Embed Size (px)
Citation preview

Estudos Genomicos de Flexibilidade e EnergiaLivre Associados a Distribuicao de SNPs
Dissertacao de mestrado
LUCIANA MARCIA DE OLIVEIRA
GERALD WEBER (DF/UFMG, ORIENTADOR)
Nucleo de Pesquisas em Ciencias Biologicas (NUPEB)Pos graduacao em Biotecnologia
Area de concentracao: Genomica e ProteomicaUniversidade Federal de Ouro Preto
Ouro Preto, junho de 2011

O482e Oliveira, Luciana Marcia deEstudos genomicos de flexibilidade e energia associada a distribuicao de SNPs
[manuscrito] / Luciana Marcia de Oliveira. -2011.xiii, 105f.: il., color; graf.; tabs.
Orientador: Prof. Dr. Gerald Weber.
Dissertacao (Mestrado) — Universidade Federal de Ouro Preto.Instituto de Ciencias Exatas e Biologicas. Nucleo de Pesquisas emCiencias Biologicas. Programa de Pos-Graduacao em Biotecnologia.
Area de concentracao: Genomica e Proteomicas
1. Genetica molecular — Teses. 2. Acido desoxirribonucleico — Teses.3. Genomas — Teses. 4. Polimorfismos de base unica (SNPs) — Teses.5. Cinetica quımica — Teses. I. Universidade Federal de Ouro Preto. II. Tıtulo.
CDU: 577.212:544.4
Catalogacao: [email protected]

Estudos Genomicos de Flexibilidade e EnergiaLivre Associados a Distribuicao de SNPs
LUCIANA MARCIA DE OLIVEIRA
GERALD WEBER (DF/UFMG, ORIENTADOR)
DISSERTACAO DE MESTRADO UNIVERSIDADE FEDERAL DE OURO PRETO COMO
PARTE DOS REQUISITOS BASICOS PARA A OBTENCAO DO GRAU DE MESTRE EM
BIOTECNOLOGIA, AREA DE CONCENTRACAO GENOMICA E PROTEOMICA.
INSTITUTO DE CIENCIAS EXATAS E BIOLOGICAS - ICEBUNIVERSIDADE FEDERAL DE OURO PRETO
Ouro Preto, junho de 2011

Dedicatoria
Este trabalho e dedicado aos meus pais que combinaram seus genes e coracoes para
que eu existisse e ao Jeco por ter lido esta dissertacao sobre os meus ombros...
I

Agradecimentos
Agradecer a todos que me apoiaram nao e uma tarefa facil assim como nao foi estetrabalho. Portanto organizarei este topico iniciando pelos meus agradecimentos profissi-onais seguido dos pessoais.
• Ao Dr. Professor Gerald Weber pela orientacao, pelo apoio e pelas oportunidades.Agradeco pelos ensinamentos, amizade e principalmente pela paciencia e confiancaquanto ao desenvolvimento deste trabalho.
• Agradeco ao Dr. Jeronimo Ruiz do Centro de Pesquisas Rene Rachou - FIOCRUZpelas discussoes, oportunidades, colaboracoes, sugestoes e por ter despertado emmim a paixao pela bioinformatica.
• Ao Dr. Guilherme Oliveira e Dra. Angela Volpine do Centro de Pesquisas ReneRachou - FIOCRUZ pelas varias oportunidades cientıficas.
• A Dra. Diana Bahia da UNIFESP pela amizade, colaboracoes e oportunidades.
• Ao grupo de biofısica computacional e fısica estatıstica da UFOP e UFMG emespecial aos colegas, amigos e companheiros Denise, Lucas e Julio pelos momentosde descontracao.
• Aos colegas do mestrado em Biotecnologia da UFOP e em especial a Dani e a Val.
• Aos colegas do Rene Rachou Fernanda Raad, Paula Santos, Maıra, Sara, Simara,Antonio Mauro, Daniela Rezende e Daniel Liarte pelas discussoes, sugestoes eagradaveis bate-papos.
• A CAPES pela bolsa.
• Ao Departamento de Fısica da UFMG pela hospitalidade durante a realizacao doprojeto e em especial ao professor Jarferson.
• A Pro-reitoria de Pos-Graduacao (PROPP/UFOP) pelo apoio dado as participacoesem conferencias.
• Aos meus pais agradeco com muito carinho pelo apoio incondicional, pelas suaspalavras sabias, por serem o tempo todo tao amorosos, carinhosos e por estaremsempre presentes na minha vida.
II

• Ao Jeco tambem agradeco carinhosamente pelo seu amor, companheirismo, pacienciae compreensao que demonstrou ao longo deste trabalho.
• As minhas irmas e amigas Sandra e Eliana por estarem ao meu lado sempre, ao meuirmao Eduardo e sobrinhos Gabi, Aninha e Davi.
• A querida amiga Denise pela companhia, por me ouvir e dividir anseios e alegriasdurante estes dois anos de trabalho em que tambem compartilhamos infinitas gar-galhadas de perder o folego.
• A todos amigos queridos, em especial Fer, Mi, Cris, Tchururu, Mickey, Cafe, Tati,Carol, Camila e Clara.
• As amigas e colegas do Izabela Hendrix Tati Generoso, Fer Silva, Magal, Caps-trano, Lele, Josi e Verinha.
• Enfim, a todos que de alguma forma contribuıram com a minha formacao pessoal eprofissional.
III

“...demasiado belo para nao ser verdade”
James Watson
IV

Resumo
Os SNPs (single nucleotide polymorphism) sao mutacoes resultantes de uma subs-tituicao, insercao ou delecao que ocorrem em uma unica base. Tais substituicoes saoperpetuadas quando ocorrem erros de pareamentos (mismatch) que nao sao corrigidos.Existem dois tipos de substituicao: transicao, quando ha trocas entre purinas (A e G) ouentre pirimidinas (C e T), e transversao quando ha a substituicao de uma purina por umapirimidina ou vice-versa.
Nos acreditamos que a ocorrencia e distribuicao desses eventos evolutivos nos geno-mas ocorrem em funcao da pressao biologica seletiva mas por outro lado tambem podemestar relacionadas as caracterısticas fısicas da microrregiao onde eles acontecem. Nestetrabalho, nos avaliamos a influencia da energia livre e da flexibilidade da microrregiaogenomica em funcao da distribuicao de SNPs em um genoma de procarioto e oito ge-nomas de eucariotos. Como as bases vizinhas tem um papel importante na ocorrenciadesses eventos, analisamos as perturbacoes locais que um mismatch promove na estruturado DNA levando em consideracao a composicao das bases imediatamente adjacentes aoerro. Para tanto, recuperamos da base de dados dbSNP (release 132) as sequencias de-positadas de nove organismos as quais foram classificadas de acordo com a presenca detransicao ou transversao nos seus genomas. A metodologia descrita na literatura para ocalculo dos valores de energia livre e flexibilidade de dois pares de bases foi extrapoladapara a avaliacao dessas propriedades em uma microrregiao composta por tres pares debases contendo um mismatch central. Nossos resultados indicam que para certos orga-nismos, como por exemplo Apis Mellifera , existe uma correlacao quase linear entre aocorrencia de SNPs e a energia livre. E tambem possıvel constatar que os SNPs ocorrempreferencialmente em regioes do DNA cujas faixas de valores de energia livre sao altas.Por outro lado, nossos resultados tambem evidenciam que a flexibilidade de uma micror-regiao interfere na ocorrencia dos SNPs, uma vez que a maioria desses eventos tendem aocorrer em regioes mais rıgidas quando comparadas as regioes de pareamentos canonicosdo DNA. Finalizando, ao correlacionar a energia livre e as flexibilidades, observamos quepara genomas como os de mamıferos as transicoes tendem a ocorrer mais frequentementeem microrregioes mais estaveis e rıgidas da molecula ao passo que as transversoes saofrequentes em microrregioes menos estaveis, porem rıgidas do DNA.
Palavras-chave: Flexibilidade, Energia livre de Gibbis, DNA, Single Nucleotide Po-limorphisms (SNPs).
V

Abstract
The single nucleotide polymorphism (SNPs) are the result of mutations from a singlesubstitution, insert, or deletion of base pair. These substitutions are perpetuated whenmismatches occur and are not corrected. There are two types of substitution: transitionswhen there are base changes between a purine (A and G) or between pyrimidine (C andT), and transversions when a purine is replaced by a pyrimidine or vice-versa.
We believe that SNPs occurence and distribuition in genomes do not occur just dueto selective biological pressure but should be related also to physical properties of regionswhere these events occur. In this work we evaluated the influence of free energy and fle-xibility in the SNP distribution for one prokaryote genome and eight eukaryote genomes.Since the adjacent bases play an important role for SNPs occurrences, we analyzed thelocal disturbance that mismatches cause in the DNA structure considering the base com-position immediately adjacent to mismatch positions. Therefore, we obtained from thedbSNP database (release 132) the available sequences for nine organisms and classifiedthem according to SNP type: transition or transversion. We use the methodology foundin the literature to calculate the values of free energy and flexibility for two base pairsand extrapolate them to evaluate the properties for a region with three base pairs contai-ning a central mismatch. Our results indicate that for some organisms there is a linearcorrelation between the SNP occurrences and free energy. It is also possible to see thatSNPs occurs preferentially in the region of DNA with a high value of free energy. On theother hand, our results also show that the flexibility of the a microregion interfere withthe occurrence of SNPs since most of these events tend to occur in a rigid microregionwhen compared with canonical DNA base pairs. Finally, when correlating free energy andflexibility we observed that for some genomes the high frequency of transitions occur inmicroregions which are more stable and rigid while transversions occur more frequentlyin microregions with low stability but which are still rigid.
VI

Sumario
Resumo V
Abstract VI
Lista de Figuras X
Lista de Tabelas XIII
1 Introducao 1
1.1 Objetivo geral do projeto . . . . . . . . . . . . . . . . . . . . . . . . . . 3
1.1.1 Objetivos especıficos . . . . . . . . . . . . . . . . . . . . . . . . 3
2 DNA e mutacoes 4
2.1 O Acido Desoxirribonucleico - DNA . . . . . . . . . . . . . . . . . . . . 4
2.2 Mutacoes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
2.2.1 Polimorfismo de base unica ou SNPs (Single Nucleotide Polymor-
phism) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
2.2.2 Causas da variacao nas taxas de substituicao dos SNPs . . . . . . 10
2.2.3 Taxas de mutacoes . . . . . . . . . . . . . . . . . . . . . . . . . 11
3 Banco de dados de sequencias biologicas 13
4 A fısica do DNA 17
4.1 Energia livre . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
4.1.1 A interferencia dos mismatches e os proximos vizinhos na estabi-lidade do DNA . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
4.2 Flexibilidade . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
4.2.1 Modelo para o calculo das flexibilidades . . . . . . . . . . . . . . 26
VII

5 Metodos 29
5.1 Mecanismo hipotetico para a formacao de um SNP a partir erros de pare-amentos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
5.2 Notacao utilizada . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5.3 Genomas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
5.4 Sequencias de SNPs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
5.5 Datamining - Mineracao de dados . . . . . . . . . . . . . . . . . . . . . 36
5.6 Dados de energia livre e flexibilidades . . . . . . . . . . . . . . . . . . . 37
5.7 Workflow computacional consolidado . . . . . . . . . . . . . . . . . . . 37
6 Resultados e discussao 42
6.1 Organismo padrao . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
6.2 Elementos para analise . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
6.3 Analise comparativa entre o organismo padrao e um genoma real . . . . . 44
6.4 Genomas . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
6.4.1 Homo sapiens . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
6.4.2 Bos taurus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
6.4.3 Mus musculus . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
6.4.4 Gallus gallus . . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
6.4.5 Danio rerio . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55
6.4.6 Apis mellifera . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
6.4.7 Plasmodium falciparum . . . . . . . . . . . . . . . . . . . . . . 59
6.4.8 Oryza sativa . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
6.4.9 Streptococcus pyogenes . . . . . . . . . . . . . . . . . . . . . . 64
6.5 Analise por regioes genomicas: resultados preliminares . . . . . . . . . . 67
6.5.1 Analise comparativa previa: Homo sapiens . . . . . . . . . . . . 67
7 Conclusao 72
VIII

8 Perspectivas futuras 74
A Apendice 75
A.1 Scripts utilizados neste trabalho . . . . . . . . . . . . . . . . . . . . . . 76
A.1.1 trimero.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76
A.1.2 fasta2summary.pl . . . . . . . . . . . . . . . . . . . . . . . . . . 77
A.1.3 trans-transv.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
A.1.4 snp.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
A.1.5 gccalc.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
A.1.6 util.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
A.1.7 histogram.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
A.1.8 padroniza.pl . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
Referencias 95
IX

Lista de Figuras
1 Os blocos fundamentais que compoem a cadeia de DNA sao chamadosde nucleotıdeos . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2 Diagrama quımico da estrutura do DNA . . . . . . . . . . . . . . . . . . 5
3 Diagrama do complexo replissomo de procariotos . . . . . . . . . . . . . 6
4 Tipos de mutacoes de ponto em regioes codificantes do DNA. . . . . . . . 8
5 Diagrama de transicoes e transversoes . . . . . . . . . . . . . . . . . . . 8
6 Estatısticas de crescimento do GenBank . . . . . . . . . . . . . . . . . . 14
7 Projetos de sequenciamento genomico . . . . . . . . . . . . . . . . . . . 15
8 Diagrama do modelo de replicacao semiconservativa do DNA . . . . . . 17
9 Conceito de proximos vizinhos e calculo de ∆Gt para uma sequenciahipotetica com pareamentos canonicos . . . . . . . . . . . . . . . . . . . 19
10 Conceito de proximos vizinhos e calculo de ∆Gt para uma sequenciahipotetica com um mismatch interno . . . . . . . . . . . . . . . . . . . . 20
11 Constante elastica k em funcao da concentracao salina . . . . . . . . . . 25
12 Constante equivalente keq para regioes promotoras . . . . . . . . . . . . . 25
13 Diagrama representando os estados de deformacao de uma mola . . . . . 27
14 Mecanismo hipotetico para a formacao de um SNP . . . . . . . . . . . . 30
15 Exemplo de arquivo tipo texto de sequencia biologica em formato fasta 31
16 Representacao da notacao estabelecida . . . . . . . . . . . . . . . . . . . 33
17 Representacao do arquivo texto no formato summary . . . . . . . . . . . 36
18 fluxograma consolidado para as etapas do datamining . . . . . . . . . . . 38
19 Expansao do fluxograma - pre processamento dos dados . . . . . . . . . 39
20 Expansao do fluxograma - processamento dos dados . . . . . . . . . . . . 40
21 Distribuicao normalizada de SNPs em funcao da energia livre (∆G) nogenoma humano. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
X

22 Distribuicao normalizada de SNPs em funcao da energia livre (∆G) paraum organismo padrao com taxa de mutacao uniforme. . . . . . . . . . . . 45
23 Frequencia relativa da distribuicao de SNPs em funcao da energia livre(∆G) no genoma humano. . . . . . . . . . . . . . . . . . . . . . . . . . 45
24 Histograma da distribuicao de SNPs em funcao da flexibilidade no ge-noma humano. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
25 Histograna da distribuicao de SNPs em funcao da flexibilidade para umorganismo padrao com taxa de mutacao uniforme. . . . . . . . . . . . . . 46
26 Frequencia relativa da distribuicao de SNPs em funcao da flexibilidade(keq) no genoma humano. . . . . . . . . . . . . . . . . . . . . . . . . . . 46
27 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de H. sapiens. . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
28 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 22 dogenoma de H. sapiens. . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
29 Distribuicao relativa de SNPs em funcao de keq no cromossomo 1 do ge-noma de H. sapiens. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
30 Distribuicao relativa de SNPs em funcao de keq no cromossomo 22 dogenoma de H. sapiens. . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
31 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de B. taurus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
32 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo X dogenoma de B. taurus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
33 Distribuicao de SNPs de keq no cromossomo 1 do genoma de B. taurus. . 51
34 Distribuicao de SNPs de keq no cromossomo X do genoma B. taurus. . . . 51
35 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de Mus musculus. . . . . . . . . . . . . . . . . . . . . . . . . . 52
36 Distribuicao relativa de SNPs em funcao de keq no cromossomo 1 do ge-noma de Mus musculus. . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
37 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de G. gallus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
XI

38 Distribuicao relativa de SNPs em funcao de keq no cromossomo 1 do ge-noma de G. gallus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
39 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de D. rerio. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
40 Distribuicao relativa de SNPs em funcao de keq no cromossomo 1 do ge-noma de D. rerio. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
41 Distribuicao relativa de SNPs em funcao de ∆G no cromossomo 1 dogenoma de A. mellifera. . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
42 Distribuicao relativa de SNPs em funcao de keq no cromossomo 1 do ge-noma de A. mellifera. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
43 Distribuicao relativa de SNPs em funcao de ∆G do genoma de P. falcipa-
rum. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
44 Distribuicao relativa de SNPs em funcao de keq do genoma de P. falciparum. 60
45 Distribuicao relativa de SNPs em funcao de ∆G do genoma de O. sativa. 63
46 Distribuicao relativa de SNPs em funcao de keq do genoma de O. sativa. . 63
47 Distribuicao relativa de SNPs em funcao de ∆G do genoma de S. pyogenes. 65
48 Distribuicao relativa de SNPs em funcao de keq do genoma de S. pyogenes. 65
49 Distribuicao relativa de SNPs em funcao de ∆G oriundos de substituicoessinonimas na regiao codificante do genoma de H. sapiens. . . . . . . . . 68
50 Distribuicao relativa de SNPs em funcao de keq oriundos de substituicoessinonimas nas regioes codificantes do genoma de H. sapiens. . . . . . . . 68
51 Distribuicao relativa de SNPs em funcao de ∆G oriundos de substituicoesnao codificantes do genoma de H. sapiens. . . . . . . . . . . . . . . . . . 69
52 Distribuicao relativa de SNPs em funcao de keq oriundos de substituicoesnas regioes nao codificantes do genoma de H. sapiens. . . . . . . . . . . 69
53 Distribuicao relativa de SNPs em funcao de ∆G oriundos de substituicoesnao sinonimas nas regioes codificantes do genoma de H. sapiens. . . . . . 70
54 Distribuicao relativa de SNPs em funcao de keq oriundos de substituicoesnao sinonimas nas regioes codificantes do genoma de H. sapiens. . . . . . 70
XII

Lista de Tabelas
1 Classes de SNPs. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
2 Parametros termodinamicos para pareamentos canonicos de bases . . . . 20
3 Parametros termodinamicos para pareamentos nao-canonicos de bases . . 21
4 Parametros termodinamicos para pareamentos nao-canonicos de bases(continuacao) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
5 Parametros de flexibilidade para pareamentos nao-canonicos de bases . . 26
6 Parametros de flexibilidade para pareamentos canonicos de bases . . . . . 27
7 Codigo dos alelos de acordo com as normas IUPAC . . . . . . . . . . . . 32
8 Tabela de estatısticas dos genomas analisados . . . . . . . . . . . . . . . 35
9 Total de sequencias de SNPs para os 9 genomas. . . . . . . . . . . . . . . 35
XIII

1 Introducao
Em 01 julho de 1858 na reuniao da Sociedade de Linnean em Londres, CharlesDarwin propos sua teoria evolucionaria atraves da selecao natural que um ano depoislevou a publicacao de “A Origem das Especies”, obra prima que revolucionou os conheci-mentos biologicos da epoca. Apesar do brilhantismo da proposta um ponto de criticismona sua teoria foi a incapacidade dela explicar as vias de transferencia das informacoesbiologicas de uma geracao para outra.
Sete anos depois da publicacao de Darwin, Gregor Johann Mendel publicou o artigoentitulado “Versuche uber Pflanzenhybriden” (“Experimentos em hibridacao de plantas”),estabelecendo a existencia dos chamados “caracteres” elementares da hereditariedade eas leis estatısticas que governam suas transmissoes de uma geracao para a proxima. Napublicacao de Mendel a natureza dos “caracteres” da hereditariedade ainda permaneciadesconhecida contudo em 1869, Johann Friederich Miescher trabalhando com celulas pu-rulentas identificou uma substancia contendo fosfato que chamou de “nucleına” e em 1889este termo foi modificado para “acido nucleico” pelo bioquımico Richard Altmann (1).
No seculo seguinte, no ano de 1953, James Watson e Francis Crick (2, 3) juntaram ascontribuicoes cientıficas de Rosalind Franklin (4, 5) e de varios outros cientistas (6–8) emontaram o quebra-cabecas da estrutura helicoidal do DNA.
Apos ter sido desvendada a estrutura do DNA varios campos da ciencia evoluıramenormemente promovendo avancos tecnicos e metodologicos significantes em diversasareas. Isso tornou possıvel responder perguntas ate entao sem respostas, entre elas, asbases hereditarias da evolucao.
A consolidacao da “Era genomica”, biotecnologica e do sequenciamento de geno-mas completos culminaram dentre varios aspectos na escrita de um “livro da vida” ondeseu conteudo e similar a um manual de instrucoes que guarda uma infinita quantidade deinformacoes vitais documentadas nas sequencias de DNA. Sem duvida, os avancos tec-nologicos e computacionais promoveram e incentivaram a busca por novas informacoesviabilizando um acesso rapido e em uma escala nunca antes imaginada.
Como marcos desses desdobramentos podemos citar o sequenciamento do genomahumano e o surgimento e crescimento de importantes bancos de dados biologicos. Den-tre esses bancos o National Center for Biotechnology Information (NCBI) (9) exem-plifica bem essa historia. O NCBI e um banco de dados primario que armazena dife-rentes informacoes como genomas completos, estruturas cristalograficas, domınios, etc
1

em varias subdivisoes. Uma dessas subdivisoes e o dbSNP que armazena dados depolimorfismos de base unica, conhecidos tambem como SNPs (Single Nucleotide Poly-
morphisms).
No genoma humano por exemplo, foram mapeadas mais de um milhao de SNPs quetem sido utilizados em estudos chamados Genome-wide association studies (GWAS). Es-ses estudos avaliam regioes codificantes do genoma e partem da premissa que variacoesgeneticas frequentes na populacao podem explicar melhor algumas doencas hereditariascomuns como o cancer de mama, prostata, pele etc (10, 11).
Os SNPs sao eventos moleculares importantes no estudo da evolucao dos organismos.Cada genoma possui padroes de variacoes especıficos que compoem o seu historico evo-lutivo. A identificacao desses padroes contribui para o entendimento de como o processode selecao natural atuou no DNA de um dado organismo ao longo da sua evolucao. Ape-sar dos modelos teoricos de evolucao utilizarem a informacao relacionada aos padroes devariacoes especıficas e a correlacao entre variacao molecular, SNPs e selecao natural tersido estabelecida, varios aspectos do papel das propriedades micromecanicas do DNA naevolucao permanecem obscuras. Fica evidente que a integracao desse conhecimento podelevar a um melhor entendimento do papel de certas propriedades fısicas nos mecanismosde evolucao molecular (12).
Atualmente, pela viabilidade e baixo custo, os projetos de ressequenciamento temse mostrado uma estrategia viavel para a deteccao de polimorfismos em organismos deimportancia medica e economica. Sequenciadores de nova geracao capazes de gerarinformacoes em larga escala como os sequenciadores 454 (13) e Solexa/Illumina short
reads (14) representam marcos na geracao desses dados, pois possuem a capacidade degerar grandes quantidades de sequencias em um intervalo de tempo relativamente baixo ea um custo acessıvel. As informacoes geradas por esses equipamentos permitem compa-rar os nıveis de diversidade genomica inter e intra-especıfica viabilizando a estimativa davariabilidade de SNPs nos genomas (15–18).
Se por um lado tais tecnologias viabilizaram esse acesso rapido ao conteudo de ge-nomas, por outro lado houve o surgimento de um problema: o processamento dessegigantesco volume de dados. Analisar, armazenar, disponibilizar e converter toda essainformacao em conhecimento biologico continua sendo um grande desafio.
E nesse momento e contexto que ocorre o surgimento de uma area de conhecimento,a bioinformatica. Esta nova ciencia se estabelece como uma linha de pesquisa teorica,multidisciplinar que transita por diferentes disciplinas (biologia, fısica, quımica, cienciasda computacao, estatıstica, matematica, dentre outras), nos campos de desenvolvimento
2

biotecnologicos industriais de importancia economica (agricultura, pecuaria, engenharia,computacao) e de saude publica (biologia forense, medicina, farmacologia,etc).
Mais do que algoritmos de analise, a bioinformatica tem viabilizado e proporcionadoos meios para o entendimento da complexidade dos sistemas biologicos. Dentro dessecontexto, utilizando dados disponıveis no dbSNP e abordagens de bioinformatica, o pre-sente trabalho tem como proposta o estudo da influencia de propriedades micromecanicasdo DNA na distribuicao de polimorfismos nos genomas.
1.1 Objetivo geral do projeto
O objetivo geral do nosso projeto e analisar a influencia da flexibilidade e energialivre na distribuicao de SNPs em diferentes genomas utilizando um modelo teorico fısico-estatıstico para avaliar uma microrregiao do DNA contendo um erro de pareamento.
1.1.1 Objetivos especıficos
1. Avaliar a influencia da flexibilidade e da energia livre na distribuicao de SNPs emdiversos genomas
2. Utilizar o metodo de proximos vizinhos para avaliar microrregioes do DNA comrelacao aos parametros fısicos
3. Desenvolver uma metodologia que facilite e viabilize analises de dados de sequenciasde SNPs em larga escala
4. contribuir para uma melhor entendimento da relacao entre mecanismos fısicos e aperpetuacao de polimorfismos (SNPs) nos genomas.
Este trabalho esta organizado da seguinte forma: secao 2 apresentamos os conceitosbasicos de DNA e mutacoes relevantes para o nosso trabalho; na secao 3 apresentamosconseitos gerais de bancos de dados; na secao 4 discutimos os conceitos de energia livree flexibilidade de DNA; secao 5 descrevemos a metodologia utilizada para o desenvol-vimento deste trabalho; secao 6 apresentamos e discutimos nossos resultados; secao 7desenvolvemos nossas conclusoes; secao 8 apresentamos as perspectivas futuras seguidado apendice e das referencias bibliograficas utilizadas.
3

2 DNA e mutacoes
2.1 O Acido Desoxirribonucleico - DNA
O DNA e uma molecula quimicamente simples, composta por quatro bases nitroge-nadas: adenina(A), timina(T), guanina(G) e citosina(C), um grupo fosfato e uma pentoseconhecida como desoxirribose. Estes componentes quımicos quando agrupados sao cha-mados de nucleotıdeos. Esses nucleotıdeos possuem uma ligacao fosfodiester do fosfatocom a hidroxila do carbono-5’ da pentose e uma ligacao covalente N- glicosıdica entre ahidroxila do carbono-1’ da pentose e a base nitrogenada. Essas bases sao classificadas deacordo com a quantidade de aneis carbonicos que possuem, as bases puricas (purinas), Ae G, sao formadas por dois aneis carbonicos e as bases pirimidicas (pirimidinas), C e Tformadas por um unico anel (2, 19–22), veja figura 1.
Figura 1Os blocos fundamentais que compoem a cadeia de DNA sao chamados de nucleotıdeos. Essasmoleculas sao compostas pelas bases puricas (A e G) e pirimidicas (T e C), por um acucar chamadodesoxirribose e um grupamento fostato (2, 21, 22). Figura retirada do livro (21)
Para formar a dupla helice, as bases nitrogenadas pareiam-se de forma complemen-tar e antiparalela atraves das ligacoes de hidrogenio. A complementaridade de bases echamada tambem de pareamento Watson-Crick ou pareamento canonico. As interacoesocorrem entre uma base purica e uma pirimidica onde A interage com T atraves deduas ligacoes de hidrogenio e G interage com C interagem atraves de tres ligacoes dehidrogenio.
A replicacao do DNA e feita de forma ordenada, semiconservativa e antiparalela.Cada fita denominada parental (original) serve de molde para a sıntese de uma nova fita.Assim as duplas helices formadas sao compostas por uma fita parental e a nova fita (fitafilha) sintetizada e orientadas de forma antiparalela ou seja, em sentidos opostos. A estru-
4

tura antiparalela do DNA ocorre em funcao da atividade enzimatica da DNA-polimeraseIII (DNA-pol III) que insere nucleotıdeos nas fitas no sentido 3′ para 5′ da fita. Essadisposicao faz com que, durante o processo de sıntese, o grupo hidroxila do carbono-3′ dapentose de um determinado nucleotıdeo se ligue a grupo fosfato do carbono 5′ da pentosedo nucleotıdeo adjacente. Essa ligacao (ligacao fosfodiester) permite que a hidroxila docarbono-5′ esteja sempre livre para formar uma nova ligacao com o proximo nucleotıdeo.Dessa forma o esqueleto acucar-fosfato determina a sıntese da dupla fita na direcao de 5′
para 3′ (12, 23), veja figura 2.
Figura 2Diagrama quımico da estrutura doDNA. As setas em azul represen-tam o arcabouco de acucar-fostatoe na porcao central em azul escuroestao representados os pares de ba-ses interagindo por ligacoes de hi-drogenio. Na molecula bifilamentardo DNA os dois arcabucos estao emorientacao oposta (21, 22). Figuraretirada do livro (21)
De forma geral, a replicacao do DNA inicia-se em uma regiao da fita denominada“origem de replicacao”. Em eucariotos encontramos varias origens e os proacariotos saocompostos geralmente por apenas uma. Esta regiao e um sıtio de ligacao para o complexonucleoproteıco replissoma que forma a maquinaria de replicacao do DNA. Essa maquina-ria molecular inclui as DNA-polimerases, proteınas estabilizadoras como as girases, to-poisomerases, as single-strand-binding (SSB) e as proteınas acessorias. Assim, na origemde replicacao o DNA e desespiralizado e a dupla fita e separada formando a forquilha dereplicacao. A fita lıder (leading) e sintetizada no sentido 5′ para 3′ continuamente semprena mesma direcao da abertura da forquilha. Ja a fita descontınua (lagging) sera sintetizadana mesma orientacao 5′ para 3′, porem o seu crescimento ocorre por insercao de pequenos
5

segmentos de nucleotıdeos no sentido contrario a abertura da forquilha. Esses segmentosde aproximadamente 1000 a 2000 nucleotıdeos sao chamados de fragmentos de Okazaki
que serao unidos pelas enzimas do complexo conhecidas como ligases, que tem a funcaode catalizar as ligacoes fosfodiesteres entre os nucleotıdeos dos fragmentos formando umsegmento unico na fita descontınua (22), veja figura 3.
Figura 3Diagrama do modelo de replicacao semicon-servativa do DNA procarioto e seu complexoproteico replissomo. Figura retirada do li-vro (22).
2.2 Mutacoes
Um processo basico na evolucao de sequencias de DNA e a substituicao de um nu-cleotıdeo por outro durante o tempo evolucionario. Assim sendo, os estudos de evolucaomolecular tratam as mutacoes como documentos historicos que nos permitem entender oprocesso evolucionario de um dado organismo (1). A partir da identificacao da ocorrenciade polimorfismos em uma populacao e possıvel, por exemplo, estimar a taxa evolutiva,reconstruir a cronologia evolucionaria e identificar as forcas que dirigem esses processosnos seres vivos (24).
As mutacoes podem ocorrer por diversos processos tais como translocacao ou in-versao cromossomica, recombinacao genica, substituicoes, insercoes ou delecoes de ba-ses, dentre outros. Esses danos podem ser causados por fatores exogenos ou externos(radiacoes, produtos quımicos) ou por fatores endogenos como erro de replicacao doDNA, danos causados por enzimas, falha da maquinaria de reparo, transposicao, alquilacao,
6

desaminacao, bases tautomericas, oxidacao, podendo ainda ocorrer de forma natural ouinduzida (25).
Vale ainda ressaltar que as sequencias de DNA sao normalmente copiadas com exa-tidao durante o processo de replicacao e essa acuracia esta associada a um eficiente sis-tema de reparo que utiliza um complexo de aproximadamente cem proteınas que desem-penham o papel de monitorar e reparar danos identificados nas moleculas de DNA. Essecomplexo proteico e tao eficiente que e capaz de detectar pequenos danos no DNA emmilhoes ou ate bilhoes de bases (20, 22).
Por outro lado, apesar da eficiencia da maquinaria de reparo nem todos os erros depareamento sao corrigidos. Alem disso, muitas vezes ao inves de reparar, o proprio meca-nismo de correcao pode inserir erros como pareamentos imperfeitos tambem conhecidoscomo mismatches. Apesar de parecerem deleterios esses erros de pareamentos podeminclusive resultar no ganho de uma determinada caracterıstica que seja importante para asobrevivencia de um organismo (26).
Apesar de pouco frequentes, as mutacoes de ponto (substituicao) tambem conhe-cidas como SNPs (Single Nucleotide Polymorphism) sao bem toleradas pela celula eresponsaveis por caracterısticas fenotıpicas individuais importantes como a propensaoa doencas do coracao, cancer, resistencia a certas drogas, etc (1, 26). Como os SNPspodem ocorrer aleatoriamente nos diferentes genomas, regioes codificantes e nao codifi-cantes podem ser afetadas. Quando a insercao de um SNP acontece em uma regiao codi-ficante temos: a) mutacoes silenciosas quando o aminoacido nao e alterado; b) mutacoesmissense conservativas quando o carater fısico-quımico do aminoacido mutado perma-nece o mesmo; c) mutacoes missense nao conservativas quando a substituicao resulta naalteracao do aminoacido; e d) non-sense quando a substituicao resulta em um codon deparada (22). Veja a figura 4.
Os SNPs podem ainda ser divididos em dois grandes grupos: transicao e transversao.A transicao representa a substituicao de uma base por outra da mesma categoria quımica,ou seja, acontece a substituicao de uma purina por outra purina (A � G) ou de umapirimidina por outra pirimidina (C � T). Ja as transversoes representam substituicoes debases de categoria quımica diferente. Tanto uma pirimidina pode ser substituıda por umapurina (C � A, C � G, T � A ou T � G) quanto uma purina pode ser substituıda poruma pirimidina (A � C, A � T, G � C, ou G � T) (22, 26), veja figura 5 e tabela 1.
7
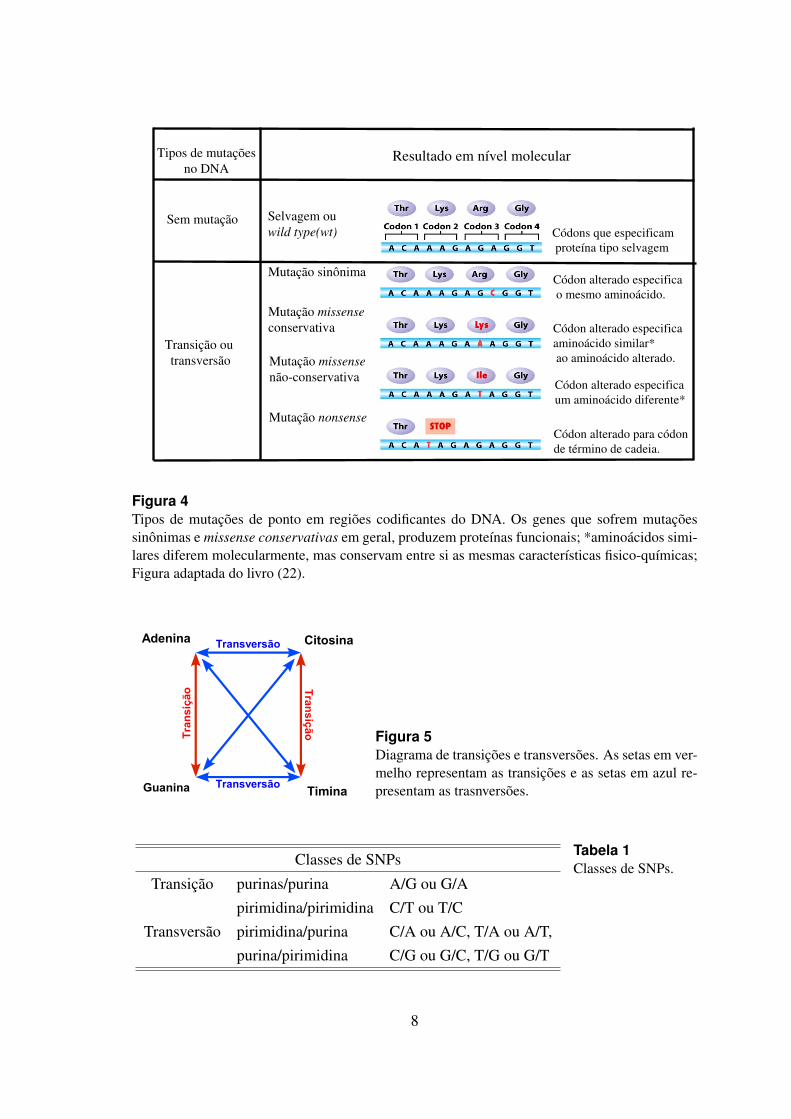
Figura 4Tipos de mutacoes de ponto em regioes codificantes do DNA. Os genes que sofrem mutacoessinonimas e missense conservativas em geral, produzem proteınas funcionais; *aminoacidos simi-lares diferem molecularmente, mas conservam entre si as mesmas caracterısticas fisico-quımicas;Figura adaptada do livro (22).
Figura 5Diagrama de transicoes e transversoes. As setas em ver-melho representam as transicoes e as setas em azul re-presentam as trasnversoes.
Classes de SNPs
Transicao purinas/purina A/G ou G/Apirimidina/pirimidina C/T ou T/C
Transversao pirimidina/purina C/A ou A/C, T/A ou A/T,purina/pirimidina C/G ou G/C, T/G ou G/T
Tabela 1Classes de SNPs.
8

