Embed Size (px)
Citation preview

September 2015
White Paper
The European Cancer Congress 2015
DISCLAIMER
While every care is taken to ensure the accuracy of the information contained in this material, the facts, estimates, and opinions
stated are based on information and sources which, while we believe them to be reliable, are not guaranteed. In particular, it
should not be relied upon as the sole source of reference in relation to the subject matter. No liability can be accepted by
Datamonitor, its directors, or employees for any loss occasioned to any person or entity acting or failing to act as a result of
anything contained in or omitted from the content of this material, or our conclusions as stated. The findings are Datamonitor's
current opinions; they are subject to change without notice. Datamonitor has no obligation to update or amend the research or
to let anyone know if our opinions change materially.
If you have questions about the research, data, and findings within this document you can put your questions directly to the analysts. Simply email your questions to [email protected]. To find out more about Datamonitor Healthcare, contact us at: email [email protected] phone +44 20 7551 9430 Visit our website: www.datamonitorhealthcare.com Or follow us on Twitter: @DatamonitorHC

White Paper
The European Cancer Congress 2015
Datamonitor Healthcare has identified the following key highlights from the 2015 European Cancer
Congress (ECC), which was held on 25–29 September 2015 in Vienna, Austria:
Late-breaking Phase II data revealed a promising future for Roche’s programmed death ligand-1 (PD-L1)
inhibitor atezolizumab in non-small cell lung cancer (NSCLC) and bladder cancer. Interim results from the
pivotal BIRCH study have placed atezolizumab as a front-runner for the first-line treatment of PD-L1-
positive (PD-L1+) NSCLC, ahead of Bristol-Myers Squibb’s Opdivo (nivolumab) and Merck & Co’s Keytruda
(pembrolizumab). In bladder cancer, atezolizumab is likely to be the first programmed death-1 (PD-1)/PD-
L1 immunotherapy to reach the market and could potentially become the new standard of care.
Six of the late-breaking abstracts (LBAs) reported data for PD-1/PD-L1 drugs other than atezolizumab
across a range of different cancer types. The most prominent development was the first look at positive
data from the Phase III CheckMate 025 trial of Opdivo in previously treated renal cell cancer (RCC). The
LBAs also contained new and updated datasets for Keytruda and Opdivo in melanoma and NSCLC, while
intriguing biomarker-stratified efficacy data for AstraZeneca’s anti-PD-L1 durvalumab in NSCLC were also
presented. Merck also communicated early-phase trial data for Keytruda in the rare skin cancer Merkel cell
carcinoma (MCC). These LBAs illustrate the continuing rich investment and expansion in PD-1/PD-L1
development, and as a result it is highly likely that we can expect intense competition between many of
these drugs in the future.
Presentations of pivotal trial results for Exelixis’s multi-tyrosine kinase inhibitor Cometriq (cabozantinib) and
Bristol-Myers Squibb’s Opdivo indicate that major change is coming in the second-line treatment of RCC.
Opdivo significantly improved overall survival (OS) in comparison to Afinitor (everolimus; Novartis) and is
now likely to become the treatment of choice in the second-line setting. Cometriq’s results were not quite
as good as Opdivo’s, but they were still positive and Exelixis will be pleased at the opportunity to expand
Cometriq’s label.
Immatics revealed that the Phase III IMPRINT study of its RCC vaccine IMA901 did not meet its primary
endpoint. Immatics had hoped that the combination of IMA901 with Sutent (sunitinib; Pfizer) would produce
a synergistic immunomodulatory effect that would extend OS compared to Sutent monotherapy, and that
this would enable IMA901 to become the first approved vaccine for the treatment of RCC. Immatics has
taken the difficult decision to shift its focus onto the Adoptive Cellular Therapies (ACT) portion of its
pipeline, effectively ending the development of IMA901.

White Paper
The European Cancer Congress 2015
Phase II data for Roche’s PD-L1 atezolizumab in NSCLC and bladder cancer were presented on the
morning of 27 September at ECC 2015. The data from the BIRCH and POPLAR studies for
atezolizumab in locally advanced and metastatic NSCLC and from the IMvigor 210 trial in metastatic
urothelial bladder cancer suggest a very bright future for atezolizumab. The anti-PD-L1 drug
demonstrated durable responses in pretreated advanced NSCLC, suggesting that therapies targeting
PD-L1 are comparable to those targeting PD-1. Indeed, interim results from the pivotal BIRCH study
have placed atezolizumab as a front-runner for the first-line treatment of PD-L1+ NSCLC, ahead of
Bristol-Myers Squibb’s Opdivo and Merck’s Keytruda. Clinical responses to atezolizumab therapy
increased significantly in patients with highly expressed PD-L1 on their tumors. In bladder cancer,
atezolizumab is likely to be the first PD-1/PD-L1 immunotherapy to reach the market and could
potentially become the new standard of care.
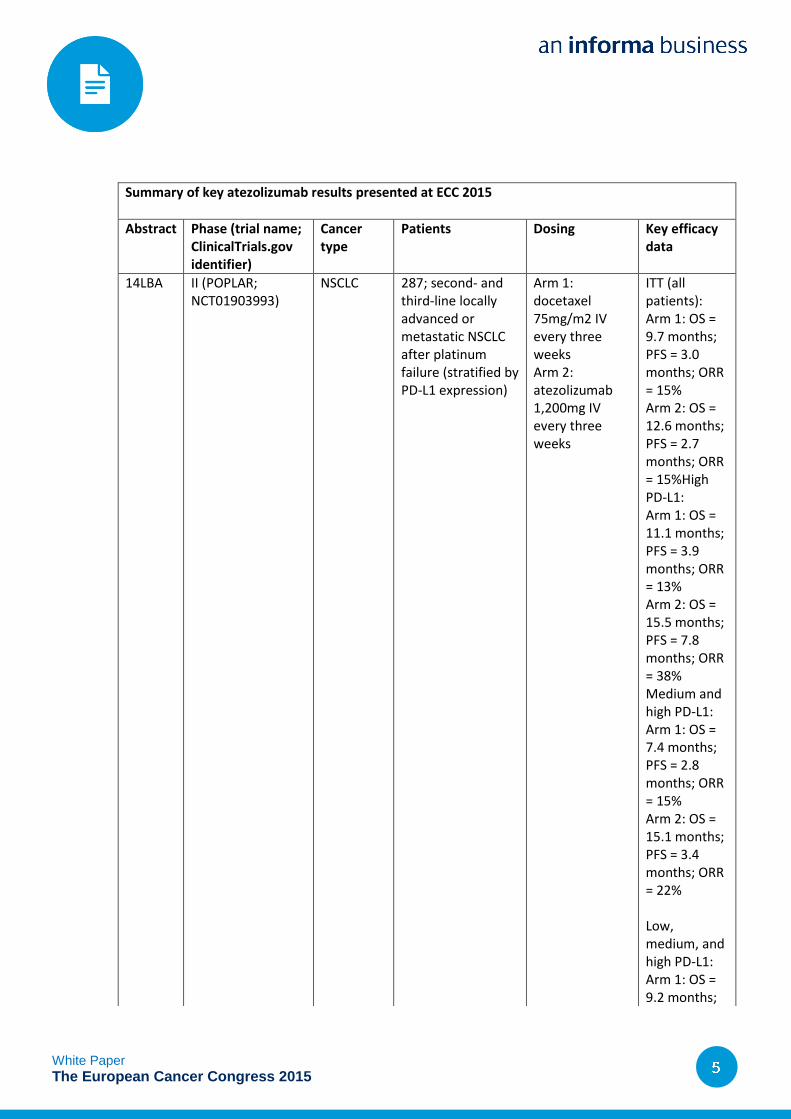
Datamonitor Healthcare has summarized the late-breaking results for atezolizumab at ECC 2015 in
the table below.
