Embed Size (px)
Citation preview

EUROPEAN
European Polymer Journal 43 (2007) 1065–1076
www.elsevier.com/locate/europolj
POLYMERJOURNAL
Effect of copolymer architecture on the response of pHsensitive fibers based on acrylonitrile and acrylic acid
Anasuya Sahoo, K.R.T. Ramasubramani, Manjeet Jassal *, Ashwini K. Agrawal *
Department of Textile Technology, Indian Institute of Technology, Hauz Khas, New Delhi 110 016, India
Received 2 February 2006; received in revised form 6 November 2006; accepted 30 November 2006Available online 26 January 2007
Abstract
Copolymers of acrylonitrile and acrylic acid with high acrylic acid feed ratio of 43 mol% were synthesized using freeradical polymerization. The architecture of copolymers was modified by regulating the dosing of more reactive comono-mer-acrylic acid. 13C NMR analysis confirmed that two copolymers – one (A) containing enriched blocks of individualmonomer-residues (architecture close to a block copolymer) and the second (B) having nearly random distribution ofthe comonomers, could be successfully synthesized. The resultant acrylic acid content was determined to be nearly50 mol% for both the copolymers. These copolymers were converted to fine fibers by solution spinning in DMF-water sys-tem, drawn in coagulation bath, and annealed at 120 �C for 2 h. The fibers were evaluated for pH response behavior,mechanical stability, and retracting stresses. The fiber A was found to have significantly higher swelling percentage(3300–3700%), faster response, and higher stability to repeated cycling compared to fiber B. Also, Fiber A showed lowerthermal shrinkage, better mechanical properties during swelling and higher retracting forces during deswelling. Theseresults indicate that the copolymer with enriched block architecture could possibly form segregated domain structure withacrylic acid domains facilitating enhanced pH response while acrylonitrile domains providing physical crosslinks for stron-ger mechanical strength. The study suggests that above approach may be more useful than chemically cross-linked gel rodsin producing artificial muscles with faster response and good mechanical properties.� 2006 Elsevier Ltd. All rights reserved.
Keywords: pH Sensitive polymers; Poly(acrylonitrile-co-acrylic acid); Architecture; Artificial muscle; Smart fibers
1. Introduction
Stimuli sensitive polymers (SSP) are beingextensively studied for the past decade due to their
0014-3057/$ - see front matter � 2006 Elsevier Ltd. All rights reserved
doi:10.1016/j.eurpolymj.2006.11.031
* Corresponding authors. Tel.: +91 11 26526154 (Ashwini K.Agrawal).
E-mail addresses: [email protected] (M. Jassal),[email protected] (A.K. Agrawal).
potential applications in areas ranging from con-trolled-delivery for functional substances (drugs,nutrients, herbicides, etc.) [1,2] to membranes formolecular separation [3–5], enzyme activity control[6,7], extraction [8], BioMEMS [9–11] such ashydrogel actuated microvalves and microfludic con-trollers in microchannels, and now recently for arti-ficial muscles [12–14]. Stimuli sensitive polymersundergo reversible transitions under various stimulisuch as pH [15], temperature [16], electric field [17],
.

1066 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
or electrolyte [18]. These environment sensitivepolymers are also called ‘‘Intelligent Polymers’’ or‘‘Smart Polymers’’ [19,20]. One of the importanttypes of SSPs is pH sensitive polymers. These poly-meric materials can sense the pH of their environ-ment as a signal, judge the magnitude of the signaland change their properties accordingly.
The pH sensitive polymers undergo hydrophobicto hydrophilic state by the change in pH [21]. Thesepolymers consist of ionizable pendant groups thatcan accept and donate protons in response to theenvironmental change in pH. As the environmentalpH changes, the degree of ionization in pendantgroups undergo dramatic change at a specific pHcalled pKa. This rapid change in the net charge ofpendant groups causes an alternation of the hydro-dynamic volume of the polymer chains. This resultsin a transition from collapsed hydrophobic state tosoluble hydrophilic state of the polymer [22]. Inorder to create structures that respond to pHchange, these polymers are crosslinked to formhydrogels. Many systems based on copolymers ofN-isopropyl acrylamide–acrylic acid [23], vinyl alco-hol–acrylic acid [24], acrylonitrile–acrylic acid [25],poly(methacrylic acid) grafted with poly(ethyleneglycol) [26] have been studied for various pH sensi-tive applications in the gel form.
The pH-sensitive hydrogel structures reported inthe literature suffer from slow response in the rangeof several hours and poor magnitude of response(i.e. extent of swelling and deswelling). Since swell-ing and deswelling is a diffusion controlled phenom-enon, thicker dimensions of hydrogels result inslower diffusion of water which is subsequentlyresponsible for the slow response. Further hydrogelshave poor mechanical properties and break downwhen subjected to repeated cycles. In order toimprove response, porous gel rods have been inves-tigated [27–29], however the porous structure givesmuch lower strength.
Recently, an approach was reported by ourgroup [30] in which linear temperature sensitivecopolymers were processed into fibers with orientedpolymer chains to impart strength. However, thesefibers were required to be chemically crosslinkedto form insoluble state. Though these fibers showedextremely fast response but they were found to befragile when in swollen state.
