Embed Size (px)
Citation preview

at SciVerse ScienceDirect
Polymer Degradation and Stability 96 (2011) 2071e2079
Contents lists available
Polymer Degradation and Stability
journal homepage: www.elsevier .com/locate /polydegstab
Enzymatic synthesis and properties of novel biodegradable and biobasedthermoplastic elastomers
Takuma Kobayashi, Shuichi Matsumura*
Faculty of Science and Technology, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan
a r t i c l e i n f o
Article history:Received 8 August 2011Received in revised form24 September 2011Accepted 2 October 2011Available online 8 October 2011
Keywords:Biodegradable polymersBiobased polymerLipaseAliphatic polyestersThermoplastic elastomersEnzymatic polymerization
* Corresponding author. Fax: þ81 45 566 1582.E-mail address: [email protected] (S. Ma
0141-3910/$ e see front matter � 2011 Elsevier Ltd. Adoi:10.1016/j.polymdegradstab.2011.10.002
a b s t r a c t
Novel biodegradable and biobased thermoplastic elastomers, poly[dodecanolide-12-hydroxystearate (12HS)],poly(pentadecanolide-12HS) and poly(hexadecanolide-12HS) with Mws of 140,000e290,000 gmol�1 wereprepared by the enzymatic copolymerization of a macrolide as the hard segment andmethyl 12HS as the softsegment. Their thermal properties, such as Tm and Tc, were measured by DSC. Physicochemical andmechanical properties, such as crystallinity, were also measured. The polymer structures were analyzed withrespect to the sequence of the two monomers by 1H NMR spectroscopy using an europium shift reagent. Therandomness of the two monomer units in the polymer chain increased with the polymerization time. BothYoung’s modulus and tensile strength decreased with increasing 12HS content in the copolymer. In contrast,elongation at break increased with increasing 12HS content, thus demonstrating the copolymers’ elastomericproperties. These copolymers showed biodegradabilities by activated sludge, which also increased withincreasing 12HS content.
� 2011 Elsevier Ltd. All rights reserved.
1. Introduction
Thermoplastic elastomers are widely used as soft materialsin both industry and medicine. A copolymer consisting of poly-ethylene and an a-olefin having a short side chain is a typicalexample of a thermoplastic elastomer [1]. In the structure of thiscopolymer, the polyethylene chains have higher crystallinity andact as hard segments, while the poly(a-olefin) moieties impart anamorphous nature caused by the side alkyl chains and thus act assoft segments of the elastomer. Specifically, a random copolymerconsisting of ethylene and 1-octene exhibits elastomeric behavior[2]. Although such a copolymer is not crosslinked via covalentbonds, elastomeric properties are afforded by pseudo crosslinkingat the crystalline moieties of the polyethylene chains as hardsegments. Because these copolymers have relatively high meltingtemperatures, reprocessing can be carried out by heating.
For next generation elastomers, environmentally benign elas-tomers are needed. Strategies to accomplish this include replacingpetrochemicals with biobased and renewable raw materials,replacing conventional metal catalysts with environmentallybenign or renewable catalysts, and incorporating chemical recy-clability and environmental biodegradability into the elastomers.
tsumura).
ll rights reserved.
Recently, much progress has been made in the replacement ofpetrochemical materials with biobased materials. Some biobasedpolymers, including polylactide, poly(butylene succinate) andpoly(trimethylene carbonate) have been reported. They possesspotential chemical recyclability and biodegradability [3e5],because their aliphatic ester or carbonate bonds can be reversiblycleaved enzymatically. These polymers can be ultimately bio-degraded into CO2 and water even when discarded as waste in theenvironment.
In order to develop next generation elastomers, the productionprocess is also an important issue. Enzymatic polymerization hasattracted attention in recent years as an environmentally benignprocess. Enzymes are renewable catalysts that exhibit high catalyticactivities under mild conditions. For example, lipases have highcatalytic activities in the synthesis of ester bonds both by poly-condensation and ring-opening polymerization (ROP) of lactones[6e8]. It is reported that lipases show higher catalytic activitiesthan metal catalysts for the ROP of macrolides [9]. Indeed, theenzymatic polymerization of macrolides, such as pentadecanolideand hexadecanolide, has been reported [10e12]. These productswere found to be highly crystalline polyesters due to the longmethylene chains between the ester bonds.We previously reportedthe synthesis of poly(12-hydroxystearate) [poly(12HS)] by lipase[13]. This polymer is considered to be a biobased material, because12HS is produced by the hydrogenation of ricinoleic acid fromcastor oil. The polymer exhibits an amorphous nature, since the

LipaseC O
O
(CH2)x
x = 11 (DDL) 14 (PDL) 15 (HDL)
MeO
OOH
CH3
4
12HS
+
x = 11 poly(DDL-12HS) 14 poly(PDL-12HS) 15 poly(HDL-12HS)
OO
O
CH3
x 4 nm
O
(CALB)
Scheme 1. Enzymatic synthesis of polyesters.
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e20792072
hexyl side chain disturbs crystallization, and also shows goodbiodegradability.
In this study, we synthesized a series of copolymers by thecopolymerization of various macrolides and methyl 12HS(12HS-Me) using lipase as a catalyst, with the aim of creating novelbiodegradable and biobased thermoplastic elastomers (Scheme 1).The hexyl side chain of 12HS imparts an amorphous nature to thecopolymer, while the long methylene chain derived from themacrolides is expected to act as a hard segment, i.e., a pseudocrosslinking point for crystallization. Thus, elastomeric propertiesare created as illustrated in Fig. 1. The physicochemical andmechanical properties and biodegradabilities of these copolymershaving various 12HS contents were measured.
2. Experimental
2.1. Materials
Pentadecanolide (PDL) and hexadecanolide (HDL) werepurchased from TCI Co., Inc. (Tokyo, Japan). Dodecanolide (DDL),12HS-Me and europium(III) tris[3-(heptafluoropropylhydroxy-methylene)-d-camphorate] [Eu(hfc)3] were purchased from Sig-maeAldrich Co. (St. Louis, MO, USA). Immobilized lipase fromCandida antarctica [CALB: Novozym 435, lipase (B) from Candidaantarctica having 10,000 PLU g�1 (propyl laurate units: lipaseactivity based on ester synthesis)] on an acrylic resin, was kindlysupplied by Novozymes Japan, Ltd. (Chiba, Japan). The enzyme wasdried under vacuum (3 mmHg) over P2O5 at 25 �C for 2 h beforeuse. Molecular sieves 4A were purchased from SigmaeAldrich Co.(St. Louis, MO, USA) and were dried at 150 �C for 2 h before use.
2.2. Measurements
The weight average (Mw) and number average (Mn) molecularweights as well as the polydispersity (Mw/Mn) of the polymer weredetermined by size exclusion chromatography (SEC) (ShodexK-Gþ K-805 columns, Showa Denko Co., Ltd., Tokyo, Japan) usinga refractive index detector. Chloroform was used as the eluent at1.0 mLmin�1. The SEC system was calibrated with polystyrene
Amorphous moiety created by 12HS
Pseudo crosslinker point created by crystallization of macrolide-derived moiety
Fig. 1. Illustration of a novel thermoplastic elastomer created by poly(macrolide-12HS).
standards having a narrow molecular weight distribution. The 1Hand 13C NMR spectra were recorded with an ECA-500 Fouriertransform spectrometer (JEOL, Ltd., Tokyo, Japan) operating at500 MHz and 125 MHz, respectively.
