Embed Size (px)
Citation preview

E F F E C T S A N D D Y N A M I C S O F I N S E R T I O N S E Q U E N C E S I N T H E E V O L U T I O N O F C Y A N O B A C T E R I A
Carl Theoden Vigil Stenman


Effects and dynamics of insertion sequences in the evolution of cyanobacteria
Carl Theoden Vigil Stenman

©Carl Theoden Vigil Stenman, Stockholm University 2015 ISBN at final page Printed in Sweden by Universitetsservice, Stockholm 2015 Distributor: Department of Ecology, Environment and Plant Sciences

To my sister Ninna, who has always been there for me.


Abstract
Cyanobacteria are globally widespread and ecologically highly significant photoautotrophic microorganisms, with diverse geno- and phenotypic char-acters unprecedented among prokaryotes. This phylum embraces representa-tives with an exclusive adaptability in highly specialized environments, from oligotrophic ocean waters to the interior of cells in symbiotic plants, the most extreme being the chloroplasts. Insertion sequences (ISs) are short (~1000 bp) mobile genetic elements prevalent in microbial genomes, poten-tially representing potent adaptive forces.
In this thesis, hypotheses tested that ISs play significant roles in both re-
ductive and adaptive evolution in physiologically versatile cyanobacteria, using two model systems. First, the genome of an obligate plant (Azolla) symbiont, the cyanobacterium ‘Nostoc azollae 0708’, was sequenced, which led to the discovery of a highly ‘eroding’ genome (5,48 Mbp), loaded with ISs covering 14% of the genome, a situation likely caused by the relaxed selection pressure within the plant. The ISs were located in close proximity to the extremely numerous pseudogenes identified, although genes with key functions in a symbiotic context escaped IS mediated erosion (e.g. nitrogen fixation and differentiation genes). Some ISs were shown to have transposed short distances within the genome (‘local hoping’), and to be likely causative agents in pseudogene formation, and thus pivotal actors in the reductive evo-lution discovered.
To widen the scope of ISs further, additionally 66 phylogenetically di-
verse microorganisms with a variety of life styles (free-living, symbionts, pathogens) were examined in regards to ISs influence. The data verified their over-all importance in shaping microbial genomes.
Finally, natural microbial populations in the Baltic Sea, a semi-enclosed
geologically young (~10,000 years) brackish water body offering steep gra-dients in salinity and nutrient loads, were examined using metatranscriptom-ics and metagenomics. A large proportion of the metagenome was devoted to ISs and most importantly a large fraction of the metatranscriptome con-sisted of IS transcripts (~1%), which may be suggestive of a high IS activity. These phenomena were most apparent in cyanobacteria in central parts of the Baltic Sea. The presence of an especially rich abundance of ISs in brackish

waters was further substantiated by their low frequency (< 0.1%) in mi-crobes of marine waters. Hence, ISs may facilitate both adaptations (short term) and adaptive evolution (long term) in microbes entering brackish wa-ter, otherwise unable to cross the distinct limnic-to-marine salinity-divide.
Together, the data reveal high genomic loads of ISs in cyanobacteria sub-ject to highly demanding conditions and stress the importance of locally migrating ISs (and pseudogenization) as important facilitators in adaptation and evolution, being a more rapid process than hitherto expected. The find-ings strongly support current theories stating a crucial role of ISs in shaping microbial genomes to render fitness.


List of publications
This thesis is based on the following publications and manuscripts:
I Ran, L, Larsson, J, Vigil-Stenman, T, Nylander, JAA, Ininbergs, K, Zheng, W-W, Lapidus, A, Lowry, S, Haselkorn, R, and Berg-man, B: Genome Erosion in a Nitrogen-Fixing Vertically Transmitted Endosymbiotic Multicellular Cyanobacterium. PLoS One 2010, 5:e11486.
II Vigil-Stenman T, Larsson J, Nylander JAA, Bergman B: Local
hopping mobile DNA implicated in pseudogene formation and reductive evolution in an obligate cyanobacteria-plant symbiosis. BMC Genomics 2015, 16:193.
III Vigil-Stenman T, Ekman M, Bergman B: High activity of inser-
tion sequences in the Baltic Sea. Manuscript, 2015 My contributions to these were as follows:
Paper I: Collaborated in isolation of samples, bioinformatic analyses and in writing the paper.
Paper II: Designed and operated the software used in the identification and analysis insertion sequences. Wrote main part of the paper.
Paper III: Identified and analysed insertion sequences. Wrote main part of the paper.

Contents
ABSTRACT ............................................................................................... VII
LIST OF PUBLICATIONS .............................................................................. X
INTRODUCTION ...................................................................................... 13 CYANOBACTERIA .......................................................................................... 13 FEATURES OF CYANOBACTERIA ........................................................................ 15 CYANOBACTERIA IN SYMBIOSIS ........................................................................ 16 THE NOAZ-‐AZOLLA SYMBIOSIS ....................................................................... 17 INSERTION SEQUENCES .................................................................................. 19
Insertion sequence abundance ............................................................ 22 Variability of insertion sequences ........................................................ 22 Local hopping ....................................................................................... 23 Insertion sequences activity ................................................................. 23 Insertion sequences in evolution .......................................................... 24
THE BRACKISH WATER BALTIC SEA ................................................................... 25 AIMS ....................................................................................................... 27
METHODS ............................................................................................... 29 RESULTS .................................................................................................. 33
IS ELEMENTS IN A CYANOBACTERIUM OF AN OBLIGATE PLANT SYMBIOSIS ................ 33 ISS IN ADDITIONAL SYMBIONTS AND OTHER CYANOBACTERIA ................................ 35
IS elements and Local hopping ............................................................. 37 IS proximity to pseudogenes ................................................................ 37
IS ELEMENTS AND ADAPTIVE EVOLUTION IN THE BALTIC SEA ................................. 38 FINAL REMARKS AND CONCLUSIONS ...................................................... 41
FUTURE PERSPECTIVES ........................................................................... 43 SWEDISH SUMMARY .............................................................................. 44
ACKNOWLEDGEMENTS ........................................................................... 46
REFERENCES ........................................................................................... 47

Abbreviations
bp base pair(s) cyanobiont Cyanobacterial symbiont endosymbiont Symbiont residing within the body of its host IS insertion sequence kbp kilo base pairs Mbp mega base pairs MGE mobile genetic element NCBI National Center for Biotechnology Information NoAz Nostoc azollae ‘0708’ ORF open reading frame Pseudogene Gene that results in a non-functioning protein Pseudogenization Process that creates a pseudogene psu Practical salinity unit transposase Protein product of IS that facilitates transposition transposition Moving DNA from one genomic position to another

13
Introduction
Cyanobacteria The history of cyanobacteria stretches back to the very origin of our discern-ible evolutionary history on Earth. From their first appearance as oxygenic photosynthesizers in an oxygen-free environment approximately 2.7 billion years ago (Lyons, 2007, Domínguez-Escobar et al., 2011), the cyanobacteria have since changed the biosphere of our planet to the green, aerobic system it is today. Being the ancestors of oxygen-evolving photosynthetic eukaryot-ic organisms (Bhattacharya et al., 2007, Rasmussen et al., 2008, Deusch et al., 2008), the progenitors of today’s cyanobacteria produced oxygen as a by-product of their photosynthesis (Mulkidjanian et al., 2006), and slowly changed the existing atmosphere from an oxygen free reducing environment to the oxygenated one we enjoy today. This simultaneously brought about the greatest environmental disaster of all times, causing the demise of count-less anaerobic species while paving the road for present day aerobic organ-isms. In present times the cyanobacterial ancestors continue to influence our planet. Firstly, they are the ancestors of all organisms performing oxygenic photosynthesis (Bhattacharya et al., 2007, Rasmussen et al., 2008, de Alda et al., 2014). Recent data suggest that in a monophyletic event about two bil-lion years ago, a differentiating filamentous cyanobacterium (Martin et al., 2002, Deusch et al., 2008) entered into an endosymbiotic relationship with a pigment-free eukaryotic organism. This led to the genesis of a new kingdom of organisms, that of embryophyte algae and eventually all land plants, the latter process likely starting about 600 million years ago (Rasmussen et al., 2008). Secondly, cyanobacteria, along with other microorganisms, to this day contribute massively to fundamental biogeochemical cycles by splitting the strong triple bond of atmospheric dinitrogen and integrating the bioavail-able nitrogen species generated (NH4
+) into pivotal cellular compounds ac-cessible to the rest of the Earth’s organisms (Berman-Frank et al., 2001, Usher et al., 2007).
The phylum Cyanobacteria consists (Oren, 2011) of a large group of or-ganisms that soon evolved from being small unicellular nano- and picobacte-ria (<2 µm) into also including larger filamentous forms (Schirrmeister et al., 2011), such as those with advanced cell differentiating capacities, the latter

14
even being discernible by the naked eye (Rippka et al., 1979, Mulkidjanian et al., 2006, Lin et al., 2011). In fact, cyanobacteria show the most advanced phenotypic complexity among the prokaryotic organisms, and are by tradi-tion morphologically divided into Sections I-V (Table 1). Their versatility is today also reflected in their unresolved phylogeny with contemporary ge-nome sizes stretching from approximately 1 up to 9 Mbp (Shi and Falkow-ski, 2008, Larsson et al., 2011).
Table 1: Cyanobacterial divisions according to the Bacteriological Code, listing common features and capabilities of the different subdivisions. Thylakoids: double membranes with pigments in subcellular
compartments for efficient photosynthesis. PBS: Phycobilisomes, thylakoid attached specialized light harvesting antenna composed of various types of phycobilin pigments. Motile: capability of movement, usually by gliding. Gas vesicles: mechanisms to regulate buoyancy. Heterocysts: cells differentiated to perform nitrogen fixation, with thickened cells walls and lack of photosystem II, in order to protect nitrogen-fixing enzymes from oxygen. Hormogonia: small celled filaments capable of gliding movement. Akinetes: resilient resting state type of cells. All sample species listed have been investigated in the
thesis, except for those in parenthesis, which are merely included as representative species of Section II and V.
Bacteriologial subsection
Samplegenera
Samplespecies
Distinguishingtraits
Features and capacities
V
Filamentous with
heterocysts,branching
morphology
FischerellaCalothrix
Stigonema
(Fischerella sp. JSC-11)
(Mastigocladopsis repens PCC 10914)
Thylakoids, PBS,motile,
heterocysts,hormogonia
IVFilamentous
with heterocysts Nostoc
Anabaena
Nodularia
Anabaena variabilis ATCC 29413
Nostoc punctiforme PCC 73102
Nostoc sp. PCC 7120
Nodularia spumigena CCY 9414
Thylakoids, PBS,motile, gas vesicles,
heterocysts,hormogonia,
akinetes
IIIArthrospira maxima CS-328
Trichodesmium erythraeum IMS101
Arthrospira platensis NIES-39FilamentousPseudoanabaena
Oscilatoria
Thylakoids, PBS, motile,
gas vesicles
II(Pleurocapsa sp. PCC 7319)
(Xenococcus sp. PCC 7305)Coccoid,
Internal fission
Dermocarpella
PleurocapsaThylakoids, PBS
I
Synechococcus
Prochlorococcus
Cyanothece
Microcystis
Acaryochloris marina MBIC11017
Crocosphaera watsonii WH 8501
Prochlorococcus marinus MIT9303
Microcystis aeruginosa NIES-843
Synechococcus sp. CB0101
Coccoid,Binary fission
Thylakoids, PBS

