Embed Size (px)
Citation preview

Research Paper
Mediators of Inflammation, 5, 196-201 (1996)
PRO-INFLAMMATORY non-pancreatic phospholipaseA2 (sPLA2) is markedly over-expressed in acutesystemic and chronic local inflammatory pro-cesses. Since in acute phase reaction sPLA2 isoften over-expressed simultaneously with acutephase proteins (APP), it is important to deter-mine whether APP interacts with sPLA2. We testedten APPs for interaction with sPLA2 using as asubstrate multilamellar Hposomes composedeither of PC:Lyso PC or PE:Lyso PE. Using PC:LysoPC substrate, CRP, lactoferrin and SAP werefound to inhibit sPLA2 activity with an ICs0 of 25g/ml, 7.5 g/ml and 50 g/ml, respectively, cor-responding to 0.21 M, 0.1 M and 0.21respectively. Using PE:Lyso PE substrate only SAPwas inhibitory, with an ICs0 of 10 g/ml (0.04M). Phosphorylcholine abolished the inhibitoryactivity of CRP but not of SAP or lactoferrin.Addition of phosphorylethanolamine or of excesscalcium had no effect on the inhibitory activity ofAPP. Limulin, lysozyme, transferrin, 2-micro-globulin, 2-macroglobulin, human and bovinealbumins had no effect on sPLA2 activity. There-fore neither the structure of pentraxins, or iron-binding, bacteriostatic property or amyloidogenicproperty preclude whether APP modulates sPLA2activity. Inhibition of pro-inflammatory sPLA2 byAPP may be one of the protective mechanisms ofthe acute phase reaction.
Key words: Acute phase proteins, Inflammation, Phospho-lipase A2
Inhibition of the activity ofpro-inflammatory secretoryphospholipase A2 by acute phaseproteins
W. Pruzanski,cA E. Stefanski and P. Vadas
Inflammation Research Group, The WellesleyHospital Research Institute, University of Toronto,Ontario, Canada, M4Y 1J3
Cacorresponding Author
Introduction
Secretory non-pancreatic phospholipase A2(sPLA2) belongs to the group of low-molecular-weight, calcium-dependent, lipolytic enzymes. Itplays an important physiological role in hostdefence participating in the destruction of Gram-negative microorganisms.2’3 sPLA was also foundto exert modulatory activity on the cellular pro-liferation and tumour formation in the intestinaltract.4’5 Excessive activity of circulating sPLA2 wasdiscovered in several systemic inflammatoryresponse syndromes (SIRS) such as clinical andexperimental sepsi6s,9multiorgan failure and sali-cylate intoxication,- and in more localized pro-cesses such as peritonitis and inflammatoryarthritis.’A pathogenetic role of sPLA2 wasimplicated by the observation that enzymaticactivity and immunoreactivity of sPLA2 in SIRScorrelated with the severity and outcome of thedisease,6-9 and by the fact that hypotensioninduced either by Gram-negative microorgan-isms7 or by infusion of sPLA26 could be at-tenuated by inhibition of the enzyme. Acute
inflammatory processes induced by sPLA2 admin-istered intracutaneously,2 into subcutaneous air
14 15pouches13 or intraarticularly, could beblocked by inhibitors of PEA2.12-15
In general terms inhibition of sPLA2 can beinduced by three mechanisms: inhibition ofsynthesis, competition for substrate, or directinhibition of sPLA2 enzymatic activity. The bestcharacterized inducers of its synthesis are cyto-kines, such as IL-1 and TNF.6 IL-6 and oncosta-tin M induce sPLA2 in cells of hepatic origin suchas Hep G217’18 and normal human liver cells (W.Pruzanski et al., unpublished). Endotoxin wasalso found to be a strong inducer of sPLA2.18The above observations were consistent with thepostulate that sPLA2 plays an important patho-genetic role in inflammatory processes and led toan extensive search for exogenous inhibitors.However, very little is known about endogenousmodulation of sPLA2. Two endogenous inhibi-tors, glucocorticoid-inducible lipocortin9 andcomplement fragments2 have been found, butneither has evolved into a useful therapeuticagent.