White Paper
The European Cancer Congress 2015
Summary of key atezolizumab results presented at ECC 2015
Abstract Phase (trial name; ClinicalTrials.gov identifier)
Cancer type
Patients Dosing Key efficacy data
14LBA II (POPLAR; NCT01903993)
NSCLC 287; second- and third-line locally advanced or metastatic NSCLC after platinum failure (stratified by PD-L1 expression)
Arm 1: docetaxel 75mg/m2 IV every three weeks Arm 2: atezolizumab 1,200mg IV every three weeks
ITT (all patients): Arm 1: OS = 9.7 months; PFS = 3.0 months; ORR = 15% Arm 2: OS = 12.6 months; PFS = 2.7 months; ORR = 15%High PD-L1: Arm 1: OS = 11.1 months; PFS = 3.9 months; ORR = 13% Arm 2: OS = 15.5 months; PFS = 7.8 months; ORR = 38% Medium and high PD-L1: Arm 1: OS = 7.4 months; PFS = 2.8 months; ORR = 15% Arm 2: OS = 15.1 months; PFS = 3.4 months; ORR = 22%
Low, medium, and high PD-L1: Arm 1: OS = 9.2 months;
White Paper
The European Cancer Congress 2015
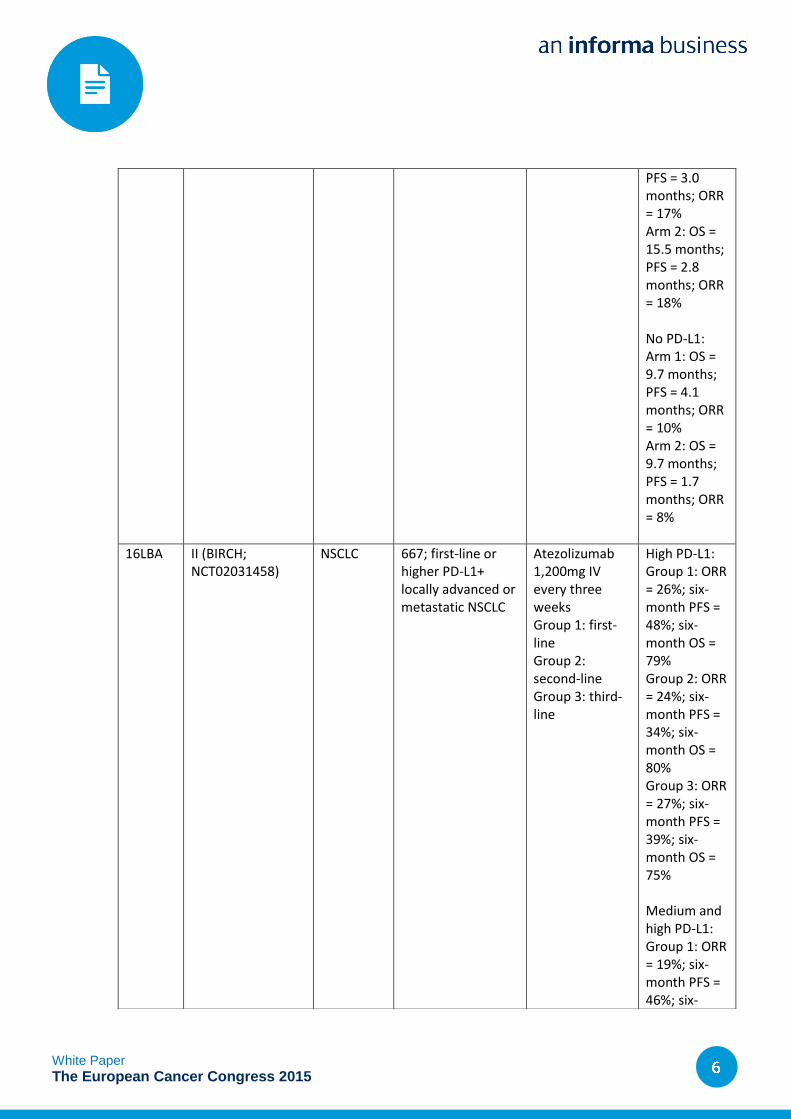
PFS = 3.0 months; ORR = 17% Arm 2: OS = 15.5 months; PFS = 2.8 months; ORR = 18%
No PD-L1: Arm 1: OS = 9.7 months; PFS = 4.1 months; ORR = 10% Arm 2: OS = 9.7 months; PFS = 1.7 months; ORR = 8%
16LBA II (BIRCH; NCT02031458)
NSCLC 667; first-line or higher PD-L1+ locally advanced or metastatic NSCLC
Atezolizumab 1,200mg IV every three weeks Group 1: first-line Group 2: second-line Group 3: third-line
High PD-L1: Group 1: ORR = 26%; six-month PFS = 48%; six-month OS = 79% Group 2: ORR = 24%; six-month PFS = 34%; six-month OS = 80% Group 3: ORR = 27%; six-month PFS = 39%; six-month OS = 75% Medium and high PD-L1: Group 1: ORR = 19%; six-month PFS = 46%; six-
White Paper
The European Cancer Congress 2015
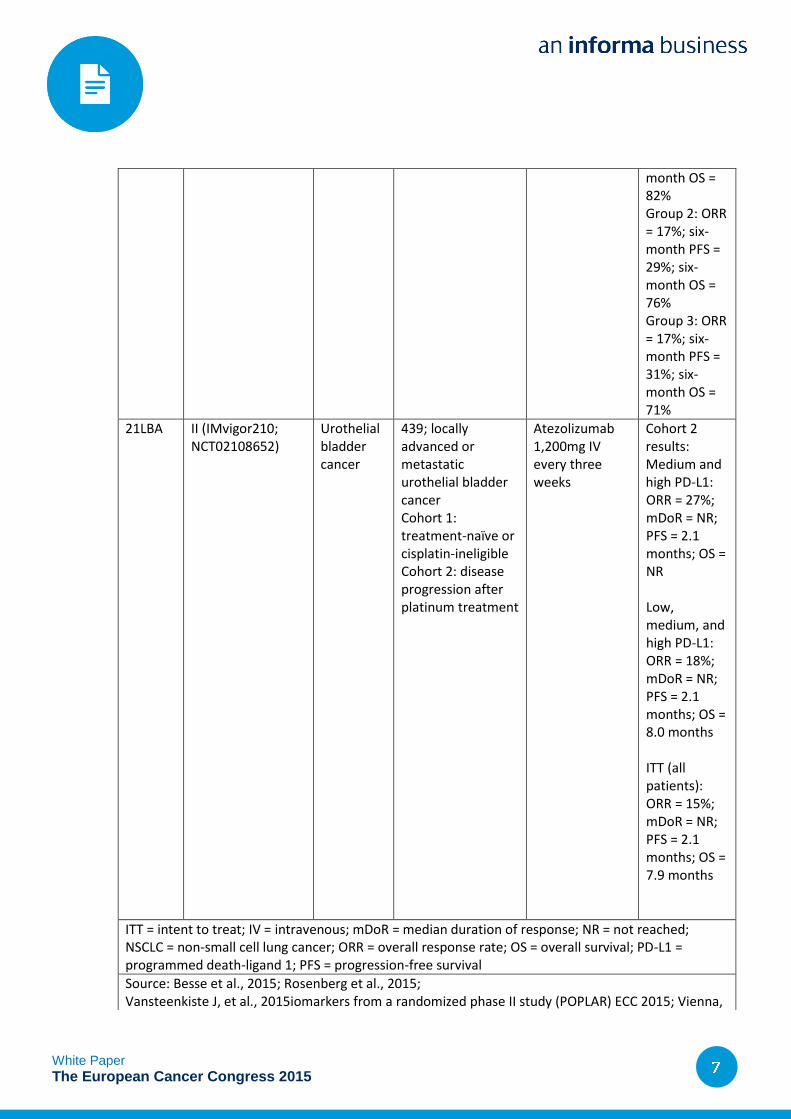
month OS = 82% Group 2: ORR = 17%; six-month PFS = 29%; six-month OS = 76% Group 3: ORR = 17%; six-month PFS = 31%; six-month OS = 71%
21LBA II (IMvigor210; NCT02108652)
Urothelial bladder cancer
439; locally advanced or metastatic urothelial bladder cancer Cohort 1: treatment-naïve or cisplatin-ineligible Cohort 2: disease progression after platinum treatment
Atezolizumab 1,200mg IV every three weeks
Cohort 2 results: Medium and high PD-L1: ORR = 27%; mDoR = NR; PFS = 2.1 months; OS = NR Low, medium, and high PD-L1: ORR = 18%; mDoR = NR; PFS = 2.1 months; OS = 8.0 months ITT (all patients): ORR = 15%; mDoR = NR; PFS = 2.1 months; OS = 7.9 months
ITT = intent to treat; IV = intravenous; mDoR = median duration of response; NR = not reached; NSCLC = non-small cell lung cancer; ORR = overall response rate; OS = overall survival; PD-L1 = programmed death-ligand 1; PFS = progression-free survival
Source: Besse et al., 2015; Rosenberg et al., 2015; Vansteenkiste J, et al., 2015iomarkers from a randomized phase II study (POPLAR) ECC 2015; Vienna,

White Paper
The European Cancer Congress 2015
Updated final results from the POPLAR study of atezolizumab show it to be an effective treatment for
pretreated NSCLC and comparable to its potential rival Keytruda. Considering Merck submitted a
supplemental Biologic License Application (sBLA) for its anti-PD-1 therapy based on comparable data
from a Phase Ib study (KEYNOTE-001), atezolizumab’s positive data are likely to support Roche’s US
Food and Drug Administration (FDA) filing in NSCLC, which is planned for early 2016.
Speaking on 27 September, Dr Johan Vansteenkiste of University Hospital Leuven presented full
results from the Phase II POPLAR study. The randomized trial assessed atezolizumab’s safety and
efficacy compared to docetaxel in patients with locally advanced or metastatic NSCLC that had
progressed after platinum-based chemotherapy. Patients were stratified by PD-L1 expression levels
as determined by Roche’s developmental immunohistochemistry assay on tumor cells and infiltrating
immune cells. The primary endpoint was OS, and secondary endpoints included progression-free
survival (PFS), overall response rate (ORR), and safety. As with interim results previously presented
at ASCO 2015, atezolizumab was well tolerated and demonstrated clinical superiority as a second-
and third-line monotherapy compared to docetaxel. Subgroup analyses showed that PD-L1 tumor
expression correlated with response to the drug.
Data from the POPLAR study in 287 previously treated advanced NSCLC patients showed
atezolizumab improved OS by almost three months in comparison to docetaxel (12.6 months versus
9.7 months). PFS in the atezolizumab monotherapy group was 2.7 months compared to a PFS of 3.0
months in the docetaxel arm. The ORR for both groups was the same at 15%. Fewer patients in the
atezolizumab arm experienced treatment-related grade 3–4 adverse events as compared to the
docetaxel arm (11% versus 39%). Patients in the treatment arm were administered atezolizumab
intravenously at a flat dose of 1,200mg every three weeks, while those in the other arm received
75mg/m2 of docetaxel intravenously every three weeks.
NSCLC accounts for over 80% of all lung cancers, and can be further divided into squamous cell
carcinoma, adenocarcinoma, and large-cell carcinoma. NSCLC is one of the most common cancers
globally, and Datamonitor Healthcare forecasts that in the US, Japan, and five major EU markets
(France, Germany, Italy, Spain, and the UK) the incidence of NSCLC will be 505,300 patients in 2014,
rising to 569,000 in 2020.
Atezolizumab is an antibody designed to target PD-L1 on tumor cells, allowing the body’s immune
system to recognize and properly respond to the tumor. PD-1 and its ligands, PD-L1 and PD-L2, are
members of the cluster of differentiation-28 and B7 family. The cell-surface molecules of the B7 family
and the cytotoxic T-lymphocyte antigen 4 family both regulate complex signaling pathways that affect
T-cell activation, tolerance, and immunopathology. The pathways have a similar effect on T-cell
immune response but are distinct from each other.