15
Features of cyanobacteria For being prokaryotes cyanobacteria show remarkable phenotypic complexi-ty. For instance, some larger cyanobacterial species exhibit cooperative mul-ticellularity (filamentous growth) and differentiation into cell types special-ized for motility and proliferation (hormogonia) and resilience (akinetes/spores) (Rippka et al., 1979, Adams and Duggan, 1999, Berman-Frank et al., 2001, Zhou and Wolk, 2002, Wolk et al., 2004, Haselkorn, 2007). In spite of lacking organelles (enclosed in double membranes), all cyanobacteria still practice compartmentalization of functions into specific subcellular structures (e.g. polyphosphate and cyanophycin granules, car-boxysomes). Some show distinct circadian rhythms (Terauchi and Kondo, 2008), while planktonic species can practice buoyancy control (Reynolds et al., 1987). Yet other cyanobacteria are capable of chromatic adaptation (De Marsac and Houmard, 1988) as well as communication between individuals (quorum sensing) (Schuster and Greenberg, 2007) and along their filaments (Haselkorn, 2007). They are also able to synthesize a wide variety of com-pounds, besides carbohydrates via oxygenic photosynthesis and nitrogen via nitrogen fixation, such as hydrogen gas (Pinto et al., 2002) and a wide range of substances that are toxic to bacteria and eukaryotes e.g. microcystin and nodularin (Sivonen and Börner, 2008, Jonasson et al., 2010, Berntzon et al., 2015). Today, cyanobacteria are widely screened for compounds of biotech-nological importance, including e.g. biofuels (Hess, 2011).
A most important event in the life cycle of certain cyanobacteria (Figure 1) is the differentiation of heterocysts (Haselkorn, 2007, Flores and Herrero, 2010). These are cells with reinforced cells walls and abolished oxygenic photosynthesis (PS II components missing) although with retained energy production (via PS I) to feed the highly energy demanding nitrogen fixation process. Under nitrogen deprivation, heterocysts develop at regular intervals (approximately 1 in 10 cells) along the interconnected cells of filamentous cyanobacteria of Section IV-V (Adams and Duggan, 1999, Flores and Herre-ro, 2010). These are the exclusive sites for the nitrogen fixation process in filamentous cyanobacteria, while unicellular cyanobacteria (Section I-II) and some filamentous non-heterocystous types (Section III) practice a temporal (day/night) separation between the two incompatible processes: oxygenic photosynthesis and nitrogen fixation (Bergman et al., 1997, Bergman et al., 2013). The specialized heterocyst cell forms are required as the enzyme cata-lysing the nitrogen fixation process, nitrogenase, is rapidly inactivated by oxygen. The nitrogen fixed is distributed to adjacent photosynthetic cells, and these in turn provide the heterocysts with photosynthetic products (car-bon skeletons and reducing power).
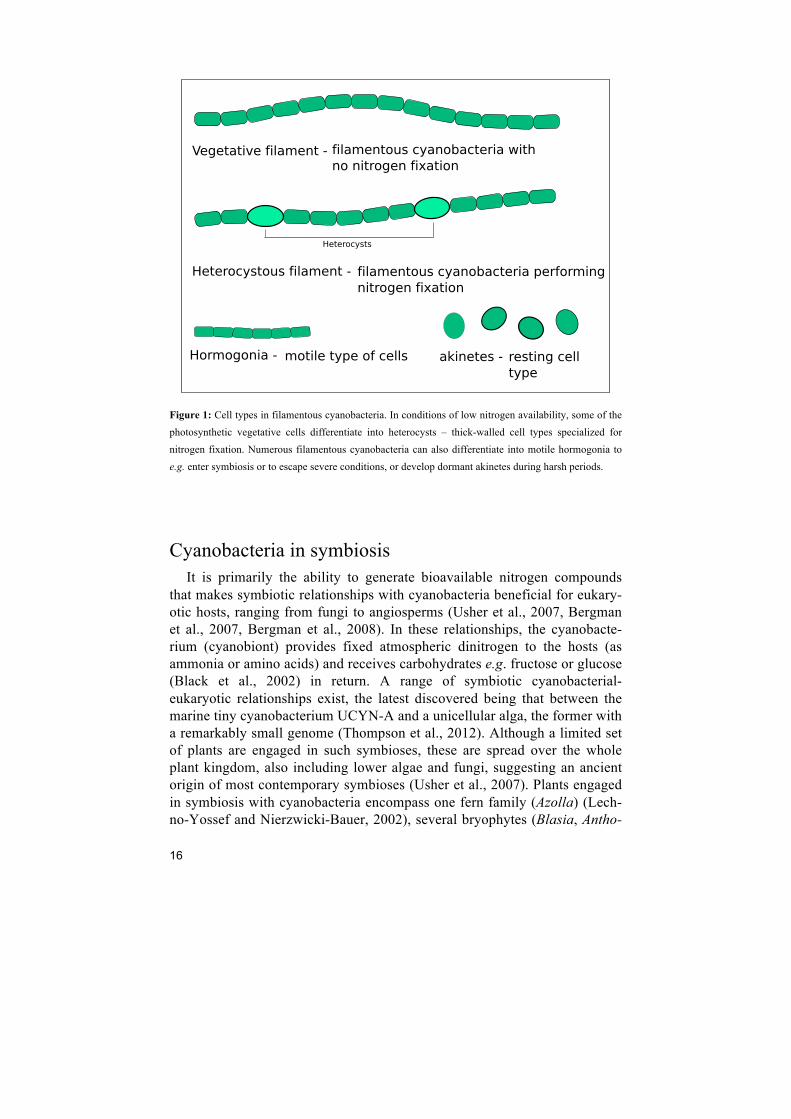
16
Figure 1: Cell types in filamentous cyanobacteria. In conditions of low nitrogen availability, some of the photosynthetic vegetative cells differentiate into heterocysts – thick-walled cell types specialized for nitrogen fixation. Numerous filamentous cyanobacteria can also differentiate into motile hormogonia to e.g. enter symbiosis or to escape severe conditions, or develop dormant akinetes during harsh periods.
Cyanobacteria in symbiosis It is primarily the ability to generate bioavailable nitrogen compounds
that makes symbiotic relationships with cyanobacteria beneficial for eukary-otic hosts, ranging from fungi to angiosperms (Usher et al., 2007, Bergman et al., 2007, Bergman et al., 2008). In these relationships, the cyanobacte-rium (cyanobiont) provides fixed atmospheric dinitrogen to the hosts (as ammonia or amino acids) and receives carbohydrates e.g. fructose or glucose (Black et al., 2002) in return. A range of symbiotic cyanobacterial-eukaryotic relationships exist, the latest discovered being that between the marine tiny cyanobacterium UCYN-A and a unicellular alga, the former with a remarkably small genome (Thompson et al., 2012). Although a limited set of plants are engaged in such symbioses, these are spread over the whole plant kingdom, also including lower algae and fungi, suggesting an ancient origin of most contemporary symbioses (Usher et al., 2007). Plants engaged in symbiosis with cyanobacteria encompass one fern family (Azolla) (Lech-no-Yossef and Nierzwicki-Bauer, 2002), several bryophytes (Blasia, Antho-
filamentous cyanobacteria with no nitrogen fixation
Hormogonia - akinetes -
Vegetative filament -
Heterocysts
Heterocystous filament - filamentous cyanobacteria performingnitrogen fixation
motile type of cells resting celltype

17
cerus) (Enderlin and Meeks, 1983), numerous gymnosperms restricted to cycads (e.g. Cycas) (Nathanielsz and Staff, 1975, Costa and Lindblad, 2002) and one angiosperm family (Gunnera) (Bergman, 2002, Osborne and Berg-man, 2009). The cyanobacterium is in the majority of cases extracellular in relation to the host, exceptions being in the marine diatoms and in the angio-sperm Gunnera. In the latter case, entire cyanobacterial filaments enter through the plant cell walls during colonization. Symbiotic cyanobacterial associations also include fungi as hosts, particularly numerous lichenized fungi (Rikkinen et al., 2002) including the ancestral lineage Geosiphon pyri-forme (Redecker et al., 2000). Additionally, several marine diatoms Rhi-zosolenia, Hemiaulus are engaged (Carpenter and Foster, 2002, Foster and Zehr, 2006), as is a unicellular alga Rhophalodia (Kneip et al., 2008), and a few marine sponges and corals (Usher et al., 2007). Although combined nitrogen is the resource provided to the host by most cyanobionts, there are also associations where the cyanobiont provides photosynthates to hetero-trophic hosts, such as in symbioses with certain fungi (e.g. the lichen Pelti-gera canina). Hence, in these cases the cyanobacteria have reassumed their ancient role as photosynthetic endosymbionts (perhaps eventually evolving into chloroplasts), providing carbon for the hosts (Herdman and Stanier, 1977).
In the symbiotic state, the nitrogen-fixing cyanobionts increase the fre-quency of heterocysts and its nitrogen-fixing ability, while restricting repro-duction to prevent the cyanobacterium from out-growing its host. These changes are likely elicited by the plant, although mechanisms are still un-known. Programmed cell death has been suggested to act as a modulator of the size of the cyanobacterial populations in symbiosis (Zheng et al., 2013). To elicit colonization, some host plants (Gunnera and the bryophytes) ex-crete chemoattractants that may also induce the differentiation of motile hormogonia, which in turn facilitate host colonization. The coevolution of cyanobionts and host plants is evident in that both partners to some extent alter their physiology to accommodate the symbiotic interactions and main-tain homeostasis (Bergman et al., 2008).
The NoAz-Azolla symbiosis In all cyanobacterial symbioses the relationship is mutualistic and in most
cases facultative, with each new plant generation requiring de novo infection by external cyanobionts. This means that the cyanobacterium must retain its capacity to live independently outside the plant between plant infection events. The only exception to this transmission style is found in the obligate symbiosis between the cyanobacterium Nostoc azollae ‘0708’ (hereafter

18
referred to as NoAz), a filamentous diazotrophic cyanobacterium of Section IV (Rippka et al., 1979), and the fern Azolla. In this relationship, the cyano-bacterium is maintained perpetually through plant generations, although it technically still resides extracellularly.
Azolla are species of small, 2-8 cm wide, ferns that grow floating on fresh water surfaces. Azolla is represented by seven species, each keeping its indi-vidual strain of cyanobiont (Plazinski et al., 1990, Zheng et al., 1999, Pa-paefthimiou et al., 2008). Azolla populates rivers, canals and ponds in south-ern Europe, Asia and America (Moore, 1969). In South East Asia and China they are utilized as a natural nitrogen fertilizer in rice paddies.
The surplus nitrogen is provided by the cyanobacterial colonies inhabiting
specialized cavities in the dorsal leaves of the Azolla fronds. The NoAz cya-nobiont has lost its capacity to grow outside the host (Peters and Meeks, 1989, Tang et al., 1990). Instead, the cyanobionts are transferred between plant generations as an inoculum inside the Azolla sporocarps, the seed-like reproductive structure of the ferns (Zheng et al., 1990). This takes place via an elegant and intricate process that apparently has co-evolved between the host and the cyanobiont (Zheng et al., 2009). A subpopulation of cyanobi-onts differentiates into motile hormogonia attracted to developing Azolla sporocarps where they subsequently enter through a narrow pore. The cya-nobionts remain in a dormant spore state (akinetes) until germination of the sporocarps (Zheng et al., 2009). Upon germination, the akinetes differentiate into hormogonia once again and make their way to the newly formed symbi-otic plant cavities where they, embedded in a mucilagous layer of nutrient rich carbohydrates, reassume their role as providers of fixed nitrogen (Moore, 1969, Lechno-Yossef and Nierzwicki-Bauer, 2002). At this stage, the small-celled hormogonia differentiate into long filaments densely inter-spersed with nitrogen-fixing heterocysts, and the nutrient exchange com-mences between the partners through specialized plant trichomes extending into the cavity (Calvert et al., 1985).
The adaptations required of both hosts and symbionts towards the maintenance of the complex Azolla symbiosis are extensive. These probably occurred less than 150 million years ago, the time point of the oldest fossil record of Azolla (Hall and Swanson, 1968). However, it is possible that the Azolla-NoAz association is only about 49 million years old. This assumption is based on the “Azolla event” (Brinkhuis et al., 2006), an enormous bloom of Azolla discovered through drilling bottom sediments in the current Arctic Ocean. From these records it is apparent that the host plant had already de-veloped the nutrient exchange finger-like structures present in the cavities, and adapted to house a foreign organism both in its vegetative body-plan (the cavities) and in its reproductive sporocarps.

