Embed Size (px)
Citation preview

Electrophysiology, Hemodynamic and Arrhythmia Efficacy Model Studies on Encainide
ALLEN W. GOMOLL, PhD, JEFFREY E. BYRNE, PhD, and MICHAEL J. ANTONACCIO, PhD
Encainide is a class IC agent possessing a broad spectrum of antiarrhythmic actions in a variety of animal models. It increases the ventricular fibrillation threshold of the perfused rabbit heart and in situ dog myocardium. Encainide suppresses atrial fibrillation resulting from topical application of aconitine in the anesthetized dog and ventricular fibrillation induced by chloroform asphyxiation in the mouse. In these latter 2 models, encainide is approximately 7 to 11 and 16 to 16 times more potent, respectively, on a milligram basis than quinidine. In anesthetized dogs encainide converts ouabain-induced tachyarrhythmias to normal sinus rhythm at a mean intravenous dose of 0.67 rng/kg. Single doses of 0.5 mg/kg intravenously or 1 mg/kg orally significantly reduced ventricular ectopy in conscious dogs 16 to 22 hours after 2-stage ligation of the left coronary artery. At doses and plasma concentrations exceeding efficacious therapeutic levels, encainide has no major negative inotropic effects and does not
compromise cardiac function or hemodynamics. It is devoid of peripheral autonomic or mediator-evoked responses and, in particular, lacks anticholinergic actions. Encainide is rapidly absorbed by all routes of administration and extensively metabolized by the liver. The major metabolites, 0-demethyl encainide and 3-methoxy-0-demethyl encainide, have been shown to have quantitatively different, but qualitatively similar, profiles of pharmacodynamic effects. Subacute and chronic administration of encainide at doses representing 11 times an effective oral human dose have produced no distinct or consistent toxicologic findings. Carcinogenicity and mutagenicity studies were negative.
The available animal data are consistent with the hypothesis that the metabolites of encainide may contribute, at least in part, to the electrophysiologic and persistent antiarrhythmic actions observed clini- cally after the chronic administration of the parent drug in patients. (Am J Cardiol 1966;56:lOC-17C)
E ncainide, 4-methoxy-2’-[Z-(1-methyl-2-piperi- dyl)ethyl]benzanilide (Fig. l), is a structurally novel antiarrhythmic compound synthesized by Mead John- son/Bristol-Myers. Its demonstrated effectiveness in several preliminary animal tests dictated the need for a broader assessment of its overall pharmacologic properties. Encainide has now been evaluated in a variety of animal models designed to quantify its phar- macokinetic, toxicologic and pharmacodynamic char- acteristics relative to its antiarrhythmic, cardiac, he- modynamic, autonomic and peripheral actions. This
From the Department of Cardiovascular Research, Bristol-My- ers Pharmaceutical Research and Development Division, Ev- ansville, Indiana.
Address for reprints: Allen W. Gomoll, PhD, Department of Cardiovascular Biology, Bristol-Myers Pharmaceutical Re- search and Development Division, 2404 Pennsylvania, Evans- ville, Indiana 47721.
article summarizes the nonclinical biologic profile of this agent.
Pharmacokinetics and Biopharmaceutics In establishing the absorption, distribution, metab-
olism and excretion characteristics of encainide, initial
N
::” AH,
=I -
,“ii ’ 0 ’ OCH3
4-Methoxy-2’-[2-(1 -methyl-2-piperidyl)ethyl]benzanilide
FIGURE 1. Chemical structure of encainide.
1oc
August 29, ;986 THE AMERICAN ,“OURNAL OF CASDlOL3GY Vok,e 58 4745
studies in rats and dogs were performed using tritium- labeled drug and its disposition was assessed on the basis of total and ether-extractable radioactivity. Sub- sequent studies in these same 2 species, as well as mice in which carbon-l&labeled encainide was adminis- tered were performed after the development of specif- ic radioimmune and high pressure liquid chromatog- raphy assay methods1 for encainide and its major me- tabolites. The initial studies indicated that after oral administration, encainide was rapidly and completely absorbed with peak plasma levels of radioactivity oc- curring within 1 hour in rats and at ‘$ to 2 hours in dogs. In rats, there was no unusual localization or excessive retention of radioactivity in any body tissue or organ system. In both species, a similar proportion of orally or intravenously administered radioactivity was ex- creted in the urine (Table I). Intravenous infusion studies of encainide indicated that the mean elimina- tion half-life was between 1.5 and 2.5 hours and the volume of distribution at steady state was 3.2 liters/kg.