2.2.1 Polimorfismo de base unica ou SNPs (Single Nucleotide Polymor-phism)
Como citado na secao anterior 2.2, o processo basico na evolucao das sequencias deDNA e a ocorrencia de mutacoes que produzem variacoes geneticas individuais, diversi-dade em uma comunidade e a evolucao das especies ao longo do tempo (27). Esse pro-cesso e lento, raro, afeta aproximadamente 1% do genoma podendo se fixar na populacaocomo um novo alelo.
A princıpio, cada base (A, T, G ou C) pode ser substituıda por um dos outros tresdiferentes nucleotıdeos, por exemplo, uma adenina (A) pode ser substituıda por uma ti-mina (T), guanina (G) ou citosina (C). Essas substituicoes levam a um erro de parea-mento tambem conhecido como mismatch que nao sendo corrigido pode levar, ao longodo tempo, ao surgimento de um SNP caso esta mutacao ocorra em celulas germinativas.Esses eventos de substituicao sao pouco frequentes, em Escherichia coli e Saccharomy-
ces cerevisiae por exemplo, a taxa mutacional estimada e de 10× 10−10 e 3.3× 10−9 porsıtio de nucleotıdeo a cada geracao, respectivamente (28, 29). Em organismos superio-res como Caenorhabditis elegans, Drosophila melanogaster e humanos a taxa estimadae de 2.1 × 10−9, 5 × 10−9 e 2 × 1010−8 por sıtio de nucleotıdeo substituıdo, respectiva-mente (30–33).
As mutacoes por substituicao podem ser divididas em duas classes: transicao e trans-versao. Essas classes possuem proporcoes diferentes relacionadas ao numero de possibi-lidades de trocas de bases, existindo quatro tipos transicoes e oito tipos de transversoes,veja tabela 1.
Esse numero de possibilidades diferentes acarreta um vies. Nos metazoarios, porexemplo, a cada transversao ocorrem duas transicoes. Uma explicacao para esse vies estaassociado a ocorrencia de altas taxas de mutacao dos resıduos de citosina (5-metilcitosina)para resıduos de timina que acontece pela perda do grupo amina do carbono 5′ do anelaromatico da base (27, 30–35). Essas mutacoes no dinucleotıdeo CpG representam apro-ximadamente 25% dos SNPs, assim sendo desvios de composicao de sequencia devemser considerados como fatores do processo mutacional (1, 35–37).
Apesar da “universalidade” da razao 1/3 de transversao para 2/3 de transicao, es-tudos atraves da exclusao de mutacoes associadas a metilacao realizados por Keller et
al. (38) no genoma de gafanhoto Podisma pedestris (genoma 100 vezes maior que ogenoma de Drosophila) demonstraram que as transicoes nao prevalecem em relacao astransversoes (27, 38–42).
9

A composicao de sequencias das regioes que flanqueiam os SNPs (neighboring nu-
cleotides NN) tambem possui uma forte influencia nas taxas e nos tipos de eventos mu-tacionais (36, 43–45). Gorjorobi et al. (46) e Majewski et al. (47) demonstraram que astaxas de mutacao por substituicao sao especıficas para cada tipo de nucleotıdeo. Alemdisso a composicao de bases nas regioes nao codificantes e nas “ilhas” CpGs do DNAnuclear contribuem diretamente com o aumento do vies que favorece as transicoes nosgenomas de vertebrados (43, 48).
Como visto, a maioria dos estudos relacionados a ocorrencia dos SNPs associam-seas modificacoes nas moleculas que compoem o DNA. Uma outra vertente, nao menosimportante, mas pouco explorada e o estudo dos parametros fısicos do DNA tais comoenergia livre e flexibilidade.
Tal importancia esta relacionada ao fato da flexibilidade e da energia livre das intera-coes das bases que flanqueiam os SNPs influenciarem a ocorrencia do evento mutacional.Interacoes intermoleculares como de DNA-proteınas, por exemplo, sao diretamente de-pendentes da dinamica fısica estrutural do DNA.
2.2.2 Causas da variacao nas taxas de substituicao dos SNPs
Para inferir as causas que definem a variacao das taxas de substituicao entre as dife-rentes regioes da molecula de DNA, devemos considerar dois fatores: a) a taxa de mutacaoe b) a probabilidade de fixacao da mutacao. Esse ultimo depende se a mutacao e vantajosa,neutra ou deleteria. Uma vez que a taxa de mutacao provavelmente nao varia dentro deum gene mas pode variar entre os genes do genoma, devemos discutir a taxa de variacaoentre diferentes regioes do gene e a variacao entre genes separadamente(1, 49).
A intensidade da selecao purificadora e determinada pelo grau de intolerancia desubstituicao de um sıtio no genoma. Essa pressao seletiva define o variacao de nu-cleotıdeos alternativos aceitaveis em um dado sıtio sem afetar negativamente a funcaoou estrutura do gene ou de seu produto (50). Regioes genomicas como por exemploregioes codificadoras de proteinas ou sequencias regulatorias, nas quais a mutacao pro-vavelmente afetara a funcao sofrem uma pressao seletiva mais estringente do que regioesnao funcionais do genoma (1).
Por outro lado a probabilidade de uma alelo particular se tornar fixo (probabilidadede fixacao da mutacao) em uma populacao depende da sua frequencia, da sua vantagemou desvantagem seletiva e do tamanho efetivo da populacao. Complementarmente, outrosfatores podem influenciar a ocorrencia das diferentes taxas de substituicao dos SNPs.
10

Pareamentos nao canonicos, por exemplo, podem ocorrer quando uma base e ionizadae tambem quando as bases apresentam tautomeria. A forma ceto e a predominante namolecula de DNA, contudo as formas imino e enol apesar de raras tambem podem serencontradas e sao decorrentes do deslocamento transitorio das ligacoes quımicas (22).
Alem dos pontos levantados acima, fatores mecanicos como uma pequena distorcaona helice do DNA pode levar a um erro de emparelhamento de um G � T por exemploque apesar de energeticamente desfavoravel tem sido constatado em estudos de difracaode raios X (49).
2.2.3 Taxas de mutacoes
Um processo basico na evolucao de sequencias de DNA e a substituicao de um nu-cleotıdeo por outro durante o tempo evolucionario. Esse processo merece uma consideracaodetalhada porque a mudanca nas sequencias de nucleotıdeos sao usadas no estudos deevolucao molecular tanto para estimar a taxa de evolucao bem como para reconstruir ahistoria evolucionaria de um organismo. Esse processo e normalmente lento e dessa formanao podem ser observados diretamente sendo necessaria a comparacao de sequencias quepossuam um ancestral comum. Tais comparacoes sao realizadas atraves de diferentesmetodos estatısticos.
Nesse contexto, varios modelos evolucionarios para estimar essas taxas foram pro-postos e aprimorados. Os primeiros modelos de substituicao do DNA foram propostosem 1969 por Jukes and Cantor (51). No seu modelo bastante simples tambem chamadode “modelo de um unico parametro” assume-se que as substituicoes ocorrem em igualprobabilidade entre os quatro tipos de nucleotıdeos. Em outras palavras nao existe umvies na direcao da mudanca.
Como o pressuposto de que as substituicoes de nucleotıdeos acontecem em proporcoesequivalentes e discutıvel, dez anos depois, Kimura (50) propos o “modelo de dois para-metros”. Esse modelo assume a existencia de diferentes taxas de mutacao para as trans-versoes e transicoes, sendo esta ultima a mais frequente.
Posteriormente varios outros metodos implementando diferentes modelos de substi-tuicao foram propostos e/ou refinados como por exemplo o algorıtimo de Felsenstein et
al. (52) que emprega o metodo verossimilhanca (Likelihood ratio tests) para estimaras taxas mutacionais e os modelos de evolucao neutra de Hasegawa, Kishino e Yano(HKY) (53) que viabilizaram a adaptacao de diferentes parametros a especificidade decada organismo (39, 54, 55).
11

Adicionalmente, diferentes abordagens e modelos evolutivos implementados em va-rios algoritmos consideram a microrregiao onde ocorre o SNP. Estes modelos nao tratamSNPs como eventos independentes que ocorrem de forma pontual e isolada no DNA umavez que ja foi comprovado que as bases adjacentes ao SNP tem uma forte influencia notipo e na taxa de eventos mutacionais que ocorrem em uma dada posicao da molecula (36,44, 56).
12

3 Banco de dados de sequencias biologicas
A bioinformatica tem desempenhado um papel mais que significante no estudo dabiologia moderna. Atualmente e impensavel a idealizacao de projetos de pesquisa ouexperimentos sem a consulta previa ou busca em alguns bancos de dados. Nesse contexto,a bioinformatica tem fornecido inumeros bancos de dados e ferramentas que ajudam avida dos pesquisadores (57, 58). Por outro lado, os dados oriundos de tecnologias dealta vazao (high throughput data) somente podem ser manipulados com algum tipo depipeline de analise que inclua metodologias que viabilizem a descoberta do conhecimentobiologico encoberto pelo grande volume de dados (57, 59).
Como a utilizacao dos termos bioinformatica e biologia computacional sao utilizadoscomo “sinonimos” faz-se necessario uma definicao. Bioinformatica e atualmente o termomais utilizado quando se refere a aplicacao de metodos computacionais e analıticos pararesolucao de um dado problema biologico. A bioinformatica se refere especificamente abusca e a utilizacao de padroes e da estrutura de bancos de dados tais como sequenciasgenomicas, bem como ao desenvolvimento de novas metodologias de acesso e extracaode informacao dos bancos de dados (a definicao completa de bioinformatica feita peloNCBI pode ser encontrada nas referencias 57 e 60).
O termo biologia computacional e mais frequentemente utilizado quando se refere asimulacao matematica e fısica de processos biologicos. Apesar disso, com o desenvolvi-mento ininterrupto de novos metodos de analise de dados de alta vazao, a linha divisoriaentre essas duas disciplinas tem se tornado tenue. Nos ultimos 50 anos, o conjunto dedados biologicos disponıveis cresceu exponencialmente (62), veja figura 6.
Projetos genoma de inumeros organismos diferentes tem produzido uma gigantescaquantidade de sequencias, veja figura 7, e nesse cenario o recurso chave e o conhecimentoe a tecnologia para a manipulacao da informacao. Os bancos de dados biologicos podemser divididos em quatro categorias gerais relacionadas a origem dos dados que contem:
1. Banco de dados primarios: contem essencialmente um tipo de informacao, porexemplo, dados de sequencia que podem ter sua origem de inumeras fontes comoprojetos de sequenciamento, submissao individual, literatura e outros bancos dedados;
2. Banco de dados secundarios: contem essencialmente dados derivados da analise deoutros conjuntos de dados, por exemplo, dados de alinhamentos de sequencias;
13
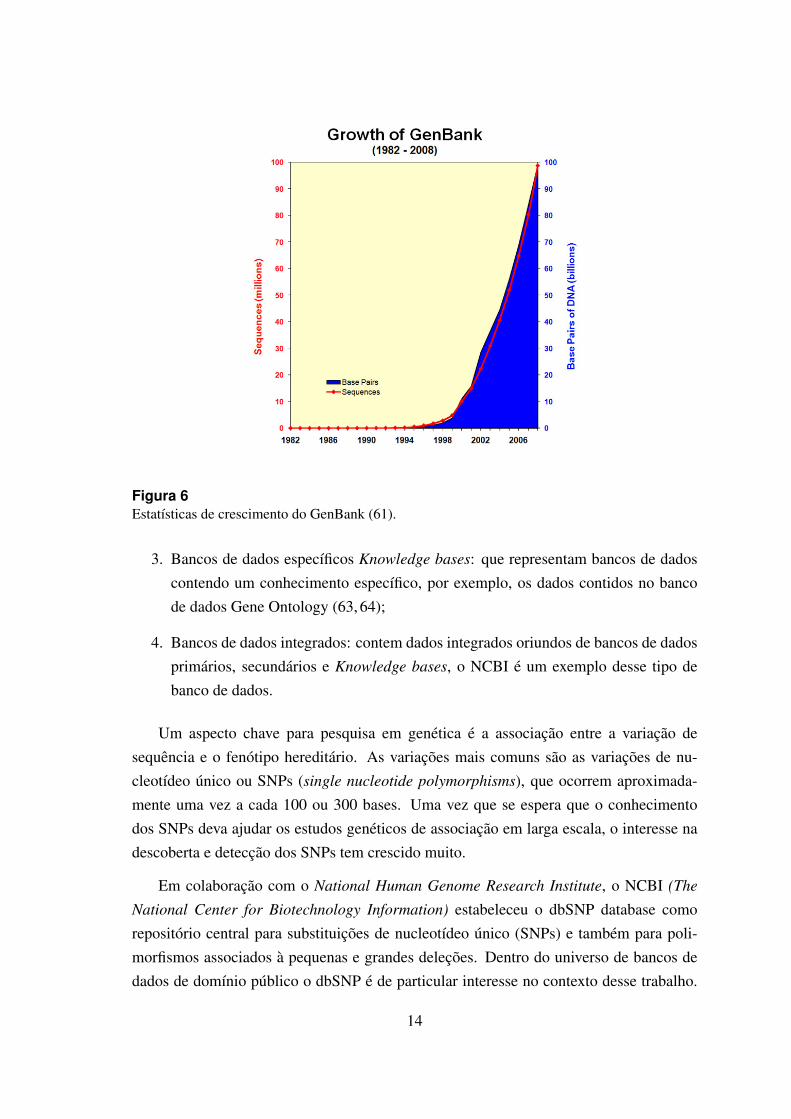
Figura 6Estatısticas de crescimento do GenBank (61).
3. Bancos de dados especıficos Knowledge bases: que representam bancos de dadoscontendo um conhecimento especıfico, por exemplo, os dados contidos no bancode dados Gene Ontology (63, 64);
4. Bancos de dados integrados: contem dados integrados oriundos de bancos de dadosprimarios, secundarios e Knowledge bases, o NCBI e um exemplo desse tipo debanco de dados.
Um aspecto chave para pesquisa em genetica e a associacao entre a variacao desequencia e o fenotipo hereditario. As variacoes mais comuns sao as variacoes de nu-cleotıdeo unico ou SNPs (single nucleotide polymorphisms), que ocorrem aproximada-mente uma vez a cada 100 ou 300 bases. Uma vez que se espera que o conhecimentodos SNPs deva ajudar os estudos geneticos de associacao em larga escala, o interesse nadescoberta e deteccao dos SNPs tem crescido muito.
Em colaboracao com o National Human Genome Research Institute, o NCBI (The
National Center for Biotechnology Information) estabeleceu o dbSNP database comorepositorio central para substituicoes de nucleotıdeo unico (SNPs) e tambem para poli-morfismos associados a pequenas e grandes delecoes. Dentro do universo de bancos dedados de domınio publico o dbSNP e de particular interesse no contexto desse trabalho.
14
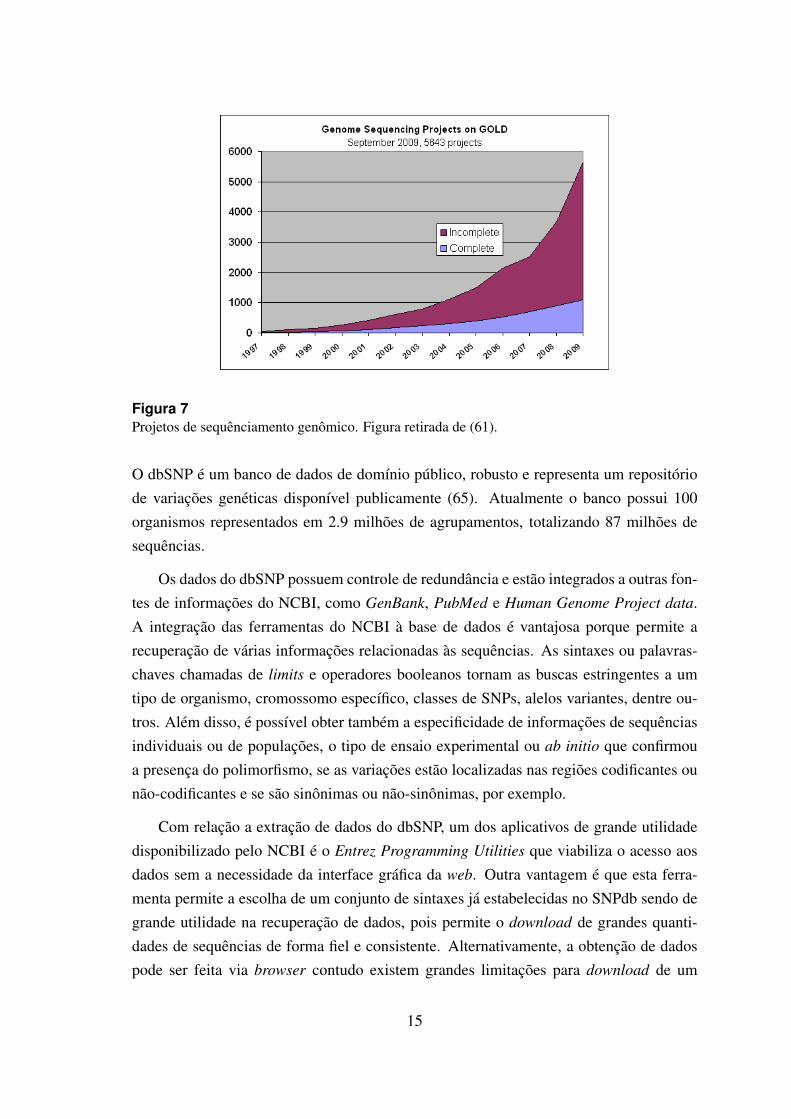
Figura 7Projetos de sequenciamento genomico. Figura retirada de (61).
O dbSNP e um banco de dados de domınio publico, robusto e representa um repositoriode variacoes geneticas disponıvel publicamente (65). Atualmente o banco possui 100organismos representados em 2.9 milhoes de agrupamentos, totalizando 87 milhoes desequencias.
Os dados do dbSNP possuem controle de redundancia e estao integrados a outras fon-tes de informacoes do NCBI, como GenBank, PubMed e Human Genome Project data.A integracao das ferramentas do NCBI a base de dados e vantajosa porque permite arecuperacao de varias informacoes relacionadas as sequencias. As sintaxes ou palavras-chaves chamadas de limits e operadores booleanos tornam as buscas estringentes a umtipo de organismo, cromossomo especıfico, classes de SNPs, alelos variantes, dentre ou-tros. Alem disso, e possıvel obter tambem a especificidade de informacoes de sequenciasindividuais ou de populacoes, o tipo de ensaio experimental ou ab initio que confirmoua presenca do polimorfismo, se as variacoes estao localizadas nas regioes codificantes ounao-codificantes e se sao sinonimas ou nao-sinonimas, por exemplo.
Com relacao a extracao de dados do dbSNP, um dos aplicativos de grande utilidadedisponibilizado pelo NCBI e o Entrez Programming Utilities que viabiliza o acesso aosdados sem a necessidade da interface grafica da web. Outra vantagem e que esta ferra-menta permite a escolha de um conjunto de sintaxes ja estabelecidas no SNPdb sendo degrande utilidade na recuperacao de dados, pois permite o download de grandes quanti-dades de sequencias de forma fiel e consistente. Alternativamente, a obtencao de dadospode ser feita via browser contudo existem grandes limitacoes para download de um
15

conjunto de dados muito extenso pois o carregamento das informacoes na pagina ficacomprometido e geralmente sao recuperadas de forma incompleta. Juntamente com umadocumentacao completa composta por FAQ (Frequently Asked Questions, ftp site e tuto-riais), essa base conta tambem com a disponibilidade de uma variedade de formatos dearquivos de sequencias flexıveis como fasta muito comum para dados de sequenciasbiologicas (veja figura 15, pagina 31).
Alem do dbSNP, o MutaDATABASE (66) tambem de domınio publico, propoe cen-tralizar e padronizar as informacoes relacionadas as variacoes do genoma humano tor-nando-se uma base de dados de referencia universal. Esse banco de dados biologicoesta sendo construıdo por um grande consorcio de laboratorios de diagnostico de doencasgeneticas humanas da Europa, Estados Unidos, Australia e Asia que pretendem disponi-bilizar um rico repositorio de variacoes do DNA com elevado grau de curadoria para todaa comunidade cientıfica.
16

4 A fısica do DNA
4.1 Energia livre
A dupla fita do DNA e formada por dois grandes polımeros de nucleotıdeos que in-teragem pelas ligacoes covalentes do esqueleto acucar-fosfato direcionando as ligacoesquımicas da estrutura e as ligacoes de hidrogenio entre as bases complementares que es-tabilizam e formam a helice dupla (12), veja figura 8.
Figura 8Diagrama do modelo de replicacao semicon-servativa do DNA (23). Figura retirada do li-vro (22).
Essa estabilidade pode ser medida atraves de parametros termodinamicos utilizadospara avaliar o comportamento e a estabilidade do DNA submetido a variacoes de tempera-tura e nesse contexto a temperatura de denaturacao (melting-Tm) e comumente utilizada.Essa medida e definida experimentalmente como a temperatura na qual metade das fitasde DNA em solucao estao em estado de dupla helice e a outra metade no estado de fitaunica (67–69). A temperatura de denaturacao e dependente do tamanho da molecula, dacomposicao da sequencia de nucleotıdeos e da concentracao salina da solucao. O em-prego da temperatura melting e importante por exemplo na avaliacao da eficiencia de uma
17

sonda para microarranjos uma vez que esse parametro potencializa a hibridacao da sondacom a amostra (template) e reduz o numero de falsos positivos e negativos melhorando aqualidade dos resultados (70).
Tecnicas experimentais sao aplicadas para medir a temperatura de melting do DNA,inclusive de suas regioes polimorficas. Como exemplo podemos citar a espectrofotome-tria e a calorimetria (67, 71, 72). Apesar de eficazes essas tecnicas possuem limitacoesassociadas a quantidade de amostras processadas e ao fato de serem laboriosas. Por ou-tro lado, modelos teoricos relativamente simples e estabelecidos ha pelo menos 30 anostem se mostrado eficientes para estimar a temperatura de hibridacao das sequencias deDNA (68, 73, 74).
Parametros termodinamicos como variacoes da entalpia (∆H), entropia (∆S) e ener-gia livre (∆G) na transicao da temperatura de hibridacao de uma microrregiao do DNAsao amplamente utilizados para calcular a estabilidade de uma pequena sequencia de oli-gonucleotıdios. Atualmente, atraves dos parametros termodinamicos e possıvel estimar aestabilidade de um genoma completo ou uma microrregiao como um duplexo (dois paresde bases) de nucleotıdeos (68). A energia livre de Gibbs (∆G) e o indicador da esponta-neidade de uma reacao quımica. Para que uma reacao ocorra de forma espontanea o ∆G
deve ser negativo o que implica que a energia livre dos produtos deve ser menor que a dosreagentes.
Esta medida e utilizada para estimar a temperatura de denaturacao do DNA em que avariacao do ∆G e dada pela equacao:
∆G = ∆H − T∆S (1)
onde ∆H e a variacao da entalpia, ∆S a variacao da entropia e T a temperatura abso-luta (75). A partir da equacao 4.1 e de medidas experimentais de calorimetria, pode-secalcular ∆H e ∆S e ajustar os modelos termodinamicos para predicao da estabilidade eda temperatura de denaturacao (Tm) da dupla fita de DNA. A temperatura de melting deuma sequencia pode entao ser estimada pela equacao:
Tm =∆H
∆S +R ln(CT/C0)(2)
onde R e a constante dos gases ideais 1,987 cal/(K ·mol) e CT e a concentracao de DNAna solucao em mol/L (C0 = 10× 10−6 mol/L).
Fica assim evidente as diferentes aplicacoes dos calculos dessas propriedades termo-
18