Austria; 27 Sept 2015; 14LBA.
White Paper
The European Cancer Congress 2015
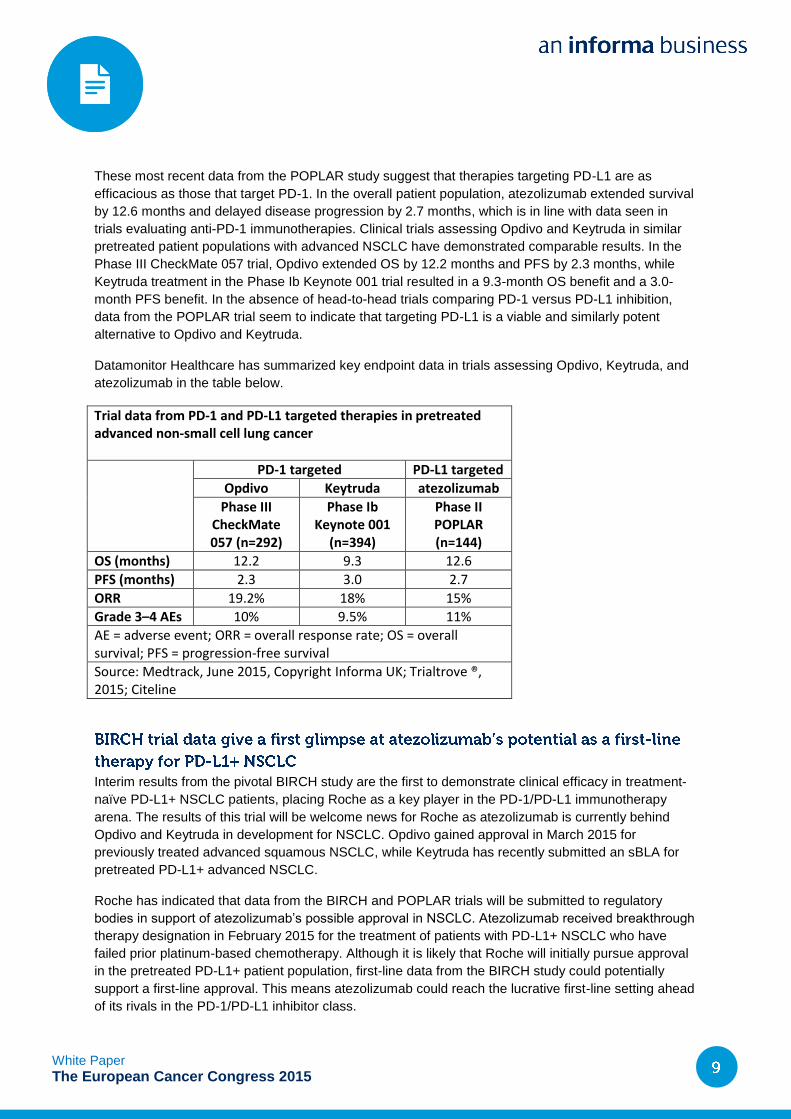
These most recent data from the POPLAR study suggest that therapies targeting PD-L1 are as
efficacious as those that target PD-1. In the overall patient population, atezolizumab extended survival
by 12.6 months and delayed disease progression by 2.7 months, which is in line with data seen in
trials evaluating anti-PD-1 immunotherapies. Clinical trials assessing Opdivo and Keytruda in similar
pretreated patient populations with advanced NSCLC have demonstrated comparable results. In the
Phase III CheckMate 057 trial, Opdivo extended OS by 12.2 months and PFS by 2.3 months, while
Keytruda treatment in the Phase Ib Keynote 001 trial resulted in a 9.3-month OS benefit and a 3.0-
month PFS benefit. In the absence of head-to-head trials comparing PD-1 versus PD-L1 inhibition,
data from the POPLAR trial seem to indicate that targeting PD-L1 is a viable and similarly potent
alternative to Opdivo and Keytruda.
Datamonitor Healthcare has summarized key endpoint data in trials assessing Opdivo, Keytruda, and
atezolizumab in the table below.
Trial data from PD-1 and PD-L1 targeted therapies in pretreated advanced non-small cell lung cancer
PD-1 targeted PD-L1 targeted
Opdivo Keytruda atezolizumab
Phase III CheckMate 057 (n=292)
Phase Ib Keynote 001
(n=394)
Phase II POPLAR (n=144)
OS (months) 12.2 9.3 12.6
PFS (months) 2.3 3.0 2.7
ORR 19.2% 18% 15%
Grade 3–4 AEs 10% 9.5% 11%
AE = adverse event; ORR = overall response rate; OS = overall survival; PFS = progression-free survival
Source: Medtrack, June 2015, Copyright Informa UK; Trialtrove ®, 2015; Citeline
Interim results from the pivotal BIRCH study are the first to demonstrate clinical efficacy in treatment-
naïve PD-L1+ NSCLC patients, placing Roche as a key player in the PD-1/PD-L1 immunotherapy
arena. The results of this trial will be welcome news for Roche as atezolizumab is currently behind
Opdivo and Keytruda in development for NSCLC. Opdivo gained approval in March 2015 for
previously treated advanced squamous NSCLC, while Keytruda has recently submitted an sBLA for
pretreated PD-L1+ advanced NSCLC.
Roche has indicated that data from the BIRCH and POPLAR trials will be submitted to regulatory
bodies in support of atezolizumab’s possible approval in NSCLC. Atezolizumab received breakthrough
therapy designation in February 2015 for the treatment of patients with PD-L1+ NSCLC who have
failed prior platinum-based chemotherapy. Although it is likely that Roche will initially pursue approval
in the pretreated PD-L1+ patient population, first-line data from the BIRCH study could potentially
support a first-line approval. This means atezolizumab could reach the lucrative first-line setting ahead
of its rivals in the PD-1/PD-L1 inhibitor class.

White Paper
The European Cancer Congress 2015
On 27 September, Dr Benjamin Besse of the Gustave Roussy Institute of Oncology and Paris Sud
University presented topline results from the Phase II BIRCH study. This single-arm study assessed
atezolizumab monotherapy in patients with PD-L1+ locally advanced or metastatic NSCLC. The
patients were divided into three groups according to the number of previous therapies they had
received, and the primary endpoint of the study was the ORR assessed by an independent review
facility per RECIST v1.1. The secondary endpoints of the study were duration of response, OS, PFS,
and safety. As a monotherapy, atezolizumab met its primary endpoint in all three cohorts and
demonstrated durable response rates while maintaining the same safety profile as observed in
previous studies.
Patients who had the highest levels of PD-L1 expression in all three cohorts of the trial had significant
response rates to single-agent atezolizumab. Advanced NSCLC patients with the highest expression
levels of PD-L1 who received atezolizumab as a first-line, second-line, and third-line therapy had
ORRs of 26%, 24%, and 27%, respectively. Additionally, the respective OS rates at six months were
79%, 80%, and 75%. Around 11% of patients experienced grade 3–4 treatment-related adverse
events.
Atezolizumab’s future commercial potential could be significant if Roche eventually gains approval for
the drug as a first-line therapy for PD-L1+ NSCLC. Datamonitor Healthcare anticipates that the drug
that penetrates the advanced/metastatic NSCLC first-line setting most effectively will be the one that
generates the highest commercial rewards. While it is highly likely that atezolizumab will be indicated
for patients who are PD-L1+, this patient population still represents a substantial portion of the market;
around 34% of NSCLC patients who were initially screened for the BIRCH trial were PD-L1+.
Datamonitor Healthcare expects that Roche’s early release of positive first-line BIRCH trial data
combined with an expedited FDA review may give atezolizumab the advantage over Opdivo and
Keytruda within the first-line PD-L1+ NSCLC treatment setting.
Results from the POPLAR and BIRCH trials presented at ECC 2015 demonstrated a strong correlation
between PD-L1 expression status and response to atezolizumab treatment, adding to the growing
body of evidence supporting PD-L1 as a viable biomarker. Datamonitor Healthcare expects that the
concurrent development of Roche’s companion diagnostic with atezolizumab is likely to facilitate the
drug’s regulatory approvals by identifying the patients who are most likely to benefit from this therapy.
In both the POPLAR and BIRCH studies, patients were stratified by their levels of PD-L1 expression,
which revealed that atezolizumab treatment favored tumors with high PD-L1 levels. The BIRCH study
indicated that 26% of patients with high levels of PD-L1 responded to first-line atezolizumab, while
only 19% of patients with medium levels of the ligand responded to the same treatment. Similar trends
were observed in the patient cohorts who received second- and third-line atezolizumab.
