Embed Size (px)
Citation preview

American Journal of Medical Genetics 39:362-366 (1991)
Familial Translocation 5;14 Resulting in an Unbalanced Offspring
Jonathan P. Park, Matthew J. Edwards, John B. Moeschler, J. Miguel Marin-Padilla, Susan Z. Berg, and Doris H. Wurster-Hill Department of Pathology (J.P.P., J.M.M.-P., D.H.W.-H.), Clinical Genetics and Child Development Center and Department of Maternal and Child Health (J.B.M., S.Z.B.), Dartmouth-Hitchcock Medical Center and Department of Biological Sciences (J.P.P.), Dartmouth College, Hanover and New Hampshire Division of Public Health, Concord, New Hampshire (S.Z.B.); Clinical Genetics and Dysmorphology, Cedars-Sinai Medical Center, Los Angeles, California (M.J.E.)
We report on an infant with multiple congeni- tal anomalies possessing a derivative 14 chro- mosome in excess of the normal comple- ment, resulting from transmission of a famil- ial t(5;14)(p13;q22). The proposita’s phe- notypically normal mother, mentally re- tarded half-brother, and fetal sib are carriers of the apparently balanced translocation. Previous cases of similar familial t(5;14) are reviewed. The proposita’s phenotype is char- acterized by failure to thrive, developmental retardation, cleft palate, congenital heart anomaly, abnormal hands and feet, unusual face with abnormal ears, and recurrent respi- ratory infections. The proposita died at age 9 months and postmortem examination showed multiple central nervous system, cardio- pulmonary, gastrointestinal, and genital mal- formations. Our proposita’s phenotype is at- tributable to contributions from both chromosomes and is consistent with the con- sequences of both the dup(5p) and dup(l4q).
KEY WORDS: agenesis of the corpus cal- losum, arrhinencephaly, chro- mosome, cleft palate, cyto- genetic, dup(5p), dup(l4qA multiple congenital anoma- liedmental retardation (MCAI MR) syndrome, translocation
Received for publication May 14,1990; revision received August 9, 1990.
Address reprint requests t,o Jonathan P. Park, CLSp(CG), De- partment of Pathology, Dartmouth-Hitchcock Medical Center, Hanover, NH 03756.
0 1991 Wiley-Liss, Inc.
INTRODUCTION Early reports of del(5p) resulting in cri-du-chat syn-
drome were often associated with translocations involv- ing chromosome 5 with other autosomes. Further studies of those kindreds occasionally showed relatives with unbalanced translocations resulting in duplication of 5p, and a karyotype: phenotype correlation emerged characterized by psychomotor retardation, hypotonia, seizures, unusual face with ear and eye anomalies, club feet, and post-natal growth retardation [Lejeune et al., 1964; Brimblecombe et al., 1977; Khodr et al., 19821. Duplication of proximal 14q is frequently associated with mental and growth retardation, abnormal crani- ofacies, recurrent respiratory infections, cleft palate, and flexion contractures [Pena et al., 19761. While other kindreds have been described with t(5;14) [Wolf et al., 1966; Borgaonkar et al., 1973; Shinno et al., 1973; Fried et al., 1977; Abeliovich et al., 19821, most probands have had either balanced karyotypes or have received a der(5) in excess. We report a unique imbalance in a female infant possessing a derivative 14 chromosome in excess of the normal complement, resulting from transmission of a familial t(5;14)(p13;q22) and associated with multi- ple congenital anomalies.
CLINICAL REPORT The proposita was born after a 36 week gestation to a
29 year-old, gravida 111, para I1 woman after an unevent- ful pregnancy; the father was 31 years old. Cleft palate was noted a t birth and the infant fed slowly and re- mained in the hospital for a month due to failure to thrive. She was readmitted 3 times with upper respira- tory tract infections.