196 Mediators of Inflammation Vol 5 1996 (C) 1996 Rapid Science Publishers

Acute phase proteins and PIA2
During the acute phase reaction the synthesis phosphatidylethanolamine (PE) with or withoutand release of sPLA2 occurs simultaneously with LysoPE (2:1) were prepared in chloroform andthat of a large number of acute phase pro- evaporated to dryness. Multilamellar liposomesteins.1’2 Experimental studies have shown that were made by dispersing the resulting lipidendotoxin, IL-1, TNF and IL-6 are the main indu- mixture in 100 mM Tris HC1 buffer, pH 8.0,cers of such release.21’22 Therefore the process followed by heating for 2 min at 41C and vor-of induction of acute phase proteins by liver cells texing for 2 min before use. Only freshly pre-seems to be similar to that of sPLA2. Little is pared liposomes were used. Assays were carriedknown about the possible interactions of acute out in a total volume of 0.2 ml of 100 mM Trisphase proteins. We reported recently that CRP, HC1, pH 8.0 containing 2.5 or 10 mM CaCl2, 0.1%one of the classical acute phase proteins is a bovine serum albumin and 5-30 nmoles of PCstrong inhibitor of sPLA2, binding competitively (or PE) vesicles (containing 2-16 nCi of [4C]to the phosphorylcholine-containing substrates2 dipalmitoyl PC per assay). The optimal con-whereas SAA, another acute phase protein, centration was found to be 20 nmoles/assay andenhances sPLA2 activity.24 Herein we report that this was used in all experiments. If acute phasetwo other acute phase proteins, serum amyloid P proteins were included, they were preincubated(SAP) and lactoferrin suppress sPLA2 activity, with 20 nmoles of liposomes for 1 h at 41CThese, previously unrecognized, interactions of before the assay. The reaction was then startedacute phase proteins with proinflammatory by addition of 20 l.tl of recombinant humansPLA2, may add a new aspect to the under- sPLA2 stock solution with final sPLA2 concentra-standing of the complex role of acute phase tions ranging between 10 and 200 ng/200 l.tlreaction, assay volume unless otherwise stated. The reac-
tion mixture was then incubated for 30 min at
Materials and Methods 41C. The reaction was stopped by the additionof 1.32 ml isopropanol/heptane/0.5 M H2SO4
1,2-Dipalmitoyl-phosphatidylcholine (dipalmi- 40:10:1 (v/v/v). The mixture was heated for 1toyl PC) was obtained from Avanti Polar Lipids min at 60C before addition of 0.66 ml H20 and(Birmingham, AL). Phosphatidylcholine I-a-dipal- 0.8 ml heptane. The two phases were allowed tomitoyl [2-palmitoyl-l-4C] (55.5 mCi/mmol) and separate and after centrifugation for 10 min atoleic acid [1-14C] (40-60 mCi/mmol) were pur- 1 500 rpm, 0.8 ml of the upper phase was addedchased from DuPont NEN Products. t-3-phospha- to 1.0 ml heptane containing 100 mg silica gel.tidylethanolamine 1-palmitoyl [2-14C linoleoyl] The mixture was spun again for 10 min at 1 500(50-60 mCi/mmol) was obtained from Amer- rpm and 1.0 ml of the supernatant was used forsham (Arlington Heights, IL). Recombinant scintillation counting of 42-1abelled free palmitichuman sPLA2 (rh-sPtA2) was a generous gift of acid. All assays were done in triplicate.Dr Jeffrey Browning, Biogen Corporation (Cam-bridge, MA). Bio-Rad protein assay reagent was Immunologic assays: Anti-human (mouse IgG2)purchased from Bio-Rad (Richmond, CA). t-t- CRP/SAP antibody (clone CRP-20) C6552 waslysophosphatidylcholine palmitoyl, -a-phospha- obtained from Sigma (St Louis, MO). It reactedtidylethanolamine-]-linoleoyl-y-palmitoyl, -a-lyso- with an epitope located on the 24 kDa subunitphosphatidylethanolamine palmitoyl, bovine of denatured and reduced CRP and it recognizedserum albumin and silica gel were purchased CRP independently of the Ca2+ binding site.from Sigma Chemical Corporation. All reagents This antibody did not recognize the calciumwere analytical grade or better, dependent phosphorylcholine binding site of
Recombinant human lysozyme, bovine serum CRP. It cross-reacted with human SAP, but notalbumin (BSA), human serum albumin (HSA), with CRP from Limulus polyphemus. Workinglactoferrin purified from human milk, transferrin, dilutions were at least 1:4 000 per btg of antigen[2-microglobulin purified from human urine, in indirect EI.ISA assay. In antibody assays, thelimulin from Limulus polyphemus and ai-macro- antigens were preincubated with appropriatelyglobulin were obtained from Sigma Chemical diluted antibodies for 60 min at room tempera-Company (St Louis, MO). CRP purified from rare. Then the liposomal substrate was addedhuman plasma was obtained from Helix Biotech and the mixture was further incubated for 60Corporation, Richmond, BC and SAP purified min at 41C. The PEA2 was finally added and thefrom human serum, from Calbiochem, San incubation was carried on for an additional 30Diego, CA. min at 41C.