In the POPLAR study, atezolizumab in previously treated advanced NSCLC showed that increasing
levels of PD-L1 correlated with improvements in clinical endpoints. Atezolizumab therapy in patients
with no PD-L1 expression gave similar results to docetaxel and extended survival by 9.7 months. In
patients with the highest levels of the ligand, atezolizumab treatment extended OS significantly by 15.5
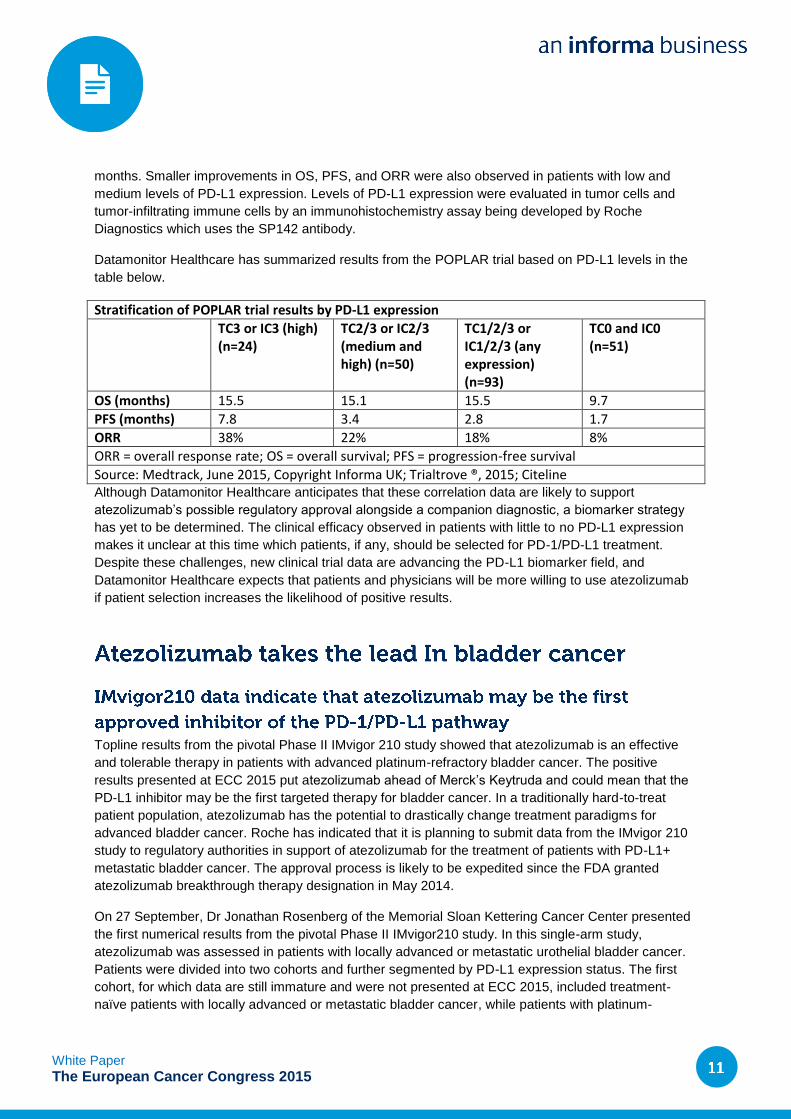
White Paper
The European Cancer Congress 2015
months. Smaller improvements in OS, PFS, and ORR were also observed in patients with low and
medium levels of PD-L1 expression. Levels of PD-L1 expression were evaluated in tumor cells and
tumor-infiltrating immune cells by an immunohistochemistry assay being developed by Roche
Diagnostics which uses the SP142 antibody.
Datamonitor Healthcare has summarized results from the POPLAR trial based on PD-L1 levels in the
table below.
Stratification of POPLAR trial results by PD-L1 expression
TC3 or IC3 (high) (n=24)
TC2/3 or IC2/3 (medium and high) (n=50)
TC1/2/3 or IC1/2/3 (any expression) (n=93)
TC0 and IC0 (n=51)
OS (months) 15.5 15.1 15.5 9.7
PFS (months) 7.8 3.4 2.8 1.7
ORR 38% 22% 18% 8%
ORR = overall response rate; OS = overall survival; PFS = progression-free survival
Source: Medtrack, June 2015, Copyright Informa UK; Trialtrove ®, 2015; Citeline Although Datamonitor Healthcare anticipates that these correlation data are likely to support
atezolizumab’s possible regulatory approval alongside a companion diagnostic, a biomarker strategy
has yet to be determined. The clinical efficacy observed in patients with little to no PD-L1 expression
makes it unclear at this time which patients, if any, should be selected for PD-1/PD-L1 treatment.
Despite these challenges, new clinical trial data are advancing the PD-L1 biomarker field, and
Datamonitor Healthcare expects that patients and physicians will be more willing to use atezolizumab
if patient selection increases the likelihood of positive results.
Topline results from the pivotal Phase II IMvigor 210 study showed that atezolizumab is an effective
and tolerable therapy in patients with advanced platinum-refractory bladder cancer. The positive
results presented at ECC 2015 put atezolizumab ahead of Merck’s Keytruda and could mean that the
PD-L1 inhibitor may be the first targeted therapy for bladder cancer. In a traditionally hard-to-treat
patient population, atezolizumab has the potential to drastically change treatment paradigms for
advanced bladder cancer. Roche has indicated that it is planning to submit data from the IMvigor 210
study to regulatory authorities in support of atezolizumab for the treatment of patients with PD-L1+
metastatic bladder cancer. The approval process is likely to be expedited since the FDA granted
atezolizumab breakthrough therapy designation in May 2014.
On 27 September, Dr Jonathan Rosenberg of the Memorial Sloan Kettering Cancer Center presented
the first numerical results from the pivotal Phase II IMvigor210 study. In this single-arm study,
atezolizumab was assessed in patients with locally advanced or metastatic urothelial bladder cancer.
Patients were divided into two cohorts and further segmented by PD-L1 expression status. The first
cohort, for which data are still immature and were not presented at ECC 2015, included treatment-
naïve patients with locally advanced or metastatic bladder cancer, while patients with platinum-

White Paper
The European Cancer Congress 2015
refractory locally advanced or metastatic bladder cancer were included in the second cohort. The
primary endpoint of the study was ORR, and secondary endpoints included duration of response, OS,
PFS, and safety. Atezolizumab met its primary endpoint in cohort 2, demonstrating an ORR of 15%
with a median PFS of 2.1 months and a median OS of 7.9 months. There were no treatment-related
deaths and atezolizumab’s safety profile remained consistent with previous safety data.
Responses to atezolizumab treatment in platinum-refractory advanced bladder cancer improved
significantly with increasing levels of PD-L1 expression. Patients who had medium to high levels of
PD-L1 expression had an ORR of 27% as compared to 18% in patients with low-to-high levels of the
protein ligand. While the median PFS at a 24-week cutoff remained unchanged between the two
patient populations (2.1 months), higher PD-L1 expression appears to confer an OS benefit. The
median OS in patients with low-to-high PD-L1 levels was eight months, while in the high PD-L1 level
patient population the OS has not yet been reached.
Overall, the pivotal data from IMvigor 210 are highly encouraging for atezolizumab’s future. The high
unmet need in advanced bladder cancer combined with the drug’s positive response rates mean that
Roche is well poised to lead and establish its presence within this indication. Dr Rosenberg
commented that there are no known agents that improve OS in metastatic urothelial bladder cancer,
and that Pierre Fabre’s Javlor (vinflunine) is the only approved therapy in Europe, which demonstrates
an ORR of only 8.6%. Atezolizumab could therefore potentially become the new standard of care and
represents a promising treatment option for patients who have had very poor prognoses. Atezolizumab
is currently in ongoing trials for treatment-naïve advanced bladder cancer as well as Phase III trials
(IMvigor 211) comparing second-line atezolizumab to chemotherapy.
White Paper
The European Cancer Congress 2015
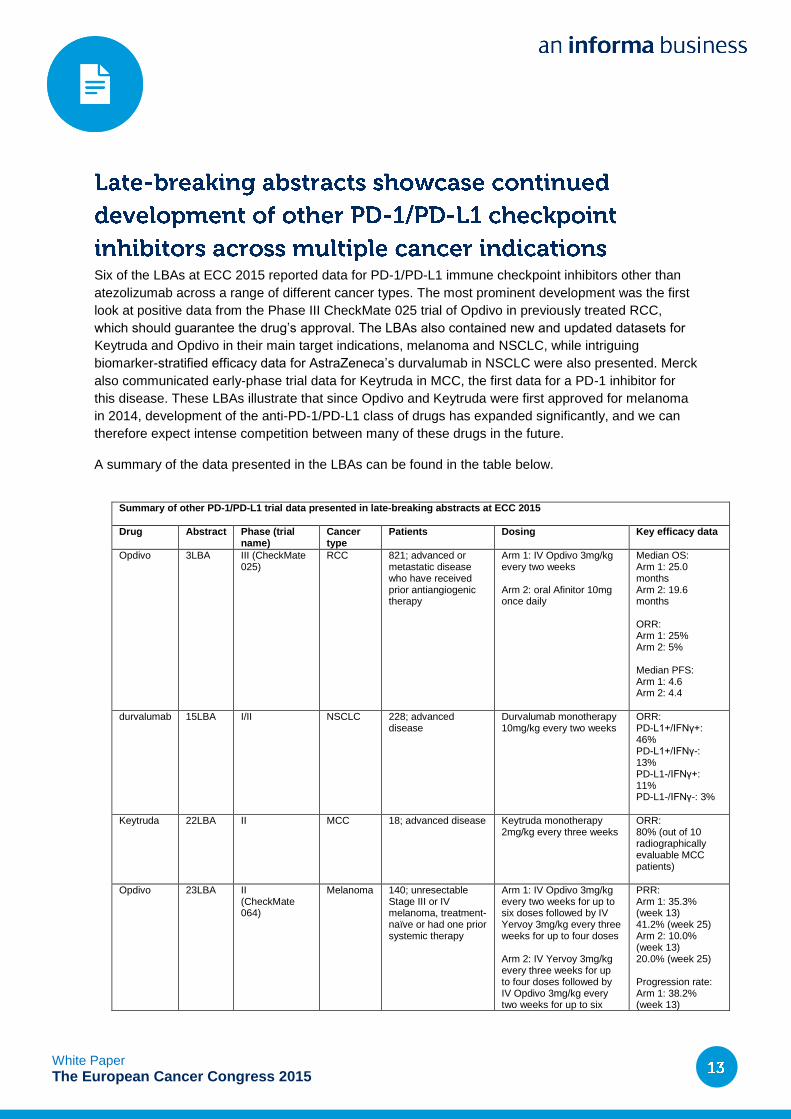
Six of the LBAs at ECC 2015 reported data for PD-1/PD-L1 immune checkpoint inhibitors other than
atezolizumab across a range of different cancer types. The most prominent development was the first
look at positive data from the Phase III CheckMate 025 trial of Opdivo in previously treated RCC,
which should guarantee the drug’s approval. The LBAs also contained new and updated datasets for
Keytruda and Opdivo in their main target indications, melanoma and NSCLC, while intriguing
biomarker-stratified efficacy data for AstraZeneca’s durvalumab in NSCLC were also presented. Merck
also communicated early-phase trial data for Keytruda in MCC, the first data for a PD-1 inhibitor for
this disease. These LBAs illustrate that since Opdivo and Keytruda were first approved for melanoma
in 2014, development of the anti-PD-1/PD-L1 class of drugs has expanded significantly, and we can
therefore expect intense competition between many of these drugs in the future.