dinamicas que passam por predicoes de estabilidade para uma sonda de um gene com-plexo, pela selecao de condicoes otimas para hibridacao, pelo ajuste de tamanho mınimode uma sonda e pela predicao da influencia das transicoes e transversoes na estabilidadede uma microrregiao do DNA (76, 77).
Ainda dentro do contexto de avaliacao de propriedades termodinamicas, os modelosque adotam o metodo de proximos vizinhos (nearest-neighbor – NN) tem se mostradoeficientes e adequados para predicao da estabilidade e da termodinamica dos pareamen-tos canonicos (match) da dupla fita do DNA. Este modelo aplicado em acidos nucleicosassume que a estabilidade de um dado par de base e dependente da composicao e daorientacao dos pares de bases vizinhos ou adjacentes (78).
Considerando as 10 possıveis interacoes canonicas entre um par de base e outro ime-diatamente adjacente, temos: AA/TT; AT/TA; TA/AT; CA/GT; GT/CA; CT/GA; GA/CT;CG/GC; GC/CG; GG/CC em que a barra separa a orientacao dos dımeros antiparalelosdo duplexo, por exemplo, o duplexo AA/TT representa o dımero 5′ −AA− 3′ e o dımero3′ − TT − 5′. Assim e possıvel estimar a estabilidade da estrutura e a temperatura dedenaturacao a partir da soma das interacoes dos seus proximos vizinhos (76–80). A fi-gura 9 esquematiza uma sequencia contendo 5 pb com os valores de ∆G obtidos a partirda tabela 2. O ∆G e calculado para cada duplexo (1 pb e seu NN). Desta forma, obtemosa energia livre total da sequencia atraves do somatorio dos ∆G de cada interacao.
Figura 9Conceito de proximos vizinhos e calculo de ∆Gt para umasequencia hipotetica de 5 pb com pareamentos canonicos; conside-rando uma janela de leitura de 2 pb e uma janela deslizante de 1 pbda esquerda para a direita, os asteriscos representam as ligacoes dehidrogenio entre as bases complementares; as setas representam osvalores de ∆G em kcal/mol para cada duplexo; ∆Gt e dado pelasoma dos ∆G entre a interacao de 1pb e seu NN (76).
4.1.1 A interferencia dos mismatches e os proximos vizinhos na estabili-dade do DNA
Para o estudo da estabilidade de sequencias polimorficas, devemos considerar que aocorrencia de um SNP tem como ponto de partida a existencia de erro de incorporacao debase ou de um mismatch na dupla fita. Este erro, se nao reparado, ira perpetuar-se a partirde uma proxima replicacao do DNA, podendo fixar-se na populacao na forma de um SNP.
19
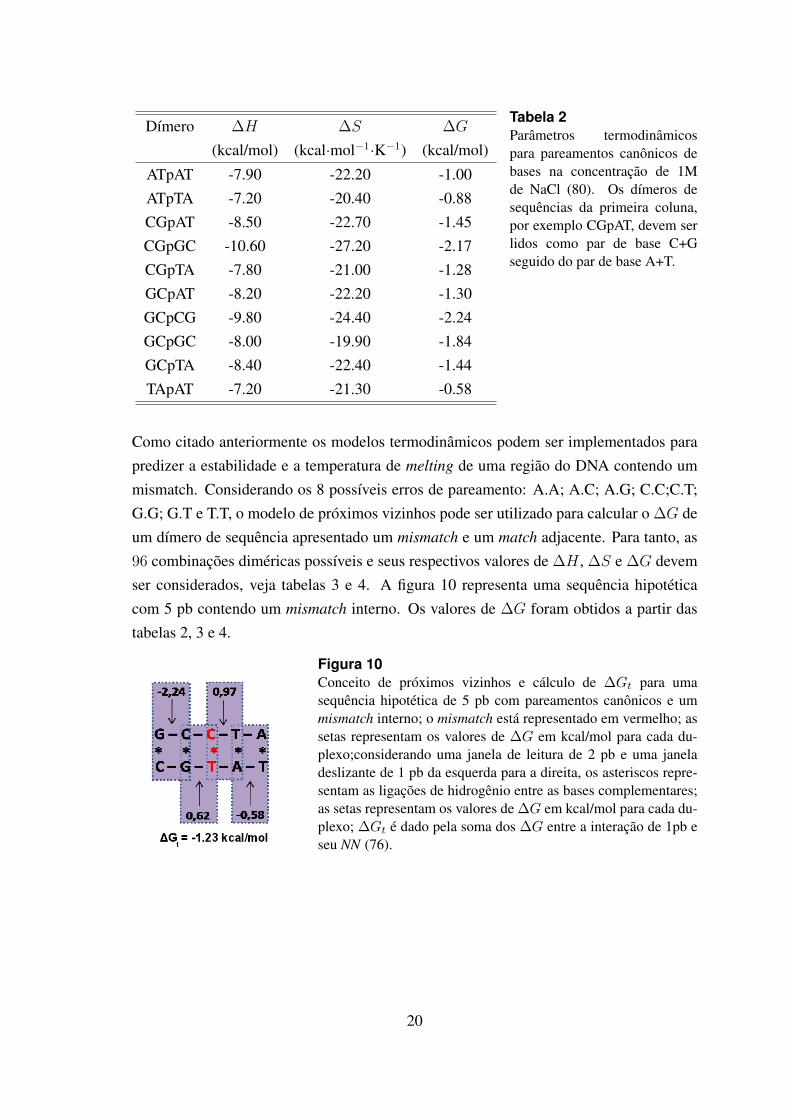
Dımero ∆H ∆S ∆G
(kcal/mol) (kcal·mol−1·K−1) (kcal/mol)
ATpAT -7.90 -22.20 -1.00ATpTA -7.20 -20.40 -0.88CGpAT -8.50 -22.70 -1.45CGpGC -10.60 -27.20 -2.17CGpTA -7.80 -21.00 -1.28GCpAT -8.20 -22.20 -1.30GCpCG -9.80 -24.40 -2.24GCpGC -8.00 -19.90 -1.84GCpTA -8.40 -22.40 -1.44TApAT -7.20 -21.30 -0.58
Tabela 2Parametros termodinamicospara pareamentos canonicos debases na concentracao de 1Mde NaCl (80). Os dımeros desequencias da primeira coluna,por exemplo CGpAT, devem serlidos como par de base C+Gseguido do par de base A+T.
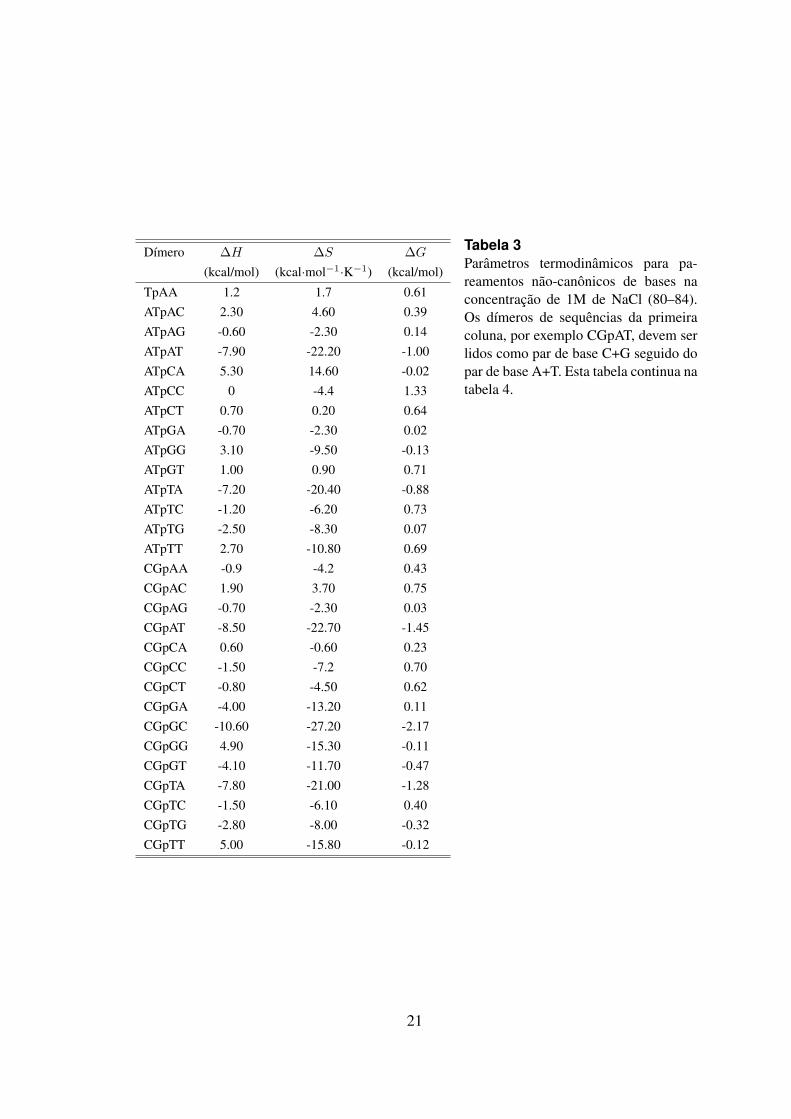
Como citado anteriormente os modelos termodinamicos podem ser implementados parapredizer a estabilidade e a temperatura de melting de uma regiao do DNA contendo ummismatch. Considerando os 8 possıveis erros de pareamento: A.A; A.C; A.G; C.C;C.T;G.G; G.T e T.T, o modelo de proximos vizinhos pode ser utilizado para calcular o ∆G deum dımero de sequencia apresentado um mismatch e um match adjacente. Para tanto, as96 combinacoes dimericas possıveis e seus respectivos valores de ∆H , ∆S e ∆G devemser considerados, veja tabelas 3 e 4. A figura 10 representa uma sequencia hipoteticacom 5 pb contendo um mismatch interno. Os valores de ∆G foram obtidos a partir dastabelas 2, 3 e 4.
Figura 10Conceito de proximos vizinhos e calculo de ∆Gt para umasequencia hipotetica de 5 pb com pareamentos canonicos e ummismatch interno; o mismatch esta representado em vermelho; assetas representam os valores de ∆G em kcal/mol para cada du-plexo;considerando uma janela de leitura de 2 pb e uma janeladeslizante de 1 pb da esquerda para a direita, os asteriscos repre-sentam as ligacoes de hidrogenio entre as bases complementares;as setas representam os valores de ∆G em kcal/mol para cada du-plexo; ∆Gt e dado pela soma dos ∆G entre a interacao de 1pb eseu NN (76).
20
Dımero ∆H ∆S ∆G
(kcal/mol) (kcal·mol−1·K−1) (kcal/mol)
TpAA 1.2 1.7 0.61ATpAC 2.30 4.60 0.39ATpAG -0.60 -2.30 0.14ATpAT -7.90 -22.20 -1.00ATpCA 5.30 14.60 -0.02ATpCC 0 -4.4 1.33ATpCT 0.70 0.20 0.64ATpGA -0.70 -2.30 0.02ATpGG 3.10 -9.50 -0.13ATpGT 1.00 0.90 0.71ATpTA -7.20 -20.40 -0.88ATpTC -1.20 -6.20 0.73ATpTG -2.50 -8.30 0.07ATpTT 2.70 -10.80 0.69CGpAA -0.9 -4.2 0.43CGpAC 1.90 3.70 0.75CGpAG -0.70 -2.30 0.03CGpAT -8.50 -22.70 -1.45CGpCA 0.60 -0.60 0.23CGpCC -1.50 -7.2 0.70CGpCT -0.80 -4.50 0.62CGpGA -4.00 -13.20 0.11CGpGC -10.60 -27.20 -2.17CGpGG 4.90 -15.30 -0.11CGpGT -4.10 -11.70 -0.47CGpTA -7.80 -21.00 -1.28CGpTC -1.50 -6.10 0.40CGpTG -2.80 -8.00 -0.32CGpTT 5.00 -15.80 -0.12
Tabela 3Parametros termodinamicos para pa-reamentos nao-canonicos de bases naconcentracao de 1M de NaCl (80–84).Os dımeros de sequencias da primeiracoluna, por exemplo CGpAT, devem serlidos como par de base C+G seguido dopar de base A+T. Esta tabela continua natabela 4.
21
Dımero ∆H ∆S ∆G
(kcal/mol) (kcal·mol−1·K−1) (kcal/mol)
GCpAA -2.9 -9.8 0.17GCpAC 5.20 14.20 -0.10GCpAG -0.60 -1.00 -0.25GCpAT -8.20 -22.20 -1.30GCpCA -0.70 -3.80 -0.33GCpCC 3.60 8.90 0.79GCpCG -9.80 -24.40 -2.24GCpCT 2.30 5.40 0.62GCpGA 0.50 3.20 -0.52GCpGC -8.00 -19.90 -1.84GCpGG 6.00 -15.80 -1.11GCpGT 3.30 10.40 0.08GCpTA -8.40 -22.40 -1.44GCpTC 5.20 13.50 0.98GCpTG -4.40 -12.30 -0.59GCpTT 2.20 -8.40 0.45TApAA 4.7 12.9 0.69TApAC 3.40 8.00 0.26TApAG 0.70 0.70 0.42TApAT -7.20 -21.30 -0.58TApCA 7.60 20.20 1.33TApCC 6.10 16.40 1.05TApCT 1.20 0.70 0.97TApGA 3.00 7.40 0.74TApGG 1.60 3.60 0.44TApGT -0.10 -1.70 0.43TApTC 1.00 0.70 0.75TApTG -1.30 -5.30 0.34TApTT 0.20 -1.50 0.68
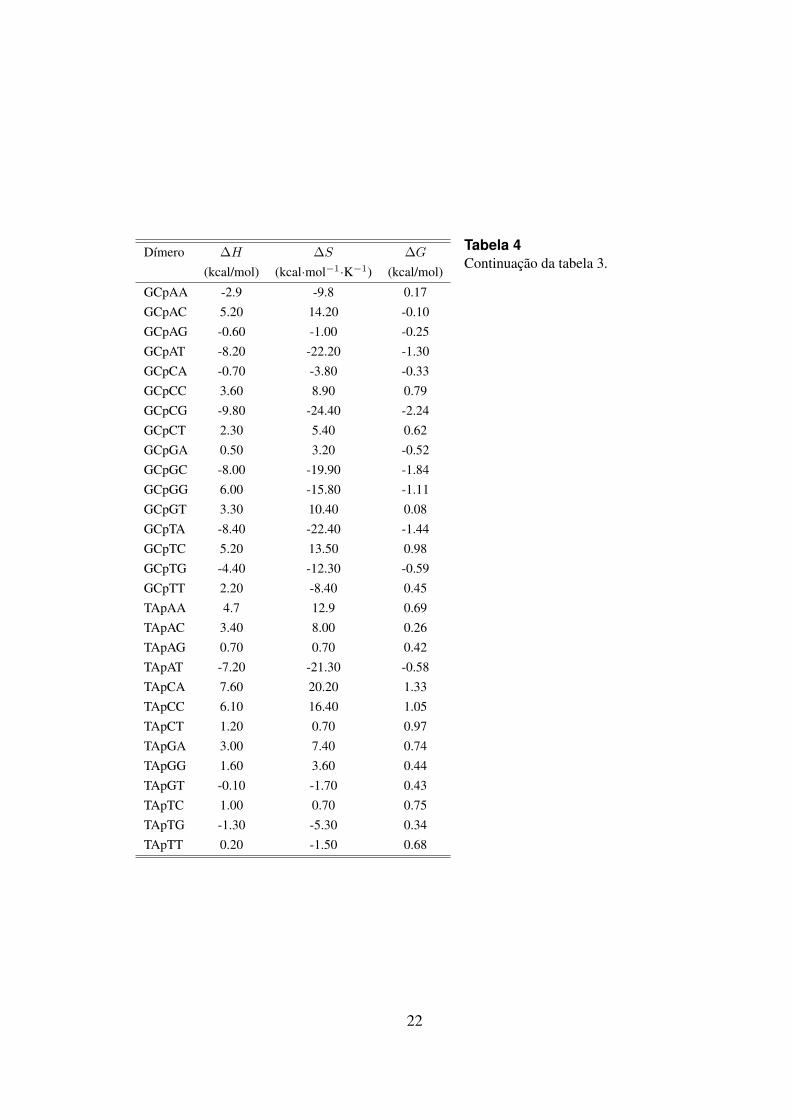
Tabela 4Continuacao da tabela 3.
22

4.2 Flexibilidade
Os modelos utilizados para representar a estrutura do DNA muitas vezes induzem auma ideia equivocada de que esta molecula e estruturalmente estatica. Ao contrario disso,a estrutura de dupla helice e dinamica apresentando movimentos torcao e dobradura. Essadinamica esta associada a flexibilidade do DNA e esta por sua vez e influenciada por doisparametros especıficos:
1. o angulo de torcao entre as bases adjacente que define a capacidade de torcao damolecula; e
2. as variacoes locais ou globais no eixo da dupla helice que definem a capacidade dedobradura do DNA (85).
Um dos parametros avaliados no presente trabalho e a influencia da capacidade de do-bra do DNA na perpetuacao de um SNP. Estudos tem demonstrado que a flexibilidade esequencia-dependente onde a flexibilidade local varia de acordo com a composicao dasbases especıficas daquela microrregiao e a flexibilidade global depende da composicaototal das bases da molecula (85).
A flexibilidade do DNA tem um papel biologico importante estando relacionado amaneira com a qual os genomas se organizam. Este tema e pouco explorado e suas pro-priedades sao complexas e pouco compreendidas. Apesar disso, varios exemplos denotama presenca dessa propriedade fısica nas atividades biologicas. Para que os processos dereplicacao e transcricao ocorram e necessario que a molecula se distorca. As interacoesDNA-proteına sao favorecidas pela capacidade de dobra de uma determinada regiao doDNA. A exemplo disso podemos citar as proteınas com domınio HMG (high mobility
group) que se ligam preferencialmente no DNA em um sıtio de 5 a 8 pb. Durante ainteracao fısica essas proteınas sao capazes de promover uma curvatura na dupla heliceque varia de 90◦ a 110◦ (85, 86).
A formacao dos nucleossomos tambem sao favorecidas pela flexibilidade da molecula.Para que haja essa formacao, o DNA deve curvar-se em torno do complexo nucleossomalproteico. Essa dobra reflete um angulo de curvatura de 47◦ a cada volta da dupla heliceao redor do complexo proteico (85, 87). Alem disso, propriedades de flexibilidade paraas regioes promotoras TATA do DNA tambem sao alvos de estudos. Essas regioes in-teragem com o complexo de proteınas de ligacao TATA-box responsaveis por iniciar atranscricao. Essa interacao e influenciada pela regiao TATA do DNA que esta localizadaem uma regiao muito flexıvel da molecula (88–91).
23

A flexibilidade de uma microrregiao do DNA possibilita o entendimento da micro-mecanica e das interacoes biologicas da molecula. Varios metodos experimentais (92)como disturbios mecanicos ou pequenas modificacoes no DNA, utilizando nanopartıculas (12,93), sao aplicados para medir a flexibilidade de longos trechos da dupla fita, mas as ca-racterısticas locais sao perdidas e nao se obtem a flexibilidade da estrutura sob condicoesnaturais. Para sequencias menores, com aproximadamente 30 pb ou menos, tecnicas ex-perimentais como microscopia de forca atomica (93) e raios X de baixo angulo (94) saoutilizadas, mas tambem necessitam perturbar mecanicamente a estrutura.
Weber et al. (91), demonstrou que e possıvel extrair informacoes detalhadas de mi-crorregioes do DNA sem pertubacoes ou modificacoes estruturais a partir de um modeloque albergue os seguintes parametros:
1. as temperaturas de denaturacao, uma vez que as propriedades termodinamicas doDNA sao dependentes da flexibilidade, como exemplo, as vibracoes molecularesoriundas do armazenamento de energia termica;
2. as propriedades de flexibilidade, como ja estabelecidos experimentalmente para asregioes TATA do DNA em solucao, veja figuras 11 e 12 e;
3. os modelos fısicos estatısticos juntamente com o metodo de proximos vizinhos quepermitem predicoes da flexibilidade para uma regiao mınima de 2 pb.
Uma das grandes vantagens desta tecnica em relacao ao metodo worm-like chain (WCL)que tem sido amplamente empregado na modelagem das flexibilidades do DNA (95),eque alem de ser um modelo relativamente simples, tem como essencia os parametros detemperatura de melting e a lei fısica de Hooke que esta relacionada a elasticidade doscorpos.
O efeito que um mismatch causa na estabilidade estrutural do DNA e no reconheci-mento do complexo proteico de reparo ainda e pouco entendido apesar de alguns estudosdemonstrarem que a flexibilidade local do DNA pode ser a responsavel pelo reconheci-mento de um mismatch pela maquinaria de reparo. Por exemplo, o reparo mais eficienteocorre em um mismatch do tipo G:T. Por ser um pareamento entre uma purina e umapirimidina, esse mismatch tende a ser estavel e perturba muito pouco a estrutura da duplafita (96, 97).
Dentro deste contexto, para avaliar a flexibilidade de uma microrregiao do DNA con-tendo um mismatch central e seus proximos vizinhos imediatamente adjacentes, nos uti-lizaremos os valores estimados de flexibilidade, veja tabela 5. Os calculos serao feitos a
24

GpG (RpR)CpG (YpR)GpC (RpY)
1
2
3
4
Ela
stic
con
stan
t k (
eV n
m-2
)
102 103
ApG (RpR)ApC (RpY)GpA (RpR)CpA (YpR)
102 103
ApT (RpY)ApA (RpR)TpA (YpR)
50
100
150
200
Stretching m
odulus S (pN
)
102 103
Salt concentration [Na+] (mM)
a b c
Figura 11Constante elastica k em funcao da concentracao salina. Os dımeros foram agrupados em ordemdecrescente em funcao da quantidade de ligacoes de hidrogenio: (a) 6 ligacoes, (b) 5 ligacoes e(c) 4 ligacoes de hidrogenio; concentracao de sal a 69 mM. Veja referencia 91.
-40 -20 0 20distance from TSS site (bp)
0.5
0.6
0.7
AG
TA
TA
GGa T7
-40 -20 0 20 40distance from TATA-box (bp)
0.6
0.7
0.8
TA
TA
AA
AAb PTMAP3
k eq
(eV
nm
-2)
Figura 12Constante equivalente keq para regioes promotoras. Concentracao de sal a 69 mM; a linha ver-melha representa a regiao TATA box considerando uma janela de 4 pb; a figura (a) representauma regiao promotora de um bacteriofago T7; (b) representa a regiao promotora do gene PT-MAP3 (GeneID:5760), da famılia proteica protimosina encontrado no cromossomo 20 do DNAde Homo sapiens,veja ref (91).
25

partir de um duplexo de DNA composto por um mismatch e um pareamento canonico le-vando em consideracao o modelo teorico fısico-estatıstico descrito por Weber et al., vejareferencia 69.
Dımero Valores de k Dımero Valores de k
(eV nm−2) (eV nm−2)
AApAT 0.0270773 TGpGT 0.0266518AApCG 0.0267305 ACpAT 0.0172037AApGC 0.0206169 ACpCG 0.042574AApTA 0.0236193 ACpGC 0.00908449ATpCC 0.0125822 ACpTA 0.0470871ATpGG 0.0290114 ATpAC 0.0269482ATpTT 0.0217041 CApAT 0.0167886CCpAT 0.0204448 CApCG 0.0409264CCpCG 0.0176158 CApGC 0.0312345CCpGC 0.0221075 AGpAT 0.0181298CGpGG 0.0226807 AGpCG 0.0237622CGpTT 0.0451272 AGpGC 0.0101556GCpGG 0.0431226 AGpTA 0.0581997GCpTT 0.0190414 ATpAG 0.0230556GGpAT 0.0222818 CGpAG 0.0129784TApTT 0.0262014 GApAT 0.031482ATpGT 0.00882912 GApCG 0.0161483ATpTG 0.0414976 ATpCT 0.0240038CGpGT 0.0755137 ATpTC 0.0738636CGpTG 0.0194157 CGpCT 0.026819GCpGT 0.0198114 CGpTC 0.0338694GCpTG 0.0457502 CTpAT 0.0211643GTpAT 0.00599592 CTpCG 0.0290512GTpGT 0.0223445 GCpCT 0.0189722GTpTG 0.02258 TApCT 0.0264967TApGT 0.00867792
Tabela 5Parametros de flexibilidade para pareamentosnao-canonicos de bases (91, 98).
4.2.1 Modelo para o calculo das flexibilidades
O DNA se comporta como uma mola linear elastica, em que a deformacao e propor-cional a forca aplicada nas suas extremidades, veja figura 13. A partir dessa comparacao,podemos usar a lei de Hooke para calcular as deformacoes elasticas da estrutura damolecula de DNA. Esta lei e dada pela equacao:
F = k∆x (3)
26

onde F e a forca produzida pelo corpo, k e a constante elastica e ∆x e a variacao dadeformacao do corpo. Valores grandes de k representam uma regiao rıgida e valorespequenos de k representam uma regiao flexıvel.
Figura 13Diagrama representando os estados de deformacao de uma mola. Figura retirada de (99).
O modelo usado neste trabalho (91) associa o DNA a molas ligadas em serie. Paramelhor entendimento desse modelo vamos considerar o seguinte exemplo que descreveuma sequencia de DNA de 3pb. Nessa situacao, ”uma unidade de mola”seria represen-tada por um duplexo e assim a sequencia seria representada por dois duplexos que seriamanalogos a duas molas ligadas. Considerando agora esses duplexos em serie e o metodode proximos vizinhos, podemos calcular a constante elastica equivalente keq final destamicrorregiao a partir dos valores de flexibilidade preditos para 2 pares de bases (um du-plexo), como mostra a tabela 6, atraves da seguinte equacao:
1
keq=
1
k1+
1
k2. . .+
1
kn(4)
Dımero Valores de k
(eV nm−2)
ATpAT 0.0248659ATpCG 0.0230182ATpGC 0.0220903ATpTA 0.0109925CGpAT 0.0315708CGpCG 0.0209859CGpGC 0.0245137GCpAT 0.0312279GCpCG 0.0359905TApAT 0.0353204
Tabela 6Parametros de flexibilidade para pare-amentos canonicos de bases (91, 98).
Tomemos agora como exemplo a sequencia de 3 pb GAG/CTC (dois duplexos) emque kCGpAT = 0.0315708 eV · nm−2 e kATpGC = 0.0220903 eV · nm−2. Para essa
27

sequencia teremos um valor de keq = 0.0129965364713 eV · nm−2. Como citado an-teriormente, quanto maior o valor de k mais dura e a mola e vice-versa. Dessa forma,para molas em serie o valor de menor k prevalece no calculo de keq. Como mostrado nafigura 12 esta propriedade influencia fortemente o perfil da flexibilidade.
O mesmo raciocınio pode ser empregado para o calculo de flexibilidade de uma mi-crorregiao de 3 pb CCA/GCA cuja base sublinhada representa um mismatch central. As-sim teremos1 kCGpCC = 0.0221075 eV · nm−2 e kCCpAT = 0.0204448 eV · nm−2, econsequentemente keq = 0.01105375 eV · nm−2.
1o dımero CGpCC e equivalente ao dımero CCpGC
28

5 Metodos
Este topico e composto pela descricao metodologica utilizada neste trabalho. Ele estadividido nas seguintes secoes: secao 5.1 descrevemos o mecanismo hipotetico para a ori-gem de um SNP; secao 5.2 explicamos a notacao adotada; secao 5.3 descreve informacoesgerais sobre os genomas analizados e o banco de dados utilizado neste trabalho; secao 5.4demonstramos como as sequencias foram obtidas a partir dos bancos de dados biologicosutilizados; secao 5.5 descrevemos o processamento dos nossos dados; secao 5.6 explica-mos a obtencao dos valores de energia livre e flexibilidade para microrregioes do DNA e;secao 5.7 explicamos o passo a passo do nosso fluxo de trabalho.
5.1 Mecanismo hipotetico para a formacao de um SNP a partirerros de pareamentos
Para auxiliar o entendimento da influencia dos parametros fısicos de energia livree flexibilidade na distribuicao dos SNPs nos propomos um mecanismo hipotetico quesimula o evento de substituicao de uma unica base no DNA. Nosso pressuposto e que aorigem de um SNP tem como ponto de partida um erro de pareamento de bases que podeocorrer em igual probabilidade em qualquer uma das fitas. Tal hipotese tem como objetivouniformizar as analises e facilitar a aplicacao do metodo de proximos vizinhos juntamentecom os calculos dos parametros de energia livre e flexibilidade descritos na secao 4.2.1.O mecanismo proposto tenta predizer quais os possıveis mismatches que deram origem aum SNP especıfico.
O esquema representado pelo diagrama 14 descreve esquematicamente como foi feitaa predicao dos mismatches. Considere uma populacao hipotetica monoalelica que, aolongo do tempo, veio a tornar-se bialelica para algumas regioes do genoma atraves demecanismos discutidos na secao 2.2, pagina 6. Assumindo que este polimorfismo surgiua partir da substituicao de uma A por uma G, que a base polimorfica e a G e que essasubstituicao ocorre em igual probabilidade em ambas as fitas, podemos inferir que:
1. se o evento de substituicao de bases ocorreu na fita contınua (5′ − 3′) o possıvelmismatch seria o pareamento de G:T em que uma A foi substituida por uma G ou;
2. se a substituicao de base tiver ocorrido na fita descontınua (3′−5′) o mismatch seriao pareamento de A:C onde uma T foi substituıda por uma C.
29

Portanto, o mecanismo assumido considera um SNP como consequencia de dois possıveismismatches.
Figura 14Mecanismo hipotetico para a formacao de um SNP a partir de um mismatch. A parte 1 demonstrao alelo selvagem (wt); a parte 2 refere-se aos possıveis mismatchs na dupla fita cujas bases estaodestacadas em vermelho; Na parte 3 e 4 esta representadas a propagacao do erro que pode originarum SNP representado pelas bases destacadas em cor de rosa.
5.2 Notacao utilizada
Nesta secao descrevemos a notacao adotada neste trabalho. Varios trabalhos na litera-tura descrevem microrregioes de DNA contendo erros de pareamentos e suas sequenciasvizinhas, mas nao existe um padrao de representacao definido para as sequencias e nempara sua notacao. Portanto estabelecemos uma notacao que melhor representa os nossosdados e facilita a nossa interpretacao. A notacao foi estabelecida a partir do mecanismohipotetico descrito na secao 5.1, pagina 29, que mostra a origem dos dois provaveis errosde pareamento de bases que dao origem a um SNP.
Utilizamos como base para a nossa notacao arquivos no formato texto contendo assequencias disponibilizadas pelo dbSNP em um formato de representacao de sequenciasbiologicas conhecido como formato fasta, veja figura 15. Este tipo de formato e larga-mente utilizado para representar sequencias biologicas. Esse formato e composto por umalinha descritiva que comeca com o sinal de maior (>) seguida por linhas consecutivas querepresentam os nucleotıdeos da sequencia. Uma ou mais sequencias podem ser represen-
30

tadas em um mesmo arquivo. A grande vantagem deste formato e sua simplicidade quepermite sua facil manipulacao pelas ferramantas de processamento de texto. Na figura 15os alelos variantes sao definidos por codigos estabelecidos pela International Union of
Pure and Applied Chemistry (IUPAC) (100, 101) que sao definidos na tabela 7 e que saoflanqueados pelas sequencias dos alelos.
1 >gnl|dbSNP|rs104894353 rs=104894353|pos=251|len=501|taxid=9606|mol="genomic"|class=snp|2 alleles="C/G"|build=1323 CCTGCCCCAG CAAGACGGAC TTCTTCAAAA ACATCATGAA CTTCATAGAC ATTGTGGCCA TCATTCCTTA TTTCATCACG4 CTGGGCACCG AGATAGCTGA GCAGGAAGGA AACCAGAAGG GCGAGCAGGC CACCTCCCTG GCCATCCTCA GGGTCATCCG5 CTTGGTAAGG GTTTTTAGAA TCTTCAAGCT CTCCCGCCAC TCTAAGGGCC TCCAGATCCT GGGCCAGACC CTCAAAGCTA6 GTATGAGAGA7 S8 CTAGGGCTGC TCATCTTTTT CCTCTTCATC GGGGTCATCC TGTTTTCTAG TGCAGTGTAC TTTGCCGAGG CGGAAGAAGC9 TGAGTCGCAC TTCTCCAGTA TCCCCGATGC TTTCTGGTGG GCGGTGGTGT CCATGACCAC TGTAGGATAC GGTGACATGT
10 ACCCTGTGAC AATTGGAGGC AAGATCGTGG GCTCCTTGTG TGCCATCGCT GGTGTGCTAA CAATTGCCCT GCCCGTACCT11 GTCATTGTGT
Figura 15Exemplo de arquivo tipo texto de sequencia biologica em formato fasta disponıvel na base dedados dbSNP. O formato fasta composto por uma unica linha descritiva que inicia-se com umsinal de > seguida dos nucleotıdeos que compoem a sequencia; o codigo S na linha 7 representa osalelos allelles=‘‘C/G’’ descritos no cabecalho de acordo com as normas da IUPAC (100,101).
31

Alelos (IUPAC) Significado
A AC CG GT TM A ou CR A ou GW A ou TS C ou GY C ou TK G ou TV A ou C ou GH A ou C ou TD A ou G ou TB C ou G ou TN G ou A ou T ou C
Tabela 7Codigo dos alelos de acordo com as normas IU-PAC (100). Os codigos dos SNPs bialelicos estaorepresentados em azul.
32

Figura 16Representacao da notacao estabelecida. A parte 1 representa o cabecalho do arquivo tipo fasta;A parte 2 representa o campo de anotacao do cabecalho fasta onde estao representados os doisalelos wt e m; A parte 3 representa a sequencia wt na direcao 5′ − 3′ e sua sequencia comple-mentar 3′ − 5′; Parte 4 demonstra os dois possıveis mismatches que poderiam levar a insercao donucleotıdeo polomorfico G, as bases em vermelho sinalizam em qual fita ocorreu a substituicaoe as bases adjacentes representam os proximos vizinhos; na parte 5 as chaves delimitam osdois dımeros contendo um pareamento canonico e um mismatch; 6 representa a conversao darepresentacao anterior (5) para a notacao adotada; a vırgula e o delimitador entre os dımeros mar-cados pelas chaves; o underline e o delimitador entre os pares de bases e representa o “p” nanotacao ATpCA ou CApCG de acordo com as tabelas 2 e 5 da secao 4.1.1.
A figura 16 demonstra o processamento da sequencia fita unica, no formato fasta,obtida da base de dados dbSNP ate a representacao de uma dupla fita, contendo um mis-
match central e um par de base adjacentes ao erro. O campo de anotacao alleles=
‘‘A/G’’ do cabecalho do arquivo tipo fasta, veja figura 15, descreve os dois alelosexistentes, que neste caso sao a A e a G. A partir do nosso mecanismo hipotetico, utili-zamos a sequencia obtida da base de dados considerando o primeiro alelo A como wt e osegundo alelo G como o alelo m. Assim podemos inferir quais mismatches ocorreram nasequencia wt NAN2 que levaram a substituicao da base A pela base G. O proximo passo
2considere N como qualquer base (A, T, G, C) de acordo com a nomenclatura estabelecida pela IUPACcomo mostra a tabela 7
33

foi gerar a sequencia complementar da sequencia wt 5′NAN3′/3′NTN5′ , separamos a
dupla fita e fazemos a predicao dos mismatches para cada uma dessas fitas. Se o erro depareamento ocorreu na fita 5′ − 3′ (NAN) o SNP NGN foi gerado a partir da troca da Apela G. Se o erro de pareamento ocorreu na fita 3′ − 5′ (NTN), o SNP NGN foi gerado apartir da substituicao da T por uma C onde em uma replicacao teremos a perpetuacao doerro na fita 5′ − 3′ representado pelo SNP NGN. Uma vez estabelecido quais mismatches
devem ser considerados para o alelo wt, convertemos as sequencias de 3 pb contendo ummismatch central e um par de bases imediatamente adjacentes. Na nossa notacao teremos5′TA AC,AC CG3′ e 3′TA GT,GT CG5′ como mostra a figura 16. E importante res-saltar que a frequencia dos alelos nao interfere na predicao dos mismatches portanto aordem de classificacao dos alelos em selvagem ou mutado nao importa.
5.3 Genomas
Para realizar este trabalho escolhemos 9 genomas, veja tabela 8. A escolha desses ge-nomas foi feita em funcao da quantidade de sequencias e das informacoes disponıveis nodbSNP. Paralelamente as sequencias do genomas completos foram obtidas via protocolode transferencia de arquivos (FTP) das bases de dados do National Center for Biotech-
nology Information (NCBI) (9) e Wellcome Trust Sanger Institute (102) . A partir dessassequencias estimamos o conteudo medio GC de cada genoma. A variacao do conteudo GCdos genomas estudados e de extrema relevancia para inferir a complexidade e a evolucaodos mesmos, alem disso pode ser utilizado para estimar a estabilidade termodinamica dassequencias (103, 104). Para extrair essas informacoes, utilizamos o programa de codigoaberto gccalc.pl escrito em Bioperl obtido do repositorio CPAN (Comprehensive Perl
Archive Network) (105), veja detalhes do script no apendice A.1.5.
5.4 Sequencias de SNPs
Para obtencao das sequencias de SNPs nos buscamos por uma base de dados quefosse a mais completa possıvel. O dbSNP representa atualmente o banco de dados publicoreferencia para submissao de dados de polimorfismos tendo representadas sequencias demais de 100 organismos, veja tabela 9 que descreve o numero de sequencias disponıveisdos organismos estudados neste trabalho.
Para extrair as milhares de sequencias de SNPs da base dbSNP nos utilizamos o script
disponıvel no NCBI eutils.pl (118, 119). Este programa e de codigo livre e utiliza o
34

