Embed Size (px)
Citation preview

Genome Assembly
Microbial Single Cell Genomics Workshop 2010
Sergey Koren – JCVI, CBCB at UMD

Introduction

Platform: 3730
Applied Biosystems 3730xl • DNA amplification in E. coli. • Sanger dye-terminator PCR. • Capillary gel electrophoresis. • 90 min run makes 96 reads. • 500 t0 800 bp/read. • Paired end range 2Kbp - 40Kbp. • Mate rate almost 100%. • Reads include vector at 5’ end. • Quality drops gradually at 3’ end. • Traces available for inspection.
Trace from 3730
ABI 3730
Model length reads bases
ABI 3730 800 96 80K

Platform: FLX
Model length reads bases
ABI 3730 800 96 80K
454 FLX 400 1M 400M
Roche 454 FLX Titanium • Successor to GS 20 and FLX Standard • Pyrosequencing: Emulsion PCR, Bead capture, Transfer to picoliter plate, sequencing by synthesis of complementary strand, Image processing • Paired-ends: Reads are cut from single circles (in lab), Size options are 3Kbp or 8Kbp or 20Kbp, Linker-positive reads cut into pairs by software, 50% of reads are linker-positive. • Run time is 9hr. • Clear ranges of 80bp-500bp. • Problems: Inaccurate counts of repeated bases, duplicate reads, lower output on paired-end.
Beads on a picotitre plate
454

Platform: Illumina
Model length reads bases
ABI 3730 800 96 80K
454 FLX 400 1M 400M
Illumina GA 100 200M 20G
Illumina Solexa Genome Analyzer • Bridge hybridization on flat surface, Cluster formation, Sequencing by synthesis, Image Processing. One 2x run in 10 days. • Lengths have grown from 35 to 2x100. • Reads have identical length initially, trimmed by software, Substitution more than indel, error accumulates at read end. • Paired ends, sequencing both ends of a fragment, range of 35bp-700bp. • Mate pairs, sequencing fragment cut from circle, range of 2Kbp-8Kbp.
Illumina
Flow cell with 8 lanes

Platform: SOLiD
Model length reads bases
ABI 3730 800 96 80K
454 FLX 400 1M 400M
Illumina GA 100 200M 20G
ABI SOLiD 50 >1G 50G
Applied Biosystems SOLiD • System 4 succeeds 1 thru 3. • Sequencing by reversible ligation, 5-mers query 2 consecutive bases per luminous interrogation, light signal is di-base encoded. • Up to 16 days per run. • Reads vary from perfect to unusable due to high error. • “Color space” output, requires special software, allows detection and correction of some errors, vendor claims higher accuracy.
Colors represent di-base encoding
ABI SOLiD

