Embed Size (px)
Citation preview

79
I.はじめに
造血システムにおいては造血幹細胞とそれを支える微小環境の双方が重要な役割を果たしている。このどちらかの機能が破綻することにより,再生不良性貧血に代表される造血不全,あるいは逆に骨髄性白血病や骨髄増殖性腫瘍などの血液腫瘍が発症する。造血微小環境は,様々な造血サイトカインや接着分子およびこれらの分子を産生する骨髄間質細胞より構成される。加えて,最近ではこれらのすべてを取り囲む物理的な環境因子として酸素濃度の変化が注目されている。骨髄内が低酸素環境にあることは古くより知られており,また酸素濃度変化が造血
機構に影響を及ぼすことも 1980年代には報告がなされている。加えて,低酸素応答転写因子HIF(Hypoxia inducible factor)の同定など,低酸素応答の分子生物学的機構が解明されるとともに,造血発生における低酸素環境の重要性についての理解が深まっている。一方,その機能破綻あるいは暴走が血液疾患にどのように関与するかについての研究は端緒についたばかりである。本稿では,我々のこれまでの研究を踏まえつつ,正常および病的な造血における,低酸素環境ならびに低酸素応答機構が果たしている役割について述べてみたい。
II.生体の低酸素応答機構
(1)HIF:低酸素応答の司令塔 生物が低酸素状態に曝された場合には,その
山梨医科学誌 27(3),79~ 89,2013
低酸素応答制御と造血システム
桐 戸 敬 太山梨大学医学部血液・腫瘍内科教室
要 旨:骨髄はヒトの造血発生の場であり,そこには造血幹細胞とそれを支持する間質細胞が存在し,接着分子を介した直接的な細胞間コミュニケーションとサイトカインやケモカインなどの液性分子による情報伝達により,秩序だった造血発生制御が行われている。骨髄はその解剖学的な特性により,中心部から骨幹端へ向かって低下する特徴的な酸素分圧勾配を示す。この酸素分圧変化も,造血制御に重要な機能を持つ事が明らかになりつつある。最も酸素分圧の低い骨幹端領域は低酸素ニッチとも呼ばれ,ここに造血幹細胞が存在する。造血幹細胞は,低酸素ニッチに存在することにより,ミトコンドリアに依存しないエネルギー産生経路を発達させている。これにより,酸化的リン酸化に伴う活性酸素への暴露を抑え,自己複製能を維持し続けている。一方,骨髄内の低酸素環境は様々な血液腫瘍の発生や治療抵抗性の獲得にも関わっている。低酸素ニッチは白血病幹細胞の聖域として機能し,低酸素応答因子HIFは多発性骨髄腫や悪性リンパ腫など多くの血液腫瘍細胞で過剰に活性化され,骨髄内での異常な血管新生や腫瘍細胞の不死化をもたらす。骨髄内の低酸素環境とその応答機構の解明は,造血発生のさらなる理解と造血系腫瘍の新たな治療へのアプローチとして重要な意義を持つ。
キーワード 低酸素ニッチ,造血幹細胞,HIF,白血病幹細胞,糖代謝
総 説
〒 409-3898 山梨県中央市下河東 1110番地 受付:2012年 10月 9 日 受理:2012年 12月 19日

80 桐 戸 敬 太
環境に適応するために様々な反応が引き起こされる。個体レベルでは,赤血球の産生を増加させ,組織レベルでは血管の新生が促進される。個々の細胞についてみると,エネルギー産生経路が酸素依存性のミトコンドリアでの酸化的リン酸化から酸素非依存性の解糖系へとシフトすることが観察される。細胞の生存を維持するための,抗アポトーシス蛋白も誘導される。このような多彩な低酸素応答は,主にHIFにより制御されている 1)。HIFはHIF-1,HIF-2,およびHIF-3からなるファミリーであり,それぞれα サブユニット (HIF-1α,2α,3α )
とβ サブユニット(HIF-β)から構成される。HIF-β は ARNT(Aryl hydrocarbon Receptor
Nuclear Translocator)とも称し,共通のユニットである。また,その発現は酸素濃度に依存していない。これに対して,α サブニットはそれぞれに特異的であり,かつその発現は酸素濃度により調節されている。
(2)HIF-αの発現調節と酸素濃度変化 HIF-α は極めて半減期が短い蛋白である。通常の酸素分圧下では,HIF-α 蛋白のプロリン残基が,プロリン水酸化酵素(PHD; Prolyl
hydroxylase)により水酸化を受ける。このプロリン残基を水酸化された HIF-α には,Von
Hippel-Lindau蛋白を含むユビキチンリガーゼ複合体が結合する。これに伴いHIF-α のユビキチン化がおこり,引続いて HIF-α はプロテアゾームで分解される。低酸素環境下では,PHDの酵素活性が低下するため,この一連の反応が停止する。結果として,HIF-α の分解が抑制され,HIF-α と HIF-β との複合体形成が促進される。HIF-α と HIF-β の複合体は低酸素応答配列(HRE; Hypoxia responsive