Organismo Tamanho Conteudo GC Haplotipo referenciado genoma* em %* (n)
Homo sapiens 3.00 Gb 41 23 (9, 106)Bos taurus 3.40 Gb 41 30 (107, 108)Mus musculus 2.80 Gb 39 20 (109, 110)Gallus gallus 1.05 Gb 42 40 (111)Danio rerio 1.70 Gb 36 25 (112)Apis mellifera 2.65 Mb 30 16 (113, 114)Plasmodium falciparum 23.3 Mb 19 14 (115)Oryza sativa 3.70 Mb 43 12 (116)Streptococcus pyogenes 1.85 Mb 38 1 (circular) (117)
Tabela 8Tabela de estatısticas dos genomas analisados. *Valores aproximados; dados obtidos em outubrode 2010 de (9, 102).
Organismos Total de SNPs Transicao TransversaoHomo sapiens 17.600.000 11.000.000 6.700.000Bos taurus 3.700.000 2.400.000 1.300.000Mus musculus 13.700.000 9.200.000 4.500.000Gallus gallus 5.200.000 3.600.00 1.600.000Danio rerio 600.000 320.000 280.000Apis mellifera 1.100.000 780.000 230.000Plasmodium falciparum 110.000 50.000 58.000Oryza sativa 3.600.000 2.100.000 1.500.000Streptococcus pyogenes 3.200 2.000 1.200
Tabela 9Total sequencias de SNPs para os 9 genomas obtidas da base de dados dbSNP (65) em outubro de2010.
35

pacote Bioperl (120). O aplicativo foi modificado para recuperar sequencias especıficasde SNPs atraves da utilizacao das sintaxes descritas no dbSNP. O nome do nosso programae util.pl e seu codigo encontra-se descrito no apendice A.1.6. As sequencias de SNPsdos 9 genomas foram obtidas separadas por cromossomos, com excecao dos SNPs dogenoma de Plasmodium falciparum que ainda nao possuem tal classificacao no dbSNP.
5.5 Datamining - Mineracao de dados
Uma vez obtidas as sequencias de SNPs, iniciamos o pre processamento dos da-dos. Primeiro convertemos o formato fasta para um formato mais simples estabelecidocomo formato de arquivo summary. Esse formato resume a sequencia em uma unicalinha considerando apenas o nome do organismo abreviado, e a microrregiao do DNA deinteresse contendo os alelos e as bases vizinhas imediatamente adjacentes, veja figura 17.Esta conversao e feita atraves do script fasta2summary.pl que tem a funcao de ve-rificar e eliminar sequencias com caracteres invalidos tais como a presenca de N flanque-ando os codigos bialelicos, sequencias incompletas e que contenham codigos alelicos quenao representam os SNPs.
Figura 17Representacao do arquivo texto no formato summary. Uma unica linha representa uma sequenciapolimorfica; rs e o termo anotador da sequencia consenso oriunda do dbSNP; entre colchetesesta o nome da especie abreviado; os dois nucleotıdeos centrais da sequencia separadas por barrarepresentam os alelos e as bases adjacentes que flanqueiam os alelos representam seus proximosvizinhos.
A proxima etapa foi separar as sequencias em transicao e transversao, considerandoo vies que favorece o numero de transicoes em relacao as transversoes, veja tabela 9na pagina 35. Esse vies para a maioria dos genomas de eucariotos e bem descrito na
36

literatura e foi discutido na secao 2.2.1, pagina 9. O script o trans-transv.pl foiutilizado nesta etapa. O arquivo tipo texto summary e utilizado como entrada (input) eprocessado, gerando dois arquivos de saıda (output) no mesmo formato que representamas sequencias separadas por transicao e transversao. O codigo desse script encontra-sedisponıvel no apendice A.1.3.
5.6 Dados de energia livre e flexibilidades
Os valores estimados de energia livre para uma microrregiao da dupla fita com pare-amentos de bases canonicos e nao canonicos foram obtidos a partir de dados validadosexperimentalmente e ja publicados da literatura, como ja discutido anteriormente. Vejatabelas 2, 3 e 4, paginas 20–22, secao 4.1.
Os valores estimados de flexibilidade para os mismatches foram obtidos pelos calculosde potenciais de Morse e constante elastica a partir de temperaturas de melting (98)tambem ja discutido anteriormente na secao 4.2 (91).
5.7 Workflow computacional consolidado
A partir dos dados pre-processados, dos valores estimados de ∆G e flexibilidade e doestabelecimento do mecanismo probabilıstico de geracao de SNPs, escrevemos um con-junto de scripts para permitir a analise dos nossos dados e a integracao das informacoes.A figura 18 define o fluxograma consolidado das etapas de obtencao das sequencias deSNPs a partir da base de dados dbSNP e a expansao deste fluxograma, etapas A e B, podeser vista nas figuras 19 e 20, respectivamente.
O script snp.pl tem a funcao de atribuir valores de flexibilidade e energia livre assequencias de trımeros contendo um mismatch central flanqueado por um par de base ad-jacente e converter as sequencias de SNPs na notacao adotada como descrito na secao 5.2.Este programa utiliza como input uma tabela contendo os valores de ∆G ou de flexibili-dade para os mismatch e um arquivo de sequencias de SNPs no formato summary.
A estatıstica dos nossos dados foi gerada pelo o script histogram.pl, veja ocodigo deste programa no apendice A.1.7. Este aplicativo tem a funcao de agrupar osvalores de flexibilidade ou energia livre em intervalos, definidos pelo usuario, calcularo mınimo e maximo desses valores e converter em porcentagem o numero absoluto decada mismatch. Este programa utiliza como input um dos arquivos de saıda do programasnp.pl que contem a somatoria dos valores de ∆G e k equivalente para 3 pb contendo
37
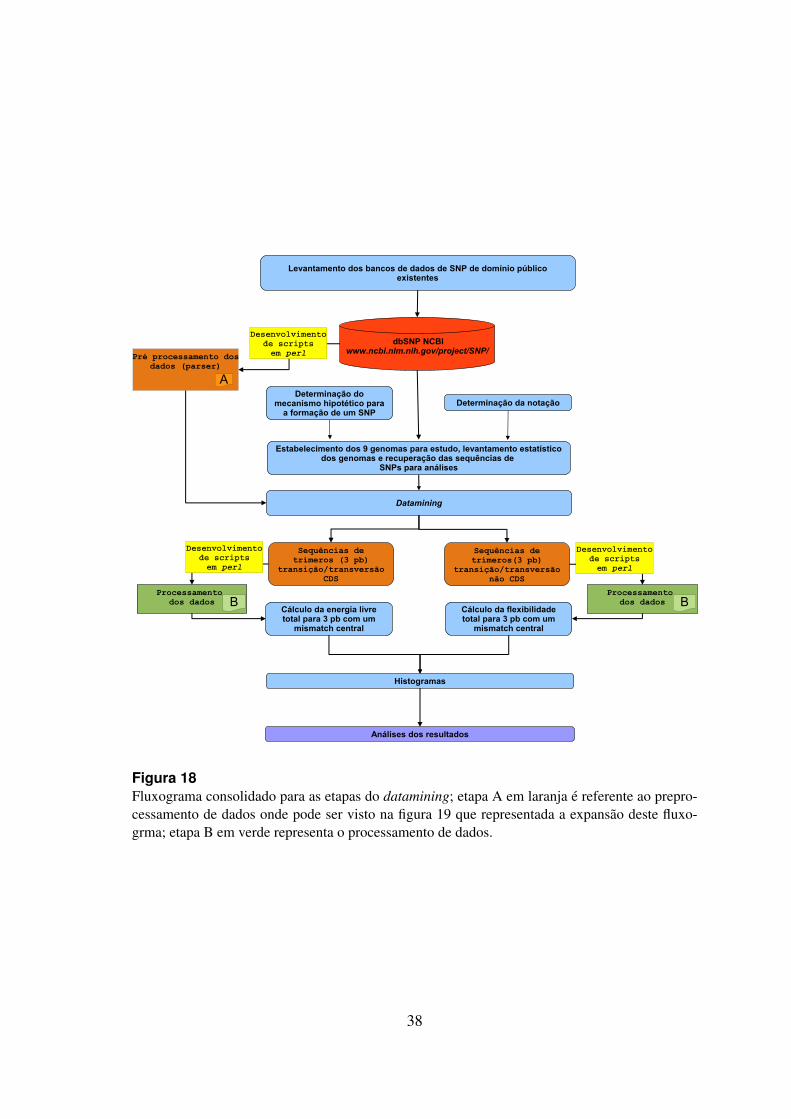
Figura 18Fluxograma consolidado para as etapas do datamining; etapa A em laranja e referente ao prepro-cessamento de dados onde pode ser visto na figura 19 que representada a expansao deste fluxo-grma; etapa B em verde representa o processamento de dados.
38

Figura 19Expansao do fluxograma - pre processamento dos dodos. A letra A a esquerda se refere a expansaodo retangulo laranja tambem demarcado pela letra A no fluxograma da figura 18.
39

Figura 20Expansao do fluxograma - processamento de dados. A letra “B” a esquerda se refere a expansaodo retangulo verde tambem demarcado pela letra “B” no fluxograma da figura 18.
40

um mismatch central.
Para uma analise comparativa nos elaboramos um organismo hipotetico onde consi-deramos que a ocorrencia de cada mismatch e igualmente provavel. Para avaliar o padraode distribuicao de ∆G e de flexibilidade esses dados foram normalizados. Para isso fize-mos a razao entre as frequencias absolutas de cada genoma e o organismo padrao. Paraeste procedimento o programa desenvolvido foi o padroniza.pl e seu codigo estadescrito no apendice A.1.8.
41

6 Resultados e discussao
Nesta secao analisamos e discutimos os nossos resultados. Utilizamos nove genomaspara avaliar a influencia da flexibilidade e energia livre no polimorfismo de uma unicabase (SNP). Estes genomas foram escolhidos em funcao da quantidade de sequenciasdisponıveis na base de dados dbSNP. Oito destes nove genomas sao de organismos euca-riotos, sendo tres genomas de mamıferos (Homo sapiens, Mus musculus e Bos taurus), umgenoma de ave (Gallus gallus), um genoma de peixe (Zebra fish), um genoma de planta(Oryza sativa), um genoma de inseto (Apis mellifera), um genoma de parasita (Plasmo-
dium falciparum) e um genoma de procarioto (bacteria Streptococcus piogenes). Alemdestes genomas nos construımos um organismo padrao para servir de referencia, ondeassumimos que a ocorrencia de cada mismatch e igualmente provavel.
A organizacao desta secao segue a seguinte linha:
1. secao 6.1 discute o organismo padrao utilizado como referencia nas nossas analises;
2. secao 6.2 relembra sucintamente alguns conceitos ja descritos anteriormente ne-cessarios as analises;
3. secao 6.3 faz uma analise comparativa entre o organismo artificial e um organismoreal (Homo sapiens);
4. secao 6.4 discute os resultados da distribuicao dos SNPs em funcao da energia livree flexibilidade para os nove genomas analisados.
5. secao 6.5 discute e faz uma analise comparativa breve dos resultados preliminaresda distribuicao de SNPs em funcao de energia livre e das flexibilidades para asregioes codificantes e nao codificantes do genoma humano.
6.1 Organismo padrao
Durante o desenvolvimento deste projeto percebemos a necessidade de ter um con-junto de dados de SNPs que pudesse servir de referencia para as nossas analises. O con-junto de dados que escolhemos possui uma unica ocorrencia de cada tipo de SNP e po-dem ser considerados como componentes um organismo artificial onde todas as mutacoesocorrem de maneira uniforme sem privilegiar qualquer tipo de SNP. Este conjunto de da-dos foi utilizado para normalizar os resultados gerados e serviu para padronizar as nossas
42

analises, assim chamamos este organismo artificial de organismo padrao ou organismo demutacao unitaria.
Para exemplificar o ganho na analise antes e depois do processo de normalizacao va-mos considerar o seguinte exemplo. Vejamos o perfil da frequencia de distribuicao dosSNPs representados pelos graficos das figuras 21a e 22a , e possıvel observar que o padraode distribuicao dos SNPs e dado por uma gaussiana onde estes SNPs sao mais frequentesna faixa de ∆G que varia entre −0.5 a 0.5 kcal/mol. Aparentemente nao existe diferencaentre o comportamento da distribuicao das substituicoes em humano e nem no organimsopadrao. O mesmo resultado pode ser observado nos graficos 21b e 22b referentes astransversoes. Verificamos tambem um perfil de distribuicao de polimorfismos entre os or-ganismo muito semelhante ocorrendo mais frequentemente nas faixas de ∆G que variamentre 0.5 e 1.5 kcal/mol. Para viabilizar a identificacao das diferencas entre os perfis de∆G e keq de cada genoma, calculamos a razao entre as frequencias de SNPs dos genomasde cada organismo e o organismo unitario, ou seja,
grafico da frequencia relativa =histograma normalizado
histograma padrao
A figura 23 explicita a frequencia relativa dos SNPs obtida atraves do calculo da razaoentre os dados apresentados na figura 21 e na figura 22. Essa razao nos permite distinguiro perfil da distribuicao entre os genomas e o organismo padrao e compara-los.
Ao avaliarmos o perfil da frequencia de distribuicao de SNPs em funcao da flexi-bilidade (keq) entre os demais genomas e o organismo padrao, observamos resultadossemelhantes, ou seja, poucas diferencas entre eles, veja figuras 24 e 25. Dessa forma,adotamos a mesma abordagem empregada para evidenciar as diferencas entre os perfis de∆G e keq de cada genoma. A razao entre as frequencias de SNPs das figuras 24 e 25 estarepresentada no grafico 26.
Atraves do emprego dessa abordagem foi possıvel avaliar como a energia livre e aflexibilidade influenciam a dinamica das transicoes e transversoes entre os diferentes ge-nomas.
6.2 Elementos para analise
Para iniciar as analises dos perfis de ∆G e flexibilidade relembramos aqui algumasconsideracoes importantes ja discutidas nas secoes 4.1.1, pagina 19, e 4.2, pagina 23.Para avaliar a influencia dos parametros fısicos do DNA na ocorrencia de SNPs devemos
43

considerar que:
1. valores de ∆G negativos conferem estabilidade a estrutura;
2. quando os valores de ∆G sao positivos a estrutura tende a se comportar de formainstavel indicando uma menor probabilidade de incorporacao e perpetuacao de ummismatch;
3. o DNA se comporta de forma flexıvel quando os valores de keq sao pequenos;
4. um keq grande denota uma microrregiao rıgida;
5. os valores estimados de ∆G para os pareamentos canonicos de DNA variam de −2.0
a −4.41 kcal/mol;
6. os valores estimados de keq para os pareamentos canonicos de DNA estao nas faixasentre 0.76 a 1.68 eV · nm−2. Estes valores estao indicados pelas faixas cinzas emnossos graficos.
6.3 Analise comparativa entre o organismo padrao e um ge-noma real
Ao comparar os valores de ∆G dos pareamentos canonicos de todos os genomas aosmismatches e possıvel observar que os SNPs ocorrem com maior frequencia em regioesmenos estaveis, com excecao de um numero muito pequeno de transversoes que incidemna faixa de ∆G = −2.5 kcal/mol, veja os histogramas da figura 22.
A figura 22a mostra que a frequencia das transicoes do organismo padrao concentra-se na faixa de energia livre (∆G) que varia de −0.5 a 0.5 kcal/mol e se comportam comouma distribuicao gaussiana. Ja o perfil da distribuicao das transversoes, representadopela figura 22b, demonstra que o maior numero de SNPs concentra-se na faixa cujo ∆G
e positivo. Esse perfil confere a essas microrregioes uma menor estabilidade quandocomparadas ao perfil de energia livre das transicoes.
Em relacao as flexibilidades a faixa de valores de keq para os mismatches esta contidadentro da mesma faixa dos pareamentos canonicos, veja os graficos da figura 25. Este re-sultado pode ser um indicativo de que alguns mismatches podem se comportar fisicamentecomo um pareamento perfeito entre as bases do DNA perturbando muito pouco a sua es-trutura e assim podemos hipotetizar que este seja um fator que favoreca a perpetuacao doerro.
44

O perfil de distribuicao das transicoes, figura 25a, mostra que estes eventos tendema ocorrer em microrregioes mais flexıveis do DNA. Podemos observar que o histogramada figura 25b mostra que as transversoes ocorrem com maior frequencia na faixa de fle-xibilidade dos pareamentos canonicos. Quando comparadas as transicoes, estes eventostendem a ocorrer em regioes mais rıgidas da molecula.
-2 0 2Mismatch free energy ∆G (kcal/mol)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
-2 0 2Mismatch free energy ∆G (kcal/mol)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
Figura 21Histograma normalizado dadistribuicao de SNPs em funcaoda energia livre (∆G) no genomahumano. Transicoes sao mostradasa esquerda e transversoes a direita.
-2 0 2Mismatch free energy ∆G (kcal/mol)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
-2 0 2Mismatch free energy ∆G (kcal/mol)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
Figura 22Histograma normalizado dadistribuicao de SNPs em funcao daenergia livre (∆G) para um orga-nismo padrao com taxa de mutacaouniforme. Transicoes sao mos-tradas a esquerda e transversoes adireita.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human ch1
Figura 23Frequencia relativa da distribuicao de SNPs em funcaoda energia livre (∆G) no genoma humano. Transicoessao mostradas a esquerda e transversoes a direita
45
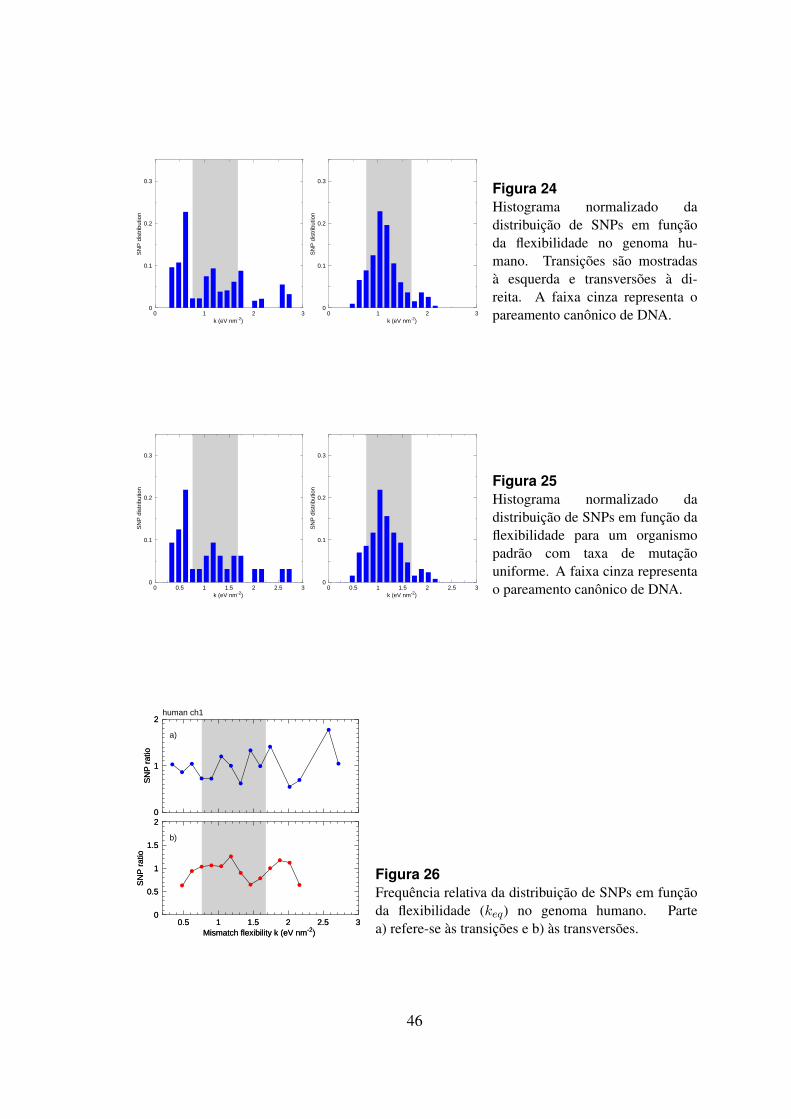
0 1 2 3k (eV nm-2)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
0 1 2 3k (eV nm-2)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
Figura 24Histograma normalizado dadistribuicao de SNPs em funcaoda flexibilidade no genoma hu-mano. Transicoes sao mostradasa esquerda e transversoes a di-reita. A faixa cinza representa opareamento canonico de DNA.
0 0.5 1 1.5 2 2.5 3k (eV nm-2)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
0 0.5 1 1.5 2 2.5 3k (eV nm-2)
0
0.1
0.2
0.3
SN
P d
istr
ibut
ion
Figura 25Histograma normalizado dadistribuicao de SNPs em funcao daflexibilidade para um organismopadrao com taxa de mutacaouniforme. A faixa cinza representao pareamento canonico de DNA.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
human ch1
Figura 26Frequencia relativa da distribuicao de SNPs em funcaoda flexibilidade (keq) no genoma humano. Partea) refere-se as transicoes e b) as transversoes.
46
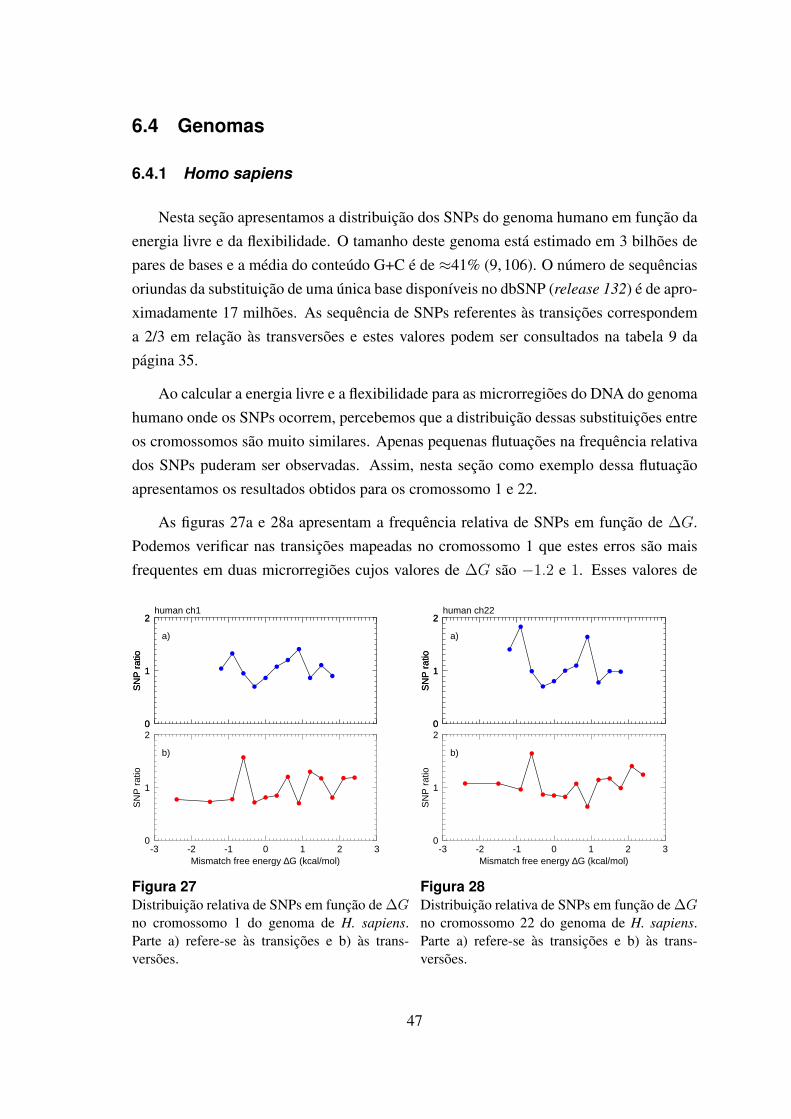
6.4 Genomas
6.4.1 Homo sapiens
Nesta secao apresentamos a distribuicao dos SNPs do genoma humano em funcao daenergia livre e da flexibilidade. O tamanho deste genoma esta estimado em 3 bilhoes depares de bases e a media do conteudo G+C e de ≈41% (9, 106). O numero de sequenciasoriundas da substituicao de uma unica base disponıveis no dbSNP (release 132) e de apro-ximadamente 17 milhoes. As sequencia de SNPs referentes as transicoes correspondema 2/3 em relacao as transversoes e estes valores podem ser consultados na tabela 9 dapagina 35.
Ao calcular a energia livre e a flexibilidade para as microrregioes do DNA do genomahumano onde os SNPs ocorrem, percebemos que a distribuicao dessas substituicoes entreos cromossomos sao muito similares. Apenas pequenas flutuacoes na frequencia relativados SNPs puderam ser observadas. Assim, nesta secao como exemplo dessa flutuacaoapresentamos os resultados obtidos para os cromossomo 1 e 22.
As figuras 27a e 28a apresentam a frequencia relativa de SNPs em funcao de ∆G.Podemos verificar nas transicoes mapeadas no cromossomo 1 que estes erros sao maisfrequentes em duas microrregioes cujos valores de ∆G sao −1.2 e 1. Esses valores de
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human ch1
Figura 27Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human ch22
Figura 28Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 22 do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
47

∆G representam uma variacao de 30 a 40% a mais que o organismo padrao.
No cromossomo 22, as transicoes tambem sao mais frequentes nas mesmas faixasde ∆G observadas no cromossomo 1. A frequencia relativa de SNPs com ∆G = −1.2
kcal/mol e aproximadamente 80% maior em relacao ao organismo padrao e 50% maiorque aquela observada no cromossomo 1. No cromossomo 22 existem duas microrregioesonde os SNPs sao mais frequentes. Tais microrregioes possuem valores aproximados de∆G igual a −1.25 kcal/mol e 1 kcal/mol o que representa uma diferenca de 20% entreestas regioes.
Com relacao as transversoes mostradas nas figuras 27b e 28b os valores de obser-vados de ∆G evidenciam que os SNPs ocorrem preferencialmente em microrregioes deaproximadamente −1.5 kcal/mol. Tal resultado mostra a preferencia da perpetuacao dosSNPs em regioes mais estaveis.
As figuras 29 e 30 mostram o perfil da frequencia relativa dos SNPs em funcao daflexibilidade. Os resultados para as transicoes do cromossomo 1, figura 29a, mostram queos mismatches ocorrem mais frequentemente em microrregioes com valor de keq proximode 2.6 eV · nm−2. Esse valor e aproximadamente 80% maior que aquele observado noorganismo padrao e representa uma microrregiao rıgida do DNA.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
human ch1
Figura 29Distribuicao relativa de SNPs em funcao de keqno cromossomo 1 do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
human ch22
Figura 30Distribuicao relativa de SNPs em funcao de keqno cromossomo 22 do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
Resultados similares foram encontrados para o cromossomo 22, contudo alem damicrorregiao de keq proximo de 2.6 eV · nm−2 existem duas outras com valores de keq
48