Emerging Platforms
• Helicos Method: Single molecule sequencing, 35bp reads Application: whole-genome resequencing Capacity: 50 channels per 8-day run, 50X coverage
of one E.coli genome per channel
• PacBio Single molecule with polymerase Light detection from small chambers Runs are fast (15m) but low-throughput Reads of 10Kbp possible Current reads are short, high-error
• Ion Torrent Electronic chip Charge detection, no photonics Machine is compact and cheap ($50K) Runs are fast (1hr) and cheap ($500) Millions of reads/run, hundreds of bases/read
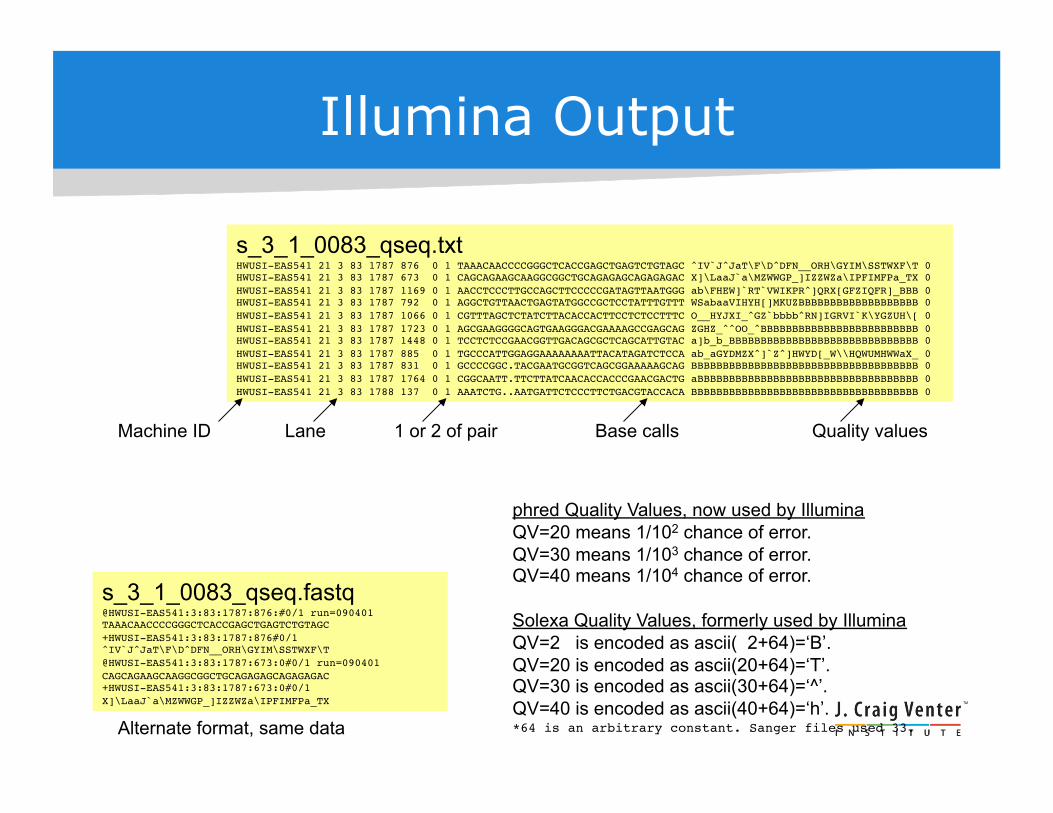
Illumina Output
s_3_1_0083_qseq.txt !HWUSI-EAS541 21 3 83 1787 876 0 1 TAAACAACCCCGGGCTCACCGAGCTGAGTCTGTAGC ^IV`J^JaT\F\D^DFN__ORH\GYIM\SSTWXF\T 0!HWUSI-EAS541 21 3 83 1787 673 0 1 CAGCAGAAGCAAGGCGGCTGCAGAGAGCAGAGAGAC X]\LaaJ`a\MZWWGP_]IZZWZa\IPFIMFPa_TX 0!HWUSI-EAS541 21 3 83 1787 1169 0 1 AACCTCCCTTGCCAGCTTCCCCCGATAGTTAATGGG ab\FHEW]`RT`VWIKPR^]QRX[GFZIQFR]_BBB 0!HWUSI-EAS541 21 3 83 1787 792 0 1 AGGCTGTTAACTGAGTATGGCCGCTCCTATTTGTTT WSabaaVIHYH[]MKUZBBBBBBBBBBBBBBBBBBB 0!HWUSI-EAS541 21 3 83 1787 1066 0 1 CGTTTAGCTCTATCTTACACCACTTCCTCTCCTTTC O__HYJXI_^GZ`bbbb^RN]IGRVI`K\YGZUH\[ 0!HWUSI-EAS541 21 3 83 1787 1723 0 1 AGCGAAGGGGCAGTGAAGGGACGAAAAGCCGAGCAG ZGHZ_^^OO_^BBBBBBBBBBBBBBBBBBBBBBBBB 0!HWUSI-EAS541 21 3 83 1787 1448 0 1 TCCTCTCCGAACGGTTGACAGCGCTCAGCATTGTAC a]b_b_BBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 0!HWUSI-EAS541 21 3 83 1787 885 0 1 TGCCCATTGGAGGAAAAAAAATTACATAGATCTCCA ab_aGYDMZX^]`Z^]HWYD[_W\\HQWUMHWWaX_ 0!HWUSI-EAS541 21 3 83 1787 831 0 1 GCCCCGGC.TACGAATGCGGTCAGCGGAAAAAGCAG BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 0!HWUSI-EAS541 21 3 83 1787 1764 0 1 CGGCAATT.TTCTTATCAACACCACCCGAACGACTG aBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 0!HWUSI-EAS541 21 3 83 1788 137 0 1 AAATCTG..AATGATTCTCCCTTCTGACGTACCACA BBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB 0
s_3_1_0083_qseq.fastq !
@HWUSI-EAS541:3:83:1787:876:#0/1 run=090401!TAAACAACCCCGGGCTCACCGAGCTGAGTCTGTAGC !+HWUSI-EAS541:3:83:1787:876#0/1!^IV`J^JaT\F\D^DFN__ORH\GYIM\SSTWXF\T!@HWUSI-EAS541:3:83:1787:673:0#0/1 run=090401!CAGCAGAAGCAAGGCGGCTGCAGAGAGCAGAGAGAC!+HWUSI-EAS541:3:83:1787:673:0#0/1!X]\LaaJ`a\MZWWGP_]IZZWZa\IPFIMFPa_TX !
Machine ID Lane 1 or 2 of pair Base calls Quality values
Alternate format, same data
phred Quality Values, now used by Illumina QV=20 means 1/102 chance of error. QV=30 means 1/103 chance of error. QV=40 means 1/104 chance of error.
Solexa Quality Values, formerly used by Illumina QV=2 is encoded as ascii( 2+64)=‘B’. QV=20 is encoded as ascii(20+64)=‘T’. QV=30 is encoded as ascii(30+64)=‘^’. QV=40 is encoded as ascii(40+64)=‘h’. *64 is an arbitrary constant. Sanger files used 33.!

