Embed Size (px)
Citation preview

Exploration and Exploitation of Synthetic Use of Oxoammonium Ions in
Alcohol Oxidation
Yoshiharu Iwabuchi
Graduate School of Pharmaceutical Sciences, Tohoku University 6-3Aoba yama, Sendai 980-8578, Japan
(Received August 25, 2008; E-mail: [email protected])
Abstract: Novel synthetic uses of oxoammonium ions enabling facile and efficient oxidation of several class-
es of alcohols to their related carbonyl compounds have been exploited on the basis of two strategies: (i)
modifying the molecular framework of the oxoammonium ion, and (ii) enhancing its reactive nature by
altering the counter ion. The reaction systems developed allow: (1) efficient oxidation of sterically crowded
secondary alcohols to ketones; (2) facile, one-pot oxidation of primary alcohols to carboxylic acids; (3)
oxidative rearrangement of tertiary allylic alcohols to ƒÀ-substituted ƒ¿,ƒÀ-unsaturated carbonyl compounds
under transition-metal-free conditions.
1. Introduction
Carbonyl compounds exhibit exceptional utility in organ-
ic chemistry, and have been described as "virtually the
back-bone of organic synthesis. " The "forces" of carbonyl
compounds will be with synthetic chemists until organic syn-
thesis finishes its role.
As a practical method of obtaining carbonyl compounds, oxidation of alcohols is the most straightforward, and, there-fore, numerous reagents and methods have been developed to enrich the chemist's toolbox: the history of alcohol oxidation reflects the-state-of-art of organic synthesis.2 What is the current situation of contemporary organic synthesis? Alco-hol oxidation, particularly on a large-scale, often suffers from problems inherent to oxidations: toxic, harmful reagents are required; rather harsh conditions are required. In addi-tion, the recent global demand for attenuating environmental stresses has motivated chemists to develop ecologically sus-tainable methods.a These situations synergistically spurred research activities directed towards the development of green-er methods for alcohol oxidation.
The evolution of catalysts capable of using environmen-
tally friendly bulk oxidants is believed to lead to ideal pro-
cesses and numerous methods have been reported to date.4f
Among them, transition-metal-free catalytic oxidation sys-
tems have been attracting great attention, owing not only to
the practical merits of avoiding toxic metals, but also from
academic interest in the electron-transfer process between
organic compounds. Here, we describe our recent develop-
ment of a novel use of oxoammonium ions as organo-redox
catalysts, which offer facile and efficient oxidation of various
classes of alcohols to their corresponding carbonyl com-
pounds.
2. Scope of TEMPO-derived Oxoammonium Ion in
Alcohol Oxidation
Since the inceptions of nitroxyl radicals as useful precur-sors of the excellent oxidants, oxoammonium ions, a stable class of organo-nitroxyl radicals, as exemplified by 2,2,6,6-tetramethylpiperidyl-l-oxyl [TEMPO (1)] have seen expanding use in oxidative synthetic transformations.
Among the reported nitroxyl radicals, the commercially avail-able TEMPO and its derivatives have enjoyed a monopoly as the reagents of choice for the expansion of their further use.6 Since the TEMPO radical undergoes reversible redox inter-conversions between oxoammonium ion and hydroxyl amine
(Scheme 1), which is the origin of its activity as a redox cata-lyst, and since methods using nitroxyl radicals as a catalytic
precursor offer facile, mild, efficient, selective oxidation of alcohols, the TEMPO-catalyzed method has now become the method of choice for alcohol oxidation in bulk pharmaceuti-cal synthesis.
The widely accepted mechanism of the TEMPO-catalyzed oxidation of alcohols is shown in Scheme 2. TEMPO (1) is
oxidized to form an active species, oxoammonium ion A, in which the NO moiety reacts with alcohol to form a cova-
lent intermediate B. B collapses via oxy-Cope-like fragmen-tation to give the corresponding ketone and a hydroxyl amine
C, which is then oxidized by a bulk oxidant to regenerate a nitroxyl radical, thereby establishing a catalytic cycle.''s
1076 (40) J. Synth. Org. Chem., Jpn.

The scope of TEMPO-based alcohol oxidation may be
described from the standpoint of classification where alcohol
oxidations are divided into four categories: (i) primary alco-
hols to aldehydes, (ii) secondary alcohols to ketones, (iii) pri-
mary alcohols to carboxylic acids, (iv) tertiary allylic alcohols
to /3-substituted a,ƒÀ-ketones (Scheme 3). The TEMPO-cat-
alyzed method is widely recognized to exhibit its best perfor-
mance in the reactions of category (i). Meanwhile, a rather
limited range of success has been reported to date for appli-
cations in categories (ii) and (iii), and no precedent is known
for category (iv).