At age 5% weeks (Fig. l), length was 46 cm (<3rd centile when corrected for prematurity), head circum- ference (OFC) 34 cm (50th centile), and weight re- mained below birthweight (2.4 kg). Abnormalities of the limbs included brachyclinodactyly of the 5th fingers and indistinct distal 5th finger creases, arches on all but the right 4th and 5th fingers which had ulnar loops, tibia1 deviation of toes 2 through 5 (especially the 2nd which
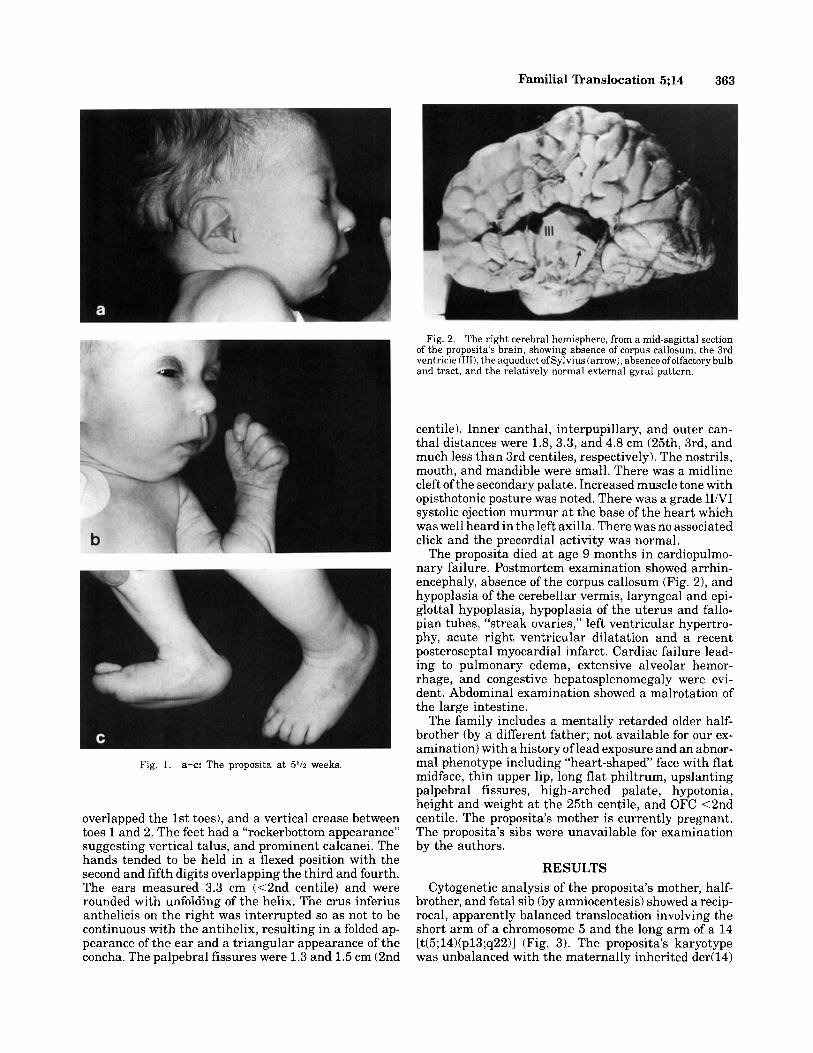
Familial Translocation 5;14 363
Fig. 1. a-e: The proposita a t 5% weeks.
overlapped the 1st toes), and a vertical crease between toes 1 and 2. The feet had a “rockerbottom appearance” suggesting vertical talus, and prominent calcanei. The hands tended to be held in a flexed position with the second and fifth digits overlapping the third and fourth. The ears measured 3.3 cm (<2nd centile) and were rounded with unfolding of the helix. The crus inferius anthelicis on the right was interrupted so as not to be continuous with the antihelix, resulting in a folded ap- pearance of the ear and a triangular appearance of the concha. The palpebral fissures were 1.3 and 1.5 cm (2nd
Fig. 2. The right cerebral hemisphere, from a mid-sagittal section of the proposita’s brain, showing absence of corpus callosum, the 3rd ventricle (1111, the aqueduct of Sylvius (arrow), absence ofolfactory bulb and tract, and the relatively normal external gyral pattern.
centile). Inner canthal, interpupillary, and outer can- thal distances were 1.8, 3.3, and 4.8 cm (25th, 3rd, and much less than 3rd centiles, respectively). The nostrils, mouth, and mandible were small. There was a midline cleft of the secondary palate. Increased muscle tone with opisthotonic posture was noted. There was a grade IIiVI systolic ejection murmur a t the base of the heart which was well heard in the left axilla. There was no associated click and the precordial activity was normal.
The proposita died a t age 9 months in cardiopulmo- nary failure. Postmortem examination showed arrhin- encephaly, absence of the corpus callosum (Fig. 2), and hypoplasia of the cerebellar vermis, laryngeal and epi- glottal hypoplasia, hypoplasia of the uterus and fallo- pian tubes, “streak ovaries,” left ventricular hypertro- phy, acute right ventricular dilatation and a recent posteroseptal myocardial infarct. Cardiac failure lead- ing to pulmonary edema, extensive alveolar hemor- rhage, and congestive hepatosplenomegaly were evi- dent. Abdominal examination showed a malrotation of the large intestine.