$ sffinfo!Usage: sffinfo [options...] [- | sfffile] [accno...]!Options:! -a or -accno Output just the accessions! -s or -seq Output just the sequences! -q or -qual Output just the quality scores! -f or -flow Output just the flowgrams! -t or -tab Output the seq/qual/flow as tab-delimited lines! -n or -notrim Output the untrimmed sequence or quality scores!
$ sffinfo FFPMSHM02.sff FFPMSHM02B382V!# of Flows: 400!# of Bases: 271!Clip Qual Left: 5!Clip Qual Right: 90 Flow Chars: TACGTACGTACGTACGTACGTACGTACGTACGTACGTACGTACGTACGTACG…!
Flowgram: 0.93 0.05 1.18 0.08 0.04 0.91 0.05 9.10 0.14 1.97… !
Bases: tcagGGGGGGGGAACCACTGTAACAAAGGAAAGTAGGTTGGTCTCGCGGTGAGTG…!
Quality Scores: 32 32 32 16 16 16 16 16 16 16 16 16 23… !
454 Output
Example of using unix utilities from 454. Shown is sffinfo. See also sfffile. !
Dump 1 read from SFF file
for half-plate #2
The first 4 bases (lower case) are the key sequence for calibration. On this slide, the 4 colors show correspondence between flows and bases.
Each flow yields 0 or more bases

Color Space
A followed by T is coded red. T followed by G is coded green. Note T is interrogated twice.
The color code.
ABI SOLiD • The reads have colors not bases.
• encoded as numbers 0 to 3 • ABI claims colors improve accuracy. • Software should align “in color space.”
• convert to bases after alignment • ABI software catalog is skimpy.