We envisioned that the reactive nature of the oxoammoni-um ion would be enhanced by (i) modifying the structural
factors hosting the catalytic site,9 and/or, (ii) modulating the reactive nature by altering the counter ion X- associated with
oxoammonium ion A10 (Scheme 2).
3. Exploitation of Novel Synthetic Use of Oxioammonium
Ions
3.1 Discovery of AZADOs: Aggressive Oxidation
Catalysts
One of the most outstanding features of TEMPO-based alcohol oxidation is its capability of conducting the selective
oxidation of primary alcohols in the presence of secondary alcohols. The rationale behind such a feature is its reac-
tion mechanism and the catalyst structure it uses, where four methyl groups flanking the nearby catalytic center play key roles in preventing bulky substrates from forming the key
intermediate B, which collapses to a carbonyl compound and the hydroxylamine C7 (Scheme 2). This scheme also explains
why TEMPO is inefficient in the oxidation of structurally
hindered secondary alcohols, posing a problem in the oxida-tion of alcohols.
We envisaged that the use of a structurally less-hindered oxoammonium ion, namely 2-azaadamantane N-oxoam-
monium ion (AZADO+; 2+), which should be obtainable
from 2-azaadamantane N-oxyl (AZADO; 2), would solve
this problem and expand the scope of the oxidation of alco-hols.
AZADO (2) was first synthesized in 1975 by Dupeyre
and Rassat as a result of their interest in its physical proper-ties as a stable radical. However, no study has been per-
formed to examine its ability as an oxidation catalyst. Thus, significant challenges remain to explore and exploit the syn-
thetic use of AZADOs. At the outset, we prepared AZADO
(2) as described in the literature 13 to clarify whether 2 gives an oxoammonium ion (2+) sufficiently robust to conduct alcohol oxidations. Unfortunately, we encountered a prob-
lem in terms of reproducibility in the crucial step for con-structing azaadamantane 6 (Scheme 4).9
After considerable experimentation, we developed a 8-step synthetic route for AZADO (2) form the diketone 5
(Scheme 5) and a 5-step synthetic route for 1-Me-AZADO
(17) starting from the ketone 4 (Scheme 6).9 In a preliminary experiment, we found that AZADO (2)
as well as 1-Me-AZADO (17) exhibited superior catalytic activities to TEMPO (1); interestingly, 2 and 17 show similar catalytic efficiencies under the standard oxidation conditions developed by Anelli et a/.6 ' (data not shown). Considering its synthetic advantage, I-Me-AZADO (17) was chosen for further investigation. As shown in Tables 1 and 2, I-Me-AZADO (17) has high catalytic efficiency under low catalytic loading conditions. 14.15
Note that 1 mol(Yo I-Me-AZADO is sufficient for obtain-ing high yield in the preparation of ketones from a variety of secondary alcohols for which TEMPO results in low yield
(Table 3, entries 1-10),9 and that I,2:4,5-di-O-isopropylidene-/3-D-fructopyranose is also efficiently oxidized on a 20 gram scale to give Shi's catalyst in 90%, yield. 15.11 Unfortunately, 1-Me-AZADO does not efficiently oxidize a substrate con-tainine an amine functionality (entry 11).3.2 SAR of AZADOs
To gain insight into the origin of the remarked catalytic efficiency of AZADO-derived oxoammonium ions, we syn-
thesized 1,3-diMe-AZADO (25)9 (Scheme 7) and examined its catalytic efficiency.
1,3-DiMe-AZADO (25) exhibited an activity for the oxi-
dation of 3-phenylpropanol comparable to those exhibited by
AZADO (2) and I-Me-AZADO (17). On the other hand, it does not efficiently oxidize 1-menthol compared with
TEMPO (1), showing a remarked difference from I-Me-AZADO and AZADO (Table 4).
Vol.66 No.11 2008 (41) 1077

Scheme 4
Table I Table 2
Cyclic voltammetric measurements revealed that AZADO
(2), 1-Me-AZADO (17), and 1,3-diMe-AZADO (25) show
well-defined redox waves, the forms of which are unchanged
after more than 100 measurement cycles, demonstrating their
high durability as oxidation catalysts.9 The E°' values of the
nitroxyl radicals are in the order of 25 (136 mV) < 17 (186
mV) < 2 (236 mV) < TEMPO (1) (294 mV).17 However, the
total efficiencies of the nitroxyl radicals as catalysts are in the
order of 1•`25 << 17•`2. These observations support the
notion that the aggressive catalytic nature of 2 and 17 is due
to kinetic factors derived from decreased steric hindrance
around the reaction center.