In another approach [21,31–34], the existingpoly(acrylonitrile) are modified to produce pH sensi-tive fibers. Since PAN fibers are known to possesskey engineering features such as excellent environ-
mental stability and high tensile strength, they makea good starting material for producing strong pHsensitive fibers. In this approach, special acrylicfibers (SAF) are thermally oxidized, where some ofthe CN groups undergo cyclization to provide theconnected structure. The remaining CN groups arehydrolyzed or saponified to convert them into car-boxylic acid groups. The cyclized acrylonitrile moie-ties remain unaltered even during hydrolysis and actas a backbone rendering strength to the fiber whilethe hydrolyzed groups impart pH sensitivity. Themodified PAN fibers, being of fine dimension (diam-eter in microns), exhibit good response to changingenvironment when activated electrically or chemi-cally. This method has several advantages. First, itis a simple process that it involves only the modifica-tion of an existing precursor fiber and essentially,does not require any new polymer to be synthesized.Though this process appears to be suitable for theproduction of artificial muscles it too has somedrawbacks.
pH Sensitive fibers produced using this methodare black in colour that makes them inappropriatefor textile applications. This colour is ascribed tothe chemical conjugation of nitrile groups to formsix-membered aromatic rings. The production rateof these fibers is considerably slow since it involvescontrolled oxidation of PAN at high temperaturefor long periods of time. Further, the parametersavailable for controlling the amount of cyclizationand crosslinking in the fiber are limited since theprecursor fiber used is of a specific microstructureand modifying the fiber to a great extent may resultin deteriorating its mechanical properties. Due tothis reason, producing fibers of varying chemicaland physical architecture for tunable response isdifficult.
Therefore, in this work, an alternate approachhas been adopted to address some of the abovedrawbacks. In this approach, a pH sensitive copoly-mers of controlled architecture based on acrylicacid and acrylonitrile have been synthesized whichwhen solution spun into fine fibers, show goodmechanical properties as well as enhanced pHresponse without the need of chemical crosslinking.It is suggested that fibers from such copolymersmay form domains of acrylonitrile and acrylic acid,where acrylonitrile domains act as physical cross-links and connect chains to allow transfer of thestresses along the fiber axis, while acrylic aciddomains swell and deswell to give the desirablepH response.

A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076 1067
2. Experimental
2.1. Materials
Acrylic acid (AA) and toluene were obtainedfrom Merck India LTD, Mumbai. Acrylonitrile(AN) was purchased from Central Drug House (p)LTD, New Delhi and initiator a,a 0-azobisisobutyro-nitrile (AIBN) from G.S.Chemical Testing Lab andAllied Industries, New Delhi. Solvents diethyl etherand dimethylformamide (DMF) were purchasedfrom Qualigens Fine Chemicals, Mumbai. Allchemicals were of minimum assay of 99% and wereused without further purification.
2.2. Copolymer synthesis
2.2.1. Copolymer with enriched segments (blocks)
AA43B
Free radical copolymerization was carried out ina four neck reactor in toluene at 65 ± 1 �C undernitrogen atmosphere. The total monomer concen-tration was fixed at 20 wt%. The final acrylic acid(AA) content in the feed was kept at 50 wt%(42.7 mol%). 200 g of toluene was taken in the reac-tion flask and degassed for 30 min. The initiatorAIBN 0.06 g (0.057 mol%) was dissolved in 20 g ofacrylonitrile (AN) and degassed for 30 min. It wasthen added to the reaction flask and the reactionwas carried out for 30 min before starting the addi-tion of acrylic acid. The more reactive comonomer(20 g of AA) was degassed, and added in small reg-ulated doses (i.e. 22 doses of �0.9 g each) to thereaction mixture over a period of 3 h with constantstirring. The reaction was allowed to continue for anadditional 30 min. The reaction mixture was cooledand then precipitated in excess diethyl ether to getwhite product. The product was washed three timeswith excess diethyl ether to remove traces of unre-acted monomers and toluene. Homopolymer ofacrylic acid was removed by thoroughly washingthe product with excess acetone. Only about 2% ofthe yield from the reaction was lost during purifica-tion with acetone. The purified copolymer was driedin a vacuum oven at 60 �C for 1 h. Gravimetric yieldof the copolymer was �37%.
2.2.2. Random copolymer AA43R
The random copolymer of acrylic acid and acry-lonitrile was also prepared using the above methodexcept that the solution of acrylic acid was addedcontinuously (drop wise) to the reaction mixture
over a period of 1 h 30 min with constant stirring.The reaction was allowed to continue for an addi-tional 30 min and precipitated and purified as men-tioned above. The gravimetric yield was found to be33%.
2.3. Copolymer characterization
2.3.1. Intrinsic viscosity
The intrinsic viscosity of the copolymers wasdetermined in DMF using Ubbelohde viscometerin a constant temperature bath at 30 ± 0.1 �C. Vis-cosity average molecular weight was determinedusing Mark–Houwink relation [g] = KMa witha = 0.75 and K = 20.9 · 10�5 dl/g [35] given forcopolymers of acrylonitrile and acrylic acid.
2.3.2. Determination of copolymer composition and
sequence length by 13C NMR spectra
The 13C NMR spectra of the copolymers wererecorded under the standard conditions at 80 �C inDMSO-d6 on a Bruker DPX-300 MHz Spectropho-tometer. The composition of the copolymers wasdetermined by using the peak areas correspondingto carbon atoms in the COOH of acrylic acid andCN groups of acrylonitrile. The composition ofthe copolymer was calculated by the formula.
Mole percent of acrylonitrile ¼ ICN
ICN þ ICO
where ICN is the intensity of CN and ICO is theintensity of CO as noted from the 13C quantitativeNMR. The sequence length of the two monomersin the copolymer was determined by evaluatingthe triads obtained for the above two carbons asper the reported approach in the literature [36].These triad fractions were obtained as normalizedareas of the respective resonance signals.