element)に結合し,転写因子として機能する 1)
(図 1)。
(3)酸素濃度以外の因子による HIFの作用調節 酸素濃度変化はHIFの機能を左右する最も大きな要因であるが,この他にも種々の細胞
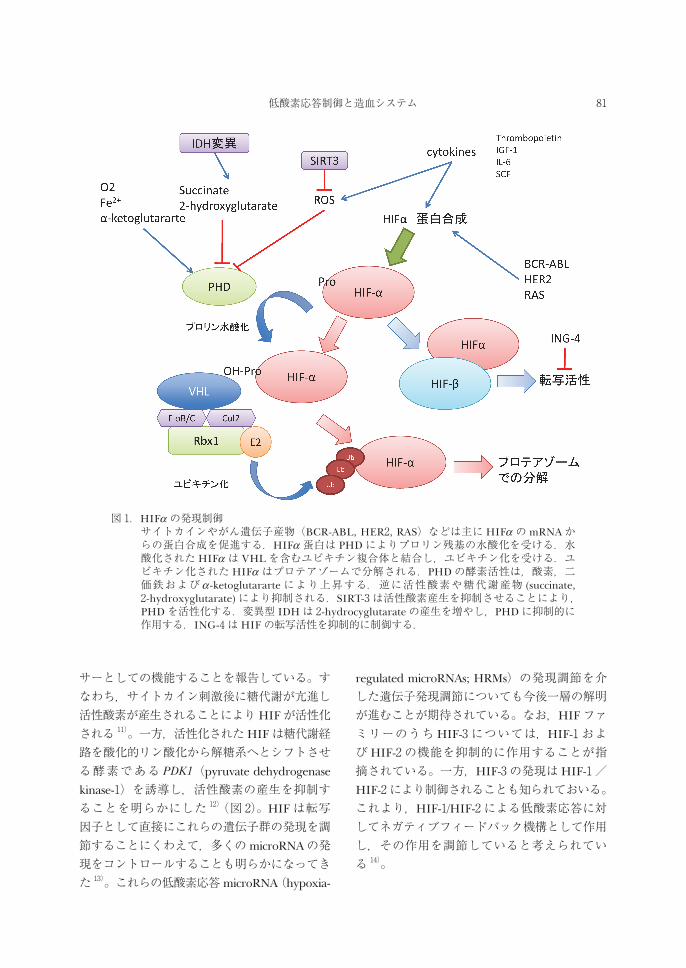
内外の因子によりHIFの機能は調節を受けている(図 1)。まず一つは,サイトカインやがん遺伝子産物などによりHIF-α のタンパク合成が亢進することである。我々も,造血因子である Thrombopoietin 2)や IL-63),さらにはInsulin like growth factor-1(IGF-1)3)により血液腫瘍細胞におけるHIF-α の発現が上昇することを報告している。この増殖因子によるHIF-α の発現誘導には細胞内の AKT/mTOR
経路や JAK/STAT経路が関与している。また,細胞内の糖代謝酵素の異常によってもHIFα の発現は大きな影響を受ける。TCA回路の中間代謝産物である Succinateは PHDの機能を抑制することによりHIF-α の発現を誘導する 4)。さらに,最近固形がんや血液腫瘍において広く遺伝子変異が存在することが明らかになっている isocitrate dehydrogenase(IDH)変異もHIFα の発現制御に関与することが報告されている。すなわち,正常な IDHは isocitrateからα -ketoglutarateへの代謝を促進するのに対して,変異型 IDHはさらにα -ketoglutararte
を 2-hydroxyglutarateへと変換する。この結果,PHDの機能が低下しHIF-α の発現が誘導される 5,6)。一方,最近 sirtuinファミリーの一つである SIRT3によるユニークなHIF-α の制御機序が明らかにされた 7)。この報告では,SIRT3
は細胞内の活性酸素の産生を抑制し,PHDの機能抑制を解除することが示された。結果的に,SIRT3はHIF-α の発現を抑制することが明らかとなった 7)。
(4)HIFはホメオスタシスを制御する 当初,HIFは低酸素状態に適応するために必要な遺伝子,例えば赤血球造血を促すerythropoietinや血管新生を促進する VEGFなどを制御する転写因子としてクローニングされた。しかし,現在では糖代謝酵素の発現制御,ミトコンドリアの生合成と分解,さらには鉄代謝酵素の発現調節 8,9)などさまざまな細胞のホメオスタシスを制御する因子であることが明らかになっている 10)。我々も,HIFが活性酸素セン
81低酸素応答制御と造血システム
サーとしての機能することを報告している。すなわち,サイトカイン刺激後に糖代謝が亢進し活性酸素が産生されることによりHIFが活性化される 11)。一方,活性化されたHIFは糖代謝経路を酸化的リン酸化から解糖系へとシフトさせる酵素である PDK1(pyruvate dehydrogenase
kinase-1)を誘導し,活性酸素の産生を抑制することを明らかにした 12)(図 2)。HIFは転写因子として直接にこれらの遺伝子群の発現を調節することにくわえて,多くのmicroRNAの発現をコントロールすることも明らかになってきた 13)。これらの低酸素応答microRNA(hypoxia-
regulated microRNAs; HRMs)の発現調節を介した遺伝子発現調節についても今後一層の解明が進むことが期待されている。なお,HIFファミリーのうちHIF-3については,HIF-1およびHIF-2の機能を抑制的に作用することが指摘されている。一方,HIF-3の発現はHIF-1/HIF-2により制御されることも知られておいる。これより,HIF-1/HIF-2による低酸素応答に対してネガティブフィードバック機構として作用し,その作用を調節していると考えられている 14)。
図 1.HIFα の発現制御 サイトカインやがん遺伝子産物(BCR-ABL, HER2, RAS)などは主にHIFα のmRNAか
らの蛋白合成を促進する.HIFα 蛋白は PHDによりプロリン残基の水酸化を受ける.水酸化されたHIFα は VHLを含むユビキチン複合体と結合し,ユビキチン化を受ける.ユビキチン化されたHIFα はプロテアゾームで分解される.PHDの酵素活性は,酸素,二価鉄およびα -ketoglutararteにより上昇する.逆に活性酸素や糖代謝産物 (succinate, 2-hydroxyglutarate)により抑制される.SIRT-3は活性酸素産生を抑制させることにより,PHDを活性化する.変異型 IDHは 2-hydrocyglutarateの産生を増やし,PHDに抑制的に作用する.ING-4はHIFの転写活性を抑制的に制御する.

