Embed Size (px)
Citation preview

oe VOL. 12, NO. 3, august 2013 15
the authors would like to dedicate this article to Dr. Robert Kyle of Rochester, Minnesota. Besides being a formidable clinician, Dr. Kyle has been intimately involved with monoclonal proteins for over 50 years. In addition to coining the terminology and accurately describing the natural history of mono-clonal gammopathy of undetermined significance,
Dr. Kyle was also the first to include descriptions of both smouldering multiple myeloma and benign Bence Jones proteinuria. Now in his 80s, Dr. Kyle still attends work at the Mayo Clinic hematology research laboratory every day and continues to lecture worldwide.
Feature
Literature-based followup recommendationsPart 7: Monoclonal gammopathy for the non-hematologist oncologistby Malcolm Brigden, MDCM, FRCPC, FACP and Christopher P. Venner MD, FRCPC
Malcolm Brigden, MDCM, FRCPC, FACP is Medical Director of the Jack ady Cancer Centre in Lethbridge, alberta, Clinical associate Professor of Medicine at the university of Calgary and associate adjunct Professor at the university of Lethbridge. Email: [email protected]. Christopher P. Venner, MD, FRCPC is a Hematologist at the Cross Cancer Institute, and Clinical assistant Professor at the university of alberta in Edmonton. Email: [email protected]
When AM I Most lIKely to enCounteR A PAtIent WIth A MonoClonAl gAMMoPAthy In My ClInICAl onCology PRACtICe?A monoclonal gammopathy is a monoclonal immunoglobulin (Ig) secreted by an abnormally expanded clone of plasma cells in an amount that can be visualized by serum protein electrophoresis (SPE) and/or immunofixation of the serum and/or urine.1,2 The monoclonal protein (M protein) can be whole (heavy and light chain Ig) or just free light chain (FLC). The incidental discovery of an M protein can occur in a wide variety of clinical practice settings.1-4 It is often discovered during routine outpatient blood testing when completing the workup for patients with an elevated sedimen-tation rate, asymptomatic rise in serum protein levels and/or hyper- or hypogammaglobulinemia. Additionally, patients who present with back pain, anemia, renal insufficiency, significant proteinuria, hypercalcemia, age-inappropriate osteopenia, osteolytic bone lesions or unexplained periph-eral neuropathy are often screened for the presence of an M protein.2-4 A list of medical conditions possibly associated with a monoclonal gammopathy is provided in Table 1, page 16.4 Very rarely, an M protein may be detected, only to completely disappear after several years of followup. This appears to be more frequent when the M protein is present only on immunofixation.6
IF My PAtIent Is FounD to hAVe An M-PRoteIn, WhAt FuRtheR InVestIgAtIon Is neeDeD?The laboratory should undertake SPE in all cases when hypo- or hypergammaglobulinemia is present, or when there are abnormally low serum levels of total immunoglobulins,
including individual Ig classes.1-3 In the case of hypergam-maglobulinemia, a first step is to identify if it is secondary to an M protein or actually polyclonal in nature, as in the context of an inflammatory state. Such screening can be effectively accomplished using SPE and immunofixation. When a localized band or spike is found on SPE, immuno-fixation is needed to confirm the presence of an M protein and also determine the associated heavy and light chain
ABstRACt