We believe that AZADO (2) and 1-Me-AZADO (17) will find complementary use with TEMPO-based methods in the oxidation of a variety of primary and secondary alcohols.3.3 Application of 1-Me-AZADO to One-Pot
Oxidation of Primary Accohols to Carboxylic Acids
The oxidation of primary alcohols to the corresponding carboxylic acids (category iii, Scheme 3) is also a fundamen-tal transformation in organic chemistry.2 The straightfor-ward appearance of this transformation, consisting of simple, two-step oxidations: primary alcohol to aldehyde, then alde-hyde to carboxylic acid, has encouraged many organic chemists to develop efficient one-pot oxidations. As a result, a number of useful methods have been developed to date, i.e., Cr03/H2SO4,18 PDC/DMF,19 Cr03/H5106,20 RuCl3/H5106,21 RuCl3/K2S208,22 Na2WO4/H2O2,4a and PhIO/KBr.23 Unfortu-nately, the task often suffers from several drawbacks, such as
1078 ( 42) J. Synth . Org . Chem . , Jpn.

Table 3 Table 4
limited substrate applicability, toxic and hazardous nature of
reagents, and harsh conditions required.
Figure 2. Cyclic voltammograms of various nitroxyl radicals (2 mM) at scan rate of 50 mV s-1(a; TEMPO, b; AZADO, c; 1-Methyl-AZADO, d; 1,3-dimethyl-AZADO)
Considering the nitroxyl radical/oxoammonium ion-based method, useful extensions have been reported as exemplified by TEMPO/NaClO,6c 6a TEMPO/PhI(OAc)2-H2O,24 and TEMPO/NaC10/NaC1022$ systems. However, further improvement is still needed to address problems asso-ciated with chemoselectivity: electron-rich alkenes and aro-matic rings are damaged underr the reaction conditions. If an oxoammonium ion requires an aldehyde to form the corre-sponding hydrate prior to oxidation, it was envisioned that the less-hindered 1-Me-AZADO+ (17+) would exhibit supe-rior activity to TEMPO, and, therefore, would enhance chemoselectivity by shortening the reaction time during which substrates were exposed to oxidative conditions.
Preliminary experiments employing the catalytic one-pot
Scheme 7
Vol.66 No.ll 2008 (43) 1079

Scheme 8
Table 5
oxidation conditions developed by Anelli et revealed
the general superiority of 1-Me-AZADO (17) to TEMPO
(1), although the substrate resisted forming the hydrate and
gave disappointing results (Table 5). As shown in Table 6, I-Me-AZADO (17) was found to make better use of
Ph1(OAc),/H,O than TEMPO, showing improved chemose-lectivity (entry 4). Investigations of bulk oxidants and co-catalysts that bring about more efficient conversion are
now in progress.
It is well recognized that tertiary allylic alcohols give rise to a characteristic oxidative rearrangement under certain conditions to furnish a-substituted a,/3-unsaturated ketones. Since the report in the mid-70s that PCC, PDC, and Collins reagent cause the one-pot allylic transposition-oxidation of a variety of tertiary allylic alcohols, oxochromium(Vl)-based reagents have been the reagents of choice and have played indispensable roles in organic synthesis (Scheme 9).262726 On the basis of speculation that the Cr=O motif plays a role in the rearrangement step,"' we focused our interest on the
potential use of organic oxoammonium ions (R,R,N+=O)30
3.4 Oxidative Rearrangement of Tertiary Allylic
Alcohols Employing Oxoammonium Salts
in promoting this particular oxidative transformation.