Each experiment was repeated at least threeLiposome assay: Aliquots of dipalmitoyl PC, [14C] times. The differences between the results anddipalmitoyl PC, with or without LysoPC (2:1) or controls were assessed by Student’s t-test.
Mediators of Inflammation Vol 5 1996 197
W. Pruzanski, E. Stefanski and P. Vadas
Results
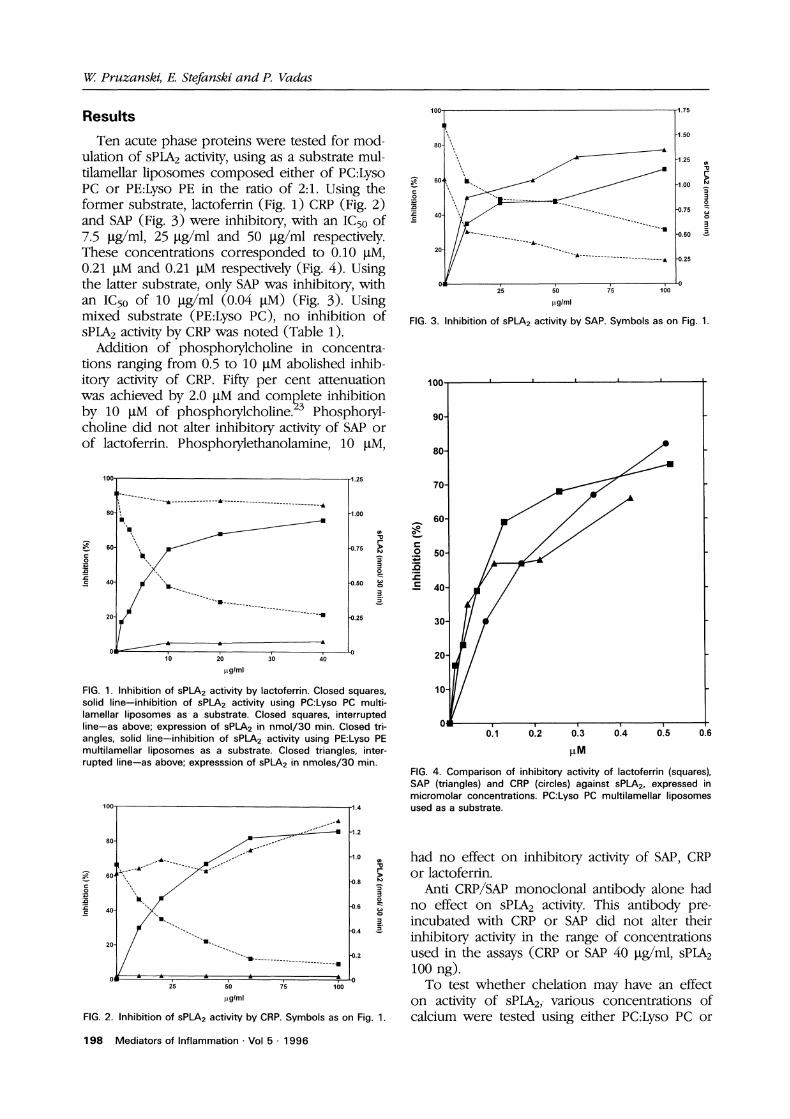
Ten acute phase proteins were tested for mod-ulation of sPLA2 activity, using as a substrate mul-tilamellar liposomes composed either of PC:LysoPC or PE:Lyso PE in the ratio of 2:1. Using theformer substrate, lactoferrin (Fig. 1) CRP (Fig. 2)and SAP (Fig. 3) were inhibitory, with an IC50 of7.5 l.tg/ml, 25 I.tg/ml and 50 Ig/ml respectively.These concentrations corresponded to 0.10 I.tM,0.21 l.tM and 0.21 l.tM respectively (Fig. 4). Usingthe latter substrate, only SAP was inhibitory, withan IC50 of 10 btg/ml (0.04 btM) (Fig. 3). Usingmixed substrate (PE:Lyso PC), no inhibition ofsPLA2 activity by CRP was noted (Table 1).