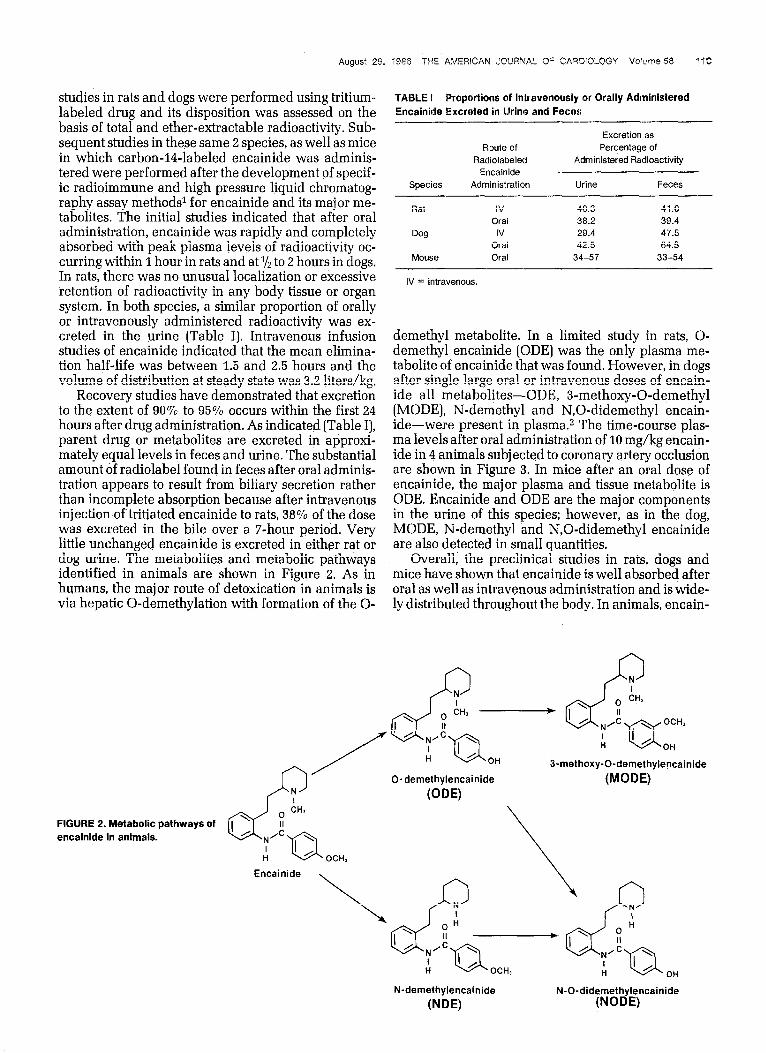
Recovery studies have demonstrated that excretion to the extent of 90% to 95% occurs within the first 24 hours after drug administration. As indicated (Table I], parent drug or metabolites are excreted in approxi- mately equal levels in feces and urine. The substantial amount of radiolabel found in feces after oral adminis- tration appears to result from biliary secretion rather than incomplete absorption because after intravenous injection of tritiated encainide to rats, 38% of the dose was excreted in the bile over a T-hour period. Very little unchanged encainide is excreted in either rat or dog urine. The metabolites and metabolic pathways identified in animals are shown in Figure 2. As in humans, the major route of detoxication in animals is via hepatic 0-demethylation with formation of the O-
TAl3LE f Proportions of Intravenously or Orally Administered Encainide Excreted in Urine and Feces
Species
Route of Radiolabeled
Encainide Administration
Excretion as Percentage of
Administered Radioactivity
Urine Feces
Rat IV 40.3 41.0 Oral 36.2 39.4
Dog IV 29.4 47.5 Oral 42.5 64.5
Mouse Oral 34-57 33-54
IV = intravenous.
demethyl metabolite. In a limited study in rats, O- demethyl encainide [ODE] was the only plasma me- tabolite of encainide that was found. However, in dogs after single large oral or intravenous doses of encain- ide all metabolites-ODE, 3-methoxy-0-demethyl (MODE), N-demethyl and N,O-didemethyl encain- ide-were present in plasma.z The time-course plas- ma levels after oral administration of 10 mg/kg encain- ide in 4 animals subjected to coronary artery occlusion are shown in Figure 3. In mice after an oral dose of encainide, the major plasma and tissue metabolite is ODE. Encainide and ODE are the major components in the urine of this species; however, as in the dog, MODE, N-demethyl and N,O-didemethyl encainide are also detected in small quantities.
Overall, the preclinical studies in rats, dogs and mice have shown that encainide is well absorbed after oral as well as intravenous administration and is wide- ly distributed throughout the body. In animals, encain-
3-methoxy-0-demethylencainide
(MODE)
FIGURE 2. Metabolic pathways of encainide in animals.
Encainide
OH
N-demethylencainide
WE)
N-0-did;r;#lencainide

1x A SYMPOSIUM: ENCAINIDE
TABLE II Encainide Toxicity Studies
Study
Type
Median tethal
dose
Duration Route
Acute IV
Dose
OwW
16.1 5.5
16.9
Species
Mouse Rabbit
Dog
Subacute
Chronic
Carcinogenicity
1-3 mo
12 mo
18 mo 24 mo
Oral
IP Oral Oral
Oral Oral Oral
8.0 Monkey 27.3 Rat 79 Mouse 54 Rat 66 Rabbit
43 Qw 1.5-10 Rat, dog
5-28 Rat, dog 7.5-30 Rat
5.5-22 Dog 15-30 Rat 15-135 Mouse
Four mutagenicity studies were also conducted. IP 4 intraperitoneal; IV = intravenous,
ide is eliminated mainly by hepatic conversion to me- tabolites that are excreted in both the urine and feces. The major identified metabolites, except for N,O-di- demethyl encainide, are the same as those formed in man. Such results provide assurance that the data from animal toxicity studies can be expected to apply to man.
Safety and Toxicology The acute toxicity ‘of encainide has been deter-
mined in a variety of species by the intravenous, intra- peritoneal and oral routes (Table II). In these evalua- tions, the interpreted oral median lethal dose values have been shown to represent 16 to 30 times the dem- onstrated clinically efficacious dose. The most signifi- cant pharmacotoxic signs were ataxia, tremors and
Encainide (10 mg/kg PO) ODE (5 mg/kg PO) (ng/mL)
loo0 I
convulsions, which occurred in most species regard- less of the route of drug administration.
Subacute and chronic administration of encainide to rats and dogs caused neither distinct nor consistent untoward findings. The upper doses administered in both species represented approximately 11 times an effective oral human dose. Long-term oral studies in 2 species demonstrated no evidence of carcinogenicity and 4 mutagenicity studies yielded similar negative findings.