The crystallization temperature (Tc) and melting temperature(Tm) were determined by differential scanning calorimetry(DSC-60, Shimadzu, Kyoto, Japan). The measurements were madewith a 10-mg sample on a DSC plate. The polymer samples wereheated to 110 �C at the rate of 10 �Cmin�1, cooled to �100 �C at therate of 30 �Cmin�1 and then scanned with heating at the rate of10 �Cmin�1 from �100 to 110 �C. The degree of crystallinity (Xc)was determined by X-ray diffraction measurements (DISCOVER,Bruker AXS K.K., Yokohama, Tokyo, Japan).
The mechanical properties (the Young’s modulus, the tensilestrength and the elongation at break) of the film samples weredetermined with an Autograph (Shimadzu, Tokyo, Japan).
The biodegradabilities of poly(DDL-12HS), poly(PDL-12HS) andpoly(HDL-12HS) were evaluated by biochemical oxygen demand(BOD) measurements. BOD was determined with a BOD tester(VELP Scientifica s.r.l., Usmate, MI, Italy) by the oxygen consump-tion method, according to the modified MITI test. The activatedsludge was obtained from a municipal sewage plant in YokohamaCity, Japan. BOD biodegradation (Biochemical Oxygen Demand/Theoretical Oxygen Demand� 100) was measured for 43 d.
2.3. General enzymatic polymerization procedure
The lipase-catalyzed copolymerization of lactone and 12HS-Mewas carried out in a dry screw-capped vial withmolecular sieves 4Aplaced at the top of the vial in the vapor phase to absorb thecondensation byproduct, such as water or methanol. As a typicalexample, the synthesis of poly[(50 mol% DDL)-co-(50 mol% 12HS)][poly(DDL-12HS)(50)] is described below. A mixture of DDL(0.185 mmol), methyl 12HS (0.185 mmol) and immobilized lipase(CALB, 40 mg) was reacted under nitrogen atmosphere in a ther-mostated oil bath at 120 �C for 120 h. The polymerization mixturewas then dissolved in chloroform (20 mL), and the insolubleenzyme was removed by filtration. The solvent was next evapo-rated under reduced pressure to obtain the polymer, which wasthen purified by reprecipitation using chloroform (good solvent) emethanol (poor solvent) to remove unreacted monomers andbyproducts. The molecular weight and the molecular weightdistribution of the polymer were determined by SEC. Themolecularstructure and monomer composition were determined by 1H and13C NMR spectroscopy. In a similar procedure, poly(PDL-12HS) andpoly(HDL-12HS) were prepared. The 1H NMR spectral data of pol-y(DDL), poly(PDL), poly(HDL), poly(12HS), poly(DDL-12HS), pol-y(PDL-12HS) and poly(HDL-12HS) are as follows.
Poly(DDL), Poly(PDL) and Poly(HDL): 1H NMR (300 MHz,CDCl3): d (ppm) 1.25e1.41 [m, 2nH, eOCH2(CH2)ne], 1.61(m, 2H, eCH2CH2COOe), 2.28 (t, 2H, J¼ 7.6 Hz, eCH2COOe), 4.05(t, 2H, J¼ 6.8 Hz, eOCH2e). 13C NMR (125MHz, CDCl3): d (ppm) 25.1,26.1, 28.8, 29.3, 29.4e29.5 [eOCH2(CH2)nþ1e], 34.5 (eCH2COOe), 64.5(eOCH2e), 174.1 (eCOOe).
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e2079 2073
Poly(12HS): 1H NMR (300MHz, CDCl3): d (ppm) 0.87 {t, 3H, J¼ 6.6Hz, eOCH[(CH2)5CH3]e}, 1.25e1.41 [m, 2nH, e(CH2)nCH2CH2COOe],1.5 {m, 4H,eOCH[CH2(CH2)4CH3]CH2e},1.61 (m, 2H,eCH2CH2COOe),2.27 (t, 2H, J¼ 7.6 Hz, eCH2CH2COOe), 4.86 [m, 1H, eOCH(C6H13)e].13C NMR (125 MHz, CDCl3): d (ppm) 14.2 (eCH3), 22.7(eCH2CH3), 25.3e29.7 {eOCH[CH2(CH2)2(CH2)2CH3]CH2(CH2)8e},31.9 (eCH2CH2CH3), 34.3 {eOCH[CH2(CH2)4CH3]CH2e}, 34.9(eCH2COOe), 74.2[(eOCH(C6H13)e], 173.8 (eCOOe).
Poly(DDL-12HS), Poly(PDL-12HS) and Poly(HDL-12HS): 1H NMR(300 MHz, CDCl3): d (ppm) 0.87 {t, J¼ 6.6 Hz, eOCH[(CH2)5CH3]e},1.25e1.41 [m, eOCH2(CH2)ne and eOCH(C6H13)CH2(CH2)7e],1.5 {m, eOCH[CH2(CH2)4CH3]CH2e}, 1.61 (m, eCH2CH2COOe),2.27 [t, J¼ 7.6 Hz, eCH2CH2COOCH(C6H13)e], 2.28 (t, 2H,J¼ 7.6 Hz, eCH2CH2COOCH2e), 4.05 (t, J¼ 6.8 Hz, eOCH2e), 4.86[m, eOCH(C6H13)e]. 13C NMR (125 MHz, CDCl3): d (ppm) 14.2(eCH3), 22.7 (eCH2CH3), 25.1e29.7 {eOCH2(CH2)nþ1e and eOCH[CH2(CH2)2(CH2)2CH3]CH2(CH2)8e}, 31.9 (eCH2CH2CH3), 34.3{eOCH[CH2(CH2)4CH3]CH2e}, 34.5 [eCH2COOCH(C6H13)e], 34.9(eCH2COOCH2e), 64.5 (eOCH2e), 74.2 [eOCH(C6H13)e], 173.8[eCOOCH(C6H13)e], 174.1(eCOOCH2e).
