Embed Size (px)
Citation preview

Biotechnology andApplied BiochemistryExpression of biologically active
human insulin-like growth factor 1 inArabidopsis thaliana seeds via oleosinfusion technology
Wei Li,1 Linguo Li,1 Kunlan Li,1 Juan Lin,1 Xiaofen Sun,1 and Kexuan Tang1,2∗
1State Key Laboratory of Genetic Engineering, School of Life Sciences, Morgan-Tan International Center for Life Sciences,Fudan–SJTU–Nottingham Plant Biotechnology R&D Center, Fudan University, Shanghai, People’s Republic of China2Plant Biotechnology Research Center, Shanghai Key Laboratory of Agrobiotechnology, School of Agriculture and Biology,Fudan–SJTU–Nottingham Plant Biotechnology R&D Center, Shanghai Jiao Tong University, Shanghai, People’s Republic of China
Abstract.Novel protein expression in plant-based systems has becomean important tool in producing and studying therapeuticproteins. Among many plant-based systems developed so far,oleosin fusion technology is one of the most cost-effective andconvenient methods. In this study, an important therapeuticprotein, human insulin-like growth factor 1 (hIGF-1), wasexpressed in Arabidopsis thaliana seeds via this technology.The plant bias codon usage-optimized hIGF-1 gene was fusedto the C-terminal of A. thaliana 18.5 kDa oleosin gene, and the
fusion gene driven by an oleosin promoter was transferred intoA. thaliana ecotype Col-0. The accumulation of oleosin–hIGF-1fusion protein in transgenic seeds was up to 0.75% of totalseed protein (TSP) and the expression level of hIGF-1 was0.17% of the TSP, which was eight times higher than previouslyreported using other plant-based hIGF-1 production systems.The biological activity of the hIGF-1 as an oleosin–hIGF-1 fusionprotein in vitro was demonstrated by using human SH-SY5Yneuroblastoma cells.
C© 2011 International Union of Biochemistry and Molecular Biology, Inc.Volume 58, Number 3, May/June 2011, Pages 139–146 •E-mail: [email protected]
Keywords: Arabidopsis thaliana, human insulin-like growth factor 1,oil body, oleosin, recombinant protein expression
1. IntroductionInsulin-like growth factor 1 (IGF-1), known as “somatomedin C”or “sulfation factor,” is a single polypeptide protein hormoneconsisting of 70 amino acids and having a molecular weightof 7,649 Da. It has three intermolecular disulfide bridgesand the molecular structure is similar to insulin [1]. IGF-1plays a major role in cell growth and differentiation [2]: it isinvolved in various physiological processes in mammals withthe regulation of somatic growth and cellular proliferationboth in vivo and in vitro [3],[4]. Because of its structuralsimilarity to insulin, IGF-1 has a high potential as a therapeuticagent for a variety of indications, including growth failure,type 1 or type 2 diabetes, amyotrophic lateral sclerosis,severe burn injury, and myotonic muscular dystrophy [5–7].
Abbreviations: PCR, polymerase chain reaction; dNTP, deoxyribonucleotidetriphosphate; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis;hIGF-1, human insulin-like growth factor 1; HmB, hygromycin; TEVP, tobacco etch virusprotease; CTAB, cetyltrimethylammonium bromide; TSP, total seed protein; DTT,dl-dithiothreitol; BSA, bovine serum albumin; RT-PCR, reverse transcriptase-PCR; CTAB,cetyltrimethylammonium bromide; TBS, Tris buffered saline; EDTA,ethylenediaminetetraacetic acid.∗Address for correspondence: Dr. Kexuan Tang, State Key Laboratory of GeneticEngineering, School of Life Sciences, Morgan-Tan International Center for Life Sciences,Fudan–SJTU–Nottingham Plant Biotechnology R&D Center, Fudan University, Shanghai200433, People’s Republic of China. Tel: 86-21-65642772; Fax: 86-21-65643552; e-mail:[email protected] 7 December 2010; accepted 7 April 2011DOI: 10.1002/bab.30Published online 4 June 2011 in Wiley Online Library(wileyonlinelibrary.com)
In 2005, two recombinant IGF-1 drugs, IncrelexTM(Tercica,Brisbane, CA, USA) (http://www.tercica.com/index.php)and IplexTM (INSMED, Monmouth Junction, NJ, USA)(http://www.insmed.com/iplex.php), were approved bythe US Food and Drug Administration (FDA) for long-termtherapy for children with growth failure with severe primaryIGF-1 deficiency (primary IGFD).
Recombinant IGF-1 has been expressed in several differentorganisms, including Escherichia coli [8–13], yeast [14], mam-malian cells [15], and transgenic rabbits [16]. The prokaryoteE. coli is the primary choice for recombinant protein expressionbecause of its facility in handling and cultivation, but directexpression of IGF-1 in E. coli may result in the formation of inclu-sion body [17],[18] or disulfide-linked aggregates [19]. A yeastexpression system has the ability of glycosylation, but the gly-cosylation is not always correct [14],[19]. A tedious in vitro pro-cess should be performed to recover its biologically active form[20],[21] or extra purification steps are needed to remove the in-correct disulfide structure. Mammalian cell culture or transgenicanimals are costly and time consuming. Recently, a transgenicplant has been widely used as an alternative for recombinantprotein expression. Numerous proteins have been successfullyexpressed in various plant species (see reviews in [22–25]). Useof transgenic plants as a vehicle for protein expression has manybenefits considering its cost-efficiency, scaling-up capacity,high-product quality, and low-contamination risks (see reviewsin [26–29]). Transgenic plants producing recombinant human
139

insulin-like growth factor-1 (hIGF-1) were recently obtained byPanahi et al. [30]; the plant-derived recombinant hIGF-1 causeddifferentiation of human neuroblastoma cell line SH-SY5Y, indi-cating its biological activity. However, the expression level wasrelatively low.
Among those strategies that have been developed forplant-derived protein expression, the oleosin fusion technol-ogy, which uses oleosins as a vehicle for recombinant proteinproduction, is one of the most attractive methods for its high-expression level, effective protein storage ability, and facile pu-rification procedure [29],[31]. Oleosins are structural proteinsfound in oil bodies, which are a type of discrete storage or-ganelle consisting of a hydrophobic triacylglycerol core, a half-unit phospholipid layer, and oil-body-related proteins such asoleosin. According to the oleosin fusion technology strategy,heterogeneous proteins are covalently attached to the N- orC-termini of the oleosin, can accumulate to high levels when ex-pressed in transgenic oil seeds, and are targeted to the oil-bodyorganelle during seed development, which can then be isolatedfrom other seed components via liquid–liquid phase separa-tion. Following the isolation process, the target protein can bereleased from oil bodies by using a site-specific protease. Thisprocess reduces the cost of purification significantly for it cutsdown the number of chromatography steps required during thepurification [31].
