Embed Size (px)
Citation preview

Over the past decade, it has become apparent that many pathogenic bacteria engage in complex crosstalk with their mammalian hosts by delivering an array of virulence ‘effector’ proteins directly into target cells1. Although often structurally divergent, these sophisti-cated effectors mimic eukaryotic functions2, and can therefore provide valuable insights into the physiological control of fundamental processes in eukaryotic cells.
Two important pathogens that encode such effectors are enteropathogenic and enterohaemorrhagic Escherichia coli (EPEC and EHEC, respectively). EPEC is a major cause of infant diarrhoea in the under developed world, and the related EHEC, particularly serotype O157:H7, is an important cause of bloody diarrhoea and renal fail-ure in North America, the United Kingdom and Japan3. Examination of intestinal samples from infected humans or experimental animals indicates that after ingestion, EPEC and EHEC intimately adhere to intestinal epi-thelial cells. Adhesion triggers localized loss of microvilli and dramatic reorganization of the host-cell actin cyto-skeleton into ‘pedestal-like’ pseudopods beneath the bound bacteria (FIG. 1). These characteristic structures, termed attaching and effacing (A/E) lesions, promote successful colonization4–6, although precisely how they do so remains unresolved7. Actin-pedestal formation might induce diarrhoea by causing tissue damage and fluid
loss, or might short-circuit phagocytic signalling by host immune cells. Also, the pedestals move along the cell surface, suggesting that pathogen ‘surfing’ might addi-tionally promote intestinal dissemination8,9. Regardless, uncovering the molecular mechanisms underlying pedestal assembly will be important to expedite the development of therapeutic strategies to combat diar-rhoeal disease caused by EPEC and EHEC. In addition, given that actin assembly is essential for diverse cellular events, EPEC and EHEC, which efficiently form pedes-tals on cultured mammalian cells10, are highly tractable experimental models that can be used to decipher fundamental molecular and cellular processes, includ-ing transmembrane -receptor-signalling pathways that regulate actin polymerization at the plasma membrane.
Cellular regulation of actin polymerizationControl of cytoskeletal dynamics is crucial for many cellular processes, including cytokinesis, intracellular trafficking and cell motility11. Cellular morphology is dic-tated by dynamic manipulation of the actin cyto skeleton in response to extracellular stimuli. Actin polymerization is essential for the uptake of extracellular material into membrane-bound vacuoles by receptor-mediated endo-cytosis and macropinocytosis. It is also necessary for the formation of projections such as ‘finger-like’ filopodia and
*University of Cambridge, Department of Pathology, Tennis Court Road, Cambridge CB2 1QP, UK.‡Department of Molecular Genetics and Microbiology, University of Massachusetts Medical School, 55 Lake Avenue North, Worcester, Massachusetts 01655, USA.§Current address: University of California, Department of Molecular and Cell Biology, Berkeley, California 94720-3200, USA.¶These authors contributed equally to this work.Correspondence to J.M.L. and V.K. e-mails: [email protected]; [email protected]:10.1038/nrmicro1391
Exploiting pathogenic Escherichia coli to model transmembrane receptor signallingRichard D. Hayward*¶, John M. Leong‡, Vassilis Koronakis*
and Kenneth G. Campellone‡ §¶
Abstract | Many microbial pathogens manipulate the actin cytoskeleton of eukaryotic target cells to promote their internalization, intracellular motility and dissemination. Enteropathogenic and enterohaemorrhagic Escherichia coli, which both cause severe diarrhoeal disease, can adhere to mammalian intestinal cells and induce reorganization of the actin cytoskeleton into ‘pedestal-like’ pseudopods beneath the extracellular bacteria. As pedestal assembly is triggered by E. coli virulence factors that mimic several host cell-signalling components, such as transmembrane receptors, their cognate ligands and cytoplasmic adaptor proteins, it can serve as a powerful model system to study eukaryotic transmembrane signalling. Here, we consider the impact of recent data on our understanding of both E. coli pathogenesis and cell biology, and the rich prospects for exploiting these bacterial factors as versatile tools to probe cellular signalling pathways.
MicrovilliSmall, finger-like projections found on the exposed surfaces of epithelial cells that maximize the surface area.
R E V I E W S
358 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group

TirEHEC: non-tyrosine-phosphorylated translocated intimin receptorEHEC EspFU: translocated adaptor proteinTirEPEC: tyrosine-phosphorylated translocated intimin receptorArg, Abl: non-receptor tyrosine kinasesNck1,2: SH2/SH3 adaptor proteins, activators of N-WASPCrkII: SH2/SH3 adaptor proteinAnnexin 2, CD44: lipid-raft-associated proteinsN-WASP: actin nucleation-promoting factorArp2/3 (active): actin nucleator, brancherCortactin: actin nucleation-promoting factor, branching regulatorVasp: regulator of filament elongation
Arp2/3 (inactive): actin brancherWIP: signalling intermediate, actin cross-linkerα-actinin, talin, vinculin: focal-adhesion proteins, actin cross-linkersGelsolin: filament capper, severerADF/cofilin: actin depolymerizerCK18: intermediate filament protein
Grb2: SH2/SH3 adaptor protein, activator of N-WASP
MacropinocytosisA form of regulated, actin-dependent endocytosis that involves the formation of large endocytic vesicles after the closure of cell-surface membrane ruffles.
FilopodiaThin, transient actin protrusions that extend out from the cell surface and are formed by the elongation of bundled actin filaments in the core.
LamellipodiaFlattened protrusions at the leading edge of a moving cell that are enriched with a branched network of actin filaments.
Tight junctionA seal between adjacent epithelial cells, just beneath the apical surface, that forms a semi-permeable diffusion barrier between individual cells.
Focal adhesionsCellular structures that link the extracellular matrix on the outside of the cell to the actin cytoskeleton inside the cell through integrin receptors.
‘fin-like’ lamellipodia at the leading edge of the cell, which can drive the plasma membrane, and consequently the cell body, forward12. Transmembrane receptors are also linked to actin filaments (F-actin) by associated adaptor proteins, and form static cell–cell connections at tight junctions and cell–substratum contacts at focal adhesions. The orchestrated cellular behaviour underlying complex biological phenomena such as immune surveillance and embryonic development requires control of the equilib-rium between actin-dependent adhesion and motility. Improper regulation of these signalling pathways is associated with pathological processes, including immu-nodeficiencies and cancer-cell metastasis12–14.
A central cellular participant in the generation of dynamic actin-filament superstructures is the ubiquitous Arp2/3 complex that accelerates the polymerization of G-actin monomers into filaments15,16. This complex both crosslinks actin filaments and drives their assembly, com-bined activities that allow newly formed filaments to be organized into diverse higher-order dendritic arrays12. As one would expect, the activity of the Arp2/3 complex is highly regulated. For example, the inefficient actin-nucleation activity of the isolated Arp2/3 complex is dra-matically stimulated by the Wiskott–Aldrich syndrome protein (WASP) family of nucleation-promoting factors (NPFs)15,17 (FIG. 2a). The founding family member, WASP, is specifically expressed in haematopoietic cells, and its mutation results in defects in actin-associated processes, including macrophage migration, phagocytosis and T-cell receptor (TCR) signalling18. Its closest homologue, neural
(N)-WASP, is expressed ubiquitously, and three related Wiskott-Aldrich verprolin homologous (WAVE) proteins are either broadly distributed or enriched in neurons19.
The nucleation-promoting function of WASP-family members resides in their conserved verprolin-connector-acidic (VCA) domain, which binds and activates the Arp2/3 complex by inducing a conformational change that facilitates incorporation of G-actin into fila-ments15,17,20. Although each WASP-family member uses this mechanism to stimulate Arp2/3, regions distinct from the VCA domain regulate their localization and NPF activities. For example, N-WASP activity can be suppressed by an intramolecular interaction between the connector and acidic (CA) segment and a central autoinhibitory region (AIR)21–24 (FIG. 2a). This inactive conformation is further stabilized by interactions with the N-terminal WASP-homology 1 (WH1) domain22 or by accessory proteins that bind to this region such as WASP-interacting protein (WIP)25,26. Aside from this inhibitory role, WIP can also regulate N-WASP localization by bind-ing adaptor proteins containing Src-homology 3 (SH3) domains such as Nck-1 (REFS 25,27) (FIG. 2a).
Stimulatory protein or lipid regulators bind other N-WASP domains and probably disrupt the auto-inhibited conformation. C-terminal to the WH1 domain is the GTPase-binding domain (GBD), which comprises the AIR, a Cdc42/Rac-interactive binding (CRIB) motif and a stretch of basic residues (B). Binding of Cdc42 to the CRIB motif unmasks the VCA domain to activate N-WASP, an effect that is synergistically enhanced by phosphatidylinositol-4,5-bisphosphate (PIP2) binding to the B-region21,22,24 (FIG. 2a). Between the GBD and VCA regions lies a proline-rich domain (PRD) that engages actin-binding proteins such as profilin28, as well as SH3-containing proteins such as the Src-family kinase (SFK) c-Fyn and the adaptor Nck-1 (REFS 29–33). Nck-1 stimulates N-WASP upon PRD binding, whereas c-Fyn phosphorylates tyrosine 256 (Y256) within the GBD to create an additional binding site for Src-homology 2 (SH2)-domain-containing adaptor proteins (FIG. 2a). The VCA domain is targeted by casein kinase-2 (CK2), which enhances Arp2/3-binding by phosphorylating serines34. Multiple signalling inputs must therefore be integrated for N-WASP to promote maximal Arp2/3 activation, especially in the presence of WIP suppressors22,29–32,35.
Pathogenic E. coli generate actin pedestalsBoth EPEC and EHEC commandeer the Arp2/3 complex and N-WASP to generate actin pedestals36,37. Not only are both of these host components localized to the tips of the pedestals where actin polymerization occurs (FIG. 1), but dominant-negative N-WASP derivatives inhibit actin assembly 37, and N-WASP-knockout cells are resistant to pedestal formation by both EPEC and EHEC36,38. However, pedestal formation by EPEC and EHEC does not require activation of Cdc42 or other Rho-family GTPases39,40, endogenous molecular switches that trigger Arp2/3-dependent actin rearrangements in response to external stimuli. As these pathogens induce intracellular actin polymerization from the extracellular surface of the plasma membrane, they must stimulate Rho-independent
Figure 1 | Protein composition of adhesion pedestals formed by enteropathogenic Escherichia coli (EPEC) and enterohaemorrhagic E. coli (EHEC). Immunofluorescence (top) and scanning electron micrograph (bottom) showing adhesion pedestals formed by EPEC on a eukaryotic cell. The triple immunofluorescence staining shows an extracellular bacterium (blue), the EPEC translocated intimin receptor (TirEPEC, green) and F-actin (red). The pedestal tip is enriched with bacterial and host components that initiate actin assembly, whereas the pedestal body contains cellular proteins that regulate the F-actin superstructure. EHEC- and EPEC-specific factors are shown to the right in red and brown shaded boxes, respectively. Proteins common to both are unshaded.
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 359
© 2006 Nature Publishing Group

WH1
WH2 NBD WBD
B CRIB AIR WH1 B CRIB AIR PRD
A C WH2 WH2
CWH2 WH2
WIP
WIP Nck Grb2
Autoinhibited Activated
PRD
A
WIPPIP2
PO4 PO4
Cdc42 c-Fyn CK2
NckGrb2
Arp2/3G-actin
Profilin
a N-WASP
b WASP-interacting adaptor proteins
Nck
SH3 SH3 SH3 SH2
pY
N-WASP, WIPN-WASP
SH3SH3 SH2
pY
N-WASP N-WASP
GBD
VCA
Pathogenicity islandA contiguous block of genes acquired by horizontal transfer in which at least a subset of the genes code for virulence factors.
Arp2/3 signalling through a host transmembrane receptor(s) and/or translocate bacterial effectors into the cell (FIG. 1). Genetic analysis revealed that EPEC and EHEC require a pathogenicity island termed the locus of entero-cyte effacement (LEE) to induce pedestal formation41–43. This encodes a specialized type III secretion system (TTSS) to translocate effector proteins directly from the bacterial cytoplasm into intestinal enterocytes44–46. Most delivered effectors are not required for pedestal assembly, but nonetheless disrupt cellular function and contribute to pathology in vivo47,48 (BOX 1).