Addition of phosphorylcholine in concentra-tions ranging from 0.5 to 10 I.tM abolished inhib-itory activity of CRP. Fifty per cent attenuationwas achieved by 2.0 btM and complete inhibitionby 10 l,tM of phosphorylcholine.2 Phosphoryl-choline did not alter inhibitory activity of SAP orof lactoferrin. Phosphorylethanolamine, 10 l.tM,
lOO 1.25
-1.00
60- .r i’.@
40- "0.50 g
20- -I 4.25
0: ,"10 20 3’0 40
tglml
FIG. 1. Inhibition of sPLA2 activity by lactoferrin. Closed squares,solid line--inhibition of sPLA2 activity using PC:Lyso PC multi-lamellar liposomes as a substrate. Closed squares, interruptedline--as above; expression of sPLA2 in nmol/30 min. Closed tri-angles, solid line--inhibition of sPLA2 activity using PE:Lyso PEmultilamellar liposomes as a substrate. Closed triangles, inter-rupted line--as above; expresssion of sPLA2 in nmoles/30 min.
80"
1.4
’s ’0 ;s lo0
g/ml
-1.2
-0.6
-0.4
-0.2
FIG. 2. Inhibition of sPLA2 activity by CRP. Symbols as on Fig. 1.
198 Mediators of Inflammation Vol 5 1996
I’,,,,, ,-1.50
o .......... ........................... -0.
2 ’o r’ 1oiglml
FIG. 3. Inhibition of sP2 activity by SAP. Symbols as on Fig. 1.
-1.25
-1.00
-0.75
80
70
60
20
0.1 0’.2 0’.3 0’.4 0’.5 0.6
FIG. 4. Comparison of inhibitory activity of lactoferrin (squares),SAP (triangles) and CRP (circles) against sPLA2, expressed inmicromolar concentrations. PC:Lyso PC multilamellar liposomesused as a substrate.
had no effect on inhibitory activity of SAP, CRPor lactoferrin.
Anti CRP/SAP monoclonal antibody alone hadno effect on sPLA2 activity. This antibody pre-incubated with CRP or SAP did not alter theirinhibitory activity in the range of concentrationsused in the assays (CRP or SAP 40 l.tg/ml, sPLA2100 ng).To test whether chelation may have an effect
on activity of sPLA2,, various concentrations ofcalcium were tested using either PC:Lyso PC or

Acute phase proteins and PIA2
Table 1. Impact of substrate on inhibitory activity of CRP
Substrate sPLA2 (nmoles/301)
PE:Lyso PE 0.99PE:Lyso PE + CRP 1.09PC:Lyso PC 0.93PC:Lyso PC + CRP 0.31PE:Lyso PC 0.93PE:Lyso PC + CRP 1.35
*sPLA 100 ng, CRP 40 ILtg/ml.
Since the increase in circulatory sPLA2 activityparallels temporally that of acute phase proteins,it was of interest to investigate whether there isan interaction between APPs and sPLA2. Of tenAPPs tested, three were found to inhibit sPLA2activity. These included CRP, SAP and lactoferrin.CRP is one of the best studied acute phase pro-teins, increasing rapidly up to 1000-fold inAPR.34 Both sPLA2 and CRP bind to the phos-pholipids of perturbed membranes of livingcells2v and to PC:Lyso PC substrate.27’35 It wasfound that CRP is a strong inhibitor of sPLA2
PE:Lyso PE as a substrate. Maximum activity of activity, acting most probably as a competitorsPLA2 was observed at the level of 0.5-1.0 l.tM for the substrate.2 The substrates used in theCa2+ In some experiments with CRP, SAP, or former2 and present study form vesicles of dif-lactoferrin, Ca2+ concentrations were increased ferent phospholipid composition. It was foundup to 20 I.tM. Excess of Ca2+ had no effect on that binding of CRP to such vesicles is pre-inhibitory activity of these proteins, ferential when they are composed of PC:Lyso PCHuman and bovine serum albumin, transferrin, in the proportion of 2:1. Most probably it is
lysozyme, 2-microglobulin, ai-macroglobulin related to optimally altered surface packingand limulin had no effect on the activity of sPLA2. density of the substrate. PE:Lyso PE and PE:Lyso
PC substrates were not susceptible to hydrolytic
Discussion activity of sPLA2.