de 1.5 eV · nm−2 e 1.7 eV · nm−2. Vale ressaltar que a microrregiao de keq igual a1.5 eV · nm−2 coincide com a faixa de flexibilidade dos pareamentos canonicos sendoeste valor 90% maior do que aquele observado no organismo padrao. Nesse contexto,podemos sugerir que os mismatches perturbam muito pouco a estrutura da molecula epode representar uma das causas do nao reconhecimento dessas regioes durante o a acaodo mecanismo de reparo. Por outro lado os resultados tambem indicam uma tendenciadas transicoes no genoma humano ocorrerem em microrregioes mais rıgidas. Diferentedas transicoes, os resultados observados para as transversoes sao muito proximos aquelesobtidos para o organismo padrao, veja as figuras 29b e 30b.
6.4.2 Bos taurus
Os bovinos sao animais de grande importancia economica. Seu genoma foi sequen-ciado tendo como um dos principais objetivos o entendimento das bases geneticas desteorganismo visando o aumento da qualidade da carne e do seu custo benefıcio, o melhora-mento de racas para gerar um rebanho mais saudavel, menos dependente de antibioticose resistente a parasitas como os carrapatos (121).
O sequenciamento do seu genoma completo foi finalizado em 2009 e aproximada-mente 2 milhoes de SNPs foram identificados durante o projeto. Em outubro de 2010 onumero de sequencias de SNPs depositadas no dbSNP ja ultrapassava o numero de 3.7milhoes de sequencias. Estima-se que este genoma tenha em torno de 2.8 bilhoes de pa-res de bases e a media do seu conteudo G+C e de 42%, similar ao conteudo do genomahumano (107, 108).
Como nossos resultados referentes a influencia da energia livre e flexibilidade nopadrao de distribuicao de SNPs nos 29 haplotipos autossomicos de B. taurus foram si-milares, nesta secao apresentaremos os resultados para o cromossomo 1. Esse e o maiorcromossomo do organismo e tambem apresenta o maior numero de SNPs (1.3 milhoes).
Com relacao ao haplotipo alossomico X (cromossomo X) o perfil de energia livre e deflexibilidade variou em relacao as transicoes e em funcao disso apresentaremos tambemestes resultados.
As figuras 31a e 32a representam o perfil da distribuicao das transicoes em funcaode ∆G e evidenciam essa diferenca. No cromossomo X podemos observar a existenciade duas faixas de ∆G onde a frequencia relativa de SNPs e elevada, 1.1 kcal/mol e1.6 kcal/mol respectivamente. Por outro lado, apesar da existencia de varias regioesfrequencia relativa de SNPs elevadas no cromossomo 1, nenhuma faixa de ∆G e tao pro-
49
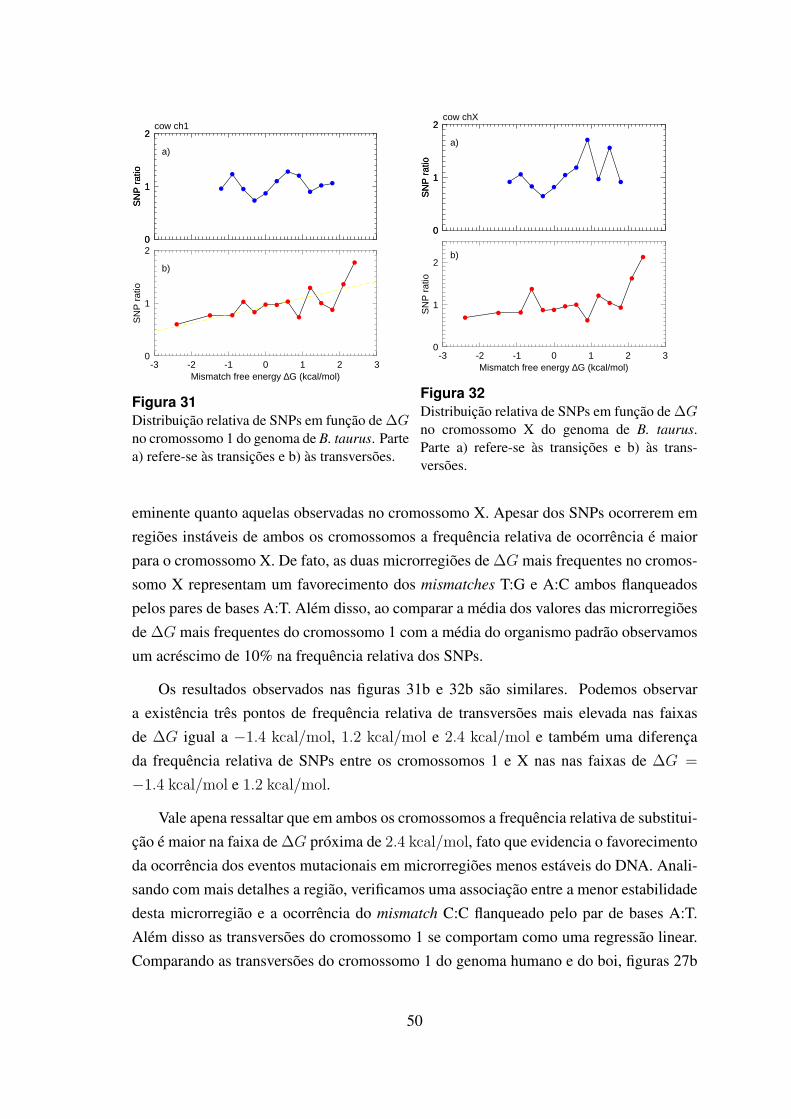
0
1
2S
NP
rat
io
0
1
2S
NP
rat
io
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
cow ch1
Figura 31Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de B. taurus. Partea) refere-se as transicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
cow chX
Figura 32Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo X do genoma de B. taurus.Parte a) refere-se as transicoes e b) as trans-versoes.
eminente quanto aquelas observadas no cromossomo X. Apesar dos SNPs ocorrerem emregioes instaveis de ambos os cromossomos a frequencia relativa de ocorrencia e maiorpara o cromossomo X. De fato, as duas microrregioes de ∆G mais frequentes no cromos-somo X representam um favorecimento dos mismatches T:G e A:C ambos flanqueadospelos pares de bases A:T. Alem disso, ao comparar a media dos valores das microrregioesde ∆G mais frequentes do cromossomo 1 com a media do organismo padrao observamosum acrescimo de 10% na frequencia relativa dos SNPs.
Os resultados observados nas figuras 31b e 32b sao similares. Podemos observara existencia tres pontos de frequencia relativa de transversoes mais elevada nas faixasde ∆G igual a −1.4 kcal/mol, 1.2 kcal/mol e 2.4 kcal/mol e tambem uma diferencada frequencia relativa de SNPs entre os cromossomos 1 e X nas nas faixas de ∆G =
−1.4 kcal/mol e 1.2 kcal/mol.
Vale apena ressaltar que em ambos os cromossomos a frequencia relativa de substitui-cao e maior na faixa de ∆G proxima de 2.4 kcal/mol, fato que evidencia o favorecimentoda ocorrencia dos eventos mutacionais em microrregioes menos estaveis do DNA. Anali-sando com mais detalhes a regiao, verificamos uma associacao entre a menor estabilidadedesta microrregiao e a ocorrencia do mismatch C:C flanqueado pelo par de bases A:T.Alem disso as transversoes do cromossomo 1 se comportam como uma regressao linear.Comparando as transversoes do cromossomo 1 do genoma humano e do boi, figuras 27b
50
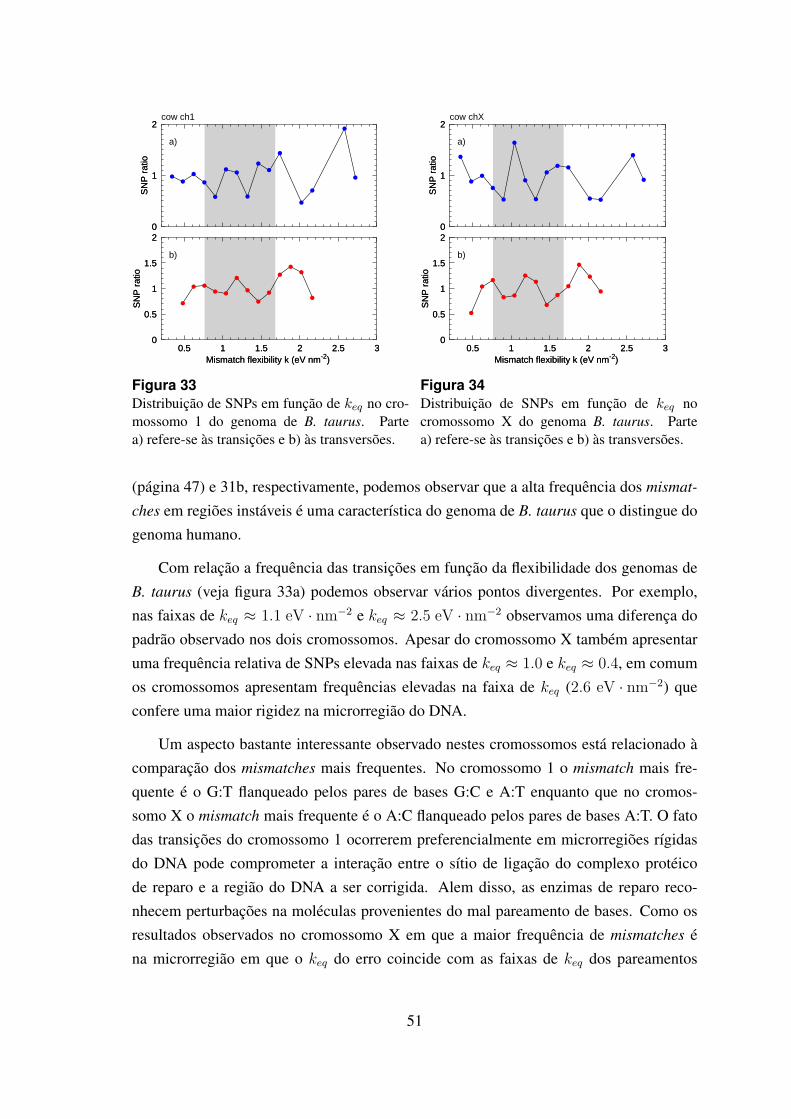
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
cow ch1
Figura 33Distribuicao de SNPs em funcao de keq no cro-mossomo 1 do genoma de B. taurus. Partea) refere-se as transicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
cow chX
Figura 34Distribuicao de SNPs em funcao de keq nocromossomo X do genoma B. taurus. Partea) refere-se as transicoes e b) as transversoes.
(pagina 47) e 31b, respectivamente, podemos observar que a alta frequencia dos mismat-
ches em regioes instaveis e uma caracterıstica do genoma de B. taurus que o distingue dogenoma humano.
Com relacao a frequencia das transicoes em funcao da flexibilidade dos genomas deB. taurus (veja figura 33a) podemos observar varios pontos divergentes. Por exemplo,nas faixas de keq ≈ 1.1 eV · nm−2 e keq ≈ 2.5 eV · nm−2 observamos uma diferenca dopadrao observado nos dois cromossomos. Apesar do cromossomo X tambem apresentaruma frequencia relativa de SNPs elevada nas faixas de keq ≈ 1.0 e keq ≈ 0.4, em comumos cromossomos apresentam frequencias elevadas na faixa de keq (2.6 eV · nm−2) queconfere uma maior rigidez na microrregiao do DNA.
Um aspecto bastante interessante observado nestes cromossomos esta relacionado acomparacao dos mismatches mais frequentes. No cromossomo 1 o mismatch mais fre-quente e o G:T flanqueado pelos pares de bases G:C e A:T enquanto que no cromos-somo X o mismatch mais frequente e o A:C flanqueado pelos pares de bases A:T. O fatodas transicoes do cromossomo 1 ocorrerem preferencialmente em microrregioes rıgidasdo DNA pode comprometer a interacao entre o sıtio de ligacao do complexo proteicode reparo e a regiao do DNA a ser corrigida. Alem disso, as enzimas de reparo reco-nhecem perturbacoes na moleculas provenientes do mal pareamento de bases. Como osresultados observados no cromossomo X em que a maior frequencia de mismatches ena microrregiao em que o keq do erro coincide com as faixas de keq dos pareamentos
51
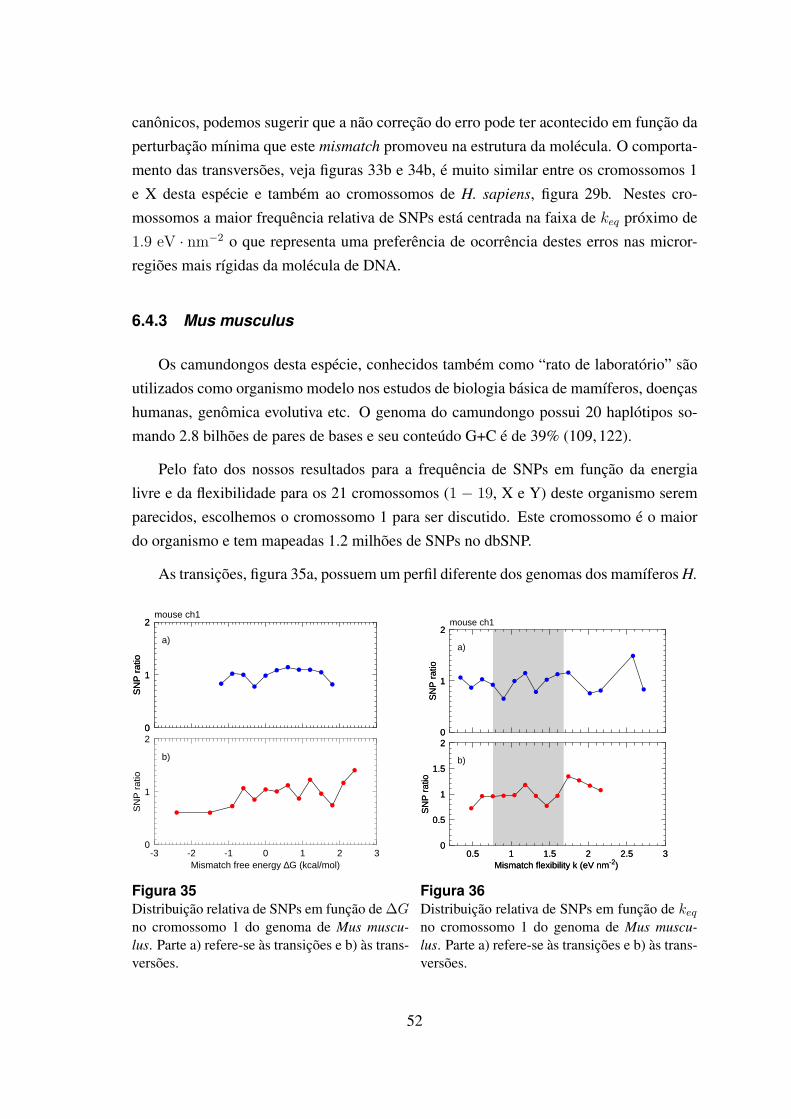
canonicos, podemos sugerir que a nao correcao do erro pode ter acontecido em funcao daperturbacao mınima que este mismatch promoveu na estrutura da molecula. O comporta-mento das transversoes, veja figuras 33b e 34b, e muito similar entre os cromossomos 1e X desta especie e tambem ao cromossomos de H. sapiens, figura 29b. Nestes cro-mossomos a maior frequencia relativa de SNPs esta centrada na faixa de keq proximo de1.9 eV · nm−2 o que representa uma preferencia de ocorrencia destes erros nas micror-regioes mais rıgidas da molecula de DNA.
6.4.3 Mus musculus
Os camundongos desta especie, conhecidos tambem como “rato de laboratorio” saoutilizados como organismo modelo nos estudos de biologia basica de mamıferos, doencashumanas, genomica evolutiva etc. O genoma do camundongo possui 20 haplotipos so-mando 2.8 bilhoes de pares de bases e seu conteudo G+C e de 39% (109, 122).
Pelo fato dos nossos resultados para a frequencia de SNPs em funcao da energialivre e da flexibilidade para os 21 cromossomos (1 − 19, X e Y) deste organismo seremparecidos, escolhemos o cromossomo 1 para ser discutido. Este cromossomo e o maiordo organismo e tem mapeadas 1.2 milhoes de SNPs no dbSNP.
As transicoes, figura 35a, possuem um perfil diferente dos genomas dos mamıferos H.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
mouse ch1
Figura 35Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de Mus muscu-lus. Parte a) refere-se as transicoes e b) as trans-versoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
mouse ch1
Figura 36Distribuicao relativa de SNPs em funcao de keqno cromossomo 1 do genoma de Mus muscu-lus. Parte a) refere-se as transicoes e b) as trans-versoes.
52

sapiens (figura27) e B. taurus (figura31) analisados anteriormente (secao 6.4.1 e 6.4.2).Esta distribuicao apresenta-se como uma flutuacao discreta em que a maioria dos SNPsestao em torno de 1 refletindo um comportamento muito similar ao do organismo padraocujos SNPs se apresentam como uma distribuicao normal.
Os resultados da frequencia relativa das transversoes do cromossomo 1 em funcao de∆G deste organismo, figura 35b, assemelham-se ao perfil das transversoes do cromos-somo 1 de B. taurus(figura31b) principalmente nos pontos de mais elevada frequenciacom valores de ∆G positivos em torno de 1.2 kcal/mol e 2.6 kcal/mol. De fato, estasduas microrregioes onde as transversoes sao mais representativas, assim como no genomado boi, neste genoma os mismatches C:C flanqueados por A:T estao tambem favorecidos.
Os graficos da figura 36 representam a frequencia relativa dos SNPs em funcao daflexibilidade. Tanto as transicoes, figura 36a, quanto as transversoes, figura 36b, pos-suem um resultado similar ao padrao de flutuacao encontrados nos graficos dos genomasanalisados anteriormente (figuras 29 e 33). Sendo assim, este resultado vem a confirmarque nos tres mamıferos analisados existe um padrao de distribuicao de SNPs em funcaoda flexibilidade comum entre eles. Neste contexto, e possıvel verificar que os SNPs destesorganismos tendem a aumentar a sua frequencia a medida que as microrregioes do DNAaumentam sua rigidez.
6.4.4 Gallus gallus
A galinha (Gallus gallus) e um organismo modelo muito utilizado na pesquisa bio-medica, alem de ser um animal de grande importancia economica pecuarista. O tamanhoestimado deste genoma e de 1 bilhao de pares de bases e a media do conteudo G+C ede 42%, conteudo similar ao dos tres mamıferos discutidos anteriormente. O cromos-somo discutido nesta secao sera tambem o cromossomo 1 pois e o de maior tamanho epossui disponıvel no dbSNP (release 132) aproximadamente 606 mil sequencias deposi-tadas (123).
O grafico 37a, mostra que a frequencia relativa das transicoes mapeadas no cromos-somo 1 tende a flutuar na frequencia proxima a um. Alem disso, este resultado e muitosemelhante ao mostrado para o genoma de B. taurus. Podemos notar que existem pontosde frequencias elevadas em diversas faixas de valores de ∆G negativo (−0, 8 kcal/mol e−1.6 kcal/mol) e positivo (0.2 kcal/mol e 0.9 kcal/mol).
Nos resultados das transversoes mostrados na figura 37b, podemos observar quatropontos de frequencia relativa de SNPs mais elevadas onde as as faixas de energia livre
53
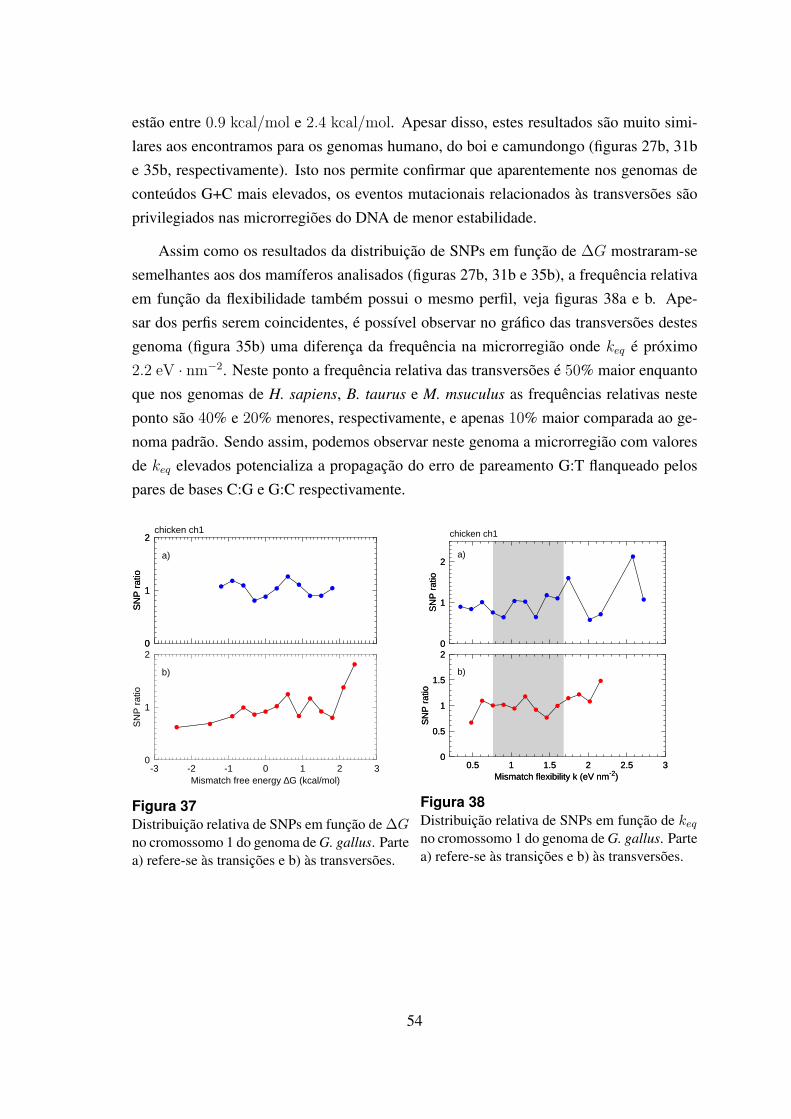
estao entre 0.9 kcal/mol e 2.4 kcal/mol. Apesar disso, estes resultados sao muito simi-lares aos encontramos para os genomas humano, do boi e camundongo (figuras 27b, 31be 35b, respectivamente). Isto nos permite confirmar que aparentemente nos genomas deconteudos G+C mais elevados, os eventos mutacionais relacionados as transversoes saoprivilegiados nas microrregioes do DNA de menor estabilidade.
Assim como os resultados da distribuicao de SNPs em funcao de ∆G mostraram-sesemelhantes aos dos mamıferos analisados (figuras 27b, 31b e 35b), a frequencia relativaem funcao da flexibilidade tambem possui o mesmo perfil, veja figuras 38a e b. Ape-sar dos perfis serem coincidentes, e possıvel observar no grafico das transversoes destesgenoma (figura 35b) uma diferenca da frequencia na microrregiao onde keq e proximo2.2 eV · nm−2. Neste ponto a frequencia relativa das transversoes e 50% maior enquantoque nos genomas de H. sapiens, B. taurus e M. msuculus as frequencias relativas nesteponto sao 40% e 20% menores, respectivamente, e apenas 10% maior comparada ao ge-noma padrao. Sendo assim, podemos observar neste genoma a microrregiao com valoresde keq elevados potencializa a propagacao do erro de pareamento G:T flanqueado pelospares de bases C:G e G:C respectivamente.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
chicken ch1
Figura 37Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de G. gallus. Partea) refere-se as transicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
chicken ch1
Figura 38Distribuicao relativa de SNPs em funcao de keqno cromossomo 1 do genoma de G. gallus. Partea) refere-se as transicoes e b) as transversoes.
54

6.4.5 Danio rerio
Conhecido tambem como o simpatico“peixe zebra” (zebra fish, paulistinha), este or-ganismo e comumente utilizado como ornamento nos diversos aquarios do mundo. Onome cientıfico dessa especie e D. rerio e e tido pela ciencia como um organismo mo-delo importante para o estudo do desenvolvimento e comportamento dos vertebrados, napesquisa de doencas e desenvolvimento de drogas. Seu genoma tem aproximadamente1.7 bilhoes de pares de bases, possui 25 haplotipos e a media do seu conteudo G+C e de36% sendo o menor conteudo em relacao aos organismos analisados anteriormente, vejasecoes 6.4.1, 6.4.2, 6.4.3, 6.4.4 (112).
Os resultados para os 25 haplotipos de D. rerio apresentaram-se de forma similarcom relacao a distribuicao de SNPs em funcao da energia livre e flexibilidade. Por isso,os resultados mostrados nesta secao, figuras 39 e 40 sao para o cromossomo 1 destaespecie.
Nos resultados referentes a frequencia relativa em funcao da energia livre e da flexibi-lidade mostrados nos graficos 39a e b, e possıvel verificar um perfil muito particular paraeste organismo quando o comparamos aos demais genomas ja analisados. A figura 39arepresenta o perfil da distribuicao das transicoes em funcao de ∆G e evidencia o padraode resultados diferentes encontrado para este organismo. Nas microrregioes que possuemo ∆G entre −1.3 kcal/mol e 0.1 kcal/mol, a ocorrencia de SNPs e aproximadamente25% menos frequentes que no organismo padrao. Por outro lado, com o aumento dosvalores de energia livre (> 0.1 kcal/mol) as frequencias de SNPs tambem tendem a au-mentar. Podemos observar que existem dois pontos de frequencia relativa de SNPs maisevidentes localizados nas microrregioes com valores de ∆G proximos de 0.6 kcal/mol
e 1.6 kcal/mol. Estes resultados sugerem que a microrregiao cujo valor de ∆G e apro-ximadamente 0.6 kcal/mol potencializa a ocorrencia do mismatch C:A juntamente comsuas bases vizinhas C:G e T:A. Ja a microrregiao que apresenta o valor mais alto de ener-gia livre (≈ 1.6 kcal/mol), o mismatch G:T flanqueado por A:T e o mais representativo.Essas analises nos levam a sugerir que neste genoma a instabilidade que estes erros depareamento promovem na estrutura pode potencializar o estabelecimento de certos poli-morfismos.
Com relacao aos resultados das transversoes mostrados no grafico 39b e possıvelobservar que exitem apenas dois pontos onde a frequencia relativa dos SNPs e elevada.O primeiro ponto incide na microrregiao onde o valor de ∆G e aproximadamente 0.6
kcal/mol sendo 30% mais representativa e o segundo ponto possui ∆G proximo de
55
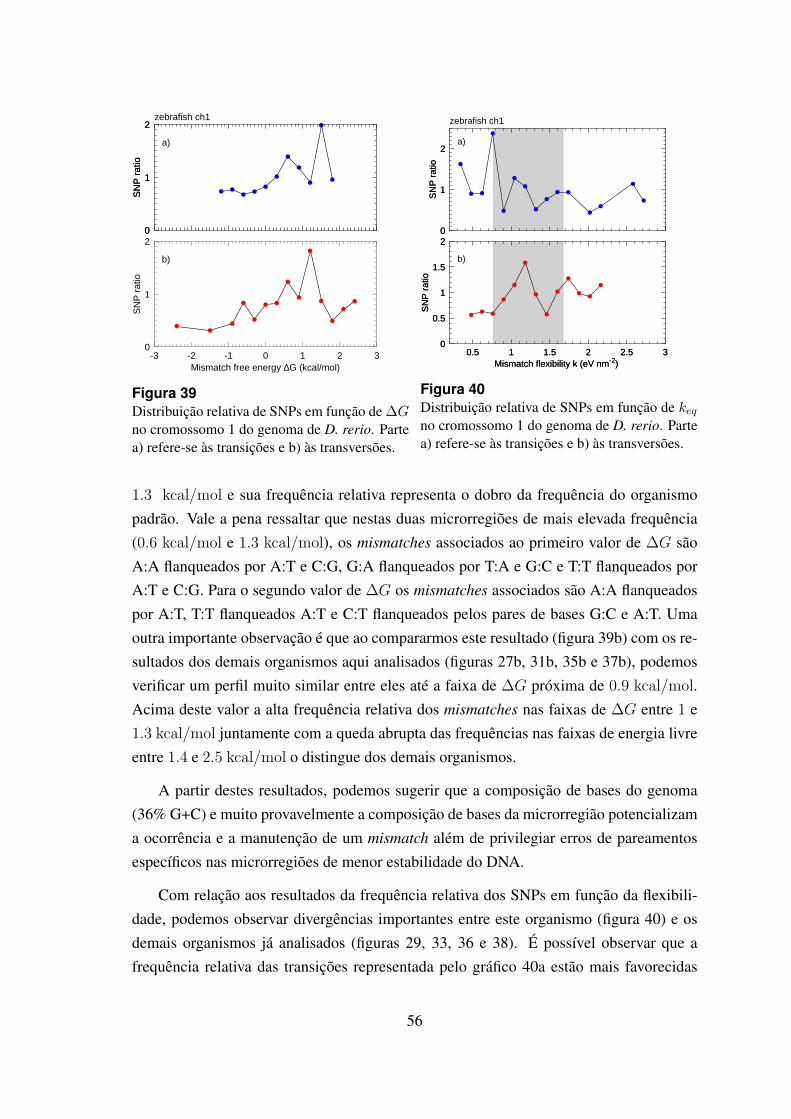
0
1
2S
NP
rat
io
0
1
2S
NP
rat
ioa)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
zebrafish ch1
Figura 39Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de D. rerio. Partea) refere-se as transicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
zebrafish ch1
Figura 40Distribuicao relativa de SNPs em funcao de keqno cromossomo 1 do genoma de D. rerio. Partea) refere-se as transicoes e b) as transversoes.
1.3 kcal/mol e sua frequencia relativa representa o dobro da frequencia do organismopadrao. Vale a pena ressaltar que nestas duas microrregioes de mais elevada frequencia(0.6 kcal/mol e 1.3 kcal/mol), os mismatches associados ao primeiro valor de ∆G saoA:A flanqueados por A:T e C:G, G:A flanqueados por T:A e G:C e T:T flanqueados porA:T e C:G. Para o segundo valor de ∆G os mismatches associados sao A:A flanqueadospor A:T, T:T flanqueados A:T e C:T flanqueados pelos pares de bases G:C e A:T. Umaoutra importante observacao e que ao compararmos este resultado (figura 39b) com os re-sultados dos demais organismos aqui analisados (figuras 27b, 31b, 35b e 37b), podemosverificar um perfil muito similar entre eles ate a faixa de ∆G proxima de 0.9 kcal/mol.Acima deste valor a alta frequencia relativa dos mismatches nas faixas de ∆G entre 1 e1.3 kcal/mol juntamente com a queda abrupta das frequencias nas faixas de energia livreentre 1.4 e 2.5 kcal/mol o distingue dos demais organismos.
A partir destes resultados, podemos sugerir que a composicao de bases do genoma(36% G+C) e muito provavelmente a composicao de bases da microrregiao potencializama ocorrencia e a manutencao de um mismatch alem de privilegiar erros de pareamentosespecıficos nas microrregioes de menor estabilidade do DNA.
Com relacao aos resultados da frequencia relativa dos SNPs em funcao da flexibili-dade, podemos observar divergencias importantes entre este organismo (figura 40) e osdemais organismos ja analisados (figuras 29, 33, 36 e 38). E possıvel observar que afrequencia relativa das transicoes representada pelo grafico 40a estao mais favorecidas
56

entre as faixas de keq que vao de 0.4 a 0.8 eV · nm−2. Esta pequena mudanca do padraomostra que estes eventos estao privilegiados nas microrregioes de maior flexibilidade,sendo esta uma caracterıstica particular deste genoma. Alem disso, os dois picos maisproeminentes (60% e 90% maiores que o padrao) mostrados neste grafico representam ofavorecimento do mismatch G:T e seus pares de bases vizinhas A:T na microrregiao comkeq ≈ 0.8 eV · nm−2 e os mismatches A:C flanqueado por A:T, G:T flanqueados por G:Ce A:T e A:C flanqueado pelo par de base G:C.
Os resultados apresentados para as transversoes, grafico 40b, mostram os SNPS saopouco frequentes nas microrregioes de maior flexibilidade (keq entre 0.4−0.9 eV · nm−2).Por outro lado, e possıvel observar uma progressao da frequencia de SNPs nas micror-regioes que coincidem com as faixas de keq dos pareamentos canonicos (faixa cinzado grafico) seguida de uma queda abrupta. Este pico mais elevado (keq ≈ 1.2) cujafrequencia dos SNPs e aproximadamento 60% maior que o padrao esta associado tambema maior frequencia dos mismatches A:A e T:T ambos flanqueados por A:T.
Os resultados das transicoes e transversoes deste genoma pode ser um indicativo deque apesar de haver erros de pareamentos nas microrregioes mais flexıveis do DNA emuito provavel que estes erros nao promovam perturbacoes mecanicas importantes aoponto de serem reconhecidas por exemplo pela maquinaria de reparo. Alem disso a maiorfrequencia das transversoes sao encontradas nas microrregioes cujas faixas de flexibi-lidade sao tambem para os pareamentos canonicos e este tambem pode ser um motivoassociado ao nao reparo destes mismatches. Uma outra observacao importante e que oconteudo de G+C deste genoma e menor (36%) com relacao aos demais genomas (tab. 8,pagina 35) e isto pode ser um forte indicativo de que um genoma com menor conteudoG+C pode privilegiar o aumento da frequencia de polimorfismos em microrregioes doDNA que tendem a ser mais flexıveis.
6.4.6 Apis mellifera
Esta especie conhecida tambem como “abelha do mel” e de grande importancia economicae agrıcola pela sua alta capacidade de polinizacao e producao de mel. Na pesquisa a A.
mellifera e um organismo modelo muito utilizado para os estudos de imunidade e dedoencas ligadas ao cromossomo X (114). Seu genoma esta estimado em 2.7 milhoes depares de bases, possui 16 haplotipos sendo o cromossomo 1 o maior deles. A media doseu conteudo G+C e aproximadamente 30%, assim A. mellifera representa o genoma demenor tamanho e de menor conteudo G+C analisada neste estudo (113).
57
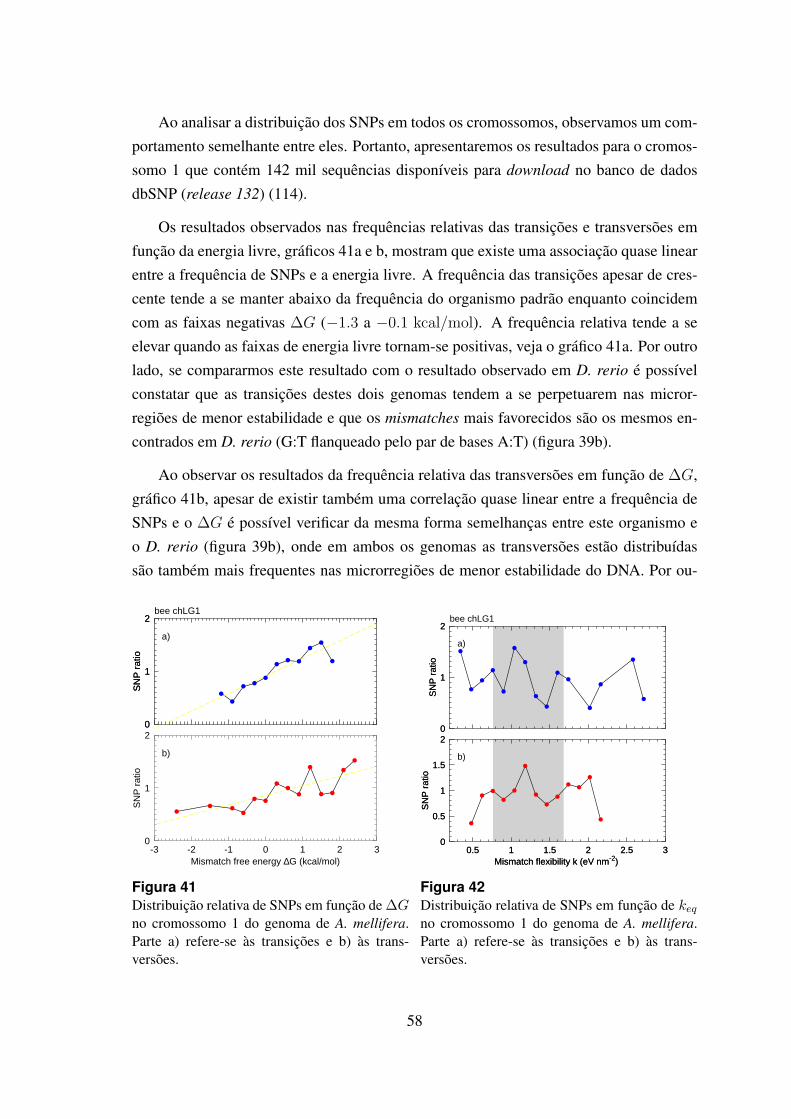
Ao analisar a distribuicao dos SNPs em todos os cromossomos, observamos um com-portamento semelhante entre eles. Portanto, apresentaremos os resultados para o cromos-somo 1 que contem 142 mil sequencias disponıveis para download no banco de dadosdbSNP (release 132) (114).
Os resultados observados nas frequencias relativas das transicoes e transversoes emfuncao da energia livre, graficos 41a e b, mostram que existe uma associacao quase linearentre a frequencia de SNPs e a energia livre. A frequencia das transicoes apesar de cres-cente tende a se manter abaixo da frequencia do organismo padrao enquanto coincidemcom as faixas negativas ∆G (−1.3 a −0.1 kcal/mol). A frequencia relativa tende a seelevar quando as faixas de energia livre tornam-se positivas, veja o grafico 41a. Por outrolado, se compararmos este resultado com o resultado observado em D. rerio e possıvelconstatar que as transicoes destes dois genomas tendem a se perpetuarem nas micror-regioes de menor estabilidade e que os mismatches mais favorecidos sao os mesmos en-contrados em D. rerio (G:T flanqueado pelo par de bases A:T) (figura 39b).
Ao observar os resultados da frequencia relativa das transversoes em funcao de ∆G,grafico 41b, apesar de existir tambem uma correlacao quase linear entre a frequencia deSNPs e o ∆G e possıvel verificar da mesma forma semelhancas entre este organismo eo D. rerio (figura 39b), onde em ambos os genomas as transversoes estao distribuıdassao tambem mais frequentes nas microrregioes de menor estabilidade do DNA. Por ou-
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
bee chLG1
Figura 41Distribuicao relativa de SNPs em funcao de ∆Gno cromossomo 1 do genoma de A. mellifera.Parte a) refere-se as transicoes e b) as trans-versoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
bee chLG1
Figura 42Distribuicao relativa de SNPs em funcao de keqno cromossomo 1 do genoma de A. mellifera.Parte a) refere-se as transicoes e b) as trans-versoes.
58