82 桐 戸 敬 太
III.造血発生と低酸素環境
生物は低酸素環境に応答できる能力を備えているが,これは受動的な環境適応のみを意味するものではない。逆に積極的に低酸素環境を利用することにより,細胞の機能を維持することが明らかになってきた。この低酸素環境を利用した細胞機能の維持は特に幹細胞において特徴的に認められる 15)。実際に,造血幹細胞をはじめ,神経幹細胞や消化管の幹細胞などは組織内でも特に酸素濃度の低い領域に存在している 15)。以下は,造血幹細胞の機能維持における低酸素環境ならびにHIFの役割について焦点をあて考えてみたい。
(1)骨髄内は低酸素環境にある: 骨髄はその中央部に血管が存在しており,酸素分圧は中心部が最も高く,末梢の骨幹端に向
かい低下するという勾配をもつ。骨幹端における酸素分圧は 0.1%程度と極めて低いことが予想されている 16)。ヒトの骨髄中の酸素分圧については,解剖学的な分布は示されていないものの,骨髄穿刺血を用いて酸素分圧を測定したところ,約 55 mmHg(酸素飽和度 85%)と低値であることが示されている 17)。
(2)造血幹細胞と低酸素環境 上述のように,骨髄内の解剖学的な特性から,骨幹端付近では特に酸素濃度が低い。この骨幹端は造血幹細胞が存在する場そのものであることが明らかにされており,幹細胞ニッチと呼ばれている。造血幹細胞の自己複製能や細胞周期を静止させるといった特性は,幹細胞がこの幹細胞ニッチに存在することにより維持されている。幹細胞ニッチでは cadherinなどの接着分子,骨芽細胞,さらには thrombopoietinなど
図 2.活性化酸素センサーとしてのHIF サイトカイン刺激により糖代謝が活性化され,引続いてミト
コンドリアでの酸化的リン酸化による ATP産生が亢進する.このさいに,活性酸素も産生される.活性酸素は PHDの機能を抑制し,HIFを活性化する.HIFは糖代謝経路の分岐点を制御する PDKを誘導し,ミトコンドリアへの糖の流入を低下させることにより過剰な活性酸素の産生を抑制する.
83低酸素応答制御と造血システム
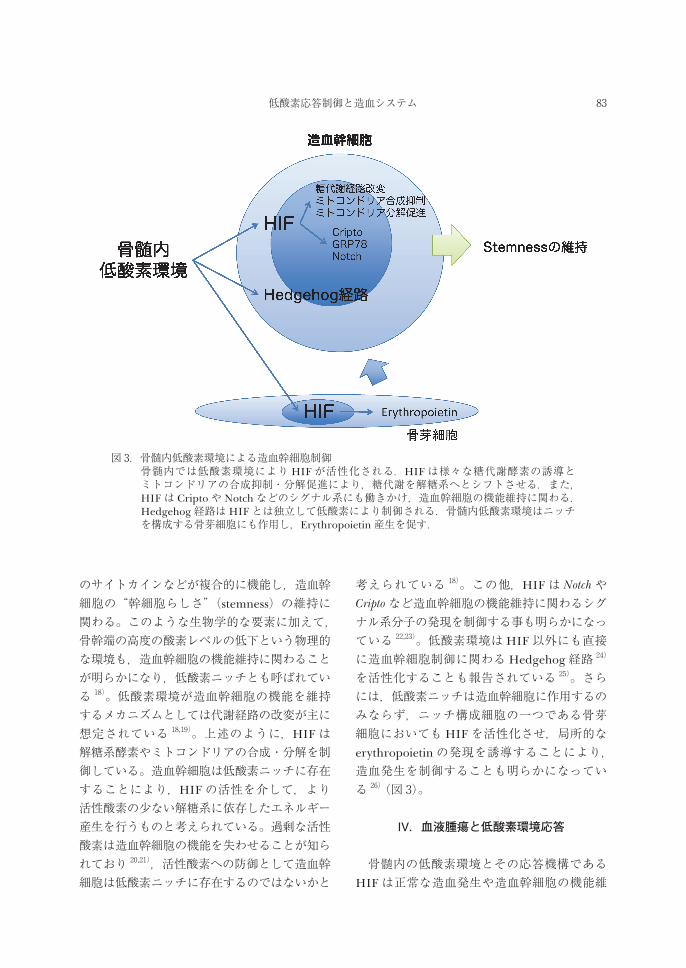
のサイトカインなどが複合的に機能し,造血幹細胞の“幹細胞らしさ”(stemness)の維持に関わる。このような生物学的な要素に加えて,骨幹端の高度の酸素レベルの低下という物理的な環境も,造血幹細胞の機能維持に関わることが明らかになり,低酸素ニッチとも呼ばれている 18)。低酸素環境が造血幹細胞の機能を維持するメカニズムとしては代謝経路の改変が主に想定されている 18,19)。上述のように,HIFは解糖系酵素やミトコンドリアの合成・分解を制御している。造血幹細胞は低酸素ニッチに存在することにより,HIFの活性を介して,より活性酸素の少ない解糖系に依存したエネルギー産生を行うものと考えられている。過剰な活性酸素は造血幹細胞の機能を失わせることが知られており 20,21),活性酸素への防御として造血幹細胞は低酸素ニッチに存在するのではないかと
考えられている 18)。この他,HIFは NotchやCriptoなど造血幹細胞の機能維持に関わるシグナル系分子の発現を制御する事も明らかになっている 22,23)。低酸素環境はHIF以外にも直接に造血幹細胞制御に関わるHedgehog経路 24)