One LEE-encoded translocated effector that is essen-tial for pedestal formation is the translocated intimin receptor (Tir). Following delivery into the host cell and subsequent plasma-membrane integration, Tir adopts a hairpin-like topology with its C and N termini in the host cytosol and its central domain exposed on the cell surface. This central domain binds the bacterial outer-membrane protein intimin, which is also encoded on the LEE. So,
EPEC and EHEC encode both the ligand and the receptor involved in pedestal formation (FIG. 3), thereby providing uniquely tractable experimental systems to investigate actin assembly in response to extracellular stimuli49–51.
Recombinant Tir derivatives bind many cellular factors that are often localized within focal adhesions, including 14-3-3τ, cortactin, α-actinin, talin and vinculin, which are also found within pedestals52–55 (FIG. 1). Other pedes-tal components such as VASP, gelsolin and ADF/cofilin, which control actin turnover, are probably recruited indi-rectly56. However, which host proteins are necessary for actin assembly and which are merely passive bystanders, autonomously recruited as a by-product of actin poly-merization, still requires further clarification. Nevertheless, bacterial intimin and Tir are clearly necessary to facilitate the assembly of a contiguous scaffold, intimately link-ing the outer membrane of the extracellular pathogen to the internal actin cytoskeleton of the target cell.
Lessons in adaptor docking from TirEPEC
EPEC forms pedestals on cultured cells more efficiently than EHEC57, and therefore has been studied more extensively. EPEC Tir (TirEPEC) is the only translocated EPEC effector required for pedestal assembly, its essen-tial activity being to recruit the redundant host adap-tor proteins Nck-1 and Nck-2 (referred to generically as Nck)36,58,59. Upon translocation, host-cell kinases phosphorylate Y474 within the cytoplasmic C-terminal domain of TirEPEC (REFS 60–62) (FIG. 3), generating a bind-ing site for the Nck SH2 domain. Nck also contains a region encompassing three SH3 domains that can acti-vate N-WASP32,63 (FIG. 2b). Consistent with a central role for host Nck, EPEC generates pedestals on Nck-knockout cells with fourfold less efficiency than on Nck-proficient cells59,64. Artificial clustering of the C terminus of TirEPEC is sufficient to generate actin pedestals in the absence of any other EPEC effector, and bacterium-sized particles coated with a 12-residue Nck-binding TirEPEC peptide trigger actin ‘comet tail’ formation in Xenopus egg extracts65, highlighting the surprising simplicity of this primary mechanism for EPEC-mediated signalling.
The Nck SH3 domains bind N-WASP PRD25 (FIG. 2), an interaction that is sufficient to stimulate Arp2/3-dependent actin nucleation in vitro32. Paradoxically, deletion analysis of N-WASP reveals that TirEPEC–Nck complexes recruit the N-terminal WH1 domain rather than the PRD38 and, although the PRD is not absolutely required for pedestal formation38, it appears to enhance the efficiency of this process (K.G.C. and J.M.L., unpub-lished data). Assuming that PRD-mediated enhancement of pedestal formation involves Nck, these observations are consistent with a model whereby direct interactions between Nck and the PRD only have a supporting role in N-WASP recruitment to TirEPEC. The major pathway might require a pivotal cellular adaptor that binds the N-WASP WH1 domain. WIP, which binds both Nck27 and the N-WASP WH1 domain25 (FIG. 2b), was an attrac-tive candidate for this second partner, but expression of dominant-negative WIP does not hinder EPEC pedestal formation. Cells expressing N-WASP variants that cannot bind WIP still generate pedestals36, and EPEC induces
Figure 2 | The structure of neural Wiskott–Aldrich syndrome protein (N-WASP) and WASP-interacting adaptor proteins. a | Schematic representation of the archetypal cellular nucleation promoting factor N-WASP in the autoinhibited (left) and activated (right) state. The regulatory domains — the WASP-homology 1 domain (WH1), the proline-rich domain (PRD) and the GTPase-binding domain (GBD) — are shown in yellow. The GBD is further subdivided into a basic region (B), Cdc42/Rac-interactive binding domain (CRIB) and autoinhibitory region (AIR). The C-terminal output domains, which together comprise the verprolin-connector-acidic (VCA) domain — the tandem G-actin-binding WASP-homology 2 (WH2) domain and the Arp2/3-binding connector (C) and acidic (A) domain — are shown in green. WASP-interacting-family proteins such as WASP-interacting protein (WIP) negatively regulate N-WASP by stabilizing the autoinhibited state. Conversely, multiple signals are integrated to activate N-WASP, including binding of phosphatidylinositol-4,5-bisphosphate (PIP2), small GTPases (for example, Cdc42) and various signalling adaptors (for example, Nck and Grb2). Post-translational modification (PO4) within the AIR and CA domains also contributes to activation. In the autoinhibited panel, dashed arrows indicate hypothetical contributors to the autoinhibited conformation that are not well-established by direct binding assays. In the activated panel, dashed arrows indicate phosphorylation rather than binding. b | Schematic representation of cellular signalling adaptors that regulate N-WASP. WIP contains a WH2 domain, in addition to a Nck-binding domain (NBD) and an N-WASP-binding domain (WBD). The signalling adaptors Nck and Grb2 contain Src-homology 2 (SH2) domains that engage phosphotyrosines (pY) of transmembrane receptors and multiple Src-homology 3 (SH3) domains that bind N-WASP and/or WIP. In both (a) and (b), black arrows represent ‘input’ interactions, red arrows represent ‘output’.
R E V I E W S
360 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group
EspFu
Cif
EspI
EspG2
EspG
EspJ SepZ
NleC NleD
Map
EspH
EspF
Tir EspB EspD
Formation of actin pedestals
Binds catenin, disrupts actin cytoskeleton
Disrupts TJs
TJ
Disrupts actin cytoskeleton
Disrupts microtubules
↑RhoA
Filopodia
? ?
Cell-cycle arrest
?
Destroys Em
Apoptosis
Binds intermediatefilaments, CK18, 14-3-3τ
Disrupts TJs, loss of TER
Golgi
Disrupts TJs
EspA
EscF
EscC
EscJEscR-V
EscN
TTSSInt
IM
OM
WxxxE
Membrane disruption
actin assembly in WIP-knockout cells (S. Snapper, unpublished data). Therefore, unidentified WIP-like adaptors or novel N-WASP-interacting factors that bind both Nck and the WH1 domain might act as crucial intermediates that link TirEPEC and N-WASP.
Although the primary pathway for actin assembly by EPEC is triggered by Nck recruitment to Y474, the
existence of a subsidiary Nck-independent cascade is indicated by the 25% residual pedestal formation on Nck-deficient cells64,66. One element of this Nck-independent pathway is mediated by phosphorylated Y474, which probably recruits other Nck-like adaptors that activate N-WASP. A second element is promoted by Y454, which is also phosphorylated upon translocation, albeit less
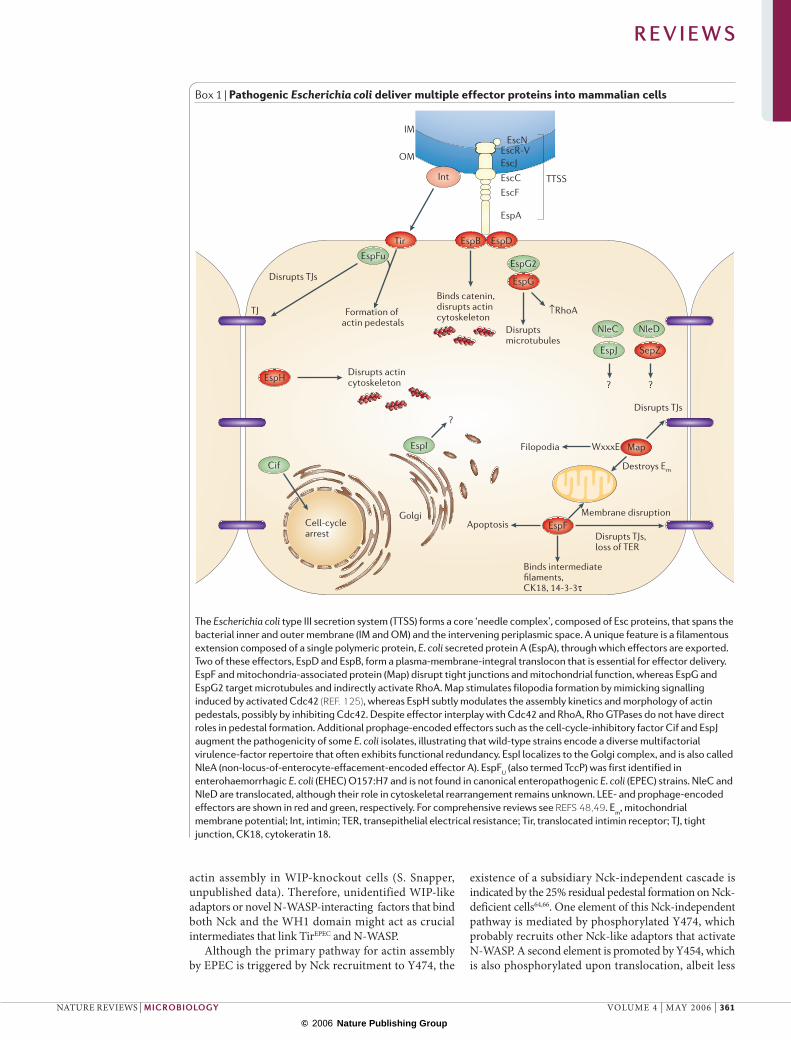
Box 1 | Pathogenic Escherichia coli deliver multiple effector proteins into mammalian cells
The Escherichia coli type III secretion system (TTSS) forms a core ‘needle complex’, composed of Esc proteins, that spans the bacterial inner and outer membrane (IM and OM) and the intervening periplasmic space. A unique feature is a filamentous extension composed of a single polymeric protein, E. coli secreted protein A (EspA), through which effectors are exported. Two of these effectors, EspD and EspB, form a plasma-membrane-integral translocon that is essential for effector delivery. EspF and mitochondria-associated protein (Map) disrupt tight junctions and mitochondrial function, whereas EspG and EspG2 target microtubules and indirectly activate RhoA. Map stimulates filopodia formation by mimicking signalling induced by activated Cdc42 (REF. 125), whereas EspH subtly modulates the assembly kinetics and morphology of actin pedestals, possibly by inhibiting Cdc42. Despite effector interplay with Cdc42 and RhoA, Rho GTPases do not have direct roles in pedestal formation. Additional prophage-encoded effectors such as the cell-cycle-inhibitory factor Cif and EspJ augment the pathogenicity of some E. coli isolates, illustrating that wild-type strains encode a diverse multifactorial virulence-factor repertoire that often exhibits functional redundancy. EspI localizes to the Golgi complex, and is also called NleA (non-locus-of-enterocyte-effacement-encoded effector A). EspFU (also termed TccP) was first identified in enterohaemorrhagic E. coli (EHEC) O157:H7 and is not found in canonical enteropathogenic E. coli (EPEC) strains. NleC and NleD are translocated, although their role in cytoskeletal rearrangement remains unknown. LEE- and prophage-encoded effectors are shown in red and green, respectively. For comprehensive reviews see REFS 48,49. Em, mitochondrial membrane potential; Int, intimin; TER, transepithelial electrical resistance; Tir, translocated intimin receptor; TJ, tight junction, CK18, cytokeratin 18.
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 361
© 2006 Nature Publishing Group
EHEC
IntiminTirEHEC
? EspFU N-WASP
Arp2/3
Actin
Nck
c-Fync-Abl
Vaccinia
Grb2
A36R
c-Srcc-Fync-Yesc-Ablc-Arg
Pedestal
Host cell
IcsA
Shigella
NckWIP
EPEC
IntiminTirEPEC
a b
c d
Y454P
Y474P
Y112P
Y132P
?