Pharmacology Electrophysiolqgy: The single cell electrophysio-
logic effects of encainide have been studied in repre- sentative tissues from all areas of the heart. The most consistent change produced by 1QW7 to 10m5 M (0.035 to 3.5 mg/liter) encainide ‘on myocardium from all spe- cies investigated was a prominent decrease in the maximum rate of rise in phase 0 depolarization.3-7 This decrease was greater at fast; compared with slow, rates of stimulation and occurred without alteration in the maximum diastolic potential and with little effect on the action potential amplitude.4 Encainide caused only minimal changes in the action potential duration de- pendent on the tissue used and the species studied. The action potential duration was decreased in Pur- kinj e fibers (dog,3,5 cow,4 sheep4), unchanged in papil- lary muscle (dog,5 guinea pig41 and increased in ven- tricular (rabbit,7 guinea pig61 and atria1 [guinea pi&) tissues.
The metabolites ODE and MODE produced changes qualitatively similar to those of encainide’on the action potential characteristics of both canine Pur- kinje and papillary muscle fibers in vitro.5.0n a molar basis, however, ODE was approximately 9 times more potent than either encainide or MODE. None of the agents suppressed the development of sloti channel- dependent action potentials, thus indicating their lack
0 60 120 180 240 300 360 0 60 120 180 2$0 300 360 Time (min) Time (min)
MODE (3.8 mg/kg PO)
_‘_ FJT---&
0 60 120 160 240 300 360 Time (mln)
FIGURE 3. Time-course plasma drug concentrations (rig/ml) after oral (PO) admjnistraiion of en- cajnide, 0-demathyl ‘encainide (ODE) or I-methox&D-demethyl qncainide (MODE) ‘In consciqus dogs. ’

August 29, ?9P6 THE AMERICAN JQLIRNA’- OF CARW'OLOGY Vobne58 13C
of effect on Ca++-mediated events in myocardial cells4s5
The overall profile of single cell electrophysiologic properties described has led to the characterization of encainide (and metabolites) as a class IC agent in the Vaughan Williams scheme as modified by Harrison.8 This classification places encainide in the same elec- trophysiologic category as flecainide and lorcainide and distinguishes it .from either the class IA drugs (quinidine, procainamide, disopyramide), which pre- dominantly prolong the action potential duration with less suppression of maximum velocity of depolariza- tion, or class IB agents (lidocaine, tocainide, mexili- tine), which shorten the action potential duration and only minimally affect maximum velocity of depolar- ization.g These distinguishing characteristics are sum- marized in Table III.
In both the isolated perfused guinea pig4 and rab- bitlo heart, encainide (0.1 to 1.4 mg/liter) slowed im- pulse conduction through the His-Purkinje system and ventricle to a greater degree than through either the atria or AV node.3 In contrast to its minimal effects on ventricular and atrioventricular (AV) nodal effective refractory periods at fixed basal cycle lengths, encain- ide produced substantial prolongation in refractori- ness after extrasystolic beats.4 This effect on recovery from extrasystolic impulses was shown to be greater in the ventricles than in the AV node, and thus suggested a potential mechanism whereby encainide might be efficacious in the suppression of ectopic arrhythmias.
The most prominent electrophysiologic effect seen with encainide after intravenous or oral administra- tion in various intact animal studies was prolongation of both the PR interval and QRS duration. A canine study by Sami et all1 demonstrated that the acute intra- venous injection of 0.9 or 2.7 (but not 0.3 mg/kg) en- cainide caused only increases in QRS duration (15% to 29%) and His-Purkinje (HV interval) conduction (31% to 48%) in the absence of changes in either AV nodal conduction or effective refractory period of the atria, AV node or ventricle. The mean plasma drug concen- trations associated with these responses were 1,300 and 4,000 rig/ml. When similar measurements were delayed until 35 to 40 minutes after drug infusion, encainide (2.7 mg/kg) administration was associated not only with an increase in the (IRS duration (24%) and HV interval (41%), but also with prolongation of the AH interval (51%) and QTc interval (7?70).12 Con- current measurements demonstrated that the mono- phasic actionfpotential at 96% recovery was increased (14%) and both the functional and effective refractory periods of the atria (26% to 31%) and ventricle (lo?70 to 13%) were prolonged after this high dose administra- tion The mean peak plasma encainide levels attained in these studies varied from 558 to 859 rig/ml. Thus, unlike other class I antiarrhythmic agents, acute ad- ministration of encainide was associated with conduc- tion slowing in the specialized His-Purkinje system without significant alterations in conduction or cefrac- toriness in other areas of the canine myocardium. Measurements made at later intervals, when metabo-
TABLE liil Electrophysiologic Efffects of Encainide, Its Metabolites and Sel,ected Anliiarrhythmic Agents in ‘Vitro (Purkinje fibers)
Class Drug MDF’ APIJ max V ERP
- IA Quinidine 011 I
1 1B Tocainide 0 O/l I 1c Encainide 0 011
ODE 0 O/l MODE 0 O/l t i
APD = action potential duration; ERP = effective refractory period; MDP = maximum diastolic potential: MODE = 3-methoxy-Odemethyl encainide; ODE = Odemethyl encalmde; V,, = maximum rate of phase 0 depolarizaiion; 0 = no change; t = increase; 1 = decrease.
lites presumably had been formed, revealed the exis- tence of a profile more comparable to that produced by quinidine.12
His-bundle observations in anesthetized dogs giv- en ODE at infusion rates to attain steady-state plasma levels comparable to those measured clinically during encainide therapy in man (149 to 230 rig/ml) support the interpretation that the differences between the electrophysiologic actions of acute intravenous and chronic oral encainide may be due to the pharmaco- logic effects of its ‘ODE metabolite.13 In addition to prolongation of the PR interval (29%) and QRS dura- tion (4270)~ ODE significantly increased the atria1 ef- fective refractory period (62%) and, to a lesser degree, ventricular refractoriness (13%) as well as AH (23%) and HV (62%) conduction time.