を活性化することも報告されている 25)。さらには,低酸素ニッチは造血幹細胞に作用するのみならず,ニッチ構成細胞の一つである骨芽細胞においてもHIFを活性化させ,局所的なerythropoietinの発現を誘導することにより,造血発生を制御することも明らかになっている 26)(図 3)。
IV.血液腫瘍と低酸素環境応答
骨髄内の低酸素環境とその応答機構であるHIFは正常な造血発生や造血幹細胞の機能維
図 3.骨髄内低酸素環境による造血幹細胞制御 骨髄内では低酸素環境により HIFが活性化される.HIFは様々な糖代謝酵素の誘導と
ミトコンドリアの合成抑制・分解促進により,糖代謝を解糖系へとシフトさせる.また,HIFは Criptoや Notchなどのシグナル系にも働きかけ,造血幹細胞の機能維持に関わる.Hedgehog経路はHIFとは独立して低酸素により制御される.骨髄内低酸素環境はニッチを構成する骨芽細胞にも作用し,Erythropoietin産生を促す.

84 桐 戸 敬 太
持において極めて重要な役割を果たすが,このシステムの異常は多くの血液腫瘍の病態にも関わっている(図 4)。以下,代表的な血液腫瘍において,低酸素環境およびHIFがその病態に関与するかについて概説する。
(1)多発性骨髄腫 多発性骨髄腫は Bリンパ系腫瘍の一つであり,抗体産生を行うレベルにまで成熟した細胞(形質細胞)の腫瘍と位置づけられる。多発性骨髄腫においては,腫瘍細胞のみならず腫瘍細胞をとりまく微小環境が病態形成に重要な役割を果たしている。すなわち,腫瘍微小環境を構成する骨髄間質細胞は様々なサイトカインやケモカインの産生,あるいは接着分子を介した直接的な細胞間相互作用により,骨髄腫細胞の増殖や生存を支持している。また,多発性骨髄腫では骨髄内の血管新生が特徴的であり,病勢の進行とも関連している。2005年に,Asosingh
らは,この多発性骨髄腫における血管新生にはHIFが関与していることを報告している 27)。さらに,Collaらは骨髄腫細胞では,ING-4
(inhibitor of growth family member 4)の発現が低下しており,これが異常なHIFの活性化を招き,血管新生を誘導することを明らかにしている 28)。我々は多発性骨髄腫細胞の増殖因子である IL-6および IGF-1が AKTの活性化を介してHIF-1α の発現を誘導することを見いだした 3)。さらに,c-mycが HIFの異常活性化を誘導するとの報告もある 29)。以上より,骨髄腫細胞では骨髄内の酸素分圧変化に関わらず,細胞内のイベント(ING-4の発現低下,mycの発現上昇)および細胞外の刺激(IL-6,
IGF-1)によりHIFの恒常的な活性化がもたらされることが明らかとなった。異常活性化されたHIFは血管新生の増強に加えて,survivin 3)
などの抗アポトーシス蛋白,あるいはケモカインの一つである CXCL12 30)を誘導すること
図 4.血液腫瘍と低酸素応答機構 血液腫瘍細胞では,骨髄内の低酸素環境,サイトカインなどの外的要因およびがん遺伝子
産物などの内的な要因によりHIFの異常な活性化が起こる.HIFは多彩な遺伝子の発現制御を通じて,腫瘍細胞の薬剤耐性獲得,生存,浸潤などに関与する.また腫瘍幹細胞としての性格の維持にもHIFは関与する.