In this study, recombinant hIGF-1 was highly accumu-lated in Arabidopsis seeds via oleosin fusion technology. Plant-derived recombinant hIGF-1 accumulates to 0.17% of total seedprotein (TSP) in transgenic seeds. The activity of the expressedhIGF-1 was also investigated.
2. Materials and methods2.1. MaterialsAll primer synthesis and sequencing were performed bySangon Biotech (Shanghai, People’s Republic of China) Com-pany. All restriction enzymes were purchased from New Eng-land Biolabs Company (Ipswich, MA, USA). Polymerase chainreaction (PCR) reagents were purchased from TaKaRa Com-pany (Kyoto, Japan). Antibodies were purchased from SantaCruz Biotechnology (Santa Cruz, CA, USA). Plasmid purificationkits, gel extraction kits, DNA and protein molecular weight mark-ers, and reagents such as IPTG, TEMED, ammonium persulfate,sodium dodecyl sulfate (SDS), fanamycin, and ampicillin werepurchased from Sangon Biotech Company.
2.2. Design and synthesis of the hIGF-1 geneThe cDNA sequence was deduced from the amino sequenceof mature hIGF-1 [1] according to the plant codon usage fre-quency table (Codon Usage Database [32], http://www.kazusa.or.jp/codon/); an additional sequence fragment encoding atobacco etch virus protease (TEVP) cleavage site (ENLYFQ|G,cleavage between Q and G) [33] was put at the 5′ end ofthe hIGF-1 gene. To facilitate the vector construction, two re-striction enzyme sites BglII (agatct) and PstI (ctgcag) wereplaced at the 5′ and 3′ end, respectively. The whole syntheticsequence was generated using the Synthetic Gene Designer[34] (http://www.evolvingcode.net/codon/sdg.php) with man-
ual optimization. Five overlapping primers (two outer and threeinner primers) were generated by the same software and wereoptimized manually, as shown in Table 1. The hIGF-1 gene wasthen synthesized via PCR-based gene synthesis [35] using thefollowing procedure.
In a 200 μL thin-wall PCR tube, the 50 μL reaction mix-ture contained 2 μL of each PCR primer (concentration of primerwork solution: 10 μmol/L for outer primers hIGF-1A and hIGF-1E, 1 μmol/L for inner primers), 2 μL of 10 mmol/L deoxyri-bonucleotide triphosphates (dNTPs), 5 μL of 10× KOD (KODDNA polymerase) buffer [1.2 mol/L Tris–HCl, 100 mM KCl, 60mM (NH4)2SO4, 1% Triton X-100, 0.01% bovine serum albumin(BSA), pH8.0], 3 μL MgSO4 (25 mM), and 1 unit of KOD plus DNApolymerase (Toyobo, Osaka, Japan).
Polymerase chain reaction was carried out on a ThermoHybaid Cycler using the following protocol: 94◦C for 2 Min fol-lowed by 10 cycles of amplification (94◦C for 15 Sec, 61◦C rampto 56◦C for 15 Sec, and 68◦C for 25 Sec) and then 25 morecycles (89◦C for 15 Sec, 56◦C for 15 Sec, 68◦C for 25 Sec). Af-ter the final cycle, 1 μL of rTaq DNA polymerase (TaKaRa) wasadded and the PCR tube was incubated for an extra 10 Min at72◦C to extend an “A” at the 3′ ends. The PCR products wereelectrophoresed on a 1.2% agarose gel; a single clear band atabout 250 bp was incised and purified using the EZ Spin ColumnDNA Gel Extraction Kit (Sangon Biotech, Shanghai, People’s Re-public of China). The fragment was then cloned into pMD 18-TVector (TaKaRa) followed by sequencing using M13 forward andreverse primers. Eight independent clones were sequenced andthree of them had the same sequence as designed. The cor-rect clone was selected for the following steps. This vector wasdesignated pMD-18-T–hIGF-1.
2.3. Cloning of the A. thaliana 18.5 kDa oleosin geneand oleosin promoterTotal RNA was extracted from mature green-stage seeds of A.thaliana seedlings grown at 22◦C in a controlled-growth cham-ber (16 H light/8 H dark) using a RNAprep pure Plant Kit with on-column DNase digestion (Tiangen, Beijing, People’s Republic ofChina) according to the manufacturer’s instructions. GenomicDNA was isolated from rosette leaves of A. thaliana using acetyltrimethylammonium bromide (CTAB) method following theprotocol of Porebski et al. [36]. The quality and concentration ofRNA and genomic DNA were all measured by agarose gel electro-phoresis and spectrophotometry analysis before later steps.
A pair of primers AtOleF1 and AtOleR1 (see Table 1) wasdesigned according to the published sequence of A. thaliana18.5 kDa oleosin gene (generally named oleo1, Genbank accessNo. X62353). The full-length cDNA was transcripted and am-plified by the One Step reverse transcriptase PCR (RT-PCR) Kit(TaKaRa) according to instruction manual using A. thaliana totalRNA as a template.
Another two primers AtOPF1 and AtOPR1 (see Table 1) wereused to amplify the oleosin promoter from A. thaliana genomicDNA by standard PCR using KOD plus DNA polymerase. PCR wasperformed using the following protocol: 94◦C for 4 Min followedby 32 cycles of amplification (94◦C for 40 Sec, 58◦C for 40 Sec,and 68◦C for 70 Sec).