Table 1Polymerization conditions and analytical data of the polymers.a
3. Results and discussion
3.1. Lipase-catalyzed copolymerization of lactone and 12HS-Me
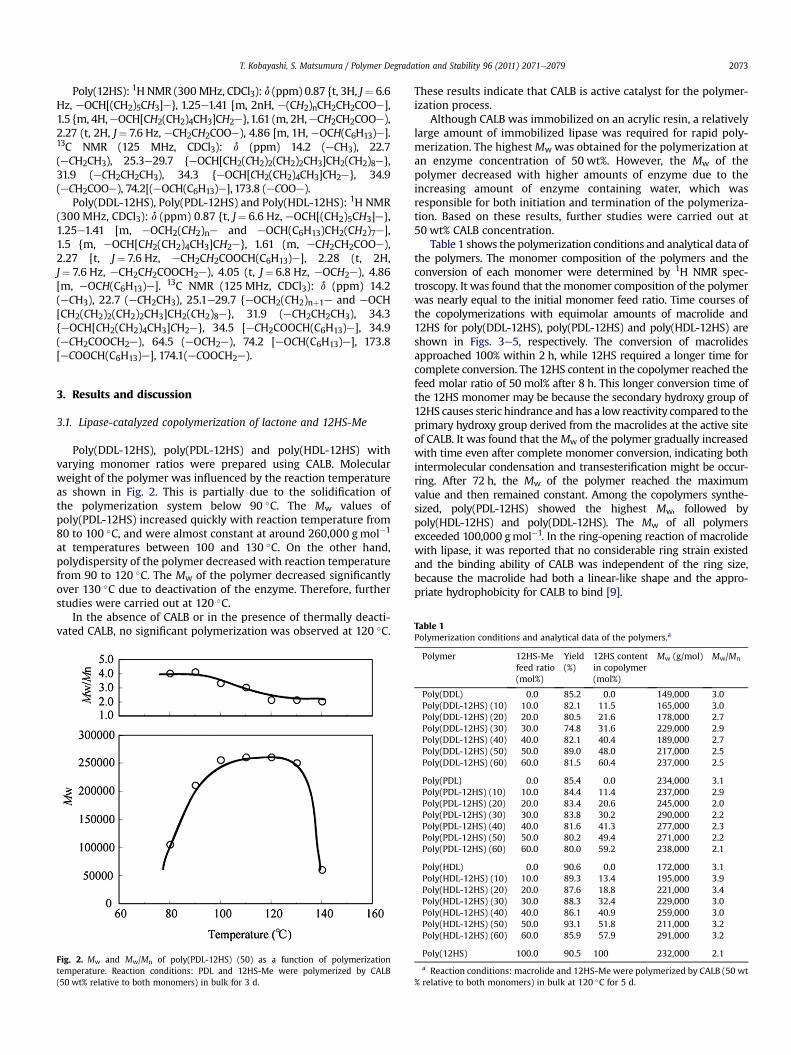
Poly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS) withvarying monomer ratios were prepared using CALB. Molecularweight of the polymer was influenced by the reaction temperatureas shown in Fig. 2. This is partially due to the solidification ofthe polymerization system below 90 �C. The Mw values ofpoly(PDL-12HS) increased quickly with reaction temperature from80 to 100 �C, and were almost constant at around 260,000 gmol�1
at temperatures between 100 and 130 �C. On the other hand,polydispersity of the polymer decreased with reaction temperaturefrom 90 to 120 �C. The Mw of the polymer decreased significantlyover 130 �C due to deactivation of the enzyme. Therefore, furtherstudies were carried out at 120 �C.
In the absence of CALB or in the presence of thermally deacti-vated CALB, no significant polymerization was observed at 120 �C.
Fig. 2. Mw and Mw/Mn of poly(PDL-12HS) (50) as a function of polymerizationtemperature. Reaction conditions: PDL and 12HS-Me were polymerized by CALB(50 wt% relative to both monomers) in bulk for 3 d.
These results indicate that CALB is active catalyst for the polymer-ization process.
Although CALB was immobilized on an acrylic resin, a relativelylarge amount of immobilized lipase was required for rapid poly-merization. The highestMw was obtained for the polymerization atan enzyme concentration of 50 wt%. However, the Mw of thepolymer decreased with higher amounts of enzyme due to theincreasing amount of enzyme containing water, which wasresponsible for both initiation and termination of the polymeriza-tion. Based on these results, further studies were carried out at50 wt% CALB concentration.
Table 1 shows the polymerization conditions and analytical data ofthe polymers. The monomer composition of the polymers and theconversion of each monomer were determined by 1H NMR spec-troscopy. It was found that the monomer composition of the polymerwas nearly equal to the initial monomer feed ratio. Time courses ofthe copolymerizations with equimolar amounts of macrolide and12HS for poly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS) areshown in Figs. 3e5, respectively. The conversion of macrolidesapproached 100% within 2 h, while 12HS required a longer time forcomplete conversion. The 12HS content in the copolymer reached thefeed molar ratio of 50 mol% after 8 h. This longer conversion time ofthe 12HS monomer may be because the secondary hydroxy group of12HS causes steric hindrance and has a low reactivity compared to theprimary hydroxy group derived from the macrolides at the active siteof CALB. It was found that theMw of the polymer gradually increasedwith time even after complete monomer conversion, indicating bothintermolecular condensation and transesterification might be occur-ring. After 72 h, the Mw of the polymer reached the maximumvalue and then remained constant. Among the copolymers synthe-sized, poly(PDL-12HS) showed the highest Mw, followed bypoly(HDL-12HS) and poly(DDL-12HS). The Mw of all polymersexceeded 100,000 gmol�1. In the ring-opening reaction of macrolidewith lipase, it was reported that no considerable ring strain existedand the binding ability of CALB was independent of the ring size,because the macrolide had both a linear-like shape and the appro-priate hydrophobicity for CALB to bind [9].
Polymer 12HS-Mefeed ratio(mol%)
Yield(%)
12HS contentin copolymer(mol%)
Mw (g/mol) Mw/Mn
Poly(DDL) 0.0 85.2 0.0 149,000 3.0Poly(DDL-12HS) (10) 10.0 82.1 11.5 165,000 3.0Poly(DDL-12HS) (20) 20.0 80.5 21.6 178,000 2.7Poly(DDL-12HS) (30) 30.0 74.8 31.6 229,000 2.9Poly(DDL-12HS) (40) 40.0 82.1 40.4 189,000 2.7Poly(DDL-12HS) (50) 50.0 89.0 48.0 217,000 2.5Poly(DDL-12HS) (60) 60.0 81.5 60.4 237,000 2.5
Poly(PDL) 0.0 85.4 0.0 234,000 3.1Poly(PDL-12HS) (10) 10.0 84.4 11.4 237,000 2.9Poly(PDL-12HS) (20) 20.0 83.4 20.6 245,000 2.0Poly(PDL-12HS) (30) 30.0 83.8 30.2 290,000 2.2Poly(PDL-12HS) (40) 40.0 81.6 41.3 277,000 2.3Poly(PDL-12HS) (50) 50.0 80.2 49.4 271,000 2.2Poly(PDL-12HS) (60) 60.0 80.0 59.2 238,000 2.1
Poly(HDL) 0.0 90.6 0.0 172,000 3.1Poly(HDL-12HS) (10) 10.0 89.3 13.4 195,000 3.9Poly(HDL-12HS) (20) 20.0 87.6 18.8 221,000 3.4Poly(HDL-12HS) (30) 30.0 88.3 32.4 229,000 3.0Poly(HDL-12HS) (40) 40.0 86.1 40.9 259,000 3.0Poly(HDL-12HS) (50) 50.0 93.1 51.8 211,000 3.2Poly(HDL-12HS) (60) 60.0 85.9 57.9 291,000 3.2
Poly(12HS) 100.0 90.5 100 232,000 2.1
a Reaction conditions: macrolide and 12HS-Me were polymerized by CALB (50 wt% relative to both monomers) in bulk at 120 �C for 5 d.
