Embed Size (px)
Citation preview

9 2 0 Clinical and laboratory observations The Journal o f Pediatrics December 1990
vitamins intravenously in a lipid emulsion: measurement of vi- tamins A, D, and E and riboflavin. J PEDIATR 1988;113:1057- 65.
4. Specker B, Gratton T, Landi T, Tsang RC. Effect of dietary calcium (Ca) to phosphorus (P) ratio on calcium homeostasis in term infants [Abstract]. Pediatr Res 1988;22:492A.
5. Pietta P, Calatroni A, Rava A. Hydrolysis of riboflavin nucle- otides in plasma monitored by high-performance liquid chro- matography. J Chromatogr 1982;229:445-9.
6. SAS Institute Inc. SAS user's guide: statistics; Version 5 ed. Cary, N.C.: SAS Institute Inc., 1985.
7. Kirshenbaum NW, Dancis J, Levitz M, Lehanka J, Young BK. Riboflavin concentration in maternal and cord blood in human pregnancy. Am J Obstet Gynecol 1987;157:748-52.
8. Jusko W J, Khanna N, Levy G, et al. Riboflavin absorption and excretion in the neonate. Pediatrics 1970;45:945-9.
9. Moore MC, Greene HL, Phillips B, et al. Evaluation of a pe- diatric multiple vitamin preparation for total parenteral nutri- tion in infants and children. 1. Blood levels of water-soluble vi- tamins. Pediatrics 1986;77:530-8.
10. Department of Health and Social Security. Report on health and social subjects. 12. The composition of mature human milk [Report of a working party of the Committee on Medical As- pects of Food Policy]. London: Her Majesty's Stationary Office, 1977.
Endocrinopathies in Cornelia de Lange syndrome
I. David Schwartz, MD, Karen J. Schwartz, BSN, RN, Boris G. Kousseff, MD, Barry B. Bercu, MD, and Allen W. Root, MD
From the Sections of Pediatric Endocrinology and Genetics, University of South Florida Col lege of Medicine, Tampa, and All Children's Hospital, St, Petersburg, Florida
Presented in part at the Annual Meeting of the Southern Society for Pediatric Research, New Orleans, La., January 1990.
Submitted for publication April 5, 1990; accepted July 18, 1990.
Reprint requests: Allen W. Root, MD, Section of Pediatric Endo- crinology, All Children's Hospital, 801 6th St. South, P.O. Box 31020, St. Petersburg, FL 33731-8920.
9/22/23849
CDLS GH GnRH H-P IGF-I
Cornelia de Lange syndrome Growth hormone Gonadotropin releasing hormone Hypothalamic-pituitary Insulin-like growth factor I
Table. Clinical and laboratory assessment of patients with Cornel ia de Lange syndrome
Age Bone Patient /(yr) Ht- BMIt age Glucose Prolactin LH FSH
No. Sex SDS ~ (kg/m z) (yr) (mg/dL) /~g/L (IU/L) (IU/L)
1 17.3/F -6.4 19.5 10.0 84/27 9.1 / 30.3 23.4/65.8 12.5/19.9
2 5.4/M -1.6 12.6 4.5 77/41 4.6/67.7 <3.0/6.4 1.4/6.0 3 6.7/F -3.8 11.6 2.8 91/43 13.5/38.0 <3.0/4.3 1.2/11.8 4 12.0/F -2.8 16.5 10.5 92/33 6.2/24.3 <3.0/24.3 0.9/4.2
5 28.5/M -2.8 20.6 Adult 97/43 14.9/137 6.8/223.3 20.8/53.2
T4, T3, and 1GF-I values are basal. Glucose values are basal/nadir; other values are basal/peak after the administration of insulin-TRH-GnRH. For conversion to Syst~me International units, multiply glucose (mmol/L) by 0.0555, DHAS (nmol/L) by 27.21, testosterone (pmol/L) by 34.67, estradiol (pmol/L) by 36.71, cortisol (nmol/L) by 27.59, T4 (nmol/L) by 12.87, and T3 (pmol/L) by 15.36. Ht-SDS, Height standard deviation score; BM1, body mass index; LH, lutenizing hormone; FSH, follicle stimulating hormone; TRH, thyrotropin releasing hor- mone; TSH, thyrotropin; DHAS, dehydroepiandrosterone sulfate; 7"4, thyroxine; T3, triiodothyronine. *Height standard deviation score =(Height - Mean height for age)/(SD of mean height for age). tNormal nonobese subject weighed 25 kg/m 2.