85低酸素応答制御と造血システム
により多発性骨髄腫の病態に関与することが示された。最近では,骨髄内の低酸素環境が骨髄腫細胞における E-cadherinの発現低下とCXCR-4の発現誘導という二つのメカニズムを介して,骨髄腫細胞の播種(dissemination)に関与するとの研究報告もなされている 31)。骨髄腫の進展に伴い,骨髄内の低酸素領域が広がり,その結果として骨髄腫細胞および骨髄間質細胞の双方が低酸素状態に晒されることが上記の変化をもたらす要因と考えられている。HIFをターゲットとした骨髄腫治療についても検討が進んでいる。我々は,株化された骨髄腫細胞のみならず臨床症例由来の骨髄腫細胞においても,HIF阻害剤である Echinomycin
が抗がん剤による骨髄腫細胞のアポトーシス誘導を促進することを明らかにした 3)。また,既存の骨髄腫の治療薬が抗HIF作用をもつことも報告されている。プロテアゾーム阻害剤bortezomibは,その作用機序より HIFα の発現を誘導することが予想されたが,HIFの発現には及ぼさない事が確認されている。逆に,その転写因子としての活性を抑制することが報告された 32)。一方,サリドマイドの誘導体レナリドマイドは HIFα の蛋白合成を低下させる 33)。これらの解析は in vitroのものであり,実際に in vivoあるいはヒトにおいて抗HIF作用が発揮されるのかについては,不明であるが,今後の新たな治療の開発に向けてのヒントの一つになるものと期待される。
(2)悪性リンパ腫 悪性リンパ腫もリンパ系の血液腫瘍であるが,その多くはリンパ節やリンパ臓器に発症するため,やや固形がん的な性格を持つ。このため,悪性リンパ腫におけるHIFの異常な活性化は,まず病理学的な解析により確認された。Stewartらは様々なリンパ腫症例において,血管新生に関わる VEGFおよび HIFの発現について免疫組織学的に解析を行い,低悪性度リンパ腫の 62%,高悪性度リンパ腫では 77%で HIF-1の発現を認めたことを報告してい
る 34)。HIF-2の発現はさらに高頻度にみられており,低悪性度リンパ腫では 76%,高悪性度リンパ腫では 94%で活性化が確認されている 34)。Evensらは悪性リンパ腫の中でも,ろ胞性リンパ腫(Follicular lymphoma; FL)とび漫性大細胞型 B細胞性リンパ腫(Diffuse large
B cell lymphoma; DLBCL)症例に焦点をしぼって解析を行った 35)。FL症例の約 11%,DLBCL症例では 44%で HIF-1および HIF-2
の中等度以上の発現が確認されている 35)。悪性リンパ腫細胞において,HIFの発現が亢進するメカニズムについても様々な解析が行われている。我々は,悪性リンパ腫細胞で高頻度に異常活性化を認める NF-κBが HIF-1α の発現誘導に関わることを明らかにした 36)。Argyriou
らはmTORC1の活性化が HIF-1α の発現に関わるとしている 37)。活性化された HIFはsurvivin 36)や Bcl-xL 38)などの抗アポトーシス蛋白の発現を誘導し,放射線治療や化学療法への抵抗獲得に関わることが推察されている。FL
および DLBCLなどの B細胞性悪性リンパ腫の治療においては,放射線治療や抗がん剤化学療法とならび,抗 CD20ヒト型モノクローナル抗体 rituximabが大きな役割を果たしている。Rituximabは,B細胞リンパ腫の細胞表面に発現している CD20分子を認識し,主に免疫学的な機序を介して B細胞性リンパ腫細胞を排除すると考えられている。最近,我々は株化された B細胞性リンパ腫細胞を用いて,rituximab
が AKTを活性化しHIF-1α の発現を誘導することを見出した 39)。また,この現象は細胞表面膜コレステロールレベルの高い細胞株において特異的に認められた。さらに,コレステロールの合成阻害剤である simvastatinを用い細胞膜表面のコレステロールレベルを下げることにより rituximabによる HIFの活性化は抑制された 39)。Rituximabにより活性化されたHIFは抗アポトーシス蛋白 survivinを誘導し,抗がん剤によるアポトーシス誘導に対して拮抗する作用を示す事も分かった。これらの結果は,リンパ腫治療の重要な柱の一つである

86 桐 戸 敬 太
rituximabが特定の細胞ではむしろ腫瘍の生存維持に関わることを示唆する。今後は,このメカニズムの詳細を明らかにするとともに,細胞膜脂質構成をもとにした悪性リンパ腫の治療効果予測システムの構築や,細胞膜脂質の改変による rituximabの治療効果改善方法の開発などに取り組む予定である。
(3)慢性骨髄性白血病 骨髄増殖性腫瘍(myeloproliferative neo-
plasms; MPN)とは,造血幹細胞に生じた異常により,一系統以上の骨髄系細胞(顆粒球系,赤血球系および血小板系)の成熟を伴った過剰な増殖を示す疾患の総称である。このカテゴリーに分類される代表的な疾患として,慢性骨髄性白血病(Chronic myelogenous leukemia;
CML)がある。CMLはフィラデルフィア染色体転座と呼ばれる染色体転座 t(9;22)を持つことが特徴であり,この染色体転座により BCR-
ABL融合遺伝子が形成される。BCR-ABL融合遺伝子産物は強力なタンパク質チロシンキナーゼ活性を有しており,AKTや STATなど様々な細胞内シグナル伝達経路を活性化する。CMLの治療成績は,BCR-ABLに対するチロシンキナーゼ阻害剤である Imatinibの登場により大きく向上している。しかしながら,一部の症例では Imatinibに抵抗性であり,またいわゆる白血病幹細胞は Imatinibにより排除できないことが指摘されている 40)。最近の研究により,CMLにおける Imatinib耐性獲得やCML幹細胞の維持に低酸素応答機構が関わることが明らかにされている。CMLにおける低酸素応答機構との関連性についての研究は古く,2002年にはすでに CML細胞ではHIF-1αの発現上昇がみられることが報告されている 41)。また,BCR-ABL陽性の白血病細胞株を低酸素環境下で培養することにより,上述の分子標的薬 Imatinibに抵抗性を持つクローンが選択されうることも示されている 42)。さらに,Imatinib耐性の獲得のメカニズムとして,低酸素環境ではHIF活性化に伴う糖代謝経路の