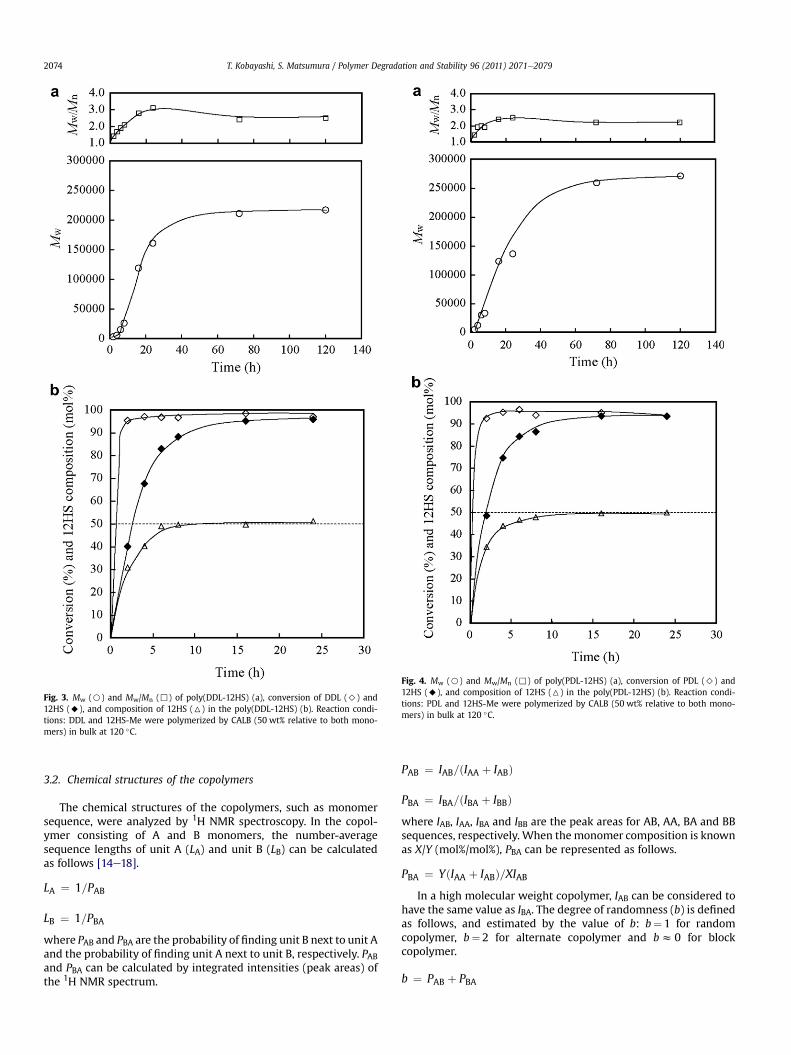
Fig. 3. Mw (B) and Mw/Mn (,) of poly(DDL-12HS) (a), conversion of DDL (>) and12HS (A), and composition of 12HS (6) in the poly(DDL-12HS) (b). Reaction condi-tions: DDL and 12HS-Me were polymerized by CALB (50 wt% relative to both mono-mers) in bulk at 120 �C.
Fig. 4. Mw (B) and Mw/Mn (,) of poly(PDL-12HS) (a), conversion of PDL (>) and12HS (A), and composition of 12HS (6) in the poly(PDL-12HS) (b). Reaction condi-tions: PDL and 12HS-Me were polymerized by CALB (50 wt% relative to both mono-mers) in bulk at 120 �C.
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e20792074
3.2. Chemical structures of the copolymers
The chemical structures of the copolymers, such as monomersequence, were analyzed by 1H NMR spectroscopy. In the copol-ymer consisting of A and B monomers, the number-averagesequence lengths of unit A (LA) and unit B (LB) can be calculatedas follows [14e18].
LA ¼ 1=PAB
LB ¼ 1=PBA
where PAB and PBA are the probability of finding unit B next to unit Aand the probability of finding unit A next to unit B, respectively. PABand PBA can be calculated by integrated intensities (peak areas) ofthe 1H NMR spectrum.
PAB ¼ IAB=ðIAA þ IABÞ
PBA ¼ IBA=ðIBA þ IBBÞwhere IAB, IAA, IBA and IBB are the peak areas for AB, AA, BA and BBsequences, respectively. When themonomer composition is knownas X/Y (mol%/mol%), PBA can be represented as follows.
PBA ¼ YðIAA þ IABÞ=XIABIn a high molecular weight copolymer, IAB can be considered to
have the same value as IBA. The degree of randomness (b) is definedas follows, and estimated by the value of b: b¼ 1 for randomcopolymer, b¼ 2 for alternate copolymer and bz 0 for blockcopolymer.
b ¼ PAB þ PBA
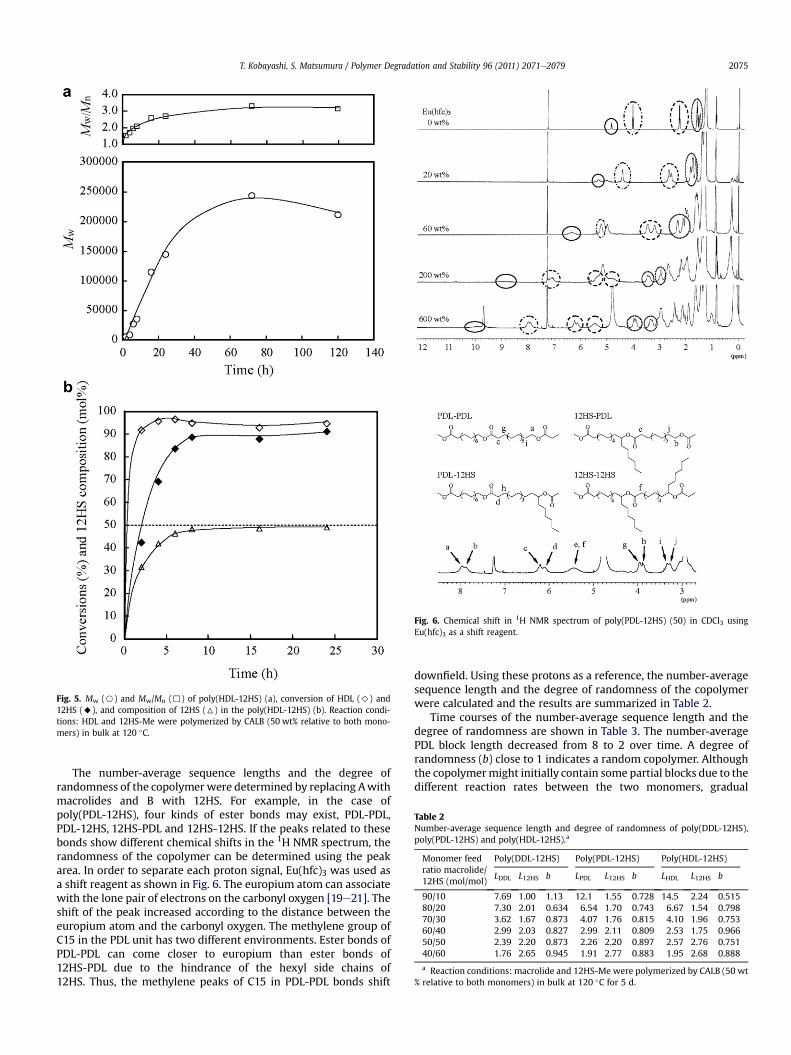
Fig. 5. Mw (B) and Mw/Mn (,) of poly(HDL-12HS) (a), conversion of HDL (>) and12HS (A), and composition of 12HS (6) in the poly(HDL-12HS) (b). Reaction condi-tions: HDL and 12HS-Me were polymerized by CALB (50 wt% relative to both mono-mers) in bulk at 120 �C.