2.3.3. Copolymer composition by acidimetric titration
The 0.5 wt% solutions of copolymers in DMFwere titrated against 0.05 N aqueous NaOH (stan-dardized) using a phenolphthalein as an indicatoras per the reported method [37].
2.4. Solution spinning of pH sensitive fibers
The dope solution of 33 wt% was prepared bystirring the purified and dried copolymer powderin DMF. The solutions were kept in vacuum atthe room temperature to allow deaeration before

1068 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
spinning. Fibers were extruded using a syringe typemonofilament extruder into a coagulation bath con-taining water at 30 �C. The extruded fibers weredrawn to a draw ratio of 2.0 before the coagulationwas complete. The coagulated fibers were driedunder taut condition in air at 30 �C.
2.5. Heat treatment of the as-spun fibers
The dried fibers were heat-treated in an air ovenat a temperature 120 �C for 2 h. The fibers wereplaced in taut condition in a wooden frame andfixed at both the ends to avoid shrinkage duringannealing.
2.6. Shrinkage above glass transition
The shrinkage percentage of fibers was deter-mined by taking a known length (IL) of the fiber,allowing it to shrink by immersing it in silicone oilbath at 100 and 130 �C. The final length (FL) ofthe fiber was determined after 10 min. The shrink-age percentage was calculated using the followingrelation.
Shrinkage% ¼ IL� FL
IL� 100:
2.7. X-ray diffraction
WAXD spectra of the fibers were recorded by X 0
Pert PRO machine of PANalytical between 2h of10–35� in the reflection mode. The fiber were cutinto small pieces and placed in powder sample stagefor the spectroscopy.
2.8. Mechanical properties
The tensile strength of the fibers was recorded onInstron tensile testing instrument (Model no. 4202)at the crosshead speed of 10 mm/min. The fiberswere conditioned by repeated swelling and deswell-ing for three cycles and then tested in both swollenand deswollen conditions. The diameter of the fiberwas measured using Lieca optical microscope at 15places along the length of the fiber samples used forthe above measurements.
2.9. Swelling behavior
The drawn and heat-set fibers were conditionedfor three cycles of swelling and deswelling before
carrying out the following measurements (exceptfor cyclability test).
2.9.1. Equilibrium swelling
The equilibrium swelling behavior of fibers wasstudied by immersing them in a slack condition ina pH 10 solution for a period of 180 min. The diam-eters of the fibers were measured in a Lieca micro-scope. The length of the fibers was also measuredbefore and after the transition. The volumetricswelling% was calculated from the changes in diam-eter and length of the fibers.
2.9.2. Hysteresis
The hysteresis during transition was studied bysubjecting the fiber samples initially to swelling byincreasing the pH of the bath, and subsequently,to the deswelling by decreasing the pH of the bath,thereby, reversing the cycle. At each pH the fiberwas placed for 30 min before the measurement wastaken.
2.9.3. Rate of transition
For determining the rate of transition, a singlefiber was placed vertically in a measuring flask witha small weight of 63 mg tied at the lower end of thefiber. The % change in length with time duringswelling cycle was measured for both fibers at apH of 10, while the rate of deswelling was evaluatedat a pH of 2.
2.9.4. Cyclability
The drawn-heat-set fibers (without conditioning)were placed on a glass slide in slack condition andthe glass slide along with fiber was immersed in apH 10 solution for determining the change indimensions. Similarly, the above slides were placedin a pH 2 solution for deswelling. At each stagesamples were kept till equilibrium was reached.The reversibility of the transition was evaluatedfor several swelling–deswelling cycles.
2.9.5. Retractive stress during deswellingFor determining the retractive stresses during
deswelling, a single fiber was hung vertically in ameasuring flask with a small weight of 63 mg tiedat the lower end of the fiber. It was allowed to swellto its maximum in pH 10. Then an additionalweight in form of several metal rings weighting8 mg each was attached to the lower end of the fiber.This fiber was hung in pH 2 for deswelling alongwith the additional weight. Subsequently, the rings

A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076 1069
were removed one at a time to determine the maxi-mum weight under which the fiber was able to comeback to its original shape and size.
3. Results and discussion
The copolymers of acrylonitrile and acrylic acidwere produced by radical polymerization. Acryloni-trile was chosen to impart fiber-forming propertiesto the copolymer while acrylic acid was selected toprovide pH response. The approach was to controlthe distribution of both acrylonitrile and acrylicacid moieties in the polymer chains in such a waythat small block segments of each are possible. Ifthis could be attained, then the segments of acrylo-nitrile moieties from different chains may cometogether (phase separate), crystallize and form vari-ous tie-points to provide strength to the formedstructure, whereas the block segments of acrylic acidmay provide pH response through ionization undersuitable environment. The proposed segregateddomain morphology of the fiber is shown schemat-
CH2 CH
CNm
AAAAAAAABBBBBBBAA
AAAAAAABBBABABBABA
A
Fig. 1. Proposed structure of the copolymers and fiber (a) chemicalschematic of fibers consisting of domain morphology.
ically in Fig. 1. In this model, the relative propor-tions of the two monomers as well as size of thesegments in polymer chains would decide thedomain structure of the copolymer, and therefore,its ultimate properties in the fiber form.