A summary of the data presented in the LBAs can be found in the table below.
Summary of other PD-1/PD-L1 trial data presented in late-breaking abstracts at ECC 2015
Drug Abstract Phase (trial name)
Cancer type
Patients Dosing Key efficacy data
Opdivo 3LBA III (CheckMate 025)
RCC 821; advanced or metastatic disease who have received prior antiangiogenic therapy
Arm 1: IV Opdivo 3mg/kg every two weeks Arm 2: oral Afinitor 10mg once daily
Median OS: Arm 1: 25.0 months Arm 2: 19.6 months ORR: Arm 1: 25% Arm 2: 5% Median PFS: Arm 1: 4.6 Arm 2: 4.4
durvalumab 15LBA I/II NSCLC 228; advanced disease
Durvalumab monotherapy 10mg/kg every two weeks
ORR: PD-L1+/IFNγ+: 46% PD-L1+/IFNγ-: 13% PD-L1-/IFNγ+: 11% PD-L1-/IFNγ-: 3%
Keytruda 22LBA II MCC 18; advanced disease Keytruda monotherapy 2mg/kg every three weeks
ORR: 80% (out of 10 radiographically evaluable MCC patients)
Opdivo 23LBA II (CheckMate 064)
Melanoma 140; unresectable Stage III or IV melanoma, treatment-naïve or had one prior systemic therapy
Arm 1: IV Opdivo 3mg/kg every two weeks for up to six doses followed by IV Yervoy 3mg/kg every three weeks for up to four doses Arm 2: IV Yervoy 3mg/kg every three weeks for up to four doses followed by IV Opdivo 3mg/kg every two weeks for up to six
PRR: Arm 1: 35.3% (week 13) 41.2% (week 25) Arm 2: 10.0% (week 13) 20.0% (week 25) Progression rate: Arm 1: 38.2% (week 13)
White Paper
The European Cancer Congress 2015
Bristol-Myers Squibb has provided the first look at data from the Phase III CheckMate 025 pivotal trial
in RCC (ClinicalTrials.gov identifier: NCT01295827). The company previously announced that the trial
had met its primary endpoint in July 2015, but the data presented at ECC 2015 now reveal the therapy
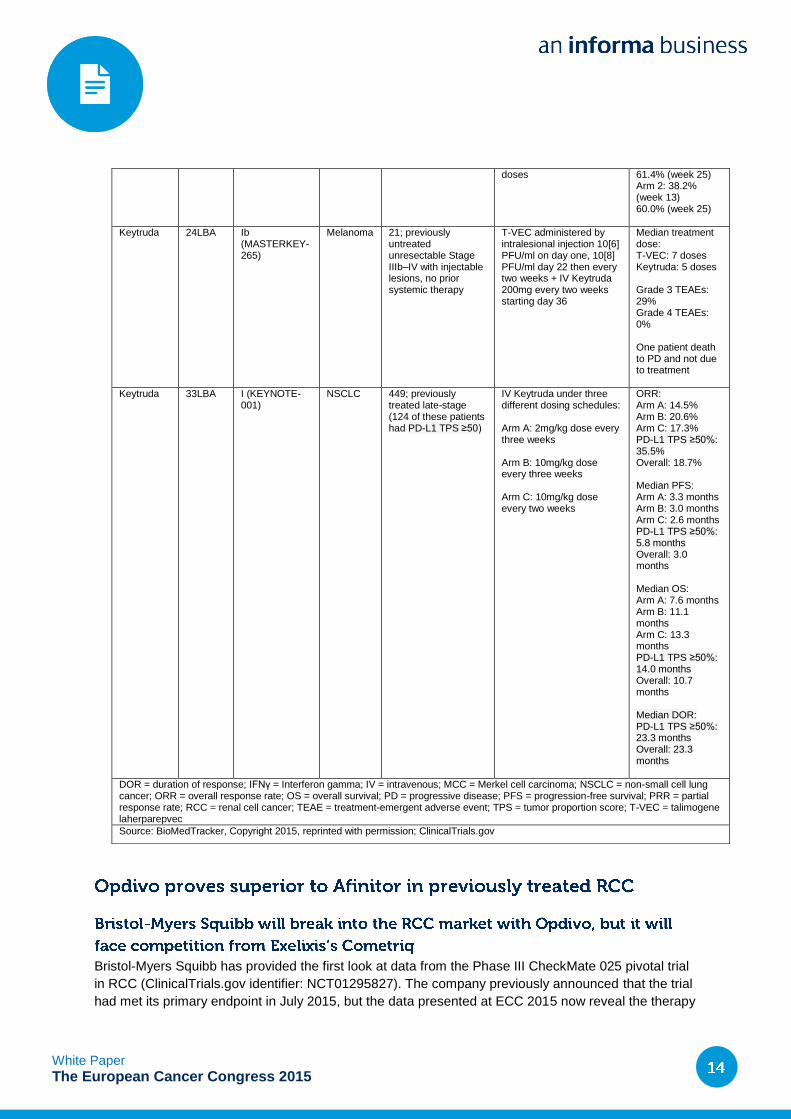
doses 61.4% (week 25) Arm 2: 38.2% (week 13) 60.0% (week 25)
Keytruda 24LBA Ib (MASTERKEY-265)
Melanoma 21; previously untreated unresectable Stage IIIb–IV with injectable lesions, no prior systemic therapy
T-VEC administered by intralesional injection 10[6] PFU/ml on day one, 10[8] PFU/ml day 22 then every two weeks + IV Keytruda 200mg every two weeks starting day 36
Median treatment dose: T-VEC: 7 doses Keytruda: 5 doses Grade 3 TEAEs: 29% Grade 4 TEAEs: 0% One patient death to PD and not due to treatment
Keytruda 33LBA I (KEYNOTE-001)
NSCLC 449; previously treated late-stage (124 of these patients had PD-L1 TPS ≥50)
IV Keytruda under three different dosing schedules: Arm A: 2mg/kg dose every three weeks Arm B: 10mg/kg dose every three weeks Arm C: 10mg/kg dose every two weeks
ORR: Arm A: 14.5% Arm B: 20.6% Arm C: 17.3% PD-L1 TPS ≥50%: 35.5% Overall: 18.7% Median PFS: Arm A: 3.3 months Arm B: 3.0 months Arm C: 2.6 months PD-L1 TPS ≥50%: 5.8 months Overall: 3.0 months Median OS: Arm A: 7.6 months Arm B: 11.1 months Arm C: 13.3 months PD-L1 TPS ≥50%: 14.0 months Overall: 10.7 months Median DOR: PD-L1 TPS ≥50%: 23.3 months Overall: 23.3 months
DOR = duration of response; IFNγ = Interferon gamma; IV = intravenous; MCC = Merkel cell carcinoma; NSCLC = non-small cell lung cancer; ORR = overall response rate; OS = overall survival; PD = progressive disease; PFS = progression-free survival; PRR = partial response rate; RCC = renal cell cancer; TEAE = treatment-emergent adverse event; TPS = tumor proportion score; T-VEC = talimogene laherparepvec
Source: BioMedTracker, Copyright 2015, reprinted with permission; ClinicalTrials.gov

White Paper
The European Cancer Congress 2015
is a significant improvement over current RCC standard of care Afinitor. Opdivo is the first PD-1
inhibitor to demonstrate improved outcomes over Afinitor in previously treated advanced/metastatic
RCC; however, positive Phase III data were also seen at ECC 2015 for Exelixis’s multi-tyrosine kinase
inhibitor Cometriq. Datamonitor Healthcare expects both of these drugs will gain approval for the
treatment of RCC, and that they will be fierce competitors in the future. For more insight into Opdivo
versus Cometriq in RCC, please see below “Late-breaking abstracts presented at ECC 2015 will have
a major impact on the second-line treatment of renal cell cancer.”
The results of the Phase III CheckMate 025 study, which were also published in the New England
Journal of Medicine, demonstrated that Opdivo improved OS in previously treated
advanced/metastatic RCC patients compared to Afinitor, which is the current standard of care for
these patients. Patients in the Opdivo cohort had a median OS of 25.0 months compared with 19.6
months for Afinitor, with clinical benefits observed regardless of PD-L1 expression status. In addition,
the ORR for Opdivo was 25% compared with 5% for Afinitor. Patients treated with Opdivo also had a
higher median PFS (4.6 months Opdivo versus 4.4 months Afinitor), although the difference was not
statistically significant. The safety profile of Opdivo in this trial was consistent with what has been
previously observed: grade 3 and 4 treatment-related adverse events were observed in 19% of
patients in the Opdivo cohort and 37% of patients in the Afinitor cohort (Motzer et al., 2015).
Opdivo and Keytruda are the first PD-1 checkpoint inhibitors to reach the oncology market, with their
approvals for melanoma occurring back in 2014. Since this time, PD-1 has continued to be a key
target for immunotherapy development across skin cancer indications. LBAs at ECC 2015 highlighted
successes in the continued development of Opdivo and Keytruda for melanoma, as well as positive
data that infer the possibility of a future label expansion for Keytruda as a therapy for Merkel cell
carcinoma, a rare and aggressive form of skin cancer.
