Embed Size (px)
Citation preview

Galactosemia screeningwhy? when? how?
Clinical Children`s Hospital “Louis Turcanu ”Timisoara, Romania
Galactosemia screeningwhy?

Galactosemia Is a genetic metabolic disease in which
there is a defect in the body's ability to use the sugar galactose.
People with galactosemia are unable to metabolize the simple sugar galactose. Galactose makes up half of the sugar called lactose that is found in milk. Lactose is a disaccharide, and is made of two sugars, galactose and glucose, bound together.

History
Galactosemia was first discovered in 1908 by the physician Von Ruess.
Publication entitled, "Sugar Excretion in Infancy," reported on a breast-fed infant with failure to thrive, enlargement of the liver and spleen, and "galactosuria".

In 1917, Goppert reported an infant with poor growth, lactose exposure, and hypergalactosuria
The first comprehensive description of the variant form of hereditary galactosemia was in 1935 by Mason and Turner of an African-American infant. It was also the first report of a patient with any form of galactosemia due to GALT deficiency in the American literature. This patient had not been placed on a lactose-restricted diet until 10 months of age.

Galactose metabolism
Normally, the body breaks down lactose into galactose and then into glucose (a sugar used for
energy). People with galactosemia are
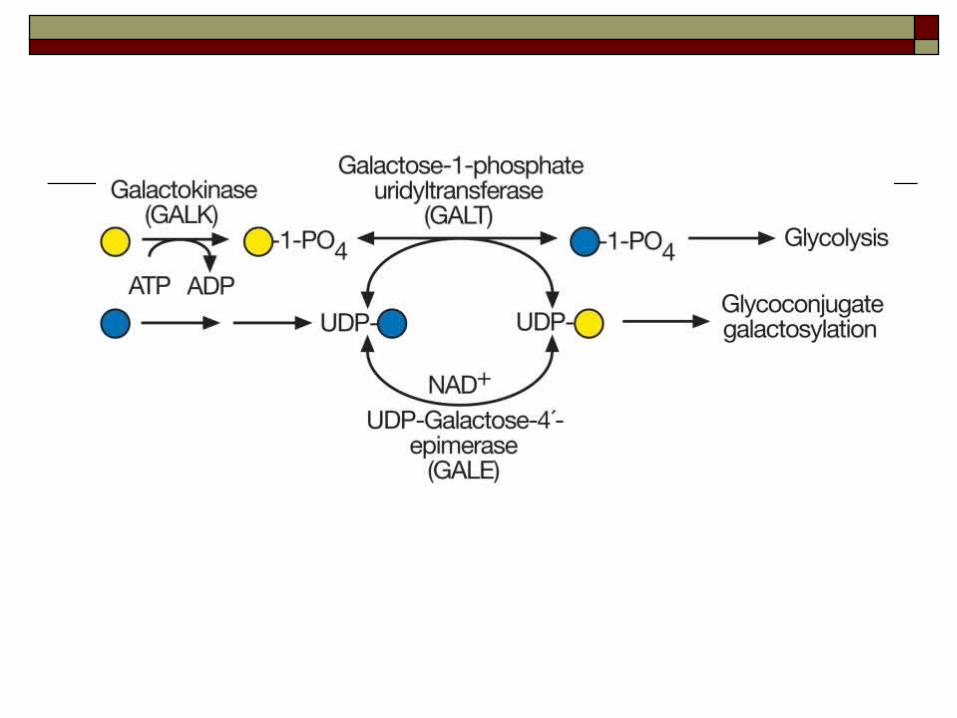
missing an enzyme called GALT , (galactose-1-phosphate uridyl transferase), GALK1 or GALE, which normally converts galactose into glucose.
Without this enzymes, harmful amounts of galactose build up in the blood.

Lactose metabolism

Galactosemia – a genetic disease
It occurs in approximately 1 out of every 60,000 births among Caucasians.
The rate is different for other groups.
•Galactosemia is an inherited disorder.
•Autosomal recessive pattern

Three forms of galactosemia
Galactose-1 phosphate uridyl transferase deficiency (classic galactosemia, the most common and most severe form)- (GALT) Type I
Deficiency of galactose kinase –
(GALK1)Type II
Deficiency of galactose-6-phosphate –epimerase (GALE)-Type III