3.1. Synthesis of pH sensitive copolymers
Ideal structure for the above copolymer would bepure block copolymer; however, it is nearly impossi-ble to achieve pure block structure in radical poly-merization. Rather the distribution of monomersin such system depends on the reactivity ratios ofthe two monomers. In our case, the reactivity ratiosof the two monomers are widely different (reactivityratio of acrylic acid is 2.502 and acrylonitrile is0.495) [38] suggesting that simple copolymerizationwould lead to formation of homopolymer of acrylicacid (AA). Therefore, it was decided to synthesizecopolymers with enriched segments (rather thanpure block) of the two monomer-moieties using reg-ulated dosing of the more reactive monomer-acrylic
CH2 CH
CO
OH
n
BAAAAABBBBBBB
ABABBABBABABBB
crylonitrile domain
Acrylic acid domains
structure (b) schematic of AA43B (c) schematic of AA43R (d)

1070 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
acid during polymerization. Acrylic acid (AA) wasadded in small doses separated by prespecified timeintervals (arrived at after several experimental trialsand subsequent characterization of resultingcopolymers). It was hoped that whenever a doseof the acrylic acid was added it would get incorpo-rated into the growing chains as a short block orenriched segment owing to its high reactivity ratio.During the interval between any two doses, scarcityof the acrylic acid monomer in the reaction mediumwould force the acrylonitrile monomer to get incor-porated as short blocks or enriched segments ofacrylonitrile moieties in the growing polymerchains.
The prepared copolymers were found to be insol-uble in acetone and water, which are solvents forpolyacrylic acid. Suggesting that approach of multi-ple dosing has successfully suppressed the polymer-ization of AA homopolymer, which had a very highprobability of formation.
In order to ascertain the effect of this approachon the properties of the resultant polymer and itsfiber, another copolymer with a near random archi-tecture was also polymerized. In this, acrylic acidwas added continuously at a very slow rate intothe polymerization medium in order to compensate
Fig. 2. (a) 13C NMR spectrum of block copolymer AA43B in DMSO-dregion.
for the high reactivity of the acrylic acid monomerwith its lower concentration compared to acryloni-trile at any time inside the reactor. The proposedstructure of the copolymers is shown in Fig. 1.
3.2. Architecture of the copolymers
The 13C NMR spectra of AA43B (block copoly-mer) and AA43R (random copolymer) in DMSO-d6
are shown in Figs. 2 and 3, respectively. In thecopolymers, the carbonyl and nitrile carbon reso-nances are around o174–o177 and o119–o123 ppm,respectively. These NMR spectra were used to findthe composition and the sequence length distribu-tion of the monomer units. The spectral regionaround o40–o20 ppm was assigned to aliphatic car-bon resonance. The methine (–CH) signals of acrylicacid (B) unit appeared around o40 ppm in both thecases and the methine (–CH) signals of acrylonitrile(A) unit appeared around o30–o20 in both thecopolymers.
The overall comonomer composition of the twocopolymers – AA43B and AA43R were very similarand was calculated to be 51.14 and 49.93 mol% ofacrylonitrile (A) moieties by 13C NMR. The compo-
6, (b) the expanded carbonyl carbon region and (c) nitrile carbon

Fig. 3. (a) 13C NMR spectrum of random copolymer AA43R in DMSO-d6, (b) the expanded carbonyl carbon region, and (c) nitrilecarbon region.
A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076 1071
sition was also confirmed by acidimetric titrationand the results are given in Table 1.
The expanded 13C NMR spectra of the nitrile(–CN) and carbonyl (–CO) carbon resonances ofthe poly(acrylonitrile(A)-b-acrylic acid(B)) areshown as insets in Figs. 2 and 3. In order to under-stand the distribution of A and B units in thecopolymers, the triads of the two regions were ana-lyzed quantitatively. The analysis was based on thesplitting resulting from the monomer distributioneffects only. In the nitrile region, only two peakswere visible in AA43B. The signals around o119–o120 and o120–o121 ppm were assigned to AAAand BAA triads, respectively.
The addition of the acrylic acid (B) to the triadcauses a downfield shift in the position of acryloni-trile carbonyl (A) in AAB. In the carbonyl carbonregion, the resonance of the signals was around
Table 1Properties of copolymers
Copolymer Molecular weight (Mv)(g/mol)
Composition (AA mol%)
By 13CNMR
Byacidimetry
AA43B 3.6 · 105 48.86 46.05AA43R 3.5 · 105 50.07 48.07
o174.0–o174.7, o174.8–o175.6, o175.7–o176.4 ppm.These change with the copolymer composition andcan be assigned to ABA, BBA and BBB triadsequence, respectively. The relative fractions of var-ious A and B centered triads were obtained forAA43B copolymer by integrating the area fromthe resonance signals.
Similarly, the triad composition of AA43R wasanalyzed and their relative fractions were evaluatedby integration of the area under the signal peaks.The composition of the triads for the two copoly-mers is given in Table 2. The table also shows theexpected triad composition of a perfectly randomcopolymer of A and B with nearly 50:50 mol%comonomer composition.
From the values of AA43B, one may infer thatthough the copolymer does not have a pure blocktype structure, it has enriched segments of A andB, and therefore, its structure may be considerednear to the block type. This is also evident fromthe fact that there is no BAB component in thenitrile region and the composition of ABA is muchless compared to that of BBB and BBA, in the car-bonyl region. This implies that the approach of con-trolled dosing used in the experiment was effective inproducing copolymers with separate enriched seg-ments of acrylonitrile and acrylic acid moieties.