140 Biotechnology and Applied Biochemistry

Table 1Primers used in the article
Name Sequence (5′->3′) NotehIGF1A 5′-agatctgaaaacctttacttccagggacctgagaccctctgtggagca-3′ Underlined is the BglII sitehIGF1B 5′-gaccctctgtggagcagaacttgttgatgctctccaattcgtgtgtggagacagaggtttctacttcaacaagccaactg-3′
hIGF1C 5′-tccggtttgaggtgctctcctagatgaagatccgtatccagttggcttgttgaagtag-3′
hIGF1D 5′-gagcgcagtacatctcaagtctcctaagatcgcatgatctgaaacagcactcatcaacgattccggtttgaggtgctc-3′
hIGF1E 5′-ggtaacctgcagttaagcagacttagctggcttaagaggagcgcagtacatctcaag-3′ Underlined is the PstI siteAtOleF1 5′-ccatggcggatacagctagag-3′ Underlined is the NcoI siteAtOleR1 5′-agatctttaagtagtgtgctggcca-3′ Underlined is the BglII siteAtOPF1 5′-gaattccctcggtcttggtcac-3′ Underlined is the EcoRI siteAtOPR1 5′-ggatccttttttgttcttgtttacta-3′ Underlined is the BamHI sitefusionF2 5′-aagttggacagtgcaagga-3′
fusionR2 5′-cgtatccagttggcttgtt-3′
All the PCR amplifications were extended for 8 Min at72◦C after the final cycle with the addition of 1 μL of rTaq DNApolymerase to add an “A” at the 3′ ends. The PCR productswere analyzed on 1% agarose gel; bands at the correspondingsize were purified using the EZ Spin Column DNA Gel Extrac-tion Kit and subsequently cloned into pMD18-T vector followedby sequencing. The plasmids were designated pMD18-T–AtOle(containing the 530 bp oleosin gene fragment) and pMD18-T–AtOP (containing the 960 bp oleosin gene promoter fragment),respectively.
2.4. Construction of pHB–hIGF-1 plant expressionvectorThe plant transformation binary vector pHB [37] containing ahygromycin (HmB)-resistant gene and phosphinothricin (PPT)-resistant gene (BAR) inside the T-DNA for the selection of trans-formants and the plasmid pBS containing the 6× myc tag (thepeptide sequence of one myc-tag unit is as follows: EQKLISEEDL)coding sequence were supplied by Prof. H. Yang (Shanghai JiaoTong University, Shanghai, People’s Republic of China). Whenthe previously mentioned vector pMD18-T–AtOP was cut withEcoRI and BamHI, the oleosin promoter was released. This frag-
ment was cloned into the pHB binary vector that was predigestedwith EcoRI and BamHI to replace the double 35S promoter, gen-erating the plasmid pHB–AtOP.
The pMD18-T–hIGF-1 plasmid was cut with BglII and PstI and the fragment of interest was subcloned into the multi-ple cloning site of a modified pCAMBIA1302 plasmid (Cambia,Australia). The resulting plasmid was designated p1302–hIGF-1. Then the plasmid p1302–hIGF-1 was cut with NcoI and BglIIto replace the GFP coding sequence with the A. thaliana 18.5kDa oleosin gene coding sequence from the plasmid pMD18-T–AtOle cut with the same enzyme, generating the plasmid p1302–AtOle–hIGF-1, in which the hIGF-1 gene was fused to the 3′ of theoleosin gene. The fused gene released by NcoI and PstI was sub-cloned into plasmid pBS to obtain the myc–oleosin–hIGF-1 fusegene, in which a myc tag coding sequence was fused to the 5′ ofthe oleosin gene. After confirmation by sequencing, the fusedgene myc–oleosin–hIGF-1 was excised with BamHI and PstI andwas subcloned into the multiple cloning site of the constructpHB–AtOP mentioned above to get the final construction. Thisfinal construct containing the A. thaliana oleosin promoter, myctag sequence, A. thaliana 18.5 kDa oleosin, the plant codon-optimized hIGF-1 gene coding sequence, and the rbsS PolyAsequence was designated as pHB–AtOP–AtOle–hIGF-1 (Fig. 1).
Fig. 1. Construction of plant expression vector pHB–Ole–hIGF-1. The myc–oleosin–hIGF-1 gene consists of a sequence of 18.5kDa Arabidopsis oleosin gene (ole1) cDNA; a myc tag was put on the 5′ end of the oleosin gene, and a sequence-encoding TEVprotease recognizing the site followed by the hIGF-1 gene with plant codon usage was fused to the 3′ end of the oleosin gene.The predicted protein expressed is 34.3 kDa; when the myc–oleosin–hIGF-1 is treated with TEV protease in vitro, the 7.6 kDahIGF-1 is released.
Expression of human insulin-like growth factor 1 in A. thaliana 141

2.5. Arabidopsis growth and transformationSeeds of A. thaliana, ecotype Columbia (Col-0) were surfacesterilized with 70% ethanol and then sown onto 1% (w/v) agarin half strength Murashige–Skoog (MS) medium (Sigma-Aldrich,St. Louis, MO, USA) with 15% (w/v) sucrose. The seeds were in-cubated at 4◦C for 3 days to break seed dormancy (stratification)and then were germinated at 22◦C under a 16 H light/8 H darkcycle at a fluence rate of 100 μmol/s/m2 of white light producedby cool-white fluorescent lamps. Seven days after germination,the seedlings were transferred onto soil for subsequent growthin the same growth conditions.
Vector pHB–AtOP–AtOle–hIGF-1 with the myc–oleosin–hIGF-1 fused gene or the vector pHB alone was introducedinto Agrobacterium tumefaciens GV3101 (pMP90) [38] usingthe freeze–thaw method [39]. Transformation of Arabidopsis(Col-0) was performed as previously described [40]. Seeds werescreened on 15 cm MS [41] basal plates supplemented with50 μg/mL HmB. Then, 30–50 independent HmB-resistant lineswere transferred to soil and screened on 1% PPT. The resis-tant lines were further confirmed by PCR using genomic DNAas template and oleosin–hIGF-1 fusion gene primers for ampli-fication. Homozygous lines were screened by PCR. Seeds fromthree independent homozygous lines (T3) were used for proteinexpression analysis.
2.6. Confirmation of transgene in recombinant T3Arabidopsis linesTotal genomic DNA was isolated from leaves of three engineeredT3 plants (independent line 7–1, 8–2, and 11–1), wild type (de-noted as WT lines), and the control lines transformed using apHB vector only (denoted as CK lines) using a CTAB method asdescribed previously. The integration of the myc–oleosin–hIGF-1 gene on the chromosome was detected by PCR using a pairof primers to amplify a 250 bp fragment in the oleosin–hIGF-1gene (Primer fusionF2/fusionR2, sequence as shown in Table 1;fusionF2 and fusion R2 are located in the coding sequence areaof oleosin and hIGF-1, respectively). In a 200 μL thin-wall PCRtube, the 50 μL reaction mixture contained 1 μL of each PCRprimer (10 μmol/L), 1 μL of 2.5 mmol/L dNTPs, 5 μL 10× PCRbuffer (Mg2 + free), 3 μL 1.5 mM MgCl2, and 0.5 units of rTaqDNA polymerase (TaKaRa) with 200 ng genomic DNA as tem-plate. PCR was carried out on a Thermo Hybaid cycler usingthe following protocol: 94◦C for 2 Min followed by 33 cycles ofamplification (94◦C for 40 Sec, 58◦C for 40 Sec, and 72◦C for40 Sec). The amplified samples were analyzed on 1% agarosegel.