The family includes a mentally retarded older half- brother (by a different father; not available for our ex- amination) with a history of lead exposure and an abnor- mal phenotype including “heart-shaped’ face with flat midface, thin upper lip, long flat philtrum, upslanting palpebral fissures, high-arched palate, hypotonia, height and weight a t the 25th centile, and OFC <2nd centile. The proposita’s mother is currently pregnant. The proposita’s sibs were unavailable for examination by the authors.
RESULTS Cytogenetic analysis of the proposita’s mother, half-
brother, and fetal sib (by amniocentesis) showed a recip- rocal, apparently balanced translocation involving the short arm of a chromosome 5 and the long arm of a 14 [t(5;14)(p13;q22)] (Fig. 3). The proposita’s karyotype was unbalanced with the maternally inherited der(l4)

364 Park et al.
Fig. 3. G-banded partial karyotypes of the t(5;14Kp13;q221 in a balanced complement. The upper row represents cells from amniocen- tesis on the proposita’s apparently balanced, fetal sib, and the lower row are stimulated lymphocytes from the proposita’s mother. The der(l41, which is present in excess in the proposita, is indicated by an open arrow in each partial karyotype while the der(5) chromosome is indi- cated by a solid arrowhead. The normal No. 5 and 14 homologues are shown for comparison.
present in excess [47,XX,16qh + , + der(14)t(5;14) (p13;q22)matl.
DISCUSSION The duplicated chromosome segments in our case are
5p13-pter and 14q22-pter. Khodr et al. [19821 reported a case of familial t(5;13) resulting in duplication 5p13- pter in a fetus with a small “triangular” face, a flat forehead, prominent occiput, abnormal ears, and pecu- liarly flexed fingers. The authors conclude in their re- view of 13 cases that dup(5)(p13) is required for obvious phenotypic effects. A case of tandem dup 5p (pl4-pter) [Chia et al., 19871 supports this contention, as clinical manifestations of the excess material were minimal, and, unlike the previous 5 cases of duplication 5p14-pter [Di Liberti et al., 1977; Stoll et al., 19751, the possible contribution of other autosomal excesses was not a fac- tor. The most common manifestations of a duplication of 5p13-pter or of the complete 5p include normal birth- weight, psychomotor retardation, hypotonia, seizures, macrodolichoscaphocephaly, unusual facial appearance including slanted palpebral fissures, eye abnormalities, depressed nasal bridge, macroglossia, micrognathia, ap- parently low-set, abnormal ears, long fingers, club feet, and, less frequently, postnatal growth failure and con- genital heart defect [Carnevale et al., 19821. Agenesis of the corpus callosum has been reported in 2 cases of dup(5p) [Brimblecombe et al., 1977; Kleczkowska et al.,
19871. The authors also concluded that in familial cases of t(5p;autosome), “partial monosomy” 5p occurs twice as often as “partial trisomy” 5p. In fact, many transloca- tion kindreds are identified after the birth of a patient with cri-du-chat syndrome, as in the first case of dupli- cation 5p ever reported [Lejeune et al., 19641.
The possible phenotypic contribution attributable to the 14q22-pter segment of the derivative chromosome is unclear, as similar cases in the literature involve addi- tional fragments of other autosomes as part of the trans- location chromosome. As with partial duplication of 5p, the breakpoint involved in 14q appears critical to the severity of the phenotype, with 14q22 at the milder end of the spectrum. Patients with duplication of only 14pter-q22 show mental and developmental retarda- tion, minor craniofacial abnormalities, microcephaly, flat nasal bridge, microphthalmia, apparently low-set ears, recurrent respiratory infections, cleft palate, and flexion contractures [Kovacs and Mihai, 1979; Johnson et al., 19791. More than two-thirds of these malforma- tions occur in every instance [Pena et al., 19761.