改変であることが示されている 43,44)。2012年には CMLの白血病幹細胞の維持にもHIF-1αは必要であることが示された。すなわち,HIF-1α を欠損させた CML細胞では,細胞周期を負に調節する p16/Ink4および p19/ARFの発現が亢進し,マウスモデルにおける CML発症能力が消失することが観察されている 45)。
(4)フィラデルフィア染色体陰性骨髄増殖性腫瘍 MPN のうち,真性赤血球増加症(Poly-
cythemia vera; PV),本態性血小板血症(Essential
thrombocythemia; ET)および原発性骨髄線維症(Primary myelofi brosis; PMF)の 3者はフィラデルフィア染色体陰性MPNと総称されている。フィラデルフィア染色体陰性MPNでは,長らくその責任遺伝子異常は不明であったが,2005年に細胞内チロシンキナーゼの一つである JAK2において,恒常活性を来す遺伝子突然変異が高頻度に見られることが明らかにされた 46–48)。この遺伝子変異により JAK2分子の617番目のバリン(V)がフェニルアラニン(F)に置換される。このため,慣習的にこの変異を JAK2V617F変異と呼ぶ事が多い。フィラデルフィア陰性MPNのうち,PMFについては VEGFレベルの上昇 49)や骨髄内での血管新生 50)の増加などが認められることより,HIF
の関与も予想される。しかしながら,PMFの病態に HIFが関与するとの報告は,現在までのところ発表されていない。JAK2V617F
と低酸素との関連については,我々が研究を進めている。現在までに,低酸素環境下ではJAK2V617Fの過剰活性(自己リン酸化)が停止し,かつ細胞周期の停止やアポトーシス誘導が認められることを発表している 51)。
V.おわりに;低酸素応答機構をターゲットとした血液腫瘍の治療戦略の展開
上述のようにHIFは様々な血液腫瘍の病態に関わることから,HIFをターゲットとした新たな治療方法の開発も精力的に進められて

87低酸素応答制御と造血システム
いる。この中でも,HIFの機能を抑制することにより,がん幹細胞を排除する試みが注目されている。Wangらは,HIFの機能阻害剤echinomycinを用いる事により,in vivoにおいてマウスのリンパ腫幹細胞および急性骨髄性白血病症例由来検体中の白血病幹細胞の機能を抑制しうることを示した 23)。さらに同じグループから,シャペロン蛋白の一つであるHSP90
(heat shock protein 90)の機能を抑制することにより,HIF1α の発現を抑制しうること,さらにはリンパ腫幹細胞の排除へと繋がることが報告された 52)。白血病幹細胞やリンパ腫幹細胞などのいわゆるがん幹細胞は,がんの治療上の最も大きな障害の一つである。HIFあるいは低酸素環境の是正により,このがん幹細胞を駆逐することができれば,がん治療全体の成績向上に貢献することも期待される 10)。
文 献
1) Greer SN, Metcalf JL, Wang Y, Ohh M: The up-dated biology of hypoxia-inducible factor. EMBO J 31: 2448–2460, 2012.
2) Kirito K, Fox N, Komatsu N, Kaushansky K: Thrombopoietin enhances expression of vascular endothelial growth factor (VEGF) in primitive hematopoietic cells through induction of HIF-1-alpha. Blood 105: 4258–4263, 2005.
3) Hu Y, Kirito K, Yoshida K, et al.: Inhibition of hypoxia-inducible factor-1 function enhances the sensitivity of multiple myeloma cells to melpha-lan. Mol Cancer Ther 8: 2329–2338, 2009.
4) Selak MA, Armour SM, MacKenzie ED, et al.: Suc-cinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 7: 77–85, 2005.
5) Zhao S, Lin Y, Xu W, et al.: Glioma-Derived Muta-tions in IDH1 Dominantly Inhibit IDH1 Catalytic Activity and Induce HIF-1α. Science 324: 261–265, 2009.
6) Kloosterhof NK, Bralten LBC, Dubbink HJ, French PJ, van den Bent MJ: Isocitrate dehydro-genase-1 mutations: a fundamentally new under-standing of diffuse glioma? The Lancet Oncology 12: 83–91, 2011.
7) Finley Lydia WS, Carracedo A, Lee J, et al.: SIRT3 Opposes Reprogramming of Cancer Cell Metabolism through HIF1α Destabilization. Can-
cer Cell 19: 416-428, 2011. 8) Salahudeen AA, Bruick RK: Maintaining Mam-
malian Iron and Oxygen Homeostasis. Annals of the New York Academy of Sciences 1177: 30–38, 2009.
9) Thompson JW, Bruick RK: Protein degradation and iron homeostasis. Biochimica et Biophys-ica Acta (BBA) - Molecular Cell Research 1823: 1484–1490, 2012.