Volume 1 Clinical and laboratory observations 9 2 1 Number 6
The Cornelia de Lange syndrome consists of prenatal and postnatal growth retardation, developmental delay, and characteristic dysmorphic features) To define the endo- crine status of patients with CDLS, we evaluated hypotha- lamic-pituitary function in five subjects with this disorder.
M E T H O D S
P a t i e n t s . The finding of an "empty" sella turcica and growth hormone deficiency in a 17-year-old girl with CDLS
(patient 1) prompted evaluation of H-P function in addi- tional patients with CDLS. After institutional review board approval was obtained, the families of 12 patients with CDLS who were being followed by the Regional Genetics Program or the Section of Pediatric Endocrinology, Uni- versity of South Florida, were invited to participate. Three families could not be contacted; four patients and their families responded positively to this request for evaluation. There were no apparent differences with respect to gender,
70
6o
50
~" 4 0
"-r" 130
20
10
insulin TRH
GnRH
.i \
�9 \ t
t
clonidinel /L..\ .
/ ,
/ +...
i \
. i // ',, ~ ~ F. . ': ". j \ \ .~ ", \ i / ""-.. ':
\\. !A\" '". ',..,k ; / "~ ~.
o . . . . . " r . . . . . _ . . . . . .
-30 0 10 20 30 40 60 90 120 150 180 210 240 El Patient 1 ~, Patient 4 + Patient 2 x Patient 5 T i m e (min)
Patient 3
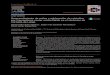
Figure. Serum growth hormone responses to insulin hypoglycemia and clonidine in five patients with Cornelia de Lange syndrome. Patients 1 and 3 had classic GH deficiency (peak GH <7 ug/L).
Table. Cont 'd
TSH (mU/k)
Testo- Estra- Asso- DHAS sterone diol Cortisol T4 T 3 IGF-I ciated
(#g/dl) (ng/dl) (ng/dl) (~g/dL) (~g/dl) (ng/dl) (U/mL) disorders
1.0/6.3
1.1/9.8 2.5/lO.3 o.4/1.9
0.7/5.3
30.2/29.7 <10/<t0 <2/<2 7.5/22.9 9.1 113 0 . 3 1 Seizures, cleft palate
13.0/14.5 <10/<10 - - 9.2/22.8 7.9 212 0.62 13.2/13.4 <10/<10 <1/<1 26.4/27.5 6.4 163 0.28 Seizures 7.0/13.7 <10/<10 <1/<1 7.5/20.5 9.3 197 1.71 Cleft palate,
essential hypernatremia, and hypodipsia
205/23 603/688 - - 13.7/23.9 8.6 171 0.24 Unilateral orchiectomy (postorchitis)

9 2 2 Clinica. ~ and laboratory observations The Journal o f Pediatrics December 1990
age, growth, and development between the patients studied and the five patients who declined evaluation.
Protocol and assays. After an 8-hour overnight fast and fluid deprivation, anterior and posterior pituitary function was evaluated by the concomitant intravenous administra- tion of insulin (0.08 to 0.1 unit/kg), thyrotropin releasing hormone (7 #g/kg), and gonadotropin releasing hormone (100 #g), followed by oral administration of clonidine (0.1 mg/m2). Serum and urine osmolalities were determined before testing. Plasma or serum concentrations of glucose, sodium, cortisol, thyrotropin, thyroxine, triiodothyronine, triiodothyronine resin uptake, growth hormone, prolactin, luteinizing hormone, follicle stimulating hormone, dehy- droepiandrosterone sulfate, testosterone, estradiol, and in- sulin-like growth factor I in unextracted plasma were mea- sured by specific and established methods. Bone age was assessed by the method of Greulich and Pyle. Pubertal de- velopment was assessed by the method of Tanner. Informed consent from parents or legal guardians was obtained before entry into the study.