Translocations involving both chromosomes 5 and 14 have been reported (Table I). Wolf et al. [1966] reported a girl with cri-du-chat syndrome and an apparently de novo translocation of most of 14q onto 5p resulting in a partial monosomy 5p. The breakpoints were similar to those in our case and the resulting translocation chro- mosome resembled the 5 found in the karyotypes of our proposita’s mother and half-brother. Shinno et al. [19731 reported 2 phenotypically abnormal children born to a mother with the presumably balanced karyotype “46,XX,t(5p + ;14q - 1”. The report was an abstract and figures are not available for the interpretaton of break- points. The 2 mentally retarded children had an extra acrocentric chromosome identified as 14q- (although more likely a derivative chromosome resulting from a reciprocal translocation). Another case of familial trans- mission was reported by Fried et al. [1977]. Their inter- pretation of the karyotype was 45,XX,-5,-14, + der(5)t (5;14)(p15;q13). Abeliovich et al. [1982] reported a sec- ond unbalanced child born to that mother. Unlike the first child where the der(l4) was lacking in the propo- sita’s karyotype resulting in partial monosomy for parts ofboth No. 5 and 14, the der(l4) was present in excess in this case.
These cases of t(5p;14q) suggest that rearrangement of these regions is not an infrequent finding. Excluding those cases of balanced transmission [Borgaonkar et al., 19731 and cases where the imbalance resulted in a deriv- ative 5 [Wolf et al., 1966; fiied et al., 19771 3 cases with excess of a derivative 14 similar to our case emerge. The 2 sibs reported by Shinno et al. [1973] are difficult to interpret owing to the absence of illustrations of the chromosomes and the uncertainty of whether the extra chromosome resulted from a deletion or a reciprocal translocation. The case of Abeliovich et al. [1982] is therefore the most similar documented case to our own. The apparent imbalance (14pter-14q13::5p15-5pter) consists of less additional material from both 14q and 5p than in our case. The additional duplicated material in our case may contribute to the more severe phenotype which we observed. The resultant unbalanced comple-

TAB
LE 1
. Cas
es o
f T
rans
loca
tions
Inv
olvi
ng 5
p an
d 14
q
App
aren
t st
atus
_
__
_-
Exc
essi
(1os
s)
Phen
otyp
e C
ase
Bre
akpo
ints
W
olf
et a
l. [1
9661
5p
13; 1
4q22
a,b
Unb
alan
ced
(5p)
C
ri-d
u-ch
at
Shin
no e
t al
. [19
731
Una
vaila
ble
Unb
alan
ced
14q,
?5p
M
icro
ceph
aly,
hyp
otel
oris
m, s
mal
l obl
ique
pal
pe-
Bor
gaon
kar
et a
l. [1
9731
5p
13; 1
4q22
” B
alan
ced
The
pro
posi
ta w
as e
mot
iona
lly d
istu
rbed
, 6 o
ther
(2
cas
es)
bra1
fiss
ures
, cl
eft p
alat
e, s
eizu
res
appa
rent
ly b
alan
ced
kin
wer
e ph
enot
ypic
ally
no
rmal
Fr
ied
et a
l. [1
9771
5p
15; 1
4q13
U
nbal
ance
d (5
~1
, (14p
), (1
4q)
Cri
-du-
chat
, alt
ered
faci
es
Abe
liovi
ch e
t al.
[198
21”
5p15
; 14q
13
Unb
alan
ced
14p,
14q
, 5p
Wei
ght
and
leng
th 5
0th
cent
ile,
OFC
3rd
cen
tile,
na
rrow
sku
ll, h
ypot
elor
ism
, “m
ongo
loid
” sl
ant
of t
he p
alpe
bral
fis
sure
s, c
onve
rgen
t str
abis
- m
us, p
tosi
s an
d bi
late
ral
iris
col
obom
ata,
ap-
pa
rent
ly l
ow-s
et p
oste
rior
ly a
ngul
ated
ear
s,
depr
esse
d na
sal
brid
ge,
incr
ease
d ph
iltr
um
leng
th, d
own-
turn
ed a
ngle
s of
the
mou
th, r
etro
- gn
athi
a, s
mal
l and
hig
hly-
arch
ed u
vula
, sub
- m
ucou
s cl
eft p
alat
e, a
nd a
sho
rt n
eck
with
so
me
skin
red
unda
ncy
Fail
ure
to t
hriv
e, c
left
pal
ate,
hea
rt m
urm
ur,
dysm
orph
ic h
ands
and
fee
t, un
usua
l fa
cies
with
ab
norm
al e
ars,
and
mul
tiple
cen
tral
ner
vous
sy
stem
, car
diop
ulm
onar
y, g
astr
oint
esti
nal,
and
geni
tal
mal
form
atio
ns e
vide
nt a
t po
stm
orte
m
Our
cas
e 5p
13; 1
4q22
U
nbal
ance
d 14
p, 1
4q, 5
p
a O
ur i
nter
pret
atio
n.
b L
ack
of b
andi
ng m
akes
int
erpr
etat
ion
equi
voca
l c
Sib
of F
ried
et
al. [
1977
1 pr
opos
ita.