19
Insertion sequences
The awareness of insertion sequences goes back to 1950, when Barbara McClintock (USA) reported on the existence of “controlling elements” in maize, i.e. genetic elements that could move in the genome and regulate gene expression, thus influencing the colour patterns in maize corncobs (No-bel Prize in 1983). However, lacking modern molecular methods and DNA sequencing facilities commonplace today, research into the area of mobile genetic elements (MGEs) progressed slowly. It took almost twenty more years for the phenomenon to be verified in bacteria (Jordan et al., 1968, Shapiro, 1969), now being termed Insertion Sequences (ISs).
Although a number of mobile genetic elements have been discovered
since then, such as the retrotransposons found in the human genome, ISs are today recognized as the most frequent type of mobile genetic element in prokaryotes. ISs are present to some degree in almost all phyla of eubacteria and archaea (Chandler and Siguier, 2013), and likewise in eukaryotes. For instance, the human genome consists of 3% ISs, although all these are inac-tive. In cyanobacteria, ISs were first discovered in 1985, in the filamentous Nostoc sp. PCC 7120 (Alam and Curtis, 1985). Next, cyanobacterial ISs were further characterized when a specific IS type, IS891, from Anabaena sp. strain M-131 was discovered (Bancroft and Wolk, 1989).
Figure 2. Structure of an insertion sequence (IS) encoding a transposase flanked by inverted repeated short genomic regions. The transposase is encoded in one or two open reading frames (ORFs). These ORFs sometimes extend into the flanking inverted repeats. In some ISs, noncoding linker regions connect
the transposase ORF with the inverted repeats.
Transposase ORF
Spacers
Inverted repeats
0-40 bp 0-3500 bp 700-3500 bp 0-3500 bp 0-40 bp

20
The genetic structure of a typical IS is straightforward: the genomic se-quence consists of one or more open reading frames (ORFs) encoding a transposase protein. The ORF is typically flanked by short inverted repeat recognition sites on both sides (Figure 2). The transposase is a specialized recombinase with the ability to excise the IS element from its location in the genome and reinsert it in another genomic position (Figure 3) (Chandler and Siguier, 2013). The entire sequence is typically 700-2500 bp in length, alt-hough truncated and probably non-functional (Lawrence et al., 1992) copies are known to exist in many bacterial genomes.
Figure 3. Simplified cartoon of a ‘cut-and-paste’ transposition event. Translation: The insertion sequence ORF is translated to produce the transposase protein. Excision: Using inverted repeats as recognition sequences, the transposase catalyses the cutting of the DNA strand. Transposition: The insertion sequence
is moved to a new insertion site. Reinsertion: The transposase catalyse the integration of the IS into a new genomic location. In reality, complete excision of the IS is not required prior to transposition.
As opposed to many other MGEs, the transposition of ISs is normally a
“cut-and-paste” reaction, i.e. the IS is excised from one location and rein-serted into another with no increase in copy number. Duplications of ISs do however occur. This may take place when they are transferred between mul-tiple chromosomes (excising from one chromosome and inserting into an-other), when the transfers occur during replication (transferring an IS ele-ment to a strand different from the donor strand), and when strands are not properly excised. This may be the case when for instance the 3’ ends of the IS element are properly excised while excision fails at the 5’ end (Mizuuchi, 1992). By these mechanisms ISs copy numbers may increase substantially in genomes (Siguier et al., 2006a, Skipper et al., 2013) and may eventually
Translation
Excision
Transposition
Reinsertion
IS
Transposase

21
come to occupy considerable portions of some prokaryotic genomes, close to 40% (Cho et al., 2007).
The recognition sites flanking the transposase consist of 10-50 bp imper-
fect inverted repeats. They act as both binding sites for the transposase and as cleavage sites. Imperfect inversions (i.e. a few nucleotides that do not adhere to the mirror pattern) provides polarity to the transposase positioning, ensuring that the IS element is reinserted in the correct direction. The part of the flanking sequences that are targeted for cleavage are often identical at both ends of the IS (Chandler and Siguier, 2013). In most cases, the ISs are found surrounded by 2-13 bp long direct repeats. These are not part of the IS, but are generated as part of the insertion process (Chandler and Siguier, 2013).
The transposase, the protein responsible for the IS motility mechanism (transposition), is most commonly encoded by one or two ORFs. When two ORFs encode a transposase, the upstream ORF encode the recognition do-main required for correct positioning of the transposase on the IS, while the downstream one encodes the catalytic domain that performs the excision and reinsertion (Zhou et al., 2008). These ORFs are commonly overlapping, making their identification problematic during automated gene calling. They can however be translated from the same start codon via programmed ribo-somal frame shifting: a 'slippery codon’, commonly A AAA AAG (Farabaugh, 1996), induces a ribosome to slip over one nucleotide, causing a frame shift that permits continuous reading of different reading frames. The promoter of the transposase ORF(s) is typically located inside the recognition sequence positioned to the left in the IS, and the stop codon inside the recognition sequence on the right (Chandler and Siguier, 2013).
Although transposases are heterogeneous in their amino acid composition, many have a strongly conserved motif representing the amino acids critical to the transposition reaction. The most common of these is the DDE motif. This consists of two aspartic acids separated by 50-90 amino acids, followed by glutamic acid 13-160 amino acids downstream. Presumably, the DDE amino acids coordinate divalent metal cations, thereby facilitating the charge distribution required for a nucleophilic attack, which initiates the strand cleavage during IS excisions. Several other motifs, connected to different methods of transposition, have been identified, such as the HUH and DEDD motifs.
The transposase motif, together with typical characteristics of the IS re-
peated sequences, IS nucleotide lengths and ORF organizations, are used to classify ISs into families. There are currently 26 such families recognized. Of these, the IS3, IS4 and IS5 families are the most common, making up one third of all annotated ISs (Siguier et al., 2014). These three IS families, and

22
15 other of the 26, all use DDE motif transposases and create direct repeats upon insertion. Other families, such as IS91 and IS110, use other types of transposases, (called HUH and DEDD), respectively, carry no flanking in-verted repeats and generate no direct repeats upon insertion. Yet others have unknown excision mechanisms (Siguier et al., 2014).
Insertion sequence abundance The IS copy numbers of bacterial genomes vary enormously in bacteria,
even between closely related bacterial species. A few bacteria contain no IS copies while others, such as the unicellular ecologically successful cyano-bacterium Microcystis aeruginosa, contain hundreds (Kaneko et al., 2007). Some data suggest that ISs are more frequent (higher copy numbers) in mi-crobes living in “extreme” ecosystems such as hot springs and acid mine drainage. However ISs are in addition abundant in the considerably more lenient environments enjoyed by pathogens and symbionts sheltered within hosts. Additionally, some organisms living freely and inhabiting presumed non-stressed environments, such as M. aeruginosa forming large blooms in lake waters, contains levels of IS elements that match or exceed those of extremophiles and symbionts.
Large genome size appears to be the only factor that statistically corre-
lates with IS abundances (Touchon and Rocha, 2007). A main factor pro-posed is that IS abundance is governed by the number of sites available where IS insertions do not reduce organism fitness. Larger genomes are like-ly to have more such sites, and to hold alternative metabolic pathways that may compensate for loss of functions through IS insertions, and these organ-isms are therefore more resilient.
Variability of insertion sequences IS copies are often found in eroded or truncated states in investigated ge-
nomes. Several reasons are proposed to account for this observation. Due to their apparently non-essential nature, there is no natural selection for func-tional ISs. In addition, some apparent truncations may be ‘artefacts’ created during gene calling by the special ORF structure of many ISs. While the high variability in IS structures and numbers creates difficulties in annota-tion and IS classification, it is a highly useful means by which to identify strains of bacteria. A feature typical of bacterial ISs is that the phylogenies of ISs are often inconsistent with the phylogenies of the host species (Tou-chon and Rocha, 2007). This phylogenetic limitation suggests that ISs are highly motile and able to cross species borders at a high rate. The variability within an IS family appears to be age correlated, and it is proposed that the most variable of IS families started to spread early in the evolutionary histo-

23
ry (Santiago et al., 2002). For instance, the hypervariable IS630 family (Lin et al., 2011) has been associated with Tc1-like and Mariner-like elements of eukaryotes (Nguyen et al., 2014). This may imply a spreading before the separation of organisms into prokaryotes and eukaryotes, although it may also be the result of later crossings of the eukaryote/prokaryote barrier.
Local hopping ISs appear to congregate in clusters in the genome (Zhou et al., 2008), in
so-called “hot spots”, that correlate with gene loss and genomic recombina-tions (Bickhart et al., 2009). In contrast, other genomic areas within the same organism can be conspicuously devoid of ISs. Even in the extremely IS rich cyanobacterium M. aeruginosa there are regions devoid of ISs, generally coinciding with essential genes such as ribosomal subunits, as well as DNA and RNA polymerase subunits (Steffen et al., 2014). The reason for the de-velopment of IS hot spots and IS free genomic areas is likely due to the fact that the range of an IS transposition is limited: instead of transposing ran-domly across the whole genome, they perform short jumps referred to as ‘local hopping’ (Boyd and Hartl, 1997).
Insertion sequences activity The rate of IS transposition is of vital importance for an impact on evolu-
tion, but complicated to analyse. The production of IS transcripts can be assayed, but the amount of transcripts does not necessarily relate directly to transposition events. IS transcripts must first be translated into active trans-posases. The reading frame shift required to overcome the frame shift pre-sent in some transposases does not always take place during translation, and truncated IS transcripts may result in non-functional transposases and even be able to repress complete transcripts. Upon successful translation of a transposase, the transposase activity may be inhibited due to lack of required co-factors, providing a further level of regulation of the transposition activi-ty. Once an active transposase is produced it may perform a high number transpositions, including of IS sequences of the same type that are unable to produce working transposases themselves. To date, the factors governing transposase expression are not well defined (Steffen et al., 2014), and esti-mates of transposition rates vary considerably, as do the experimental ap-proaches used. Moreover, IS transpositions appear to occur in ‘bursts’ rather than at an even rate. For instance, transposition speeds in strains of Esche-richia coli were estimated to be ~0.01 transpositions/IS element/year for the IS5 family, and this rate was comparable to that of several other IS families (Green et al., 1984). Later, mutational changes were noted to be time-dependent (over a 30 year period) and mutation rates in E. coli were estimat-ed to be roughly 10-5 DNA rearrangements per chromosome per hour of

24
storage, translating to almost 1 transposition/year (Naas et al., 1995). Other investigators have arrived at rates of 10-7-10-9 transpositions per generation, which translates to roughly 0.001-0.00001 transpositions per year (Chandler and Siguier, 2013).
The IS activity may also depend on environmental conditions experienced by the IS host. Many organisms with recently active ISs were found to origi-nate from hot springs (Zhou et al., 2008), while drastic changes in transposa-se transcription have been discovered in the cyanobacterium M. aeruginosa subject to changes in the availability of key-nutrients such as nitrogen and phosphorous (Steffen et al., 2014).
Insertion sequences in evolution Although some authors have perceived ISs as being detrimental for or-
ganisms, ultimately leading to the eradication (Wagner, 2006), their role as important facilitators of adaptive change is in recent years increasingly acknowledged. Their role as facilitators is clearly reflected in the genetic adaptations imposed on a bacterium that enters into a symbiotic or parasitic relationship with eukaryotic organisms. Genomic changes found in obligate microbial symbionts appear to be universal, and have been observed in sev-eral gut bacteria (Gil Garcia et al., 2008), in pathogens (Fuxelius et al., 2008) and in the ‘spheroid bodies’ of Rhophalodia gibber, among others. These genomic changes are characterized by a decrease in the genomic GC content, a shrunken genome, numerous pseudogenes and the presence of MGEs (Kneip et al., 2008, Andersson and Kurland, 1998), and the process is re-ferred to as ‘reductive evolution’.
The driving force behind reductive evolution in symbiosis is likely to be
the extreme environmental differences between a free-living life style and that required inside a host organism. Biological functions essential in an autonomous organism, such as production of energy and cellular building blocks, may in symbiotic relationships be provided by the host. This together with protection inside a host from predation and competition for resources as well as environmental stresses, offer an environment with highly relaxed selection pressures (Fuxelius et al., 2008). These circumstances decrease the need for genes that are required in autonomous life styles, making the loss of such genes trivial to fitness value in the sheltered environment. Mechanisms underlying genome degradation in microbes include replication errors, loss of repair mechanisms, bottleneck effects and reduced repair and recombina-tion events (Andersson and Kurland, 1998). However, it can be argued that IS proliferation is the most powerful known mechanism known today in relation to genome reduction. In a highly relaxed (symbiotic) environment, with many redundant bacterial genes, the detrimental effects of IS prolifera-tion is reduced. The interruption or elimination of such genes do not reduce