Fig. 6. Chemical shift in 1H NMR spectrum of poly(PDL-12HS) (50) in CDCl3 usingEu(hfc)3 as a shift reagent.
Table 2Number-average sequence length and degree of randomness of poly(DDL-12HS),poly(PDL-12HS) and poly(HDL-12HS).a
Monomer feedratio macrolide/12HS (mol/mol)
Poly(DDL-12HS) Poly(PDL-12HS) Poly(HDL-12HS)
LDDL L12HS b LPDL L12HS b LHDL L12HS b
90/10 7.69 1.00 1.13 12.1 1.55 0.728 14.5 2.24 0.51580/20 7.30 2.01 0.634 6.54 1.70 0.743 6.67 1.54 0.79870/30 3.62 1.67 0.873 4.07 1.76 0.815 4.10 1.96 0.75360/40 2.99 2.03 0.827 2.99 2.11 0.809 2.53 1.75 0.96650/50 2.39 2.20 0.873 2.26 2.20 0.897 2.57 2.76 0.75140/60 1.76 2.65 0.945 1.91 2.77 0.883 1.95 2.68 0.888
a Reaction conditions: macrolide and 12HS-Me were polymerized by CALB (50 wt% relative to both monomers) in bulk at 120 �C for 5 d.
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e2079 2075
The number-average sequence lengths and the degree ofrandomness of the copolymer were determined by replacing Awithmacrolides and B with 12HS. For example, in the case ofpoly(PDL-12HS), four kinds of ester bonds may exist, PDL-PDL,PDL-12HS, 12HS-PDL and 12HS-12HS. If the peaks related to thesebonds show different chemical shifts in the 1H NMR spectrum, therandomness of the copolymer can be determined using the peakarea. In order to separate each proton signal, Eu(hfc)3 was used asa shift reagent as shown in Fig. 6. The europium atom can associatewith the lone pair of electrons on the carbonyl oxygen [19e21]. Theshift of the peak increased according to the distance between theeuropium atom and the carbonyl oxygen. The methylene group ofC15 in the PDL unit has two different environments. Ester bonds ofPDL-PDL can come closer to europium than ester bonds of12HS-PDL due to the hindrance of the hexyl side chains of12HS. Thus, the methylene peaks of C15 in PDL-PDL bonds shift
downfield. Using these protons as a reference, the number-averagesequence length and the degree of randomness of the copolymerwere calculated and the results are summarized in Table 2.
Time courses of the number-average sequence length and thedegree of randomness are shown in Table 3. The number-averagePDL block length decreased from 8 to 2 over time. A degree ofrandomness (b) close to 1 indicates a random copolymer. Althoughthe copolymermight initially contain some partial blocks due to thedifferent reaction rates between the two monomers, gradual
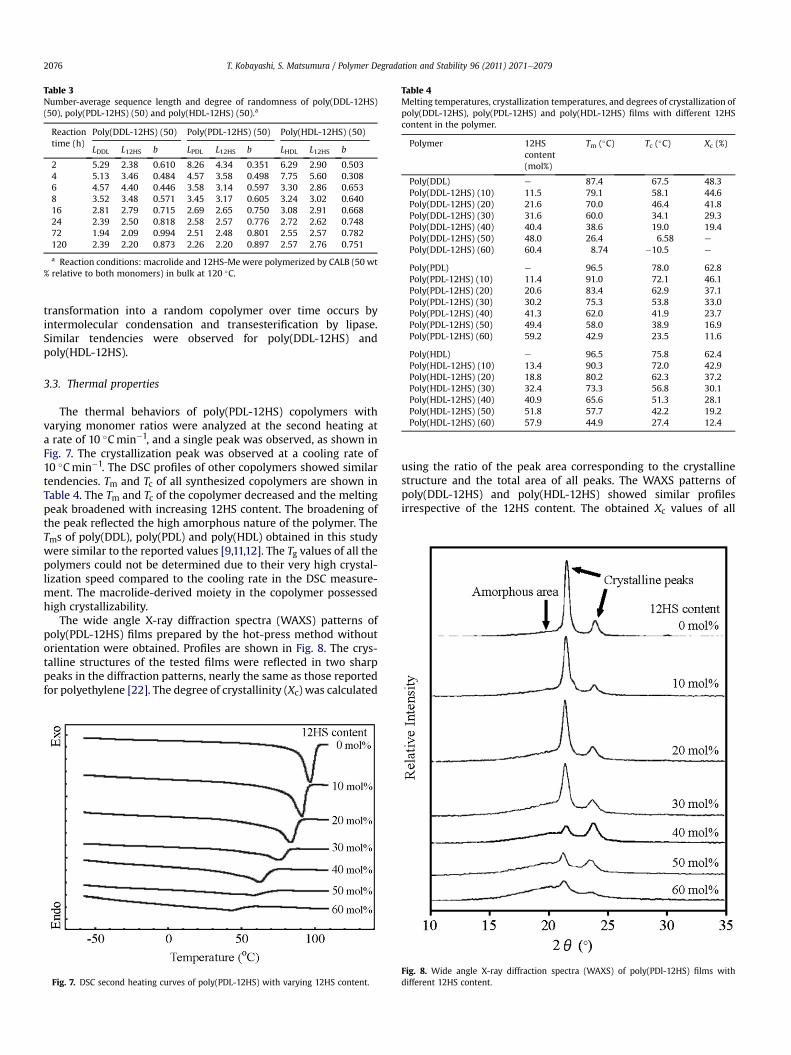
Table 3Number-average sequence length and degree of randomness of poly(DDL-12HS)(50), poly(PDL-12HS) (50) and poly(HDL-12HS) (50).a
Reactiontime (h)
Poly(DDL-12HS) (50) Poly(PDL-12HS) (50) Poly(HDL-12HS) (50)
LDDL L12HS b LPDL L12HS b LHDL L12HS b
2 5.29 2.38 0.610 8.26 4.34 0.351 6.29 2.90 0.5034 5.13 3.46 0.484 4.57 3.58 0.498 7.75 5.60 0.3086 4.57 4.40 0.446 3.58 3.14 0.597 3.30 2.86 0.6538 3.52 3.48 0.571 3.45 3.17 0.605 3.24 3.02 0.64016 2.81 2.79 0.715 2.69 2.65 0.750 3.08 2.91 0.66824 2.39 2.50 0.818 2.58 2.57 0.776 2.72 2.62 0.74872 1.94 2.09 0.994 2.51 2.48 0.801 2.55 2.57 0.782120 2.39 2.20 0.873 2.26 2.20 0.897 2.57 2.76 0.751
a Reaction conditions: macrolide and 12HS-Me were polymerized by CALB (50 wt% relative to both monomers) in bulk at 120 �C.
Table 4Melting temperatures, crystallization temperatures, and degrees of crystallization ofpoly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS) films with different 12HScontent in the polymer.
