH, Hurwitz N, Mayr AC, Krause PH, Mihatsch MJ:Immunotactoid-like glomerulopathy with massive fibrillary de-posits in liverandbonemarrow inmonoclonal gammopathy.AmJNephrol 16: 523–528, 1996
57. Strati P, Shanafelt TD: Monoclonal B-cell lymphocytosis andearly-stage chronic lymphocytic leukemia: Diagnosis, naturalhistory, and risk stratification. Blood 126: 454–462, 2015
138 Clinical Journal of the American Society of Nephrology

58. Fogo AB, Lusco MA, Najafian B, Alpers CE: AJKD atlas of renalpathology: Immunotactoid glomerulopathy. Am J Kidney Dis 66:e29–e30, 2015
59. Nasr SH, Valeri AM, Cornell LD, Fidler ME, Sethi S, Leung N,Fervenza FC: Fibrillary glomerulonephritis: A report of 66 casesfrom a single institution.Clin J AmSocNephrol 6: 775–784, 2011
60. Javaugue V, Karras A, Glowacki F, McGregor B, Lacombe C,Goujon JM, Ragot S, Aucouturier P, TouchardG, Bridoux F: Long-term kidney disease outcomes in fibrillary glomerulonephritis: Acase series of 27 patients. Am J Kidney Dis 62: 679–690, 2013
61. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF,Offord JR,Dispenzieri A, Katzmann JA,Melton 3rd LJ: Prevalenceof monoclonal gammopathy of undetermined significance. NEngl J Med 354: 1362–1369, 2006
62. Strati P,NasrSH,LeungN,HansonCA,ChaffeeKG,SchwagerSM,Achenbach SJ, Call TG, Parikh SA,DingW,KayNE, Shanafelt TD:Renal complications in chronic lymphocytic leukemia andmonoclonal B-cell lymphocytosis: The mayo clinic experience.Haematologica 100: 1180–1188, 2015
63. Vos JM, Gustine J, Rennke HG, Hunter Z, Manning RJ, Dubeau TE,MeidK,MinnemaMC,KerstenMJ, TreonSP,Castillo JJ: Renal diseaserelated to Waldenstrom macroglobulinaemia: Incidence, pathologyand clinical outcomes. Br J Haematol 175: 623–630, 2016
64. IvanyiB:Frequencyof light chaindepositionnephropathy relativeto renal amyloidosis and Bence Jones cast nephropathy in anecropsy study of patients with myeloma. Arch Pathol Lab Med114: 986–987, 1990
65. Nasr SH, Valeri AM, Sethi S, FidlerME, Cornell LD,GertzMA, LacyM, Dispenzieri A, Rajkumar SV, Kyle RA, Leung N: Clinicopatho-logiccorrelations inmultiplemyeloma:Acase series of 190patientswith kidney biopsies. Am J Kidney Dis 59: 786–794, 2012
66. Paueksakon P, Revelo MP, Horn RG, Shappell S, Fogo AB:Monoclonal gammopathy: Significance and possible causality inrenal disease. Am J Kidney Dis 42: 87–95, 2003
67. KyleRA,BensonJT,LarsonDR,TherneauTM,DispenzieriA,KumarS,Melton 3rd LJ, Rajkumar SV: Progression in smolderingWaldenstrommacroglobulinemia:Long-termresults.Blood119:4462–4466,2012
68. Morel-Maroger L, Basch A, Danon F, Verroust P, Richet G: Pa-thology of the kidney in Waldenstrom’s macroglobulinemia.Study of sixteen cases. N Engl J Med 283: 123–129, 1970
69. Strati P, Chaffee KG, Achenbach SJ, Slager SL, Leung N, Call TG,DingW,ParikhSA,KayNE, Shanafelt TD:Renal insufficiency is anindependent prognostic factor in patients with chronic lympho-cytic leukemia. Haematologica 102: e22–e25, 2017
70. Poitou-Verkinder AL, Francois A, Drieux F, Lepretre S, LegallicierB, Moulin B, Godin M, Guerrot D: The spectrum of kidney pa-thology in B-cell chronic lymphocytic leukemia / small lym-phocytic lymphoma: A 25-year multicenter experience. PLoSOne 10: e0119156, 2015