25
fitness, and the decreased need for a rapid growth of a symbiont means that even IS transpositions that outright kill individual bacteria may not decrease the symbiont population to a critically low size (Badawi et al., 2014). Hence the IS copy number are allowed to increase under such circumstances, accel-erating an adaptation to the new symbiotic life style.
Initially, the size of the genome thus increases when the IS elements pro-
liferate and their copy numbers rise. At later stages of the symbiotic estab-lishment, a decrease in genome size starts. The initial stages are character-ized by large genomic deletions mediated by recombination between ISs or other repeated elements (Moran and Plague, 2004, Walker and Langridge, 2008, Badawi et al., 2014). Such recombinations can lead to both duplica-tions and deletions (Krawiec and Riley, 1990). However, duplications can be reversed, but once a nucleotide sequence is deleted, the information cannot be recovered by another recombination event (Andersson and Kurland, 1998). Newly formed pseudogenes via replication errors or IS insertions are ultimately lost, along with the mobile genetic elements (Mira et al., 2001, Fuxelius et al., 2008).
In the later stages of reductive evolution, most redundant genes and IS el-
ements have been eliminated, along with the pathway for homologous re-combination, leading to a new genomic stability observed in various ancient endosymbiont genomes (Badawi et al., 2014). The membrane bound orga-nelles of extant eukaryotes represent the ultimate extremes of this process. Mitochondrial genomes derive from much larger genomes originating from a free-living ancestor of Rickettsiaceae (Andersson and Kurland, 1998), while the chloroplasts, today having a genome size of 150-200 kbp, most likely arose from multicellular cyanobacteria capable of differentiation (de Alda et al., 2014, de Cambiaire et al., 2006). It is noteworthy that members of the Rickettsiaceae family and the Cyanobacterial phylum to this day include representatives prone to establish symbiosis or function as parasites.
The brackish water Baltic Sea The Baltic Sea is a large body of brackish water enclosed by the Swedish
east coast, Finland, the Baltic countries, Poland and Germany. In a north-south direction, it stretches from the Bothnian Bay in the north (66° N) to the Danish islands in the south (55° N) where it connects to fully marine waters (Skagerrak and the North Sea) through narrow sounds. The limited inflow through these sounds is the reason for the brackish salinity of the Baltic Sea. This allows limited mixing with saline waters, while fresh water is continu-ously added through inflow from voluminous rivers in the north. While the salinity of marine environments is 35 psu (practical salinity units), the salini-

26
ty of the Baltic Sea varies between 2-32 psu, increasing from north to south. In most parts, the Baltic Sea salinity is within the ‘Remanes Artenminimum zone’ of 5-8 psu, a salinity in which both fresh water and marine adapted organisms experience difficulties in coping with prevailing osmolarities, while the few brackish water adapted organisms existing apparently thrive in this less competitive environment (Telesh and Khlebovich, 2010, Whitfield et al., 2012).
The Baltic Sea is to a high degree affected by human activities, among
them phosphate and nitrate land runoffs originating primarily from agricul-tural fertilizers. The phosphate, and to some extent nitrogen (Dolman et al., 2012), is believed to promote unwanted cyanobacterial blooms, as these on death and decay cause expanding oxygen depletion bottom zones, which further exacerbate the living conditions in the Baltic Sea. The Baltic Sea was formed after the last glaciation (10,000–15,000 years ago) and is a geologi-cally and evolutionary young sea (Feistel et al., 2008), containing few multi-cellular organisms. However, its long water retention time (between 3 and 30 years) and fast evolutionary pace may have allowed brackish-adapted line-ages to establish and evolve (Herlemann et al., 2011, Dupont et al., 2014a, Ininbergs et al., 2015).

27
Aims
Based on this background knowledge, the following hypotheses were pro-posed and tested:
o Insertion sequences are important facilitators in the evolution of mi-crobes, and their presence and activity in a genome are responses to external cues, such as those experienced entering tight interactions with eukaryotes (reductive evolution) or when being subjected to stressful environments (adaptive evolution).
o Insertion sequences cause severe pseudogenization via insertions
and a ‘local hopping’ mechanism. The IS transposition into and out of a gene is not always obvious but may leave pseudogenization as a ‘scar’ at its former insertion site.
o Contemporary sequenced genomes may be “snapshots”, representing
genomic configurations optimized for the current environment, but amenable to rapid IS induced change when this environment chang-es. The distribution of IS copies in the genomes are likely to indicate genes or genomic regions that are fitness neutral in their current en-vironment.
To test the hypotheses, investigations were performed in a series of gradual-ly expanding scopes. Initially, the genome of a unique plant cyanobacterial symbiont lacking autonomous growth, indicative of a far stretched co-evolution between the partners, was sequenced and examined to explore the concept of reductive evolution. This scope was further expanded through comparative genomic analyses including a large set of other sequenced cya-nobacteria and bacterial symbionts/pathogens. Furthermore, the role of inser-tion sequences as mediators or drivers in adaptive evolution in large natural microbial populations, dwelling in a geologically recent aquatic environ-ment, was tested through metagenomic and metatranscriptomic analyses (Figure 4).

28
Figure 4: Flow and lay out of the research in the Thesis in regards to insertion sequences (ISs) in microbes: from ISs identified through sequencing of an individual genome of a cyanobacterium (cyanobi-ont) residing extracellularly in leaf cavities of the Azolla plant (Paper I); through comparative genomic analyses including a wide range of sequenced microbes of differing life styles (Paper II), and to analyses
of the IS biogeography, organismal distribution and IS activity (by proxy of transcription) in microbial populations in the Baltic Sea, revealed through metagenomic and metatranscriptomic analyses (Paper III).
��������� ��
�
Insertion sequencesin the genome of an
obligate plantcyanobiont
Insertion sequencesin 67 sequenced
microbial genomes:- symbionts-pathogens-free-living
Insertion sequencesin natural
populations of microbes in
the Baltic Sea
Paper I Paper II Paper III

29
Methods
To test the above-presented hypothesis in regards to the presence and role of IS elements in the evolution and adaptation of microorganisms, several experimental approaches at the genomic level were used.
First, the role of ISs in the reductive evolution of the symbiotic cyanobac-
terium inhabiting the floating fern Azolla was examined. To sequence the genome of the cyanobiont of Azolla (designated as ‘Nostoc azollae’ 0708, or NoAz), this cyanobacterium was isolated from the host plants as detailed in Paper I. The multi-leaved fronds of a high number of small Azolla filicu-loides plants were gently pressed to eject the cyanobiont from the leaf cavi-ties. The cell suspension obtained was then thoroughly purified from con-taminants and debris through a density centrifugation procedure. The NoAz cell population was next sequenced, followed by automated annotation by standardized methods (Department of Energy, Joint Genome Institute, Oak Ridge National Laboratory, USA) and analysed as detailed in Paper I.
The automated data cleaning and annotation procedure revealed high
loads of pseudogenes in the NoAz genome. This verified our hypothesis of a shrinking cyanobacterial genome and hence on-going reductive evolution in the NoAz cyanobiont, the first of its kind discovered. Next, the annotated genome was screened for IS elements by comparison to known amino acid sequences. To identify additional unannotated ISs, regions with flanking inverted repeats were identified using the bioinformatics toolkit UGENE (Okonechnikov et al., 2012) and examined for similarities with known ISs. To ensure that fragmented IS copies were not counted as multiple ISs, ISs with split reading frames were manually annotated as single insertions.
Secondly, to obtain a more comprehensive understanding of the role of
ISs in microbes in general and in reductive evolution in particular, 66 addi-tional microorganisms with available sequenced genomes (NCBI non-redundant genomic database, USA), were examined for the presence of ISs (Paper II). These organisms were selected to represent a wide spectrum of both free-living and symbiotically competent cyanobacteria, as well as nine phylogenetically widely separated symbiotic or pathogenic bacteria, some in severe/late stages of reductive evolution. These selected representatives were derived from a wide range of eukaryotic hosts: Bordetella pertussis, a human

30
pathogen and causative agent of whooping cough; Candidatus Amoebophilus asiaticus, endosymbionts of free-living amoebae Acanthamoeba spp.; Frankia strains ACN 14a, Eu1c, EAN1pec, ccI3 and Dg1, living in symbio-sis with actinorhizal plants; Mycoplasma mycoides, a pathogen found in cattle and goats; Onion yellows phytoplasma, a plant pathogen living en-docellularly in plant phloem cells; Orientia tsutsugamushi, responsible for scrub typhus in humans; Shigella flexneri, a human pathogen causing dysen-tery and diarrhoea; Yersinia pestis Angola and Antiqua, and finally Wolbachia symbionts from the fruit fly Drosophila annanasae, D. melano-gaster, Culex quinquefasciatus and C. quinquefasciatus JHB as well as Wolbachia sp. wRi and sp. TRS of Brugia malayi. An automated pipeline for the identification of ISs was constructed to analyse these genomes, using the scripting language Python. In addition to subjecting all investigated genomes to comparisons with already identified ISs, Repeatscout (Kurtz et al., 2000) was used to identify repeated sequences, which were then manually curated by comparing their nucleotide and amino acid sequences to those of known ISs, phage proteins and enzymes involved in DNA interaction. Furthermore, automated methods were developed to compute distances between IS copies, and between IS copies and pseudogenes, with the purpose of investigating local hopping and IS proximity to pseudogenes statistically.
Finally, the scope of the thesis was further widened by examining the
presence of ISs in the bacterial phyla outside the realm of sequenced micro-organisms, namely in the massive natural (mostly unculturable and un-known) microbial populations inhabiting the vast brackish water body of the Baltic Sea (Paper III). This ecosystem offers highly unusual conditions, e.g. a salinity gradient increasing more than tenfold from the north (2 psu) to the south (32 psu), and suffers from anthropogenically imposed eutrophication. As these are highly stressful conditions, to which a limited number of organ-isms can adapt, this system was selected as a model to search for signs of on-going ‘adaptive evolution’, again based on IS elements in the microbial ge-nomes. In order to capture information about the biogeography, organismal identity and activity of microbial IS elements, our approach was to examine the massive genomic datasets obtained from a representatively dispersed (non-biased) sampling expedition of microbes (including size fractionation of the populations) in the Baltic Sea, and adjacent waters with opposing sa-linities (limnic and fully marine). These data sets were obtained from the European part of the Global Ocean Sampling Expedition (June/July 2009) (performed in collaboration with J. Craig Venter Institute (CA, USA)). The comprehensive metagenomic and metatranscriptomic data sets derived via high-through-put sequencing (JCVI, USA) of the microbial nucleotides formed the basis for our IS element screenings. For further details of the experimental procedures related to sampling and sequencing see (Dupont et al., 2014b, Larsson et al., 2014 and Ininbergs et al., 2015). To get perspec-

31
tives on the IS data obtained in the highly specific Baltic Sea ecosystem, a comparative study was performed on microbes off the fully marine west coast of USA using comparable metagenomic and metatranscriptomic ana-lytical and bioinformatics protocols (the CalCOFI, California Cooperative Oceanic Fisheries Investigations; see (Dupont et al., 2014a).
In order to retrieve and analyse ISs in the genomic and transcriptomic da-
ta sets retrieved from the three investigations referred to above, similarity searches to already known ISs, stored in the ISfinder database (Siguier et al., 2006b) and in the NCBI non-redundant nucleotide and protein databases, were performed. The primary tool for identifying ISs was BLAST (Basic Local Alignment Search Tool) (Ye et al., 2006), an efficient algorithm for identifying similarities between DNA, RNA or amino acid sequences (Lin et al., 2011). The versatility and easy access to web-based BLAST engines such as NCBI and ISfinder were additional benefits of using BLAST, as were the support provided for scripted searches and results parsing by the Biopython package (Cock et al., 2009). The BlastX function, which com-pares nucleic sequences against amino acids, was particularly useful. This version of the algorithm translates the query nucleic acids into amino acids in all six possible reading frames, and compares these to a database of amino acid sequences. For example, the latter approach was frequently used to identify the numerous ISs mutated to such a degree that an identification using a nucleotide sequence similarity search was prevented. In addition, BlastX identifies frame shifts and stop codons, a useful feature when identi-fying potential pseudogenes. Another search algorithm using Hidden Mar-kov Models (HMM) was considered in the IS screening due to its ability to identify amino acid combinations, such as those typical for the catalytic mo-tifs of many IS transposase proteins. However, HMM was later rejected as it would have required the assembly of numerous transposase motif models and extensive human based curations, including also the translation of frame shifted nucleotide sequences into amino acids prior to the search. Moreover, HMM models lack the web support, and support in Biopython, provided by BLAST.
The methods used in the Thesis are among the most potent currently
known for sequencing and analysing genomes and cellular phenomena, of both individual genomes of cultured often well-known microorganisms as well as of microorganisms captured directly from comprehensive natural aquatic populations/assemblies, the latter representing the large non-culturable ‘hidden majority’.
The introduction of metagenomics/metatranscriptomics in the analyses of natural populations in fact revolutionized our knowledge as they revealed huge biomasses of highly active microorganisms with an astonishing biodi-versity, from phytoplankton to viruses, and in globally spread oligotrophic

32
oceanic surface water masses, up till then considered as ‘deserts’ unable to sustain any life (Venter et al., 2004).
In spite of the enormously efficient output- and identification-machinery
of the metagenomic and metatranscriptomic technologies, it should be noted that these do not capture every individual organisms and that there are still too few and often a biased (culturable microorganisms) collections of micro-bial genomes available in databases, causing non-optimized identifications as well as poor phylogenies.