Since CRP belongs to the family of pentrax-An insult to an organism’s homeostasis caused ins (PEP), two other pentraxins, SAP and
by injury, infection or inflammation, leads to a limulin were also tested. The former was foundswift systemic response called the acute phase to inhibit sPLA2 whereas the latter did not. SAPreaction (APR) (reviewed in References 21, 25- is a major APP in mice27 but a minor one in27). In the case of chronic or recurrent man,6 increasing in APR only three-fold frominflammation, APR may become quite pro- the physiological level of 30-40 I.tg/ml to nolonged.25 An integral part of APR is a rapid more than 90 l.tg/ml.27’36 It shares 60% homo-synthesis and extracellular release of a large logy in amino acid sequence with CRP.2 SAPnumber of acute phase proteins (APP), and circulates in the form of two pentameric mole-simultaneous decrease in some other proteins, cules bound ’face to face’ and, in contrast tothe so called negative APP. Liver is the major CRP, is glycosylated.v The gene for SAP issource of APP synthesis, but other cells partici- located on the long arm of chromosome 1pate in the synthesis of APP as well.22’26’28 APR is (1q12-1q23), close to the gene coding for CRP.orchestrated by a group of inflammatory media- It was suggested that both are products of antors, including glucocorticoids, cytokines, ana- ancestral duplication event.8 Whereas CRP
21 25phylotoxins and growth factors. The group binds mainly to phosphorylcholine,6’9 SAP hasof cytokines which induce APP synthesis high affinity to phosphorylethanolamine.6 Inincludes, but is not limited to, IL-1, TNF, IL-6 and our study, SAP inhibited sPLA2 activity whenoncostatin.21’22’26’29’3 either PE:Lyso PE or PC:Lyso PC were used asThe same group of cytokines was also found substrates; however, much lesser concentrations
to induce the synthesis and release of the pro- of SAP were needed to achieve IC50 wheninflammatory enzyme, secretory non-pancreatic PE:Lyso PE was employed. In human serum,
16 18phospholipase A2 (sPLA2). sPLA2, first dis- SAP binds to C4-binding protein (C4BP) andcovered in experimental peritonitis and in to various types of phospholipid vesicles. Thesefluids draining inflammatory sites,2 was found reactions were found to be calcium dependentto raise rapidly in the circulation in systemic and can be disrupted by phosphorylethanol-inflammatory response syndromes such as septic amine. In our study phosphorylethanolamineshock,7 salicylate poisoning9 and malaria and did not block the inhibitory activity of SAP.in the milieu of more localized inflammatory Limulin in Limulus polyphemus is analogous
11sites such as arthritis. In the former group, cir- to CRP in humans. It shares only 25-30% aminoculating sPLA2 correlated with both complica- acid homology with CRP40’41 and SAP41 and,tions and the outcome of the disease,7 whereas similarly to CRP, binds avidly to phosphorylcho-in the latter it correlated with the disease line.34’42’43 Limulin shares with human CRP twoactivity. regions of preserved residues, 52-67, respon-
Mediators of Inflammation Vol 5 1996 199

W. Pruzanski, E. Stefanski and P. Vadas
Table 2. Acute phase proteins as inhibitors of sPLA2
Group Inhibitors Non-inhibitors
Pentraxins CRP, SAP LimulinIron binders Lactoferrin TransferrinBacteriostatic Lactoferrin LysozymeAmyloidogenic substances SAP SAA*, 2-mNegative APP HDL* BSA, HSAProteinase inhibitors N/D 2-Macroglobulin
ferrin, and enhancers such as SiR24 of sPLA2activity, emphasizes the complexity of the organ-ism’s response to injury. Since, in vivo, variousacute phase proteins are over-expressed to verydifferent orders of magnitude, the end resultregarding their impact on proinflammatory activ-ity of sPLA2 cannot be predicted.