10) Semenza GL: Hypoxia-inducible factors: media-tors of cancer progression and targets for can-cer therapy. Trends Pharmacol Sci 33: 207–214, 2012.
11) Yoshida K, Kirito K, Yongzhen H, Ozawa K, Kaushansky K, Komatsu N: Thrombopoietin (TPO) regulates HIF-1alpha levels through gen-eration of mitochondrial reactive oxygen species. Int J Hematol 88: 43–51, 2008.
12) Kirito K, Hu Y, Komatsu N: HIF-1 prevents the overproduction of mitochondrial ROS after cy-tokine stimulation through induction of PDK-1. Cell Cycle 8: 2844–2849, 2009.
13) Kulshreshtha R, Davuluri RV, Calin GA, Ivan M: A microRNA component of the hypoxic response. Cell Death Differ 15: 667–671, 2008.
14) Maynard MA, Evans AJ, Hosomi T, Hara S, Jew-ett MAS, Ohh M: Human HIF-3α4 is a domi-nant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. The FASEB Journal 19: 1396–1406, 2005.
15) Mohyeldin A, Garzón-Muvdi T, Quiñones-Hino-josa A: Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 7: 150–161, 2010.
16) Guitart AV, Hammoud M, Dello Sbarba P, Ivanovic Z, Praloran V: Slow-cycling/quiescence balance of hematopoietic stem cells is related to physiological gradient of oxygen. Exp Hematol 38: 847–851, 2010.
17) Harrison JS, Rameshwar P, Chang V, Bandari P: Oxygen saturation in the bone marrow of healthy volunteers. Blood 99: 394, 2002.
18) Takubo K, Goda N, Yamada W, et al.: Regulation of the HIF-1α Level Is Essential for Hematopoi-etic Stem Cells. Cell Stem Cell 7: 391–402, 2010.
19) Suda T, Takubo K, Semenza Gregg L: Metabolic Regulation of Hematopoietic Stem Cells in the Hypoxic Niche. Cell Stem Cell 9: 298–310, 2011.
20) Ito K, Hirao A, Arai F, et al.: Regulation of oxida-tive stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431: 997–1002, 2004.
21) Ito K, Hirao A, Arai F, et al.: Reactive oxygen spe-cies act through p38 MAPK to limit the lifespan of

88 桐 戸 敬 太
hematopoietic stem cells. Nat Med 12: 446–451, 2006.
22) Miharada K, Karlsson G, Rehn M, et al.: Cripto Regulates Hematopoietic Stem Cells as a Hypox-ic-Niche-Related Factor through Cell Surface Re-ceptor GRP78. Cell Stem Cell 9: 330–344, 2011.
23) Wang Y, Liu Y, Malek SN, Zheng P, Liu Y: Tar-geting HIF1alpha eliminates cancer stem cells in hematological malignancies. Cell Stem Cell 8: 399–411, 2011.
24) Irvine DA, Copland M: Targeting hedgehog in hematologic malignancy. Blood 119: 2196–2204, 2012.
25) Onishi H, Kai M, Odate S, et al.: Hypoxia ac-tivates the hedgehog signaling pathway in a ligand-independent manner by upregulation of Smo transcription in pancreatic cancer. Cancer Sci 102: 1144–1150, 2011.
26) Rankin EB, Wu C, Khatri R, et al.: The HIF sig-naling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell 149: 63–74, 2012.
27) Asosingh K, De Raeve H, de Ridder M, et al.: Role of the hypoxic bone marrow microenvironment in 5T2MM murine myeloma tumor progression. Haematologica 90: 810–817, 2005.
28) Colla S, Tagliaferri S, Morandi F, et al.: The new tumor-suppressor gene inhibitor of growth fam-ily member 4 (ING4) regulates the production of proangiogenic molecules by myeloma cells and suppresses hypoxia-inducible factor-1 alpha (HIF-1alpha) activity: involvement in myeloma-induced angiogenesis. Blood 110: 4464–4475, 2007.
29) Zhang J, Sattler M, Tonon G, et al.: Targeting an-giogenesis via a c-Myc/hypoxia-inducible factor-1alpha-dependent pathway in multiple myeloma. Cancer Res 69: 5082–5090, 2009.
30) Martin SK, Diamond P, Williams SA, et al.: Hy-poxia-inducible factor-2 is a novel regulator of aberrant CXCL12 expression in multiple my-eloma plasma cells. Haematologica 95: 776–784, 2010.
31) Azab AK, Hu J, Quang P, et al.: Hypoxia promotes dissemination of multiple myeloma through ac-quisition of epithelial to mesenchymal transition-like features. Blood 119: 5782–5794, 2012.
32) Shin DH, Chun YS, Lee DS, Huang LE, Park JW: Bortezomib inhibits tumor adaptation to hypoxia by stimulating the FIH-mediated repression of hypoxia-inducible factor-1. Blood 111: 3131–3136, 2008.
33) Lu L, Payvandi F, Wu L, et al.: The anti-cancer drug lenalidomide inhibits angiogenesis and me-
tastasis via multiple inhibitory effects on endothe-lial cell function in normoxic and hypoxic condi-tions. Microvasc Res 77: 78–86, 2009.