2.7. Analysis of the expression of hIGF-1 andpurification of seed oil bodiesTotal seed protein extraction and expression level measure-ment of hIGF-1 were performed as described previously withminor modifications [42]: 5 mg wild-type (Col-0) or transgenicA. thaliana mature seeds were ground in a centrifuge tube witha pestle in 200 μL of 50 mM Tris–HCl, pH 8.0. SDS (10%; 50 μL)was then added to the slurry and mixed by vortex briefly. Afterboiled for 10 Min followed by centrifugation, supernatant was
taken for protein determination and electrophoresis. The totalprotein content was determined by the bicinchoninic acid (BCA)protein assay [43] using BCA Protein Assay Kit (Pierce, Rockford,IL, USA, http://www.piercenet.com/) according to the manu-facturer’s instrument. For negative controls, total proteins fromwild-type Col-0 Arabidopsis seeds were prepared in the samemanner. Samples containing equal protein content (20 μg) werethen mixed with 1/5 volume of 6× SDS sample buffer [0.35 MTris–HCl, pH 6.8, 30% glycerol, 4% SDS, 0.02% bromophenolblue, and 300 mM dl-dithiothreitol (DTT)], boiled for 4 Min,and subjected to discontinuous 15% sodium dodecyl sulfatepolyacrylamide gel electrophoresis (SDS-PAGE) gels [44]. Gelswere subsequently Coomassie stained or transferred to 0.45 μmpolyvinylidene difluoride (PVDF) membranes (Millipore, Biller-ica, MA, USA, http://www.millipore.com/) for Western blottinganalysis, respectively.
For Western blotting, the membranes were washed twicein Tris buffered saline (TBS) buffer [20 mM Tris–HCl, pH 7.5,150 mM NaCl, 0.05% (v/v) Tween-20] and then incubated inblocking buffer (3% BSA in TBS buffer) for 1 H at 25◦C, fol-lowed by incubation with antibodies for 1 H combined withwashing twice in TBST [50 mM Tris–HCl, pH 7.5, 500 mMNaCl, 0.1% (v/v) Tween-20, 0.1% (v/v) Triton X-100] and oncein TBS. For hIGF-1 conformation, rabbit anti-hIGF-1 polyclonalantibody (dilution, 1:1000) was used as the primary antibodyand alkaline phosphatase-conjugated goat anti-rabbit IgG (di-lution, 1:2000) as the secondary antibody. Although for myctag detection, mouse anti-myc monoclone antibody (dilution,1:2000) and alkaline phosphatase-conjugated goat anti-mouseIgG (dilution, 1:2000) were used as primary and secondaryantibodies, respectively. All antibodies were purchased fromSanta Cruz Biotechnology (http://www.scbt.com/). Antibodydetection was performed using Western Blue R© stabilized sub-strate for alkaline phosphatase (Promega, Madison, WI, USA;http://www.promega.com/).
Preparation of seed oil bodies was performed accordingto Nykiforuk et al. [42]; transgenic and wild-type seeds (100 mg)were grounded in 1,000 μL oil-body extraction buffer I (0.6 Msucrose, 0.5 M NaCl, 50 mM Tris–HCl, pH 8.0). Samples werecentrifuging at 10,000g for 10 Min. The fat pad fraction was col-lected to a clean microfuge tube and resuspended in 500 μLof buffer II (0.5 M NaCl, 20 mM Tris–HCl, pH 8.0), followed bycentrifuging again at 10,000g for 10 Min. The procedure was re-peated three times, and an additional two washes with buffer IIIwere done (20 mM Tris–HCl, pH 8.0). The final fat pad wasresuspended in 200 μL of buffer III. A 5-μL aliquot (10 μg)was taken for protein measurement, SDS-PAGE analysis, andWestern blotting as mentioned above.
2.8. TEVP cleavageThe oil bodies containing the myc–oleosin–hIGF-1 protein wereprepared and the protein content was determined as describedabove. TEVP was a generous gift from Prof. Yu Ding (FudanUniversity, People’s Republic of China). For TEVP digestion [33],in a 1.5 mL tube, the 750 μL reaction mixture contained 75μL 10× protease buffer (5 mM ethylenediaminetetraacetic acid[EDTA], 0.5 M Tris–HCl, pH 8.0), 200 μL oil bodies, 7.5 μL TEVP
142 Biotechnology and Applied Biochemistry

(1 μg/μL), DTT to a final concentration of 1 mM, and distilledwater added up to 750 μL.
Digestion was performed at 4◦C, 16◦C, and 30◦C, and a50-μL aliquot was taken at 0, 2, 4, 8, and 16 H intervals to seekthe best condition; sarkosyl and triton were added to improvethe digestive efficiency. Samples were mixed with 6× SDS sam-ple buffer, boiled for 4 Min, and subjected to tricine–SDS-PAGEon discontinuous 10% polyacrylamide gels according to Schag-ger [45], followed by staining with Coomassie blue or Westernblotting.