An exploratory experiment was started by screening the reactivity of readily available TEMPO-derived oxoammonium salts using I-phenylcyclohex-2-en-l-ol as the substrate (Table 7).31 It was found that TEMPO+species carrying bulky, poorly nucleophilic anions, such as BF4 la or SbF( lb, exhibit excellent reactivity to furnish 3-phenylcyclohex-2-en-l-one in 957 yield within 3 min at room temperature (entries 1, 2). On the other hand, [TEMPO+][Br3-] (lc) and [TEMPO+][CI ] (ld) are completely ineffective in the same reaction (entries 3, 4). Typical TEMPO oxidation conditions using NaOCI, Phl(OAc)2, or Oxone as the bulk oxi-dant did not afford the product, although they are com-
petent for generating oxoammonium species. Experiments for probing the range of tertiary allylic
alcohols that undergo oxidative transformation are summarized in Table 8. The aryl- and alkyl-substituted six-membered substrates were smoothly converted to the corresponding transposed products in high yields
(entries 1-3). For medium-sized to macrocyclic sub-strates, the reactions suffered from the generation of several by-products including dimeric ethers. We found that H2O addition in some cases greatly improves the
productivity to allow spot-to-spot conversion in high yields (entries 4-7). We conjecture that H,O aids the reactions in part to proceed via the SN2' pathway (vide infra). The tricyclic substrate also effectively yielded the desired product (entry 8). The steroid afforded the sec-ondary allylic alcohol in moderate yield due to consid-erable steric hindrance. Acyclic substrates also smooth-ly underwent oxidative rearrangement in high yield
(entry 10). Unfortunately, the substrate did not yield the desired product; instead an ene-like adduct was obtained in moderate yield as the major product (entry 11).
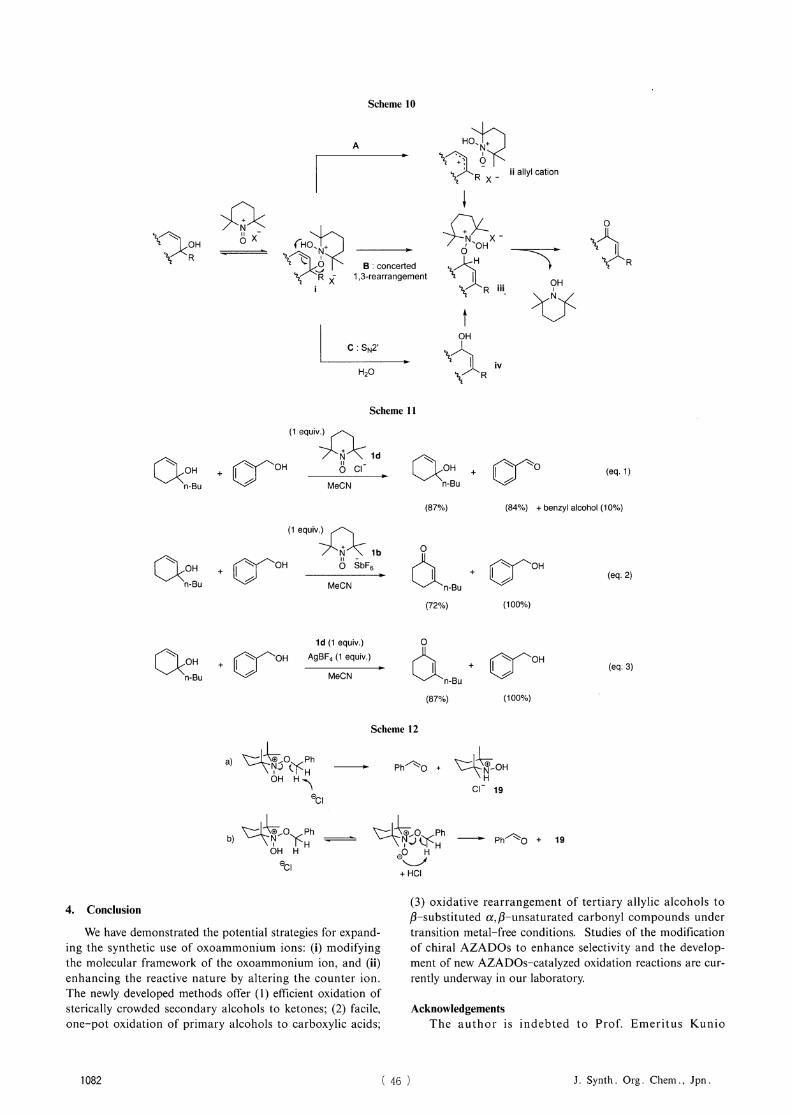
Plausible reaction pathways are depicted in Scheme 10. Considering the steric and electronic effects of the
anion, it is reasonable to expect that the oxoammonium salt carrying a bulkier counter anion, such as BF4 and
SbF6 , should be more electrophilic, owing to the enhanced electrostatic potential, to allow formation of the crucial
adduct i under equilibrium conditions. Once formed, i could facilely proceed to give a rearranged product iii either via
allylic cation formation (path A) or concerted intramolecular rearrangement (path B), which has been proposed for PCC and IBX In the case in which H,O addition plays a pro-
ductive role, we believe that mechanism C, where H,O attacks intermediate i in the SN2' mode, operates in part.