Polymer 12HScontent(mol%)
Tm (�C) Tc (�C) Xc (%)
Poly(DDL) e 87.4 67.5 48.3Poly(DDL-12HS) (10) 11.5 79.1 58.1 44.6Poly(DDL-12HS) (20) 21.6 70.0 46.4 41.8Poly(DDL-12HS) (30) 31.6 60.0 34.1 29.3Poly(DDL-12HS) (40) 40.4 38.6 19.0 19.4Poly(DDL-12HS) (50) 48.0 26.4 6.58 e
Poly(DDL-12HS) (60) 60.4 8.74 �10.5 e
Poly(PDL) e 96.5 78.0 62.8Poly(PDL-12HS) (10) 11.4 91.0 72.1 46.1Poly(PDL-12HS) (20) 20.6 83.4 62.9 37.1Poly(PDL-12HS) (30) 30.2 75.3 53.8 33.0Poly(PDL-12HS) (40) 41.3 62.0 41.9 23.7Poly(PDL-12HS) (50) 49.4 58.0 38.9 16.9Poly(PDL-12HS) (60) 59.2 42.9 23.5 11.6
Poly(HDL) e 96.5 75.8 62.4Poly(HDL-12HS) (10) 13.4 90.3 72.0 42.9Poly(HDL-12HS) (20) 18.8 80.2 62.3 37.2Poly(HDL-12HS) (30) 32.4 73.3 56.8 30.1Poly(HDL-12HS) (40) 40.9 65.6 51.3 28.1Poly(HDL-12HS) (50) 51.8 57.7 42.2 19.2Poly(HDL-12HS) (60) 57.9 44.9 27.4 12.4
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e20792076
transformation into a random copolymer over time occurs byintermolecular condensation and transesterification by lipase.Similar tendencies were observed for poly(DDL-12HS) andpoly(HDL-12HS).
3.3. Thermal properties
The thermal behaviors of poly(PDL-12HS) copolymers withvarying monomer ratios were analyzed at the second heating ata rate of 10 �Cmin�1, and a single peak was observed, as shown inFig. 7. The crystallization peak was observed at a cooling rate of10 �Cmin�1. The DSC profiles of other copolymers showed similartendencies. Tm and Tc of all synthesized copolymers are shown inTable 4. The Tm and Tc of the copolymer decreased and the meltingpeak broadened with increasing 12HS content. The broadening ofthe peak reflected the high amorphous nature of the polymer. TheTms of poly(DDL), poly(PDL) and poly(HDL) obtained in this studywere similar to the reported values [9,11,12]. The Tg values of all thepolymers could not be determined due to their very high crystal-lization speed compared to the cooling rate in the DSC measure-ment. The macrolide-derived moiety in the copolymer possessedhigh crystallizability.
The wide angle X-ray diffraction spectra (WAXS) patterns ofpoly(PDL-12HS) films prepared by the hot-press method withoutorientation were obtained. Profiles are shown in Fig. 8. The crys-talline structures of the tested films were reflected in two sharppeaks in the diffraction patterns, nearly the same as those reportedfor polyethylene [22]. The degree of crystallinity (Xc) was calculated
Fig. 7. DSC second heating curves of poly(PDL-12HS) with varying 12HS content.
using the ratio of the peak area corresponding to the crystallinestructure and the total area of all peaks. The WAXS patterns ofpoly(DDL-12HS) and poly(HDL-12HS) showed similar profilesirrespective of the 12HS content. The obtained Xc values of all
Fig. 8. Wide angle X-ray diffraction spectra (WAXS) of poly(PDl-12HS) films withdifferent 12HS content.
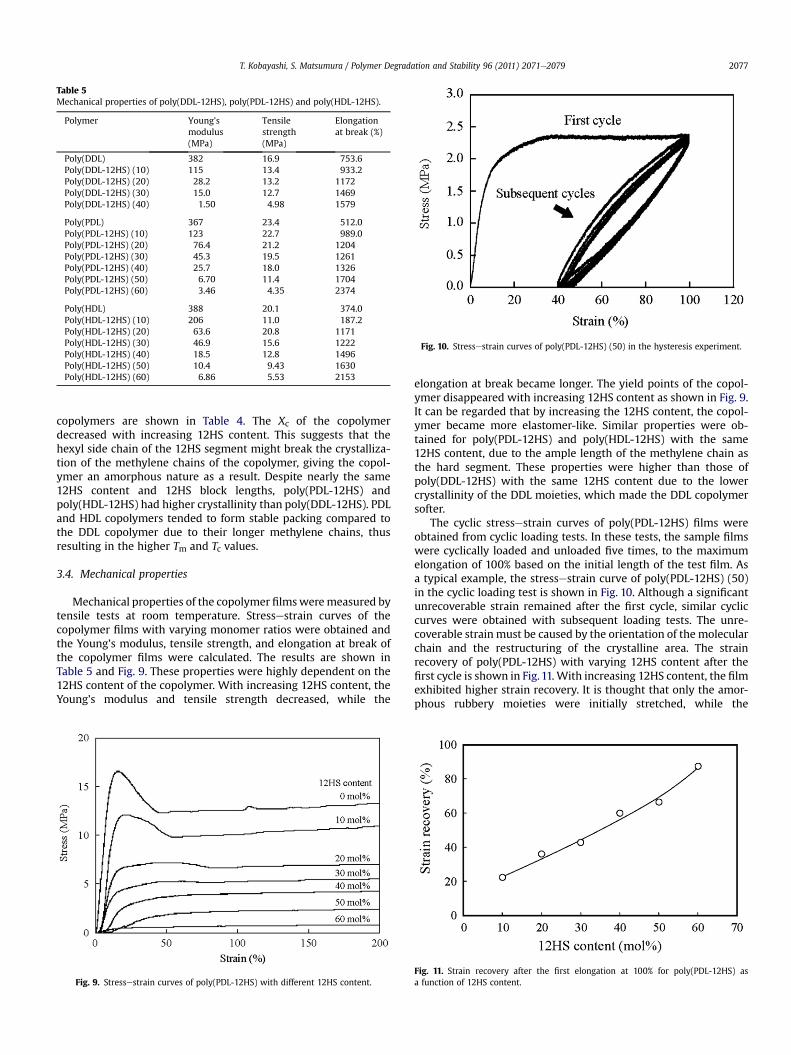
Table 5Mechanical properties of poly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS).
Polymer Young’smodulus(MPa)
Tensilestrength(MPa)
Elongationat break (%)
Poly(DDL) 382 16.9 753.6Poly(DDL-12HS) (10) 115 13.4 933.2Poly(DDL-12HS) (20) 28.2 13.2 1172Poly(DDL-12HS) (30) 15.0 12.7 1469Poly(DDL-12HS) (40) 1.50 4.98 1579
Poly(PDL) 367 23.4 512.0Poly(PDL-12HS) (10) 123 22.7 989.0Poly(PDL-12HS) (20) 76.4 21.2 1204Poly(PDL-12HS) (30) 45.3 19.5 1261Poly(PDL-12HS) (40) 25.7 18.0 1326Poly(PDL-12HS) (50) 6.70 11.4 1704Poly(PDL-12HS) (60) 3.46 4.35 2374
Poly(HDL) 388 20.1 374.0Poly(HDL-12HS) (10) 206 11.0 187.2Poly(HDL-12HS) (20) 63.6 20.8 1171Poly(HDL-12HS) (30) 46.9 15.6 1222Poly(HDL-12HS) (40) 18.5 12.8 1496Poly(HDL-12HS) (50) 10.4 9.43 1630Poly(HDL-12HS) (60) 6.86 5.53 2153
Fig. 10. Stressestrain curves of poly(PDL-12HS) (50) in the hysteresis experiment.