tro lado, vale a pena ressaltar que existem dois pontos divergentes entre estes genomas.Em A. mellifera alem frequencia relativa na microrregiao onde ∆G e ≈ 1.3, podemosobservar mais dois pontos de frequencias elevadas entre as faixas de energia livre 2.1 e2.4 kcal/mol que estao correlacionadas ao mismatch C:C flanquedo pelos pares de basesG:C e A:T ou apenas por A:T.
Nos resultados dos SNPs oriundos das transicoes em funcao da flexibilidade (fi-gura 42a) e possıvel identificar flutuacoes abruptas entre as frequencias relativas. Apesardisso, e possıvel observar que a maioria dos picos cujas frequencias relativas sao maiselevadas, concentram-se nas microrregioes flexıveis. Por outro lado, e possıvel ver queexiste uma frequencia de SNPs elevada na microrregiao onde o keq e alto, mostrando quealem dos sıtios flexıveis, exite um sıtio rıgido que tambem potencializa a manutencao dopolimorfismo neste genoma.
O grafico 42b descreve o perfil da distribuicao das transversoes em funcao da fle-xibilidade. Um resultado similar entre este organismo e D. rerio (figura 40b) pode serobservado no sıtio onde o valor de keq e proximo de 1.2 eV · nm−2(mismatches A:A eT:T ambos flanqueados por A:T). Por outro lado, e possıvel observar que exitem doispontos cuja frequencia relativa de SNPs e aproximadamente 60% menor que o padrao.A microrregiao de menor keq (0.5 eV · nm−2), ou seja, mais flexıvel, esta associada abaixa frequencia do mismatch A:G e seus pares de bases adjacentes G:C. Ja a micror-regiao de maior keq (2.3 eV · nm−2), ou seja, a mais rıgida, esta correlacionada a menorfrequencia de dois mismatches: G:G e TT flanqueados pelos pares de bases G:C. Maisuma vez, estes resultados nos levam a acreditar que e muito provavel que a composicaodas bases adjacentes interferem no estabelecimento de um polimorfismo oriundo de umatransversao uma vez que os mismatches mais frequentes deste organismo estao associ-ados as bases adjacentes A e T e os menos frequentes sao flanqueados pelas bases C eG. Vale a pena ressaltar que as semelhancas entre os resultados deste genoma e o de ze-bra fish (figura 40) podem estar associados ao baixo conteudo G+C destes genomas. Epossıvel observar que a medida que o conteudo G+C diminui, o perfil das transicoes e dastransversoes em funcao da flexibilidade tendem a se manter de forma similar entre eles ediferente dos perfis encontrados para os mamıferos e a ave (figuras 29b, 33b, 36b, 38b)aqui analisados.
6.4.7 Plasmodium falciparum
A malaria e uma doenca parasitologica provocada pelo protozoario do genero Plas-
modium e a forma grave desta doenca e provocada pela especie Plasmodium falciparum.
59
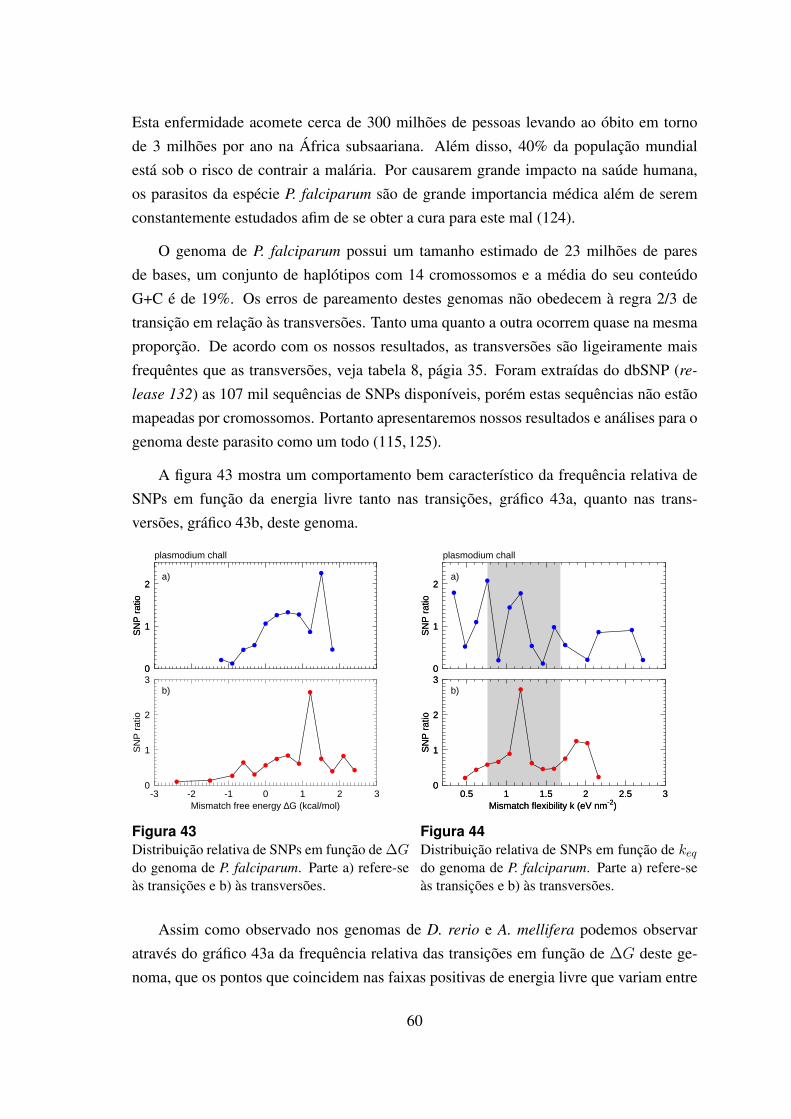
Esta enfermidade acomete cerca de 300 milhoes de pessoas levando ao obito em tornode 3 milhoes por ano na Africa subsaariana. Alem disso, 40% da populacao mundialesta sob o risco de contrair a malaria. Por causarem grande impacto na saude humana,os parasitos da especie P. falciparum sao de grande importancia medica alem de seremconstantemente estudados afim de se obter a cura para este mal (124).
O genoma de P. falciparum possui um tamanho estimado de 23 milhoes de paresde bases, um conjunto de haplotipos com 14 cromossomos e a media do seu conteudoG+C e de 19%. Os erros de pareamento destes genomas nao obedecem a regra 2/3 detransicao em relacao as transversoes. Tanto uma quanto a outra ocorrem quase na mesmaproporcao. De acordo com os nossos resultados, as transversoes sao ligeiramente maisfrequentes que as transversoes, veja tabela 8, pagia 35. Foram extraıdas do dbSNP (re-
lease 132) as 107 mil sequencias de SNPs disponıveis, porem estas sequencias nao estaomapeadas por cromossomos. Portanto apresentaremos nossos resultados e analises para ogenoma deste parasito como um todo (115, 125).
A figura 43 mostra um comportamento bem caracterıstico da frequencia relativa deSNPs em funcao da energia livre tanto nas transicoes, grafico 43a, quanto nas trans-versoes, grafico 43b, deste genoma.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
3
SN
P r
atio
b)
plasmodium chall
Figura 43Distribuicao relativa de SNPs em funcao de ∆Gdo genoma de P. falciparum. Parte a) refere-seas transicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3
SN
P r
atio
plasmodium chall
Figura 44Distribuicao relativa de SNPs em funcao de keqdo genoma de P. falciparum. Parte a) refere-seas transicoes e b) as transversoes.
Assim como observado nos genomas de D. rerio e A. mellifera podemos observaratraves do grafico 43a da frequencia relativa das transicoes em funcao de ∆G deste ge-noma, que os pontos que coincidem nas faixas positivas de energia livre que variam entre
60

0.1 e 1.4 kcal/mol possuem a frequencia relativa elevada. Por outro lado, apesar de exis-tirem varios pontos onde a frequencia relativa e elevada, nenhuma outra e tao evidentequanto frequencia da microrregiao com valor de ∆G proximo de 1.4 kcal/mol cujo mis-
match associado e o G:T flanqueado pelo par de bases A:T. E importante ressaltar que osresultados da frequencia relativa das transicoes em funcao de ∆G para os genomas de D.
rerio, A. mellifera e P. falciparum cujo conteudo G+C e baixo (36%, 30% e 19% respecti-vamente), nos permite observar que estas microrregioes juntamente com sua composicaode bases podem favorecer a preferencia e a manutencao de um mismatch especıfico (G:T).Alem disso, e possıvel identificar que alguns erros de pareamentos neste genoma sao rarosem funcao da baixa frequencia relativa (< 10%) dos SNPs nas microrregioes com valoresde ∆G entre −1.2 e −0.9 kcal/mol. Os mismatches associados a esta microrregiao saoA:C e G:T ambos flanqueados pelos pares de bases G:C. Porem, apesar de existir doiserros de pareamentos iguais (G:T) com frequencias e valores de ∆G diferentes, podemosobservar que os pares de bases que os flanqueiam sao divergentes (G:T flanqueado porG:C; G:T flanqueado por A:T) sugerindo que a energia livre local aparentemente deter-mina a frequencia e a preferencia de um SNPs.
Com relacao aos resultados referentes a distribuicao das transversoes em funcao de∆G e possıvel verificar um unico ponto de frequencia relativa de SNPs mais elevada, vejao grafico 43. As transversoes se mostram expressivas na microrregiao que possui valorde ∆G proximo de 1.2 kcal/mol, enquanto as demais transversoes sao pouco frequentes.Podemos observar que nesta faixa de energia livre a elevada frequencia de SNPs e comumem todos os genomas analisados (figuras 27b, 31b, 35b, 37b, 39b, 41b), porem nestegenoma esta frequencia e a mais proeminente. Alem disso, assim como observado nastransicoes deste genoma, existem erros de pareamentos que tambem sao raros (< 10%)nas transversoes como o mismatch G:G flanqueado pelo par de bases G:C, fato que nosleva a inferir mais uma vez que a energia livre local aparentemente determina a frequenciae a preferencia de um SNPs uma vez que as transversao C:A flanqueadas pelos pares debases A:T sao os erros mais frequentes neste genoma. Vale a pena ressaltar que o fato dasbases adjacentes A e T estarem flanqueando o mismatch mais frequente pode ser explicadapelo baixo conteudo de G+C (19%) deste genoma.
Analisando os resultados da distribuicao das transicoes em funcao da flexibilidade,grafico 44a, observamos cinco pontos cujas frequencias relativas sao elevadas entre as fai-xas de keq que vao de 0.3 a 1.25 eV · nm−2seguidas de uma reducao abrupta da frequenciarelativa nas microrregioes mais rıgidas. Alem disso tres pontos de elevada frequencia po-dem ser observados dentro da faixa de keq dos pareamentos canonicos. Como ja discutidoanteriormente, o fato de existir uma grande frequencia de erros de pareamentos com valo-
61

res de flexibilidade proximos aos dos pareamentos canonicos, reafirma que estes eventospodem promover pouca perturbacao na estrutura do DNA podendo ser uma das causas doseu nao reconhecimento pelo mecanismo de reparo. Por outro lado, resultados similaresforam encontrados nos genomas de D. rerio e de A. mellifera o que pode ser um indicativode que o baixo conteudo G+C destes genomas pode potencializar atraves dos sıtios maisflexıveis a manutencao e a perpetuacao do polimorfismo nestas especies.
O grafico 44b de P. falciparum mostra os resultados da frequencia relativa das trans-versoes em funcao da flexibilidade. Podemos verificar que apesar de existirem tres pon-tos cujas frequencias sao elevadas, a frequencia de SNPs mais evidente (3 vezes maisfrequente que o padrao) concentra-se na microrregiao com valores de keq em torno de1.3 eV · nm−2 e que coincide tambem com a mesma faixa de keq dos pareamentos canonicos(faixa cinza do grafico). Resutados similares foram encontrados tambem nos demais ge-nomas analisados (figuras 29b, 33b, 36b, 38b, 40b, 42b) porem neste genoma e possıvelnotar que a frequencia relativa concentra-se nesta faixa de flexibilidade (1.3 eV · nm−2).O grupo de mismatches mais representativos sao A:A e T:T ambos flanqueados pelospares de bases A:T que faz deste sıtio uma microrregiao flexıvel e com comportamentoestrutural similar aos pareamentos canonicos.
6.4.8 Oryza sativa
A Oryza sativa e uma das duas sub-especies de arroz mais cultivadas em climas quen-tes, principalmente no sul da Asia. De grande importancia economica agrıcola, o arroze um alimento basico nas refeicoes da maioria da populacao mundial alem de ser larga-mente utilizado como organismo modelo em projetos de pesquisas (126).
A sub especie O. sativa pertence ao domınio Eukaryota porem esta inserida no reinoViridiplantae, diferente dos demais genomas ate agora descritos que se encontram classi-ficados no reino Metazoa. O tamanho do genoma do arroz esta estimado em 3.7 milhoesde pares de bases, e composto por 12 haplotipos e a media do conteudo G+C e de 42%.Como nossos resultados referentes a influencia da energia livre e flexibilidade no padraoda distribuicao de SNPs nos 12 cromossomos deste genoma foram semelhantes entre si,apresentaremos os resultados para o cromossomo 1. Este cromossomo e o maior emnumero de pares de bases e em numero de sequencias de SNPs sendo (547 mil) dis-ponıveis no dbSNP (release 132).
Inciamos a nossa discussao com as analises dos resultados da frequencia relativadas transicoes em funcao de ∆G nesta especie, veja figura 45a. Podemos verificar que
62

0
1
2S
NP
rat
io
0
1
2S
NP
rat
ioa)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
rice ch1
Figura 45Distribuicao relativa de SNPs em funcao de ∆Gdo genoma de O. sativa. Parte a) refere-se astransicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
rice ch1
Figura 46Distribuicao relativa de SNPs em funcao de keqdo genoma de O. sativa. Parte a) refere-se astransicoes e b) as transversoes.
este tipo de SNP se distribui sob a forma de uma flutuacao discreta cujas frequencias semantem proximas de 1 sendo semelhante a distribuicao gaussiana encontrada no orga-nismo padrao (figura 22a).
Avaliando o perfil da frequencia relativa das transversoes em funcao de ∆G, grafi-co45b, percebemos que este perfil e muito semelhante aos perfis dos demais genomasate agora discutidos (figuras 27b, 31b, 35b, 37b, 39b, 41b e 43b), principalmente nasmicrorregioes em que os valores de ∆G variam entre 0.1 a 1.8 kcal/mol. Tendo em vistaeste ponto em comum entre os organismos, podemos afirmar a existencia de um padraona distribuicao das transversoes em funcao de ∆G pode indicar que este sıtio especıficopode favorecer a incorporacao de erros bem como sua propagacao.
Podemos verificar que a frequencia relativa das transicoes em funcao da flexibili-dade mostrada na figura 46a, assemelha-se mais aos perfis encontrados nos resultadosdas transicoes dos genomas dos mamıferos e da ave (figuras 29b, 33b, 36b, 38b), cujosconteudos G+C sao similares do que aos resultados encontrados nos genomas D. rerio,A. mellifera e P. falciparum (figuras 40b, 42b e 44b respectivamente). Sendo assim, estesresultados tambem nos confirmam que as transicoes sao mais favorecidas nas micror-regioes mais rıgidas deste genoma e dos genomas cujo conteudo G+C sao semelhantes(H. sapiens, B. taurus, M. musculus e G. gallus).
Os resultados mostrados para a frequencia relativa das transversoes em funcao da fle-xibilidade, grafico 46b, assemelham-se tambem com o perfil dos graficos dos mamıferos
63

e da ave confirmando que alem da microrregiao que possui valores de keq proximos dosvalores dos pareamentos canonicos (faixa cinza do grafico), as microrregioes que con-ferem maior rigidez na estrutura do DNA, cujas faixas de keq vao de 1.7 a 2.2, tambempotencializam a propagacao dos mismatches neste genoma e nos genomas de H. sapiens,B. taurus, M. musculus, G. gallus e A. mellifera, veja figuras 29b, 33b, 36b, 38b e 46b.
6.4.9 Streptococcus pyogenes
A bacteria patogenica Streptococcus pyogenes (estreptococo do grupo A) esta as-sociada a varias manifestacoes clınicas importantes como a sındrome do choque toxicoestreptococica, febre reumatica, escarlatina, dentre outras. Esses aspectos fazem desteorganismo um importante patogeno alvo de varias pesquisas para o desenvolvimentode farmacos alem de ser um organismo bastante interessante para o estudo de meca-nismos evolutivos relacionados a sua capacidade de escape do sistema imunologico hu-mano (117).
O genoma de S. pyogenes e circular e tem um tamanho medio de 1.85 milhoes depares de bases com conteudo G+C de aproximadamente 38% (127). Existem aproxima-damente 4 mil sequencias de SNPs de bacterias disponıveis para download no dbSNP(release 132). Dentre estas apenas 3100 sequencias estao disponıveis para o organismoS. pyogenes. Como um dos nossos objetivos e comparar o perfil da distribuicao dos SNPsem funcao da energia livre e da flexibilidade entre os diferentes genomas, resolvemos uti-lizar um organismo procarioto para verificar se alguns padroes encontrados nos resultadosdos eucariotos sao tambem reincidentes para este genoma.
Os resultados apresentados nos graficos da figura 47a mostram que a frequencia re-lativa das transicoes em funcao da energia livre deste genoma possui um padrao comumaos demais genomas analisados. Podemos observar que a baixa frequencia de SNPs nasfaixas em que ∆G e negativo tambem esta presente neste organismo. Por outro lado,tambem e possıvel verificar que a frequencia relativa das transicoes tende a aumentar nasmicrorregioes menos estaveis deste genoma.
Ja as transversoes deste organismo, grafico 47b, possuem um padrao de distribuicaode SNPs diferentes dos genomas de H. sapiens e B. taurus (figuras 29b e 33b respectiva-mente) mas similar ao resultados encontrados nos genomas de M. musculus e G. gallus,D. rerio, A. mellifera, P. falciparum e O. sativa (figuras 36b, 38b, 40b, 42b, 44b e 46brespectivamente). De uma forma geral, estes resultados nos levam a acreditar que de fatoum erro de incorporacao de base esta associado a estabilidade de uma microrregiao e que
64
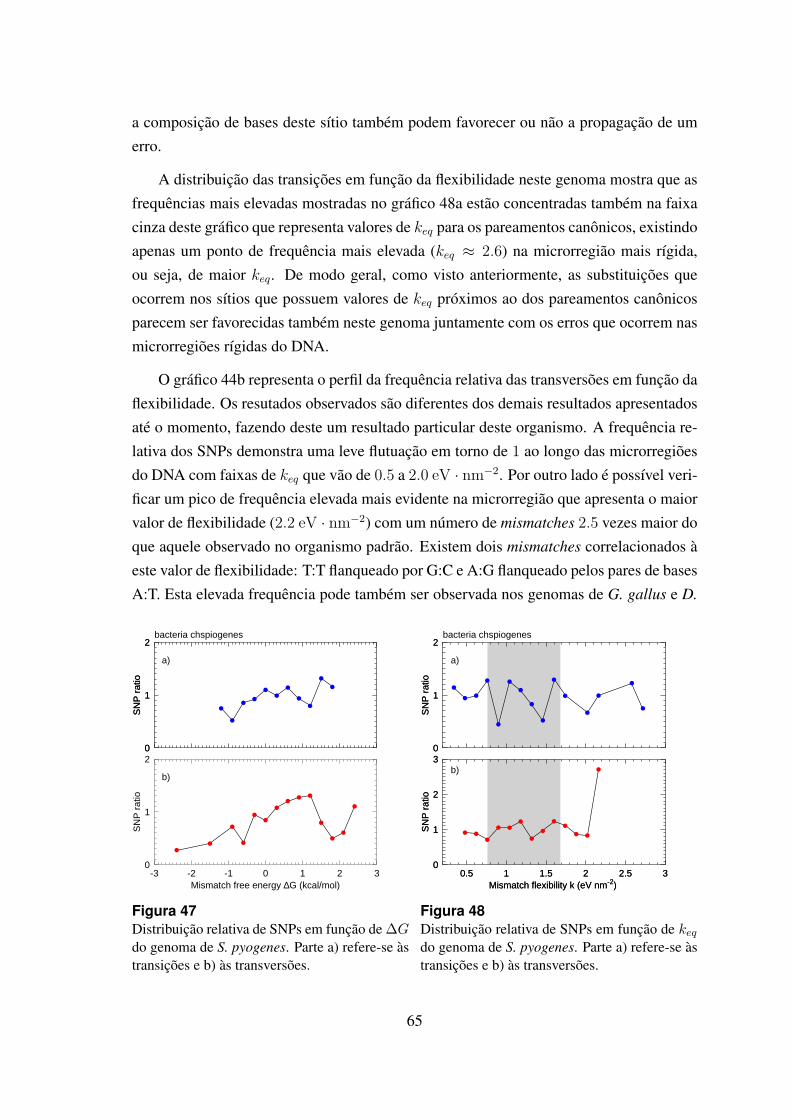
a composicao de bases deste sıtio tambem podem favorecer ou nao a propagacao de umerro.
A distribuicao das transicoes em funcao da flexibilidade neste genoma mostra que asfrequencias mais elevadas mostradas no grafico 48a estao concentradas tambem na faixacinza deste grafico que representa valores de keq para os pareamentos canonicos, existindoapenas um ponto de frequencia mais elevada (keq ≈ 2.6) na microrregiao mais rıgida,ou seja, de maior keq. De modo geral, como visto anteriormente, as substituicoes queocorrem nos sıtios que possuem valores de keq proximos ao dos pareamentos canonicosparecem ser favorecidas tambem neste genoma juntamente com os erros que ocorrem nasmicrorregioes rıgidas do DNA.
O grafico 44b representa o perfil da frequencia relativa das transversoes em funcao daflexibilidade. Os resutados observados sao diferentes dos demais resultados apresentadosate o momento, fazendo deste um resultado particular deste organismo. A frequencia re-lativa dos SNPs demonstra uma leve flutuacao em torno de 1 ao longo das microrregioesdo DNA com faixas de keq que vao de 0.5 a 2.0 eV · nm−2. Por outro lado e possıvel veri-ficar um pico de frequencia elevada mais evidente na microrregiao que apresenta o maiorvalor de flexibilidade (2.2 eV · nm−2) com um numero de mismatches 2.5 vezes maior doque aquele observado no organismo padrao. Existem dois mismatches correlacionados aeste valor de flexibilidade: T:T flanqueado por G:C e A:G flanqueado pelos pares de basesA:T. Esta elevada frequencia pode tambem ser observada nos genomas de G. gallus e D.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
bacteria chspiogenes
Figura 47Distribuicao relativa de SNPs em funcao de ∆Gdo genoma de S. pyogenes. Parte a) refere-se astransicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3
SN
P r
atio
bacteria chspiogenes
Figura 48Distribuicao relativa de SNPs em funcao de keqdo genoma de S. pyogenes. Parte a) refere-se astransicoes e b) as transversoes.
65

rerio (figuras 38b e 40b respectivamente) porem a mais proeminente encontra-se nestegenoma.
Os resultados obtidos para este genoma mostram um comportamento misto quandoos perfis de energia livre e flexibilidades sao comparados aos demais organismos ana-lisados sugerindo que um o conteudo G+C menor ou maior que 38% pode ser um dosdeterminantes na preferencia e na distribuicao de polimorfismos nos genomas.
66

6.5 Analise por regioes genomicas: resultados preliminares
A evolucao do DNA esta associada ao acumulo de substituicoes no genoma ao longodas geracoes. As taxas de mutacoes variam e sao dependentes das regioes do DNA ondeelas ocorrem. Isto esta associado a pressao seletiva que atua de forma nao uniforme aolongo genoma, por exemplo em regioes codificadoras e nao codificadoras do genoma.
Alteracoes nas regioes codificantes do DNA promovidas por polimorfismos, podemimplicar em alteracoes na sequencia de aminoacido e consequentemente na alteracao es-trutura e funcao de uma proteına (1, 16). Como discutido na secao 2.2, pagina 6, asmutacoes podem ser silenciosas, conservativas ou nao conservativas. Neste contexto,apresentamos nesta secao alguns resultados prelimares das analises comparativas feitaspara o genoma humano que teve as suas sequencias de SNPs separadas em funcao da suaocorrencia em regioes codificadoras de proteınas (CDS) e nao codificantes de proteınas(introns).
6.5.1 Analise comparativa previa: Homo sapiens
A partir destas analises a primeira informacao importante que estes resultados nosmostram esta associado ao perfil de ocorrencia da frequencia relativa dos SNPs em relacaoa energia e a flexibilidade que sao diferentes entre as regioes codificantes e nao codifican-tes de proteınas.
Os resultados da frequencia relativa das transicoes em funcao de ∆G que ocorremnas regioes codificantes de proteınas possuem perfis similares independente do tipo demutacao ocorrida (sinonima ou nao sinonima), veja figuras 49a e 53a. E possıvel observara existencia de seis pontos de frequencia relativa mais elevada, porem os tres picos maisproeminentes concentram-se nas faixas de ∆G proximos de −0.9, 0.9 e 1.8 kcal/mol. Osmismatches associados a estes valores de energia livre sao G:T flanqueado pelos pares debases G:C e A:C e A:C flanqueado pelos pares de bases G:C e A:T.
Estes resultados mostram que a maioria dos erros de pareamentos encontram-se entreas bases G e C no genoma e que apesar de conferirem uma maior estabilidade entresuas ligacoes potencializam a ocorrencia destes mismatches. Por outro lado e possıvelobservar que a frequencia relativa dos SNPs diminui progressivamente entre os tres picosdestacados, indicando que os sıtios das regioes codificantes compostos por G e C podemfavorecer os erros de incorporacao de bases em microrregioes mais estaveis da molecula.
Comparando estes resultados com as transicoes nas regioes intronicas do DNA, grafico 51a,
67
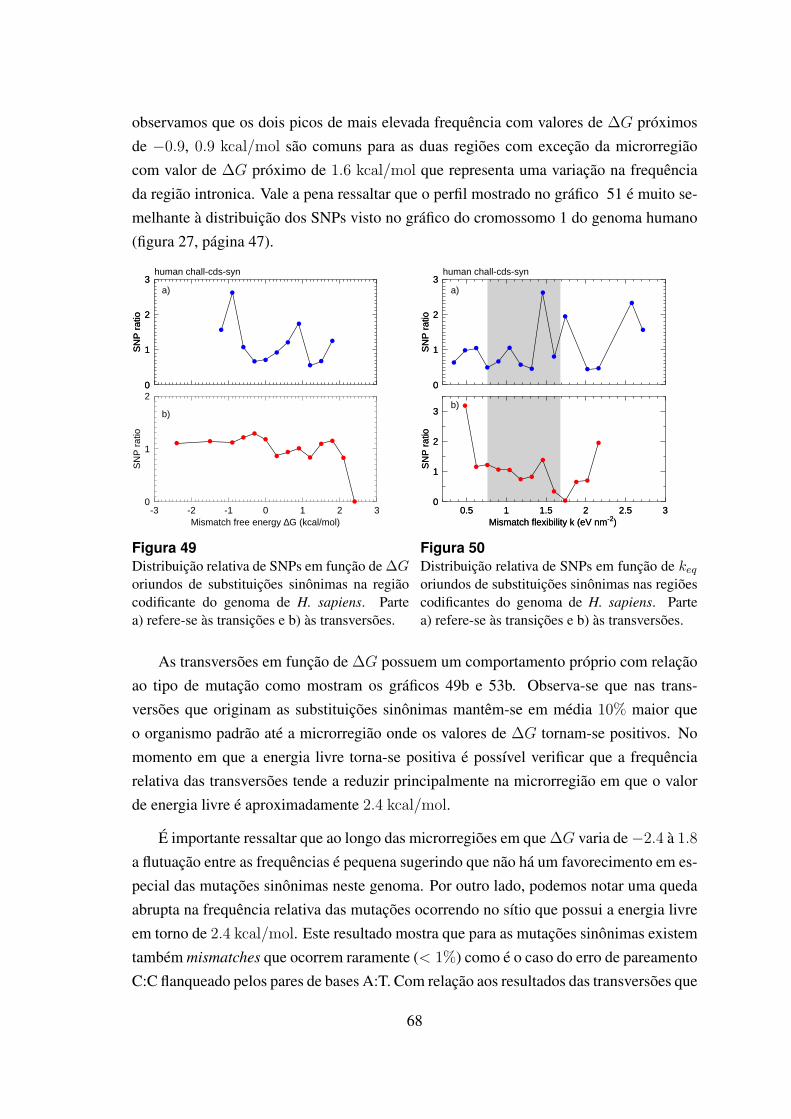
observamos que os dois picos de mais elevada frequencia com valores de ∆G proximosde −0.9, 0.9 kcal/mol sao comuns para as duas regioes com excecao da microrregiaocom valor de ∆G proximo de 1.6 kcal/mol que representa uma variacao na frequenciada regiao intronica. Vale a pena ressaltar que o perfil mostrado no grafico 51 e muito se-melhante a distribuicao dos SNPs visto no grafico do cromossomo 1 do genoma humano(figura 27, pagina 47).
0
1
2
3
SN
P r
atio
0
1
2
3
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human chall-cds-syn
Figura 49Distribuicao relativa de SNPs em funcao de ∆Goriundos de substituicoes sinonimas na regiaocodificante do genoma de H. sapiens. Partea) refere-se as transicoes e b) as transversoes.
0
1
2
3
SN
P r
atio
0
1
2
3
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3S
NP
rat
io
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
1
2
3S
NP
rat
io
human chall-cds-syn
Figura 50Distribuicao relativa de SNPs em funcao de keqoriundos de substituicoes sinonimas nas regioescodificantes do genoma de H. sapiens. Partea) refere-se as transicoes e b) as transversoes.
As transversoes em funcao de ∆G possuem um comportamento proprio com relacaoao tipo de mutacao como mostram os graficos 49b e 53b. Observa-se que nas trans-versoes que originam as substituicoes sinonimas mantem-se em media 10% maior queo organismo padrao ate a microrregiao onde os valores de ∆G tornam-se positivos. Nomomento em que a energia livre torna-se positiva e possıvel verificar que a frequenciarelativa das transversoes tende a reduzir principalmente na microrregiao em que o valorde energia livre e aproximadamente 2.4 kcal/mol.
E importante ressaltar que ao longo das microrregioes em que ∆G varia de −2.4 a 1.8a flutuacao entre as frequencias e pequena sugerindo que nao ha um favorecimento em es-pecial das mutacoes sinonimas neste genoma. Por outro lado, podemos notar uma quedaabrupta na frequencia relativa das mutacoes ocorrendo no sıtio que possui a energia livreem torno de 2.4 kcal/mol. Este resultado mostra que para as mutacoes sinonimas existemtambem mismatches que ocorrem raramente (< 1%) como e o caso do erro de pareamentoC:C flanqueado pelos pares de bases A:T. Com relacao aos resultados das transversoes que
68
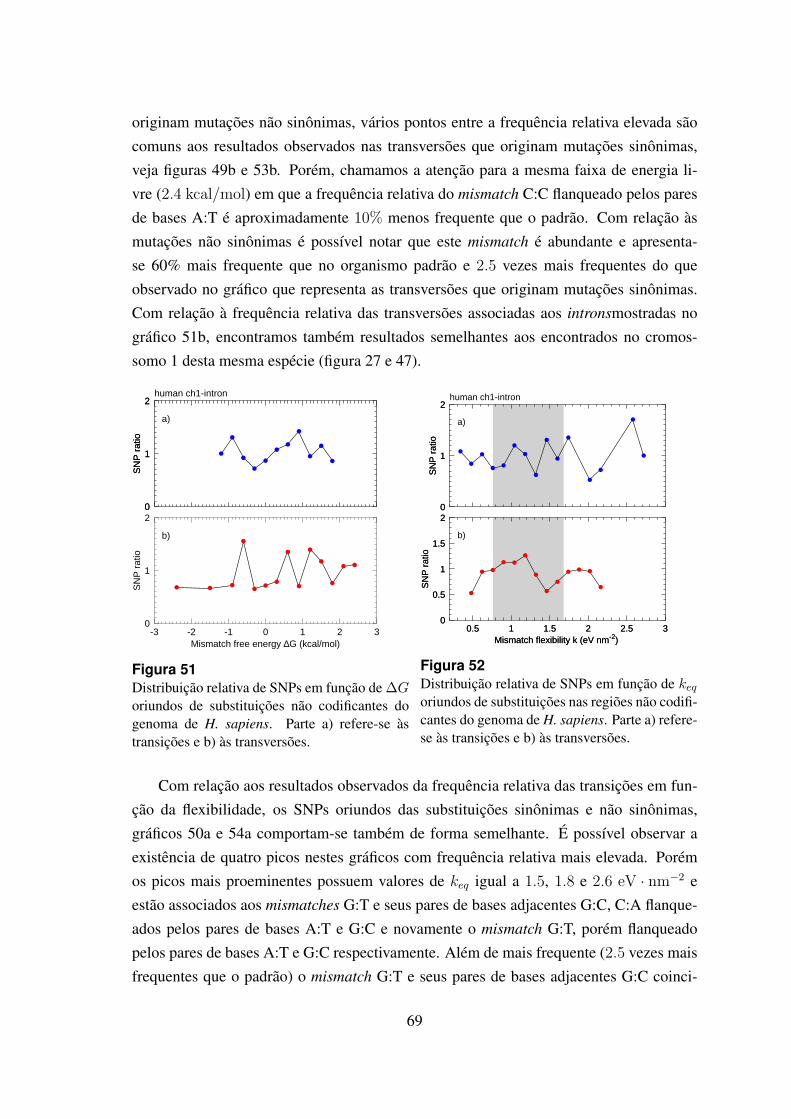
originam mutacoes nao sinonimas, varios pontos entre a frequencia relativa elevada saocomuns aos resultados observados nas transversoes que originam mutacoes sinonimas,veja figuras 49b e 53b. Porem, chamamos a atencao para a mesma faixa de energia li-vre (2.4 kcal/mol) em que a frequencia relativa do mismatch C:C flanqueado pelos paresde bases A:T e aproximadamente 10% menos frequente que o padrao. Com relacao asmutacoes nao sinonimas e possıvel notar que este mismatch e abundante e apresenta-se 60% mais frequente que no organismo padrao e 2.5 vezes mais frequentes do queobservado no grafico que representa as transversoes que originam mutacoes sinonimas.Com relacao a frequencia relativa das transversoes associadas aos intronsmostradas nografico 51b, encontramos tambem resultados semelhantes aos encontrados no cromos-somo 1 desta mesma especie (figura 27 e 47).
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human ch1-intron
Figura 51Distribuicao relativa de SNPs em funcao de ∆Goriundos de substituicoes nao codificantes dogenoma de H. sapiens. Parte a) refere-se astransicoes e b) as transversoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
human ch1-intron
Figura 52Distribuicao relativa de SNPs em funcao de keqoriundos de substituicoes nas regioes nao codifi-cantes do genoma de H. sapiens. Parte a) refere-se as transicoes e b) as transversoes.
Com relacao aos resultados observados da frequencia relativa das transicoes em fun-cao da flexibilidade, os SNPs oriundos das substituicoes sinonimas e nao sinonimas,graficos 50a e 54a comportam-se tambem de forma semelhante. E possıvel observar aexistencia de quatro picos nestes graficos com frequencia relativa mais elevada. Poremos picos mais proeminentes possuem valores de keq igual a 1.5, 1.8 e 2.6 eV · nm−2 eestao associados aos mismatches G:T e seus pares de bases adjacentes G:C, C:A flanque-ados pelos pares de bases A:T e G:C e novamente o mismatch G:T, porem flanqueadopelos pares de bases A:T e G:C respectivamente. Alem de mais frequente (2.5 vezes maisfrequentes que o padrao) o mismatch G:T e seus pares de bases adjacentes G:C coinci-
69
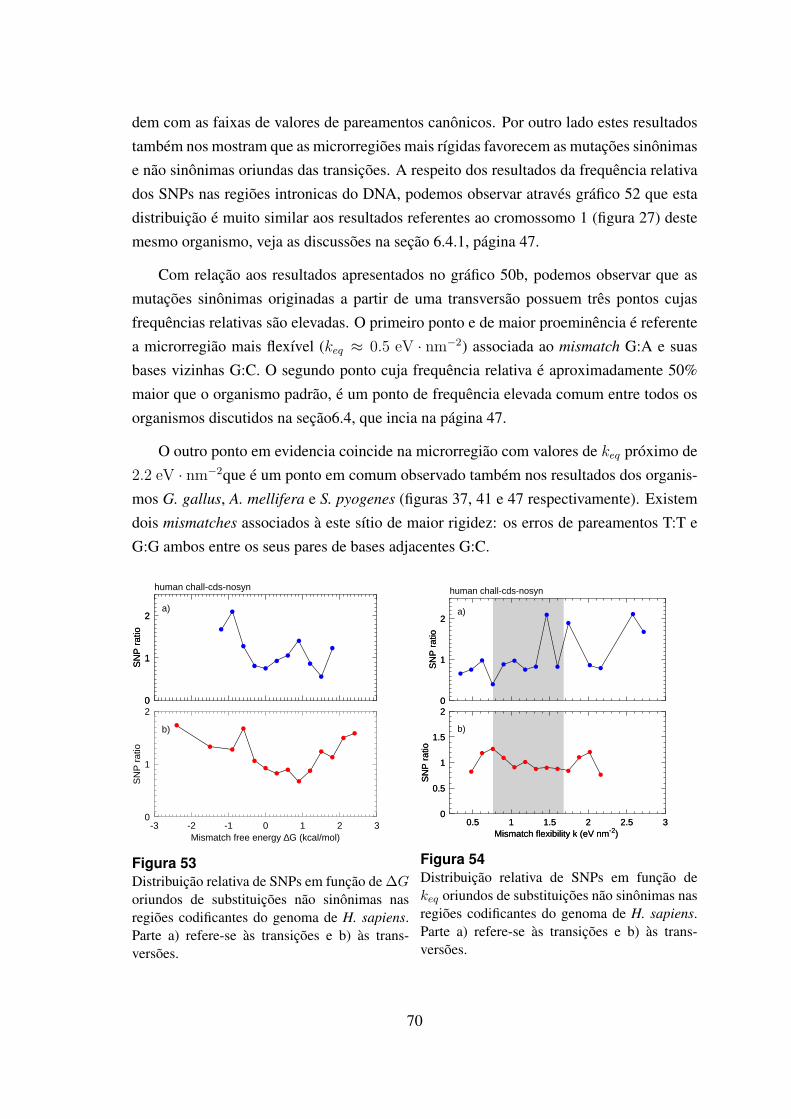
dem com as faixas de valores de pareamentos canonicos. Por outro lado estes resultadostambem nos mostram que as microrregioes mais rıgidas favorecem as mutacoes sinonimase nao sinonimas oriundas das transicoes. A respeito dos resultados da frequencia relativados SNPs nas regioes intronicas do DNA, podemos observar atraves grafico 52 que estadistribuicao e muito similar aos resultados referentes ao cromossomo 1 (figura 27) destemesmo organismo, veja as discussoes na secao 6.4.1, pagina 47.
Com relacao aos resultados apresentados no grafico 50b, podemos observar que asmutacoes sinonimas originadas a partir de uma transversao possuem tres pontos cujasfrequencias relativas sao elevadas. O primeiro ponto e de maior proeminencia e referentea microrregiao mais flexıvel (keq ≈ 0.5 eV · nm−2) associada ao mismatch G:A e suasbases vizinhas G:C. O segundo ponto cuja frequencia relativa e aproximadamente 50%maior que o organismo padrao, e um ponto de frequencia elevada comum entre todos osorganismos discutidos na secao6.4, que incia na pagina 47.
O outro ponto em evidencia coincide na microrregiao com valores de keq proximo de2.2 eV · nm−2que e um ponto em comum observado tambem nos resultados dos organis-mos G. gallus, A. mellifera e S. pyogenes (figuras 37, 41 e 47 respectivamente). Existemdois mismatches associados a este sıtio de maior rigidez: os erros de pareamentos T:T eG:G ambos entre os seus pares de bases adjacentes G:C.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
-3 -2 -1 0 1 2 3Mismatch free energy ∆G (kcal/mol)
0
1
2
SN
P r
atio
b)
human chall-cds-nosyn
Figura 53Distribuicao relativa de SNPs em funcao de ∆Goriundos de substituicoes nao sinonimas nasregioes codificantes do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
0
1
2
SN
P r
atio
0
1
2
SN
P r
atio
a)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
b)
0.5 1 1.5 2 2.5 3Mismatch flexibility k (eV nm-2)
0
0.5
1
1.5
2
SN
P r
atio
human chall-cds-nosyn
Figura 54Distribuicao relativa de SNPs em funcao dekeq oriundos de substituicoes nao sinonimas nasregioes codificantes do genoma de H. sapiens.Parte a) refere-se as transicoes e b) as trans-versoes.
70