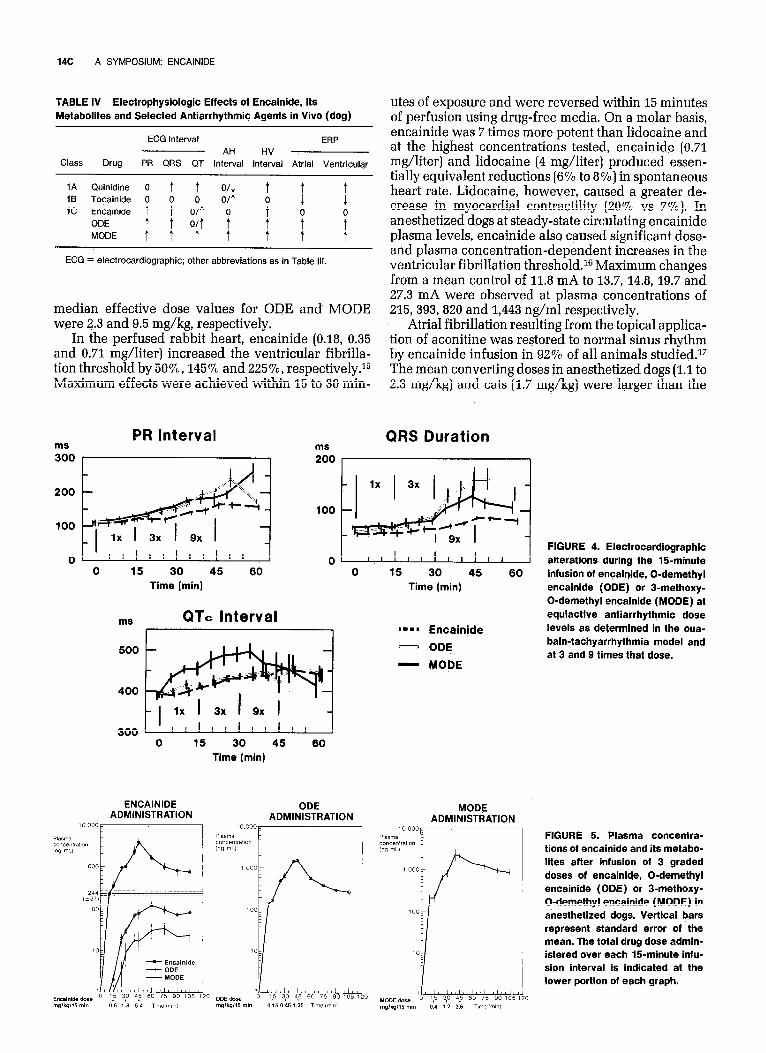
Following the determination of the comparative antiarrhythmic activities of encainide and its metabo- lites, ODE and MODE (to be discussed), the qualita- tively similar but quantitatively different acute elec- trocardiographic profiles of these agents at 1,3, and 9 times the equiactive doses were assessed (Fig. 4). As indicated, ODE, and to a’lesser extent MODE, caused more conduction slowing’as assessed from the PR in- terval and QRS duration than encainide. At 3 times the equiactive dose, for example, the PR interval and the QRS duration were increased 41% and 43% vs XWO,
and 54% and 24% vs 20%, respectively, by ODE and MODE vs encainide. The recovery phase interpreted from the QTc interval, in contrast, was slowed to a greater degree by MODE (25%) than by either encain- ide (9%) or ODE (1170) at equiactive doses. The plasma drug levels associated with these temporal responses are shown in Figure 5.
To summarize so far, the electrophysiologic alter- ations recorded in anesthetized (and conscious) ani- mals after encainide and its metabolites (Table IV), as with other class I agents9 have been predictive of the PR, QRS and slight QTc changes observed clinically.14
Antiarrhythmic actions: Pretreatment of mice with encainide by the intraperitoneal route prevented the development of ventricular fibrillation induced by chloroform asphyxiation.2 The interpreted median ef- fective dose was 5.3 mg/kg. In this model, encainide was 16 to 18 times more potent than quinidine and 5 times more potent than disopyramide. Comparable
14c A SYMPOSIUM: ENCAINIDE
TABLE IV Electrophysiologic Effects of Encainide, Its Metabolites and Selected Antiarrhythmic Agents in Vivo (dog)
Class Drug
ECG Interval ERP AH HV
PR QRS QT Interval Interval Atrial Ventricular
1A Quinidine 0 It3 Tocainide 0 + + 4 + 0 0 0 I IC Encainide
ODE r T :;I ‘; !
0 0
MODE tttt t T ?
ECG = electrocardiographic; other abbreviations as in Table Ill.
median effective dose values for ODE and MODE were 2.3 and 9.5 mg/kg, respectively.
In the perfused rabbit heart, encainide (0.18, 0.35 and 0.71 mg/liter) increased the ventricular fibrilla- tion threshold by 50%, 145% and 22570, respectively.15 Maximum effects were achieved within 15 to 30 min-
ms PR Interval
ms
utes of exposure and were reversed within 15 minutes of perfusion using drug-free media. On a molar basis, encainide was 7 times more potent than lidocaine and at the highest concentrations tested, encainide (0.71 mg/liter) and lidocaine (4 mg/liter) produced essen- tially equivalent reductions (6% to 8%) in spontaneous heart rate. Lidocaine, however, caused a greater de- crease in myocardial contractility (20% vs 7%). In anesthetized dogs at steady-state circulating encainide plasma levels, encainide also caused significant dose- and plasma concentration-dependent increases in the ventricular fibrihation threshold.16 Maximum changes from a mean control of 11.8 mA to 13.7, 14.8, 19.7 and 27.3 mA were observed at plasma concentrations of 215, 393, 820 and 1,443 rig/ml respectively.