71. Nasr SH,Shanafelt TD,HansonCA,FidlerME,Cornell LD,Sethi S,Chaffee KG, Morris J, Leung N: Granulomatous interstitial ne-phritis secondary to chronic lymphocytic leukemia/small lym-phocytic lymphoma. Ann Diagn Pathol 19: 130–136, 2015
72. LeungN,BridouxF,HutchisonCA,Nasr SH,Cockwell P, FermandJP, Dispenzieri A, Song KW, Kyle RA; International Kidney andMonoclonal Gammopathy Research Group: Monoclonalgammopathy of renal significance: When MGUS is no longerundetermined or insignificant. Blood 120: 4292–4295, 2012
73. Kourelis TV, Kumar SK, Gertz MA, Lacy MQ, Buadi FK, HaymanSR, Zeldenrust S, Leung N, Kyle RA, Russell S, Dingli D, Lust JA,LinY,KapoorP,RajkumarSV,McCurdyA,DispenzieriA:Coexistentmultiple myeloma or increased bone marrow plasma cells defineequally high-risk populations in patients with immunoglobulinlight chain amyloidosis. J Clin Oncol 31: 4319–4324, 2013
74. Kourelis TV, Nasr SH, Dispenzieri A, Kumar SK, Gertz MA,Fervenza FC, Buadi FK, LacyMQ, Erickson SB, Cosio FG, KapoorP, Lust JA, Hayman SR, Rajkumar V, Zeldenrust SR, Russell SJ,Dingli D, Lin Y, GonsalvesW, Lorenz EC, Zand L, Kyle RA, LeungN:Outcomes of patientswith renalmonoclonal immunoglobulindeposition disease. Am J Hematol 91: 1123–1128, 2016
75. Fermand JP, Bridoux F, Kyle RA, Kastritis E, Weiss BM, Cook MA,Drayson MT, Dispenzieri A, Leung N; International Kidney andMonoclonal Gammopathy Research Group: How I treat mono-clonal gammopathy of renal significance (MGRS). Blood 122:3583–3590, 2013
76. Chauvet S, Fremeaux-Bacchi V, Petitprez F, Karras A, Daniel L,Burtey S, Choukroun G, Delmas Y, Guerrot D, Francois A, LeQuintrecM, JavaugueV,RibesD,VrigneaudL,ArnulfB,Goujon JM,Ronco P, Touchard G, Bridoux F: Treatment of B-cell disorder im-proves renal outcome of patients with monoclonal gammopathy-associated C3 glomerulopathy. Blood 129: 1437–1447, 2017
77. MohtyD,Damy T, Cosnay P, Echahidi N, Casset-SenonD, Virot P,Jaccard A: Cardiac amyloidosis: Updates in diagnosis and man-agement. Arch Cardiovasc Dis 106: 528–540, 2013
78. Gertz MA: How to manage primary amyloidosis. Leukemia 26:191–198, 2012
79. GertzMA,LacyMQ,DispenzieriA,Kumar SK,DingliD, LeungN,HoganWJ,Buadi FK,HaymanSR:Refinement inpatient selectionto reduce treatment-related mortality from autologous stem celltransplantation inamyloidosis.BoneMarrowTransplant48:557–561, 2013
80. Nasr SH, Sethi S, Cornell LD, FidlerME, BoelkinsM, Fervenza FC,Cosio FG, D’Agati VD: Proliferative glomerulonephritis withmonoclonal IgG deposits recurs in the allograft. Clin J Am SocNephrol 6: 122–132, 2011
81. Lonial S, Boise LH: Current advances in novel proteasomeinhibitor-based approaches to the treatment of relapsed/refractory multiple myeloma. Oncology (Williston Park)25[Suppl 2]: 25–31, 2011
82. PalladiniG,PerfettiV,Obici L,CaccialanzaR, SeminoA,AdamiF,Cavallero G, Rustichelli R, Virga G, Merlini G: Association ofmelphalan and high-dose dexamethasone is effective and welltolerated in patients with AL (primary) amyloidosis who are in-eligible for stem cell transplantation. Blood 103: 2936–2938, 2004