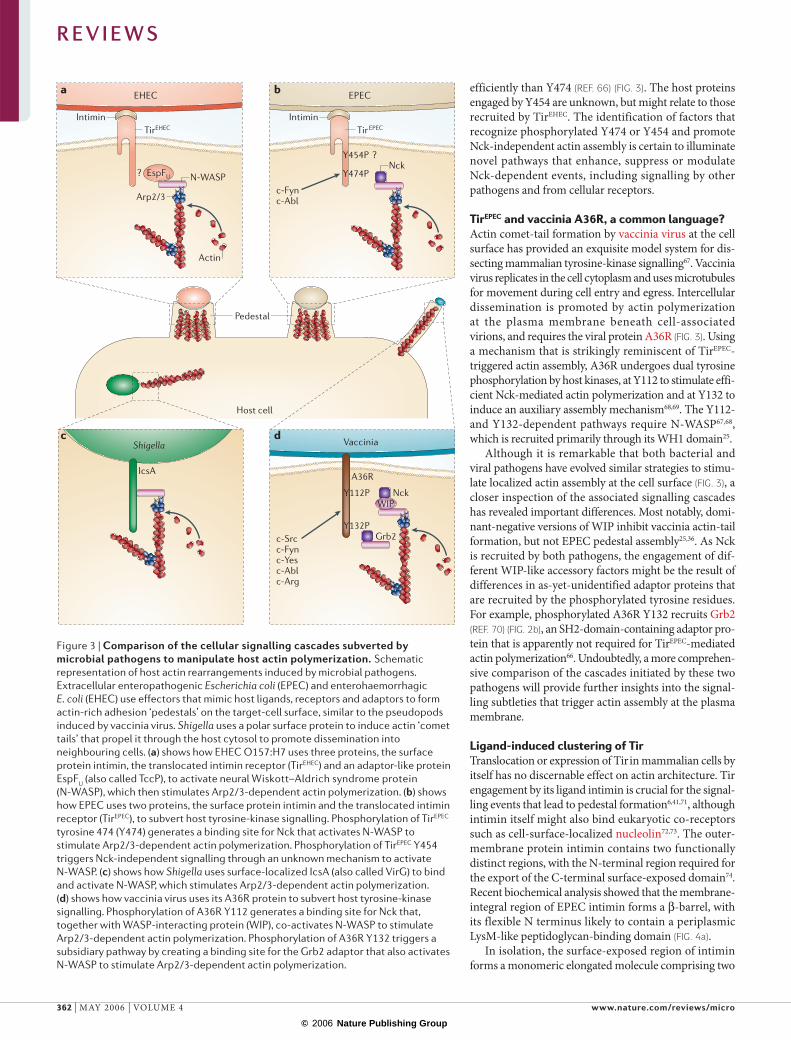
efficiently than Y474 (REF. 66) (FIG. 3). The host proteins engaged by Y454 are unknown, but might relate to those recruited by TirEHEC. The identification of factors that recognize phosphorylated Y474 or Y454 and promote Nck-independent actin assembly is certain to illuminate novel pathways that enhance, suppress or modulate Nck-dependent events, including signalling by other pathogens and from cellular receptors.
TirEPEC and vaccinia A36R, a common language?Actin comet-tail formation by vaccinia virus at the cell surface has provided an exquisite model system for dis-secting mammalian tyrosine-kinase signalling67. Vaccinia virus replicates in the cell cytoplasm and uses microtubules for movement during cell entry and egress. Intercellular dissemination is promoted by actin poly merization at the plasma membrane beneath cell-associated virions, and requires the viral protein A36R (FIG. 3). Using a mechanism that is strikingly reminiscent of TirEPEC-triggered actin assembly, A36R undergoes dual tyrosine phosphorylation by host kinases, at Y112 to stimulate effi-cient Nck-mediated actin polymerization and at Y132 to induce an auxiliary assembly mechanism68,69. The Y112- and Y132-dependent pathways require N-WASP67,68, which is recruited primarily through its WH1 domain25.
Although it is remarkable that both bacterial and viral pathogens have evolved similar strategies to stimu-late localized actin assembly at the cell surface (FIG. 3), a closer inspection of the associated signalling cascades has revealed important differences. Most notably, domi-nant-negative versions of WIP inhibit vaccinia actin-tail formation, but not EPEC pedestal assembly25,36. As Nck is recruited by both pathogens, the engagement of dif-ferent WIP-like accessory factors might be the result of differences in as-yet-unidentified adaptor proteins that are recruited by the phosphorylated tyrosine residues. For example, phosphorylated A36R Y132 recruits Grb2 (REF. 70) (FIG. 2b), an SH2-domain-containing adaptor pro-tein that is apparently not required for TirEPEC-mediated actin polymerization66. Undoubtedly, a more comprehen-sive comparison of the cascades initiated by these two pathogens will provide further insights into the signal-ling subtleties that trigger actin assembly at the plasma membrane.
Ligand-induced clustering of TirTranslocation or expression of Tir in mammalian cells by itself has no discernable effect on actin architecture. Tir engagement by its ligand intimin is crucial for the signal-ling events that lead to pedestal formation6,41,71, although intimin itself might also bind eukaryotic co-receptors such as cell-surface-localized nucleolin72,73. The outer-membrane protein intimin contains two functionally distinct regions, with the N-terminal region required for the export of the C-terminal surface-exposed domain74. Recent biochemical analysis showed that the membrane-integral region of EPEC intimin forms a β-barrel, with its flexible N terminus likely to contain a periplasmic LysM-like peptidoglycan-binding domain (FIG. 4a).
In isolation, the surface-exposed region of intimin forms a monomeric elongated molecule comprising two
Figure 3 | Comparison of the cellular signalling cascades subverted by microbial pathogens to manipulate host actin polymerization. Schematic representation of host actin rearrangements induced by microbial pathogens. Extracellular enteropathogenic Escherichia coli (EPEC) and enterohaemorrhagic E. coli (EHEC) use effectors that mimic host ligands, receptors and adaptors to form actin-rich adhesion ‘pedestals’ on the target-cell surface, similar to the pseudopods induced by vaccinia virus. Shigella uses a polar surface protein to induce actin ‘comet tails’ that propel it through the host cytosol to promote dissemination into neighbouring cells. (a) shows how EHEC O157:H7 uses three proteins, the surface protein intimin, the translocated intimin receptor (TirEHEC) and an adaptor-like protein EspFU (also called TccP), to activate neural Wiskott–Aldrich syndrome protein (N-WASP), which then stimulates Arp2/3-dependent actin polymerization. (b) shows how EPEC uses two proteins, the surface protein intimin and the translocated intimin receptor (TirEPEC), to subvert host tyrosine-kinase signalling. Phosphorylation of TirEPEC tyrosine 474 (Y474) generates a binding site for Nck that activates N-WASP to stimulate Arp2/3-dependent actin polymerization. Phosphorylation of TirEPEC Y454 triggers Nck-independent signalling through an unknown mechanism to activate N-WASP. (c) shows how Shigella uses surface-localized IcsA (also called VirG) to bind and activate N-WASP, which stimulates Arp2/3-dependent actin polymerization. (d) shows how vaccinia virus uses its A36R protein to subvert host tyrosine-kinase signalling. Phosphorylation of A36R Y112 generates a binding site for Nck that, together with WASP-interacting protein (WIP), co-activates N-WASP to stimulate Arp2/3-dependent actin polymerization. Phosphorylation of A36R Y132 triggers a subsidiary pathway by creating a binding site for the Grb2 adaptor that also activates N-WASP to stimulate Arp2/3-dependent actin polymerization.
R E V I E W S
362 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group

EPEC intimin Yersinia invasin
Signal
LysM-likedomain
β-barrel ?
D0
D1
D2
D3
D1
D2
Oligomerization OM
D3
D4
D5
Tir binding
Integrin binding
Extracellular
Periplasm
Intimin 559–939
Tir-IBD
N
Tir PM
OM
N
C C
NN
C C
NN
C C
PeriplasmNLysM
Ig
CTLDC
β
D0
D1
D2
D3
Host-cell cytosol
a b
GG
Oligomerization
C-type lectinCalcium-dependent carbohydrate-binding protein.
immunoglobulin (Ig)-like domains and a C-type lectin-like domain (CTLD)75,76. The surface-distal Ig domain and the CTLD are sufficient for Tir binding74, although only the latter interacts directly with Tir75. Intimin archi-tecture closely corresponds to the analogous region of the intimin homologue invasin, which is encoded by the enteric pathogen Yersinia pseudotuberculosis. In contrast to intimin, invasin is sufficient to promote actin-dependent bacterial entry into non-phagocytic cells, as it binds cellular β1-integrin receptors77 (FIG. 4a).
Although extracellular ligands often transduce transmembrane signals by inducing conformational changes in their receptors that permit the intracellular engagement or disassociation of cytoplasmic proteins, binary ligand–receptor interactions are seldom suf-ficient to trigger a robust cellular response. Rather, sig-nalling is usually amplified by receptor aggregation or clustering by multivalent ligand binding or homotypic receptor–receptor interactions78,79. In this regard, the
β-barrel-forming region of intimin is not only essen-tial for localization and export of the extracellular domain, it also directs intimin homodimerization, which is predicted to influence the distribution of Tir in the membrane80. Indeed, given that the extracellular intimin-binding domain of Tir formed crystallographic homodimers even in the absence of intimin (FIG. 4b, upper panel)75, intimin homo-oligomerization is pre-dicted to generate a reticular array-like superstructure of clustered Tir dimers (FIG. 4b, lower panel)80. These intimin–Tir arrays are not formed when the extra-cellular domain of intimin is artificially dimerized, suggesting that the β-barrel-forming region imposes a specific geometry on the ligand–receptor complex, a structural constraint necessary for ordered clustering80. Intriguingly, the invasin D2 Ig-domain, which is absent from intimin (FIG. 4a), promotes homo multimerization and bacterial internalization by clustering cellular β1-integrins81. Considering that invasin also contains
Figure 4 | Intimin-induced clustering of the translocated intimin receptor (Tir) in the host plasma membrane. a | Structure of enteropathogenic Escherichia coli (EPEC) intimin (left) and Yersinia pseudotuberculosis invasin (right). The crystal structure of the surface-exposed C-terminal residues (559–939) of EPEC intimin revealed a monomeric rod-shaped molecule comprising three adjacent globular domains: two immunoglobulin (Ig)-like domains (D1 and D2) and a C-type lectin-like domain (CTLD, D3). The surface-proximal D0 domain is predicted to adopt an Ig-like fold similar to D1 and D2 and to contain a flexible glycine linker (GG). This architecture corresponds to the extracellular domain of Y. pseudotuberculosis invasin, which contains four Ig-like domains (D1–D4) and a CTLD (D5). The N-terminal domains remain structurally uncharacterized but are functionally interchangeable, as either is sufficient to promote the surface exposure of the C-terminal target-binding domains. The intimin N-terminal domain includes a flexible region likely to contain a periplasmic LysM-like peptidoglycan-binding domain (residues 40–188) and a membrane-integral β-barrel required for membrane localization, surface assembly and homodimerization. The invasin Ig-like D2 domain that is absent from intimin promotes invasin homo-oligomerization and drives clustering of cellular integrins. Intimin dimerization suggests a new model for intimin–Tir interaction. (b) The upper panel shows a binary interaction between the minimal intimin-binding fragment (IBD) of TirEPEC (red) and the dissociated intimin C-terminal Tir-binding fragment (blue) suggested by the crystal structure, whereas the lower panel shows a reticular array resulting from interaction between full-length dimeric intimin (blue) localized in the EPEC outer membrane (OM), and plasma membrane (PM)-integral TirEPEC (red).
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 363
© 2006 Nature Publishing Group

Immunological synapseA large junctional structure formed at the cell surface between a T cell and an antigen-presenting cell, also known as the supramolecular activation cluster. Important molecules that are involved in T-cell activation — including the T-cell receptor, numerous signal-transduction molecules and molecular adaptors — accumulate in an orderly manner at this site. Immunological synapses are now known to also form between other types of immune cells, for example, between dendritic cells and natural killer cells.