this article continues a series on literature-based followup recommendations for conditions commonly encoun-tered in general oncology practice. The accidental dis-
covery of a monoclonal gammopathy (monoclonal protein or M protein, M spike) becomes increasingly common with advanced patient age, affecting approximately 3% to 4% of those over age 50, 5% over age 65 and almost 10% of those 85 or older.1-4 Since adult oncology practice is largely con-cerned with individuals 60 years and older, all oncologists require some familiarity with the investigation, natural history, followup and timing of subspecialist referral of patients with monoclonal gammopathy of undetermined significance (MGUS). Earlier studies suggest that in over
50% of patients in whom an M protein was accidentally detected, no subsequent followup was actually undertaken.5 In order to facilitate an orderly approach, this article utilizes a question-based format to address the most common concerns that arise when oncologists are confronted with a monoclonal protein. Virtually no randomized data exist on this topic, so references are primarily literature-based consensus guidelines and recommendations.1-3
Key wordsMGUS, monoclonal gammopathy, M spike, M protein, Waldenström macroglobulinemia, multiple myeloma, smouldering multiple myeloma, systemic amyloidosis
16 oe VOL. 12, NO. 3, august 2013
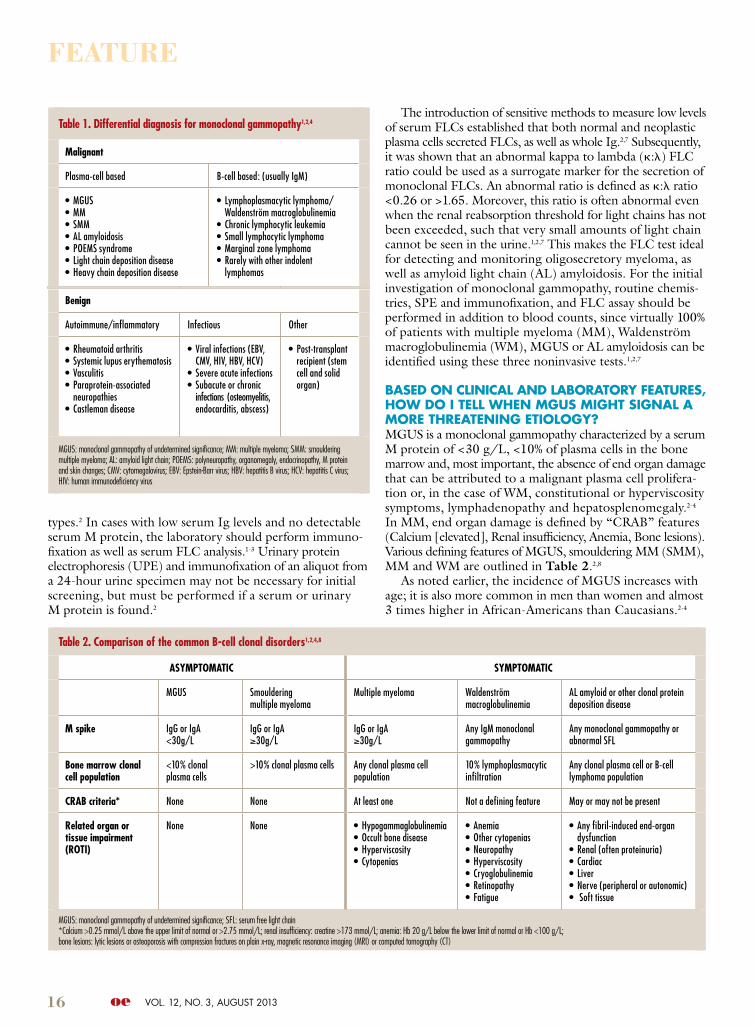
Table 2. Comparison of the common B-cell clonal disorders1,2,4,8
AsympTomATiC sympTomATiC
MGUS Smoulderingmultiple myeloma
Multiple myeloma Waldenströmmacroglobulinemia
AL amyloid or other clonal protein deposition disease
m spike IgG or IgA<30g/L
IgG or IgA≥30g/L
IgG or IgA≥30g/L
Any IgM monoclonal gammopathy
Any monoclonal gammopathy or abnormal SFL
Bone marrow clonal cell population
<10% clonal plasma cells
>10% clonal plasma cells Any clonal plasma cell population
10% lymphoplasmacytic infiltration
Any clonal plasma cell or B-cell lymphoma population
CRAB criteria* None None At least one Not a defining feature May or may not be present
Related organ or tissue impairment (RoTi)
None None • Hypogammaglobulinemia• Occult bone disease• Hyperviscosity• Cytopenias
• Anemia• Other cytopenias• Neuropathy• Hyperviscosity• Cryoglobulinemia• Retinopathy• Fatigue
• Any fibril-induced end-organ dysfunction
• Renal (often proteinuria)• Cardiac• Liver• Nerve (peripheral or autonomic)• Soft tissue
MGUS: monoclonal gammopathy of undetermined significance; SFL: serum free light chain*Calcium >0.25 mmol/L above the upper limit of normal or >2.75 mmol/L; renal insufficiency: creatine >173 mmol/L; anemia: Hb 20 g/L below the lower limit of normal or Hb <100 g/L; bone lesions: lytic lesions or osteoporosis with compression fractures on plain x-ray, magnetic resonance imaging (MRI) or computed tomography (CT)
Feature
types.2 In cases with low serum Ig levels and no detectable serum M protein, the laboratory should perform immuno-fixation as well as serum FLC analysis.1-3 Urinary protein electrophoresis (UPE) and immunofixation of an aliquot from a 24-hour urine specimen may not be necessary for initial screening, but must be performed if a serum or urinary M protein is found.2