Galactose-1-phosphate uridyltransferase defect causes accumulation of galactose (appears in blood, urine, reduced to galactitol in lens).
Galactose enters eye, aldose reductase changes it to galactitol. Galactitol accumulates in lens and causes infantile cataracts.
Jaundice, vomiting, poor feeding, infections
Failure to thrive, hepatomegaly Speech disabilities, mental retardation
Classic galactosemia – type I
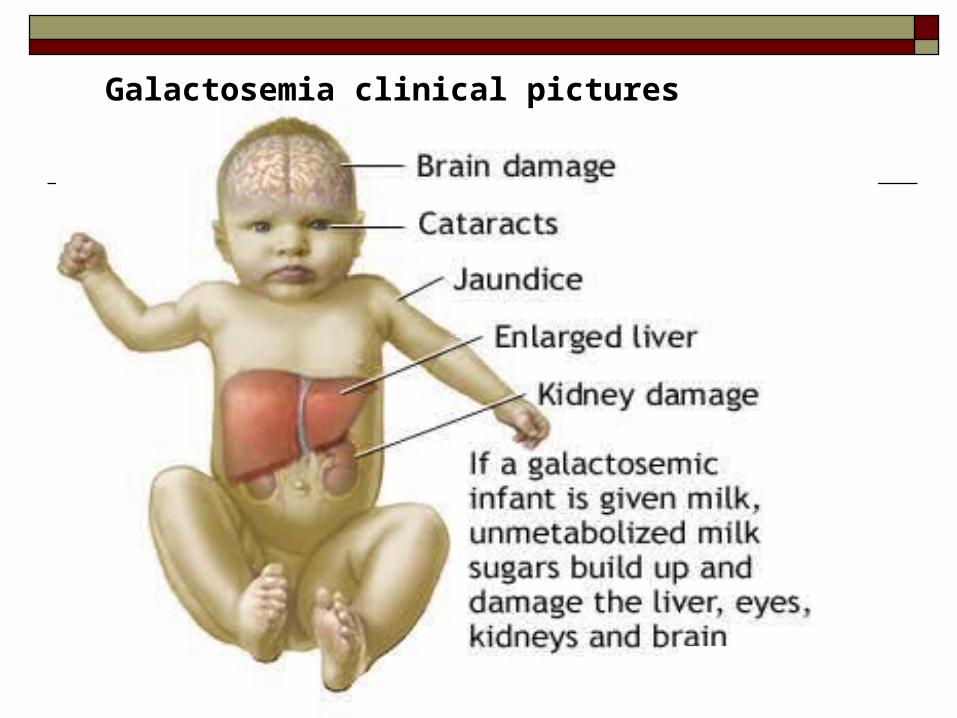
Galactosemia clinical pictures
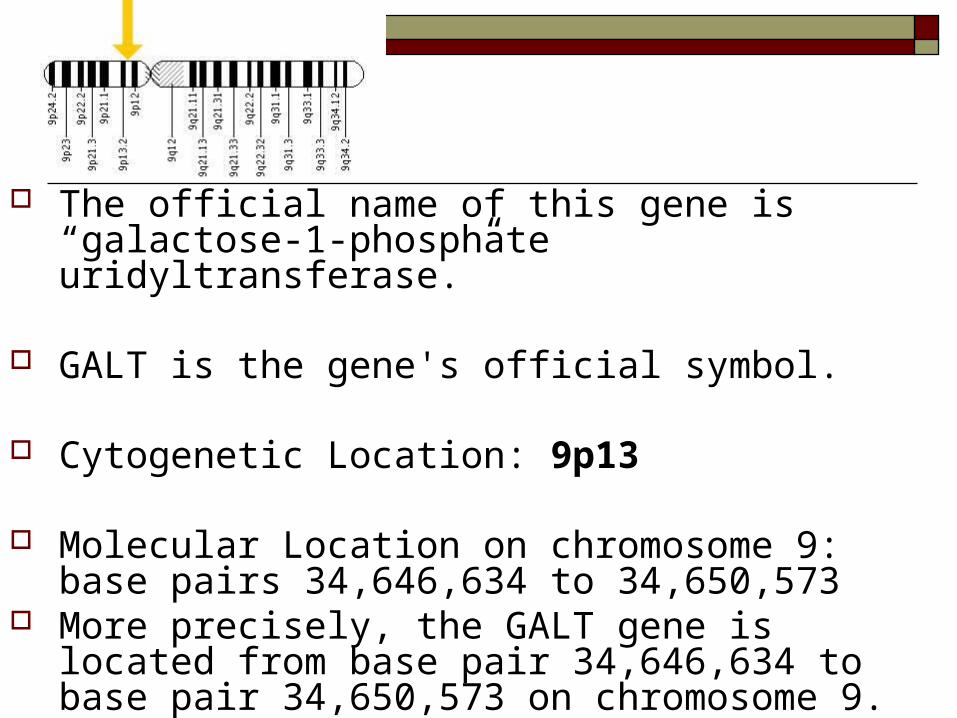