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e2079 2077
copolymers are shown in Table 4. The Xc of the copolymerdecreased with increasing 12HS content. This suggests that thehexyl side chain of the 12HS segment might break the crystalliza-tion of the methylene chains of the copolymer, giving the copol-ymer an amorphous nature as a result. Despite nearly the same12HS content and 12HS block lengths, poly(PDL-12HS) andpoly(HDL-12HS) had higher crystallinity than poly(DDL-12HS). PDLand HDL copolymers tended to form stable packing compared tothe DDL copolymer due to their longer methylene chains, thusresulting in the higher Tm and Tc values.
3.4. Mechanical properties
Mechanical properties of the copolymer filmsweremeasured bytensile tests at room temperature. Stressestrain curves of thecopolymer films with varying monomer ratios were obtained andthe Young’s modulus, tensile strength, and elongation at break ofthe copolymer films were calculated. The results are shown inTable 5 and Fig. 9. These properties were highly dependent on the12HS content of the copolymer. With increasing 12HS content, theYoung’s modulus and tensile strength decreased, while the
Fig. 9. Stressestrain curves of poly(PDL-12HS) with different 12HS content.
elongation at break became longer. The yield points of the copol-ymer disappeared with increasing 12HS content as shown in Fig. 9.It can be regarded that by increasing the 12HS content, the copol-ymer became more elastomer-like. Similar properties were ob-tained for poly(PDL-12HS) and poly(HDL-12HS) with the same12HS content, due to the ample length of the methylene chain asthe hard segment. These properties were higher than those ofpoly(DDL-12HS) with the same 12HS content due to the lowercrystallinity of the DDL moieties, which made the DDL copolymersofter.
The cyclic stressestrain curves of poly(PDL-12HS) films wereobtained from cyclic loading tests. In these tests, the sample filmswere cyclically loaded and unloaded five times, to the maximumelongation of 100% based on the initial length of the test film. Asa typical example, the stressestrain curve of poly(PDL-12HS) (50)in the cyclic loading test is shown in Fig. 10. Although a significantunrecoverable strain remained after the first cycle, similar cycliccurves were obtained with subsequent loading tests. The unre-coverable strain must be caused by the orientation of the molecularchain and the restructuring of the crystalline area. The strainrecovery of poly(PDL-12HS) with varying 12HS content after thefirst cycle is shown in Fig.11. With increasing 12HS content, the filmexhibited higher strain recovery. It is thought that only the amor-phous rubbery moieties were initially stretched, while the
Fig. 11. Strain recovery after the first elongation at 100% for poly(PDL-12HS) asa function of 12HS content.
0 5 10 15 20 25 30 35 40 45Time (day)
0
20
40
60
80
100
BO
D-b
iode
grad
atio
n (%
)
12HS content
Aniline
100 mol%40 mol%20 mol%
0 mol%
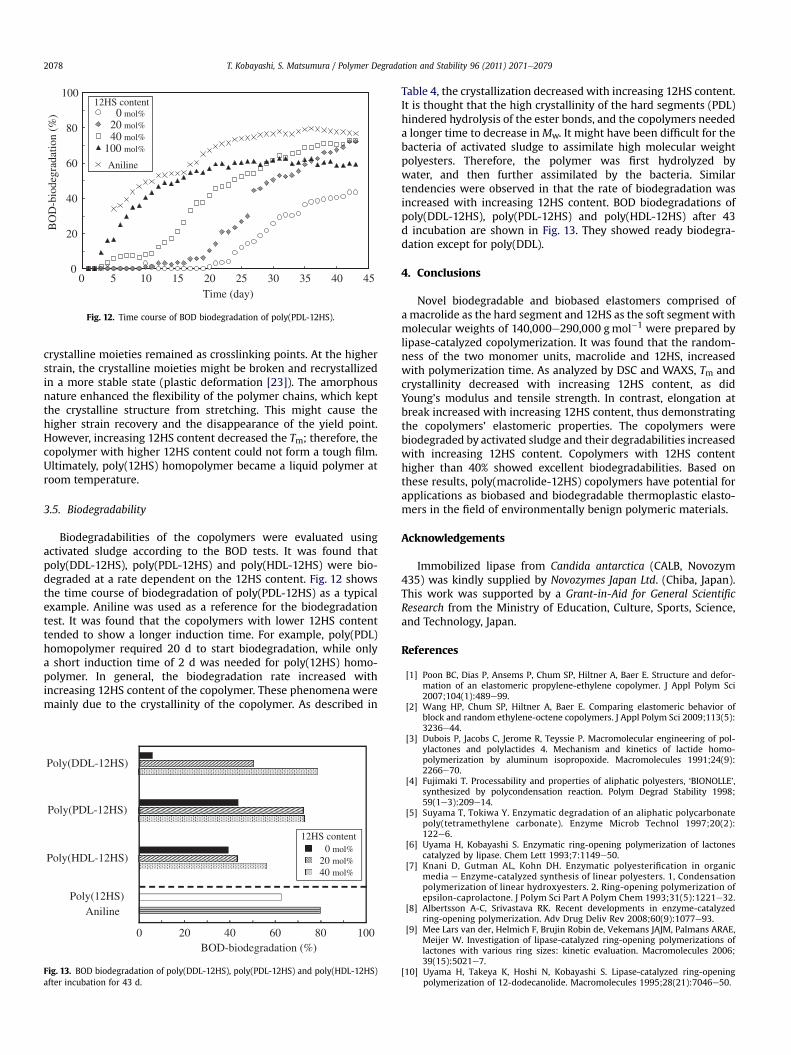
Fig. 12. Time course of BOD biodegradation of poly(PDL-12HS).
T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e20792078
crystalline moieties remained as crosslinking points. At the higherstrain, the crystalline moieties might be broken and recrystallizedin a more stable state (plastic deformation [23]). The amorphousnature enhanced the flexibility of the polymer chains, which keptthe crystalline structure from stretching. This might cause thehigher strain recovery and the disappearance of the yield point.However, increasing 12HS content decreased the Tm; therefore, thecopolymer with higher 12HS content could not form a tough film.Ultimately, poly(12HS) homopolymer became a liquid polymer atroom temperature.
3.5. Biodegradability
Biodegradabilities of the copolymers were evaluated usingactivated sludge according to the BOD tests. It was found thatpoly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS) were bio-degraded at a rate dependent on the 12HS content. Fig. 12 showsthe time course of biodegradation of poly(PDL-12HS) as a typicalexample. Aniline was used as a reference for the biodegradationtest. It was found that the copolymers with lower 12HS contenttended to show a longer induction time. For example, poly(PDL)homopolymer required 20 d to start biodegradation, while onlya short induction time of 2 d was needed for poly(12HS) homo-polymer. In general, the biodegradation rate increased withincreasing 12HS content of the copolymer. These phenomena weremainly due to the crystallinity of the copolymer. As described in
AnilinePoly(12HS)
Poly(DDL-12HS)
Poly(PDL-12HS)
Poly(HDL-12HS)
0 20 40 60 80 100BOD-biodegradation (%)
12HS content
40 mol%20 mol%
0 mol%
Fig. 13. BOD biodegradation of poly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS)after incubation for 43 d.