As mutacoes nao sinonimas tambem originadas pelas de transversoes possuem umperfil particular muito diferente do resultado observado para as mutacoes sinonimas. Ografico 54b representa a frequencia relativa das mutacoes nao sinonimas originadas dastransversoes em funcao da flexibilidade. Este resultado mostra que essas mutacoes temum perfil diferente ao perfil observado das mutacoes sinonimas (figura 50b) mas se as-semelha com o perfil da frequencia relativa dos SNPs encontrados nas regioes intronicas(figura 52b) e tambem no cromossomo 1 (figura 48b, pagina 65) deste organismo.
71

7 Conclusao
Nesta secao, tecemos as nossas conclusoes a partir dos resultados mostrados e discu-tidos na secao 6. As nossas analises relacionam a influencia da flexibilidade e da energialivre na distribuicao de polimorfismos (SNPs) em nove genomas.
Verificamos que a frequencia das substituicoes, tanto para transicoes como para trans-versoes, sao dependentes da composicao das bases vizinhas em acordo com o que ja foidescrito na literatura (26, 41–43). Os nossos resultados indicam que a ocorrencia dos po-limorfismos nos diversos genomas e influenciada pela energia livre e pela flexibilidadeda microrregiao do DNA em que ele ocorre. Portanto, existe uma razao fısica para apredominancia de certas bases vizinhas sobre outras na frequencia de SNPs.
Na comparacao entre os cromossomos de cada especie notamos uma grande uniformi-dade nos resultados. Ou seja, nao ha diferencas importantes na frequencia de SNPs entreos cromossomos de uma mesma especie com a excecao das transicoes do cromossomosexual de Bos taurus.
A analise comparativa entre os 9 genomas analisados mostrou que o conteudo G+Cdo genoma influencia a frequencia dos SNPs tanto em relacao a energia livre quanto emrelacao a flexibilidade. Em particular, o conteudo G+C privilegia o aumento da frequenciade polimorfismos em microrregioes do DNA que sao pouco estaveis e pouco flexıveis.No entanto, o conteudo G+C nao e suficiente para explicar todas as diferencas entre osdiversos genomas. Quais sao os fatores que poderiam levar a uma diferenca na ocorrenciade SNPs, por exemplo quando analisados em funcao de flexibilidade? Uma possibilidadeenvolve as proteınas responsaveis pelo mecanismo de reparo que sao diferentes entre osorganismos, possivelmente com eficiencias de reparo diferentes (56). Estas proteınasusam a flexibilidade do DNA na regiao do mismatch para identificar seus alvos. Nossosresultados indicam uma dependencia importante da ocorrencia de SNPs com flexibilidade,em especial encontramos frequentemente que regioes mais rıgidas promovem uma maiorocorrencia de SNPs em varios dos organismos estudados. Embora isto seja indicador daimportancia da flexibilidade, nossos resultados ainda nao sao conclusivos se existe ou naouma relacao com o mecanismo de reparo.
Claramente, para responder a estas perguntas de maneira mais definitiva, precisamosdetalhar mais a nossa analise, principalmente distinguindo entre regioes diferentes dosgenomas. Aqui, o nosso grande limitador foi a falta de algumas informacoes referentesaos genomas analisados nao disponibilizadas no dbSNP (release 132), como por exem-
72

plo a disponibilidade das sequencias de SNPs separadas por regioes codificantes e naocodificantes e pelo tipo de mutacao (sinonima, conservativa e nao sinonima) que so esta-vam disponıveis para o genoma humano. Os resultados para este genoma mostram queas mutacoes oriundas das transicoes encontradas nas regioes codificantes em funcao daenergia livre e das flexibilidades se distribuem de forma semelhante com relacao ao tipode mutacao (sinonima e nao sinonima) mas possuem um perfil muito particular quandocomparadas ao cromossomo 1 e com as regioes nao codificantes (introns) deste mesmoorganismo. Verificamos tambem que existem mismatches que originam mutacoes nao-sinonimas que estao favorecidas nas microrregioes rıgidas do DNA. Em contrapartidaestes mesmos mismatches originam mutacoes sinonimas extremamente raras na mesmamicrorregiao do DNA.
73

8 Perspectivas futuras
Os resultados obtidos neste trabalho levantaram um grande numero de perguntas queensejam novas e mais detalhadas analises. Por exemplo, temos uma clara indicacao deque o conteudo C+G e importante na distribuicao de SNPs. Portanto um dos primeirospassos para a continuacao deste trabalho e o levantamento detalhado da frequencia daocorrencia de cada trımero ou seja cada base mutada e as bases vizinhas que a flanqueiampara cada organismo e substituir o nosso modelo de organismo padrao uniforme por umque reflita a composicao de bases de cada organismo. Alem disto, a nossa presente analisetrata transicoes e transversoes de maneira independente. Poderıamos portanto usar um“modelo de dois parametros” como o de Kimura (50) para comparar as frequencias entretransicoes e transversoes.
Neste trabalho, a analise em funcao da regiao genomica so foi realizada para o ge-noma humano por limitacoes do dbSNP. Nossa intencao e buscar alguma estrategia quenos permita separar as sequencias por regioes codificantes, nao codificantes e pelo tipo demutacao para os demais genomas analisados neste trabalho independente da classificacaodo dbSNP. A partir destas sequencias separadas um outro passo sera ampliar o numero debases adjacentes afim de analisar o conteudo das bases vizinhas e confirmar se o vies doconteudo G+C do genoma de fato interfere na frequencia dos SNPs.
Uma outra limitacao importante e que nos nao sabemos a origem dos SNPs presentesno dbSNP, apenas sabemos que foram incorporados ao longo da evolucao dos organis-mos. Seria interessante obtermos dados de SNPs que foram gerados de forma contro-lada, como por exemplo criados intencionalmente ao irradiar organismos por radiacaoionizante. Alternativamente, poderıamos procurar por SNPs que ocorreram sob pressaoseletiva controlada em laboratorio.
74

A Apendice
75

A.1 Scripts utilizados neste trabalho
A.1.1 trimero.pl
#!/bin/perl
##########################################################################
#Luciana Oliveira 22/11/2010
4
if (scalar(@ARGV) < 1)#help do programa
{
print "este programa conta os possiveis trimeros existentes (% e inteiros)
em uma sequencia em
9 formato fasta.\nUSAGE: perl <program.pl> <file.fasta> <out>\nObs.:quantidade
em porcentagem na
segunda coluna e em numero inteiro na terceira coluna\n";
exit;
}
14 ##########################################################################
use strict;
use Bio::SeqIO;#modulo de bioperl SeqIO;
my $file = $ARGV[0];#arquivo de entrada
19 my $out_file = $ARGV[1];#arquivo de saida
my $seqio_obj = Bio::SeqIO->new(-file => "$file", -format => "fasta" );
#cria objeto sequencia
open(OUT,">$out_file-trimeros.dat");
24 my $seq_obj;
my %tri; #cria hash vazio
my $conta;#contador para os trimeros;
my $nao_conta;
# print ’trimeros’,"\t",’%’,"\t ",’qtdade’,"\n";
29 while ($seq_obj = $seqio_obj->next_seq)
{
my $seq = $seq_obj->seq;#atribui a variavel $seq o objeto sequencia
my $len = $seq_obj->length; # tamanho da sequencia
#print $seq_obj->id, "\n";
34 for (my $i=0; $i < $len-2; $i++)
{
my $trio=substr($seq,$i,3);# pega os trios com janela de 1
#print $trio, "\n";
if ($trio =˜ /[ACGT]{3}/)
39 {
76

$tri{$trio}++;$conta++;
#adiciona a hash os 256 trimeros possiveis e conta o total de trimeros
}
else
44 {$nao_conta++;
}
}
}
for my $trio(sort keys %tri) #ordena a hash
49 {i
print OUT "$trio\t",$tri{$trio}/$conta,"\t",$tri{$trio},"\n";
}
close(OUT);
print "Total de nucleotideos=$conta naotraduzidos=$nao_conta\n";
54 exit;
A.1.2 fasta2summary.pl
1 #!/usr/bin/perl -w
##########################################################################
#este programa converte um arquivo em formato fasta em um arquivo com
#formato summary estabelecidos por nos.
#arquivo de entrada em formato fasta estraido do SNPdb
6 #arquivo de saida: rs123 [HS] R A/M A (com 25pb de cada lado)
##########################################################################
use strict;
my @seq;
my $snp;
11 my $next_line;
my $seq3;
my $seq4;
my $seq5;
my $mais_line;
16
my $in_file = $ARGV[0];
if ($in_file =˜ /\.gz/)
{open(FL,"zcat $in_file|") or die "nao pude abrir o arquivo $in_file!\n";}
else
21 {open(FL,"$in_file") or die "nao pude abrir o arquivo $in_file!\n";}
#open(FL,$in_file) or die "nao pude abrir o arquivo $in_file!\n";
my $line = <FL>;
while(not eof(FL))
26 {
77

if ($line =˜ /alleles="[ATGC]\/[ATGC]"/)
{
$line =˜ /alleles="(.*)\/(.*)"/;
$snp = "$1/$2";
31 #print "$snp\n";
$line = <FL>;
chomp($line);
while (not $line =˜ /ˆ>/)
36 {
$next_line = <FL>;
chomp($next_line);
while (length($next_line) == 0) {$next_line = <FL>;chomp($next_line);}
if (eof(FL)) {exit;} #eof ->end of file
41 chomp ($next_line);
if (length($next_line)==1)
{
$mais_line = <FL>;
46 chomp($mais_line);
while (length($mais_line) == 0) {$mais_line = <FL>;chomp($mais_line);}
if (eof(FL)) {exit;}
# print $line,"\n",$snp,"\n",$next_line,"\n$mais_line\n";
$line =˜ s/\s//g;
51 $mais_line =˜ s/\s//g;
print "rs1 [H s] ",substr($line,length($line)-1,1)," ",$snp," ",
substr($mais_line,0,1),"\n";
# print "rs123 [Homo sapiens] ",$line," ",$snp," ",$mais_line,"\n";
last;
56 }
else
{
$line = $next_line;
}
61 }
}
else
{
$line = <FL>;
66 }
}
78

A.1.3 trans-transv.pl
#!/usr/bin/perl -w
##########################################################################
3 #Este programa usa um arquivo.summary e separa as sequencias de transicao
# e transversao.
#USAGE: programa.pl infile.summary
#Luciana Oliveira 21/05/2010
##########################################################################
8 use strict;
my $file=$ARGV[0];
open(FL,$file)or die "nao pude abrir $file!";
13 while(my $line=<FL>)
{
system("grep -E ’(A/G|G/A|T/C|C/T)’ $file >transicao-$file");
}
close (FL);
18 exit;
open(FL,$file)or die "nao pude abrir $file!";
while(my $line=<FL>)
{
23 system("grep -v -E ’(A/G|G/A|T/C|C/T)’ $file >transversao-$file");
}
close (FL);
exit;
A.1.4 snp.pl
#!/usr/bin/perl -w
##########################################################################
3 #Este programa atribui valores de flexibilidade e energia livre aos
#trimeros de sequencias normais e com um unico snp.
#Luciana Oliveira e Gerald Weber 09/02/10
##########################################################################
use strict;
8
if (scalar(@ARGV) <=0)#help do programa
{
print "USAGE: program_name.pl <d*-trimeros-pm-mm.dat> <file.summary>
<outfile_name>\ninfile1 format=(d*-trimeros-pm-mm.dat):
13 ex.: AT_AA,AA_AT = 1.3, infile2=summary formart e outfile=name-*.dat
79

out files:
name-mis.dat AT_AA,AA_AT enlivre quantidade
name-valor.dat: valor da energia livre e quantidade
name-conv.dat: trimero:seq normal
18 mis1 (snp1)
seq nor comp
mis2 (snp2)
seq???
name-dupla.dat os trimeros com seus valores de energia livre dos
23 dois snps
name-mis-d.dat seq direta do mis
name-mis-c.dat seq complem do mis
name--snpid.dat AT/AG quantidade -lista dos snps com a quantidade\n";
exit;
28 }
sub conv #converte AAA\nTTT para AT_AT,AT_AT
{
my $trimero=$_[0];
33 my @trim=split(’’,$trimero);
my $converte="$trim[0]$trim[4]_$trim[1]$trim[5],$trim[1]
$trim[5]_$trim[2]$trim[6]";
return $converte;
}
38
my %k;
my %kval;
my $in_file = $ARGV[0];#arq .dat gerado dos freeenergy.pl
43 if ($in_file =˜ /\.gz/)
{open(EN,"zcat $in_file|")or die "Nao deu para abrir $in_file!\n";}
else
{open(EN,"$in_file")or die "Nao deu para abrir $in_file!\n";}
48
while(my $line=<EN>)
{
my @fields=split(’ ’,$line);
$k{$fields[0]}=$fields[2];
53 $kval{$fields[2]}=0;
}
my $line;
my @string;
my @db;
80

58 my $base_n1;
my $base_n3;
my $seq_db;
my $comp_n;
my $substitui;
63
my %stat; #tabela de statistica - quantidade de trimeros
my %stat_sep; #tabela de statistica - quantidade de trimeros
my %dup;
my %tabela_conversao;
68 my %snp_id;
my $in = $ARGV[1];
if ($in =˜ /\.gz/)
{open(FL,"zcat $in|")or die "Nao deu para abrir $in!\n";}
73 else
{open(FL,"$in")or die "Nao deu para abrir $in!\n";}
while ($line = <FL>)
78 {
if ((not $line =˜ /ˆrs0\s/) and (not $line =˜ /ˆ$/) and (not $line =˜ /\/.\//))
{
@string = split(" ",$line);
#rs74310592 [Homosapiens] TCCCAGCACTTTGGGAGGCCGAGGCG C/G GAAAGTATTCCTTTT
83 if (scalar(@string) < 5) {next;}
my $seq1 = $string[3];
#$seq1=prim-seq (TCCCAGCACTTTGGGAGGCCGAGGCG)
$seq1 =˜ tr/acgt/ACGT/;
88 my @array1 = split("",$seq1);
my $base1 = pop @array1;
#adiciona o ultimo elemento do array a uma variavel=G
my $seq2 = $string[4];#$seq2=C/G
93 $seq2 =˜ tr/acgt/ACGT/;
my @array2 = split(" ",$seq2);
my $base2 = $array2[0]; #$base2=C/G
my $seq3 = $string[5];#$seq3 = GAAAGTATTCCTTTT-final da seq
98 $seq3 =˜ tr/acgt/ACGT/;
my @array3 = split("",$seq3);
my $base3 = shift @array3;
#adiciona o primeiro elemento do array a uma variavel=G
81

# print "\nseq_DB=[$base1][$base2][$base3]\n";
103
$seq_db = "$base2"; #C/G
my $snp="$base1$base2$base3";
if ($snp =˜ /[ACGT]{2}\/[ACGT]{2}/) {$snp_id{$snp}{’count’}++};
@db = split("/",$base2);
108 my $base_n = $db[0];#base do meio do trimero - normal
(segunda base - variante)=C
$base_n1 = $base1;#primeira base (posicao fixa)=G
$base_n3 = $base3;#ultima base (posicao fixa)=G
my $trim_n = $base_n1 . $base_n . $base_n3;#direta normal=GCG
113 my $mis_d = $base_n1 . $db[1] . $base_n3;#snpd-direta=GGG
$comp_n = $trim_n;
$comp_n =˜ tr/ACGT/TGCA/;#complemnt-normal=CGC
my $mis_c = $mis_d;
$mis_c =˜ tr/ACGT/TGCA/;#snp-compl=CCC
118 my $mis1="$mis_d\n$comp_n";
$snp_id{$snp}{’mis1’}=$mis1;
my $mis2="$trim_n\n$mis_c";
$snp_id{$snp}{’mis2’}=$mis2;
# print "Od=$trim_n\nOc=$comp_n\n";
123 #Od=fita original direta, Oc=fita original complementar
# print "Mismatch1\n=$mis1\n";
#Od=fita original direta, Oc=fita original complementar
# print "Mismatch2\n=$mis2\n";#Od=fita original direta, Oc=fita original
128 $stat{conv($mis1)}++;
$stat{conv($mis2)}++;
$stat_sep{’d’}{conv($mis1)}++;
$stat_sep{’c’}{conv($mis2)}++;
133 if ((exists $k{conv($mis1)}) and (exists $k{conv($mis2)}))
{$dup{$trim_n}=$k{conv($mis1)} . " " . $k{conv($mis2)};}
$tabela_conversao{$trim_n}[0]=$mis1;
$tabela_conversao{$trim_n}[1]=$mis2;
138
}
}
my $out_file = $ARGV[2];
open(RES,">$out_file-mis.dat");
143
for my $mis (sort keys %stat)
{
82

if (exists $k{$mis})
{
148 print RES "$mis $k{$mis} $stat{$mis}\n";
$kval{$k{$mis}}+=$stat{$mis};
}
}
close(RES); #RES-->resultado com a redundancia
153
open(RESD,">$out_file-mis-d.dat");
for my $mis (sort keys %{$stat_sep{’d’}})
{
158 if (exists $k{$mis})
{
print RESD "$mis $k{$mis} $stat_sep{’d’}{$mis}\n";
}
}
163 close(RESD); #RES-->resultado com a redundancia
open(RESC,">$out_file-mis-c.dat");
for my $mis (sort keys %{$stat_sep{’c’}})
{
168 if (exists $k{$mis})
{
print RESC "$mis $k{$mis} $stat_sep{’c’}{$mis}\n";
}
}
173 close(RESC); #RES-->resultado com a redundancia
open(RESK,">$out_file-valor.dat");
for my $ks (sort keys %kval)
178 {
print RESK "$ks $kval{$ks}\n";
}
close(RESK);
#RESK resultado com valores aproximedos sendo considerados
183 #como unicos
open (DUP,">$out_file-dupla.dat");
for my $d (sort keys %dup)
{
188 print DUP "$d $dup{$d}\n";
}
83

close(DUP);open (DUP,">$out_file-dupla.dat");
for my $d (sort keys %dup)
{
193 print DUP "$d $dup{$d}\n";
}
close(DUP);#seq com redundancia: valores duplicados
open (CNV,">$out_file-conv.dat");
198 for my $trim (sort keys %tabela_conversao)
{
print CNV "trimero=$trim \n mis1=$tabela_conversao{$trim}[0]\n
mis2=$tabela_conversao{$trim}[1]\n";
}
203 close(CNV);
open (SNPID,">$out_file-snpid.dat");
#print SNPID scalar(%snp_id);
for my $snp (sort keys %snp_id)
208 {
if (exists $snp_id{$snp}{’count’})
{
my $mis1=$snp_id{$snp}{’mis1’};
my $k1=$k{conv($mis1)};
213 $mis1 =˜ s/\n/\//g;
my $mis2=$snp_id{$snp}{’mis2’};
my $k2=$k{conv($mis2)};
$mis2 =˜ s/\n/\//g;
print SNPID "$snp $snp_id{$snp}{’count’} $mis1 $k1 $mis2 $k2\n";
218 }
}
close(SNPID);
A.1.5 gccalc.pl
#!perl -w
# $Id: gccalc.PLS,v 1.5 2006/07/04 22:23:29 mauricio Exp $
4 use strict;
use Bio::SeqIO;
use Bio::Tools::SeqStats;
use Getopt::Long;
9 my $format = ’fasta’;
my $file;
84

my $help =0;
GetOptions(
’f|format:s’ => \$format,
14 ’i|in:s’ => \$file,
’h|help|?’ => \$help,
);
19 my $USAGE = "usage: gccalc.pl -f format -i filename\n";
if( $help ) {
die $USAGE;
}
24 $file = shift unless $file;
my $seqin;
if( defined $file ) {
print "Could not open file [$file]\n$USAGE" and exit unless
-e $file;
29 $seqin = new Bio::SeqIO(-format => $format,
-file => $file);
} else {
$seqin = new Bio::SeqIO(-format => $format,
-fh => \*STDIN);
34 }
while( my $seq = $seqin->next_seq ) {
next if( $seq->length == 0 );
if( $seq->alphabet eq ’protein’ ) {
39 warn("gccalc does not work on amino acid sequences
...skipping this seq");
next;
}
44 my $seq_stats = Bio::Tools::SeqStats->new(’-seq’=>$seq);
my $hash_ref = $seq_stats->count_monomers();
# for DNA sequence
print "Seq: ", $seq->display_id, " ";
print $seq->desc if $seq->desc;
49 print " Len:", $seq->length, "\n";
printf "GC content is %.4f\n", ($hash_ref->{’G’} +
$hash_ref->{’C’}) /
$seq->length();
54 foreach my $base (sort keys %{$hash_ref}) {
85

print "Number of bases of type ", $base, "= ",
$hash_ref->{$base},"\n";
}
print "--\n";
59 }
# alternatively one could use code submitted by
64 sub calcgc {
my $seq = $_[0];
my @seqarray = split(’’,$seq);
my $count = 0;
foreach my $base (@seqarray) {
69 $count++ if $base =˜ /[G|C]/i;
}
my $len = $#seqarray+1;
return $count / $len;
74 }
__END__
79 =head1 NAME
gccalc - GC content of nucleotide sequences
=head1 SYNOPSIS
84
gccalc [-f/--format FORMAT] [-h/--help] filename
or
gccalc [-f/--format FORMAT] < filename
or
89 gccalc [-f/--format FORMAT] -i filename
=head1 DESCRIPTION
This scripts prints out the GC content for every nucleotide
94 sequence from the input file.
=head1 OPTIONS
The default sequence format is fasta.
86

99
The sequence input can be provided using any of the three
methods:
=over 3
104
=item unnamed argument
gccalc filename
109 =item named argument
gccalc -i filename
=item standard input
114
gccalc < filename
=back
119 =head1 FEEDBACK
=head2 Mailing Lists
User feedback is an integral part of the evolution of this
124 and other Bioperl modules. Send your comments and suggestions
preferably to the Bioperl mailing list. Your participation is
much appreciated.
[email protected] - General discussion
129 http://bioperl.org/wiki/Mailing_lists - About the mailing lists
=head2 Reporting Bugs
Report bugs to the Bioperl bug tracking system to help us keep
134 track of the bugs and their resolution. Bug reports can be submitted via the
web:
http://bugzilla.open-bio.org/
139 =head1 AUTHOR - Jason Stajich
Email [email protected]
87

=head1 HISTORY
144
Based on script code (see bottom) submitted by [email protected]
Submitted as part of bioperl script project 2001/08/06
149 =cut
A.1.6 util.pl
#!/usr/bin/perl
#Define o help do programa
if (scalar(@ARGV) < 1)#help do programa
5 {
print ’USAGE: <util.pl> <arg1=[ORGAN]> <arg2=num de [CHR]>
<arg3=[Function_Class]>’,"\n",
’Para detalhes mais detalhes de sintaxe ver
http://www.ncbi.nlm.nih.gov/snp’;
10 exit;
}
# ===========================================================================
#
# PUBLIC DOMAIN NOTICE
15 # National Center for Biotechnology Information
#
# This software/database is a "United States Government Work" under the
# terms of the United States Copyright Act. It was written as part of
# the author’s official duties as a United States Government employee and
20 # thus cannot be copyrighted. This software/database is freely available
# to the public for use. The National Library of Medicine and the U.S.
# Government have not placed any restriction on its use or reproduction.
#
# Although all reasonable efforts have been taken to ensure the accuracy
25 # and reliability of the software and data, the NLM and the U.S.
# Government do not and cannot warrant the performance or results that
# may be obtained by using this software or data. The NLM and the U.S.
# Government disclaim all warranties, express or implied, including
# warranties of performance, merchantability or fitness for any particular
30 # purpose.
#
# Please cite the author in any work or product based on this material.
#
# ===========================================================================
88

35 #
# Author: Oleg Khovayko
# Modificado por Luciana Oliveira e Gerald Weber em 02/12/2010.
#
# File Description: eSearch/eFetch calling example
40 #
# ---------------------------------------------------------------------------
# Subroutine to prompt user for variables in the next section
# sub ask_user {
45 # print "$_[0] [$_[1]]: ";
# my $rc = <>;
# chomp $rc;
# if($rc eq "") { $rc = $_[1]; }
# return $rc;
50 # }
# ---------------------------------------------------------------------------
# Define library for the ’get’ function used in the next section.
# $utils contains route for the utilities.
# $db, $query, and $report may be supplied by the user when prompted;
55 # if not answered, default values, will be assigned as shown below.
use LWP::Simple;
my $db;
60 my $query;
my $report;
my $organism = $ARGV[0];#define o organismo
my @chrlist = split(’,’,$ARGV[1]);#define o numero de cromossomos
65 my @add;
foreach my $chr (@chrlist)
{
if ($chr =˜ /(\d+)-(\d+)/) #1-22 vira $1=1 e $2=22
{
70 for (my $c=$1; $c <= $2; $c++) {push(@add,$c);}
}
}
@chrlist=grep(!/(\d+)-(\d+)/,(@chrlist,@add)); # ! e equivalente a -v
75
print "Cromossomos=",join(’|’,@chrlist),"\n";
my $fc = $ARGV[2];#define o "function class"
89