33
Results
IS elements in a cyanobacterium of an obligate plant symbiosis Sequencing the genome of the cyanobacterium (NoAz), living in an obligate symbiosis with the small free-floating Azolla plant, revealed a 5,486,145 bp genome, highly decorated by ISs and suggestive of extensive genome ero-sion (Paper I), the first discovered in any cyanobiont. This and the exquisite transfer procedure between plant generations (Zheng et al., 2009), suggests that an extensive co-evolution between the partners have already taken place. Notably, pseudogenes were apparent in not less than 52% of the genes in NoAz, a larger percentage than in any other cyanobacterium sequenced. This is consistent with the finding of massive numbers of IS elements, or truncat-ed remains of IS elements, in more than 600 genomic locations (paper I). Together these discoveries verify our hypothesis that the cyanobiont lost autonomy due to an on-going ‘reductive evolution’ in its genome. However, the reductive evolutionary process is probably still in its infancy, i.e. this symbiosis represents a relatively recent association with a symbiotic host. This is for instance suggested by its relatively large genome size (5,49 Mbp), even comparable to some free-living close relatives, and the high frequency of ISs still present. The enhanced level and widespread occurrence of the ISs in the NoAz genome is likely permitted by the presence of the numerous genomic spaces being redundant under the sheltered conditions offered by the host plant. Once inside the host, certain functions required by the free-living cyanobacterial ancestor are successively taken over by the host (e.g. carbon fixation via photosynthesis). This in turn opens up for ISs to colonize more relaxed genes, those no longer critical for survival. These extra ISs copies merely pose a low threat to the fitness of the ‘newly’ established cya-nobiont. The reduced threat of ISs is primarily reflected in the high frequen-cy of pseudogenes. Genes made redundant in association with a physiologi-cally fully competent eukaryotic host may be colonized by ISs, while all cyanobacterial core genes (Gil et al., 2004, Shi and Falkowski, 2008) still remain intact.
For example, pseudogenes were lacking in the crucial category “Nucleo-tide transport and Metabolism”. Most importantly, the nif operon, containing a multitude of genes (>20) encoding components of the nitrogen fixation

34
process, was fully intact, although it was flanked by ISs on both sides (Paper I). It is still possible however, that nif operons (encoding the key function in all cyanobacterial-plant symbioses) of countless individual cyanobacteria were ‘attacked’ by ISs during the co-evolution of the partners, removing individuals from the gene pool that were unable to supply the host with fixed nitrogen, the key function in all cyanobacterial-plant symbiosis. Genes en-coding proteins necessary for the heterocyst differentiation, the exclusive site for the nitrogen fixation process, also remain intact. In contrast, the loss of the patS gene in the NoAz genome, encoding a short (13 or 17 aa) sup-pressor of the heterocyst development, may rather represent an advantage as it generates the multi-heterocyst phenotype typical for NoAz (Lechno-Yossef and Nierzwicki-Bauer, 2002), thereby stimulating the delivery of fixed nitrogen to the host.
Likewise, genes for photosystem I and II, cytochrome b6/f and a complete
set of genes encoding the biosynthesis of the light-harvesting biliproteins were intact. As these provide reducing equivalents to the nitrogen fixation they may in symbiosis rather be crucial in this function in symbiosis than in photosynthesis considering the dim light offered in plants.
Pseudogenized genes included for instance a set of genes required for
carbohydrate transport and metabolism (pfkA, gapA, pykA, gpmA, ldh, melB). This is consistent with the fact that NoAz in symbiosis may be specialized in utilizing a different set of carbohydrates, now delivered by the host. Indeed, a function in the carbohydrate metabolism in the preceding free-living state may have been to respond to signalling compounds secreted by the hosts (e.g. arabinose, glucose and galactose) known to be needed during the initia-tion of the symbiosis (Bergman et al., 2008, Rasmussen et al., 1994) which is redundant in the perpetual NoAz-Azolla symbiosis. In line with the flow of nutrients provided by the plant in symbiosis, genes underpinning the import and utilization of alternative nitrogen sources (nitrate and urea) were also impaired.
Notably, about 56% of the genes in the “Replication, Recombination and Repair” category were pseudogenized in NoAz. Interestingly, ISs belong to this category, which highlights the fact that even ISs genes can be safely pseudogenized. The genes recD and dnaA were also found to be pseudogenes. In fact, recD is missing in many endosymbionts and decrease genomic recombinations which otherwise may increase due to the effect of multicopy ISs. The lack of the canonical initiator of DNA replication in cya-nobacteria, dnaA, may be yet another example of a gained advantage for NoAz in symbiosis, in particular during periods of increasing IS copy num-bers in the genome. Given that some ISs transpose primarily during replica-tion, the lack of dnaA may lead to diminished transposition events.

35
Together the data suggest that the NoAz genome is still in a phase of ge-nome expansion, a phase that (at least) theoretically precedes the genome reductions seen in truly ancient endosymbionts. In its current configuration of 5.49 Mbp, with a large contingent of pseudogenes and IS elements, the NoAz genome is for instance considerably larger than that of its closest rela-tives Raphidiopsis brookii (3.16 Mbp) and Cylindrospermopsis raciborskii (3.69 Mbp), while smaller than the likewise symbiotically competent cyano-bacterium of numerous other facultative plant symbioses. However, remov-ing all ISs and pseudogenes (1670) from the genome, it approaches 3.17 Mbp, a genome size close to that of the two closest relatives.
In spite of having the full sequence of the NoAz genome, it is difficult to
predict the current stage in the reductive evolution. The copy number of ISs may still be increasing in redundant genes still present, leading to a further genome expansion. If on the other hand a point has been reached where most remaining genes are crucial, the NoAz genome will then have entered the genome reduction phase, with remaining ISs and pseudogenes slowly erod-ing and eventually being lost. If a second round of increased intimacy and co-evolution between partners is elicited, a new burst of IS mediated evolu-tion may commence. An even higher degree of intimacy may well be facili-tated by the ISs. This may not only be restricted to their ability to affect ge-netic change, but perhaps more importantly to provide a mechanism for the transfer of genes to the host nuclei, as seen in contemporary chloroplast hosts.
ISs in additional symbionts and other cyanobacteria To provide a perspective on the discoveries in regards to IS elements in the NoAz genome, a thorough search for ISs was performed on NoAz and total-ly 66 other cyanobacteria and bacterial symbionts (Paper II). The results of this comparative investigation further highlights the unique nature of NoAz as an organism in a very early stage of genomic adaptation ‘trapped’ in a symbiotic and highly secluded environment. The frequency of ISs and pseudogenes was higher than in the majority of the investigated bacteria, both free-living and symbiotic. These findings further substantiate our con-clusion that the detainment of NoAz in the symbiotic environment is recent, while that of the other bacterial symbionts is considerably older. As many as 970 genomic locations with sequences similar to ISs were discovered in NoAz, covering 14.3% of its genome. This frequency is merely slightly low-er than that found in the genome of O. tsutsugamushi (formerly known as Rickettsia tsutsugamushi), previously noted for its extremely high IS content (Cho et al., 2007). According to Cho et al. the ISs of O. tsutsugamushi cover close to 40% of the genome. In contrast, the coverage of ISs in O. tsutsuga-

36
mushi was 14.7% using our search approach and identification criteria for ISs. In the former study, repeated sequences with a size above 200 bp in length were considered, while only sequences 500 bp or longer were consid-ered in our analyses, as this was regarded as the lowest length required for transposase-like proteins.
Moreover, through our analyses it was evident that IS elements were far from extensively catalogued in databases. This is not surprising given the extreme variability of IS elements. For instance, among the 1108 repeated sequences being >500 bp in the 67 genomes here investigated, merely 578 matched sequences catalogued in the ISfinder website (Siguier et al., 2006b) with a BlastX e-value of 10-6 or less. Up to 419 repeat sequences lacked suf-ficiently similar relatives, but were clearly of IS origin, as ascertained by comparisons to transposases in NCBI’s non-redundant protein database. Twenty-four previously unannotated species of ISs were discovered in No-Az.
In all bacterial genomes analysed, ISs represented the main constituent of the total repeated contingent present. Other, less frequent, categories of re-peats were prophage remains (50 sequences), followed by uncategorized repeats noted due to their high copy numbers or functions involved in DNA manipulation (61 sequences). The comparatively low number of phage re-mains indicates that IS duplications occur far more often than prophage in-sertions. The uncategorized repeats may represent not yet categorized types of MGEs. Our data also showed that IS fragmentation is a common phenom-enon in bacterial genomes. Among previously annotated ISs, full length-copies were found for 210 ISs, while another 362 were found as fragments being <95% of their reference sequence lengths. This is probably due to the fact that ISs tend to insert into and interrupt other ISs, either because these provide ‘safe’ insertion sites or contain sequences especially amenable to insertions.
Of the total number of IS copies found in the 67 genomes examined, 31%
extended over 95% of the reference sequence, while 37% were shorter than 15%. Among bacteria with a relatively high repeat density (>3% of the ge-nome), the majority represented full-length IS copies, while bacteria with a low IS density had a larger proportion of fragmented copies. In NoAz, IS copies with lengths ≥ 95% constituted up to 40% of the ones identified, again verifying that the Azolla symbiosis is still in a rather initial evolutionary stage. It is likely that full length IS copies are able to proliferate in the NoAz genome, while truncated versions will not be able to produce active trans-posases.
The ISs in NoAz were most similar to the previously identified ISs from
the close multi-cellular relatives, Nostoc punctiforme and Anabaena varia-bilis, or to other IS-rich cyanobacteria such as the unicellular Acaryochloris

37
and C. watsonii. One NoAz IS showed an 86% identity to the nucleotide sequence of the previously annotated ISAva8 (identified in the genome of A. variabilis). As this constitutes a high level of identity a recent cross-species transposition of an IS is implied.
IS elements and Local hopping Some ISs are known to perform ‘local hopping’ within the genomes, i.e. they are subject to short rather than genome-wide transpositions. Short distance transpositions may be less likely to negatively affect other genes in adjacent operons or genomic regions and are therefore less harmful.
The number of IS elements and pseudogenes in NoAz were high enough to be discerned by visual inspection of its genomic map (Paper II). It was also apparent that copies of the same IS elements tended to cluster in the NoAz genome, and that ISs were lacking all together from regions of the genome. Our analyses also showed that IS copies in close proximity to each other showed a higher nucleotide similarity than IS copies located more dis-tantly from each other. The most proximal IS copies on the chromosome of NoAz also tended to be closest in terms of sequence identity (uncorrected p-distance). To evaluate the propensity for local hopping as a valid mechanism in reductive evolution in NoAz and other genomes, the similarities in the uncorrected p-distance between two IS copies was compared to their dis-tance from each other on the genome. Statistical evidence was obtained showing that local hopping may be a general behaviour of the IS elements, although the signal for local hopping must have been severely affected by mutations and genomic rearrangements. On average, the uncorrected p-distance between two IS elements decreased with 0.1-1 percentage unit per Mbp as their distance from each other increased. This means that 1-10 base pairs had mutated for every Mbp of separation (disregarding reversions). When the 49 757 pairs of IS elements from the 100 most frequently found ISs were tested together, an average change in uncorrected p-distance of -0.48 percentage units per Mbp of distance was found using linear regression (p-value of 2.35e-25), further strengthening the evidence for general local hopping.
IS proximity to pseudogenes The propensity for local hopping implies that genes in proximity to ISs
are at risk to be pseudogenized more often than those at distance from ISs. This was found to be the case in NoAz, except for genes critical to symbiotic functions (such as the nif genes), which were observed intact despite their proximity to presumably active ISs. To test this hypothesis, the number of pseudogenes and intact genes in relation to distances from IS elements were