Atria1 fibrillation resulting from the topical applica- tion of aconitine was restored to normal sinus rhythm by encainide infusion in 92% of all animals studied.17 The mean converting doses in anesthetized dogs (1.1 to 2.3 mg/kg) and cats (1.7 mg/kg) were larger than the
QRS Duration 300 200
200 - +ik$
,oo wfqy
,00
_ 1 lx I 3x 9x
:w;:
0 . I 1 I 1 I I I I I I I 0 1 I I, I I I I I I I 0 15 30 45 60 0 15 30 45 tjo
Time (min) Time (min)
ms QTc Interval ‘=-I Encainide
- ODE
- MODE 500 -
400
ff-+++d
#++tld ~
1 lx I 3x 9x I I I 300 1 1 I I I I I II,,
0 15 30 45 60 Time (min)
ENCAINIDE ODE ADMINISTRATION ADMINISTRATION
10000~ /
FIGURE 4. Electrocardiographic alterations during the %-minute infusion of encainide, 0-demethyl encainide (ODE) or 3-methoxy- 0-demethyl encainide (MODE) at equiactive antiarrhythmic dose levels as determined in the oua- bain-tachyarrhythmia model and at 3 and g times that dose.
FIGURE 5. Plasma concentra- tions of encainide and its metabo- lites after infusion of 3 graded doses of encainide, 0-demethyl encainide (ODE) or 3-methoxy- 0-demethyl encainide (MODE) in anesthetized dogs. Vertical bars represent standard error of the mean. The total drug dose admin- istered over each 15-minute infu- sion interval is indicated at the lower portion of each graph.

effective dose in squirrel monkeys (0.7 mg/kg). Com- parative assessments in dogs demonstrated that en- cainide was approximately 7 to 11 times more potent on a milligram basis than quinidine. Encainide was also effective in protecting anesthetized rats against ventricular arrhythmias induced by the intravenous infusion of aconitine.18 A 25% increase in the time to aconitine-induced ventricular tachycardia was pro- duced by 0.16 mg/kg encainide. ODE (0.068 mg/kg) was significantly more and procainamide (3.1 mg/kg] substantially less active in producing the same delay in the appearance of an arrhythmia.
Encainide abolished ventricular arrhythmias in- duced by administration of ouabain in anesthetized dog.szJ7Jg A return to normal sinus rhythm occurred without adverse effects on blood pressure or electro- cardiogram when compared with measurements be- fore the induction of tachycardia. The mean effective dose when infused (0.25 or 0.5 mg/kg/min) varied from 1.7 to 2.3 mg/kg.*’ On a relative milligram basis, encainide was 6 times more potent than quinidine and approximately 20 times more potent than procaina- mide. When encainide or its metabolites were admin- istered in fixed unit doses at sequential 5-minute inter- vals, the range of converting doses of encainide (0.3 to 1.7 mg/kg), ODE (0.05 to 0.45 mg/kg] and MODE (0.2 to 1.2 mg/kg] were associated with plasma levels of the administered agent of 65 to 448,33 to 172 and 74 to 232 rig/ml, respectively.2 In 17 of 17 responding animals, the mean converting dose of encainide was 0.67 mg/ kg. This intravenous dose was significantly higher than that determined for ODE (0.15 mg/kg] in 10 of 10 re- sponsive dogs but not different from MODE, which was effective in 8 of 10 animals at 0.39 mg/kg. Of these 3 agents, encainide produced sustained cardioversion in 16 of 17 dogs for the entire go-minute postdrug observation. Antiarrhythmic effects persisted for 90 minutes in only 4 of 10 responding animals given ODE. MODE had the shortest duration of action by the intra- venous route with only 2 of 8 dogs remaining in normal sinus rhythm for the same interval, The mean plasma concentrations of the administered drug at the onset and go-minute termination point in responding dogs were 244 and 78, 77 and 24 and 164 and 46 rig/ml for encainide, ODE and MODE, respectively.z
Assessments of encainide efficacy in conscious dogs with 2-stage coronary artery ligation-induced ar- rhythmias (Harris model) were comparable in studies conducted beforezO and after2Jg elucidation of the met- abolic pathway. Single intravenous doses of 0.5,1,2 or 5 mg/kg of encainide slowed overall heart rate and decreased ventricular ectopic activity by 23%, 29%, 38% and 86%, respectively.2 Antiarrhythmic effects were seen within 2 to 4 minutes of injection and a significant decrease in the incidence of abnormal beats was observed in most animals for up to 4 hours. Injection of 0.25, 0.5 or 1 mg/kg of ODE produced maximum decreases of 22%, 47% and 72% in the incidence of ectopic activity. MODE had the least pre- dictable efficacy and the shortest duration of action given intravenously in this model. Doses of 0.18 to 0.76 mg/kg of MODE were associated with a range of maxi-
mum decreases in ectopic activity of only 25% to 55% and the response was not improved by increasing the dose to 1.52 mg/kg. In these trials, 3 of 3 dogs given 10 mg/kg of encainide, 4 of 4 given 2 mg/kg of ODE and 1 of 6 given 1.52 mg/kg of MODE died within 1 to 8 minutes after intravenous injection.2
After oral administration of 0.5,1,2.5,5 or 10 mg/kg of encainide, suppression of ectopic activity was ob- served within 15 to 30 minutes and continued for 4 to >6 hoursZJO The mean percentage decreases in ven- tricular ectopic activity at these dose levels were 18%, 40%, 52%,57% and 7770, respectively. ODE (0.5,1,2.5 and 5 mg/kg) caused maximum decreases in ectopy of 15%, 24%, 33% and 63%. These responses were es- sentially equivalent to the 2270, 2470, 45% and 65% decreases in ectopic activity produced by MODE (0.38, 0.76, 1.91 and 3.85 mg/kg). Oral doses of 25, 50 or 80 mg/kg quinidine slowed ectopic rate to a lesser extent than did encainide. Based on either or both the degree of drug-induced reduction in ventricular ectopy and the number of dogs converted to normal sinus rhythm, encainide on a milligram basis was at least 8 times more potent than quinidine.