2.9. hIGF-1 bioactivity assayThe SH-SY5Y cells were grown as suspension cultures in RPMI1640 medium (Gibco, Langley, OK, USA) supplemented with 10%fetal bovine serum (Gibco). The human neuroblastoma SH-SY5Ycells were taken from liquid nitrogen into a 37◦C water bath withgentle shaking for rapid resuscitation. Cells were then trans-ferred into a 15 mL centrifuge tube containing 9 mL prewarmedRPMI1640 medium (37◦C, supplemented with 10% fetal bovineserum). Cells were collected by centrifuge at 800g for 3 Min, re-suspended with 5 mL medium, and transferred into 25 cm2 cellculture dishes; the conditions of cultivation were 37◦C, 5% CO2.The medium was changed every 24 H to remove dead cells andcell debris. When cell growth covered the dishes, the cells werewashed twice with 1× PBS, digested in 0.5 mL trypsin–EDTAsolution (0.25% trypsin and 0.02% EDTA) at 37◦C for 2 Min andwere mixed with 2.5 mL of medium. The cells were collectedby centrifuge at 800g for 3 Min and resuspended with 1 mLfresh RPMI 1640 medium containing 10% fetal bovine serum.The live cells were counted using trypan blue exclusion, andequal cells (5 × <104) were transferred to the wells of a tis-sue culture plate. After 12 H culture, an aliquot of 0.5 mL RPMImedium with 10% fetal bovine serum containing one of the fol-lowing samples at a time was added to each of the wells: 50ng/mL E. coli-derived hIGF-1 (Shanghai PrimeGene Bio-Tech,Shanghai, People’s Republic of China), transgenic Arabidopsisseed oil-body extract containing 50 ng/mL of hIGF-1, wild-typeArabidopsis seed oil-body extract at equivalent protein concen-tration, protein extraction buffer alone. The experiments weredone in triplicate and under sterile conditions. Cellular differen-tiation was monitored at 24, 48, and 72 H intervals after sampleswere appended.
3. Results and discussion3.1. Designing and construction of hIGF-1-containingvectorThe construction used for A. thaliana transformation encodesa fusion protein comprising recombinant hIGF-1 with an N-terminal TEVP cleavable propeptide fused to the C-terminusof the A. thaliana 18.5 kDa oleosin and a myc tag fused to theN-terminal of the oleosin. The expression of the myc–oleosin–hIGF-1 fusion gene was under the control of the A. thaliana 18.5kDa oleosin promoter and the rbsS polyA terminator. For pos-itive transgenic plant selection, a HmB-resistance gene underthe control of a CaMV 35S promoter and NOS terminator was
set within the T-DNA. The core structure of the construction isshown in Fig. 1.
On the basis of oleosin fusion technology, heterogeneousprotein was fused to the N- or C-terminal of oleosin and segre-gated with a site-specific protease recognition sequence, whichhas the ability to recognize a specific amino sequence in pro-tein and to make a cleavage [29],[31]. Site-specific protease iswidely used for removal of the tag from the protein of interest orfor separation of two units of a protein. Some commonly usedproteases are Factor Xa, thrombin, and enterokinase [46].
The TEVP is a novel site-specific cysteine protease that isfound in the tobacco etch virus. The sequence specificity of thisenzyme is far more stringent than that of factor Xa, thrombin,or enterokinase. It is also easy to be expressed in E. coli and bepurified via a polyhistidine tag to a high degree of purity, so it isan ideal choice for removal of tags from fusion proteins [46],[33].The most commonly used recognition site for this enzyme isthe sequence Glu–Asn–Leu–Tyr–Phe–Gln–Gly (ENLYFQ|G); thecleavage occurs between the Gln and Gly residues, resulting ina Gly at the amino terminus of the protein of interest. In thisstudy, Gly corresponds to the first amino acid of hIGF-1, andtherefore correct proteolytic digestion with TEV would result inan authentic N-terminus. The use of TEVP will not induce extraamino residues, which may cause safety problems in clinicalusage, so we choose the TEVP as a tool for the release of hIGF-1from the oil bodies after the initial purification.
Oleosins and oil bodies were found in various “oil plants,”and those oleosins are correctly targeted to the oil bodies. Re-search has shown that the ability to target oil bodies is theproperty of oleosin and it has less effect with species [47]. Sev-eral promoters were demonstrated to induce specific expressionin seeds, for instance, napin promoter [47] from Brassica napusand β-phaseolin promoter from Phaseolus vulgaris [48],[42]. Inthis study, we choose the Arabidopsis 18.5 kDa oleosin as afusion partner and its own promoter to control the expressionbecause the 18.5 kDa oleosin is the most abundant oleosin inArabidopsis seeds.
3.2. Plant transformation and molecularcharacterization of transgenic ArabidopsisArabidopsis was transformed with the pHB–AtOP–AtOle–hIGF-1binary construct containing both a myc–oleosin–hIGF-1 fusedgene (driven by oleosin promoter) and two selectable markergenes (HmB and bar, driven by 35S promoter) within the sameT-DNA fragment. We selected the primary transformants (T0) onthe basis of resistance to the antibiotic HmB (on 1
2 MS mediumplates contain 50 μg/mL HmB) and PPT (on solid culture sprayedusing 1% PPT) and established transgenic lines by single seeddescent, with a selection maintained to the third generation(T3). For all three T3 plants analyzed, the presence of oleosin–hIGF-1 fusion gene was confirmed using a pair of primers (theforward primer is located in the coding sequence area of oleosingene and the reverse primer is located in the coding sequencearea of hIGF-1 gene). An expected 250 bp fragment was observedin agarose analysis of PCR products; no such product was ob-served when wild-type DNA was used as the template (data notshown).
Expression of human insulin-like growth factor 1 in A. thaliana 143

Fig. 2. Expression of myc–oleosin–hIGF-1 fusion proteins intransgenic Arabidopsis T3 seeds. Oil-body proteins fromthree transgenic A. thaliana lines (7–1, 8–2, 11–1) andwide-type A. thaliana were analyzed on sodium dodecylsulfate polyacrylamide gel (SDS-PAGE) and for Westernblotting. (a) Coomassie-stained SDS-PAGE. Arrowsindicating the 34.3-kDa fusion proteins. (b) Duplicated gelsof (a) transferred to PVDF membrane for Western blottingusing antibodies against hIGF-1 as primary antibody. WTrefers to wide-type A. thaliana seeds. P refers to a purified14.2 kDa protein containing the hIGF-1 amino sequencederived from E. coli (a 14.2 kDa His-tagged hIGF-1 fusionprotein overexpressed in E. coli and purified via Ni-NTAagarose, the E. coli-derived hIGF-1 has the same aminosequence with the plant-derived hIGF-1). M1 refers to thelow-range protein marker (six bands from top to bottom: 97,66, 43, 29, 20, and 14.4 kDa) and M2 refers to theprestained protein marker (5 bands from top to bottom: 175,87, 47.5, 25, and 16.5 kDa).