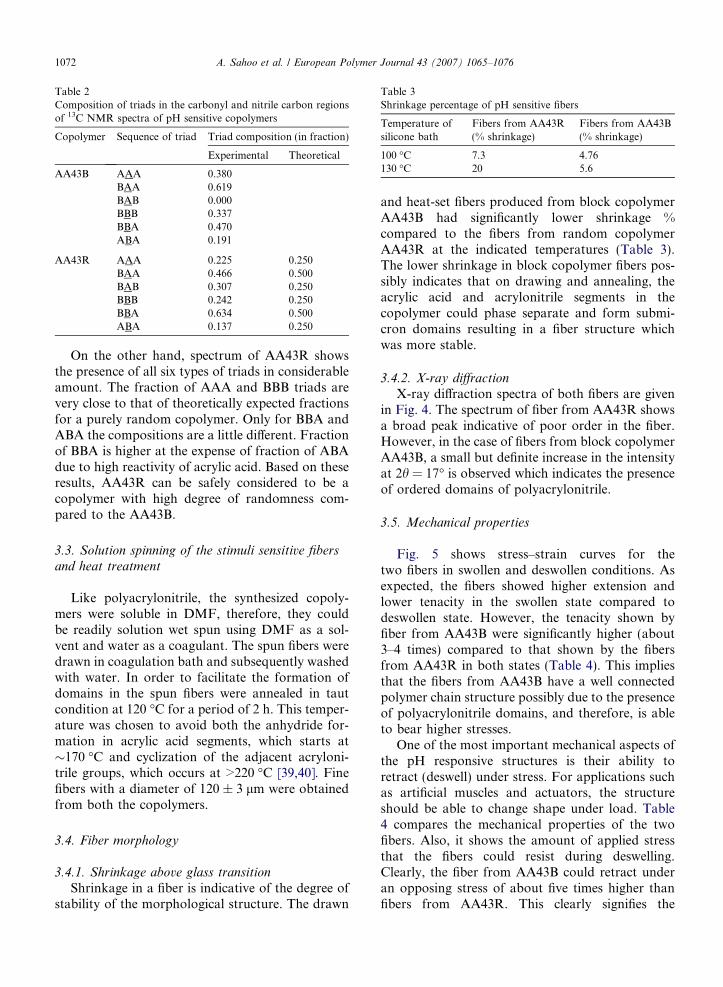
Table 3Shrinkage percentage of pH sensitive fibers
Temperature ofsilicone bath
Fibers from AA43R(% shrinkage)
Fibers from AA43B(% shrinkage)
100 �C 7.3 4.76130 �C 20 5.6
Table 2Composition of triads in the carbonyl and nitrile carbon regionsof 13C NMR spectra of pH sensitive copolymers
Copolymer Sequence of triad Triad composition (in fraction)
Experimental Theoretical
AA43B AAA 0.380BAA 0.619BAB 0.000BBB 0.337BBA 0.470ABA 0.191
AA43R AAA 0.225 0.250BAA 0.466 0.500BAB 0.307 0.250BBB 0.242 0.250BBA 0.634 0.500ABA 0.137 0.250
1072 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
On the other hand, spectrum of AA43R showsthe presence of all six types of triads in considerableamount. The fraction of AAA and BBB triads arevery close to that of theoretically expected fractionsfor a purely random copolymer. Only for BBA andABA the compositions are a little different. Fractionof BBA is higher at the expense of fraction of ABAdue to high reactivity of acrylic acid. Based on theseresults, AA43R can be safely considered to be acopolymer with high degree of randomness com-pared to the AA43B.
3.3. Solution spinning of the stimuli sensitive fibersand heat treatment
Like polyacrylonitrile, the synthesized copoly-mers were soluble in DMF, therefore, they couldbe readily solution wet spun using DMF as a sol-vent and water as a coagulant. The spun fibers weredrawn in coagulation bath and subsequently washedwith water. In order to facilitate the formation ofdomains in the spun fibers were annealed in tautcondition at 120 �C for a period of 2 h. This temper-ature was chosen to avoid both the anhydride for-mation in acrylic acid segments, which starts at�170 �C and cyclization of the adjacent acryloni-trile groups, which occurs at >220 �C [39,40]. Finefibers with a diameter of 120 ± 3 lm were obtainedfrom both the copolymers.
3.4. Fiber morphology
3.4.1. Shrinkage above glass transition
Shrinkage in a fiber is indicative of the degree ofstability of the morphological structure. The drawn
and heat-set fibers produced from block copolymerAA43B had significantly lower shrinkage %compared to the fibers from random copolymerAA43R at the indicated temperatures (Table 3).The lower shrinkage in block copolymer fibers pos-sibly indicates that on drawing and annealing, theacrylic acid and acrylonitrile segments in thecopolymer could phase separate and form submi-cron domains resulting in a fiber structure whichwas more stable.
3.4.2. X-ray diffractionX-ray diffraction spectra of both fibers are given
in Fig. 4. The spectrum of fiber from AA43R showsa broad peak indicative of poor order in the fiber.However, in the case of fibers from block copolymerAA43B, a small but definite increase in the intensityat 2h = 17� is observed which indicates the presenceof ordered domains of polyacrylonitrile.
3.5. Mechanical properties
Fig. 5 shows stress–strain curves for thetwo fibers in swollen and deswollen conditions. Asexpected, the fibers showed higher extension andlower tenacity in the swollen state compared todeswollen state. However, the tenacity shown byfiber from AA43B were significantly higher (about3–4 times) compared to that shown by the fibersfrom AA43R in both states (Table 4). This impliesthat the fibers from AA43B have a well connectedpolymer chain structure possibly due to the presenceof polyacrylonitrile domains, and therefore, is ableto bear higher stresses.