38
examined. In NoAz there were on average >30% more pseudogenes within 5 kbp of an IS element, compared to the number of pseudogenes in proximity to non-IS genes. This pseudogene enrichment dropped to below 5% at 200 kbp distance and became undetectable at a 500 kbp distance from ISs. Simi-lar patterns were apparent in the majority of the investigated bacterial ge-nomes holding a sufficient number of IS elements and pseudogenes to make statistical differences (p < 0.05) reliable. The ISs presently observed close to a pseudogene might represent the ISs that once caused the pseudogeniza-tions, but that have now moved on.
IS elements and adaptive evolution in the Baltic Sea
To further investigate the effects of ISs on microbial genomic adaptations, the metagenome and metatranscriptome of natural populations of bacteria inhabiting the geologically young Baltic Sea was examined (Paper III). To validate the significance of our findings these were compared to data from bacterial populations in the geographically distant but considerably more stable Pacific Ocean.
Baltic Sea bacteria may have had a relatively short time to adapt to the
brackish water environment in the geologically young sea (approximately 10,000 years) compared to in for instance oceans. The brackish salinity con-ditions offered in the Baltic Sea are known to be a distinct obstacle for both limnic and marine microorganisms (Herlemann et al., 2011). A high number of ISs, compared to more stable marine and limnic environments, was there-fore hypothesized to exist in the Baltic Sea. The IS content of the Baltic Sea, and the neighbouring water bodies (the limnic Lake Torne Träsk and the marine west coast of Sweden), varied from 0.7-6.1 ‰ of the total meta-genomic contigs. The higher proportions were invariably found in the Baltic Proper (central Baltic Sea) while distinctly lower proportions where apparent in the fully marine waters of the west coast and the Pacific Ocean. This may indicate that 1) bacteria in the Baltic Sea are species that excelled in adapta-bility when colonizing these waters, 2) the Baltic strains were present in the marine pre-Baltic waters, i.e. before the change into the contemporary brack-ish salinity, and increased their IS content as a response to the changed salin-ity, or 3) Baltic Sea bacteria have increased there IS content due to some other more recently imposed environmental change, such as eutrophication (primarily nitrogen and phosphorous).
Although the fraction of transcribed ISs were hypothesized to be high in
the Baltic Sea ecosystem due to its recently formed brackish water environ-

39
ment, the levels of ISs identified here exceeded these expectations. In some samples ISs made up more than 1% of the metatranscriptome. This suggests that a considerable part of the transcriptional machinery of Baltic Sea pro-karyotes is devoted to IS transcription. IS transcripts in samples from marine environments outside the Baltic Sea, both from the neighbouring west coast waters and from samples from the Pacific Sea outside the Californian coast were considerably fewer, reaching from 0.1% in the Swedish west coast to a few parts per million in the Pacific Sea by the Californian coast.
The IS fraction of Baltic Sea Synechococcus strains frequently made up 1-
3% of all Synechococcus transcripts, while the same fraction was close to zero in Synechococcus inhabiting the fully marine waters west of the Baltic Sea. This suggests that Synechococcus in the Baltic Sea belong to highly adaptable strains, or strains in early stages of adaptation. This finding is sub-stantiated by recent discovery of the flexible genomic ‘behaviour’ in Baltic Sea Synechococcus strains sp. CB 0101 and sp. CB 0205 (Larsson et al. 2014). Conversely, the marine Synechococcus strains are likely species with small, specialized genomes, which are generally devoid of ISs (Vigil-Stenman et al., 2015, Lin et al., 2011).
Although several mechanisms exist to repress IS activity after the tran-
scriptional stage, high IS transcription have been correlated with a high IS proteome content in some investigations (Kleiner et al., 2013, Steffen et al., 2014). While the transposases produced by ISs must pass additional regula-tory processes to effect transpositions, the high IS transcriptional levels re-vealed may challenge the traditional view of IS transpositions as rare events.

40

41
Final remarks and Conclusions
The main conclusions of the thesis are:
• An astonishingly high level of ISs and pseudogenes were discovered in the cyanobacterial symbiont NoAz. Since it participates in an ob-ligatory symbiosis where it provides nitrogen to the host, this finding verifies the hypothesis that this symbiosis may be at an early to in-termediary point of reductive evolution, potentially ending in a ge-nome with all genes not vital to symbiosis abolished, including ISs
• Several of the IS elements in NoAz perform ‘local hopping’, moving
across short distances in the genome rather than randomly.
• As IS copies appear in proximity to pseudogenes, these may be the results of local IS copies’ recent transpositions into and then out of the gene, leaving a ‘scars’ that resulted in pseudogenization.
• Genetically proximal IS elements pose a risk to genes, while vital
genes distant from ISs confer a fitness advantage.
• The activity of ISs may be higher than previously assumed, as sug-gested by the high ratio of IS transcripts found in Baltic Sea bacteria. Since most IS transpositions likely affect local genomic regions with little fitness value, and because IS ‘cut-and-paste’ events are likely much more frequent than the ‘copy-and-paste’ events discernable as additional copies, IS activity may have been underestimated.
From being regarded as mere curiosities and occasional examples of self-ish genes, a picture of ISs as important effectors in evolution is emerging. The high transcript levels and evolutionary recent activities observed suggest that they are more active than previously assumed. However, ISs do not spread indiscriminately throughout a genome. Every local hop that brings an IS element closer to currently advantageous genes decreases the fitness of the host and its progeny. This forces the ISs to congregate in areas of low fitness importance where their concentration can lead to IS copies inactivat-

42
ing each other and to deletions through recombinations. The main factor leading to IS proliferation, as suggested by (Touchon and Rocha, 2007) is apparently the amount of non-vital regions available in a genome. Such re-gions are frequent in two kinds of organisms: those with large genomes, and those that have recently lost the need for portions of their genomes (symbi-onts). Typical occasions when genes rather suddenly become redundant are when a bacterium gets access to vital functions of another organism, i.e. during entry into a symbiotic of pathogenic life style. Sequencing the ge-nome of NoAz therefore provided an unusual genomic ‘snapshot’ of ge-nomic erosion. In most symbiotic genomes investigated, the proliferation and subsequent eradication of ISs have already run its course, leaving small genomes with few traces of their presence and of mechanisms in action. Furthermore, the NoAz data obtained not only illustrated an increased IS copy number, but also that numerous genes were pseudogenized and with numerous IS copies commonly proximal to these.
Together, these findings lend strong credence to current theories of both reductive and adaptive evolution as mechanisms shaping microbial genomes to gain fitness, and add a widened understanding of the connection between IS elements and pseudogenization, as shown for the symbiotic systems.

43
Future perspectives
The field of IS research still has numerous unanswered questions that could be resolved using currently available techniques and information. Among these are:
o How frequent are IS transpositions? With today’s low costs of genome sequencing, it is feasible to carry out frequent sequencing of DNA and RNA inn microbes, and single-cell sequencing techniques make it possible to find rare transposition events in individual microbes. However, it is still not possible to identify or sequence lethal insertions in dying cells.
o What governs the speed of IS transpositions?
Comparisons between DNA, transcriptome, proteome and IS trans-positions during controlled conditions would make it possible to elu-cidate during which conditions ISs are transcribed, the ratios be-tween IS transcripts, IS translation and IS transposition events, and the factors that influence and regulate these processes.
o Which are the common characteristics of the IS families?
Although substantial efforts have been made to classify IS families, their ever increasing numbers, and advances in computing power and techniques, make it more feasible to find common traits among IS sequences, the circumstances of their transposition and their char-acteristics of the genomes in which they proliferate.

44
Swedish Summary
Cyanobakterier är välspridda och ekologiskt viktiga fotosyntetiskt självförsörjande mikroorganismer med många skepnader och förmågor. Det Cyanobakteriella fylumet innefattar arter från varierande miljöer, från näringsfattiga hav till insidan av symbiotiska växter, inklusive existensen som kloroplaster, vilka är extremt förenklade cyanobakterier som inkluderats i nästan alla växtceller.
Insertionssekevenser är korta (ca 1000 nukleotider i längd) mobila ge-
netiska element som kan fungera som kraftiga evolutionära verktyg, eft-ersom de innehåller den genetiska koden för ett protein med förmågan att flytta insertionssekvensen från en plats i arvsmassa till en annan.
I denna doktorsavhandling prövas hypoteser där insertionssekvenser har
en central roll i anpassningen till nya miljöer och i storleksminskningen av symbiotiska bakteriers arvsmassa, med hjälp av flera modellsystem:
Först undersöktes den permanent symbiotiska bakterien i ormbunken Az-olla. Detta är en cyanobakterie benämnd ‘Nostoc azollae 0708’ (NoAz). När NoAz-bakterien sekvenserades avslöjades en krympande arvsmassa (5.48 miljoner baspar) som utgjordes till minst 14% av insertionssekvenser. Den höga mängden insertionssekvenser kan vara skadlig, men upprätthålls troligtvis på grund av de gynnsamma levnadsförhållandena för NoAz, som skyddas och förses med näring av sin symbiotiska värd. Insertionssekven-serna var ofta belägna i närheten av de åtskilliga trasiga gener som hittades i NoAz’ arvsmassa. Gener som var viktiga i det symbiotiska förhållandet, t.ex. de gener som omvandlar atmosfäriskt kväve till näringsämnen av värde för Azolla (värden i symbiosen), var fortfarande intakta, medan gener som kan tänkas viktigare för en frilevande organism var skadade. Insertionssekvenser verkade förflytta sig i små steg genom arvsmassan och hittades ofta i närheten av trasiga gener, vilket gjorde det sannolikt att insertionssekven-serna skulle medverkat i störningen av dessa gener. Detta gjorde det troligt att insertionssekvenserna har medverkat i den gradvisa minskningen av arvsmassans storlek.
Efter dessa upptäcker utvidgades forskningen genom att undersöka inser-
tionssekvenser i 66 ytterligare bakterier, både frilevande, symbionter och patogener. De bakterier som levde inuti en värdorganism hade i allmänhet

45
fler insertionssekvenser i sin arvsmassa än frilevande bakterier, och dessa insertionssekvenser var påfallande ofta belägna i närheten av trasiga gener.
Slutligen undersöktes en samlad bakteriell arvsmassa från bakterier i Östersjön, och de genetiska avskrifter från arvsmassan som utgör ett mel-lansteg till proteintillverkning. Proteintillverkning utifrån en avskrift från arvsmassan är nödvändig för att tillverka det protein som förflyttar inser-tionssekvenser. Östersjön var intressant i detta sammanhang eftersom den utgör en geologiskt ung miljö med brackvatten, till skillnad från marina miljöer (saltvatten) som ofta är betydligt äldre. Östersjön var således lämplig för att leta efter spår av organismers anpassning till nya miljöer, i likhet med att symbiotiska bakterier upplevde förändringar när de påbörjade sin symbi-otiska livsstil. Östersjöns bakterier visade sig innehålla betydligt större ande-lar insertionssekvenser än bakterier från äldre marina vatten, som haft lång tid på sig att anpassa sig till sin miljö. Insertionssekvenser skrevs också av för proteintillverkning i mycket högre grad i Östersjön än i marina vatten, vilket gör det sannolikt att insertionssekvenser förflyttas oftare i Östersjön. Detta antyder att insertionssekvenser kan vara behjälpliga i anpassning till den nya miljön som brackvattnet i Östersjön utgör.
Sammanlagt antyder våra upptäckter att insertionssekvenser fungerar som
katalysatorer för bakteriell anpassning till flera olika nya miljöer, både de som upplevs av symbiotiska bakterier och de i ett hav med “ny” miljö (brackvatten). Den stora andelen insertionssekvenser som skrivits av som ett steg inför proteintillverkning antyder också att insertionssekvenser kan utöva denna effekt med oväntad hastighet.