3.3. hIGF-1 expression and oil-body purificationTo evaluate the accumulation of the myc–oleosin–hIGF-1 fu-sion protein, TSPs from transgenic and wild-type plants werescreened via Western blotting using antibodies anti-myc tag andanti-hIGF-1, respectively. Of the 18 independent lines analyzed,15 gave positive signals based on the Western blotting analy-sis; of these 15 positive lines, three transformed lines showedthe highest expression of myc–oleosin–hIGF-1; 7–1, 8–2, and11–1 were used for further analysis. TSP and purified oil-bodyprotein were analyzed on SDS-PAGE; a band indicated the pres-ence of myc–oleosin–hIGF-1 fusion protein at the predicted size34.3 kDa from transgenic seeds. No band at this position wasobserved either in the TSP or oil-body protein from wild-typeseed (Fig. 2a). The Western blotting analysis using antibodyagainst hIGF-1 established that the band mentioned above con-tained the hIGF-1 sequence (Fig. 2b). The expression levels weredetermined by densitometry using the native 18 kDa oleosinband as an internal standard (equivalent to 1.5% TSP) accord-ing to Nykiforuk et al. [42] (Table 2). The expression of hIGF-1 intransformed A. thaliana seeds was approximately seven timeshigher than previously reported in plants. In an earlier report, re-combinant hIGF-1 was expressed in transgenic rice and tobacco[30]; the expression of hIGF-1 in transgenic rice and tobaccowas 113 ± 24 and 70 ± 13 ng of rthIGF-1/mg total protein,respectively.
Table 2Expression levels of oleosin–hIGF-1 fusion protein wasdetermined by densitometry of Coomassie-stained sodiumdodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE) gels according to Nykiforuk et al. [42]
Fusion protein hIGF-1(oleosion–hIGF-1) expression
Line expression (%TSP) (%TSP)7-1 0.51 0.118-2 0.69 0.1511-1 0.75 0.17Wide type
(negative control)0 0
3.4. TEVP cleavage of myc–oleosin–hIGF-1Reaction time and temperature were tested for TEVP cleavage inour study. Cleavage efficiency was checked on SDS-PAGE or byWestern blotting, but no significant cleavage was observed evenovernight at 30◦C. The incomplete cleavage of oleosin fusionprotein was reported previously [49]. This may be due to theparticular environment at the surface of oil bodies. The interfaceof oil and water might have disturbed the action of protease.Furthermore, other proteins on the surface of oil bodies mighthave caused steric hindrance and thus prevented the oleosinfusion from the protease.
Detergents such as sarkosyl or triton were used to reducethe interfacial tension between oil and water on the surface ofoil bodies. The cleavage efficiency was improved, as shown inFig. 3; 7.6 kDa hIGF-1 was released from oleosin fusion in thepresence of 0.5% sarkosyl or 1% triton X-100, but the cleavagewas not yet complete.
Linkers were commonly placed between subunits of fusionprotein to reduce the interaction between the two parts [50]. Forthe oleosin fusion technology, linkers may also be put betweenthe oleosin and the target protein to weaken the influence ofsteric hindrance during the cleavage. Meanwhile, special pro-tease with a valid activity on the oil–water interface may bedeveloped to facilitate the cleavage of oleosin fusion proteinand further cut down the cost for protein purification.
3.5. Biological activity assay of plant-derivedrecombinant hIGF-1For the biological assay, the human neuroblastoma cell lineSH-SY5Y, which is known to proliferate and differentiate in thepresence of hIGF-1 or insulin [51–53], was used. Cells were cul-tured in tissue culture plate and incubated in the presence orabsence of E. coli-derived hIGF-1 or aliquots of oil bodies ex-tracted from transgenic or nontransgenic A. thaliana seeds. Inthis experiment, seed oil bodies containing hIGF-1 from trans-genic A. thaliana line 8–2 or E. coli-derived hIGF-1 were usedto supplement the assay medium to a final concentration of50 ng/mL; equal aliquots of oil bodies from wild-type A. thalianaand oil-body extraction buffer alone were tested in the samemanner.
144 Biotechnology and Applied Biochemistry
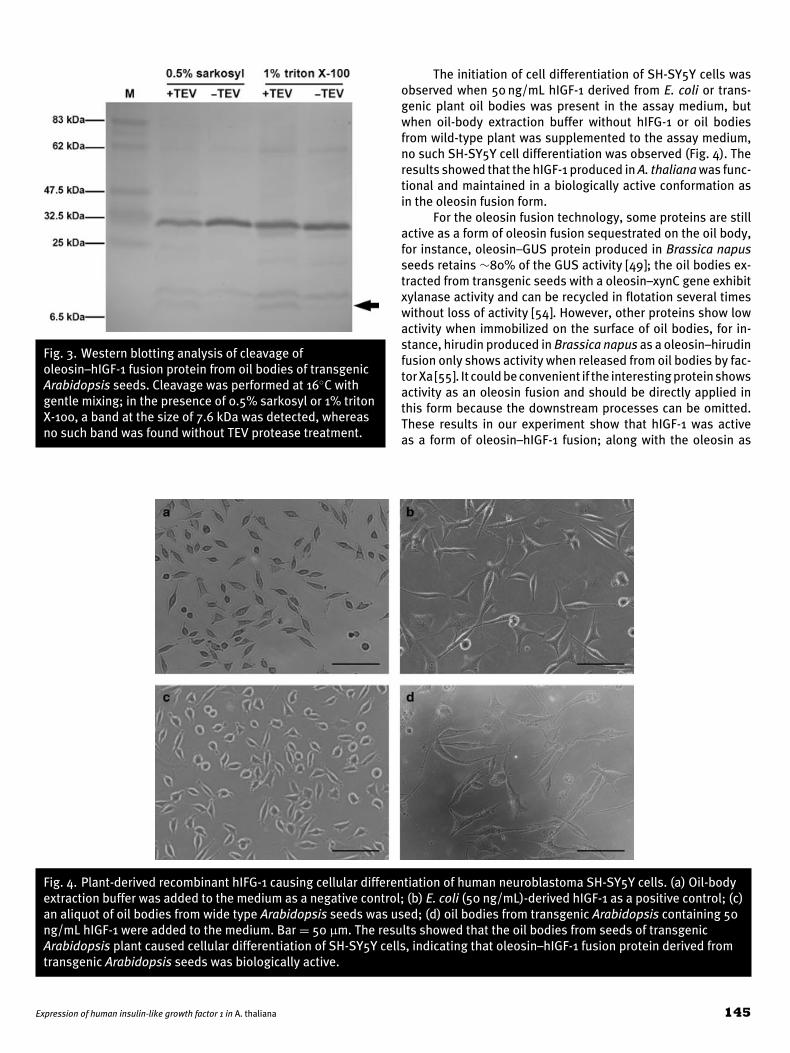
Fig. 3. Western blotting analysis of cleavage ofoleosin–hIGF-1 fusion protein from oil bodies of transgenicArabidopsis seeds. Cleavage was performed at 16◦C withgentle mixing; in the presence of 0.5% sarkosyl or 1% tritonX-100, a band at the size of 7.6 kDa was detected, whereasno such band was found without TEV protease treatment.