366 Part et al.
ment in our proposita is therefore unusual and uniquely informative in a karyotypiciphenotypic correlation of the chromosomal segments involved. Our case supports the assertion that regions 5p13 and 14q22 are independ- ently contributory to the more severe phenotype associ- ated with duplication of their respective chromosomal segments and that our proposita’s phenotype is consis- tent with contributions from both critical regions.
ACKNOWLEDGMENTS The authors gratefully acknowledge the contributions
of Renee Bauer in the preparation of this manuscript. This study was supported in part by NIH grant
REFERENCES NS-22897 (J.M.M.-P.).
Abeliovich D, Yagupsky P, Bashen N (1982): 3: 1 meiotic disjunction in a mother with a balanced translocation, 46,XX,t(5;14)(p15;ql3) resulting in tertiary trisomy and tertiary monosomy offspring. Am J Med Genet 12:83-89.
Borgaonkar DS, Blair SM, Lutz JB, Kelly T, Tice RR, Delaaey NV, Hutchinson JR, Bias WB (1973): Cytogenetic study of a 5;14 trans- location in man. J Hered 64:299, 300.
Brimblecombe FSW, Lewis FJ, Vowles M (1977): ‘Complete 5p’trisomy: 1 case and 19 translocation carriers in 6 generations. J Med Genet 14:271-275.
Carnevale A, Hernandez M, Lim6n-Toledo I, Frias S, Castillo J, Del Castillo V (1982): A clinical syndrome associated with dup(5p). Am J Med Genet 13:277-283.
Chia NL, Bousfield LR, Johnson BH (1987): A case report of a de novo tandem duplication (5p)(pl4-pter). Clin Genet 31:65-69.
Di Liberti JH , McKean R, Webb MJ, Williams G (1977): Trisomy 5p: Delineation of clinical features. Birth Defects 19:185-194.
Fried K, Tieder M, Beer S, Rosenblatt M, Krespin HI (1977): Mental retardation with 45 chromosomes 45,XX, - 5, - 14, + der(5)t(5;14) (p15;q13)mat due to familial balanced reciprocal translocation. J Med Genet 14:68-72.
Johnson VP, Aceto T, Likness C (1979): Trisomy 14 mosaicism: case report and review. Am J Med Genet 3:331-339.
Khodr GS, Cadena G, Le KL, Kagen-Hallet KS (1982): Duplication (5p13-pter): Prenatal diagnosis and review of the literature. Am J Med Genet 12:43-49.
Kleczkowska A, Fryns JP, Moerman Ph, Vandenberghe K, Van den Berghe H (1987): Trisomy of the short arm of chromosome 5: au- topsy data in a malformed newborn with inv dup (5Kp13.1-~15.3). Clin Genet 32:49-56.
Kovacs G, Mihai C (1979): Tertiary trisomy 14q-, due to paternal balanced translocation 46,XY,t(1;14)(q44;q22). Hum Genet 49:
Lejeune J, Lafourcade J, Berger R, Turpin R (1964): Segregation fami- hale d’une translocation 5-13 determinant une monosomie et une trisomie partielle du bras court du chromosome 5: maladie du “cri du chat” et sa “reciproque.” C R Seances Acad Sci [III]
175-178.
258:5767-5770. Pena SDJ, Ray M, McAlpine PJ, Ducasse C, Biggs J, Hamerton JL
(1976): Tertiary trisomy 14: Is there a syndrome? Birth Defects XII(5): 113-1 18.
Shinno NW, Wilson MG, Towner JW, Siris E (1973): Partial trisomy in a family with an inherited translocation involving chromosomes no. 5 and 14. Clin Res 21:297.
Stoll C, Rethore MO, Laurent C, Lejeune J (1975): Le contretype de la maladie du cri du chat la trisomie 5p. Arch Fr Pediatr 32:551-561.
Wolf U, Reinwein H, Gey W, Klose J (1966): Cri-du-chat-Syndrom mit Translokation 5iD2. Humangenetik 2:63-77.





