R E S U L T S
For patient 4, essential hypernatremia and hypodipsia had previously been documented. All patients were prepu- bertal except patient 5, who was at Tanner genital stage IV. In all patients the H-P-adrenocortical and H-P-thyroid axes were intact (Table). Patients 1 and 3 had classic hyposo- matotropism (peak GH concentrations were 2.4 and 4.5 #g/L, respectively [Figure]). For a well-nourished adult, patient 5 had a low plasma concentration of IGF-I. Patients 2 to 4 were clinically prepubertal; they had low serum de- hydroepiandrosterone levels, appropriate prepubertal sex hormone concentrations, and appropriate gonadotropin secretory responses to GnRH. Patient 1 was also prepuber- tal, but the basal LH concentration was slightly elevated and GnRH administration induced pubertal gonadotropin secretory responses. In patient 5 the basal follicle stimulat- ing hormone concentration was elevated and the gonado- tropin secretory responses to GnRH were exaggerated. Pa- tient 5 also had an increased prolactin response to thyrotro- pin releasing hormone. 2 Serum sodium concentrations or osmolalities or both were normal in subjects 1, 2, 3, and 5, as was renal concentrating ability in those tested. Central nervous system imaging of patient 1 by computed tomogra- phy demonstrated an "empty" sella; imaging studies of the other patients were not performed.
D I S C U S S I O N
Investigations of the endocrine system in patients with CDLS have been infrequent. 39 Schlesinger et al. 3 studied insulin counterregulatory mechanisms after the adminis- tration of insulin, 0.125 to 0.25 U/kg, and demonstrated
increased insulin sensitivity in four CDLS patients. McArthur and Edwards 4 recorded fasting GH concentra- tions of 10 and 37 #g/L, respectively, in two patients. Hill- man et al. 5 measured fasting GH concentrations in three patients with CDLS; blood glucose concentrations were <3.3 mmol/L (<60 mg/dl) in all, and plasma GH concen- trations were 3.6 to 22.0 #g/L. Abraham and Russell 6 studied GH concentrations after insulin-induced hypogly- cemia in two patients and during "reactive" hypoglycemia after a glucose load in seven. In the former study, peak GH concentrations were >--14 #g/L; in the latter, peak GH val- ues were consistently >--_8 ~zg/L. Two of our five subjects (40%) had GH deficiency. These patients and patient 5 had low plasma concentrations of IGF-I . The IGF-I levels and GH responses to insulin and clonidine in patient 5 were dis- cordant; this subject may have some degree of end-organ resistance to GH.
Adrenal and thyroid function were essentially normal in our patients with CDLS and in those reported by other investigators. 3-9 Beer et al. 8 described a patient with thyroid ectopia and evidence of TSH deficiency.
Hypospadias, cryptorchidism, and genital hypoplasia are present in 94%, 58%, and 24%, respectively, of boys with CDLS, 1 suggesting dysfunction of the H-P-testicular axis. However, in the series of McArthur and Edwards, 4 all girls more than 10 years of age had normal secondary sexual char/ttcteristics and two girls more than 14 years of age were menstruating. Pregnancy in patients with CDLS has been described, l~ Based on the present data, the gonadotropin and sex steroid responses after stimulation by GnRH sug- gest variable dysregulation of the H-P-gonadal axis in CDLS. The extent to which hemiorchiectomy contributed to the abnormal GnRH test result in patient 5 is indeter- minable.
Similarly to the patients of Abraham and Russell, 6 our patients had normal renal concentrating ability or normal serum sodium concentrations and osmolalities or both, ex- cept patient 4, who had essential hypernatremia and hypo- dipsia. Ptacek et al. 7 observed diminished renal concentrat- ing ability in CDLS. Skeletal maturation in CDLS has been either commensurate with chronologic age or delayed. 49, 11
Histologic features of the pituitary gland of patients with CDLS have been both normal and abnormal. 3, 6, 9 Atrophy of the hypophysis because of pressure by a cyst of the Rathke cleft was noted in one case. 9 Dysfunction of H-P mechanisms in CDLS was also suggested by the pathologic findings of thyroid and adrenal hypoplasia 3, 9, ~ l and by the small size of the ovaries. 7, 9
We conclude that a spectrum of endocrinopathies may be seen in patients with CDLS. Postnatal growth retardation in these patients may be an intrinsic feature of CDLS or may be related to abnormalities of GH secretion or to IGF-

Volume 117 Clinical and laboratory observations 9 2 3 Number 6
I response to G H or both. These patients may also be at risk
for dysfunction of gonadotropin and prolactin secretion and
of osmoregulatory mechanisms. In our opinion the admin-
istration of G H to C D L S patients with hyposomatotropism
is not warranted unless there is significant hypoglycemia
unresponsive to conventional therapy or unless developmen-
tal progress exceeds that commonly present in patients with
CDLS.
We thank Mr. Gregory Duekett, Ms. Margaret Sweetland, Mr. Jack Strzelecki, and Ms. Lorraine Forson for technical assistance, and Ms. Becky DeCroteau and the clinical laboratory at All Chil- dren's Hospital.