Immunoreceptor tyrosine-based activation motif(ITAM). A sequence found in the cytoplasmic domains of the invariant chains of various cell-surface immune receptors, such as the T-cell receptor. Following phosphorylation of their tyrosine residue, ITAMs function as docking sites for Src-homology-2-domain-containing tyrosine kinases and adaptor molecules, thereby facilitating intracellular-signalling cascades.
a potential intra-membranous homodimerization domain similar to that of intimin74, invasin might require multiple domains that promote higher-order oligomers to induce a signal sufficient to sustain the actin rearrangements that drive Yersinia uptake. Members of the intimin/invasin family of bacterial ligand mimics therefore induce multivalent oligomerization and clus-tering of host or pathogen-delivered receptors to initiate cellular signalling cascades.
TirEPEC: modelling tyrosine-kinase signallingPhosphorylation of TirEPEC Y474 by a host-cell tyrosine kinase(s) is an essential control point during pedestal assembly, as beads coated with a Y474-spanning peptide are sufficient to trigger phosphotyrosine-dependent actin assembly in cell-free extracts65. The precedent of cellular tyrosine-kinase signalling suggests that intimin–TirEPEC clustering might act as an essential recruitment signal for mammalian kinases. EPEC mutants lacking intimin were exploited to investigate this possibility, as they deliver TirEPEC and other effectors into cultured cells without forming pedestals, which are only induced sub-sequently when ‘Tir-primed’ cells are challenged with E. coli expressing intimin, intimin proteoliposomes or intimin-coated particles49,74,80. By segregating these steps, intimin-induced TirEPEC clustering was demonstrated to trigger TirEPEC Y474 phosphorylation and actin poly-merization61. The sequence context of TirEPEC Y474 closely resembles the consensus c-Src tyrosine-kinase target motif 61,67. Correspondingly, SFK inhibitors block TirEPEC Y474 phosphorylation and pedestal formation in the ‘priming and challenge’ model. Using knockout cell lines, in vitro phosphorylation assays and siRNA-mediated knockdown, the SFK c-Fyn was recently identified as a kinase that specifically phosphorylates Y474 downstream of TirEPEC clustering61. However, unlike other essential host factors such as Nck and N-WASP, c-Fyn does not appear to be a stable pedestal component (N. Phillips, R.D.H. and V.K., unpublished data), suggesting that the Tir–c-Fyn interaction is probably transient, as might be expected given the potential for mutually exclusive c-Fyn and Nck binding to the residues flanking Y474 (REF. 61).
As c-Fyn is enriched in cholesterol-rich plasma-membrane microdomains (‘rafts’)82, intimin-induced clus-tering might recruit TirEPEC to rafts, allowing juxtaposition of cellular c-Fyn and its prokaryotic substrate. Indeed, CD44, a hyaluronic-acid receptor that co-partitions with c-Fyn in rafts83, is recruited to EPEC contact sites together with cholesterol and glycosyl phosphoinositol-anchored proteins84 (FIGS 1,5). The c-Fyn-dependent phosphoryla-tion of TirEPEC Y474 has clear mechanistic parallels in mammalian signalling pathways. One notable example is the immunological synapse, a well defined instance of cell–cell communication, in which ligand–receptor inter-actions drive actin reorganization by receptor phospho-rylation14,85 (FIG. 5). In resting T cells, the raft-associated SFK c-Lck is in a ‘closed’ inactive conformation, owing to an intramolecular association between its SH2 domain and a C-terminal phosphotyrosine, which is modified by C-terminal Src kinase (Csk)86. TCR clustering and raft aggregation induces spatial segregation of c-Lck from Csk
and subsequent c-Lck activation by trans-autophosphor-ylation of a separate regulatory tyrosine. Activated c-Lck phosphorylates tandem immuno receptor tyrosine-based activation motifs (ITAMs) within the CD3ξ subunits and allows subsequent recruitment of the specialized tyrosine kinase ZAP-70, which in turn phosphorylates dual tyro-sines of the adaptor protein SLP-76 (REFS 85,87). Along with additional adaptors including Nck, phosphorylated SLP-76 recruits WASP, leading to Arp2/3 activation and actin assembly88.
So, immunological-synapse and EPEC actin-pedes-tal formation both potentially involve transmembrane-receptor recruitment into lipid rafts, SFK activation, tyrosine phosphorylation of receptors or associated pro-teins to generate binding sites for Nck, and subsequent activation of WASP-family NPFs and the Arp2/3 complex. Reinforcing these similarities, the sequence encompassing TirEPEC Y474 (Y474XXV-X5-Y483XXI) resembles an ITAM motif (YXXL/I-X8-10-YXXL/I)61. Continued comparison of actin-pedestal formation and TCR signalling is likely to provide insights into both processes. For example, just as integrins and the TCR complex engage growth-factor receptors and CD4/CD8 (REFS 89,90), respectively, as co-receptors to finetune signalling responses, TirEPEC might associate with endo genous mammalian membrane proteins such as CD44 to modulate pedestal assembly and persistence. In addition, c-Abl-family kinases have recently been implicated in both processes62,91.
A second example of host signalling with similarity to TirEPEC-directed actin-pedestal assembly is the generation of focal adhesions, connections between the extracellular matrix and the internal actin cytoskeleton, the formation of which is mediated by heterodimeric trans membrane integrin receptors, which also use SFKs92 (FIG. 5). However, unlike TCRs, integrins constitutively associate with a cognate SFK, but the kinase is in an inactive state. These stable integrin–SFK complexes are excluded from membrane rafts. Extracellular-matrix binding induces receptor clustering and concomitant recruitment into microdomains, which increases the local concentration of the associated SFK93. This triggers kinase activation by autophosphorylation, and tyrosine phosphorylation of multiple downstream effectors including focal-adhesion kinase (FAK), cortactin and paxillin, which in turn stim-ulate Arp2/3-dependent actin polymerization beneath the plasma membrane94.
An apparently distinct strategy for SFK activation is used by vaccinia virus, which deploys a second viral protein, B5R, to activate SFKs. Remarkably, it engages ‘outside-in’ signalling, presumably by triggering SFK activation through an as-yet-unidentified host trans-membrane protein or complex. B5R activity switches viral motility from a microtubule-based to an actin-based mode, by triggering kinesin dissociation from A36R to allow SFK-dependent phosphorylation of Y112 and Y132 (REF. 95) (FIG. 5). The precise mechanisms by which EPEC manipulates host kinases remain unknown, but given the remarkable kinase specificity in the ‘priming and challenge’ model61, deciphering how TirEPEC recruits and activates c-Fyn in this context will not only yield important insights into molecular dynamics early in
R E V I E W S
364 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group
IEV
PP
PP
P P P P P P
PP P P
PPPP PP
a Immunological synapse b Focal adhesion c Actin pedestal d Poxvirus pseudopod
TCRαβ
α βδε εγζζ
PM
ITAM
CD3
c-Lck
c-Lck
c-Src
c-Src
c-Fyn
c-Fyn
c-Abl?
c-Abl?
c-Abl
CD4/CD8
Lipid raft
GFR CD44? ?Tir
Integrin
MTSFK
c-AblSFK
A36R
A36R
CEV
B5R
B5R
IntiminECM
Kinesin
MHC engagement Substrate-induced clustering
Substrate-induced clustering
Viral egress
SLP-76Nck
WASP
Arp2/3-dependentactin polymerization
NckGrb2
N-WASP
Arp2/3-dependentactin polymerization
Nck
N-WASP
Arp2/3-dependentactin polymerization
Rac Paxillin
Focal-adhesion kinase
Cortactin
Arp2/3-dependentactin polymerization
ZAP-70
+
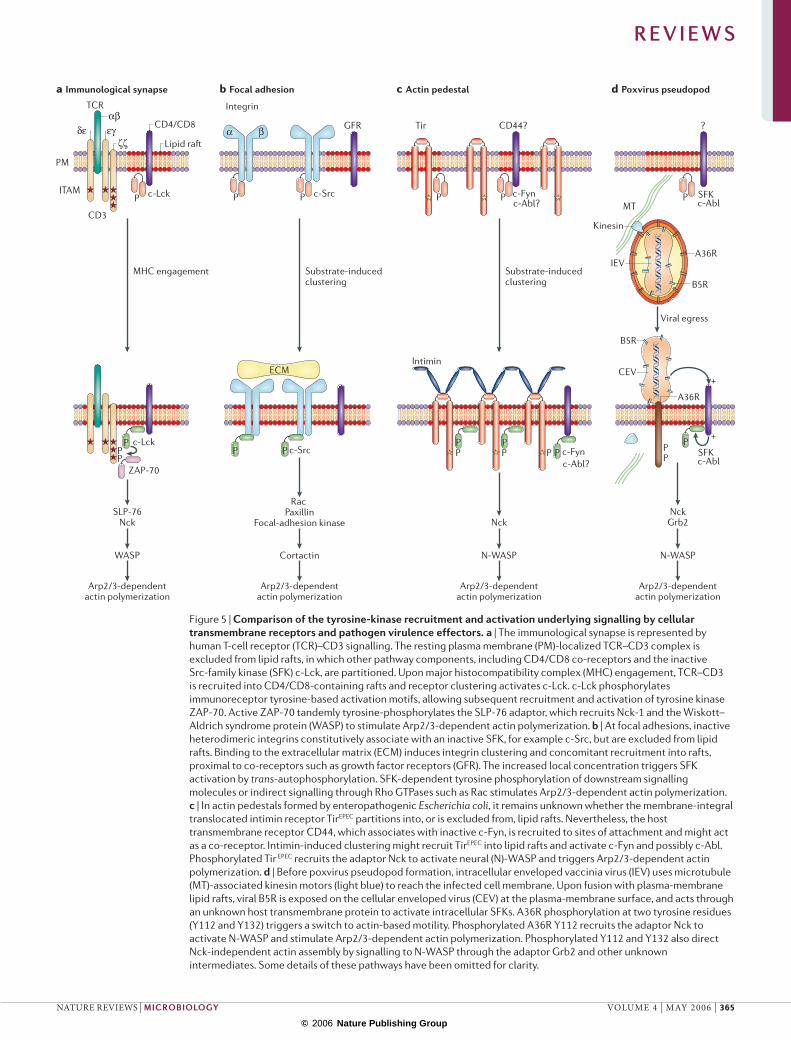
+
Figure 5 | Comparison of the tyrosine-kinase recruitment and activation underlying signalling by cellular transmembrane receptors and pathogen virulence effectors. a | The immunological synapse is represented by human T-cell receptor (TCR)–CD3 signalling. The resting plasma membrane (PM)-localized TCR–CD3 complex is excluded from lipid rafts, in which other pathway components, including CD4/CD8 co-receptors and the inactive Src-family kinase (SFK) c-Lck, are partitioned. Upon major histocompatibility complex (MHC) engagement, TCR–CD3 is recruited into CD4/CD8-containing rafts and receptor clustering activates c-Lck. c-Lck phosphorylates immunoreceptor tyrosine-based activation motifs, allowing subsequent recruitment and activation of tyrosine kinase ZAP-70. Active ZAP-70 tandemly tyrosine-phosphorylates the SLP-76 adaptor, which recruits Nck-1 and the Wiskott–Aldrich syndrome protein (WASP) to stimulate Arp2/3-dependent actin polymerization. b | At focal adhesions, inactive heterodimeric integrins constitutively associate with an inactive SFK, for example c-Src, but are excluded from lipid rafts. Binding to the extracellular matrix (ECM) induces integrin clustering and concomitant recruitment into rafts, proximal to co-receptors such as growth factor receptors (GFR). The increased local concentration triggers SFK activation by trans-autophosphorylation. SFK-dependent tyrosine phosphorylation of downstream signalling molecules or indirect signalling through Rho GTPases such as Rac stimulates Arp2/3-dependent actin polymerization. c | In actin pedestals formed by enteropathogenic Escherichia coli, it remains unknown whether the membrane-integral translocated intimin receptor TirEPEC partitions into, or is excluded from, lipid rafts. Nevertheless, the host transmembrane receptor CD44, which associates with inactive c-Fyn, is recruited to sites of attachment and might act as a co-receptor. Intimin-induced clustering might recruit TirEPEC into lipid rafts and activate c-Fyn and possibly c-Abl. Phosphorylated Tir EPEC recruits the adaptor Nck to activate neural (N)-WASP and triggers Arp2/3-dependent actin polymerization. d | Before poxvirus pseudopod formation, intracellular enveloped vaccinia virus (IEV) uses microtubule (MT)-associated kinesin motors (light blue) to reach the infected cell membrane. Upon fusion with plasma-membrane lipid rafts, viral B5R is exposed on the cellular enveloped virus (CEV) at the plasma-membrane surface, and acts through an unknown host transmembrane protein to activate intracellular SFKs. A36R phosphorylation at two tyrosine residues (Y112 and Y132) triggers a switch to actin-based motility. Phosphorylated A36R Y112 recruits the adaptor Nck to activate N-WASP and stimulate Arp2/3-dependent actin polymerization. Phosphorylated Y112 and Y132 also direct Nck-independent actin assembly by signalling to N-WASP through the adaptor Grb2 and other unknown intermediates. Some details of these pathways have been omitted for clarity.