Sanger-era Paired Ends
1. Randomly shear copies of the genome 2. Build libraries of similar-length fragments 3. Amplify individual fragments 4. Sequence two ends of each fragment
1. 2. 3. 4.
Oriented reads with distance estimate.
Insert size estimate given as µ ± σ per library.
µ ± σ
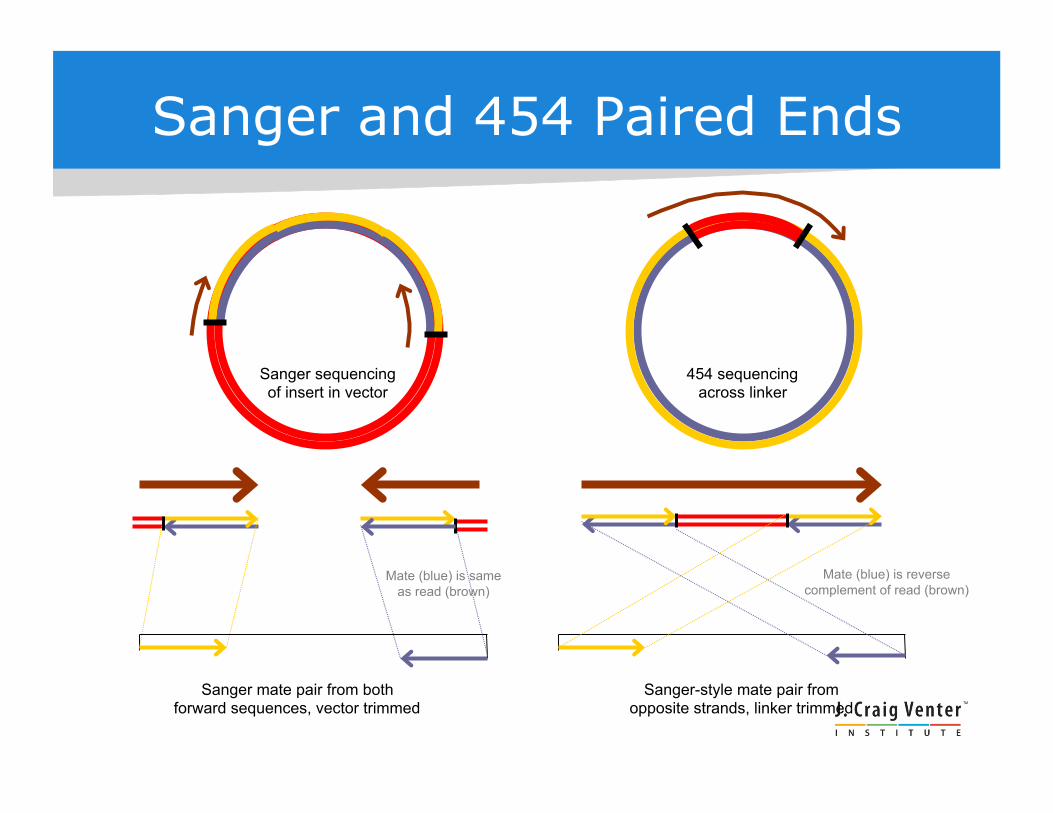
Sanger sequencing of insert in vector
454 sequencing across linker
Sanger mate pair from both forward sequences, vector trimmed
Sanger-style mate pair from opposite strands, linker trimmed
Mate (blue) is reverse complement of read (brown)
Mate (blue) is same as read (brown)
Sanger and 454 Paired Ends

Sequencer Summary
• Sequencing Machines ABI 3730xl 454 FLX with Titanium: variable-length reads Illumina GA (Solexa): pairs of 100bp reads ABI SOLiD system 4: reads of 50 colors Helicos Heliscope: 35bp from single molecule Pacific Biosciences: variable-length reads from single molecule Ion Torrent: not yet available
• Sequencing Approaches Whole-genome shotgun Mate pair and paired end Bar coding Contract sequencing

de novo Assemblers
• Greedy Graph Algorithms for short reads SSAKE, SHARCGS, VCAKE
• Traditional Overlap/Layout/Consensus Algorithms Developed for Sanger reads Celera Assembler (maintained at JCVI, for reads of 100bp+) Newbler (proprietary from Roche, tuned for 454 reads) Others: TIGR, phrap, Arachne, PCAP, Phusion, Atlas, Jazz
• de Bruijn Graph Algorithms EULER: originally developed for Sanger reads Velvet: the first widely used assembler for short reads AllPaths: version 3 may scale up to human genome ABySS: distributed memory, distributed processing, first assembly
of human from short (<100bp) reads SOAP de novo: best assembly of human from short reads, running
on large-RAM computer CLC: commercial assembler designed for mixed platform fragments,
uses many processors and low memory