The CheckMate 064 data presented at ECC 2015 provide insight into additional treatment strategies
for the combination of Opdivo and Yervoy in advanced melanoma (ClinicalTrials.gov identifier:
NCT01783938). Bristol-Myers Squibb is already seeking approval for an Opdivo-Yervoy combination
therapy regimen for the first-line treatment of advanced melanoma (filed with the FDA in September
2015). The CheckMate 064 data demonstrate that the sequential use of Opdivo and Yervoy in
previously treated melanoma might provide patients with clinical benefit, suggesting that the company
can successfully introduce even more flexibility into the use of its two immune checkpoint drugs in the
future.
In the CheckMate 064 study, patients were either given Opdivo followed by Yervoy or Yervoy followed
by Opdivo during the induction therapy period. Following induction treatment, both cohorts received
Opdivo until disease progression or unacceptable toxicity. At the end of the 25-week induction therapy
period, 41.2% of patients who received Opdivo first in the sequence exhibited a partial response
compared to 20.0% of patients who received Yervoy first. In addition, 38.2% of patients who received
Opdivo first demonstrated disease progression versus 60.0% of patients who received Yervoy first.
During the induction periods, the incidence of high-grade adverse events was higher in patients who
received Opdivo initially, with 50% of patients exhibiting grade 3–5 adverse events, compared with
42.9% patients who were given Yervoy initially. No drug-related deaths were reported in either cohort.

White Paper
The European Cancer Congress 2015
Data from the Phase Ib/II MASTERKEY-265 trial revealed that the combination of Keytruda and
Amgen’s investigational oncolytic virus T-VEC (talimogene laherparepvec) was well tolerated in
treatment-naïve advanced melanoma patients, although no efficacy data have yet been reported
(ClinicalTrials.gov identifier: NCT02263508). The MASTERKEY-265 data mark the first step in what is
a novel combination in PD-1 development so far, and could be a key point of differentiation for Merck
from competitor combination regimens in the future. T-VEC is the first oncolytic virus to be tested in
combination with a PD-1 for melanoma, and is also the first oncolytic virus to demonstrate efficacy in a
Phase III clinical trial for melanoma. Amgen previously stated in May 2015 that it would advance the T-
VEC and Keytruda combination into a Phase III pivotal trial.
On 27 September, Dr Georgina Long presented safety data from the Phase Ib portion of the
MASTERKEY-265 trial investigating the combination of Keytruda with T-VEC. Patients in this study
were initially given intralesional injections of T-VEC, with Keytruda administration beginning at day 36
of treatment and administered biweekly thereafter. T-VEC and Keytruda combination therapy was
shown to have favorable and non-overlapping adverse event profiles. Of the 21 patients who were
enrolled in the study, 29% exhibited grade 3 treatment-emergent adverse events, and no patients
exhibited grade 4 treatment-emergent adverse events. Although one patient died on study, the cause
of death was deemed to be related to progressive disease and not to treatment. No patients
discontinued therapy due to adverse events. These data demonstrate the safety of T-VEC in
combination with Keytruda at full dose.
Further developments of Keytruda for skin malignancies at ECC 2015 also included interim data from
a Phase II study investigating the therapeutic potential of Keytruda monotherapy in advanced
unresectable MCC (ClinicalTrials.gov identifier: NCT02267603). Although these are very early data
from only a small patient population, they suggest promising efficacy and will raise hopes that
Keytruda can meet some of the high unmet need in this very rare and aggressive form of skin cancer.
Of the 18 patients who received at least one dose of Keytruda, 10 had undergone at least one
radiologic and clinical response assessment. Of these 10, eight showed evidence of response to PD-1
pathway blockade (ORR = 80%). Merck speculates that the high response rate observed thus far may
in part be due to the immune response to antigens from the polyomavirus that often drives MCC.
Since the mechanism of action here is not well understood, future investigations have been planned
and follow-up results are to be presented at a later date.
MCC is a rare and highly aggressive form of skin cancer linked to ultraviolet exposure and Merkel cell
polyomavirus. The difficulty in diagnosing MCC often leads to confirmed diagnoses following
metastasis. Responses to chemotherapy in metastatic MCC are typically not durable, and up to 50%
of patients that are initially determined to be disease-free suffer disease relapse.
Late-breaking abstracts at ECC 2015 also highlighted promising PD-1 immunotherapy developments
in the NSCLC treatment space with Keytruda and durvalumab, as Merck and AstraZeneca look to
challenge Opdivo’s current position as the only PD-1/PD-L1 inhibitor approved for NSCLC.

White Paper
The European Cancer Congress 2015
Dr Jean-Charles Soria presented an updated dataset from KEYNOTE-001, the study which formed the
basis of the sBLA that Merck filed for Keytruda in NSCLC in April 2015. Data from this study were last
seen in the New England Journal of Medicine in April 2015 (Garon et al., 2015). The update at ECC
2015 relates to a larger population of previously treated patients enrolled in the study and provided
more information about the drug’s efficacy at different dose levels. Datamonitor Healthcare believes
Keytruda will be approved for the treatment of patients with EGFR mutation-negative and ALK
rearrangement-negative NSCLC whose disease has progressed on or following platinum-containing
chemotherapy. These updated data should solidify Keytruda’s dosing regimen and help to ensure that
the drug can compete effectively with Opdivo, which is currently being reviewed by the FDA for
licensing in non-squamous cell NSCLC (it is already marketed for the smaller squamous-cell NSCLC
population). The FDA has granted priority review for Keytruda with a target decision date of 2 October
2015.
In KEYNOTE-001, patients were placed into one of three dosing schedules (see table above) until
confirmed progression, intolerable toxicity, or investigator decision. The results update at ECC 2015
conveyed that there is a lack of significant exposure-response relationship, and showed similar
efficacy across doses and schedules, supporting the use of 2mg/kg every three weeks for NSCLC.
However, Keytruda demonstrated particularly robust antitumor activity in patients with PD-L1 tumor
proportion score ≥50%, with ORR, median PFS, and median OS higher in this subgroup in comparison
with the total study group. Grade 3 and 4 treatment-related adverse events occurred in 10.5% of
patients, and there were three treatment-related deaths due to cardiorespiratory arrest, interstitial lung
disease, and respiratory arrest.
Phase I/II data from a trial of AstraZeneca’s anti-PD-L1 durvalumab demonstrated promising efficacy
in a very specific population of NSCLC patients (ClinicalTrials.gov identifier: NCT01693562).
Durvalumab development in NSCLC is a long way behind key rivals Opdivo and Keytruda, and
AstraZeneca has sought to differentiate the drug by looking for biomarkers other than PD-L1 for
patient selection. The data presented at ECC 2015 show that interferon-gamma (IFN-gamma) may be
a promising biomarker candidate to be used in combination with the PD-L1 biomarker for patient
selection for durvalumab in NSCLC.
On 27 September, data from a Phase I/II clinical trial of durvalumab monotherapy were presented,
looking at whether treatment response in NSCLC is correlated with high tumoral IFN-gamma mRNA,
PD-L1 protein, and combined IFN-gamma/PD-L1 protein expression. IFN-gamma-negative/PD-L1-
negative patients demonstrated the lowest ORR to durvalumab monotherapy at 3%, while IFN-
gamma-positive/PD-L1-positive patients demonstrated the highest ORR at 46%. While this
demonstrates a significant increase in response over IFN-gamma-negative/PD-L1-negative patients,
the IFN-gamma-positive/PD-L1-positive patient subgroup had the second smallest sample size of the
four subgroups, while IFN-gamma-negative/PD-L1-negative patients were the most common. Patients
expressing either PD-L1 or IFN-gamma, but not both, had similar ORRs, of 13% and 11%.
AstraZeneca is looking to further investigate the effect of IFN-gamma and PD-L1 expression on
durvalumab efficacy in NSCLC, but the data presented suggest that NSCLC patients whose tumors
have elevated IFN-gamma mRNA expression, PD-L1 protein expression, or a combination of both
may be more likely to benefit from durvalumab therapy.

White Paper
The European Cancer Congress 2015
The results of pivotal Phase III trials for Exelixis’s Cometriq and Bristol-Myers Squibb’s Opdivo look set
to change the treatment algorithm for previously treated RCC patients. Late-breaking abstracts for
Cometriq’s METEOR trial and Opdivo’s CheckMate 025 trial were presented on 26 September at ECC
2015. Both trials had a positive outcome, and both Opdivo and Cometriq are likely to gain approvals
for the second-line treatment of RCC. Opdivo significantly improved OS in comparison to Afinitor and
is now likely to become the treatment of choice in this setting. Meanwhile, Cometriq’s results were not
quite as good as Opdivo’s, but they were still positive and Exelixis will be pleased at the opportunity to
expand Cometriq’s label.
Cometriq significantly improved PFS in patients with advanced or metastatic RCC in the pivotal
METEOR trial (ClinicalTrials.gov identifier: NCT01865747). This was an open-label Phase III trial
comparing Cometriq to Afinitor in patients with advanced or metastatic RCC with a clear-cell
component that had progressed after treatment with vascular endothelial growth factor receptor
(VEGFR)-targeting tyrosine kinase inhibitors (TKIs). Patients were stratified based on previous
treatments with VEGFR-targeting TKIs and prognostic risk category. The trial met the primary endpoint
of improved PFS with a median of 7.4 months for patients receiving Cometriq compared to 3.8 months
for patients receiving Afinitor, corresponding to a 42% reduction in rate of disease progression or
death. A significant increase in OS was not seen at this interim analysis, but there was a positive trend
of 33% lower death rates for Cometriq (HR: 0.67, p=0.005). The full analysis of OS is expected in
2016. Adverse events observed during the trial were similar for both arms, but 60% of patients
receiving Cometriq had a dose reduction following adverse events compared to 25% of patients
receiving Afinitor. The most common grade 3 or 4 adverse events for patients receiving Cometriq were
hypertension (15%), diarrhea (11%), and fatigue (9%).
Cometriq is a TKI with multiple targets including VEGFR, AXL receptor tyrosine kinase, and
hepatocyte growth factor receptors (c-Met). It is currently approved for the treatment of progressive,
unresectable locally advanced or metastatic medullary thyroid cancer in the US and EU.