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 365
© 2006 Nature Publishing Group

pedestal biogenesis, but also clarify how cellular receptors recognize and activate specific SFKs.
Although the ‘priming and challenge’ model defines a specific requirement for c-Fyn, facilitates the capture and characterization of intermediates and provides a valuable experimental system to study kinase recruitment and acti-vation61, the situation during infection in vivo is potentially more complex. A recent report suggests that SFKs, along with c-Abl and c-Arg kinases, might have functionally redundant roles in EPEC pedestal formation, and showed c-Abl and c-Arg to be stable pedestal components. Using engineered kinase derivatives that are resistant to a c-Abl family inhibitor, either c-Arg or c-Abl were apparently suf-ficient for EPEC pedestal assembly 62. Remarkably, SFKs and c-Abl also have redundant and/or sequential functions in poxvirus actin polymerization and virion release69,96,97. However, whereas vaccinia A36R is a substrate for SFKs and c-Arg/c-Abl96, in vitro TirEPEC is specifically a substrate for c-Fyn and is not directly phosphorylated by recom-binant c-Abl or other SFKs (N. Phillips, R.D.H. and V.K., unpublished data). As c-Abl itself is activated by SFK-dependent tyrosine phosphorylation98, it remains possible that c-Fyn acts upstream of c-Abl in the phosphorylation of TirEPEC, as it does in TCR91 and platelet-derived growth factor (PDGF) receptor signalling98.
These studies raise the possibility that EPEC uses compensatory and redundant mechanisms, possibly influenced by other effectors, to maximize its host range and colonize different intestinal cell types. The extent and nature of this diversity remains largely unknown, and might vary with the EPEC strain. Nevertheless, evidence for the existence of redundant signalling is already emerging from studies of TirEPEC, which induces actin assembly through at least two independent pathways66, and from the analysis of the EHEC Tir, which generally functions independently of tyrosine phosphorylation64,99. The precise interplay between SFKs and c-Abl-family kinases during pedestal formation remains to be fully deciphered. Further stud-ies will undoubtedly reveal important new insights into how these interrelated kinases function in cells.
The EHEC missing link: EspFULike EPEC, EHEC requires N-WASP to generate actin pedestals36. However, EHEC O157:H7 Tir (TirEHEC) is functionally divergent from TirEPEC, as it lacks a tyrosine residue equivalent to TirEPEC Y474 (REF. 60), is not detect-ably tyrosine-phosphorylated in cells51,100 and does not bind Nck58,59. Whereas clustering of TirEPEC in the plasma membrane of transfected eukaryotic cells is sufficient to induce pedestal formation65, equivalent manipulation of TirEHEC is not, although expressed TirEHEC, but not TirEPEC, can apparently be co-sedimented with F-actin101. Correspondingly, an engineered EPEC that translocates TirEHEC in the absence of other EHEC effectors stimulates actin polymerization only weakly58,102,103.
Recent findings have provided exciting new insights into the mechanism of pedestal formation by EHEC. Remarkably, EHEC O157:H7 translocates a second effec-tor, EspFU (also termed TccP), which can recruit N-WASP to TirEHEC (REFS 64,99) (FIG. 3). EspFU is 25% identical to
the LEE-encoded effector EspF but is located within defective prophage 933U, which is present in EHEC but not EPEC. Although EspF and EspFU share the ability to disassemble cellular tight junctions104 (BOX 1), only EspFU can stimulate pedestal formation by EHEC64,99. This unique EspFU activity is probably linked to diver-gence in its proline-rich C terminus, a region comprising six nearly identical 47-residue peptide repeats (FIG. 6). Indeed, far-western and yeast two-hybrid analyses showed that this region of EspFU interacts directly with the N-WASP GBD64. Consistent with the hypothesis that EspFU recruits N-WASP directly, the GBD is suffi-cient to promote N-WASP localization to sites of EHEC adherence36, and inhibitors of Rho-family GTPases do not prevent EHEC pedestal formation57. EspFU not only recruits but also activates N-WASP, as it stimulates Arp2/3-dependent actin polymerization in vitro99.
TirEHEC, EspFU and N-WASP are probably part of a plasma-membrane-localized complex, as all three co-precipitate from EHEC-infected mammalian cells64. Interestingly, neither EspFU nor N-WASP appears to bind to TirEHEC directly, raising the possibility that an unidentified factor(s) connects TirEHEC and EspFU. Co-transfection studies indicate that TirEHEC and EspFU are the only two EHEC effectors required for pedestal formation (K.G.C. and J.M.L., unpublished data), so this putative factor is likely to be of host origin (FIG. 6). It is tempting to speculate that this unidentified factor(s) might also be central to actin assembly at some cellular transmembrane receptors.
N-WASP activation by EspFU and IcsAAs with TirEPEC and vaccinia A36R, intriguing mecha-nistic parallels between actin-pedestal formation by EHEC O157:H7 and the actin-based intracellular motil-ity of Shigella are emerging. Once inside mammalian cells, Shigella (like Listeria, Rickettsia and Burkholderia) escapes into the cytosol and uses actin-dependent motil-ity to promote intercellular spread105. Shigella encodes the critical IcsA surface protein (also called VirG) that usurps the signalling cascade upstream of the Arp2/3 complex by binding and activating N-WASP to stimulate actin polymerization, an activity reminiscent of the Rho GTPase Cdc42 (REF. 106) (FIG. 3).
Like EHEC EspFU, Shigella IcsA is sufficient to acti-vate N-WASP directly and stimulate Arp2/3 -dependent actin polymerization in vitro106. EspFU and IcsA share no sequence similarity, but both use repetitive sequences to recruit N-WASP. Whereas in EspFU these are proline-rich64, IcsA contains glycine-rich repeats107. Both proteins recruit the N-WASP GBD25,36,38. Similar to EHEC ped-estal assembly (S. Snapper, unpublished data), Shigella comet-tail formation occurs in the absence of Cdc42 (REFS 108,109), although Cdc42 might contribute to the initiation of Shigella motility 110. Importantly, the expres-sion of N-WASP derivatives that retain the GBD but lack the VCA segment inhibits both Shigella actin-dependent motility 25,107 and EHEC pedestal assembly (K.G.C. and J.M.L., unpublished data). Overall, these results suggest that EspFU and IcsA both mimic Cdc42 by binding the GBD of N-WASP (FIG. 6).
R E V I E W S
366 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group
PM PM
a TirEHEC c Mammalian transmembrane receptor
NPY458
N Pro-repeats
SH3?
PIP2
b Shigella IcsA
Gly-repeats
WH1 B CRIB AIR PRD CWH2 WH2 AWH1 B CRIB AIR PRD CWH2 WH2 A WH1 B CRIB AIR PRD CWH2 WH2 A
Cdc42GEF SFKP
YN-WASP
EspFU
OM
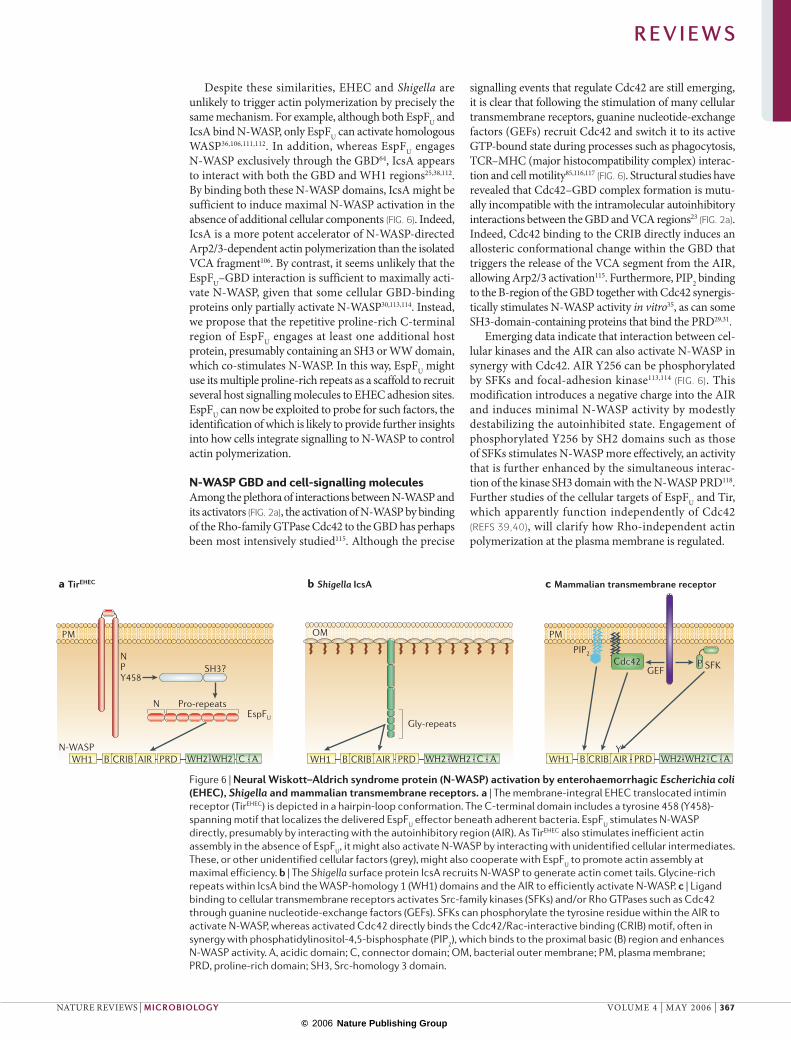
Despite these similarities, EHEC and Shigella are unlikely to trigger actin polymerization by precisely the same mechanism. For example, although both EspFU and IcsA bind N-WASP, only EspFU can activate homologous WASP36,106,111,112. In addition, whereas EspFU engages N-WASP exclusively through the GBD64, IcsA appears to interact with both the GBD and WH1 regions25,38,112. By binding both these N-WASP domains, IcsA might be sufficient to induce maximal N-WASP activation in the absence of additional cellular components (FIG. 6). Indeed, IcsA is a more potent accelerator of N-WASP-directed Arp2/3-dependent actin polymerization than the isolated VCA fragment106. By contrast, it seems unlikely that the EspFU–GBD interaction is sufficient to maximally acti-vate N-WASP, given that some cellular GBD-binding proteins only partially activate N-WASP30,113,114. Instead, we propose that the repetitive proline-rich C-terminal region of EspFU engages at least one additional host protein, presumably containing an SH3 or WW domain, which co-stimulates N-WASP. In this way, EspFU might use its multiple proline-rich repeats as a scaffold to recruit several host signalling molecules to EHEC adhesion sites. EspFU can now be exploited to probe for such factors, the identification of which is likely to provide further insights into how cells integrate signalling to N-WASP to control actin polymerization.