The time-course plasma drug levels associated with oral administration of encainide, ODE or MODE at the highest dose are shown in Figure 3. Unlike observa- tions in patients after chronic encainide therapy, sin- gle dose oral [or intravenous) administration of encain- ide to conscious dogs did not result in plasma metabo- lite concentrations exceeding those of the parent drug.2 As indicated in dogs given encainide, plasma concentrations of ODE approached, but neither reached nor exceeded, the levels associated with an antiarrhythmic effect when the metabolite itself was administered (Fig. 3).
Preclinical antiarrhythmic data are thus consistent with an interpretation that the metabolites of encain- ide are capable of contributing to its favorable actions; however, the fact that encainide itself has electro- physiologic effects in vitro and antiarrhythmic actions in vivo, which appear to be more consistent than those of either metabolite, would suggest that it possesses substantial inherent efficacy.
Cardiac and hemodynamic actions: Studies in 2 in vitro models and 1 in vivo canine preparation indicat- ed that encainide evoked only minimal negative ino- tropic effects at and above therapeutic levels.
Encainide at bath concentrations from 0.1 to 10 mg/ liter (3 X 10e7 to 3 X 1O-5 M) did not alter mechanical performance of isolated cat papillary muscle prepara- tions during either the contraction or relaxation phase.21 Comparatively, lidocaine (1 to 5 mg/liter) caused statistically significant depression of myocardi- al performance in terms of force-velocity-length rela- tions Like encainide, lidocaine failed to affect either the zero load clamp or the load dependence of relax- ation of this cardiac preparation.
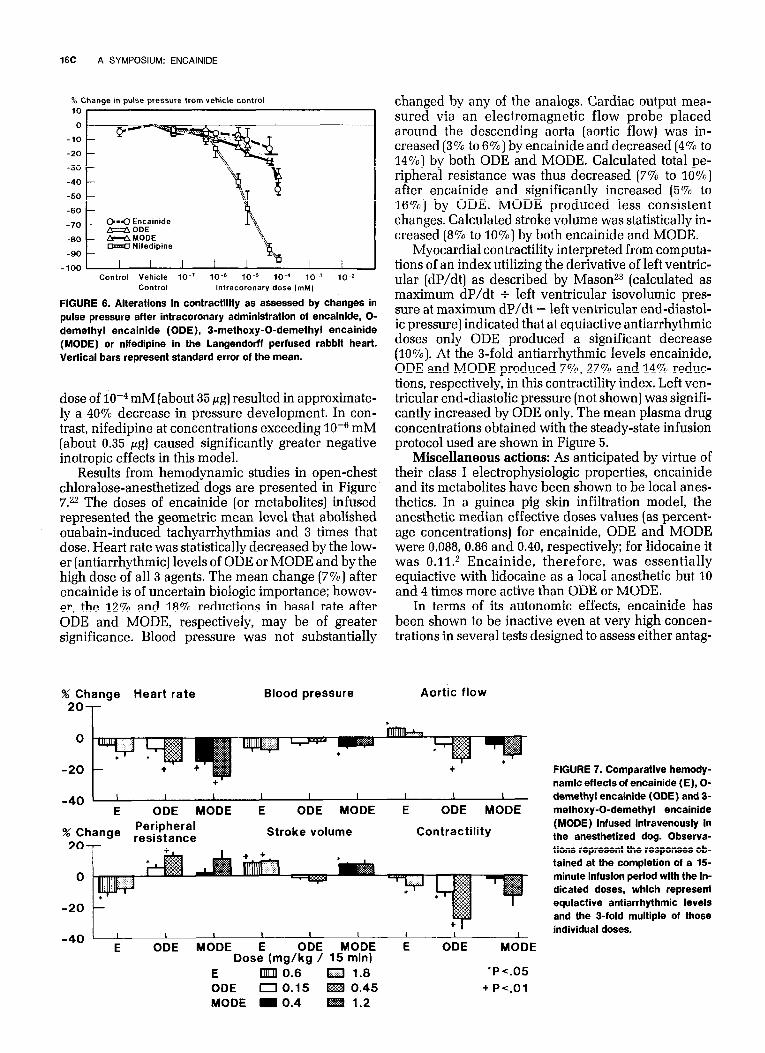
In the isolated perfused rabbit heart, negative ef- fects on performance were noted with encainide or its metabolites only after administration of doses exceed- ing 2 X 10M5 mM (about 7 pg) had been injected into the coronary artery (Fig. 6). As shown, an intracoronary
16C A SYMPOSIUM: ENCAINIDE
% Change in pulse pressure from vehicle control
Control Vehicle lo-’ 10-e 10-s 1om4 10~3 10-z Control lntracoronary dose ImM)
FIGURE 6. Alterations in contractility as assessed by changes in pulse pressure after intracoronary administration of encainide, O- demethyl encainide (ODE), 3-methoxy-0-demethyl encainide (MODE) or nifedipine in the Langendorff perfused rabbit heart. Vertical bars represent standard error of the mean.
dose of 10m4 mM (about 35 fig] resulted in approximate- ly a 40% decrease in pressure development. In con- trast, nifedipine at concentrations exceeding 10m6 mM (about 0.35 pg] caused significantly greater negative inotropic effects in this model.