The official name of this gene is “galactose-1-phosphate uridyltransferase.”
GALT is the gene's official symbol.
Cytogenetic Location: 9p13
Molecular Location on chromosome 9: base pairs 34,646,634 to 34,650,573
More precisely, the GALT gene is located from base pair 34,646,634 to base pair 34,650,573 on chromosome 9.
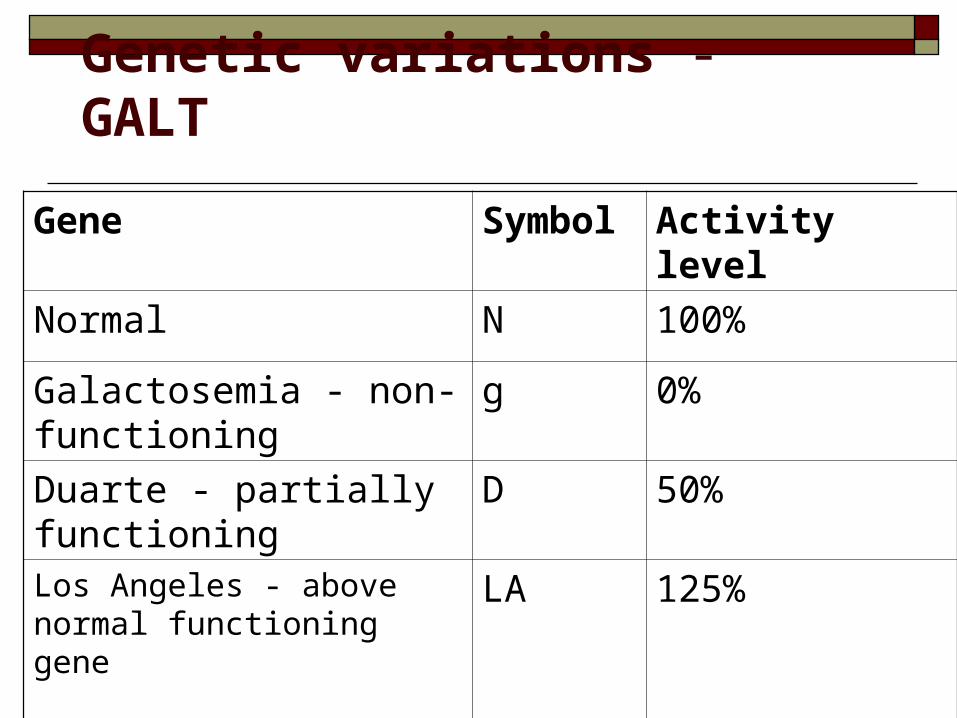
Genetic variations - GALT
Gene Symbol Activity level
Normal N 100%
Galactosemia - non-functioning
g 0%
Duarte - partially functioning
D 50%
Los Angeles - above normal functioning gene
LA 125%