The introduction of sensitive methods to measure low levels of serum FLCs established that both normal and neoplastic plasma cells secreted FLCs, as well as whole Ig.2,7 Subsequently, it was shown that an abnormal kappa to lambda (k:l) FLC ratio could be used as a surrogate marker for the secretion of monoclonal FLCs. An abnormal ratio is defined as k:l ratio <0.26 or >1.65. Moreover, this ratio is often abnormal even when the renal reabsorption threshold for light chains has not been exceeded, such that very small amounts of light chain cannot be seen in the urine.1,2,7 This makes the FLC test ideal for detecting and monitoring oligosecretory myeloma, as well as amyloid light chain (AL) amyloidosis. For the initial investigation of monoclonal gammopathy, routine chemis-tries, SPE and immunofixation, and FLC assay should be performed in addition to blood counts, since virtually 100% of patients with multiple myeloma (MM), Waldenström macroglobulinemia (WM), MGUS or AL amyloidosis can be identified using these three noninvasive tests.1,2,7
BAseD on ClInICAl AnD lABoRAtoRy FeAtuRes, hoW Do I tell When Mgus MIght sIgnAl A MoRe thReAtenIng etIology?MGUS is a monoclonal gammopathy characterized by a serum M protein of <30 g/L, <10% of plasma cells in the bone marrow and, most important, the absence of end organ damage that can be attributed to a malignant plasma cell prolifera-tion or, in the case of WM, constitutional or hyperviscosity symptoms, lymphadenopathy and hepatosplenomegaly.2-4 In MM, end organ damage is defined by “CRAB” features (Calcium [elevated], Renal insufficiency, Anemia, Bone lesions). Various defining features of MGUS, smouldering MM (SMM), MM and WM are outlined in Table 2.2,8
As noted earlier, the incidence of MGUS increases with age; it is also more common in men than women and almost 3 times higher in African-Americans than Caucasians.2-4
Table 1. Differential diagnosis for monoclonal gammopathy1,2,4
malignant
Plasma-cell based B-cell based: (usually IgM)
• MGUS• MM• SMM• AL amyloidosis• POEMS syndrome• Light chain deposition disease• Heavy chain deposition disease
• Lymphoplasmacytic lymphoma/Waldenström macroglobulinemia
• Chronic lymphocytic leukemia• Small lymphocytic lymphoma• Marginal zone lymphoma• Rarely with other indolent
lymphomas
Benign
Autoimmune/inflammatory Infectious Other
• Rheumatoid arthritis• Systemic lupus erythematosis• Vasculitis• Paraprotein-associated
neuropathies• Castleman disease
• Viral infections (EBV, CMV, HIV, HBV, HCV)
• Severe acute infections• Subacute or chronic
infections (osteomyelitis, endocarditis, abscess)
• Post-transplant recipient (stem cell and solid organ)
MGUS: monoclonal gammopathy of undetermined significance; MM: multiple myeloma; SMM: smouldering multiple myeloma; AL: amyloid light chain; POEMS: polyneuropathy, organomegaly, endocrinopathy, M protein and skin changes; CMV: cytomegalovirus; EBV: Epstein-Barr virus; HBV: hepatitis B virus; HCV: hepatitis C virus; HIV: human immunodeficiency virus

oe VOL. 12, NO. 3, august 2013 17
Table 3. Natural history with risk of progression of mGUs and smouldering multiple myeloma
Type Category Risk of progression up to 20 years
References
IgG, IgA, MGUS
Entire group
Subclassification by # of risk factors*
1% per year risk of progression to multiple myeloma, AL amyloidosis or related disorder.
Low (0) 5%Low-Intermediate (1) 21%Intermediate (2) 37%High (3) 58%
2,3,10
IgM, MGUS Entire group 1.5% per year risk of progression to Waldenström macroglobulinemia; rare patients can progress to IgM multiple myeloma.
9,10
Smouldering multiple myeloma
Entire group 10% per year risk of progression to multiple myeloma or related disorder for the first 5 years. Risk decreases to 3% per year for the next 5 years and 1% per year thereafter.