Results from hemodynamic studies in open-chest chloralose-anesthetized dogs are presented in Figure 7.22 The doses of encainide (or metabolites) infused represented the geometric mean level that abolished ouabain-induced tachyarrhythmias and 3 times that dose. Heart rate was statistically decreased by the low- er (antiarrhythmic] levels of ODE or MODE and by the high dose of all 3 agents. The mean change (7%) after encainide is of uncertain biologic importance; howev- er, the 12% and 18% reductions in basal rate after ODE and MODE, respectively, may be of greater significance. Blood pressure was not substantially
% Change Heart rate
*OT Blood pressure Aortic flow
changed by any of the analogs. Cardiac output mea- sured via an electromagnetic flow probe placed around the descending aorta (aortic flow) was in- creased (3% to 6%) by encainide and decreased (4% to 14%) by both ODE and MODE. Calculated total pe- ripheral resistance was thus decreased (7% to 10%) after encainide and significantly increased (5% to 16%) by ODE. MODE produced less consistent changes, Calculated stroke volume was statistically in- creased (8% to 10700) by both encainide and MODE.
Myocardial contractility interpreted from computa- tions of an index utilizing the derivative of left ventric- ular (dP/dt) as described by Masonz3 (calculated as maximum dP/dt + left ventricular isovolumic pres- sure at maximum dP/dt - left ventricular end-diastol- ic pressure) indicated that at equiactive antiarrhythmic doses only ODE produced a significant decrease (10%). At the s-fold antiarrhythmic levels encainide, ODE and MODE produced 7%, 27% and 14% reduc- tions, respectively, in this contractility index. Left ven- tricular end-diastolic pressure (not shown] was signifi- cantly increased by ODE only. The mean plasma drug concentrations obtained with the steady-state infusion protocol used are shown in Figure 5.
Miscellaneous actions: As anticipated by virtue of their class I electrophysiologic properties, encainide and its metabolites have been shown to be local anes- thetics. In a guinea pig skin infiltration model, the anesthetic median effective doses values (as percent- age concentrations) for encainide, ODE and MODE were 0,088, 0.86 and 0.40, respectively; for lidocaine it was 0.11n2 Encainide, therefore, was essentially equiactive with lidocaine as a local anesthetic but 10 and 4 times more active than ODE or MODE.
In terms of its autonomic effects, encainide has been shown to be inactive even at very high concen- trations in several tests designed to assess either antag-
-40 ’ ’ I I I I I I I I
E ODE MODE E ODE MODE E ODE MODE
% Change Peripheral
20 resistance Stroke volume Contractility
0
-40 ’ ’ I I I I I 1 I
E ODE MODE E ODE MODE E ODE MODE Dose (mg/kg / 15 min)
E UULU 0.6 Cl 1.8 ‘PC.05 ODE 0 0.15 0.45 + Ps.01 MODE m 0.4 1.2
FIGURE 7. Comparative hemody- namic effects of encainide (E), O- demethyl encainide (ODE) and 3- methoxy-0-demethyl encainide (MODE) infused intravenously in the anesthetized dog. Observa- tions represent the responses ob- tained at the completion of a 15- minute infusion period with the in- dicated doses, which represent equiactive antiarrhythmic levels and the 3-fold multiple of those individual doses.

August 29, 1986 THE AMERICAN JOURNAL OF CARDIOLOGY Volume58 17c
TABLE V Autonomic Effects
Median Effective Concentration (Kg/ml)
Receptor Encainide Reference Agent
Alpha agonist Alpha antagonist Beta agonist Beta antagonist Anticholinergic Cholinergic Antihistamine Histamine
Inactive 41 f-5 944 13 Inactive 56 f 8 Inactive 3 f 0.6
Inactive
0.024 f 0.004’ (phentolamine) 0.006 f 0.0009+ (isoproterenol)
0.48 f 0.1 I+ (sotalol) 0.0002 f 0.00002+ (atropine)
. 0.001 f 0.0004+ (pyrilamine)
aprindine, mexilitine, lorcainide, flecainide or diso- pyramide, each an agent with known local anesthetic properties.
Overall, the activity profiles generated in animals appear to be predictive of the efficacy, pharmacody- namic, pharmacokinetic and safety characteristics ob- served clinically.
Acknowledgment: We wish to thank our colleagues K. Dungan and Drs. L. Gillespie, D. Gallo and R. John- son for their substantive contributions to this manu- script.
Antibradykinin Antiserotonin
39 f 6 6 f 0.4t 0.006 f 0.0003 0.009 f 0.0002* References
* = rat seminal vesicle. + = GP trachea. t = rat uterus.
onist or agonist potency as an adrenergic, cholinergic and histaminergic agent (Table V). In only 1 property, as an in vitro serotonin antagonist, was encainide (me- dian effective concentration 6 rig/ml] more potent than the accepted standard reference, methysergide (median effective concentration 9 rig/ml). However, when tested in the serotonin rat pedal edema model, encainide with a subcutaneous median effective dose of 12.5 mg/kg was only very weakly active in compari- son to methysergide (0.048 mg/kg, subcutaneous). This property is thus felt to be of no significance in vivo in either animals or man.