Most commonly seen are Duarte (a low enzyme producing gene) and Los Angeles (an enhanced enzyme producing gene).
These variants and combination of genes (gN, DN, DD, LAN, LAg, LAD) do not require treatment or dietary restriction of galactose.

The Duarte variant is found in 1 in 20 persons.
GALT enzyme (10-25% to 50% activity ) and do not commonly have any symptoms.
The children with Duarte galactosemia who continue to receive breast milk in the first year of life might have higher than normal galactose-1-phosphate levels, but they do not have any symptoms or health problems related to the Duarte variant.

Galactokinase – deficiencyor galactosemia Type II
In 1965, galactokinase deficiency was first identified in a patient who presented with cataracts and galactosuria that developed upon drinking milk.
This presentation differed from that of classic
galactosemia in many important aspects; neither hepatosplenomegaly nor signs of mental retardation were present.
When the researchers realized that the patient did not accumulate galactose-1-phosphate despite the accumulated galactose, the patient's underlying defect was deduced as the lack of the enzyme mediating 1-phosphorylation of galactose.
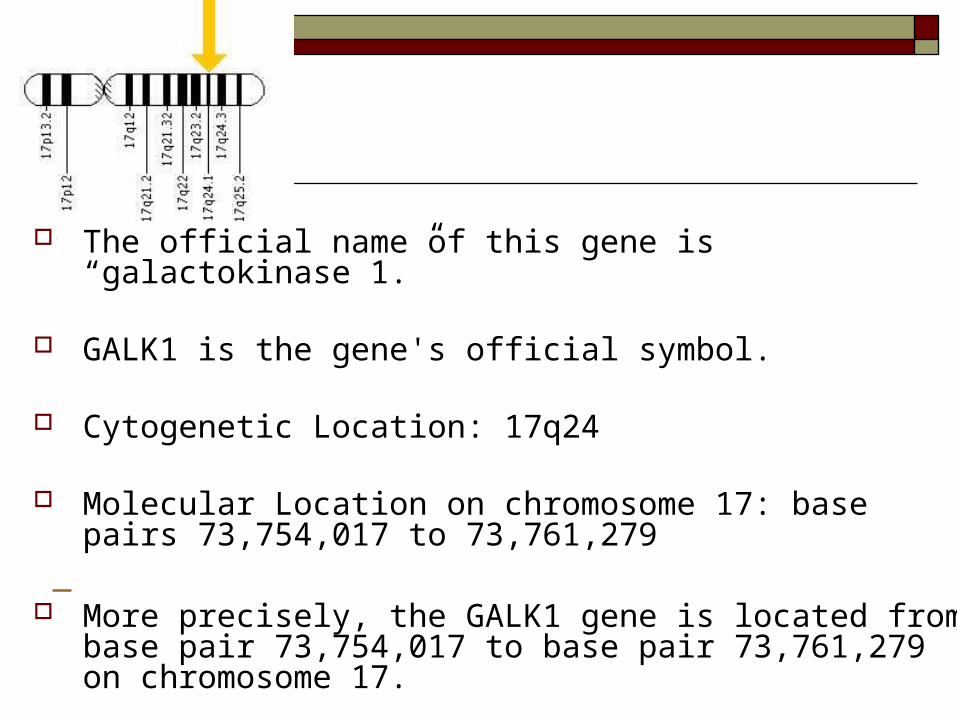
The official name of this gene is “galactokinase 1.”
GALK1 is the gene's official symbol.
Cytogenetic Location: 17q24
Molecular Location on chromosome 17: base pairs 73,754,017 to 73,761,279
More precisely, the GALK1 gene is located from base
pair 73,754,017 to base pair 73,761,279 on chromosome 17.
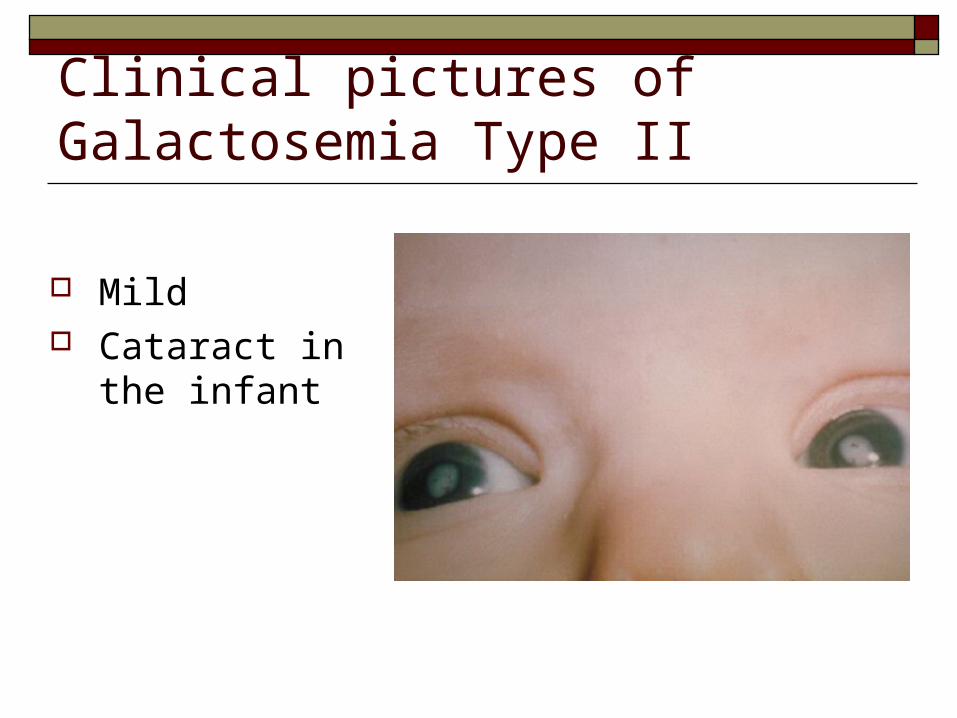
Clinical pictures of Galactosemia Type II
Mild Cataract in the
infant