8,10,19
*Potential risk factors: (1) quantity of M protein >15g/L; (2) non-IgG monoclonal protein; (3) abnormal serum free light chain ratio
Feature
While the precise etiology of MGUS remains unknown, as in the case of MM, there is a higher incidence in agricultural workers and it is associated with exposure to insecticides, herbicides and fungicides.2-4 Radiation exposure also repre-sents a risk factor and there is familial tendency with a 3.3-fold increased risk in relatives of a propositus.1-3 Interestingly, although conventional cytogenetics rarely detect MGUS with fluorescence in situ hybridization (FISH) techniques, the same cytogenetic abnormalities typical of MM are pres-ent in a high proportion of patients. This suggests that a multistep process is required, with the accumulation of “genetic hits” and progressive alteration and deregulation of the bone marrow microenvironment, leading to a more clonally permissive niche.2,3,8
WhAt ClInICAl FeAtuRes WIll AlloW Me to PRognostICAte oR RIsK-stRAtIFy My PAtIents WIth Mgus?Longitudinal studies have demonstrated that virtually all cases of MM are preceded by MGUS, with a median duration of more than 10 years.3,4,8 Patients were found to be at risk for progression even after 25 years of followup.2,3 However, it has also been recognized that, despite an average overall risk for progression of 1% per year, not all patients with MGUS ultimately evolve to a symptomatic B-cell clonal disorder.1-3 This finding prompted considerable research to discover which factors might help stratify the risk for progression. The Spanish group identified a preponderance of phenotypically abnormal plasma cells on bone marrow flow cytometry as being asso-ciated with significant risk for progression.4,8 Such an approach requires both invasive and adequate bone marrow sampling.
In North America, after many years of detailed followup, Kyle and associates identified 3 noninvasive, easily quantifi-able variables that allowed stratification of patients into 4 groups for predicting yearly risk of progression: size of M protein >15 g/L, presence of IgA or IgM protein (i.e. non-IgG), and presence of abnormal FLC ratio.3,8 Other studies have defined similar data for SMM and IgM MGUS.2-4 The presence of more than 5% plasma cells on bone marrow aspiration biopsy was also identified as an independent risk factor for progression, but this measurement requires an invasive procedure.3,4 Interestingly, neither the type nor pres-ence of urinary light chain nor the presence of reduced levels of the noninvolved immunoglobulins (each abnormality pre-senting in approximately 30% of newly diagnosed MGUS) turned out to be predictive for later progression.2-4
Low-risk patients, with none of the 3 risk factors, have an absolute risk of progression to a plasma proliferative dis-order at 20 years of only 5% (Table 3). Approximately 70% of new incidentally discovered MGUS patients fall into this category. The low-intermediate group with one abnormal variable has a progression risk of 21%. Intermediate MGUS patients with 2 abnormal variables have a progression risk of 37%. The high-risk group, possessing all 3 parameters, has a 58% risk of progressing within 20 years.3,4