*Published in Biochem J 1995; 309:461-464.+N/D: not done.
sible for the binding site to phosphorylcholineand 139-153 that binds Ca2+.1’44 The fact that,in contrast to human CRP, limulin does notinhibit sPLA2 activity, may mean that either theabove two binding sites are not responsible forinhibition of sPLA2 activity, or that the affinity oflimulin to the substrate is much weaker thanthat of CRP. Therefore, pentraxin structure doesnot automatically confer anti-sPLA2 activity, sincein contrast to CRP and SAP, limulin was notinhibitory. Neither am.yloidogenic property ofSAP can be linked to the inhibition of sPLA2,since SiR24 and [2-microglobulin, both knownparticipants in amyloidogenesis, did not inhibitsPLA2 activity (Table 2).Of three sPLA2 inhibitors, lactoferrin was the
most active. Lactoferrin is one of the iron-carry-ing proteins, produced mainly by polymorpho-nuclear cells.45’46 Its synthesis is induced byTNF45’46 and in sepsis it behaves like a classicalacute phase protein46’47 with the potential toincrease in the circulation ten or more fold from
46 48its physiological level of 0.2-2.8 mg(.ml.-Infusions of LPS to healthy volunteers46 or topiglets,48 or of Escherichia coli to piglets47 leadto a marked increase in circulating lactoferrin. Inturn, lactoferrin interacts with LPS, preventin.iron-catalysed formation of hydroxyl radicals.Lactoferrin protects mice against a lethal dose ofE. coli in vivo,47 acting as bacteriostatic glycopro-tein and inducing damage to the outer mem-brane of Gram-negative bacteria.49
It seems, therefore, that lactoferrin acts uponGram-negative bacteria similarly to bacterialpermeability increasing protein (BPI).2 SincesPLA2 hydrolyses membrane phospholipids of
killed by BPI, the fact thatmicroorganisms 2
lactoferrin inhibits sPLA2 activity may mean thatit acts as a limiting factor in hydrolytic activityof the latter. The property of iron chelationby lactoferrin was not related to inhibitoryanti-sPLA2 activity, since transferrin, another ironchelator did not inhibit sPLA2.
The fact that during the acute phase there issimultaneous co-induction and over-expressionof both inhibitors such as CRP, SAP and lacto-
References1. Dennis EA. Diversity of group types, regulation and function of phospho-
lipase A. J Biol Chem 1994; 269: 13057-13060.2. Wright GW, Ooi CE, Weiss J, Elsbach P. Purification of a cellular (granu-
locyte) and an extracellular (serum) phospholipase A2 that participate inthe destruction of Escherichia coli in rabbit inflammatory exudate. J BiolChem 1990; 265: 6675-6681.
3. Harwig SSL, Tan L, Qu X-D, Cho Y, Eisenhauer PB, Lehrer RI. Bactericidalproperties of murine intestinal phospholipase A2. J Clin Invest 1995; 95:603-610.
4. MacPhee M, Chepenik KP, Liddell RA, Nelson KK, Siracusa LD, BuchbergAM. The secretory phospholipase A2 gene is a candidate for the Momllocus, a major modifier of Apc>Linduced intestinal neoplasia. Cell 1995;81: 957-966.
5. Kennedy BP, Payette P, Mudgett J, et al. A natural disruption of thesecretory group II phospholipase A2 gene in inbred mouse strains. J BiolChem 1995; 2"/0: 22378-22385.
6. Vadas P, Hay JB. Involvement of circulating phospholipase A2 in thepathogenesis of the hemodynamic changes in endotoxin shock. Can JPhysiol Pharmaco11983; 61: 561-566.
7. Vadas P, Pruzanski W. Induction of group II phospholipase A2 expres-sion and the pathogenesis of the sepsis syndrome. Circulatory Shock1993; 39: 160-167.
8. Vadas P, Pruzanski W, Stefanski E, et al. The pathogenesis of hypoten-sion in septic shock: correlation of circulating phospholipase A2 levelswith circulatory collapse. Crit Care Med 1988; 16: 1-7.
9. Vadas P, Schouten B, Stefanski E, Scott E, Pruzanski W. The associationof hyperphospholipasemia A2 with multisystem organ failure due to sali-cylate intoxication. Crit Care Med 1993; 21: 1087-1091.
10. Vadas P, Pruzanski W, Stefanski E, et al. Phospholipase A in acutebacterial peritonitis in man. In: Dennis EA, Hunter T, eds. Cell Activationand Signal Initiation: receptor and phospholipase control of inositolphosphate, PAF, and eicosanoidproduction. Alan R Liss, 1989; 311-316.