Bristol-Myers Squibb presented impressive results from the Phase III CheckMate 025 trial
(ClinicalTrials.gov identifier: NCT01668784). Treatment with the PD-1 inhibitor Opdivo significantly
increased OS and the drug had a favorable safety profile compared to Afinitor. The Phase III trial
enrolled patients with advanced or metastatic RCC with a clear cell component, previously treated with
one or two antiangiogenic therapies. Exclusion criteria included more than three prior treatments with
systemic therapy, brain metastases, and prior treatment with an mTOR inhibitor. Following advice from
an objective independent data monitoring committee, the study was terminated early (in June 2015)
because the primary endpoint of OS had been met. The median OS for patients treated with Opdivo
was 25.0 months compared to 19.6 months for patients treated with Afinitor. Grade 3 or 4 treatment-
related adverse events occurred in 19% of the patients treated with Opdivo compared to 37% treated
with Afinitor, and the most common grade 3 or 4 adverse event associated with treatment with Opdivo
was fatigue (2%). In this study no correlation between PD-1 expression and response to treatment
with Opdivo was seen.
Opdivo is a fully human immunoglobulin G4 PD-1 immune checkpoint inhibitor that binds to the
checkpoint receptor PD-1 expressed on activated T-cells. It is approved for the treatment of
White Paper
The European Cancer Congress 2015
unresectable or metastatic melanoma in the US and EU, and for the treatment of metastatic squamous
cellNSCLC in the US.
A summary of Cometriq’s METEOR trial and Opdivo’s CheckMate 025 trial can be found in the table
below.
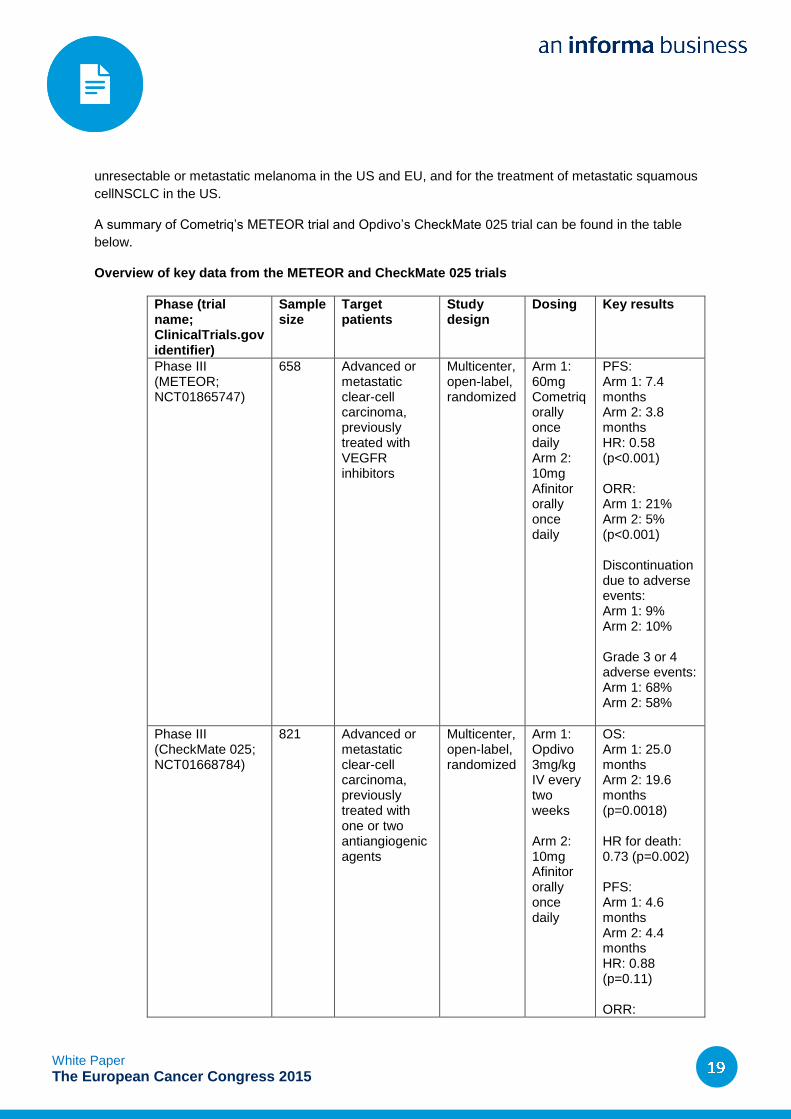
Overview of key data from the METEOR and CheckMate 025 trials
Phase (trial name; ClinicalTrials.gov identifier)
Sample size
Target patients
Study design
Dosing Key results
Phase III (METEOR; NCT01865747)
658 Advanced or metastatic clear-cell carcinoma, previously treated with VEGFR inhibitors
Multicenter, open-label, randomized
Arm 1: 60mg Cometriq orally once daily Arm 2: 10mg Afinitor orally once daily
PFS: Arm 1: 7.4 months Arm 2: 3.8 months HR: 0.58 (p<0.001) ORR: Arm 1: 21% Arm 2: 5% (p<0.001) Discontinuation due to adverse events: Arm 1: 9% Arm 2: 10% Grade 3 or 4 adverse events: Arm 1: 68% Arm 2: 58%
Phase III (CheckMate 025; NCT01668784)
821 Advanced or metastatic clear-cell carcinoma, previously treated with one or two antiangiogenic agents
Multicenter, open-label, randomized
Arm 1: Opdivo 3mg/kg IV every two weeks Arm 2: 10mg Afinitor orally once daily
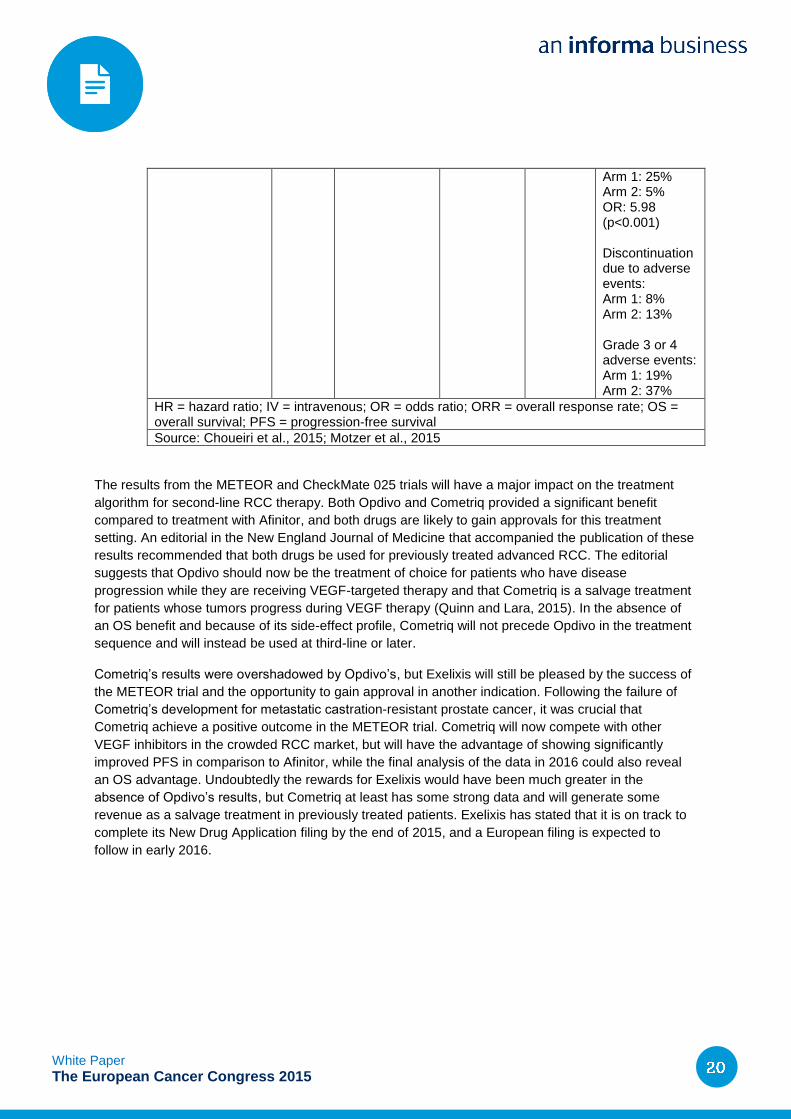
OS: Arm 1: 25.0 months Arm 2: 19.6 months (p=0.0018) HR for death: 0.73 (p=0.002) PFS: Arm 1: 4.6 months Arm 2: 4.4 months HR: 0.88 (p=0.11) ORR:
White Paper
The European Cancer Congress 2015
Arm 1: 25% Arm 2: 5% OR: 5.98 (p<0.001) Discontinuation due to adverse events: Arm 1: 8% Arm 2: 13% Grade 3 or 4 adverse events: Arm 1: 19% Arm 2: 37%
HR = hazard ratio; IV = intravenous; OR = odds ratio; ORR = overall response rate; OS = overall survival; PFS = progression-free survival
Source: Choueiri et al., 2015; Motzer et al., 2015
The results from the METEOR and CheckMate 025 trials will have a major impact on the treatment
algorithm for second-line RCC therapy. Both Opdivo and Cometriq provided a significant benefit
compared to treatment with Afinitor, and both drugs are likely to gain approvals for this treatment
setting. An editorial in the New England Journal of Medicine that accompanied the publication of these
results recommended that both drugs be used for previously treated advanced RCC. The editorial
suggests that Opdivo should now be the treatment of choice for patients who have disease
progression while they are receiving VEGF-targeted therapy and that Cometriq is a salvage treatment
for patients whose tumors progress during VEGF therapy (Quinn and Lara, 2015). In the absence of
an OS benefit and because of its side-effect profile, Cometriq will not precede Opdivo in the treatment
sequence and will instead be used at third-line or later.