Galactose epimerase deficiency –galactosemia -Type III
The official name of this gene is “UDP-galactose-4-epimerase.”
More than 20 mutations in the GALE gene have been identified in people with a form of galactosemia known as type III or galactose epimerase deficiency.
Most of these genetic changes alter a single protein building block (amino acid) in UDP-galactose-4-epimerase, which makes the enzyme unstable or disrupts its usual function.

Cytogenetic Location: 1p36-p35
Molecular Location on chromosome 1: base pairs 24,122,088 to 24,127,293

Galactosemia screening
when? how?
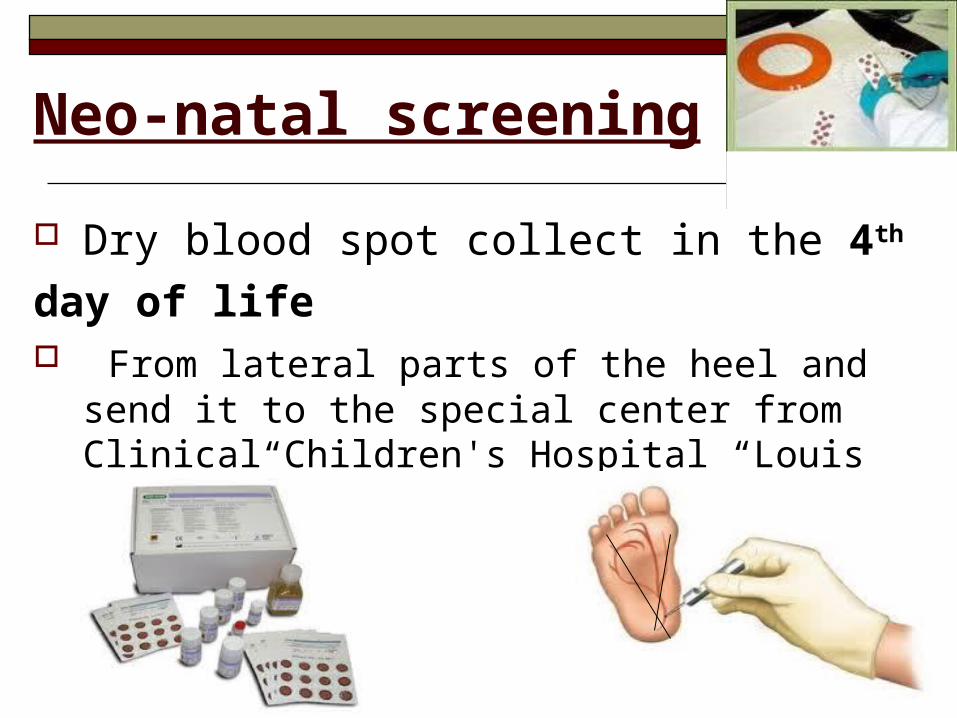
Neo-natal screening
Dry blood spot collect in the 4th day of life From lateral parts of the heel and send it to
the special center from Clinical Children's Hospital “Louis Turcanu”

A positive newborn screen test must be followed by a quantitative erythrocyte galactose-1-phosphate uridyltransferase (GALT) analysis
A GALT isoelectric-focusing electrophoresis test helps distinguish variant forms such as the Duarte defect.
GALT genotyping may provide a specific molecular diagnosis

How is treated How is treated galactosemia?galactosemia?
The only way to treat galactosemia is through dietary restrictions from the first days of life
No brest feeding
Soya- based formula
• The newborn with questionable results on newborn screening should continue to be treated with soy-based formula pending definitive results of confirmatory testing.

Treatment
The mainstay of medical care in the postnatal period is to immediately discontinue ingestion of lactose-containing formula.
Totally eliminating galactose is difficult because it is present in a wide variety of foods (eg, infant foods, fruits, vegetables)
This ameliorates the acute toxicity associated with the neonatal period but does not prevent all long-term complications.

Galactosemia screening Why? If unscreened and untreated,
galactosemia is a life-threatening disorder. When? Neonatal period: 4th day
How? Screening of every neonates, followed by confirming tests. Thereby, affected infants are treated before they become ill.
Thank you !





