N-WASP GBD and cell-signalling moleculesAmong the plethora of interactions between N-WASP and its activators (FIG. 2a), the activation of N-WASP by binding of the Rho-family GTPase Cdc42 to the GBD has perhaps been most intensively studied115. Although the precise
signalling events that regulate Cdc42 are still emerging, it is clear that following the stimulation of many cellular transmembrane receptors, guanine nucleotide-exchange factors (GEFs) recruit Cdc42 and switch it to its active GTP-bound state during processes such as phagocytosis, TCR–MHC (major histocompatibility complex) interac-tion and cell motility85,116,117 (FIG. 6). Structural studies have revealed that Cdc42–GBD complex formation is mutu-ally incompatible with the intramolecular autoinhibitory interactions between the GBD and VCA regions23 (FIG. 2a). Indeed, Cdc42 binding to the CRIB directly induces an allosteric conformational change within the GBD that triggers the release of the VCA segment from the AIR, allowing Arp2/3 activation115. Furthermore, PIP2 binding to the B-region of the GBD together with Cdc42 synergis-tically stimulates N-WASP activity in vitro35, as can some SH3-domain-containing proteins that bind the PRD29,31.
Emerging data indicate that interaction between cel-lular kinases and the AIR can also activate N-WASP in synergy with Cdc42. AIR Y256 can be phosphorylated by SFKs and focal-adhesion kinase113,114 (FIG. 6). This modification introduces a negative charge into the AIR and induces minimal N-WASP activity by modestly destabilizing the autoinhibited state. Engagement of phosphorylated Y256 by SH2 domains such as those of SFKs stimulates N-WASP more effectively, an activity that is further enhanced by the simultaneous interac-tion of the kinase SH3 domain with the N-WASP PRD118. Further studies of the cellular targets of EspFU and Tir, which apparently function independently of Cdc42 (REFS 39,40), will clarify how Rho-independent actin polymerization at the plasma membrane is regulated.
Figure 6 | Neural Wiskott–Aldrich syndrome protein (N-WASP) activation by enterohaemorrhagic Escherichia coli (EHEC), Shigella and mammalian transmembrane receptors. a | The membrane-integral EHEC translocated intimin receptor (TirEHEC) is depicted in a hairpin-loop conformation. The C-terminal domain includes a tyrosine 458 (Y458)-spanning motif that localizes the delivered EspFU effector beneath adherent bacteria. EspFU stimulates N-WASP directly, presumably by interacting with the autoinhibitory region (AIR). As TirEHEC also stimulates inefficient actin assembly in the absence of EspFU, it might also activate N-WASP by interacting with unidentified cellular intermediates. These, or other unidentified cellular factors (grey), might also cooperate with EspFU to promote actin assembly at maximal efficiency. b | The Shigella surface protein IcsA recruits N-WASP to generate actin comet tails. Glycine-rich repeats within IcsA bind the WASP-homology 1 (WH1) domains and the AIR to efficiently activate N-WASP. c | Ligand binding to cellular transmembrane receptors activates Src-family kinases (SFKs) and/or Rho GTPases such as Cdc42 through guanine nucleotide-exchange factors (GEFs). SFKs can phosphorylate the tyrosine residue within the AIR to activate N-WASP, whereas activated Cdc42 directly binds the Cdc42/Rac-interactive binding (CRIB) motif, often in synergy with phosphatidylinositol-4,5-bisphosphate (PIP2), which binds to the proximal basic (B) region and enhances N-WASP activity. A, acidic domain; C, connector domain; OM, bacterial outer membrane; PM, plasma membrane; PRD, proline-rich domain; SH3, Src-homology 3 domain.
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 367
© 2006 Nature Publishing Group

As EHEC EspFU and Shigella IcsA seemingly both engage the GBD to activate N-WASP64,106, their mecha-nism has been compared to that of Cdc42 (FIG. 6). However, there are several crucial differences. Recent findings indicate that both EHEC and Shigella trigger actin polymerization in the absence of the N-WASP B-region and CRIB motif, but recruit derivatives contain-ing the AIR alone36,38. Therefore, EspFU, and perhaps IcsA, might activate N-WASP by interacting directly with the AIR in the absence of tyrosine-kinase signalling, as phos-photyrosine immunostaining is not detected in either EHEC actin pedestals or Shigella actin comet tails57,67,105. It is possible that EspFU or IcsA bind the AIR with higher affinity than the VCA segment of N-WASP, displacing the latter to activate the Arp2/3 complex. Structural studies of N-WASP complexes with these microbial factors are now required to investigate this hypothesis.
Unexpected parallels between TirEHEC and TirEPEC
As investigators began to uncover the mechanisms underlying actin-pedestal formation by EPEC and EHEC O157:H7, a surprising finding was the fundamental dif-ference between these pathways: TirEPEC Y474 is modi-fied by host tyrosine kinases to generate a binding site for Nck, whereas TirEHEC generates pedestals independently of detectable tyrosine phosphorylation and requires EspFU to assemble pedestals efficiently64,99. However, the recent identification of the Nck-independent pathway of TirEPEC that requires phosphorylated Y454 (REF. 66), along with further characterization of functional elements of TirEHEC signalling, suggests significant parallels between pedestal formation by EPEC and EHEC O157:H7.
First, analysis of TirEHEC deletion derivatives indicates that a peptide encompassing residues 452–463 is required for actin polymerization (FIG. 6) (REF.119; R. DeVinney, unpublished data). An essential residue in this segment is TirEHEC Y458, which is contextually analogous to TirEPEC Y454, a crucial residue for the Nck-independent EPEC pathway66. Second, TirEHEC is sufficient to trigger low-level actin assembly in the absence of EspFU, because EHEC espFU mutants generate actin pedestals at a low but detectable level. In fact, the actin-assembly efficiency of this EHEC mutant is comparable to that of an EPEC tirY474F mutant in which the major Nck-dependent path-way is disrupted64,66. Third, TirEPEC Y454-dependent actin assembly is substantially stimulated when EspFU is artificially introduced into cells (M. Brady, K.G.C and JM.L., unpublished data). Taken together, these findings suggest that mechanistically related signalling occurs from TirEPEC Y454 and TirEHEC Y458. This suggests that the putative host molecules linking TirEPEC Y454 to the host actin-assembly machinery and those connecting TirEHEC Y458 to EspFU and N-WASP are related or identical. If true, then TirEPEC Y454 and TirEHEC Y458 would appear to provide a common signalling function that has been augmented by EspFU in EHEC O157:H7 and superseded by the Y474/Nck-dependent pathway in EPEC. One might speculate that a primordial strain might have encoded a Tir molecule containing only a Y454/Y458-like motif. EPEC and EHEC subsequently independently acquired the ability to enhance actin assembly by either
evolving a version of Tir capable of tyrosine phosphoryla-tion and Nck recruitment or by acquiring EspFU, which compensates for tyrosine phosphorylation, respectively. By extension, it might be predicted that pathogen fitness would be further increased where both pathways coexist. In fact, Frankel and co-workers have recently identified an O119:H6 EPEC encoding both EspFU and a Tir protein harbouring Y474 (REF. 120).
Concluding remarksEPEC and EHEC manipulate the actin cytoskeleton with remarkable sophistication, which is reflected by their ability to mimic eukaryotic signalling processes by encoding both components of a ligand–receptor inter-action. Exciting recent data have additionally revealed that TirEPEC subverts eukaryotic tyrosine-kinase signal-ling to stimulate both a highly efficient Nck-dependent pathway and a subsidiary Nck-independent pathway, and EHEC delivers an adaptor-like protein that triggers efficient actin polymerization, perhaps using a signalling cascade related to the EPEC Nck-independent pathway. As EPEC and EHEC are such accomplished manipulators and imitators of mammalian actin-signalling pathways from transmembrane receptors, future work is likely to provide unique insights into important features of host-cell signalling, and to establish new experimental systems to probe cellular processes using E. coli effectors.
Many important questions remain to be investigated. For example, what is the relationship between c-Fyn and c-Abl/c-Arg kinases during EPEC pedestal formation? What factors link TirEHEC to EspFU and N-WASP, and what are their functions in cells? Are other N-WASP reg-ulators involved in TirEPEC and TirEHEC signalling? More generally, do E. coli or any other microbial pathogens exploit WAVE NPFs121 or non-Arp2/3 actin nucleators to promote actin assembly122,123? The continued study of E. coli-triggered actin assembly will require careful fusion of cell biology, innovative models, structural approaches, stepwise biochemical analyses and reconstitution of con-tributory pathways. Undoubtedly, future work will not only provide further insights into the remarkable nature of microbial pathogenesis but also uncover how our own cells remodel their cytoskeleton.
Note added in proofRecently, Pawson and colleagues124 showed that the trans-membrane receptor nephrin selectively binds Nck adap-tor proteins. Nephrin functions as an adhesion molecule and signalling hub at kidney epithelial-cell junctions, which assembles specialized actin-based foot processes called podosomes that maintain the glomerular filtration barrier. Just like TirEPEC and vaccinia A36R, clustered nephrin is tyrosine-phosphorylated by Src-family kinases on multiple YDxV motifs, which form binding sites for the Nck SH2 domain. In turn, Nck recruits N-WASP and triggers Arp2/3-dependent actin polymerization. These findings reveal the first physiological signalling pathway from a transmembrane receptor to the actin cytoskeleton directly analogous to that downstream of TirEPEC Y474 and A36R Y112, fittingly illustrating the value of pathogen effectors as relevant models for eukaryotic processes.
R E V I E W S
368 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group

1. Cossart, P. & Sansonetti, P. J. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248 (2004).
2. Stebbins, C. E. & Galan, J. E. Structural mimicry in bacterial virulence. Nature 412, 701–705 (2001).
3. Nataro, J. P. & Kaper, J. B. Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201 (1998).
4. Dean-Nystrom, E. A., Bosworth, B. T., Moon, H. W. & O’Brien, A. D. Escherichia coli O157:H7 requires intimin for enteropathogenicity in calves. Infect. Immun. 66, 4560–4563 (1998).
5. Donnenberg, M. S. et al. The role of the eae gene of enterohemorrhagic Escherichia coli in intimate attachment in vitro and in a porcine model. J. Clin. Invest. 92, 1418–1424 (1993).
6. Donnenberg, M. S. et al. Role of the eaeA gene in experimental enteropathogenic Escherichia coli infection. J. Clin. Invest. 92, 1412–1417 (1993).
7. Deng, W., Vallance, B. A., Li, Y., Puente, J. L. & Finlay, B. B. Citrobacter rodentium translocated intimin receptor (Tir) is an essential virulence factor needed for actin condensation, intestinal colonization and colonic hyperplasia in mice. Mol. Microbiol. 48, 95–115 (2003).
8. Shaner, N. C., Sanger, J. W. & Sanger, J. M. Actin and α-actinin dynamics in the adhesion and motility of EPEC and EHEC on host cells. Cell Motil. Cytoskeleton 60, 104–120 (2005).
9. Sanger, J. M., Chang, R., Ashton, F., Kaper, J. B. & Sanger, J. W. Novel form of actin-based motility transports bacteria on the surfaces of infected cells. Cell Motil. Cytoskeleton 34, 279–287 (1996).
10. Knutton, S., Baldwin, T., Williams, P. H. & McNeish, A. S. Actin accumulation at sites of bacterial adhesion to tissue culture cells: basis of a new diagnostic test for enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 57, 1290–1298 (1989).
11. Nicholson-Dykstra, S., Higgs, H. N. & Harris, E. S. Actin dynamics: growth from dendritic branches. Curr. Biol. 15, R346–R357 (2005).
12. Pollard, T. D. & Borisy, G. G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 112, 453–465 (2003).
13. Carragher, N. O. & Frame, M. C. Focal adhesion and actin dynamics: a place where kinases and proteases meet to promote invasion. Trends Cell Biol. 14, 241–249 (2004).
14. Thrasher, A. J. WASP in immune-system organization and function. Nature Rev. Immunol. 2, 635–646 (2002).
15. Welch, M. D. & Mullins, R. D. Cellular control of actin nucleation. Annu. Rev. Cell. Dev. Biol. 18, 247–288 (2002).
16. Millard, T. H., Sharp, S. J. & Machesky, L. M. Signalling to actin assembly via the WASP (Wiskott–Aldrich syndrome protein)-family proteins and the Arp2/3 complex. Biochem. J. 380, 1–17 (2004).