34) Stewart M, Talks K, Leek R, et al.: Expression of angiogenic factors and hypoxia inducible factors HIF 1, HIF 2 and CA IX in non-Hodgkin’s lym-phoma. Histopathology 40: 253-260, 2002.
35) Evens AM, Schumacker PT, Helenowski IB, et al.: Hypoxia inducible factor-alpha activation in lym-phoma and relationship to the thioredoxin fam-ily. British Journal of Haematology 141: 676–680, 2008.
36) Qiao Q, Nozaki Y, Sakoe K, Komatsu N, Kirito K: NF-kappaB mediates aberrant activation of HIF-1 in malignant lymphoma. Exp Hematol 38: 1199–1208, 2010.
37) Argyriou P, Papageorgiou S, Panteleon V, et al.: Hypoxia-inducible factors in mantle cell lympho-ma: implication for an activated mTORC1→HIF-1α pathway. Annals of Hematology 90: 315–322, 2011.
38) Hernandez-Luna MA, Rocha-Zavaleta L, Vega MI, Huerta-Yepez S: “HIF-1α induces a chem-oresistance phenotype in a Non-Hodgkin’s Lym-phoma cell line via up-regulation of Bcl-xL”. Leukemia & Lymphoma 1–17, 2012.
39) Nozaki Y, Mitsumori T, Komatsu N, Kirito K: Rituximab Activation of AKT and HIF Pathways Is Dependent on Membrane Lipid Raft Choles-terol Levels in Lymphoma Cells. ASH Annual Meeting Abstracts 118: 1571, 2011.
40) Corbin AS, Agarwal A, Loriaux M, Cortes J, Dei-ninger MW, Druker BJ: Human chronic myeloid leukemia stem cells are insensitive to imatinib de-spite inhibition of BCR-ABL activity. The Journal of Clinical Investigation 121: 396–409, 2011.
41) Mayerhofer M, Valent P, Sperr WR, Griffi n JD, Sillaber C: BCR/ABL induces expression of vascular endothelial growth factor and its tran-scriptional activator, hypoxia inducible factor-1α, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamy-cin. Blood 100: 3767–3775, 2002.
42) Giuntoli S, Rovida E, Barbetti V, Cipolleschi MG, Olivotto M, Dello Sbarba P: Hypoxia suppresses BCR//Abl and selects imatinib-insensitive progen-itors within clonal CML populations. Leukemia 20: 1291–1293, 2006.
43) Zhao F, Mancuso A, Bui TV, et al.: Imatinib re-sistance associated with BCR-ABL upregulation is dependent on HIF-1[alpha]-induced metabolic reprograming. Oncogene 29: 2962–2972, 2010.
44) Giuntoli S, Tanturli M, Di Gesualdo F, Barbetti V, Rovida E, Dello Sbarba P: Glucose availability

89低酸素応答制御と造血システム
in hypoxia regulates the selection of chronic my-eloid leukemia progenitor subsets with different resistance to imatinib-mesylate. Haematologica 96: 204–212, 2011.
45) Zhang H, Li H, Xi HS, Li S: HIF1alpha is re-quired for survival maintenance of chronic my-eloid leukemia stem cells. Blood 119: 2595–2607, 2012.
46) Baxter EJ, Scott LM, Campbell PJ, et al.: Ac-quired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 365: 1054–1061, 2005.
47) James C, Ugo V, Le Couedic JP, et al.: A unique clonal JAK2 mutation leading to constitutive sig-nalling causes polycythaemia vera. Nature 434: 1144–1148, 2005.
48) Levine RL, Wadleigh M, Cools J, et al.: Activat-ing mutation in the tyrosine kinase JAK2 in poly-cythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofi brosis. Cancer Cell 7: 387–397, 2005.
49) Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A: Circulating Interleukin (IL)-8, IL-2R, IL-12, and IL-15 Levels Are Independ-ently Prognostic in Primary Myelofi brosis: A Comprehensive Cytokine Profi ling Study. Jour-nal of Clinical Oncology 29: 1356–1363, 2011.
50) Medinger M, Skoda R, Gratwohl A, et al.: Angio-genesis and vascular endothelial growth factor-/receptor expression in myeloproliferative neo-plasms: correlation with clinical parameters and JAK2-V617F mutational status. British Journal of Haematology 146: 150–157, 2009.
51) Mitsumori T, Nozaki Y, Komatsu N, Kirito K: Hy-poxia Blocks Activation of JAK2V617F Through Suppression of SHP-2 Expression in MPN Cells. ASH Annual Meeting Abstracts 118: 126, 2011.
52) Newman B, Liu Y, Lee HF, Sun D, Wang Y: HSP90 Inhibitor 17-AAG Selectively Eradicates Lymphoma Stem Cells. Cancer Res 72: 4551–4561, 2012.



![Title シトクロムb5の役割を中心としたCMP-N-アセチルノイ …...薪一制1 コ く ] 京大附図 シトクロムb5の役割を中心としたCMP-N」 アセチルノイラミン酸水酸化機構に関する研究シトクロムbの役割を中心としたCMP-N」](https://img.pdfslide.net/doc/110x75/60ce05f5650e135f021f6b86/title-fffb5fcmp-n-fff-e1.jpg)

