WhAt FolloWuP testIng shoulD Be unDeR-tAKen AnD hoW oFten shoulD sKeletAl suRVeys Be PeRFoRMeD DuRIng FolloWuP?In studying the patterns and rates of progression of MGUS over 40 years, Kyle and colleagues made a number of inter-esting observations.3,10 First, risk for progression persisted even after 30 to 40 years of followup. Among those who progressed, median time to progression was around 5 years and overall progression risk was significantly higher in the first 5 years.3,8,10 However, there was no uniformly predictable pattern for progression and only 50% of patients ultimately developed MM, superimposed on a steady yearly progressive rise in monoclonal spike. In fact, 4 different modes of evo-lution were identified: rapid initial increase, initial stability followed by rapid increase, slow but steady increase from baseline, or apparent stability for a number of years followed by the development of lytic lesions, anemia or renal insuffi-ciency/progressive M protein proteinuria. The identification of this last group makes it essential for MGUS followup to include not only laboratory testing but also periodic history and physical exam.3,10 In a series of patients who received optimal followup, changes in laboratory parameters led to the diagnosis of MM or WM in only 16%, while de novo typical clinical presentations of myeloma-related complications (bone pain/fractures, anemia/fatigue, hypercalcemia, renal failure) led to diagnosis in 45%, and 25% were diagnosed following workup for less serious symptoms.11
With regards to serial M protein SPE, UPE or FLC deter-minations, it is important for clinicians to realize that inter-laboratory variation can be as high as 25%, while the biologic coefficient of variation is around 8% for SPE but approaches 30% for FLC or UPE. This means that, for a change to be truly significant between 2 serial observations in SPE, a 30%
18 oe VOL. 12, NO. 3, august 2013
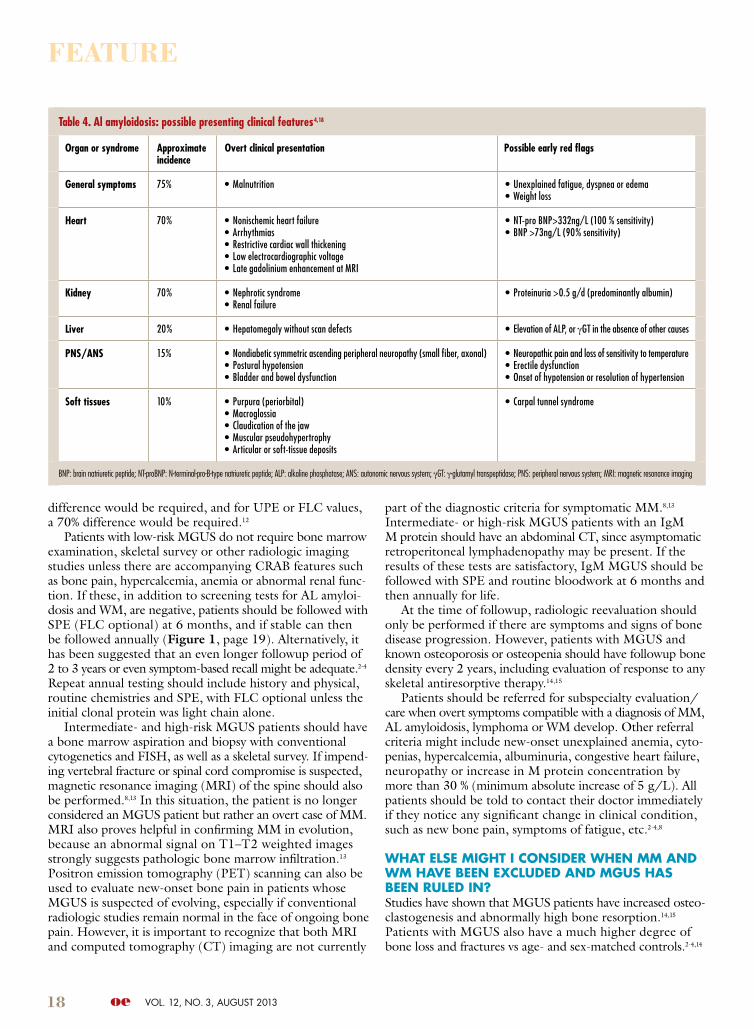
Table 4. Al amyloidosis: possible presenting clinical features4,18
organ or syndrome Approximate incidence
overt clinical presentation possible early red flags
General symptoms 75% • Malnutrition • Unexplained fatigue, dyspnea or edema• Weight loss
Heart 70% • Nonischemic heart failure• Arrhythmias• Restrictive cardiac wall thickening• Low electrocardiographic voltage• Late gadolinium enhancement at MRI
• NT-pro BNP>332ng/L (100 % sensitivity)• BNP >73ng/L (90% sensitivity)
Kidney 70% • Nephrotic syndrome• Renal failure
• Proteinuria >0.5 g/d (predominantly albumin)
Liver 20% • Hepatomegaly without scan defects • Elevation of ALP, or γGT in the absence of other causes
pNs/ANs 15% • Nondiabetic symmetric ascending peripheral neuropathy (small fiber, axonal)• Postural hypotension• Bladder and bowel dysfunction
• Neuropathic pain and loss of sensitivity to temperature• Erectile dysfunction• Onset of hypotension or resolution of hypertension
soft tissues 10% • Purpura (periorbital)• Macroglossia• Claudication of the jaw• Muscular pseudohypertrophy• Articular or soft-tissue deposits
• Carpal tunnel syndrome
BNP: brain natriuretic peptide; NT-proBNP: N-terminal-pro-B-type natriuretic peptide; ALP: alkaline phosphatase; ANS: autonomic nervous system; γGT: γ-glutamyl transpeptidase; PNS: peripheral nervous system; MRI: magnetic resonance imaging
Feature
difference would be required, and for UPE or FLC values, a 70% difference would be required.12
Patients with low-risk MGUS do not require bone marrow examination, skeletal survey or other radiologic imaging studies unless there are accompanying CRAB features such as bone pain, hypercalcemia, anemia or abnormal renal func-tion. If these, in addition to screening tests for AL amyloi-dosis and WM, are negative, patients should be followed with SPE (FLC optional) at 6 months, and if stable can then be followed annually (Figure 1, page 19). Alternatively, it has been suggested that an even longer followup period of 2 to 3 years or even symptom-based recall might be adequate.2-4 Repeat annual testing should include history and physical, routine chemistries and SPE, with FLC optional unless the initial clonal protein was light chain alone.