Table 4, the crystallization decreased with increasing 12HS content.It is thought that the high crystallinity of the hard segments (PDL)hindered hydrolysis of the ester bonds, and the copolymers neededa longer time to decrease inMw. It might have been difficult for thebacteria of activated sludge to assimilate high molecular weightpolyesters. Therefore, the polymer was first hydrolyzed bywater, and then further assimilated by the bacteria. Similartendencies were observed in that the rate of biodegradation wasincreased with increasing 12HS content. BOD biodegradations ofpoly(DDL-12HS), poly(PDL-12HS) and poly(HDL-12HS) after 43d incubation are shown in Fig. 13. They showed ready biodegra-dation except for poly(DDL).
4. Conclusions
Novel biodegradable and biobased elastomers comprised ofa macrolide as the hard segment and 12HS as the soft segment withmolecular weights of 140,000e290,000 gmol�1 were prepared bylipase-catalyzed copolymerization. It was found that the random-ness of the two monomer units, macrolide and 12HS, increasedwith polymerization time. As analyzed by DSC and WAXS, Tm andcrystallinity decreased with increasing 12HS content, as didYoung’s modulus and tensile strength. In contrast, elongation atbreak increased with increasing 12HS content, thus demonstratingthe copolymers’ elastomeric properties. The copolymers werebiodegraded by activated sludge and their degradabilities increasedwith increasing 12HS content. Copolymers with 12HS contenthigher than 40% showed excellent biodegradabilities. Based onthese results, poly(macrolide-12HS) copolymers have potential forapplications as biobased and biodegradable thermoplastic elasto-mers in the field of environmentally benign polymeric materials.
Acknowledgements
Immobilized lipase from Candida antarctica (CALB, Novozym435) was kindly supplied by Novozymes Japan Ltd. (Chiba, Japan).This work was supported by a Grant-in-Aid for General ScientificResearch from the Ministry of Education, Culture, Sports, Science,and Technology, Japan.
References
[1] Poon BC, Dias P, Ansems P, Chum SP, Hiltner A, Baer E. Structure and defor-mation of an elastomeric propylene-ethylene copolymer. J Appl Polym Sci2007;104(1):489e99.
[2] Wang HP, Chum SP, Hiltner A, Baer E. Comparing elastomeric behavior ofblock and random ethylene-octene copolymers. J Appl Polym Sci 2009;113(5):3236e44.
[3] Dubois P, Jacobs C, Jerome R, Teyssie P. Macromolecular engineering of pol-ylactones and polylactides 4. Mechanism and kinetics of lactide homo-polymerization by aluminum isopropoxide. Macromolecules 1991;24(9):2266e70.
[4] Fujimaki T. Processability and properties of aliphatic polyesters, ‘BIONOLLE’,synthesized by polycondensation reaction. Polym Degrad Stability 1998;59(1e3):209e14.
[5] Suyama T, Tokiwa Y. Enzymatic degradation of an aliphatic polycarbonatepoly(tetramethylene carbonate). Enzyme Microb Technol 1997;20(2):122e6.
[6] Uyama H, Kobayashi S. Enzymatic ring-opening polymerization of lactonescatalyzed by lipase. Chem Lett 1993;7:1149e50.
[7] Knani D, Gutman AL, Kohn DH. Enzymatic polyesterification in organicmedia e Enzyme-catalyzed synthesis of linear polyesters. 1, Condensationpolymerization of linear hydroxyesters. 2. Ring-opening polymerization ofepsilon-caprolactone. J Polym Sci Part A Polym Chem 1993;31(5):1221e32.
[8] Albertsson A-C, Srivastava RK. Recent developments in enzyme-catalyzedring-opening polymerization. Adv Drug Deliv Rev 2008;60(9):1077e93.
[9] Mee Lars van der, Helmich F, Brujin Robin de, Vekemans JAJM, Palmans ARAE,Meijer W. Investigation of lipase-catalyzed ring-opening polymerizations oflactones with various ring sizes: kinetic evaluation. Macromolecules 2006;39(15):5021e7.
[10] Uyama H, Takeya K, Hoshi N, Kobayashi S. Lipase-catalyzed ring-openingpolymerization of 12-dodecanolide. Macromolecules 1995;28(21):7046e50.

T. Kobayashi, S. Matsumura / Polymer Degradation and Stability 96 (2011) 2071e2079 2079
[11] Focarete ML, Scandola M, Kumar A, Gross RA. Physical characterizationof poly(omega-pentadecalactone) synthesized by lipase-catalyzed ring-opening polymerization. J Polym Sci Part B Polym Phys 2001;39(15):1721e9.
[12] Meulen Inge van der, de Geus Matthijs, Antheunis H, Deumens R,Joosten EAJC, Koning E, et al. Polymers from functional macrolactones aspotential biomaterials: enzymatic ring opening polymerization, biodegrada-tion, and biocompatibility. Biomacromolecules 2008;9(12):3404e10.
[13] Ebata H, Toshima K, Matsumura S. Lipase-catalyzed synthesis and propertiesof poly(12-hydroxydodecanoate-co-12-hydroxystearate) directed towardsnovel green and sustainable elastomer. Macromol Biosci 2008;8(1):38e45.
[14] Miller RL, Nielsen LE. On the characterization of stereoregular polymers. 1.Theory J Polym Sci 1960;46(148):303e16.
[15] Yamadera R, Murano M. Determination of randomness in copolyesters byhigh resolution nuclear magnetic resonance. J Polym Sci Part A 1967;5(9PA1):2259.
[16] Chen MS, Chang SJ, Chang RS, Kuo WF, Tsai HB. Copolyesters. 1. Sequencedistribution of poly(butylene terephthalate-co-adipate) copolyesters deter-mined by 400 MHz NMR. J Appl Polym Sci 1990;40(5e6):1053e7.
[17] Lorenzetti C, Finelli L, Lotti N, Vannini M, Gazzano M, Berti C, et al.Synthesis and characterization of poly(propylene terephthalate/2,6-naphthalate) random copolyesters. Polymers 2005;46(12):4041e51.
[18] Soccio M, Finelli L, Lotti N, Gazzano M, Munari A. Novel random poly(propyleneisophthalate/adipate) copolyesters: synthesis and characterization. Eur Polym J2006;42(11):2949e58.
[19] Carroll FI, Blackwell JT. Structure and conformation of cis and trans-3,5-dimethylvalerolactones. Tetrahedron Lett 1970;11(48):4173.
[20] Katritzky AR, Smith A. Application of contact shift reagents to NMR spectra ofpolymers. Tetrahedron Lett 1971;12(21):1765.
[21] Cheng HN, Lee GH. The combined use of two-dimensional NMR and lantha-nide shift-reagents for the characterization of ethylene vinyl-acetate copoly-mers. Polym Bull 1988;19(1):89e96.
[22] Caminiti R, Pandolfi L, Ballirano P. Structure of polyethylene from X-raypowder diffraction: influence of the amorphous fraction on data analysis.J Macromol Sci Phys Part B 2000;B39(4):481e92.
[23] Luyt AS, Hato MJ. Thermal and mechanical properties of linear low-densitypolyethylene/low-density polyethylene/low-density polyethylene/waxternary blends. J Appl Polym Sci 2005;96(5):1748e55.





