REFERENCES
1. Jones KL. De Lange syndrome. In: Jones KL, ed. Smith's recognizable patterns of human malformations. 4th ed. Philadelphia: WB Saunders, 1988:80.
2. Shulman DI, Hu C-S, Root AW, Bercu BB. Pooled prolactin measurements in the evaluation of short children. J Clin En- docrinol Metab 1989;69:1261-7.
3. Schlesinger B, Clayton B, Bodian M, Jones KV. Typus degen- erativus amstelodamensis. Arch Dis Child 1963;38:349-57.
4. McArthur RG, Edwards JH. De Lange syndrome: report of 20 cases. Can Med Assoc J 1967;96:1185-98.
5. Hillman JC, Hammond J, Noe O, Reiss M. Endocrine inves- tigations in De Lange's and Seckel's syndromes. Am J Ment Defic 1968;3:30-3.
6. Abraham JM, Russell A. De Lange syndrome: a study of nine examples. Acta Paediatr Scand 1968;57:339-53.
7. Ptacek L J, Opitz JM, Smith DW, Gerritsen T, Waisman HA. The Cornelia de Lange syndrome. J PEDIATR 1963;63:1000-20.
8. Beer S, Wallis K, Czerniak P. Two cases of De Lange's syn- drome (typus degenerativus amstelodamensis). J Ment Defic Res 1968;12:128-37.
9. Bj6rkl6f K, Brundelet PJ. Typus degenerativus amsteloda- mensis (Cornelia de Lange first syndrome): congenital hypop- ituitarism due to a cyst of Rathke's cleft? Aeta Paediatr Scand 1965;54:275-87.
10. Mosher GA, Schulte RL, Kaplan PA, Buehler BA, Sanger WG. Pregnancy in a woman with the Brachmann-de Lange syndrome. Am J Med Genet 1985;22:103-7.
11. Filippi G. The de Lange syndrome: report of 15 cases. Clin Genet 1989;35:343-63.
Prenatal treatment of a patient with vitamin B 2-responsive methylmalonic acidemia
S. B. van der Meer, MD, L. J, M. Spaapen , PhD, B. Fowler, PhD, C. Jakobs, PhD, W. J. Kleijer, PBD, a n d U. Wende l , MD, PhD
From the Department of Pediatrics, Academic Hospital Maastricht, Maastricht, The Nether- lands, the Department of Genetics and Cell Biology, University of Limburg, Maastricht, The Netherlands, Willink Biochemical Genetics Unit, Royal Manchester Children's Hospital, Manches- ter, United Kingdom, the Department of Pediatrics, Free University Hospital, Amsterdam, The Netherlands, the Department of Clinical Genetics, Erasmus University, Rotterdam, The Nether- lands, and Kinderklinik der Heinrich-Heine UniversitOt, DOsseldorf, Federal Republic of Germany
The clinical presentation of methylmalonic acidemia con-
sists of lethargy, recurrent vomiting, dehydration, respira-
tory distress, and muscular hypotonia. The biochemical
abnormalities include life-threatening ketoaeidosis, hyper-
ammonemia, and hyperglycinemia. Methylmal0nic acid
accumulates either because of a defect in methylmalonyl-
coenzyme A mutase or in the biosynthesis of 5 ' -deoxyade-
Presented in part as a poster at the 27th Annual Symposium of the Society for the Study of Inborn Errors of Metabolism, Munich, Federal Republic of Germany, September 1989.
Submitted for publication May 29, 1990; accepted July 18, 1990.
Reprint requests: S. B. van der Meer, MD, Department of Pediat- rics, Academic Hospital Maastricht, P.O. Box 1918, 6201 BX Maastricht, The Netherlands.
9/22/23846
nosylcobalamin. Complementation studies have revealed
the existence of seven distinct mutant classes.1 Prenatal di-
agnosis has been achieved by measuring M M A in amniotic
fluid and maternal urine, 2 by quantifying methylcitrie acid
in amniotic fluid, 3 and by measuring the incorporation of
propionate labeled with carbon 14 into protein 4 or by
CoA GC-MS MMA OLCFA
Coenzyme A Gas chromatograph-mass spectrometer Methylmalonic acid Odd-numbered long-chain fatty acids
assaying methylmalonyl-CoA mutase activity in chorionic
villi or amniocytes, s
In 1975, Ampola et al. 6 reported on the prenatal treat-
ment of a fetus with methylmalonic acidemia caused by a
defect in 5 '-deoxyadenosylcobalamin synthesis. During the