Intermediate- and high-risk MGUS patients should have a bone marrow aspiration and biopsy with conventional cytogenetics and FISH, as well as a skeletal survey. If impend-ing vertebral fracture or spinal cord compromise is suspected, magnetic resonance imaging (MRI) of the spine should also be performed.8,13 In this situation, the patient is no longer considered an MGUS patient but rather an overt case of MM. MRI also proves helpful in confirming MM in evolution, because an abnormal signal on T1–T2 weighted images strongly suggests pathologic bone marrow infiltration.13 Positron emission tomography (PET) scanning can also be used to evaluate new-onset bone pain in patients whose MGUS is suspected of evolving, especially if conventional radiologic studies remain normal in the face of ongoing bone pain. However, it is important to recognize that both MRI and computed tomography (CT) imaging are not currently
part of the diagnostic criteria for symptomatic MM.8,13 Intermediate- or high-risk MGUS patients with an IgM M protein should have an abdominal CT, since asymptomatic retroperitoneal lymphadenopathy may be present. If the results of these tests are satisfactory, IgM MGUS should be followed with SPE and routine bloodwork at 6 months and then annually for life.
At the time of followup, radiologic reevaluation should only be performed if there are symptoms and signs of bone disease progression. However, patients with MGUS and known osteoporosis or osteopenia should have followup bone density every 2 years, including evaluation of response to any skeletal antiresorptive therapy.14,15
Patients should be referred for subspecialty evaluation/care when overt symptoms compatible with a diagnosis of MM, AL amyloidosis, lymphoma or WM develop. Other referral criteria might include new-onset unexplained anemia, cyto-penias, hypercalcemia, albuminuria, congestive heart failure, neuropathy or increase in M protein concentration by more than 30 % (minimum absolute increase of 5 g/L). All patients should be told to contact their doctor immediately if they notice any significant change in clinical condition, such as new bone pain, symptoms of fatigue, etc.2-4,8
WhAt else MIght I ConsIDeR When MM AnD WM hAVe Been exCluDeD AnD Mgus hAs Been RuleD In?Studies have shown that MGUS patients have increased osteo-clastogenesis and abnormally high bone resorption.14,15 Patients with MGUS also have a much higher degree of bone loss and fractures vs age- and sex-matched controls.2-4,14

oe VOL. 12, NO. 3, august 2013 19
Figure 1. management of patient with monoclonal protein on serum protein electophresis, positive immunofixation or abnormal serum free light chain ratio1-4
CBC, Ca2+, creatine, albumin, LFTs; serum immunoglobulins;
serum immunofixation; SFLC; urinalysis (protein/creatine ratio)
MGUS
CRAB features absent*
Assess patient for any symptoms or signs of AL amyloidosis.
If absent, consider M protein class.
M protein < 30 g/L, abnormal SFLC ratio or positive immunofixation
Low-intermediate, intermediate and high risk
BM aspirate and biopsy;skeletal survey
On repeat visit: history, physical exam, CBC, Ca2+, creatine, SPE
CRAB features present*
Bone imaging studies; BM aspirate & biopsy.
Evaluate for MM, WM and subspecialist referral
Lymphadenopathy; hepatosplenomegally;
constitutional or hyperviscosity symptoms present
WM: Waldenström macroglobulinemia; MGUS monoclonal gammopathy
of undetermined significance; MM: multiple myeloma;
BM: bone marrow; SFLC: serum free light chain;
SMP: serum monoclonal protein; SPE: serum protein
electrophoresis.
*CRAB features: hypercalcemia, renal failure, anemia,
bone lesions.‡Repeat visits at more frequent
intervals if M protein value is progressively increasing.
SMP ≥ 15 g/L and/or FLC
ratio abnormal
Repeat visit in 6 months and yearly thereafter‡
SMP < 15 g/L and FLC ratio
normal
SMP < 15 g/L and FLC ratio
normal
Repeat visit in 6 months and 2-3 years thereafter‡
IgG, IgA, M protein
IgG
IgM, M protein
IgA
Low risk
Feature
There is an increased likelihood of developing fractures, espe-cially in the vertebral column or hips.14,15 For this reason, both the UK Myeloma Forum and Nordic Myeloma Study Group have recommended that anyone with age-inappro-priate bone loss or vertebral compression fractures without a definitive known risk factor should undergo plasma cell dyscrasia screening.1-3 Similarly, MGUS patients with T scores <–1.0 should maintain a daily oral intake of 1000 IU vita-min D as well as 1000–1500 mg of calcium.2,3 Those with frank osteoporosis should be treated with bisphosphonates and/or other appropriate therapy.14,15
There is an increased risk of peripheral neuropathy with MGUS and various expert consensus groups have also sug-gested that those presenting with idiopathic peripheral neu-ropathy should be screened for MGUS. The etiology varies and includes AL amyloidosis, anti-myelin-associated glyco-protein (anti-MAG) antibodies in the context of WM and potentially as bystander clonal expansion of autoimmunity. With screening, approximately 10% of patients presenting with peripheral neuropathy will be found to have an M protein.2-4 Nevertheless, population-based cohort studies have suggested that MGUS patients may not have a higher risk of developing peripheral neuropathy, and most associated cases are unlikely to be caused by the MGUS itself.16 In the case of IgM MGUS, patients with IgM-related neuropathy often possess serum anti-MAG antibodies and may respond to appropriate ther-apies such as rituximab, plasmapheresis or cytotoxic agents.16 If this is present, however, the patient is no longer considered to have MGUS and a more complete workup for lympho-plasmatic lymphoma, including a bone marrow examination, should be initiated.