17. Higgs, H. N. & Pollard, T. D. Regulation of actin filament network formation through ARP2/3 complex: activation by a diverse array of proteins. Annu. Rev. Biochem. 70, 649–676 (2001).
18. Burns, S., Cory, G. O., Vainchenker, W. & Thrasher, A. J. Mechanisms of WASP-mediated hematologic and immunologic disease. Blood 104, 3454–3462 (2004).
19. Stradal, T. E. et al. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 14, 303–311 (2004).
20. Goley, E. D., Rodenbusch, S. E., Martin, A. C. & Welch, M. D. Critical conformational changes in the Arp2/3 complex are induced by nucleotide and nucleation promoting factor. Mol. Cell 16, 269–279 (2004).
21. Prehoda, K. E., Scott, J. A., Mullins, R. D. & Lim, W. A. Integration of multiple signals through cooperative regulation of the N-WASP–Arp2/3 complex. Science 290, 801–806 (2000).
22. Rohatgi, R., Ho, H. Y. & Kirschner, M. W. Mechanism of N-WASP activation by CDC42 and phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 1299–1310 (2000).
23. Kim, A. S., Kakalis, L. T., Abdul-Manan, N., Liu, G. A. & Rosen, M. K. Autoinhibition and activation mechanisms of the Wiskott–Aldrich syndrome protein. Nature 404, 151–158 (2000).
24. Miki, H., Sasaki, T., Takai, Y. & Takenawa, T. Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 391, 93–96 (1998).
25. Moreau, V. et al. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nature Cell Biol. 2, 441–448 (2000).
26. Martinez-Quiles, N. et al. WIP regulates N-WASP-mediated actin polymerization and filopodium formation. Nature Cell Biol. 3, 484–491 (2001).
27. Anton, I. M., Lu, W., Mayer, B. J., Ramesh, N. & Geha, R. S. The Wiskott–Aldrich syndrome protein-interacting protein (WIP) binds to the adaptor protein Nck. J. Biol. Chem. 273, 20992–20995 (1998).
28. Suetsugu, S., Miki, H. & Takenawa, T. The essential role of profilin in the assembly of actin for microspike formation. EMBO J. 17, 6516–6526 (1998).
29. Ho, H. Y. et al. Toca-1 mediates Cdc42-dependent actin nucleation by activating the N-WASP–WIP complex. Cell 118, 203–216 (2004).
30. Suetsugu, S. et al. Sustained activation of N-WASP through phosphorylation is essential for neurite extension. Dev. Cell 3, 645–658 (2002).
31. Carlier, M. F. et al. GRB2 links signaling to actin assembly by enhancing interaction of neural Wiskott–Aldrich syndrome protein (N-WASP) with actin-related protein (ARP2/3) complex. J. Biol. Chem. 275, 21946–21952 (2000).
32. Rohatgi, R., Nollau, P., Ho, H. Y., Kirschner, M. W. & Mayer, B. J. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP–Arp2/3 pathway. J. Biol. Chem. 276, 26448–26452 (2001).
33. Martinez-Quiles, N., Ho, H. Y., Kirschner, M. W., Ramesh, N. & Geha, R. S. Erk/Src phosphorylation of cortactin acts as a switch on–switch off mechanism that controls its ability to activate N-WASP. Mol. Cell Biol. 24, 5269–5280 (2004).
34. Cory, G. O., Cramer, R., Blanchoin, L. & Ridley, A. J. Phosphorylation of the WASP-VCA domain increases its affinity for the Arp2/3 complex and enhances actin polymerization by WASP. Mol. Cell 11, 1229–1239 (2003).
35. Rohatgi, R. et al. The interaction between N-WASP and the Arp2/3 complex links Cdc42-dependent signals to actin assembly. Cell 97, 221–231 (1999).
36. Lommel, S., Benesch, S., Rohde, M., Wehland, J. & Rottner, K. Enterohaemorrhagic and enteropathogenic Escherichia coli use different mechanisms for actin pedestal formation that converge on N-WASP. Cell. Microbiol. 6, 243–254 (2004).Both EPEC and EHEC require N-WASP for pedestal assembly, but each uses different mechanisms of recruitment and activation.
37. Kalman, D. et al. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nature Cell Biol. 1, 389–391 (1999).
38. Lommel, S. et al. Actin pedestal formation by enteropathogenic Escherichia coli and intracellular motility of Shigella flexneri are abolished in N-WASP-defective cells. EMBO Rep. 2, 850–857 (2001).
39. Ebel, F., von Eichel-Streiber, C., Rohde, M. & Chakraborty, T. Small GTP-binding proteins of the Rho- and Ras-subfamilies are not involved in the actin rearrangements induced by attaching and effacing Escherichia coli. FEMS Microbiol. Lett. 163, 107–112 (1998).
40. Ben-Ami, G. et al. Agents that inhibit Rho, Rac, and Cdc42 do not block formation of actin pedestals in HeLa cells infected with enteropathogenic Escherichia coli. Infect. Immun. 66, 1755–1758 (1998).
41. Jerse, A. E., Yu, J., Tall, B. D. & Kaper, J. B. A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl Acad. Sci. USA 87, 7839–7843 (1990).
42. McDaniel, T. K. & Kaper, J. B. A cloned pathogenicity island from enteropathogenic Escherichia coli confers the attaching and effacing phenotype on E. coli K-12. Mol. Microbiol. 23, 399–407 (1997).
43. Elliott, S. J. et al. The complete sequence of the locus of enterocyte effacement (LEE) from enteropathogenic Escherichia coli E2348/69. Mol. Microbiol. 28, 1–4 (1998).
44. Ebel, F. et al. Initial binding of Shiga toxin-producing Escherichia coli to host cells and subsequent induction of actin rearrangements depend on filamentous EspA-containing surface appendages. Mol. Microbiol. 30, 147–161 (1998).
45. Knutton, S. et al. A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17, 2166–2176 (1998).
46. Sekiya, K. et al. Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl Acad. Sci. USA 98, 11638–11643 (2001).
47. Dean, P., Maresca, M. & Kenny, B. EPEC’s weapons of mass subversion. Curr. Opin. Microbiol. 8, 28–34 (2005).
48. Garmendia, J., Frankel, G. & Crepin, V. F. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect. Immun. 73, 2573–2585 (2005).
49. Rosenshine, I. et al. A pathogenic bacterium triggers epithelial signals to form a functional bacterial receptor that mediates actin pseudopod formation. EMBO J. 15, 2613–2624 (1996).
50. Kenny, B. et al. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520 (1997).The first demonstration that a bacterium could transfer its own receptor into a mammalian host cell. This paper also defined a link between actin nucleation and TirEPEC.
51. Deibel, C., Kramer, S., Chakraborty, T. & Ebel, F. EspE, a novel secreted protein of attaching and effacing bacteria, is directly translocated into infected host cells, where it appears as a tyrosine-phosphorylated 90 kDa protein. Mol. Microbiol. 28, 463–474 (1998).
52. Patel, A. et al. Host protein interactions with enteropathogenic Escherichia coli (EPEC): 14-3-3τ binds Tir and has a role in EPEC-induced actin polymerization. Cell. Microbiol. 8, 55–71 (2006).
53. Goosney, D. L. et al. Enteropathogenic E. coli translocated intimin receptor, Tir, interacts directly with α-actinin. Curr. Biol. 10, 735–738 (2000).
54. Freeman, N. L. et al. Interaction of the enteropathogenic Escherichia coli protein, translocated intimin receptor (Tir), with focal adhesion proteins. Cell Motil. Cytoskeleton 47, 307–318 (2000).
55. Cantarelli, V. V. et al. Cortactin is necessary for F-actin accumulation in pedestal structures induced by enteropathogenic Escherichia coli infection. Infect. Immun. 70, 2206–2209 (2002).
56. Goosney, D. L., DeVinney, R. & Finlay, B. B. Recruitment of cytoskeletal and signaling proteins to enteropathogenic and enterohemorrhagic Escherichia coli pedestals. Infect. Immun. 69, 3315–3322 (2001).
57. Campellone, K. G. & Leong, J. M. Tails of two Tirs: actin pedestal formation by enteropathogenic E. coli and enterohemorrhagic E. coli O157:H7. Curr. Opin. Microbiol. 6, 82–90 (2003).
58. Campellone, K. G., Giese, A., Tipper, D. J. & Leong, J. M. A tyrosine-phosphorylated 12-amino-acid sequence of enteropathogenic Escherichia coli Tir binds the host adaptor protein Nck and is required for Nck localization to actin pedestals. Mol. Microbiol. 43, 1227–1241 (2002).
59. Gruenheid, S. et al. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nature Cell Biol. 3, 856–859 (2001).Identified Nck adaptor proteins as critical host-cell determinants of EPEC virulence that engage phosphorylated Y474 of TirEPEC.
60. Kenny, B. Phosphorylation of tyrosine 474 of the enteropathogenic Escherichia coli (EPEC) Tir receptor molecule is essential for actin nucleating activity and is preceded by additional host modifications. Mol. Microbiol. 31, 1229–1241 (1999).
61. Phillips, N., Hayward, R. D. & Koronakis, V. Phosphorylation of the enteropathogenic E. coli receptor by the Src-family kinase c-Fyn triggers actin pedestal formation. Nature Cell Biol. 6, 618–625 (2004).Used the ‘priming and challenge’ model to demonstrate that intimin-induced clustering of membrane-integral TirEPEC induces Y474 phosphorylation by the host SFK c-Fyn.
62. Swimm, A. et al. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol. Biol. Cell 15, 3520–3529 (2004).Demonstrated that wild-type EPEC can use redundant host SFKs and Abl-family kinases to form pedestals on cultured cells. This study also highlighted pyrido[2,3-d]pyrimidine compounds as potential inhibitors of EPEC infection.
63. Rivera, G. M., Briceno, C. A., Takeshima, F., Snapper, S. B. & Mayer, B. J. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization. Curr. Biol. 14, 11–22 (2004).
64. Campellone, K. G., Robbins, D. & Leong, J. M. EspFU is a translocated EHEC effector that interacts with Tir and N-WASP and promotes Nck-independent actin assembly. Dev. Cell 7, 217–228 (2004).Identified an effector (EspFU/TccP) encoded outside the LEE that links TirEHEC to N-WASP independently of tyrosine phosphorylation, which triggers localized actin assembly. This study was independent of reference 99.
R E V I E W S
NATURE REVIEWS | MICROBIOLOGY VOLUME 4 | MAY 2006 | 369
© 2006 Nature Publishing Group

65. Campellone, K. G. et al. Clustering of Nck by a 12-residue Tir phosphopeptide is sufficient to trigger localized actin assembly. J. Cell Biol. 164, 407–416 (2004).Determined the minimal requirements for EPEC-mediated actin assembly by demonstrating that Nck clustering by a 12-residue Y474-spanning phosphopeptide triggered actin comet-tail formation in Xenopus egg extracts.
66. Campellone, K. G. & Leong, J. M. Nck-independent actin assembly is mediated by two phosphorylated tyrosines within enteropathogenic Escherichia coli Tir. Mol. Microbiol. 56, 416–432 (2005).
67. Frischknecht, F. & Way, M. Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30–38 (2001).
68. Frischknecht, F. et al. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature 401, 926–929 (1999).
69. Reeves, P. M. et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nature Med. 11, 731–739 (2005).
70. Scaplehorn, N. et al. Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 12, 740–745 (2002).
71. Tzipori, S. et al. The role of the eaeA gene in diarrhea and neurological complications in a gnotobiotic piglet model of enterohemorrhagic Escherichia coli infection. Infect. Immun. 63, 3621–3627 (1995).
72. Frankel, G. et al. Intimin and the host cell — is it bound to end in Tir(s)? Trends Microbiol. 9, 214–218 (2001).
73. Sinclair, J. F. & O’Brien, A. D. Cell surface-localized nucleolin is a eukaryotic receptor for the adhesin intimin-γ of enterohemorrhagic Escherichia coli O157:H7. J. Biol. Chem. 277, 2876–2885 (2002).