46
Acknowledgements
In my work with cyanobacteria and ISs I have had the benefit of a mag-
nificent academic environment at the Department of Ecology, Environment and Plant Sciences (formerly Department of Botany) at Stockholm Universi-ty, and have enjoyed the education, support and company from more people than I can enumerate.
My deep gratitude goes especially to professor Birgitta Bergman, my
helpful and supportive supervisor, to my co-supervisor Sofia Carlson, and to my colleagues John Larsson, Ran Liang, Johan Nyberg, Johannes Asplund-Samuelsson, Martin Ekman and Karolina Ininbergs. I would also like to especially thank professor Lisbeth Jonsson for her help and advise during the writing of this Thesis.
This work was supported financially by the Swedish Energy Agency, the Swedish Research Council Formas, the Baltic Sea 2020 foundation and Stiftelsen Olle Engkvist, Byggmästare foundation. The metagenomic and metatranscriptomic data (paper III) was obtained via a cooperation with the J Craig Venter Institute and the Global Ocean Sampling expedition in the Bal-tic Sea project, the latter funded by JCVI internal funds and the US Depart-ment of Energy, Office of Science, Office of Biological and Environmental Research (DE-FC02-02ER63453).

47
References
Adams, D.G., and Duggan, P.S. (1999). Tansley Review No. 107. Hetero-cyst and akinete differentiation in cyanobacteria. New Phytol. 144, 3–33.
Alam, J., and Curtis, S.E. (1985). First International Congress on Plant Mo-lecular biology (Savannah, GA).
De Alda, J.A.O., Esteban, R., Diago, M.L., and Houmard, J. (2014). The plastid ancestor originated among one of the major cyanobacterial lineages. Nat. Commun. 5.
Andersson, S.G., and Kurland, C.G. (1998). Reductive evolution of resident genomes. Trends Microbiol. 6, 263–268.
Badawi, M., Giraud, I., Vavre, F., Grève, P., and Cordaux, R. (2014). Signs of Neutralization in a Redundant Gene Involved in Homologous Recombina-tion in Wolbachia Endosymbionts. Genome Biol. Evol. 6, 2654–2664.
Bancroft, I., and Wolk, C.P. (1989). Characterization of an insertion se-quence (IS891) of novel structure from the cyanobacterium Anabaena sp. strain M-131. J. Bacteriol. 171, 5949–5954.
Bergman, B. (2002). The Nostoc-Gunnera symbiosis. In Cyanobacteria in Symbiosis, (Springer), pp. 207–232.
Bergman, B., Gallon, J.R., Rai, A.N., and Stal, L.J. (1997). N2 fixation by non-heterocystous cyanobacteria. FEMS Microbiol. Rev. 19, 139–185.
Bergman, B., Rai, A.., and Rasmussen, U. (2007). Cyanobacterial Associa-tions. In Associative and Endophytic Nitrogen-Fixing Bacteria and Cyano-bacterial Associations, C. Elmerich, and W. Newton, eds. (Springer Nether-lands), pp. 257–301.
Bergman, B., Ran, L., Adams, D.G., Herrero, A., and Flores, E. (2008). Cy-anobacterial-plant Symbioses: signalling and development. Cyanobacteria Mol. Biol. Genomics Evol. 447–473.
Bergman, B., Sandh, G., Lin, S., Larsson, J., and Carpenter, E.J. (2013). Trichodesmium–a widespread marine cyanobacterium with unusual nitrogen fixation properties. FEMS Microbiol. Rev. 37, 286–302.

48
Berman-Frank, I., Lundgren, P., Chen, Y.-B., Küpper, H., Kolber, Z., Berg-man, B., and Falkowski, P. (2001). Segregation of nitrogen fixation and ox-ygenic photosynthesis in the marine cyanobacterium Trichodesmium. Sci-ence 294, 1534–1537.
Berntzon, L., Ronnevi, L.O., Bergman, B., and Eriksson, J. (2015). Detec-tion of BMAA in the human central nervous system. Neuroscience 292, 137–147.
Bhattacharya, D., Archibald, J.M., Weber, A.P., and Reyes‐Prieto, A. (2007). How do endosymbionts become organelles? Understanding early events in plastid evolution. Bioessays 29, 1239–1246.
Bickhart, D., Gogarten, J., Lapierre, P., Tisa, L., Normand, P., and Benson, D. (2009). Insertion sequence content reflects genome plasticity in strains of the root nodule actinobacterium Frankia. BMC Genomics 10, 468.
Black, K.G., Parsons, R., and Osborne, B.A. (2002). Uptake and metabolism of glucose in the Nostoc–Gunnera symbiosis. New Phytol. 153, 297–305.
Boyd, E.F., and Hartl, D.L. (1997). Nonrandom location of IS1 elements in the genomes of natural isolates of Escherichia coli. Mol. Biol. Evol. 14, 725–732.
Brinkhuis, H., Schouten, S., Collinson, M.E., Sluijs, A., Damsté, J.S.S., Dickens, G.R., Huber, M., Cronin, T.M., Onodera, J., and Takahashi, K. (2006). Episodic fresh surface waters in the Eocene Arctic Ocean. Nature 441, 606–609.
Calvert, H.E., Pence, M.K., and Peters, G.A. (1985). Ultrastructural ontoge-ny of leaf cavity trichomes inAzolla implies a functional role in metabolite exchange. Protoplasma 129, 10–27.
De Cambiaire, J.-C., Otis, C., Lemieux, C., and Turmel, M. (2006). The complete chloroplast genome sequence of the chlorophycean green alga Scenedesmus obliquus reveals a compact gene organization and a biased distribution of genes on the two DNA strands. BMC Evol. Biol. 6, 37.
Carpenter, E.J., and Foster, R.A. (2002). Marine cyanobacterial symbioses. In Cyanobacteria in Symbiosis, (Springer), pp. 11–17.
Chandler, M., and Siguier, P. (2013). Insertion Sequences. In Brenner’s En-cyclopedia of Genetics (Second Edition), S.M. Hughes, ed. (San Diego: Ac-ademic Press), pp. 86–94.
Cho, N.-H., Kim, H.-R., Lee, J.-H., Kim, S.-Y., Kim, J., Cha, S., Kim, S.-Y., Darby, A.C., Fuxelius, H.-H., and Yin, J. (2007). The Orientia tsutsugamu-

49
shi genome reveals massive proliferation of conjugative type IV secretion system and host–cell interaction genes. Proc. Natl. Acad. Sci. 104, 7981–7986.
Cock, P.J., Antao, T., Chang, J.T., Chapman, B.A., Cox, C.J., Dalke, A., Friedberg, I., Hamelryck, T., Kauff, F., and Wilczynski, B. (2009). Biopy-thon: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423.
Costa, J.-L., and Lindblad, P. (2002). Cyanobacteria in symbiosis with cy-cads. In Cyanobacteria in Symbiosis, (Springer), pp. 195–205.
Deusch, O., Landan, G., Roettger, M., Gruenheit, N., Kowallik, K.V., Allen, J.F., Martin, W., and Dagan, T. (2008). Genes of Cyanobacterial Origin in Plant Nuclear Genomes Point to a Heterocyst-Forming Plastid Ancestor. Mol. Biol. Evol. 25, 748–761.
Dolman, A.M., Rücker, J., Pick, F.R., Fastner, J., Rohrlack, T., Mischke, U., and Wiedner, C. (2012). Cyanobacteria and cyanotoxins: the influence of nitrogen versus phosphorus. PLoS One 7, e38757.
Domínguez-Escobar, J., Beltrán, Y., Bergman, B., Díez, B., Ininbergs, K., Souza, V., and Falcón, L.I. (2011). Phylogenetic and molecular clock infer-ences of cyanobacterial strains within Rivulariaceae from distant environ-ments. FEMS Microbiol. Lett. 316, 90–99.
Dupont, C.L., McCrow, J.P., Valas, R., Moustafa, A., Walworth, N., Goode-nough, U., Roth, R., Hogle, S.L., Bai, J., Johnson, Z.I., et al. (2014a). Ge-nomes and gene expression across light and productivity gradients in eastern subtropical Pacific microbial communities. ISME J.
Dupont, C.L., Larsson, J., Yooseph, S., Ininbergs, K., Goll, J., Asplund-Samuelsson, J., McCrow, J.P., Celepli, N., Allen, L.Z., Ekman, M., et al. (2014b). Functional Tradeoffs Underpin Salinity-Driven Divergence in Mi-crobial Community Composition. PLoS ONE 9, e89549.
Enderlin, C.S., and Meeks, J.C. (1983). Pure culture and reconstitution of the Anthoceros-Nostoc symbiotic association. Planta 158, 157–165.
Farabaugh, P.J. (1996). Programmed translational frameshifting. Annu Rev Genet 30, 507–528.
Feistel, R., Nausch, G., and Wasmund, N. (2008). State and evolution of the Baltic Sea, 1952-2005: a detailed 50-year survey of meteorology and cli-mate, physics, chemistry, biology, and marine environment (John Wiley & Sons).

50
Flores, E., and Herrero, A. (2010). Compartmentalized function through cell differentiation in filamentous cyanobacteria. Nat. Rev. Microbiol. 8, 39–50.
Foster, R.A., and Zehr, J.P. (2006). Characterization of diatom–cyanobacteria symbioses on the basis of nifH, hetR and 16S rRNA sequenc-es. Environ. Microbiol. 8, 1913–1925.
Fuxelius, H.-H., Darby, A., Cho, N.-H., and Andersson, S. (2008). Visuali-zation of pseudogenes in intracellular bacteria reveals the different tracks to gene destruction. Genome Biol. 9, R42.
Gil, R., Silva, F.J., Peretó, J., and Moya, A. (2004). Determination of the core of a minimal bacterial gene set. Microbiol. Mol. Biol. Rev. 68, 518–537.
Gil Garcia, R., Belda Cuesta, E., Gosalbes Soler, M.J., Delaye, L., Vallier, A., Vincent-Monégat, C., Heddi, A., Silva Moreno, F.J., Moya Simarro, A., and Latorre Castillo, D. (2008). Massive presence of insertion sequences in the genome of SOPE, the primary endosymbiont of the rice weevil Sitophi-lus oryzae.
Green, L., Miller, R.D., Dykhuizen, D.E., and Hartl, D.L. (1984). Distribu-tion of DNA insertion element IS5 in natural isolates of Escherichia coli. Proc. Natl. Acad. Sci. 81, 4500–4504.
Hall, J.W., and Swanson, N.P. (1968). Studies on fossil Azolla: Azolla mon-tana, a Cretaceous megaspore with many small floats. Am. J. Bot. 1055–1061.
Haselkorn, R. (2007). Heterocyst differentiation and nitrogen fixation in cyanobacteria. In Associative and Endophytic Nitrogen-Fixing Bacteria and Cyanobacterial Associations, (Springer), pp. 233–255.
Herdman, M., and Stanier, R.Y. (1977). The cyanelle: chloroplast or endo-symbiotic prokaryote? FEMS Microbiol. Lett. 1, 7–11.
Herlemann, D.P., Labrenz, M., Jurgens, K., Bertilsson, S., Waniek, J.J., and Andersson, A.F. (2011). Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J 5, 1571–1579.
Hess, W.R. (2011). Cyanobacterial genomics for ecology and biotechnology. Curr. Opin. Microbiol. 14, 608–614.
Ininbergs, K., Bergman, B., Larsson, J., and Ekman, M. (2015). Microbial metagenomics in the Baltic Sea: Recent advancements and prospects for environmental monitoring. (in press). AMBIO DOI 10.1007/s13280-015-0663-7.