One of the most important mechanical aspects ofthe pH responsive structures is their ability toretract (deswell) under stress. For applications suchas artificial muscles and actuators, the structureshould be able to change shape under load. Table4 compares the mechanical properties of the twofibers. Also, it shows the amount of applied stressthat the fibers could resist during deswelling.Clearly, the fiber from AA43B could retract underan opposing stress of about five times higher thanfibers from AA43R. This clearly signifies the

10 20 302515 35
500
1000
1500
AA43R
AA43B
10 20 302515 35
500
1000
1500
10 20 302515 35
500
1000
1500
Position [ 2 theta] 0
Cou
nts
Fig. 4. X-ray diffraction of the heat-set pH sensitive fibers.
0
5
10
15
20
25
30
35
40
0 20 40 60 80 100 120Strain (%)
Ten
acity
(M
Pa)
AA43B swollen AA43R swollen
AA43R deswollen
AA43B deswollen
Fig. 5. Stress–strain curves of the pH sensitive fibers in bothswollen and deswollen states.
A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076 1073
improvement obtained by controlling the chemicalarchitecture and physical morphology of the fibers.
Table 4Mechanical properties of pH sensitive fibers
Copolymers Retractive stress during deswelling(MPa)
Breaking stress d(MPa)
AA43B 0.26 3.31AA43R 0.058 0.78
These results also suggest that the block typecopolymers have significant presence of both AAand AN monomer moieties and do not have largecompositional variation among various chains.Had the compositional variation been large, the var-ious domains would have had a poor connectivity,and therefore, significantly lower mechanical prop-erties than the fibers from random copolymers.
3.6. Evaluation of transition properties
The pH sensitive fibers showed swelling anddeswelling in alkaline and acidic pH solutions,respectively. The variation in diameter of the fiberwith change in external pH solution is shown inFig. 6. The pKa value of polyacrylic acid is 4.28
uring swollen condition Breaking stress in deswollen condition(MPa)
34.2913.55
0
50
100
150
200
250
300
350
400
450
500
0 2 4 6 8 10 12 14pH
Dia
in m
icro
ns
AA43B
AA43R
Deswelling
Swelling
Fig. 6. Diameter of pH sensitive fibers as a function of pH of theexternal solution during swelling and deswelling cycles: (1)AA43B, (2) AA43R.
0
50
100
150
200
0 1 2 3 4 5 6 7 8 9 10 11 12 13Time (min)
Ch
ang
e in
len
gth
(%
)
AA43B
AA43R
Fig. 8. Rate of deswelling of the pH sensitive fibers under a stressof 0.05 MPa.
0
20
40
60
80
100
120
140
160
180
200
0 10 20 30 40 50 60 70 80 90 100 110 120 130 140 150 160Time in min
Ch
ang
e in
len
gth
(%
) AA43B
AA43R
Fig. 7. Rate of transition (swelling) of pH sensitive fibers under astress of 0.05 MPa.
1074 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
[41]. Therefore, below pH 4.28 the H+ ion concen-tration in the solution is very high. This effectivelysuppresses the ionization of carboxylic acid groupsand the carboxylate groups change to COOHgroups. The polymeric chains in the fiber remainin the coiled state resulting in deswelling. As thepH is increased above the pKa value, the fiber swellsbecause the concentration of negatively charged car-boxylate ions increases. When the carboxylategroups in the polymer chain are fully ionized theeffect of electrostatic repulsion is the highest andthe gel fibers expand significantly at about a pH of10. Similarly the complete collapse of the fiber wasachieved at a pH of 2.
A hysteresis was observed in fiber from AA43Bimplying that the swelling did not start until thepH was increased above 6, and similarly in thereverse cycle, the deswelling did not begin untilthe pH was decreased below 5.5. However, the tran-sition was sharp and was complete in a narrow pHrange. On the other hand, in the case of fibers fromAA43R the hysteresis was significantly higher. Theswelling started at a pH 8 and continued till a pHof 12 while the deswelling started at a pH 5 andthe fiber did not achieve its original shape even ata pH of 2.
The fibers from AA43B achieved equilibriumswelling in slack condition within 45 min with a vol-umetric change of about 3890% (change in lengthwas 227% and diameter was 414%), whereas, theequilibrium swelling of the fibers from AA43Rwas about 1730% (length: 168%; diameter: 321%).
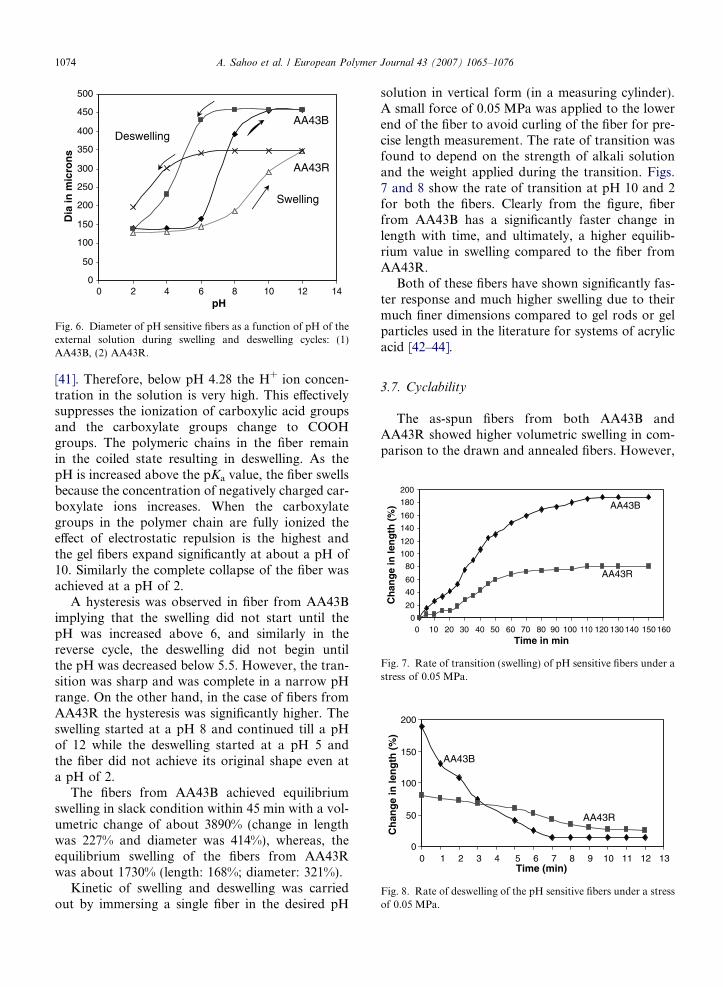
Kinetic of swelling and deswelling was carriedout by immersing a single fiber in the desired pH
solution in vertical form (in a measuring cylinder).A small force of 0.05 MPa was applied to the lowerend of the fiber to avoid curling of the fiber for pre-cise length measurement. The rate of transition wasfound to depend on the strength of alkali solutionand the weight applied during the transition. Figs.7 and 8 show the rate of transition at pH 10 and 2for both the fibers. Clearly from the figure, fiberfrom AA43B has a significantly faster change inlength with time, and ultimately, a higher equilib-rium value in swelling compared to the fiber fromAA43R.