The initiation of cell differentiation of SH-SY5Y cells wasobserved when 50 ng/mL hIGF-1 derived from E. coli or trans-genic plant oil bodies was present in the assay medium, butwhen oil-body extraction buffer without hIFG-1 or oil bodiesfrom wild-type plant was supplemented to the assay medium,no such SH-SY5Y cell differentiation was observed (Fig. 4). Theresults showed that the hIGF-1 produced in A. thaliana was func-tional and maintained in a biologically active conformation asin the oleosin fusion form.
For the oleosin fusion technology, some proteins are stillactive as a form of oleosin fusion sequestrated on the oil body,for instance, oleosin–GUS protein produced in Brassica napusseeds retains ∼80% of the GUS activity [49]; the oil bodies ex-tracted from transgenic seeds with a oleosin–xynC gene exhibitxylanase activity and can be recycled in flotation several timeswithout loss of activity [54]. However, other proteins show lowactivity when immobilized on the surface of oil bodies, for in-stance, hirudin produced in Brassica napus as a oleosin–hirudinfusion only shows activity when released from oil bodies by fac-tor Xa [55]. It could be convenient if the interesting protein showsactivity as an oleosin fusion and should be directly applied inthis form because the downstream processes can be omitted.These results in our experiment show that hIGF-1 was activeas a form of oleosin–hIGF-1 fusion; along with the oleosin as
Fig. 4. Plant-derived recombinant hIFG-1 causing cellular differentiation of human neuroblastoma SH-SY5Y cells. (a) Oil-bodyextraction buffer was added to the medium as a negative control; (b) E. coli (50 ng/mL)-derived hIGF-1 as a positive control; (c)an aliquot of oil bodies from wide type Arabidopsis seeds was used; (d) oil bodies from transgenic Arabidopsis containing 50ng/mL hIGF-1 were added to the medium. Bar = 50 μm. The results showed that the oil bodies from seeds of transgenicArabidopsis plant caused cellular differentiation of SH-SY5Y cells, indicating that oleosin–hIGF-1 fusion protein derived fromtransgenic Arabidopsis seeds was biologically active.
Expression of human insulin-like growth factor 1 in A. thaliana 145

a natural emulsifying agent [56–58], the product may be useddirectly for external application in some area such as skin careand beauty treatment.
In summary, we have successfully expressed hIGF-1 in A.thaliana seeds as an oleosin–hIGF-1 fusion and have demon-strated that the oleosin–hIGF-1 expressed in plant was biologi-cally active. By using a strong oleosin promoter, the most abun-dant oleosin gene in A. thaliana as a fusion partner, and propercodon usage for hIGF-1 gene, we achieved high accumulationof hIGF-1 in transgenic A. thaliana seeds. This study provides abasis and useful information for using a plant oleosin system toexpress and produce hIGF-1 in large scale in the future.
AcknowledgementsThis work was supported by China National High-Tech “863”Program (grant number 2007AA100503), China “973” Program(grant number 2010AA1000691007), and China Transgenic Re-search Program (2008ZX08002-001). We thank Dr. Yu Ding at Fu-dan University for providing the TEVP and technical assistance,and Dr. Grace H. Wireman at George Washington CommunitySchool for comments on the manuscript.
References[1] Rinderknecht, E. and Humbel, R. E. (1978) J. Biol. Chem. 253, 2769–2776.[2] D’Ercole, A. J. (1996) Endocrinol. Metab. Clin. North Am. 25, 573–590.[3] Ferry Jr, R. J., Katz, L. E., Grimberg, A., Cohen, P., and Weinzimer, S. A. (1999)
Horm. Metab. Res. 31, 192.[4] Rajaram, S., Baylink, D. J., and Mohan, S. (1997) Endocr. Rev. 18, 801–831.[5] Cotterill, A. M. (1992) Clin. Endocrinol. 37, 11–16.[6] Canalis, E. (1992) J. Clin. Endocrinol. Metab. 75, 1–4.[7] Flyvbjerg, A. (1997) Kidney Int. Suppl. 60, S12.[8] Moks, T., Abrahmsen, L., Osterlof, B., Josephson, S., Ostling, M., Enfors,
S. O., Persson, I., Nilsson, B., and Uhlen, M. (1987) Nat. Biotechnol. 5,379–382.
[9] Moks, T., Abrahmsen, L., Holmgren, E., Bilich, M., Olsson, A., Pohl, G.,Sterky, C., Hultberg, H., and Josephson, S. (1987) Biochemistry 26, 5239–5244.
[10] Wong, E. Y., Seetharam, R., Kotts, C. E., Heeren, R. A., Klein, B. K., Braford,S. R., Mathis, K. J., Bishop, B. F., Siegel, N. R., Smith, C. E., Tacon, W. C.(1988) Gene 68, 193–203.
[11] Forsberg, G., Palm, G., Ekebacke, A., Josephson, S., and Hartmanis, M.(1990) Biochem. J. 271, 357.
[12] Kim, S. and Lee, Y. I. (1996) J. Biotechnol. 48, 97–105.[13] Chung, B., Choi, Y., Yoon, S., Lee, S., and Lee, Y. (2000) J. Ind. Microbiol.
Biotechnol. 24, 94–99.[14] Gellerfors, P., Axelsson, K., Helander, A., Johansson, S., Kenne, L., Lindqvist,
S., Pavlu, B., Skottner, A., and Fryklund, L. (1989) J. Biol. Chem. 264, 11444–11449.
[15] Bovenberg, W. A., Dauwerse, J. G., Pospiech, H. M., Van Buul-Offers, S. C.,Van Den Brande, J. L., and Sussenbach, J. S. (1990) Mol. Cell. Endocrinol.74, 45.
[16] Wolf, E., Jehle, P. M., Weber, M. M., Sauerwein, H., Daxenberger, A., Breier,B. H., Besenfelder, U., Frenyo, L., and Brem, G. (1997) Endocrinology 138,307–313.