51
Jonasson, S., Eriksson, J., Berntzon, L., Spáčil, Z., Ilag, L.L., Ronnevi, L.-O., Rasmussen, U., and Bergman, B. (2010). Transfer of a cyanobacterial neurotoxin within a temperate aquatic ecosystem suggests pathways for hu-man exposure. Proc. Natl. Acad. Sci. 107, 9252–9257.
Jordan, E., Saedler, H., and Starlinger, P. (1968). O and strong-polar muta-tions in the gal operon and insertions. Mol. Gen. Genet. MGG 102, 353–363.
Kaneko, T., Nakajima, N., Okamoto, S., Suzuki, I., Tanabe, Y., Tamaoki, M., Nakamura, Y., Kasai, F., Watanabe, A., Kawashima, K., et al. (2007). Complete Genomic Structure of the Bloom-forming Toxic Cyanobacterium Microcystis aeruginosa NIES-843. DNA Res. 14, 247–256.
Kleiner, M., Young, J.C., Shah, M., VerBerkmoes, N.C., and Dubilier, N. (2013). Metaproteomics reveals abundant transposase expression in mutual-istic endosymbionts. MBio 4, e00223–13.
Kneip, C., Voβ, C., Lockhart, P.J., and Maier, U.G. (2008). The cyanobacte-rial endosymbiont of the unicellular algae Rhopalodia gibba shows reductive genome evolution. BMC Evol. Biol. 8, 30.
Krawiec, S., and Riley, M. (1990). Organization of the bacterial chromo-some. Microbiol. Rev. 54, 502.
Kurtz, S., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2000). Computation and visualization of degenerate repeats in complete genomes. Proc Int Conf Intell Syst Mol Biol 8, 228–238.
Larsson, J., Nylander, J.A., and Bergman, B. (2011). Genome fluctuations in cyanobacteria reflect evolutionary, developmental and adaptive traits. BMC Evol. Biol. 11, 187.
Larsson, J., Celepli, N., Ininbergs, K., Dupont, C.L., Yooseph, S., Bergman, B., and Ekman, M. (2014). Picocyanobacteria containing a novel pigment gene cluster dominate the brackish water Baltic Sea. ISME J.
Lawrence, J.G., Ochman, H., and Hartl, D.L. (1992). The evolution of inser-tion sequences within enteric bacteria. Genetics 131, 9–20.
Lechno-Yossef, S., and Nierzwicki-Bauer, S.A. (2002). Azolla-Anabaena symbiosis. In Cyanobacteria in Symbiosis, (Springer), pp. 153–178.
Lin, S., Haas, S., Zemojtel, T., Xiao, P., Vingron, M., and Li, R. (2011). Genome-wide comparison of cyanobacterial transposable elements, potential genetic diversity indicators. Gene 473, 139–149.

52
Lyons, T.W. (2007). Palaeoclimate: Oxygen’s rise reduced. Nature 448, 1005–1006.
De Marsac, N.T., and Houmard, J. (1988). [34] Complementary chromatic adaptation: Physiological conditions and action spectra. Methods Enzymol. 167, 318–328.
Martin, W., Rujan, T., Richly, E., Hansen, A., Cornelsen, S., Lins, T., Leis-ter, D., Stoebe, B., Hasegawa, M., and Penny, D. (2002). Evolutionary anal-ysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. 99, 12246–12251.
Mira, A., Ochman, H., and Moran, N.A. (2001). Deletional bias and the evo-lution of bacterial genomes. Trends Genet. 17, 589–596.
Mizuuchi, K. (1992). Transpositional recombination: mechanistic insights from studies of Mu and other elements. Annu. Rev. Biochem. 61, 1011–1051.
Moore, A.W. (1969). Azolla: Biology and agronomic significance. Bot. Rev. 35, 17–34.
Moran, N.A., and Plague, G.R. (2004). Genomic changes following host restriction in bacteria. Curr. Opin. Genet. Dev. 14, 627–633.
Mulkidjanian, A.Y., Koonin, E.V., Makarova, K.S., Mekhedov, S.L., So-rokin, A., Wolf, Y.I., Dufresne, A., Partensky, F., Burd, H., Kaznadzey, D., et al. (2006). The cyanobacterial genome core and the origin of photosynthe-sis. Proc. Natl. Acad. Sci. 103, 13126–13131.
Naas, T., Blot, M., Fitch, W.M., and Arber, W. (1995). Dynamics of IS-related genetic rearrangements in resting Escherichia coli K-12. Mol Biol Evol 12, 198–207.
Nathanielsz, C.P., and Staff, I.A. (1975). On the occurrence of intracellular blue-green algae in cortical cells of the apogeotropic roots of Macrozamia communis. L. Johnson. Ann. Bot. 39, 363–368.
Okonechnikov, K., Golosova, O., and Fursov, M. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167.
Oren, A. (2011). Cyanobacterial systematics and nomenclature as featured in the international bulletin of bacteriological nomenclature and taxono-my/international journal of systematic bacteriology/international journal of systematic and evolutionary microbiology. Int. J. Syst. Evol. Microbiol. 61, 10–15.

53
Osborne, B., and Bergman, B. (2009). Why Does Gunnera Do It and Other Angiosperms Don’t? An Evolutionary Perspective on the Gunnera–Nostoc Symbiosis. In Prokaryotic Symbionts in Plants, (Springer), pp. 207–224.
Papaefthimiou, D., Van Hove, C., Lejeune, A., Rasmussen, U., and Wil-motte, A. (2008). Diversity and host specificity of Azolla cyanobionts. J. Phycol. 44, 60–70.
Peters, G.A., and Meeks, J.C. (1989). The Azolla-Anabaena symbiosis: basic biology. Annu. Rev. Plant Biol. 40, 193–210.
Pinto, F.A.L., Troshina, O., and Lindblad, P. (2002). A brief look at three decades of research on cyanobacterial hydrogen evolution. Int. J. Hydrog. Energy 27, 1209–1215.
Plazinski, J., Zheng, Q., Taylor, R., Croft, L., Rolfe, B.G., and Gunning, B.E. (1990). DNA probes show genetic variation in cyanobacterial symbi-onts of the Azolla fern and a closer relationship to free-living Nostoc strains than to free-living Anabaena strains. Appl. Environ. Microbiol. 56, 1263–1270.
Rasmussen, B., Fletcher, I.R., Brocks, J.J., and Kilburn, M.R. (2008). Reas-sessing the first appearance of eukaryotes and cyanobacteria. Nature 455, 1101–1104.
Rasmussen, U., Johansson, C., and Bergman, B. (1994). Early communica-tion in the Gunnera-Nostoc symbiosis: plant-induced cell differentiation and protein synthesis in the cyanobacterium. Mol. Plant Microbe Interact. 7, 696–702.
Redecker, D., Morton, J.B., and Bruns, T.D. (2000). Ancestral lineages of arbuscular mycorrhizal fungi (Glomales). Mol. Phylogenet. Evol. 14, 276–284.
Reynolds, C.S., Oliver, R.L., and Walsby, A.E. (1987). Cyanobacterial dom-inance: The role of buoyancy regulation in dynamic lake environments. N. Z. J. Mar. Freshw. Res. 21, 379–390.
Rikkinen, J., Oksanen, I., and Lohtander, K. (2002). Lichen guilds share related cyanobacterial symbionts. Science 297, 357–357.
Rippka, R., Deruelles, J., Waterbury, J.B., Herdman, M., and Stanier, R.Y. (1979). Generic assignments, strain histories and properties of pure cultures of cyanobacteria. J. Gen. Microbiol. 111, 1–61.
Schirrmeister, B.E., Antonelli, A., and Bagheri, H.C. (2011). The origin of multicellularity in cyanobacteria. BMC Evol. Biol. 11, 45.

54
Schuster, M., and Greenberg, E.P. (2007). Early activation of quorum sens-ing in Pseudomonas aeruginosa reveals the architecture of a complex regu-lon. BMC Genomics 8, 287.
Shapiro, J.A. (1969). Mutations caused by the insertion of genetic material into the galactose operon of Escherichia coli. J. Mol. Biol. 40, 93–105.
Shi, T., and Falkowski, P.G. (2008). Genome evolution in cyanobacteria: the stable core and the variable shell. Proc. Natl. Acad. Sci. 105, 2510–2515.
Siguier, P., Filée, J., and Chandler, M. (2006a). Insertion sequences in pro-karyotic genomes. Curr. Opin. Microbiol. 9, 526–531.
Siguier, P., Pérochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006b). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36.
Siguier, P., Gourbeyre, E., and Chandler, M. (2014). Bacterial insertion se-quences: their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38:865-891.
Sivonen, K., and Börner, T. (2008). Bioactive compounds produced by cya-nobacteria. Cyanobacteria Mol. Biol. Genomics Evol. 159–197.
Skipper, K., Andersen, P., Sharma, N., and Mikkelsen, J. (2013). DNA transposon-based gene vehicles - scenes from an evolutionary drive. J. Bio-med. Sci. 20, 92.
Steffen, M.M., Dearth, S.P., Dill, B.D., Li, Z., Larsen, K.M., Campagna, S.R., and Wilhelm, S.W. (2014). Nutrients drive transcriptional changes that maintain metabolic homeostasis but alter genome architecture in Microcyst-is. ISME J 8, 2080–2092.
Tang, L.F., Watanabe, I., and Liu, C.C. (1990). Limited multiplication of symbiotic cyanobacteria of Azolla spp. on artificial media. Appl. Environ. Microbiol. 56, 3623–3626.
Telesh, I.V., and Khlebovich, V.V. (2010). Principal processes within the estuarine salinity gradient: a review. Mar. Pollut. Bull. 61, 149–155.
Terauchi, K., and Kondo, T. (2008). The cyanobacterial circadian clock and the KaiC phosphorylation cycle. Cyanobacteria Mol. Biol. Genomics Evol. 199–216.
Thompson, A.W., Foster, R.A., Krupke, A., Carter, B.J., Musat, N., Vaulot, D., Kuypers, M.M., and Zehr, J.P. (2012). Unicellular cyanobacterium sym-biotic with a single-celled eukaryotic alga. Science 337, 1546–1550.

55
Touchon, M., and Rocha, E.P.C. (2007). Causes of Insertion Sequences Abundance in Prokaryotic Genomes. Mol. Biol. Evol. 24, 969–981.
Usher, K.M., Bergman, B., and Raven, J.A. (2007). Exploring cyanobacteri-al mutualisms. Annu. Rev. Ecol. Evol. Syst. 255–273.
Venter, J.C., Remington, K., Heidelberg, J.F., Halpern, A.L., Rusch, D., Eisen, J.A., Wu, D., Paulsen, I., Nelson, K.E., and Nelson, W. (2004). Envi-ronmental genome shotgun sequencing of the Sargasso Sea. Science 304, 66–74.
Vigil-Stenman, T., Larsson, J., and Bergman, B. (2015). Local hopping mo-bile DNA implicated in pseudogene formation and reductive evolution in an obligate cyanobacteria-plant symbiosis. BMC Genomics 16, 193.
Wagner, A. (2006). Periodic extinctions of transposable elements in bacterial lineages: evidence from intragenomic variation in multiple genomes. Mol. Biol. Evol. 23, 723–733.
Walker, A., and Langridge, G. (2008). Does my genome look big in this? Nat. Rev. Microbiol. 6, 878–879.
Whitfield, A.K., Elliott, M., Basset, A., Blaber, S.J.M., and West, R.J. (2012). Paradigms in estuarine ecology – A review of the Remane diagram with a suggested revised model for estuaries. Estuar. Coast. Shelf Sci. 97, 78–90.
Wolk, C.P., Ernst, A., and Elhai, J. (2004). Heterocyst metabolism and de-velopment. In The Molecular Biology of Cyanobacteria, (Springer), pp. 769–823.
Ye, J., McGinnis, S., and Madden, T.L. (2006). BLAST: improvements for better sequence analysis. Nucleic Acids Res 34, W6–W9.
Zheng, W., Bergman, B., Chen, B., Zheng, S., Xiang, G., and Rasmussen, U. (2009). Cellular responses in the cyanobacterial symbiont during its vertical transfer between plant generations in the Azolla microphylla symbiosis. New Phytol. 181, 53–61.
Zheng, W., Rasmussen, U., Zheng, S., Bao, X., Chen, B., Gao, Y., Guan, X., Larsson, J., and Bergman, B. (2013). Multiple Modes of Cell Death Discov-ered in a Prokaryotic (Cyanobacterial) Endosymbiont. PloS One 8, e66147.
Zheng, W.W., Lin, Y.H., Lu, P.J., and Liu, Z.Z. (1990). Electron microscop-ic observations on symbiotic relationship between Azolla and Anabaena during the megaspore germination and spore development of Azolla. Acta Bot. Sin. 32, 514–520.

56
Zheng, W.W., Nilsson, M., Bergman, B., and Rasmussen, U. (1999). Genetic diversity and classification of cyanobacteria in different Azolla species by the use of PCR fingerprinting. Theor. Appl. Genet. 99, 1187–1193.
Zhou, R., and Wolk, C.P. (2002). Identification of an akinete marker gene in Anabaena variabilis. J. Bacteriol. 184, 2529–2532.
Zhou, F., Olman, V., and Xu, Y. (2008). Insertion Sequences show diverse recent activities in Cyanobacteria and Archaea. BMC Genomics 9, 36.
ISBN 978-91-7649-191-1





