Both of these fibers have shown significantly fas-ter response and much higher swelling due to theirmuch finer dimensions compared to gel rods or gelparticles used in the literature for systems of acrylicacid [42–44].
3.7. Cyclability
The as-spun fibers from both AA43B andAA43R showed higher volumetric swelling in com-parison to the drawn and annealed fibers. However,

0
500
1000
1500
2000
2500
3000
3500
4000
0 1 2 3 4 5 6Cycles
Vo
lum
etri
c sw
ellin
g (
%)
AA43B
AA43R
Fig. 9. Cycles of swelling and deswelling of the pH sensitivefibers.
A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076 1075
the as-spun fibers were not able to withstandrepeated cycling and were disintegrated. The fiberson heat-setting at 120 �C were stable to repeatedcycling and did not show noticeable deteriorationin strength even after �15 cycles for AA43B. Thecyclic swelling/deswelling behavior of the two fibersfor the first four cycles is shown in Fig. 9. Theannealed fiber from AA43B exhibited excellentreversibility, however, the fibers from AA43Rshowed incomplete reversibility. During the firsttwo cycles in fibers from AA43B, the swelling wasin the range of �1200–1300%. Subsequently, theswelling % increased to �3300–3669%. This maybe due to the fact that the fibers underwent condi-tioning in the first two cycles and then in the subse-quent cycles showed a stable response. In the case offibers from AA43R, initial two cycles showed swell-ing in the range of �700–1000%, which increasedonly to �1500% in the subsequent cycles.
4. Conclusions
pH sensitive polymers, poly(acrylonitrile-co-acrylic acid:50:50), were synthesized using freeradical polymerization. The architecture of thecopolymers could be successfully controlled usingthe approach of regulated dosing of the more reac-tive comonomer-acrylic acid. Using quantitative 13CNMR, copolymer (AA43B) prepared with intermit-tent dosing of acrylic acid was found to have theblock-type architecture, while the one (AA43R) pre-pared with continuous dosing of acrylic acid wasfound to have comparatively more random distribu-tion of comonomer moieties.
The copolymers were converted to fibers by solu-tion spinning and annealed at 120 �C to get a stablemorphology. The shrinkage and X-ray diffractionstudies indicated that enriched/block segments of
monomer moieties in copolymer AA43B, on conver-sion to fiber and annealing, could phase separateand form segregated domain morphology, whileno such structure could be formed in fibers fromAA43R.
The chemical architecture and physical morphol-ogy of the fibers were found to have a significantimpact on their mechanical properties, transitionbehavior and retractive forces during the transition.The fibers from AA43B showed a stable, reversibletransition with an equilibrium volumetric swellingof 3300–3600% at the pH 10, however, the fibersfrom AA43R showed volumetric swelling of only1200–1500% and did not show complete reversibil-ity of the transition. Also, the fibers from AA43Bshowed significantly higher strength (3–4 times),extensibility (up to 1.5 times), and retractive stresses(five times) during the transition compared to thefibers from AA43R. The rate of transition was alsofound to be higher for the former fibers.
The study suggests that the control of the chem-ical architecture and physical morphology of the pHresponsive structure may be the key in producingpH responsive fibers with desirable mechanicalproperties and pH response that is suitable for theirapplications in artificial muscles and actuators.
Acknowledgements
The authors acknowledge the partial financialsupport provided for this work under the researchGrant RP01429 from the Ministry of Human Re-source Development, Government of India.
References
[1] Byrne ME, Park K, Pappas NA. Adv Drug Delivery Rev2002;54:149–61.
[2] Serres A, Baudys M, Kim SW. Pharmaceut Res 1996;13(2):196–201.
[3] Fiel G, Bae YH, Feijin J, Kim SW. J Membr Sci 1991;64:283.
[4] Oak MS, Kobayashi T, Fukaya T, Fujii N, Wang H. JMembr Sci 1997;123:185–95.
[5] Kobayashi T, Fukaya T, Fujii N. J Membr Sci 2000;164:157–66.
[6] Lin F, Tao GL, Zhu RX. Polym J 1993;25:561.[7] Park TG, Hoffman AS. Biotechnol Bioeng 1990;24:21.[8] Peng T, Cheng YL. Polymer 2001;42:2091.[9] Beebe DJ, Moore J, Bauer JM. Nature 2000;404:588–90.