11. Pruzanski W, Koo Seen Lin M, Vadas P. Secretory phospholipase A2 inrheumatic diseases. In: Glaser KB, Vadas P, eds. Phospholipae Aa in Clin-ical Inflammation. Molecular approaches to pathophysiology. CRC Press,1995; 127-147.
12. Pruzanski W, Vadas P, Fomasier V. Inflammatory effect of intradermaladministration of soluble phospholipase A in rabbits. J Invest Dermatol1986; 86: 380-383.
13. Cirino G, Cicala C, Sorrentino L, Maiello FM, Browning JL. Recombinantsecreted nonpancreatic phospholipase A induces a synovitis-like inflam-mation in the rat air pouch. J Rheumato11994; 21: 824-829.
14. Vadas P, Pruzanski W, Kim J, Fomasier V. The proinflammatory effect ofintra-articular injections of soluble human and venom phospholipase A2.AmJ Patho11989; 134: 807-811.
15. Bomalaski JS, Clark MA. Phospholipase A2 and arthritis. Arth Rheum1993; 36: 190-198.
16. Vadas P, Pruzanski W, Stefanski E, et al. Extracellular phospholipase &secretion is a common effector pathway of interleukin-1 and tumournecrosis factor action. Immunolog Letters 1991; 28: 187-194.
17. Crowl RM, Stoller TJ, Conroy RR, Stoner CR. Induction of phospholipaseA gene expression in human hepatoma cells by mediators of the acutephase response. J Biol Chem 1991; 266: 2647-2651.
18. Vadas P, Browning J, Edelson J, Pruzanski W. Extracellular phospholipase& expression and inflammation: the relationship with associated diseasestates. J Lipid Mediators 1993; 8: 1-30.
19. Goulding NJ, Guyre PM. Glucocorticoids, lipocortins and the immuneresponse. Current Opinion Immuno11993; 5: 108-113.
20. Suwa Y, Kudo I, Imaizumi A, et al. Proteinaceous inhibitors of phospho-lipase A2 purified from inflammatory sites in rats. Proc Natl Acad Sci USA1990; 8"/: 2395-2399.
21. Baumann H, Gauldie J. The acute phase response. Immunol Today 1994;15: 74-80.
22. Ramadori G, Sipe JD, Dinarello CA, Mizel SB, Colten HR. Pretranslationalmodulation of acute phase hepatic protein synthesis by murine recombi-nant interleukin-i (IL-1) and purified human IL-1. J Exp Med 1985; 162:930-942.
23. Vadas P, Stefanski E, Grouix B, Schouten BD, Pruzanski W. Inhibition ofhuman group II phospholipase A by C-reactive protein in vitro. J LipidMediat Cell Signalling 1995; 11: 187-200.
24. Pruzanski W, de Beer FC, de Beer MC, Stefanski E, Vadas P. Serum
200 Mediators of Inflammation Vol 5 1996

Acute phase proteins and PIA2
amyloid A protein enhances the activity of secretory non-pancreatic phos-pholipase A2. BiochemJ 1995; 309: 461-464.
25. Steel DM, Whitehead AS. The major acute phase reactants. C-reactiveprotein, serum amyloid P component and serum amyloid A protein.Immunol Today 1994; 15: 81-88.
26. Richards C, Gauldie J, Baumann H. Cytokine control of acute phaseprotein expression. Eur Cytokine Net 1991; 2: 89-98.
27. Pepys MB, Baltz ML. Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid Aprotein. Adv Immuno11983; 34: 141-212.
28. Courtoy PJ, Lombart C, Feldmann G, Moguilevsky N, Rogier E. Synchro-nous increase of four acute phase proteins synthesized by the samehepatocytes during the inflammatory reaction. Lab Invest 1981; 44:105-115.
29. Heinrish PC, Castell JV, Andus T. Interleukin-6 and the acute phaseresponse. BiochemJ 1990; 265: 621-636.
30. Richards CD, Shoyab M. The role of oncostatin M in the acute phaseresponse. In: Mackiewicz A, Kushner I, Baumann H, eds. Acute PhaseProteins: molecular biology, biochemistry, clinical applications. BocaRaton: CRC Press, 1993; 321-327.
31. Franson R, Dobrow R, Weiss J, Elsbach P, Weglicki WB. Isolation andcharacterization of a phospholipase A2 from an inflammatory exudate. JLipid Res 1978; 19: 18-23.