To probe the reactive nature of oxoammonium salts, we conducted intermolecular competition experiments with a tertiary allylic alcohol and benzyl alcohol (Scheme I I). On treatment with [TEMPO+][Cl ] (ld), benzaldehyde was rapid-ly produced and the tertiary allylic alcohol was recovered (eq. 1). On the other hand, treatment with [TEMPO+][SbF, I (lb) selectively converted the tertiary allylic alcohol into the corre-sponding P-substituted a,/3-unsaturated ketone (eq. 2). The treatment of the tertiary allylic alcohol with Id in the pres-ence of an equimolar amount of AgBF4 afforded almost the same result as the reaction using lb (eq. 3).32
The observed chemoselectivity may be rationalized by
1080 (44) J. Synth. Org. Chem., Jpn.

Table 6 Table 7
Table 8
Scheme 9
considering the basicity of the counteranions.33 Thus, in the case of [TEMPO+][CV] (1d), chloride anions can act as a base that abstracts either a benzylic (path a) or OH proton (path b) giving an ammonium oxide to bring about the generation of benzaldehyde and TEMPOH HCl 19. On the other hand, the less basic BF4-does not abstract a proton, thus slowing the oxidation (Scheme 12).
Studies toward the development of a catalytic version of this process are under way.
Vol.66 No.11 2008 (45) 1081
Scheme 10
Scheme 11
Scheme 12
4. Conclusion
We have demonstrated the potential strategies for expand-ing the synthetic use of oxoammonium ions: (i) modifying the molecular framework of the oxoammonium ion, and (ii) enhancing the reactive nature by altering the counter ion. The newly developed methods offer (1) efficient oxidation of sterically crowded secondary alcohols to ketones; (2) facile, one-pot oxidation of primary alcohols to carboxylic acids;
(3) oxidative rearrangement of tertiary allylic alcohols to
ƒÀ -substituted a, ƒÀ-unsaturated carbonyl compounds under
transition metal-free conditions. Studies of the modification
of chiral AZADOs to enhance selectivity and the develop-
ment of new AZADOs-catalyzed oxidation reactions are cur-
rently underway in our laboratory.
Acknowledgements•@
The author is indebted to Prof. Emeritus Kunio
1082 (46) J. Synth. Org. Chem., Jpn.

Ogasawara of Tohoku University for his encouragement and useful discussions. The author wishes to thank his co-work-ers whose names are cited in the references. This work was supported in part by a Grant-in-Aid for Scientific Research
(B) (No. 17390002) from Japan Society for the Promotion of Science, Grant-in-Aid for the Global COE Program for "International Center of Research & Educati on for Molecu-lar Complex Chemistry" from Ministry of Education, Culure, Sports, Science, and Technology, Japan, and a research grant from Uehara Memorial Fundation for the Promotion of Life Science.
References and Notes
1) Seebach, D.; Weidmann, B.; Wilder, L. In Modern Synthetic Methods 1983; Scheffold, R., Ed.; Otto Salle Verlag: Frankfurt, 1983; Vol 7, p 324.
2) (a) Schlecht, M. F. In Comprehensive Organic Synthesis; Trost, B. M.; Fleming, I.; Ley, S. V., Eds.; Pergamon: Oxford, 1991; Vol 7, pp 251-327. (b) Modern Oxidation Methods, Backvall, J.-E., Ed; Willey-VCH. Weinheim, Germany, 2004.
3) (a) Carey, J. S.; Laffan, D.; Thomson, C.; Williams, M. T. Org. Biomol. Chem. 2006, 4, 2337. (b) Caron, S.; Dugger, R. W; Rug-geri, S. G.; Ragan, J. A.; Ripin, D. F. B. Chem. Rev. 2006, 106, 2943.
4) (a) Noyori, R.; Aoki, M.; Sato, K. Chem. Commun. 2003, 1977. (b) Mallat, T.; Baiker, A. Chem. Rev. 2004, 104, 3037. (c) Uozu-mi, Y.; Nakao, R. Angew. Chem. Int. Ed. 2003, 42, 194. (d) Nishide, K.; Patra, P. K.; Matoba, M.; Shanmugasundaram, K.; Node, M. Green Chem. 2004, 6, 142. (e) Drugger, R., W; Ragan, J. A.; Ripin, D. H. B. Org. Process Res. Dev. 2005, 9, 253. (f) Matsumoto, T.; Ueno, M.; Wang, N.; Kobayashi, S. Chem. Asian J. 2008, 3, 196. (g) Piera, J.; Backvsll, J.-E. Angew. Chem. Int. Ed. 2008, 47, 3506.