74. Liu, H., Magoun, L., Luperchio, S., Schauer, D. B. & Leong, J. M. The Tir-binding region of enterohaemorrhagic Escherichia coli intimin is sufficient to trigger actin condensation after bacterial-induced host cell signalling. Mol. Microbiol. 34, 67–81 (1999).
75. Luo, Y. et al. Crystal structure of enteropathogenic Escherichia coli intimin-receptor complex. Nature 405, 1073–1077 (2000).The crystal structures of an EPEC intimin fragment alone, and in complex with the TirEPEC intimin-binding domain, provided the first insights into the molecular mechanisms underlying intimate adhesion.
76. Batchelor, M. et al. Structural basis for recognition of the translocated intimin receptor (Tir) by intimin from enteropathogenic Escherichia coli. EMBO J. 19, 2452–2464 (2000).Used NMR and mutagenesis approaches to resolve the structure of the extracellular domain of EPEC intimin and to delineate the Tir binding site.
77. Hamburger, Z. A., Brown, M. S., Isberg, R. R. & Bjorkman, P. J. Crystal structure of invasin: a bacterial integrin-binding protein. Science 286, 291–295 (1999).
78. Thomason, P. A., Wolanin, P. M. & Stock, J. B. Signal transduction: receptor clusters as information processing arrays. Curr. Biol. 12, R399–R401 (2002).
79. Cochran, J. R., Aivazian, D., Cameron, T. O. & Stern, L. J. Receptor clustering and transmembrane signaling in T cells. Trends Biochem. Sci. 26, 304–310 (2001).
80. Touze, T., Hayward, R. D., Eswaran, J., Leong, J. M. & Koronakis, V. Self-association of EPEC intimin mediated by the β-barrel-containing anchor domain: a role in clustering of the Tir receptor. Mol. Microbiol. 51, 73–87 (2004).
81. Dersch, P. & Isberg, R. R. A region of the Yersinia pseudotuberculosis invasin protein enhances integrin-mediated uptake into mammalian cells and promotes self-association. EMBO J. 18, 1199–1213 (1999).
82. Harder, T. & Simons, K. Clusters of glycolipid and glycosyl-phosphatidylinositol-anchored proteins in lymphoid cells: accumulation of actin regulated by local tyrosine phosphorylation. Eur. J. Immunol. 29, 556–562 (1999).
83. Foger, N., Marhaba, R. & Zoller, M. Involvement of CD44 in cytoskeleton rearrangement and raft reorganization in T cells. J. Cell Sci. 114, 1169–1178 (2001).
84. Zobiack, N. et al. Cell-surface attachment of pedestal-forming enteropathogenic E. coli induces a clustering of raft components and a recruitment of annexin 2. J. Cell Sci. 115, 91–98 (2002).
85. Fuller, C. L., Braciale, V. L. & Samelson, L. E. All roads lead to actin: the intimate relationship between TCR signaling and the cytoskeleton. Immunol. Rev. 191, 220–236 (2003).
86. Palacios, E. H. & Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 23, 7990–8000 (2004).
87. Lowell, C. A. Src-family kinases: rheostats of immune cell signaling. Mol. Immunol. 41, 631–643 (2004).
88. Sasahara, Y. et al. Mechanism of recruitment of WASP to the immunological synapse and of its activation following TCR ligation. Mol. Cell 10, 1269–1281 (2002).
89. ffrench-Constant, C. & Colognato, H. Integrins: versatile integrators of extracellular signals. Trends Cell Biol. 14, 678–686 (2004).
90. Drevot, P. et al. TCR signal initiation machinery is pre-assembled and activated in a subset of membrane rafts. EMBO J. 21, 1899–1908 (2002).
91. Zipfel, P. A., Zhang, W., Quiroz, M. & Pendergast, A. M. Requirement for Abl kinases in T cell receptor signaling. Curr. Biol. 14, 1222–1231 (2004).
92. Playford, M. P. & Schaller, M. D. The interplay between Src and integrins in normal and tumor biology. Oncogene 23, 7928–7946 (2004).
93. Leitinger, B. & Hogg, N. The involvement of lipid rafts in the regulation of integrin function. J. Cell Sci. 115, 963–972 (2002).
94. Wiesner, S., Legate, K. R. & Fassler, R. Integrin-actin interactions. Cell Mol. Life Sci. 62, 1081–1099 (2005).
95. Newsome, T. P., Scaplehorn, N. & Way, M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 306, 124–129 (2004).
96. Newsome, T. P., Weisswange, I., Frischknecht, F. & Way, M. Abl collaborates with Src family kinases to stimulate actin-based motility of vaccinia virus. Cell. Microbiol. 8, 233–241 (2006).
97. Yang, H. et al. Antiviral chemotherapy facilitates control of poxvirus infections through inhibition of cellular signal transduction. J. Clin. Invest. 115, 379–387 (2005).
98. Hernandez, S. E., Krishnaswami, M., Miller, A. L. & Koleske, A. J. How do Abl family kinases regulate cell shape and movement? Trends Cell Biol. 14, 36–44 (2004).
99. Garmendia, J. et al. TccP is an enterohaemorrhagic Escherichia coli O157:H7 type III effector protein that couples Tir to the actin-cytoskeleton. Cell. Microbiol. 6, 1167–1183 (2004).Identified an effector (EspFU/TccP) encoded outside the LEE that links TirEHEC to N-WASP independently of tyrosine phosphorylation, which triggers localized actin assembly. This study was independent of reference 64.
100. DeVinney, R. et al. Enterohemorrhagic Escherichia coli O157:H7 produces Tir, which is translocated to the host cell membrane but is not tyrosine phosphorylated. Infect. Immun. 67, 2389–2398 (1999).
101. Chuang, C. H., Chiu, H. J., Hsu, S. C., Ho, J. Y. & Syu, W. J. Comparison of Tir from enterohemorrhagic and enteropathogenic Escherichia coli strains: two homologues with distinct intracellular properties. J. Biomed. Sci. 13, 73–87 (2006).
102. DeVinney, R., Puente, J. L., Gauthier, A., Goosney, D. & Finlay, B. B. Enterohaemorrhagic and enteropathogenic Escherichia coli use a different Tir-based mechanism for pedestal formation. Mol. Microbiol. 41, 1445–1458 (2001).
103. Kenny, B. The enterohaemorrhagic Escherichia coli (serotype O157:H7) Tir molecule is not functionally interchangeable for its enteropathogenic E. coli (serotype O127:H6) homologue. Cell. Microbiol. 3, 499–510 (2001).
104. Viswanathan, V. K. et al. Comparative analysis of EspF from enteropathogenic and enterohemorrhagic Escherichia coli in alteration of epithelial barrier function. Infect. Immun. 72, 3218–3227 (2004).
105. Stevens, J. H., Galyov, E. E. & Stevens, M. P. Actin-dependent movement of bacterial pathogens. Nature Rev. Microbiol. 4, 91–101 (2006).
106. Egile, C. et al. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J. Cell Biol. 146, 1319–1332 (1999).
107. Suzuki, T., Miki, H., Takenawa, T. & Sasakawa, C. Neural Wiskott–Aldrich syndrome protein is implicated in the actin-based motility of Shigella flexneri. EMBO J. 17, 2767–2776 (1998).
108. Mounier, J. et al. Rho family GTPases control entry of Shigella flexneri into epithelial cells but not intracellular motility. J. Cell Sci. 112, 2069–2080 (1999).
109. Shibata, T., Takeshima, F., Chen, F., Alt, F. W. & Snapper, S. B. Cdc42 facilitates invasion but not the actin-based motility of Shigella. Curr. Biol. 12, 341–345 (2002).
110. Suzuki, T. et al. Rho family GTPase Cdc42 is essential for the actin-based motility of Shigella in mammalian cells. J. Exp. Med. 191, 1905–1920 (2000).
111. Snapper, S. B. et al. N-WASP deficiency reveals distinct pathways for cell surface projections and microbial actin-based motility. Nature Cell Biol. 3, 897–904 (2001).
112. Suzuki, T. et al. Neural Wiskott–Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell. Microbiol. 4, 223–233 (2002).
113. Wu, X., Suetsugu, S., Cooper, L. A., Takenawa, T. & Guan, J. L. Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 279, 9565–9576 (2004).
114. Torres, E. & Rosen, M. K. Contingent phosphorylation/dephosphorylation provides a mechanism of molecular memory in WASP. Mol. Cell 11, 1215–1227 (2003).
115. Abdul-Manan, N. et al. Structure of Cdc42 in complex with the GTPase-binding domain of the ‘Wiskott–Aldrich syndrome’ protein. Nature 399, 379–383 (1999).
116. Niedergang, F. & Chavrier, P. Regulation of phagocytosis by Rho GTPases. Curr. Top. Microbiol. Immunol. 291, 43–60 (2005).
117. Raftopoulou, M. & Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 265, 23–32 (2004).
118. Torres, E. & Rosen, M. K. Protein tyrosine kinase and GTPase signals cooperate to phosphorylate and activate WASP/N-WASP. J. Biol. Chem. 281, 3513–3520 (2006).
119. Campellone, K. G. et al. Enterohaemorrhagic Escherichia coli Tir requires a C-terminal 12-residue peptide to initiate EspFU-mediated actin assembly and harbors N-terminal sequences that influence pedestal length. Cell. Microbiol. (in the press).
120. Garmendia, J. et al. Distribution of TccP in clinical enterohemorrhagic and enteropathogenic Escherichia coli isolates. J. Clin. Microbiol. 43, 5715–5720 (2005).Identification of an O119:H6 EPEC strain encoding both EspFU (TccP) characteristic of EHEC O157:H7 and a Tir protein harbouring Y474.
121. Bompard, G. & Caron, E. Regulation of WASP/WAVE proteins: making a long story short. J. Cell Biol. 166, 957–962 (2004).
122. Quinlan, M. E., Heuser, J. E., Kerkhoff, E. & Mullins, R. D. Drosophila Spire is an actin nucleation factor. Nature 433, 382–388 (2005).
123. Wallar, B. J. & Alberts, A. S. The formins: active scaffolds that remodel the cytoskeleton. Trends Cell Biol. 13, 435–446 (2003).
124. Jones, N. et al. Nck adaptor proteins link nephrin to the actin cytoskeleton of kidney podocytes. Nature 8 Mar 2006 (doi:10.1038/nature04662).
125. Alto, N. M. et al. Identification of a bacterial type III effector family with G protein mimicry functions. Cell 124, 133–145 (2006).
AcknowledgementsWe thank N. Phillips, D. Tipper and E. Koronakis for discus-sions and critique of the manuscript, and M. Brady, N. Phillips, S. Snapper, R. DeVinney, T. Newsome and M. Way for discussion and communication of unpublished results. Our work is supported by a Wellcome Trust programme grant, a Medical Research Council project grant and Biotechnology and Biological Research Council Studentship to V.K., and a National Institutes of Health (NIH) grant to J.M.L. K.G.C. vis-ited the Koronakis laboratory with support from a Human Frontier Science Program international fellowship.
Competing interests statementThe authors declare no competing financial interests.
DATABASESThe following terms in this article are linked online to:Entrez Genome: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genomevaccinia virusEntrez Genome Project: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=genomeprjO157:H7 | Yersinia pseudotuberculosisUniProtKB: http://ca.expasy.org/sprot14-3-3τ | α-actinin | A36R | B5R | c-Abl | CD3ξ | CD4 | CD44 | Cdc42 | cofilin | cortactin | Csk | EspF | gelsolin | Grb2 | IcsA | intimin | invasin | Nck-1 | Nck-2 | nucleolin | N-WASP | paxillin | SLP-76 | talin | Tir | VASP | vinculin | WASP | WIP | ZAP-70
FURTHER INFORMATIONVassilis Koronakis’ homepage: http://www.path.cam.ac.uk/pages/koronakisJohn M. Leong’s homepage: http://www.umassmed.edu/gsbs/faculty/show.cfm?faculty=288Access to this interactive links box is free online.
R E V I E W S
370 | MAY 2006 | VOLUME 4 www.nature.com/reviews/micro
© 2006 Nature Publishing Group





