Other studies have shown that MGUS patients are also more likely to develop thromboembolic events such as deep vein thrombosis (DVT).17 One investigation determined that the relative risk for DVT with MGUS was 3.3 and that the greatest risk existed during the first year post diagnosis.2,3,17 Despite this finding, there is no evidence that any prophy-lactic measures should be taken.
When shoulD I ConsIDeR the PossIBIlIty oF Al systeMIC AMyloIDosIs In A PAtIent WIth An M PRoteIn, AnD WhAt FuRtheR testIng shoulD Be unDeRtAKen In susPeCt CAses?In AL amyloidosis, the abnormal monoclonal light chain (frequently l) causes systemic proteotoxicity by misfolding, aggregating in fibrils, and interacting with and depositing in various tissues.4,8,18 Definitive diagnosis requires the pathologic identification of amyloid deposition.18 Surrogate tissue that is easier to obtain (e.g. bone marrow biopsy or fat aspirate) can often be used, but if testing for these proves negative, tissue from the affected organ(s) is required. As part of a workup for newly discovered M protein, we strongly encourage routine Congo Red staining on any bone marrow biopsy. Importantly, since AL amyloidosis is commonly associated with a low-level monoclonal gammo-pathy, it is imperative that symptoms and signs typically associated with AL amyloidosis be actively pursued with M proteins of any level.18 In addition, the clonal protein
most often present is FLC alone. Thus, if AL amyloidosis is suspected, an FLC assay should be ordered in addition to UPE and SPE. Finally, it is important to confirm the fibril subtype to ensure AL amyloid is the culprit once Congo Red positivity is demonstrated. Up to 10% of patients with amyloidosis and a monoclonal gammo pathy will not have AL amyloidosis. In these cases, the fibril is unrelated to any clonal disorder (and thus the MGUS) and chemotherapy is contraindicated.
The clinical presentation in amyloidosis depends on the organ system predominantly involved, and the differential diagnosis is based on early recognition of organ damage (Table 4, page 18). For instance, in any patient with low-level

20 oe VOL. 12, NO. 3, august 2013
References1. Berenson JR, Anderson KC, Audell RA et al. Monoclonal gammopathy of undetermined
significance: a consensus statement. Br J Haematol 2010;150:28-38.2. Bird J, Behrens J, Westin J et al. UK Myeloma Forum (UKMF) and Nordic Myeloma
Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undermined significance (MGUS). Br J Haematol 2009;147:22-42.
3. Kyle RA, Buadi F, Rajkumar SV. Management of monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM). Oncology 2011;25:578-86.
4. Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undeter-mined significance. Hematology Am Soc Hematol Educ Program 2012;2012:595-603.
5. Vladutiu A. Prevalence of M-proteins in serum of hospitalized patients. Ann Clin Lab Sci 1987;17:157-61.
6. Murray DL, Seningen JL, Dispenzieri A et al. Laboratory persistence and clinical progression of small monoclonal abnormalities. Am J Clin Pathol 2012;138:609-13.
7. Tosi P, Tomassetti S, Merli A, Polli V. Serum free light-chain assay for the detection and monitoring of multiple myeloma and related conditions. Ther Adv Hem 2013;4:37-41.
8. Bianchi G, Ghobrial I. Does my patient with a serum monoclonal spike have multiple myeloma? Hematol Oncol Clin North Am 2012;26:383-93.
9. Kyle RA, Therneau TM, Dispenzieri A et al. Immunoglobulin M monoclonal gammopathy of undetermined significance and smoldering waldenström macroglobulinemia. Clin Lymphoma Myeloma Leuk 2013;13:184-6.
10. Kyle RA, Rajkumar SV. Monoclonal gammopathies of undetermined significance: a review. Immunol Rev 2003;194:112-39.
11. Bianchi G, Kyle RA, Colby CL et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019-25.
12. Katzmann JA, Snyder MR, Rajkumar SV et al. Long-term biological variation of serum protein electrophoresis M-spike, urine M-spike, and monoclonal serum free light chain quantification: implications for monitoring monoclonal gammopathies. Clin Chem 2011;57:1687-92.