5) (a) Golubev, V. A.; Rozantsev, E. G.; Neiman, M. B. Izv. Akad. Nauk USSR, Ser. Khim. 1965, 1898. (b) Golubev, V. A.; Rozant-sev, E. G.: Neiman, M. B. Bull. Acad. Sci. USSR, Div. Chem. Sci. 1965, 1927.
6) (a) Cella, J. A.; Kelly, J. A.; Kenehan, E. F. J. Org. Chem. 1975, 40, 1860. (b) Semmelhack, M. E; Schmid, C. R.; Cortes, D. A.; Chou, C. S. J. Am. Chem. Soc. 1984, 106, 3374. (c) Anelli, P. L.; Banfi, C.; Montanari, F.; Quici, S. J. Org. Chem. 1987, 52, 2559. (d) Anelli, P. L.; Banfi, S.; Montanari, F.; Quici. J. Org. Chem. 1989, 54, 2970. (e) De Mico, A.; Margarita, R.; Parlanti, L.; Vescovi, A.; Piancatelli, G. J. Org. Chem. 1997, 62, 6974. (f) Biom, C.; Magus, A. S.; Hildebrand, J. P. Org. Lett. 2000, 2, 1173. (g) Bjorsvik, H.; Liguori, L.; Costantino, F.; Minisci, F. Org. Process Res. Dev. 2002, 6, 197. (h) Miller, R. A.; Hoerrner, R. S. Org. Lett. 2003, 5, 285. (i) Liu, R.; Liang, X.; Dong, C.; Hu, X. J. Am. Chem. Soc. 2004, 126, 4112.
7) (a) de Nooy, A. E.; Besemer, A. C.; van Bekkum, H. Synthesis 1996, 1153. (b) Adam, W; Saha-Moller, C. R.; Ganeshpure, P. A. Chem. Rev. 2001, 101, 3499. (c) Sheldon, R. A.; Arends, I. W. C. E. Adv. Synth. Catal. 2004, 346, 1051. (d) Sheldon, R. A.; Andres, I. W. C. E. J. Mol. Cat. A: Chem. 2006, 251, 200.
8) (a) Siedlecka, R.; Skarzewski, J.; Michowski, J. Tetrahedron Lett. 1990, 31, 2177. (b) de Nooy, A. E. J.; Besemer, A. C.; van Bekkum, H. Tetrahedron 1995, 51, 8023-8032.
9) Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi. Y. J. Am. Chem. Soc. 2006, 128, 8412.
10) Shibuya, M.; Tomizawa, M.; Iwabuchi, Y. J. Org. Chem. 2008, 73, 4750.
11) (a) Mandal, M.; Yun, H.; Dudley, G. B.; Lin, S.; Tan, D. S.; Danishefsky, S. J. J. Org. Chem. 2005, 70, 10619. (b) Paterson, I.; Delgado, 0.; Florence, G. J.; Lyothier, I.; Scott, J. P.; Sere-inig, N. Org. Lett. 2003, 5, 35.
12) Dupeyre, R. M.; Rassat, A. Tetrahedron 1978, 34, 1901.13) (a) Stetter, H.; Tacke, P.; Garner, J. J. Chem. Ber.1964, 97, 3480.
(b) Henkel, J. G.; Faith, W. C. J. Org. hem.1981, 46, 4953.14) Representative procedure for oxidation of alcohols under Anelli's con-
ditions.6C To a stirring mixture of 3-phenyl propanol (200 mg, 1.47 mmol), 1-Me-AZADO (3) (0.244 mg, 1.47 umol) in CH2C12 (3.9 ml) and aqueous sat. NaHCO3 (2 ml) containing KBr (17.5 mg, 0.074 M) and Bu4NBr (23.7 mg, 0.037 M) was added dropwise a premixed solution of aqueous NaOCI (8%
Cl) and aqueous sat. NaHCO3 (3.3 ml, 1:1.4 v/v) at 0 •Ž over 6
min. The mixture was vigorously stirred for 20 min at 0 •Ž,
then quenched with aqueous sat. Na2S203 (4 ml). The aqueous
layer was separated and extracted with Et20. The combined
organic layers were washed with brine, dried over MgSO4, and
concentrated. The residue was purified by flash column chro-
matography (SiO2, 1:6 Et20:hexane) to give 3-phenylpropanal
(177 mg, 1.32 mmol, 90%) as a colorless oil.