80
my $utils = "http://www.ncbi.nlm.nih.gov/entrez/eutils";
foreach my $chr (@chrlist)
85 {
$db = ’snp’;#define o banco de dados
$query = $organism . "[ORGN] AND " . $chr . "[CHR] AND
(" . $fc . "[Function_Class])";#define a query
$report = ’fasta’;#define o formato do arquivo
90
print "$query\n";
# ---------------------------------------------------------------------------
# $esearch contains the PATH & parameters for the ESearch call
95 # $esearch_result containts the result of the ESearch call
# the results are displayed and parsed into variables
# $Count, $QueryKey, and $WebEnv for later use and then displayed.
my $esearch = "$utils/esearch.fcgi?" .
100 "db=$db&retmax=1&usehistory=y&term=";
my $esearch_result = get($esearch . $query);
print "\nESEARCH RESULT: $esearch_result\n";
105
$esearch_result =˜
m|<Count>(\d+)</Count>.*<QueryKey>(\d+)</QueryKey>.*<WebEnv>(\S+)
</WebEnv>|s;
110 print "Count $1\n";#exit;
my $Count = $1;
my $QueryKey = $2;
my $WebEnv = $3;
115
print "Count = $Count; QueryKey = $QueryKey; WebEnv = $WebEnv\n";
# ---------------------------------------------------------------------------
# this area defines a loop which will display $retmax citation
120 #results from Efetch each time the the Enter Key is pressed,
#after a prompt.
90

my $retstart;
my $retmax=1000;
125
my $outfile="$organism-chr$chr-$fc.fas";
$outfile =˜ s/\s/-/g;
130 print "Escrevendo para $outfile\n";
open(RES,">$outfile");
for($retstart = 0; $retstart < $Count; $retstart += $retmax) {
my $efetch = "$utils/efetch.fcgi?" .
135 "rettype=$report&retmode=text&retstart=$retstart
&retmax=$retmax&" .
"db=$db&query_key=$QueryKey&WebEnv=$WebEnv";
#print "\nEF_QUERY=$efetch\n";
140
my $efetch_result = get($efetch);
print RES "$efetch_result";
}
145 close(RES);
}
A.1.7 histogram.pl
#!/usr/bin/perl -w
##########################################################################
3 #Este script gera uma tabela para a construcao de um histograma e calcula
#os valors minimos e maximos para a geracao da curva normal
#23/04/2010 - Luciana Oliveira e Gerald Weber
##########################################################################
use strict;
8
if (scalar(@ARGV) < 1)#help do programa
{
print "USAGE: [program_name.pl] [in_file1-name]=*-mis.dat [min_value]
[max_value] or [program_name.pl] [calcule] [in_file1-name]\n
13 OBS:OS VALORES SAO DADOS EM PORCENTAGENS\n";
exit;
}
my @valor;
my $result=0.0;
91

18 my $quant=0.0;
my $min=1000;
my $max=0;
my $total;
my $intervalos=20;
23 my $soma=0;
my @x; #valor
my @y; #quantide
my $file = $ARGV[0];
my $out = $file;
28 $out =˜ s/\.dat/.histo/;
open(FL,$file) or die "nao pude abrir o arquivo $file!";
# arquivo de entrada: *-mis.dat
open(OUT,">$out") or die "nao pude abrir o arquivo de saida";
33 while (my $line=<FL>)
{
@valor = split(" ",$line);
$result = $valor[1];
$quant = $valor[2];
38 $soma+=$quant;
push(@x,$result);
#empurra valor por valor - adiciona um valor por vez no array
push(@y,$quant);
#empurra quantidade por quantidade
43 if($result<=$min)
{
$min=$result;
}
48 if($result>=$max)
{
$max=$result;
}
}
53 close(FL);
if ($ARGV[1] eq ’calcule’)
{print "minimo=$min maximo=$max\n"; exit;}
#especificando os intervalos arbitrarios
if (exists $ARGV[1]) {$min=$ARGV[1];}
58 if (exists $ARGV[2]) {$max=$ARGV[2];}
my $deltax=($max-$min)/$intervalos;
#deltaX e o tamanho de cada intervalo
my @histograma;
92

my $i;
63 for($i=0; $i < scalar(@x); $i++)
{
my $pos=($x[$i]-$min)/$deltax;
#define a posicao do valor no intervalo
$histograma[$pos]+=$y[$i];
68 #faz a soma da quantidade de valores y dentro do intervalo x.
}
for($i=0; $i < $intervalos; $i++)
{
if (not exists $histograma[$i]) {$histograma[$i]=0;}
73 #se encontrar intervalo vazio, atribui 0 ao resultado.
$histograma[$i] /= $soma;#faz a porcentagem dos valores absolutos.
print OUT $i*$deltax+$min," $histograma[$i]\n";
#multiplica a posicao do array pelo deltaX+minimo(intervalo)
}
78 close (OUT);
exit;
A.1.8 padroniza.pl
1 #!/usr/bin/perl -w
##########################################################################
#este programa faz a normalizacao dos valores de flexibilidade e de energia
#livre dividindo a media das quantidades de cada valor(flex ou de energia)
#pela media das quantidades das sequencias controles, fazendo a normalizacao
6 #dos valores.
#Luciana Oliveira e Gerald Weber 29/09/2010.
##########################################################################
#USAGE: normaliza.pl <file-mis.histo> <file-controle.histo>
11 #outfile-mis-norm.histo
use strict;
if (scalar(@ARGV) < 1)#help do programa
16 {
print "USAGE: normaliza.pl <file-mis.histo> <file-controle.histo>
...OUT=outfile-mis.histon\n";
exit;
}
21 my $in1=$ARGV[0];
my $in2=$ARGV[1];
my $out=$in1;
93

my $line;
my $line1;
26 my $i=1;
my $j=0;
my @histo;
my @histo1;
my $valor;
31 my $valor1;
open (IN,$in1) or die "nao pude abrir $in1";
#abre o arquivo.histo das especies
36 open (CON,$in2) or die "nao pude abrir $in2";
#abre o arquivo.histo controle
$out=˜s/\.histo/-padrao.histo/;
#substitui no nome do arquivo de saida file.histo por file.histon
open(OUT,">$out") or die"nao pude abrir $out";
41 #abre o arquivo de saida
while($line=<IN>) #le o arquivo.histo com os valores das especies
{
@histo=split(’ ’,$line);
46 $valor=$histo[$i];
$line1=<CON>;#le os valores brutos do arquivo-controle.histo
@histo1=split(’ ’,$line1);
$valor1=$histo1[$i];
if ($valor1 != 0)
51 {
print OUT $histo[0],’ ’, $valor/$valor1,"\n";
#divide a quantidade total das especies pelo quantidade do organismo
#padrao.
}
56 elsif (($valor1) ==0 and ($valor != 0))
{
print "Problema: SNP fantasma\n"; exit;
}
}
94

Referencias
1 GRAUR, D.; LI, W. Fundamentals of molecular evolution. [S.l.]: Sinauer AssociatesSunderland, MA, 2000.
2 WATSON, J. D.; CRICK, F. H. C. Molecular structure of nucleic acids: A structurefor deoxyribose nucleic acid. Nature, v. 171, p. 737–738, 1953.
3 WATSON, J.; CRICK, F. Genetic implications of the structure of DNA. Nature,v. 171, p. 964–967, 1953.
4 FRANKLIN, R.; GOSLING, R. Evidence for 2-chain helix in crystalline structure ofsodium deoxyribonucleate. Nature, Nature Publishing Group, v. 172, 1953.
5 FRANKLIN, R.; GOSLING, R. The structure of sodium thymonucleate fibres. II.The cylindrically symmetrical Patterson function. Acta Crystallographica, InternationalUnion of Crystallography, v. 6, n. 8-9, p. 678–685, 1953. ISSN 0365-110X.
6 MCCARTY, M.; AVERY, O. Studies on the chemical nature of the substanceinducing transformation of pneumococcal types. The Journal of Experimental Medicine,Rockefeller Univ Press, v. 83, n. 2, p. 97, 1946. ISSN 0022-1007.
7 CHARGAFF, E. Chemical specificity of nucleic acids and mechanism of theirenzymatic degradation. Cellular and Molecular Life Sciences, Springer, v. 6, n. 6, p.201–209, 1950. ISSN 1420-682X.
8 WILKINS, M.; STOKES, A.; WILSON, H. Molecular structure of deoxypentosenucleic acids. Nature, v. 171, n. 4356, p. 738, 1953.
9 PRUITT, K. D.; TATUSOVA, T.; MAGLOTT, D. R. NCBI reference sequences(RefSeq): a curated non-redundant sequence database of genomes, transcripts andproteins. Nucleic Acids Research, Oxford Univ Press, v. 35, p. D61–D65, 2007. ISSN0305-1048.
10 MYLES, S. et al. Wide population differentiation at disease-associated SNPs. BMC
Medical Genomics, BioMed Central Ltd, v. 1, n. 1, p. 22, 2008. ISSN 1755-8794.
11 MOORE, J.; ASSELBERGS, F.; WILLIAMS, S. Bioinformatics challenges forgenome-wide association studies. Bioinformatics, Oxford Univ Press, v. 26, n. 4, p. 445,2010. ISSN 1367-4803.
95

12 MARKO, J.; COCCO, S. The micromechanics of DNA. Phys. World, v. 16, p.37–41, 2003.
13 GREEN, R. et al. Analysis of one million base pairs of Neanderthal DNA. Nature,Nature Publishing Group, v. 444, n. 7117, p. 330–336, 2006. ISSN 0028-0836.
14 METZKER, M. Sequencing technologies—the next generation. Nature Reviews
Genetics, Nature Publishing Group, v. 11, n. 1, p. 31–46, 2009. ISSN 1471-0056.
15 1000 GENOMES, A DEEP CATALOG OF HUMAN GENETIC VARIATION.Disponıvel em: <http://www.1000genomes.org/>. Acesso em: 09-2010.
16 CHARLESWORTH, B.; CHARLESWORTH, D. Elements of evolutionary genetics.[S.l.]: CSIRO, 2010. ISBN 0981519423.
17 HUANG, X. et al. Genome-wide association studies of 14 agronomic traits in ricelandraces. Nature Genetics, Nature Publishing Group, v. 42, 2010. ISSN 1061-4036.
18 BARBAZUK, W. et al. SNP discovery via 454 transcriptome sequencing. The Plant
Journal, Wiley Online Library, v. 51, n. 5, p. 910–918, 2007. ISSN 1365-313X.
19 ANASTASSIOU, D. Genomic signal processing. IEEE Signal Processing Mag., jul.,p. 8–20, 2001.
20 ALBERTS, B. Molecular Biology of the Cell - Fifth Edition. [S.l.]: Taylor & Francis,Inc., 2007. ISBN 0815341105.
21 PIERCE, B. Genetics: A conceptual approach. [S.l.]: WH Freeman & Co, 2007.ISBN 0716779285.
22 GRIFFITHS, A. Introduction to genetic analysis. [S.l.]: WH Freeman, 2009. ISBN9788527714976.
23 MESELSON, M.; STAHL, F. The replication of DNA in Escherichia coli.Proceedings of the National Academy of Sciences of the United States of America,National Academy of Sciences, v. 44, n. 7, p. 671, 1958.
24 ZUCKERKANDL, E.; PAULING, L. Molecules as documents of evolutionaryhistory. Journal of Theoretical Biology, Elsevier, v. 8, n. 2, p. 357–366, 1965. ISSN0022-5193.
96

25 FRIEDMAN, J.; STIVERS, J. Detection of Damaged DNA Bases by DNAGlycosylase Enzymes. Biochemistry, ACS Publications, v. 49, n. 24, p. 4957–4967,2010. ISSN 0006-2960.
26 BROOKS, A. The essence of SNPs. Gene, v. 234, n. 2, p. 177–86, 1999.
27 ZHAO, H. et al. The study of neighboring nucleotide composition andtransition/transversion bias. Science in China Series C: Life Sciences, Springer, v. 49,n. 4, p. 395–402, 2006. ISSN 1006-9305.
28 DRAKE, J. et al. Rates of spontaneous mutation. Genetics, Genetics Soc America,v. 148, n. 4, p. 1667, 1998.
29 LYNCH, M. et al. A genome-wide view of the spectrum of spontaneous mutations inyeast. Proceedings of the National Academy of Sciences, National Acad Sciences, v. 105,n. 27, p. 9272, 2008.
30 DENVER, D. et al. A genome-wide view of Caenorhabditis elegans base-substitutionmutation processes. Proceedings of the National Academy of Sciences, National AcadSciences, v. 106, n. 38, p. 16310, 2009. ISSN 0027-8424.
31 HAAG-LIAUTARD, C. et al. Direct estimation of per nucleotide and genomicdeleterious mutation rates in Drosophila. Nature, Nature Publishing Group, v. 445,n. 7123, p. 82–85, 2007. ISSN 0028-0836.
32 KEIGHTLEY, P. et al. Analysis of the genome sequences of three Drosophilamelanogaster spontaneous mutation accumulation lines. Genome Research, Cold SpringHarbor Lab, v. 19, n. 7, p. 1195, 2009. ISSN 1088-9051.
33 NACHMAN, M.; CROWELL, S. Estimate of the mutation rate per nucleotide inhumans. Genetics, Genetics Soc America, v. 156, n. 1, p. 297, 2000.
34 COLLINS, D.; JUKES, T. Rates of transition and transversion in coding sequencessince the human-rodent divergence. Genomics, Elsevier, v. 20, n. 3, p. 386–396, 1994.ISSN 0888-7543.
35 WANG, D. G. et al. Large-Scale Identification, Mapping, andGenotyping of Single-Nucleotide Polymorphisms in the Human Ge-nome. Science, v. 280, n. 5366, p. 1077–1082, 1998. Disponıvel em:<http://www.sciencemag.org/cgi/content/abstract/280/5366/1077>.
97

36 HESS, S.; BLAKE, J.; BLAKE, R. Wide variations in neighbor-dependentsubstitution rates* 1. Journal of Molecular Biology, Elsevier, v. 236, n. 4, p. 1022–1033,1994. ISSN 0022-2836.
37 RAZIN, A.; RIGGS, A. DNA methylation and gene function. Science, AAAS,v. 210, n. 4470, p. 604, 1980.
38 KELLER, I.; BENSASSON, D.; NICHOLS, R. Transition-transversion bias is notuniversal: a counter example from grasshopper pseudogenes. PLoS Genet., v. 3, n. 2,p. e22, 2007.
39 TAMURA, K. Estimation of the number of nucleotide substitutions when thereare strong transition-transversion and G+ C-content biases. Molecular Biology and
Evolution, SMBE, v. 9, n. 4, p. 678, 1992. ISSN 0737-4038.
40 WAKELEY, J. Substitution-Rate Variation among Sites and the Estimation ofTransition Bias. Mol. Biol. Evol., Elsevier, v. 20, n. 3, p. 386–396, 1994. ISSN0888-7543.
41 LERCHER, M.; HURST, L. Human SNP variability and mutation rate are higher inregions of high recombination. Trends in genetics, Elsevier, v. 18, n. 7, p. 337–340, 2002.ISSN 0168-9525.
42 DURET, L.; ARNDT, P. The impact of recombination on nucleotide substitutions inthe human genome. PLoS Genet., v. 4, n. 5, p. e1000071, 2008.
43 KRAWCZAK, M.; BALL, E.; COOPER, D. Neighboring-nucleotide effects on therates of germ-line single-base-pair substitution in human genes. The American Journal
of Human Genetics, Elsevier, v. 63, n. 2, p. 474–488, 1998. ISSN 0002-9297.
44 ARNDT, P.; BURGE, C.; HWA, T. DNA sequence evolution with neighbor-dependent mutation. Journal of Computational Biology, Mary Ann Liebert, Inc., v. 10,n. 3-4, p. 313–322, 2003. ISSN 1066-5277.
45 MORTON, B. et al. Variation in mutation dynamics across the maize genome as afunction of regional and flanking base composition. Genetics, Genetics Soc America,v. 172, n. 1, p. 569, 2006.
46 GOJOBORI, T.; LI, W.; GRAUR, D. Patterns of nucleotide substitution inpseudogenes and functional genes. Journal of Molecular Evolution, Springer, v. 18, n. 5,p. 360–369, 1982. ISSN 0022-2844.
98

47 MAJEWSKI, J. Dependence of mutational asymmetry on gene-expression levels inthe human genome. The American Journal of Human Genetics, Elsevier, v. 73, n. 3, p.688–692, 2003. ISSN 0002-9297.
48 ZHAO, Z.; BOERWINKLE, E. Neighboring-nucleotide effects on single nucleotidepolymorphisms: a study of 2.6 million polymorphisms across the human genome.Genome Research, Cold Spring Harbor Lab, v. 12, n. 11, p. 1679, 2002. ISSN 1088-9051.
49 MOUNT, D. Bioinformatics: sequence and genome analysis. [S.l.]: CSHL press,2004. ISBN 0879697121.
50 KIMURA, M. A simple method for estimating evolutionary rates of basesubstitutions through comparative studies of nucleotide sequences. Journal of Molecular
Evolution, Springer, v. 16, n. 2, p. 111–120, 1980. ISSN 0022-2844.
51 JUKES, T.; CANTOR, C. Evolution of protein molecules. Mammalian Protein
Metabolism, New York, v. 3, p. 21–132, 1969.
52 FELSENSTEIN, J. Evolutionary trees from DNA sequences: a maximum likelihoodapproach. Journal of Molecular Evolution, Springer, v. 17, n. 6, p. 368–376, 1981. ISSN0022-2844.
53 HASEGAWA, M.; KISHINO, H.; YANO, T. Dating of the human-ape splitting by amolecular clock of mitochondrial DNA. Journal of Molecular Evolution, Springer, v. 22,n. 2, p. 160–174, 1985. ISSN 0022-2844.
54 TAMURA, K.; NEI, M. Estimation of the number of nucleotide substitutions in thecontrol region of mitochondrial DNA in humans and chimpanzees. Molecular Biology
and Evolution, SMBE, v. 10, n. 3, p. 512, 1993. ISSN 0737-4038.
55 HALPERN, A.; BRUNO, W. Evolutionary distances for protein-coding sequences:modeling site-specific residue frequencies. Molecular Biology and Evolution, SMBE,v. 15, n. 7, p. 910, 1998. ISSN 0737-4038.
56 NAG, N.; RAO, B.; KRISHNAMOORTHY, G. Altered dynamics of DNA basesadjacent to a mismatch: a cue for mismatch recognition by MutS. Journal of Molecular
Biology, Elsevier, v. 374, n. 1, p. 39–53, 2007. ISSN 0022-2836.
57 YU, U. et al. Bioinformatics in the post-genome era. Journal of biochemistry and
molecular biology, BIOCHEMICAL SOCIETY OF THE REPUBLIC OF KOREA,v. 37, n. 1, p. 75–82, 2004. ISSN 1225-8687.
99

58 LESK, A. Introduction to bioinformatics. [S.l.]: Oxford University Press Oxford,2008. ISBN 0199251967.
59 BAKER, P.; BRASS, A. Recent developments in biological sequence databases.Current Opinion in Biotechnology, Elsevier, v. 9, n. 1, p. 54–58, 1998. ISSN 0958-1669.
60 NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. Disponıvel em:<http://www.ncbi.nlm.nih.gov/About/primer/bioinformatics.
html>. Acesso em: 03-2011.
61 LIOLIOS, K. et al. The Genomes On Line Database (GOLD) in 2009: statusof genomic and metagenomic projects and their associated metadata. Nucleic Acids
Research, Oxford Univ Press, v. 38, n. suppl 1, p. D346, 2010. ISSN 0305-1048.
62 NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. NCBI. Dis-ponıvel em: <http://www.ncbi.nlm.nih.gov/genbank/genbankstats.html>. Acesso em: 01-2011.
63 ASHBURNER, M. et al. Gene ontology: tool for the unification of biology. Nature
genetics, NIH Public Access, v. 25, n. 1, p. 25, 2000.
64 HARRIS, M. et al. The gene ontology (go) database and informatics resource.Nucleic acids research, Nucleic Acids Res, v. 32, n. Database issue, p. D258, 2004.
65 SHERRY, S. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids
Research, Oxford Univ Press, v. 29, n. 1, p. 308, 2001. ISSN 0305-1048.
66 BALE, S. et al. MutaDATABASE: a centralized and standardized DNA variationdatabase. Nature Biotechnology, Nature Publishing Group, v. 29, n. 2, p. 117–118, 2011.ISSN 1087-0156.
67 RAZLUTSKII, I.; SHLYAKHTENKO, L.; LYUBCHENKO, Y. The effect ofnucleotide substitution on DNA denaturation profiles. Nucleic Acids Research, OxfordUniv Press, v. 15, n. 16, p. 6665, 1987. ISSN 0305-1048.
68 MANYANGA, F. et al. Origins of the “Nucleation” Free Energy in the HybridizationThermodynamics of Short Duplex DNA†. The Journal of Physical Chemistry B, ACSPublications, v. 113, n. 9, p. 2556–2563, 2009. ISSN 1520-6106.
69 WEBER, G. et al. Thermal equivalence of DNA duplexes for probe design. J. Phys.
Condens. Matter, v. 21, p. 034106, 2009.
100

70 OKONIEWSKI, M.; MILLER, C. Hybridization interactions between probesets inshort oligo microarrays lead to spurious correlations. BMC Bioinformatics, v. 7, n. 1,p. 276, 2006. ISSN 1471-2105. Disponıvel em: <http://www.biomedcentral.com/1471-2105/7/276>.
71 FISCHER, S.; LERMAN, L. Separation of random fragments of DNA according toproperties of their sequences. Proceedings of the National Academy of Sciences of the
United States of America, National Acad Sciences, v. 77, n. 8, p. 4420, 1980.
72 FISCHER, S.; LERMAN, L. DNA fragments differing by single base-pairsubstitutions are separated in denaturing gradient gels: correspondence with meltingtheory. Proceedings of the National Academy of Sciences of the United States of America,National Acad Sciences, v. 80, n. 6, p. 1579, 1983.
73 LYUBCHENKO, Y.; VOLOGODSKII, A.; FRANK-KAMENETSKII, M. Directcomparison of theoretical and experimental melting profiles for RFII ΦX174 DNA.Nature, Nature Publishing Group, v. 271, 1978.
74 LYUBCHENKO, Y. et al. A comparison of experimental and theoretical meltingmaps for replicative form of φX174 DNA. Nucleic Acids Research, Oxford Univ Press,v. 10, n. 6, p. 1867, 1982. ISSN 0305-1048.
75 CROWE, J. et al. Chemistry for the Biosciences. [S.l.]: Oxford University Press,2006. ISBN 0199280975.
76 BRESLAUER, K. J. et al. Predicting DNA duplex stability from the base sequence.Proc. Natl. Acad. Sci. USA, v. 83, n. 11, p. 3746–3750, 1986.
77 SANTALUCIA JR., J.; ALLAWI, H. T.; SENEVIRATNE, P. A. Improvednearest-neighbour parameters for predicting DNA duplex stability. Biochem., v. 35, p.3555–3562, 1996.
78 SANTALUCIA JR., J. A unified view of polymer, dumbbell, and oligonucleotideDNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA, v. 95, n. 4, p. 1460–1465, 1998. Disponıvel em: <http://www.pnas.org/cgi/content/abstract/95/4/1460>.
79 VOLOGODSKII, A. et al. Allowance for heterogeneous stacking in the DNAhelix-coil transition theory. Journal of biomolecular structure & dynamics, v. 2, n. 1,p. 131, 1984.
80 ALLAWI, H. T.; JR, J. S. Thermodynamics and NMR of internal G·T mismatches inDNA. Biochemistry, v. 36, n. 34, p. 10581–10594, 1997.
101

81 ALLAWI, H.; SANTALUCIA J, J. Thermodynamics of internal C.T mismatchesin DNA. Nucl. Acids. Res., v. 26, n. 11, p. 2694–2701, 1998. Disponıvel em:<http://nar.oupjournals.org/cgi/content/abstract/26/11/2694>.
82 ALLAWI, H. T.; SANTALUCIA JR., J. Nearest-neighbor thermodynamics ofinternal A·C mismatches in DNA: Sequence dependence and pH effects. Biochem., v. 37,p. 9435–9444, 1998.
83 ALLAWI, H.; JR, J. S. Nearest neighbor thermodynamic parameters for internalG·A mismatches in DNA. Biochemistry, v. 37, n. 8, p. 2170–2179, 1998.
84 PEYRET, N. et al. Nearest-neighbour thermodynamics and NMR of DNA sequenceswith internal A·A, C·C G·G and T·T mismatches. Biochem., v. 38, n. 12, p. 3468–3477,1999.
85 TRAVERS, A. The structural basis of DNA flexibility. Philosophical Transactions
of the Royal Society of London. Series A: Mathematical, Physical and Engineering
Sciences, The Royal Society, v. 362, n. 1820, p. 1423, 2004. ISSN 1364-503X.
86 CHURCHILL, M. et al. HMG-D is an architecture-specific protein that preferentiallybinds to DNA containing the dinucleotide TG. The EMBO Journal, Nature PublishingGroup, v. 14, n. 6, p. 1264, 1995.
87 RICHMOND, T.; DAVEY, C. The structure of DNA in the nucleosome core. Nature,v. 423, n. 6936, p. 145–150, 2003. ISSN 0028-0836.
88 STARR, D. B.; HOOPES, B. C.; HAWLEY, D. K. DNA bending is an importantcomponent of site-specific recognition by the TATA binding protein. J. Mol. Biol., v. 250,p. 434–446, 1995.
89 LEBRUN, A.; SHAKKED, Z.; LAVERY, R. Local DNA stretching mimics thedistortion caused by the TATA box-binding protein. Proc. Natl. Acad. Sci. USA, v. 94, p.2993–2998, 1997.
90 SOUZA, O. de; ORNSTEIN, R. Inherent DNA curvature and flexibility correlatewith TATA box functionality. Biopolymers, John Wiley & Sons, v. 46, n. 6, p. 403–415,1998. ISSN 1097-0282.
91 WEBER, G.; ESSEX, J. W.; NEYLON, C. Probing the microscopic flexibility ofDNA from melting temperatures. Nature Physics, v. 5, p. 769–773, 2009.
102

92 RITORT, F. Single-molecule experiments in biological physics: methods andapplications. J. Phys. Condens. Matter, v. 18, p. R531–R583, 2006.
93 WIGGINS, P. A. et al. High flexibility of DNA on short length scales probed byatomic force microscopy. Nature Nanotech., v. 1, p. 137–141, 2006.
94 MATHEW-FENN, R.; DAS, R.; HARBURY, P. Remeasuring the double helix.Science, AAAS, v. 322, n. 5900, p. 446, 2008.
95 YAMAKAWA, H.; STOCKMAYER, W. H. Statistical mechanics of wormlikechains. II. Excluded volume effects. J. Chem. Phys., v. 57, n. 7, p. 2843–3854, 1972.
96 SCHOFIELD, M.; HSIEH, P. DNA M ISMATCH R EPAIR: Molecular Mechanismsand Biological Function*. Annual Reviews in Microbiology, Annual Reviews 4139 ElCamino Way, PO Box 10139, Palo Alto, CA 94303-0139, USA, v. 57, n. 1, p. 579–608,2003. ISSN 0066-4227.
97 KUNKEL, T.; ERIE, D. DNA mismatch repair. Biochemistry, v. 74, 2005.
98 WEBER, G. Calculation of DNA mismatch Morse potentials and elastic constantsfrom melting temperatures. Unpublished data.
99 BLOOM, K. Beyond the code: the mechanical properties of DNA as they relate tomitosis. Chromosoma, Springer, v. 117, n. 2, p. 103–110, 2008. ISSN 0009-5915.
100 MCNAUGHT, A. et al. Compendium of chemical terminology: IUPAC
recommendations. [S.l.]: Blackwell Science, 1997. ISBN 0865426848.
101 IUPAC. Disponıvel em: <http://www.iupac.org/>. Acesso em: 03-2010.
102 WELLCOME TRUST SANGER INSTITUTE. Disponıvel em: <http:
//www.sanger.ac.uk/>. Acesso em: 09-2010.
103 BERNARDI, G. Isochores and the evolutionary genomics of vertebrates. Gene,Elsevier, v. 241, n. 1, p. 3–17, 2000. ISSN 0378-1119.
104 YAKOVCHUK, P.; PROTOZANOVA, E.; FRANK-KAMENETSKII, M.Base-stacking and base-pairing contributions into thermal stability of the DNA doublehelix. Nucleic acids research, Oxford Univ Press, v. 34, n. 2, p. 564, 2006. ISSN0305-1048.
105 PATWARDHAN, S.; PEDERSEN, T. The cpan wordnet:: similarity package.2003. Disponıvel em: <http://www.cpan.org/>. Acesso em: 09-2010.
103

106 VENTER, J. C. et al. The Sequence of the Human Genome.Science, v. 291, n. 5507, p. 1304–1351, 2001. Disponıvel em:<http://www.sciencemag.org/cgi/content/abstract/291/5507/1304>.
107 LIU, Y. et al. Bos taurus genome assembly. BMC Genomics, BioMed Central Ltd,v. 10, n. 1, p. 180, 2009. ISSN 1471-2164.
108 ECK, S. et al. Whole genome sequencing of a single Bos taurus animal for singlenucleotide polymorphism discovery. Genome Biology, BioMed Central Ltd, v. 10, n. 8,p. R82, 2009.
109 FRAZER, K. et al. A sequence-based variation map of 8.27 million SNPs in inbredmouse strains. Nature, Nature Publishing Group, v. 448, n. 7157, p. 1050–1053, 2007.ISSN 0028-0836.
110 GNERRE, S. et al. High-quality draft assemblies of mammalian genomes frommassively parallel sequence data. Proceedings of the National Academy of Sciences,National Acad Sciences, v. 108, n. 4, p. 1513, 2011. ISSN 0027-8424.
111 HILLIER, L. et al. Sequence and comparative analysis of the chicken genomeprovide unique perspectives on vertebrate evolution. Nature, Nature Publishing Group,v. 432, n. 7018, p. 695–716, 2004. ISSN 0028-0836.
112 EMBL-EBI. Ensembl versao 61 de fev 2011. E!ensembl. Disponıvel em:<http://www.ensembl.org/Danio\_rerio/Info/Index>. Acesso em:02-2011.
113 HUBBARD, T. et al. Ensembl 2007. Nucleic Acids Research, Oxford Univ Press,v. 35, n. suppl 1, p. D610, 2006. ISSN 0305-1048.
114 NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. NCBI.Disponıvel em: <http://www.ncbi.nlm.nih.gov/mapview/stats/
BuildStats.cgi?taxid=7460\&build=4\&ver=1>. Acesso em: 09-2010.
115 GARDNER, M. J. et al. Genome sequence of the human malaria parasitePlasmodium falciparum. Nature, Nature Publishing Group, v. 419, n. 6906, p. 498–511,2002.
116 SASAKI, T.; BURR, B. International Rice Genome Sequencing Project: the effortto completely sequence the rice genome. Current opinion in plant biology, Elsevier, v. 3,n. 2, p. 138–142, 2000. ISSN 1369-5266.
104

117 FERRETTI, J. et al. Complete genome sequence of an M1 strain of Streptococcuspyogenes. Proceedings of the National Academy of Sciences of the United States of
America, National Acad Sciences, v. 98, n. 8, p. 4658, 2001.
118 SAYERS, E.; WHEELER, D.; (US), N. C. for B. I. Building Customized Data
Pipelines Using the Entrez Programming Utilities (eUtils). [S.l.]: NCBI, 2004.
119 KHOVAYKO, O. NCBI. Disponıvel em: <http://www.ncbi.nlm.nih.gov/corehtml/query/static/eutils\_example.pl>. Acesso em:03-2011.
120 STAJICH, J. et al. The Bioperl toolkit: Perl modules for the life sciences. Genome
Research, Cold Spring Harbor Lab, v. 12, n. 10, p. 1611, 2002. ISSN 1088-9051.
121 NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. NCBI.Disponıvel em: <http://www.ncbi.nlm.nih.gov/projects/genome/
guide/cow/>. Acesso em: 09-2010.
122 NATIONAL CENTER FOR BIOTECHNOLOGY INFORMATION. NCBI.Disponıvel em: <http://www.ncbi.nlm.nih.gov/genome/guide/
mouse/>. Acesso em: 09-2010.
123 EMBL-EBI. Ensembl, versao no 50 de Jul 2008. E!ensembl. Disponıvel em:<http://ensembl.fugu-sg.org/Gallus\_gallus/index.html>.Acesso em: 09-2010.
124 WHO, WORLD HEALTH ORGANIZATION. Disponıvel em: <http:
//www.who.int/>. Acesso em: 03-2011.
125 WELLCOME TRUST SANGER INSTITUTE. Disponıvel em:<http://www.sanger.ac.uk/resources/downloads/protozoa/
plasmodium-falciparum.html>. Acesso em: 09-2010.
126 OHYANAGI, H. et al. The Rice Annotation Project Database (RAP-DB): hub forOryza sativa ssp. japonica genome information. Nucleic acids research, Oxford UnivPress, v. 34, n. suppl 1, p. D741, 2006. ISSN 0305-1048.
127 GENOME BIOINFORMATICS GROUP OF UC SANTA CRUZ - UNIVERSITYOF CALIFORNIA. Streptococcus pyogenes M1 GAS - Genome Browser Gateway.Disponıvel em: <http://microbes.ucsc.edu/cgi-bin/hgGateway?db=strePyog_M1_GAS>. Acesso em: 09-2010.
105





