Side effects: Characterizations of the untoward ef- fects of encainide and its metabolites during pharma- codynamic testing were limited to those experimental situations in which animals were studied in the con- scious state. At antiarrhythmic dose levels, the side effects with encainide [or metabolites) given either orally or intravenously were unremarkable. Emesis and ataxia were observed in a limited number of dogs given encainide whereas hyperventilation, vocaliza- tion and muscular weakness were more commonly reported after administration of ODE and especially MODE.2 These responses would be predictive of the nausea, tremor, muscular weakness or malaise that have been encountered clinically after high drug doses. Encainide should not evoke the anticholinergic side effects, i.e., drying of the mouth, urinary retention and constipation, which have been observed after ad- ministration of other class I antiarrhythmic agents. No observations in animals were predictive of the occur- rence of blurred vision that has been reported clinical- ly. In the absence of a possible mechanism via an anticholinergic action, it would appear that this latter side effect may in some way be related to the local anesthetic properties of the compound. This interpre- tation is based on the analogous episodes of diplopia or visual disturbances that have been reported after the administration of antiarrhythmic doses of lidocaine,
1. Mayo1 RF, Gammans RE. Analysis of encainide in plasma by radioimmuno- assay and high pressure liquid chromatography. Ther Drug Monit 1979;1:567- 524. 2. Gomoll AW, Byrne JE, Mayo1 RF. Comparative antiarrhythmic actions of encainide and its major metabolites. Arch Int Pharmacodyn Ther, in press. 3. Gibson JK, Somani P, Bassett AL. Electrophysiologic effects of encainide (MJ 9067) an canine Purkinje fibres. Eur J PharmacoI1978;52:161-169. 4. Carmeliet E. Electrophysiologic effects of encainide on isolated cardiac muscle and Purkinje fibers and on the Lagendorff-perfused guinea pig heart. Eur 1 Pharmacof 1989;61:247-262. 5. Elharrar V, Zipes DP. Effects of encainide and metabolites (MJ 14030 and MJ 9444) on canine cardiac Purkinje and ventricular fibers. J Pharmacol Exp Ther 1982;220:440-447. 6. Campbell TJ. Resting and rate-dependent depression of maximum rate of depolarization (Vmax] in guinea pig ventricular action potentials by mexile- tine, disopyramide, and encainide. J Cardiovasc PharmacoI1983;5:291-296. 7. Wong SS. Myerburg RJ, Erzin AM, Gelband H, Bassett AL. Electrophysio- logic effects of encainide on acutely ischemic rabbit myocardial cells. Eur J Pharmacol1982;80:323-329. 8. Harrison DC. Antiarrhythmic drug classification: new science and practi- ca1 appfications. Am J CardioI1985;56:185-187. 9. Caillard CG, Louis JC. Assessment ofantiarrhythmic drugs in experimental pharmacology. Methods Find Exp CIin PharmacoI1980;2:223-252. 10. Dresel PE. Effect of encainide and its two major metabolites on cardiac conduction. J Pharmacol Exp Ther 1984;228:180-186. 11. Sami M, Mason JW, Oh G, Harrison DC. Canine electrophysiology of encainide, a new antiarrhythmic drug. Am J CardioIl979;43:1149-1154. 12. Samuelsson RG, Harrison DC. Electrophysiologic evaluation of encainide with use of monophasic action potential recording. Am J CardioI1981;48:871- 876. 13. Duff HJ, Dawson AK, Roden DM, Oates JA, Smith RF, Woosley RL. Electrophysiologic actions of 0-demethyf encainide: an active metabolite. CircuIation 1983;68:385-391. 14. Jackman WN, Zipes DP, Naccarelli GV, Rinkenberger RL, Heger JJ, Prystowsky EN. Electrophysiology of oral encainide. Am J Cardiol 1982;49: 1270-1278. 15. Almotrefi AA, Baker JBE. AntifibriIIatory efficacy of encainide, Iorcain- ide and ORG 6001 compared with Iignocaine in isolated hearts of rabbits and guinea-pigs. Br J PharmacoIl981;73:373-377. 16. Jaillon P, de la Rosa S, Griffin JC, Winkle RA. Harrison DC. Effects of encainide (MJ9067) on the ventricular fibrillation threshold in anesthetized dogs. J Cardiovasc PharmacoI1980;2:517-526. 17. Byrne JE, Gomoll AW, McKinney GR. Antiarrhythmic properties of MJ 9067 in acute animal models. J. Pharmacol Exp Ther 1977;200:147-154. 18. Roden DM, Duff HJ, Altenbern D, Woosley RL. Antiarrhythmic activity of the 0-demethyl metabolite of encainide. J Pharmacol Exp Ther 1982;221: 552-557. 19. Gomoll AW, Byrne JE, Mayo1 RF. Comparative antiarrhythmic and local anesthetic actions ofencainide and its two major metabolites. Pharmacologist 1981;23:209. 20. Byrne JE, Gomoll AW. Antiarrhythmic action of encainide versus ventric- ular arrhythmias in the conscious dog following coronary artery ligation. Can J Physiol PharmacoI1982:60:369-375. 21. Brutsaert DL. Effect of encainide on myocardial contractility of cat papiI- Iary muscle. Eur J PharmacoI1981;76:267-269. 22. Gomoll AW, Mayo1 RF. Comparative hemodynamic effects of encainide and its two major metabolites in anesthetized dogs. Pharmacologist 1982; 24:233. 23. Mason DT. Usefulness and limitations of the rate of rise of intraventricu- lar pressure (dP/dtJ in the evaluation ofmyocardial contractility in man. Am J Cardiol 1969;23:516-527.





