15) Representative procedure for oxidation of alcohols under Margarita's
conditions. PhI(OAc)2 (720 mg, 2.24 mmol) was added to a
solution of cinnamyl alcohol (200 mg, 1.49 mmol) and
1-Me-AZADO (3) (2.47 mg, 14.9 ƒÊmol) in CH2C12 (1.5 ml).
The reaction mixture was stirred for 40 min; it was then diluted
with Et20 and quenched with aqueous sat. NaHCO3 (4 ml),
followed by aqueous sat. Na2S203 (4 ml). The layers were sep-
arated and the aqueous layer was extracted with Et20. The
combined organic layers were washed with brine, dried over
MgSO4, and concentrated. The residue was purified by flash
column chromatography (SiO2, 1:9 Et20:hexane) to give cin-
namaldehyde (183 mg, 1.39 mmol, 93%) as a colorless oil.
16) (a) Gonsalvi, L.; Arends, I. W. C. E.; Sheldon, R. A. Org. Lett. 2002, 4, 1659. (b) Tu, Y.; Frohn, M.; Wang, Z.; Shi, Y. Org. Synth. 2003, 80, L (c) Wang. Z.; Shu, L.; Frohn, M.; Tu, Y.;Shi, Y. Org. Synth. 2003, 80, 9.
17) (a) Bobbitt, J M.; Flores, M. C. Heterocycles 1988, 27, 509. (b) Rychnovsky, S. D.; Vaidyanathan, R.; Beauchamp, T.; Lin, R.; Farmer, P. J. J. Org. Chem. 1999, 64, 6745-6749.
18) Bowden, K.; Heibron, I. M.; Jones, E. R. H.; Weedon, B. C. L. J. Chem. Soc. 1946, 39.
19) Corey, E. J.; Schmidt, G. Tetrahedron Lett.1979, 5, 399.20) Zhao, M.; Song, J.; Li, Z.; Desmond, R.; Tschaen, D. M.;
Grabowski, E. J J; Reider, P. J. Tetrahedron Lett.1998, 39, 5323.21) Carlsen, P. H. J.; Katsuki, T.; Martin, V. S.; Sharpless, K. B. J.
Org. Chem. 1981, 46, 3936.22) Schroder, M.; Griffith, W. P. J. Chem. Soc. Chem. Common.
1979, 58.23) (a) Tohma, H.; Takizawa, S.; Maegawa, T.; Kita, Y. Angew.
Chem. Int. Ed. 2000, 39, 1306. (b) Tohma, H.; Maegawa, T.; Takizawa, S.; Kita, Y. Adv. Synth. Catal. 2002, 344, 328.
24) Epp, J. B.; Widlanski, T. S. J. Org. Chem. 1999, 64, 293.25) (a) Zhao, M.; Mano, J. Li, E.; Song, Z.; Tschaen, D. M.;
Grabowski, E. J. J.; Reider, P. J. J. Org. Chem. 1999, 64, 2564. (b) Zhao, M.; Li, J.; Mano, E.; Song, Z. J.; Tschaen, D. M. Org. Synth. 2004, 81, 195. (c) Zanka, A. Chem. Pharm. Bull. 2003, 51, 888.
26) (a) Babler, J. H.; Coghlan, M. J. Synth. Commun. 1976, 6, 469. (b) Dauben, W. G.; Michno, D. M. J. Org. Chem. 1977, 42, 682. (c) Sundararaman, P.; Herz, W. J. Org. Chem. 1977, 42.813.
27) For a recent review on an oxochromium(VI)-based oxidant, see: (a) Luzzio, F. A. Org. React. 1998, 53, 1. (b) Wietzerbin, K.; Bernadou, J.; Meunier, B. Eur. J. Inorg. Chem. 2000, 1391.