32. Vadas P, Wasi S, Movat HZ, Hay JB. A novel, vasoactive product and plas-minogen activator from afferent lymph cells draining chronic inflamma-tory lesions. Proc Soc Exp Biol Med 1979; 161: 82-85.
33. Vadas P, Taylor T, Molyneux M, Stefanski E, Pruzanski W. Serum phos-pholipase A2 and disease severity in children with falciparum malaria. AmJ Trop Med Hyg 1993; 49: 455-459.
34. Schultz DR, Arnold PI. Properties of four acute phase proteins: C-reactiveprotein, serum amyloid A protein, 0tl-acid glycoprotein and fibrinogen.Sem Arthr Rheum 1990; 20: 129-147.
35. Nagpurkar A, Saxena U, Mookerjea S. Interaction of rat serum phosphor-ylcholine-binding protein with phospholipid-containing liposomes. J BiolChem 1983; 258: 10518-10523.
36. Schwalbe RA, Dahlback B, Coe JE, Nelsestuen GL. Pentraxin family ofproteins interact specifically with phosphorylcholine and/orphosphorylethanolamine. Biochem 1992; 31: 4907-4915.
37. Swanson SJ, Christner RB, Mortensen RF. Human serum amyloid P-component (SAP) selectively binds to immobilized or bound formsof C-reactive protein (CRP). Biochim iBiophys Acta 1992; 1160: 309-316.
38. Dowton SB, Colten HR. Acute phase reactants in inflammation andinfection. Sem Hemato11988; 25: 84-90.
39. Volanakis JE, Xu Y, Macon KJ. Human C-reactive protein and hostdefence. In: Marchalonis J, Reinisch C, eds. Defense Molecules. (UCIASymposia on Molecular and Cellular Biology. New Series). Alan R. Liss,1990; 121: 161-175.
40. Ying S-C, Marchalonis JJ, Gewurz AT, et al. Reactivity of anti-human C-reactive protein (CRP) and serum amyloid P component (SAP) mono-clonal antibodies with limulin and pentraxins of other species. Immunol1992; ’76: 324-330.
41. Vasta GR. Invertebrate lectins, C-reactive proteins and serum amyloid.Structural relationships and evolution. In: Marchalonis J, Reinisch C, eds.Defense Molecules. (UCIA Symposia on Molecular and Cellular Biology.New Series). Alan R. Liss, 1990; 121: 183-199.
42. Tennent GA, Butler PJG, Hutton T, et al. Molecular characterization ofLimulus polyphemus C-reactive protein. I. Subunit composition. Eur JBiochem 1993; 214: 91-97.
43. Robey FA, Liu T-Y. Limulin: a C-reactive protein from Limulus poly-phemus. J Biol Chem 1981; 256: 969-975.
44. Nguyen NY, Suzuki A, Cheng SM, Zon G, Liu TY. Isolation and character-ization of Limulus C-reactive protein genes. J Biol Chem 1986; 261:10450-10455.
45. Cohen MS, Mao J, Rasmussen GT, Serody JS, Britigan BE. Interaction oflactoferrin and lipopolysaccharide (LPS): effects on the antioxidant prop-erty of lactoferrin and the ability of LPS to prime human neutrophils forenhanced superoxide formation. J Infec Dis 1992; 166: 1375-1378.
46. Nuijens JH, Abbink JJ, Wachtfogel YT, et al. Plasma elastase alpha 1-anti-
trypsin and lactoferrin in sepsis: evidence for neutrophils as mediators infatal sepsis. J Lab Clin Med 1992; 119: 159-168.
47. Zagulski T, Lipinski P, Zagulska A, Broniek S, Jarzabek Z. tactoferrin canprotect mice against a lethal dose of Escherichia coli in experimentalinfection in vivo. BrJ Exp Path 1989; ’70: 697-704.
48. Gutteberg TJ, Rokke O, Andersen O, Jorgensen T. Early fall of circulatingiron and rapid rise of lactoferrin in septicemia and endotoxemia: an earlydefence mechanism. ScandJ Infect Dis 1989; 21: 709-715.
49. Ellison Ill RT, Giehl TJ. Killing of Gram-negative bacteria by lactoferrinand lysozyme. J Clin Invest 1991; 88: 1080-1091.
Received 24 January 1996;accepted 22 February 1996
Mediators of Inflammation Vol 5 1996 201





