[10] Shahinpoor M. J Intell Mater Syst Struct 1995;6:307–14.[11] Kaneko D, Gong JP, Osada Y. J Mater Chem 2002;12(8):
2169.[12] Kajiwara K, Ross Murphy SB. Nature 1992;355:208–9.[13] Osada Y, Okuzaki H, Hori H. Nature 1992;355:242–4.

1076 A. Sahoo et al. / European Polymer Journal 43 (2007) 1065–1076
[14] Shahinpoor M. Artificial muscles. Encyclopedia of bioma-terials and biomedical engineering, vol. 1. New York(NY): Marcel Dekker Inc; 2004. p. 43–52.
[15] Siegal RA, Firestone BA. Macromolecules 1988;21(11):3254.[16] Bae YH, Tokano RH, Kim SW. Macromol Rapid Commun
1987;8:481.[17] Tanaka T, Nishio I, Sung ST, Nishio SU. Science 1982;218:
467.[18] Ohmine I, Tanaka T. J Chem Phys 1982;77:5725.[19] Hoffman AS et al. J Biomed Mater Res 2000;52:577–86.[20] Tao X. Smart fibres and fabrics and clothing. 1st
ed. England: Woodhead Publishing Limited; 2000. p. 93–107.
[21] Schreyer HB, Gebhart N, Kim KJ, Shahinpoor M. Biomac-romolecules 2000;1:642–7.
[22] Tonge SR, Tighe BJ. Adv Drug Delivery Rev 2001;53:109–22.
[23] Zareie HM, Bulmus EV, Gunning AP, Hoffman AS, PiskinE, Morris VJ. Polymer 2000;41:6723–7.
[24] Jianqi F, Lixia G. Eur Polym J 2002;38(8):1653–8.[25] Huang J, Gongneng MZ. Gaofenzi Xuebao 2002;15(2):
177–80.[26] Nakamura K, Peppas NA. J Control Release 1999;61:329–35.[27] Marsano E, Bianchi E, Sciutto L. Polymer 2003;44:6835–41.[28] Kabiri K, Omidian H, Zohuriaan-Mehr M. J Polym Int
2003;52(7):1158–64.[29] Kabiri K, Omidian H, Zohuriaan-Mehr MJ, Hashemi SA.
Eur Polym J 2003;39(7):1341–8.[30] Jassal M, Agrawal AK, Vishnoi A, Save NS. J Appl Polym
Sci 2005;95(3):681–8.
[31] Umemoto S, Matsumura T, Sakai T, Okui N. Polym GelsNetworks 1993;1(2):115–26.
[32] Guo X, Gong Z, Liu D, Hua J, Wang Q. Ying Yong KexueXuebao 1999;17(4):439–44.
[33] Schreyer HB, Shahinpoor M, Kim KJ. Proc SPIE Int SocOpt Eng (Electroactive Polymer Actuators and Devices)1999;3669:192–8.
[34] Jassal M, Agrawal AK, Ghosh AK, Ramasubramani KRT,Sahoo A. Stimuli responsive fibres from acrylic fibres: Effectsof modification conditions on transition properties. In:Jassal M, Agrawal AK, editors. Proceedings of the interna-tional conference on emerging trends in polymers andtextiles. Delhi: IIT; 2005. p. 114–9.
[35] Bandrup J, Immergut EH, Grulke EA. Polymer handbook.4th ed. New York: Wiley; 1999 [chapter VII-11].
[36] Brar AS, Dutta K. Eur Polym J 1998;34(11):1585–97.[37] Bajaj P, Sreekumar TV, Sen K. J Appl Polym Sci 2001;79:
1640–52.[38] Bajaj P, Sen K, Bahrami SH. J Appl Polym Sci 1996;59:
1539–50.[39] Jin X, Hsieh YL. Polymer 2005;46:5149–60.[40] Bajaj P, Sen K, Bahrami SH. J Appl Polym Sci 2003;88:
685–98.[41] Jianqi F, Lixia G. J Appl Polym Sci 2002;85:2423–30.[42] Bajpai SK. J Appl Polym Sci 2001;80:2782–9.[43] Moolee Y, Kim SH, Cho CS. J Appl Polym Sci 1996;62:
301–11.[44] Bajpai SK, Saxena S. J Appl Polym Sci 2004;92:3630–43.


![Index [application.wiley-vch.de]...acrylonitrile–styrene–acrylic ester, 264 activation stress, 237 actual temperature, 55 Adam–Gibbs model, 14, 31, 36 additives, 232 – value,](https://img.pdfslide.net/doc/110x75/613be29df8f21c0c82694050/index-acrylonitrileastyreneaacrylic-ester-264-activation-stress-237.jpg)



















![Controlled polymers for pigment dispersants€¦ · pigment dispersants [6]. In this contribution acrylic block copolymer type "controlled pigment dispersants" are presented based](https://img.pdfslide.net/doc/110x75/5ea9e9ef0447ea48144fa6b6/controlled-polymers-for-pigment-pigment-dispersants-6-in-this-contribution-acrylic.jpg)
![Glycidyl Azide Polymer and its Derivatives-Versatile Binders ......Non-energetic polymers, such as hydroxyl-terminated polybutadiene (HTPB, NB1 ) [3] and butadiene-acrylonitrile-acrylic](https://img.pdfslide.net/doc/110x75/60e59159c5aba964866e5bb5/glycidyl-azide-polymer-and-its-derivatives-versatile-binders-non-energetic.jpg)