13. Walker R, Barlogie B, Haessler J et al. Magnetic resonance imaging in multiple myeloma: diagnostic and clinical implications. J Clin Oncol 2007;25:1121-8.
14. Kristinsson SY, Tang M, Pfeiffer RM et al. Monoclonal gammopathy of undetermined significance and risk of skeletal fractures; a population based study. Blood 2010;116:2651-5.
15. Berenson JR, Yellin O, Boccia RV et al. Zoledronic acid markedly improves bone mineral density for patients with monoclonal gammopathy of undetermined significance and bone loss. Clin Cancer Res 2008;14:6289-95.
16. Rajabally YA. Neuropathy and paraproteins: review of a complex association. Eur J Neurol 2011;18:1291-8.
17. Cohen AL, Sarid R. The relationship between monoclonal gammopathy of undetermined significance and venous thromboembolic disease. Thromb Res 2010;125:216-9.
18. Rosenzweig M, Landau H. Light chain (AL) amyloidosis: update on diagnosis and management. J Hematol Oncol 2011;10:4-47.
19. Korde N, Kristinsson SY, Landgren O. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering multiple myeloma (SMM): novel biological insights and development of early treatment strategies. Blood 2011;117:5573-81.
20. Mateos MV, López-Corral L, Hernández M et al. Smoldering multiple myeloma (SMM) at high-risk of progression to symptomatic disease: a phase III, randomized, multicenter trial based on lenalidomide-dexamethasone (len-dex) as induction therapy followed by maintenance therapy with len alone vs no treatment. Blood (ASH Annual Meeting Abstracts) 2011;118:Abstract 911.
Feature
proteinuria and renal failure, or alternatively, heavy proteinuria and nephrotic syndrome, renal biopsy should be strongly considered. Hepatitis and liver failure related to amyloid deposition can initially present with nonspecific symptoms such as asthenia and unintentional weight loss with eleva-tions in alkaline phosphatase. Gastrointestinal (GI) tract involvement can present with gut motility problems, mal-absorption, diarrhea or recurrent GI bleeding.4,18 Peripheral neuropathy including bilateral carpal tunnel syndrome can frequently occur with amyloidosis and is typically sensory in nature. Autonomic neuropathy manifested by postural hypotension or abnormal gut motility is also not infrequent.18 A high index of suspicion/early diagnosis is especially important with cardiac amyloidosis since by the time symp-toms are evident, irreversible cardiac damage has frequently occurred.4,18 With cardiac amyloidosis, both conduction system and pump function may be affected, producing arrhythmias or heart failure. Troponin T and the N-terminal fragment of pronatriuretic peptide (NT-Pro-BNP) represent sensitive markers of cardiac dysfunction in AL amyloidosis, with increases seen several months before symptoms appear or classical echocardiographic features are detected.4 Unfor-tunately, elevations may not be specific and may also be seen with other cardiac diseases. Since almost all patients with cardiac amyloidosis also have an abnormal FLC ratio, a higher index of suspicion for occult amyloidosis has been suggested for all MGUS patients with this abnormality.7
WhAt FutuRe tRenDs RegARDIng MonoClonAl gAMMoPAthy AnD Mgus WIll Be ReleVAnt to the PRACtIsIng ClInICAl onCologIst?Since over 70% of patients with incidentally discovered MGUS present in the lowest-risk category and the condition itself occurs predominantly in older individuals, it is probable that more than 90% of MGUS patients diagnosed today will never develop MM or a malignant plasmaproliferative disorder.2-4 Thus, the recent literature justifies a risk-adapted approach to followup of MGUS using the 3 easily quantifiable estab-lished risk factors: non-IgG type M protein; serum mono-clonal spike >15 g/L; and abnormal FLC ratio.4,19 In addition, the majority of low-risk category MGUS patients may not require annual followup and can be seen at less frequent intervals.19 Alternatively, increasing precision in defining a high-risk MGUS population may allow for future chemopre-vention or complication prevention trials, especially if more sensitive predictive biomarkers are discovered.2,19 A number of candidate agents for investigation in patients with high-risk MGUS or especially SMM have already been identified. A recent trial with lenalidomide and dexamethasone in SMM has yielded positive results.20 Studies with prophylactic zoledronic acid have demonstrated increases in bone den-sity in MGUS and a reduction in skeletal-related events at progression in MM, but without influence yet shown on the natural history of either disease.15
Disclosure: Dr. Brigden and Dr. Venner have previously received honoraria from Celgene and Janssen.





