Cometriq’s results were overshadowed by Opdivo’s, but Exelixis will still be pleased by the success of
the METEOR trial and the opportunity to gain approval in another indication. Following the failure of
Cometriq’s development for metastatic castration-resistant prostate cancer, it was crucial that
Cometriq achieve a positive outcome in the METEOR trial. Cometriq will now compete with other
VEGF inhibitors in the crowded RCC market, but will have the advantage of showing significantly
improved PFS in comparison to Afinitor, while the final analysis of the data in 2016 could also reveal
an OS advantage. Undoubtedly the rewards for Exelixis would have been much greater in the
absence of Opdivo’s results, but Cometriq at least has some strong data and will generate some
revenue as a salvage treatment in previously treated patients. Exelixis has stated that it is on track to
complete its New Drug Application filing by the end of 2015, and a European filing is expected to
follow in early 2016.

White Paper
The European Cancer Congress 2015
At ECC 2015, Immatics revealed that the Phase III IMPRINT study of its RCC vaccine IMA901 did not
meet its primary endpoint. Immatics had hoped that the combination of IMA901 with Sutent would
produce a synergistic immunomodulatory effect that would extend OS compared to Sutent
monotherapy, and that this would enable IMA901 to become the first approved vaccine for the
treatment of RCC. However, the negative outcome of IMPRINT has driven Immatics to make a difficult
decision to shift its focus onto the ACT portion of its pipeline, effectively ending the development of
IMA901. The company is likely to now quickly advance its ACT candidates from preclinical to clinical
development in a bid to not get left behind by other companies that are already highly active in this
space.
On 27 September at ECC 2015, Dr Brian Rini, professor of medicine at the Cleveland Clinic Taussig
Cancer Center, presented the results from the pivotal Phase III IMPRINT trial (ClinicalTrials.gov
identifier: NCT01265901) of IMA901 in advanced or metastatic RCC. The study failed to meet its
primary endpoint of OS: median OS was 33.1 months in the IMA901 vaccination plus Sutent group,
but median OS in the control group (Sutent alone) had not yet been reached (HR: 1.34; p=0.08). In the
favorable-risk patient subpopulation, OS was comparable between the IMA901 and control arms (33.7
months vs not reached, HR: 0.82; p=0.59). Intermediate-risk patients displayed a longer OS benefit in
the control group (not yet reached) versus the IMA901 vaccination group (27.8 months; HR: 1.52;
p<0.05). A blinded independent central review found that PFS was similar between treatment and
control groups (15.1 vs 15.1 months; HR: 1.05; p=0.62), but an investigator-initiated assessment found
PFS to be longer in the control group compared to the IMA901 vaccination group (17.9 vs 15.1
months; HR: 1.18; p=0.19). This led to a higher median exposure of Sutent in the control group (13.7g
control vs 11.2g IMA901). IMA901-specific CD8 T-cell responses from treatment with the IMA901 and
Sutent combination were reduced by a factor of three compared to the response observed in IMA901’s
Phase II monotherapy trial. IMA901 displayed a favorable safety profile, with mild injection site
adverse events accounting for the most common treatment-related side effects.
In the open-label, randomized, controlled Phase III IMPRINT study, 339 human leukocyte antigen A2-
positive metastatic RCC patients were randomized 3:2 to receive either 10 intradermal vaccinations of
IMA901 plus 75µg of granulocyte-macrophage colony-stimulating factor and 50mg of Sutent, or Sutent
alone. A single injection of cyclophosphamide at 300mg/m2 was administered three days prior to the
first vaccination of IMA901 to reduce regulatory T-cell levels. The patients were stratified by risk group
(Heng risk criteria), nephrectomy status, and region (US, Western EU, and Central Eastern EU).
IMA901 is a fully synthetic cancer vaccine consisting of 10 tumor-associated peptides (TUMAPS) that
are found to be overexpressed on the tumor cells of RCC patients. TUMAPS work by activating
cytotoxic T-cells (class I TUMAPS) and helper T-cells (class II TUMAPS) to target tumor cells. The
helper T-cells assist cytotoxic T-cells by secreting cytokines, and the cytotoxic T-cells directly kill tumor
cells. Thus, IMA901 acts to prime a patient’s own T-cells so that they can recognize TUMAPS
presented on tumor cells, which may otherwise have evaded the immune system.
Early-phase data presented at the 2010 ESMO Cancer Congress suggested IMA901 had promising
efficacy in previously treated RCC patients. The Phase II Study 202 trial (ClinicalTrials.gov identifier:
NCT00523159) tested IMA901 in second-line advanced/metastatic RCC patients who relapsed after
previous treatment with cytokines or kinase inhibitors. Median OS was 19.8 months in all second-line

White Paper
The European Cancer Congress 2015
patients (previously treated with cytokines) who received IMA901. The disease control rates at six
months were 31% for post-cytokine patients and 14% in post-TKI patients.
In a press release following the presentation of the IMPRINT data, Dr Carsten Reinhardt, chief medical
officer of Immatics, expressed his disappointment at the failure of the study to meet its primary
endpoint (Immatics, 2015). He stated that the company will continue to evaluate the data, but that it
will now shift its focus towards developing its ACT technology, in partnership with the MD Anderson
Cancer Center. This signifies that development of IMA901 is likely finished, and investors and market
commentators will now be left to worry about the fate of the company’s other pipeline cancer vaccines.
Immatics is developing two other cancer vaccines for colorectal cancer and glioma, which are in
Phase II and Phase I clinical trials, respectively. It is also unclear what effect this will have on
Immatics’ partnership with Roche for TUMAP vaccines in NSCLC, gastric cancer, and prostate cancer.
These vaccines are still presently in preclinical stages.
By choosing to focus on its ACT program, Immatics is joining a host of other biotechnology companies
that are pioneering various cell-based techniques in the immuno-oncology space. The most
developmentally advanced ACT therapies are the personalized chimeric antigen receptor T-cell (CAR-
T) therapies. Juno’s JCAR015 and Novartis’s CTL019 have both achieved promising results in early-
phase clinical trials in acute lymphoblastic leukemia. Both drugs have also received breakthrough
therapy designation for this indication from the FDA. Another CAR-T therapy, Kite’s KTE-C19, has
demonstrated preliminary efficacy in diffuse large B-cell lymphoma (a subtype of non-Hodgkin’s
lymphoma) and chronic lymphocytic leukemia. Pfizer and Cellectis are also partnering on the
development of next-generation allogeneic ACT therapies, representing an “off-the-shelf” approach to
cellular immunotherapy. Immatics’ ACT program will focus on developing both autologous and
allogeneic ACT therapies.

White Paper
The European Cancer Congress 2015
Besse B, et al. (2015) Phase II, single-arm trial (BIRCH) of atezolizumab as first-line or subsequent
therapy for locally advanced or metastatic PD-L1-selected non-small cell lung cancer (NSCLC). ECC
2015; Vienna, Austria; 27 September 2015; 16LBA.
Choueiri TK et al. (2015) Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. The
New England Journal of Medicine, 25 September 2015 <DOI>10.1056/NEJMoa1510016</DOI>.
Garon EB, et al. (2015) Pembrolizumab for the treatment of non-small-cell lung cancer. The New
England Journal of Medicine, 372(21), 2018–28 <DOI>10.1056/NEJMoa1501824</DOI>.
Immatics (2015) Immatics announces results of IMPRINT phase 3 clinical trial investigating the
addition of IMA901 to standard first-line therapy with sunitinib for advanced/metastatic RCC. Available
from: http://immatics.com/immatics-announces-results-of-imprint-phase-3-clinical-trial-investigating-
the-addition-of-ima901-to-standard-first-line-therapy-with-sunitinib-for-advancedmetastatic-rcc/
[Accessed September 27 2015].
Motzer RJ et al. (2015) Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. The New
England Journal of Medicine, 25 September 2015 <DOI>10.1056/NEJMoa1510665</DOI>.
Quinn DI, Lara PN (2015) Renal-Cell Cancer — Targeting an Immune Checkpoint or Multiple Kinases.
The New England Journal of Medicine, 25 September 2015 <DOI>10.1056/NEJMe1511252</DOI>.
Rosenberg J, et al. (2015) Atezolizumab in patients (pts) with locally-advanced or metastatic urothelial
carcinoma (mUC): Results from a pivotal multicenter phase II study (IMvigor 210). ECC 2015; Vienna,
Austria; 27 September 2015; 21LBA.
Vansteenkiste J, et al. (2015) Atezolizumab monotherapy vs docetaxel in 2L/3L non-small cell lung
cancer: Primary analyses for efficacy, safety and predictive biomarkers from a randomized phase II
study (POPLAR). ECC 2015; Vienna, Austria; 27 September 2015; 14LBA.
White Paper
The European Cancer Congress 2015
Bringing you a clearer, richer and more responsive view of the pharma & healthcare market.
Complete market coverage
Our independent research and analysis provides extensive coverage of major disease areas,
companies and strategic issues, giving you the perspective to identify opportunities and
threats arising from shifting market dynamics and the insights to respond with faster, more
effective decision-making.
Unique expert capabilities
With teams located across developed and emerging pharma markets, we are uniquely
placed to understand local healthcare trends and provide accurate and reliable
recommendations. By working closely with our partners at MedTrack, Citeline, SCRIP
Intelligence and Informa Healthcare, our experts are able to share data and resources to
produce the most authoritative and robust market intelligence.
Cutting-edge delivery
Available through single reports or via subscription to our state-of-the art online intelligence
service that features intuitive design and interactive capabilities, our analysis offers the
definitive platform to enhance your product management, market assessment and strategic
planning.
To find out more about Datamonitor Healthcare, please contact us at:
email [email protected]
phone +44 20 7551 9430
website: www.datamonitorhealthcare.com
Twitter: @DatamonitorHC





