83. Skinner M, Sanchorawala V, Seldin DC, Dember LM, Falk RH,Berk JL, Anderson JJ, O’Hara C, Finn KT, Libbey CA, Wiesman J,Quillen K, Swan N, Wright DG: High-dose melphalan and au-tologous stem-cell transplantation in patients with AL amyloid-osis: An 8-year study. Ann Intern Med 140: 85–93, 2004
84. Mikhael JR, Schuster SR, Jimenez-Zepeda VH, Bello N, SpongJ, Reeder CB, Stewart AK, Bergsagel PL, Fonseca R:Cyclophosphamide-bortezomib-dexamethasone (CyBorD)produces rapid and complete hematologic response inpatients with AL amyloidosis. Blood 119: 4391–4394, 2012
85. Venner CP, Lane T, Foard D, Rannigan L, Gibbs SD, Pinney JH,WhelanCJ, LachmannHJ,Gillmore JD,HawkinsPN,WechalekarAD: Cyclophosphamide, bortezomib, and dexamethasone ther-apy in AL amyloidosis is associated with high clonal responserates and prolonged progression-free survival. Blood 119: 4387–4390, 2012
86. Palladini G, Dispenzieri A, Gertz MA, Kumar S, Wechalekar A,Hawkins PN, Schonland S, Hegenbart U, Comenzo R, Kastritis E,Dimopoulos MA, Jaccard A, Klersy C, Merlini G: New criteria forresponse to treatment in immunoglobulin light chain amyloidosisbased on free light chain measurement and cardiac biomarkers:Impact on survival outcomes. J Clin Oncol 30: 4541–4549, 2012
87. Palladini G, Hegenbart U, Milani P, Kimmich C, Foli A, Ho AD,Vidus Rosin M, Albertini R, Moratti R, Merlini G, Schonland S: Astaging system for renal outcome and early markers of renal re-sponse to chemotherapy in AL amyloidosis. Blood 124: 2325–2332, 2014
88. Herrmann SM, Gertz MA, Stegall MD, Dispenzieri A, Cosio FC,Kumar S, LacyMQ,Dean PG, Prieto M, Zeldenrust SR, Buadi FK,Russell SJ, Nyberg SL, Hayman SR, Dingli D, Fervenza FC, LeungN: Long-term outcomes of patients with light chain amyloidosis(AL) after renal transplantation with or without stem cell trans-plantation. Nephrol Dial Transplant 26: 2032–2036, 2011
89. Ziogas DC, Kastritis E, Terpos E, Roussou M, Migkou M,Gavriatopoulou M, Spanomichou D, Eleutherakis-PapaiakovouE, Fotiou D, Panagiotidis I, Kafantari E, Psimenou E, Boletis I,Vlahakos DV, Gakiopoulou H, Matsouka C, Dimopoulos MA:Hematologic and renal improvement of monoclonal immuno-globulin deposition disease after treatment with bortezomib-based regimens. Leuk Lymphoma 58: 1832–1839, 2017
Received: January 16, 2017 Accepted: September 20, 2017
Published online ahead of print. Publication date available at www.cjasn.org.
Clin J Am Soc Nephrol 13: 128–139, January, 2018 Dysproteinemias and Glomerular Disease, Leung et al. 139




![Dysproteinemias and Glomerular Disease - Loyola Medicine · Patients present with mild renal impairment (median serum creatinine [Scr] of 1.2 mg/dl) and nephrotic-range proteinuria](https://img.pdfslide.net/doc/110x75/5e4a71a71f8eca231e509ca4/dysproteinemias-and-glomerular-disease-loyola-medicine-patients-present-with-mild.jpg)






![Glomerular Function and Structure in Living Donors ... · glomerular filtration rate (SNGFR) and glomerular capillary hydraulic pressure (P GC)[3]. Further insights into glomerular](https://img.pdfslide.net/doc/110x75/5ed58c3d3f40d10acd516aa6/glomerular-function-and-structure-in-living-donors-glomerular-filtration-rate.jpg)

