28) (a) Bohno, M.; Imase, H.; Chida, N. Chem. Commun. 2004, 1086. (b) Dominguez, M.; Alvarez, R.; Martras, S.; Farres, J.; Pares, X.; de Lera, A. R. Org. Biomol. Chem. 2004, 2, 3368. (c) Mandal, M.; Yun, H.; Dudley, G. B.; Lin, S.; Tan, D. S.; Dan-ishefsky, S. J. J. Org. Chem. 2005, 70, 10619. (d) Fernandez-Mateos, A.; Silvo, A. I. R.; Gonzalez, R. R.; Sim-monds, M. S. J. Tetrahedron 2006, 62, 7809. (e) Li, C.-C.; Wang, C.-H.; Liang, B.; Zang, X.-H.; Deng, L.-J.; Liang, S.; Chen, J.-H.; Wu, Y.-D.; Yang, Z. J. Org. Chem. 2006, 71, 6892. (f) Chavan, S. P.; Thakkar, M.; Jogdand, G. E; Kalkote, U. R. J. Org. Chem. 2006, 71, 8986. (g) Shimizu, N.; Mizaguehi, A.; Murakami, K.; Noge, K.; Mori, N.; Nishida, R. Kuwahara, Y. J. Pestic. Sci., 2006, 31, 311. (h) Tanimoto, H.; Kato, T.; Chida, N. Tetrahedron Lett. 2007, 48, 6267.
29) Shibuya, M.; Ito, S.; Takahashi, M. Iwabuchi, Y. Org. Lett. 2004, 6, 4303.
30) (a) Kagiya, T.; Kumuro, C.; Sakano, K.; Nishimoto, S. Chem. Lett. 1983, 365. (b) Miyazawa, T.; Endo, T.; Shiihashi, S.; Okawara, M. J. Org. Chem. 1985, 50, 1332. (c) Bobbitt, J. M.; Flores, M. C. L. Heterocycles 1988, 27, 509. (d) Liu, Y.-C.; Liu, Z.-L.; Wu, L.-M.; Chen, P. Tetrahedron Lett. 1985. 26, 4201. (e) Miyazawa, T.; Endo, T. J. Org. Chem. 1985, 50, 3930. (f) Bob-bitt, J. M.; Guttermuth, M. C. F.; Ma, Z.; Tang, H. Heterocy-cles 1990, 30, 1131. (g) Ma, Z.; Bobbitt. J. M. J. Org. Chem. 1991, 56, 6110. (h) Ren, T.; Liu, Y.-C.; Guo, Q.-X. Bull. Chem. Soc. Jpn. 1996, 69, 2935. (i) Bobbitt, J. M. J. Org. Chem. 1998, 63, 9367. (j) Takata, T.; Tsujino, Y.; Nakanishi, S.; Nakamura,

K.; Yoshida, E.; Endo, T. Chem. Lets. 1999, 937. (k) Kernag, C. A.; Bobbitt, J. M.; McGrath, D. V. Tetrahedron Lett. 1999, 40, 1635. (1) Merbouh, N.; Bobbitt, J. M.; Bruckner, C. J. Org. Chem. 2004, 69, 5116. (m) Zakrzewski, J.; Grodner, J.; Bobbitt, J. M.; Karpiriska, M. Synthesis 2007, 2491.
31) (a) Golubev, V. A.; Rozantsev, E. G.; Neiman, M. B. Bull. Acad. Sri. U. S. S. R., Chem. Sci. 1965, 11, 1898. (b) Zhdanov, R. I.; Golubev, V. A.; Rozantsev, E. G. Bull. Acad. Sci. U.S.S.R.,
Chain. Set. 1970, 19, 186. (c) Golubev, V. A.; Zhdanov, R. I.; Rozantsev, E. G. Bull. Acad. Sci. U.S.S.R., Chem. Ser. 1970, 19, 188.
32) Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395.33) (a) Olah, G. A.; Prakash, G. K. S. Superacids; John Wiley and
Sons: New York, 1973. (b) Olah, G. A. J. Org. Chem. 2005, 70, 2413.
34) Bailey, W. F.; Bobbitt, J. M.; Wiberg, K. B. J. Org. Chem. 2007, 72, 4504.
35) If substrate possesses sufficiently acidic protons (cf. allylic methine proton in cyclohexenol), the proton would be abstract-
ed by the solvent to afford oxidized products.
PROFILE
Yoshiharu Iwabuchi is a Professor of the Graduate School of Pharmaceutical Sci-ences, Tohoku University. He received his Ph.D. from Tohoku University in 1991 under the direction of Prof. S. Takano. After a year of postdoctoral study in Professor K. C. Nicolaou's group at Scripps, he joined the Protein Engineering Research Institute in Osaka as a researcher. He moved to Nagasaki University as Associate Professor in 1997. Since 2002, he has been Professor at Tohoku University. He received 2006 Award for Excellence of The Japanese Society for Process Chemistry. His research interests include development of synthetic methodology, asymmetric synthesis, and natural product synthesis.
1084 (48) J. Synth. Org. Chem., Jpn.





