[17] Saito, Y., Yamada, H., Niwa, M., and Ueda, I. (1987) J. Biochem. 101, 123.[18] Schulz, M. F., Buell, G., Schmid, E., Movva, R., and Selzer, G. (1987)
J. Bacteriol. 169, 5385–5392.[19] Elliott, S., Fagin, K. D., Narhi, L. O., Miller, J. A., Jones, M., Koski, R., Peters,
M., Hsieh, P., Sachdev, R., Rosenfeld, R. D., Rohde, M. F., Arakawa, T. (1990)J. Protein Chem. 9, 95–104.
[20] Samuelsson, E., Wadensten, H., Hartmanis, M., Moks, T., and Uhlen, M.(1991) Biotechnology 9, 363–366.
[21] Samuelsson, E., Moks, T., Uhlen, M., and Nilsson, B. (1994) Biochemistry33, 4207–4211.
[22] Giddings, G., Allison, G., Brooks, D., and Carter, A. (2000) Nat. Biotechnol.18, 1151–1155.
[23] Larrick, J. W. and Thomas, D. W. (2001) Curr. Opin. Biotechnol. 12, 411–418.
[24] Ma, J. K. C., Drake, P. M. W., and Christou, P. (2003) Nat. Rev. Genet. 4,794–805.
[25] Ma, J. K., Chikwamba, R., Sparrow, P., Fischer, R., Mahoney, R., and Twyman,R. M. (2005) Trends Plant Sci. 10, 580–585.
[26] Twyman, R. M., Stoger, E., Schillberg, S., Christou, P., and Fischer, R. (2003)Trends Biotechnol. 21, 570–578.
[27] Fischer, R., Stoger, E., Schillberg, S., Christou, P., and Twyman, R. M. (2004)Curr. Opin. Plant Biol. 7, 152–158.
[28] Hood, E. E. and Jilka, J. M. (1999) Curr. Opin. Biotechnol. 10, 382–386.[29] Boothe, J., Nykiforuk, C., Shen, Y., Zaplachinski, S., Szarka, S., Kuhlman,
P., Murray, E., Morck, D., Moloney, M. M. (2010) Plant Biotechnol. J. 8,588–606.
[30] Panahi, M., Alli, Z., Cheng, X., Belbaraka, L., Belgoudi, J., Sardana, R.,Phipps, J., and Altosaar, I. (2004) Transgenic Res. 13, 245–259.
[31] Stoger, E., Ma, J. K. C., Fischer, R., and Christou, P. (2005) Curr. Opin.Biotechnol. 16, 167–173.
[32] Nakamura, Y., Gojobori, T., and Ikemura, T. (2000) Nucleic Acids Res. 28,292.
[33] Parks, T. D., Leuther, K. K., Howard, E. D., Johnston, S. A., and Dougherty,W. G. (1994) Anal. Biochem. 216, 413–417.
[34] Wu, G., Bashir-Bello, N., and Freeland, S. J. (2006) Protein Expr. Purif. 47,441–445.
[35] Xiong, A. S., Yao, Q. H., Peng, R. H., Duan, H., Li, X., Fan, H. Q., Cheng, Z. M.,and Li, Y. (2006) Nat Protoc. 1, 791–797.
[36] Porebski, S., Bailey, L., and Baum, B. (1997) Plant Mol. Biol. Rep. 15, 8–15.
[37] Mao, J., Zhang, Y. C., Sang, Y., Li, Q. H., and Yang, H. Q. (2005) Proc. Natl.Acad. Sci. USA 102, 12270–12275.
[38] Koncz, C. and Schell, J. (1986) Mol. Gen. Genet. 204, 383–396.[39] Hofgen, R. and Willmitzer, L. (1988) Nucleic Acids Res. 16, 9877.[40] Clough, S. J. and Bent, A. F. (1998) Plant J. 16, 735–743.[41] Murashige, T. and Skoog, F. (1962) Physiol. Plant 15, 473–497.[42] Nykiforuk, C. L., Boothe, J. G., Murray, E. W., Keon, R. G., Goren, H. J.,
Markley, N. A., and Moloney, M. M. (2006) Plant Biotechnol. J. 4, 77–85.
[43] Smith, P. K. et al. (1985) Anal. Biochem. 150, 76–85.[44] Laemmli, U. K. (1970) Nature 227, 680–685.[45] Schagger, H. (2006) Nat Protoc. 1, 16–22.[46] Polayes, D. A., Goldstein, A., Ward, G., and Hughes, A. J. (1994) Focus 16,
2–5.[47] Lee, W. S., Tzen, J. T. C., Kridl, J. C., Radke, S. E., and Huang, A. H. C. (1991)
Proc. Natl. Acad. Sci. USA 88, 6181–6185.[48] Slightom, J. L., Sun, S. M., and Hall, T. C. (1983) Proc. Natl. Acad. Sci. USA
80, 1897–1901.[49] Van Rooijen, G. J. H. and Motoney, M. M. (1995) Nat. Biotechnol. 13, 72–
77.[50] Arai, R., Ueda, H., Kitayama, A., Kamiya, N., and Nagamune, T., (2001)
Protein Eng. 14, 529–532.[51] Pahlman, S., Meyerson, G., Lindgren, E., Schalling, M., and Johansson, I.
(1991) Proc. Natl. Acad. Sci. USA 88, 9994–9998.[52] Zeidman, R., Pettersson, L., Sailaja, P. R., Truedsson, E., Fagerstrom, S.,
Pahlman, S., and Larsson, C. (1999) Int. J. Cancer 81, 494–501.[53] Zumkeller, W. and Schwab, M. (1999) Horm. Metab. Res. 31, 138.[54] Liu, J. H., Selinger, L. B., Cheng, K. J., Beauchemin, K. A., and Moloney, M. M.
(1997) Mol. Breed. 3, 463–470.[55] Parmenter, D. L., Boothe, J. G., Rooijen, G. J. H., Yeung, E. C., and Moloney,
M. M. (1995) Plant Mol. Biol. 29, 1167–1180.[56] Tzen, J. T. C., Chuang, R. L. C., Chen, J. C. F., and Wu, L. S. H. (1998)
J. Biochem. 123, 318.[57] Tzen, J. and Huang, A. (1992) J. Cell Biol. 117, 327–335.[58] Beisson, F., Ferte, N., Voultoury, R., and Arondel, V. (2001) Plant Physiol.
Biochem. 39, 623–630.
146 Biotechnology and Applied Biochemistry