Algorithms: Basic Steps
• Read processing Filter erroneous reads and trim erroneous bases
• Graph construction Reads and their pair-wise overlaps, or K-mers and their K-1 overlaps
• Graph reduction Remove spurs (erroneous read ends) Collapse bubbles (minor polymorphisms) Tease apart collapsed repeats (use reads or mates) Form contigs but break at repeats
• Scaffold with mate pairs • Consensus from multiple sequence alignment
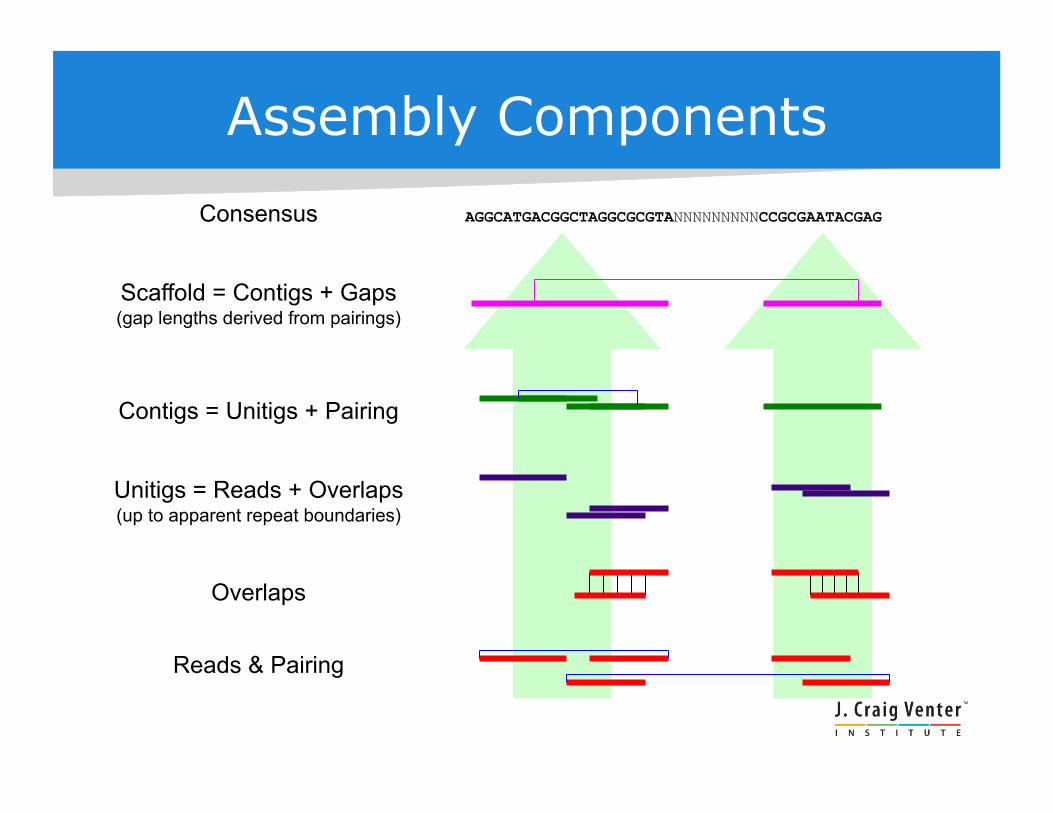
Assembly Components
AGGCATGACGGCTAGGCGCGTANNNNNNNNNCCGCGAATACGAG Consensus
Scaffold = Contigs + Gaps (gap lengths derived from pairings)
Contigs = Unitigs + Pairing
Reads & Pairing
Unitigs = Reads + Overlaps (up to apparent repeat boundaries)
Overlaps

Algorithms: Techniques
• Allowing for sequencing error complicates everything Use sequence similarity as guide and paired-ends as constraint
• Techniques for extra speed Use indexes to avoid all-against-all pair-wise alignment K-mers (exact match substrings) are suitable for fast computes
– In De Bruijn Graph assembly (Velvet), identical K-mers collapse to graph nodes – In traditional assembly (Celera Assembler) K-mers seed overlaps
Arbitrary thresholds – Minimum overlap length or percent identity – Maximum repetitiveness tolerated in a read
Sub-optimal algorithms – Greedy algorithms (keep enlarging my contig without concern for global picture) – Heuristic algorithms (use coverage ratios that seem to work well on real data)
Save consensus for last – Multiple pair-wise alignments is faster than one multiple sequence alignment
• Must choose a strategy for reads from genomic repeats Report 0 reads/repeat (leave gap or provide consensus with 0X coverage) Report 1 read/repeat (chosen randomly for even coverage) Report all reads/repeat (repeat reads are multiply placed)

aaccggaaccaccgccgg
ccggttcggtggtt
Algorithms: Graphs
Assembly algorithms construct graphs, which are nodes connected by edges. In traditional assemblers (left) nodes are reads, edges are overlaps. In de Bruijn assemblers (right) nodes are K-mers, edges are exact matches of K-1.
Assembly algorithms reduce graph complexity. Boxes are reads or K-mers. Edges are overlaps or matches. Edge thickness can indicate amount of support in reads.
Left: A spur is induced by bad sequence at a read end or low coverage and polymorphism. Middle: A bubble is induced by polymorphism or sequencing error. Right: A collapsed repeat might get teased apart or isolated or multiply placed.

Coverage: Impact on Assembly
Sequence this genome by the random “shotgun” method.
1. Initially, all new reads make contigs.
2. Later, new reads fill gaps. Contigs enlarge and coalesce.
3. Eventually, new reads add little to nothing. Gaps remain due to chance or bias.
4. At very high coverage, error accumulates, assemblies fracture (not shown).
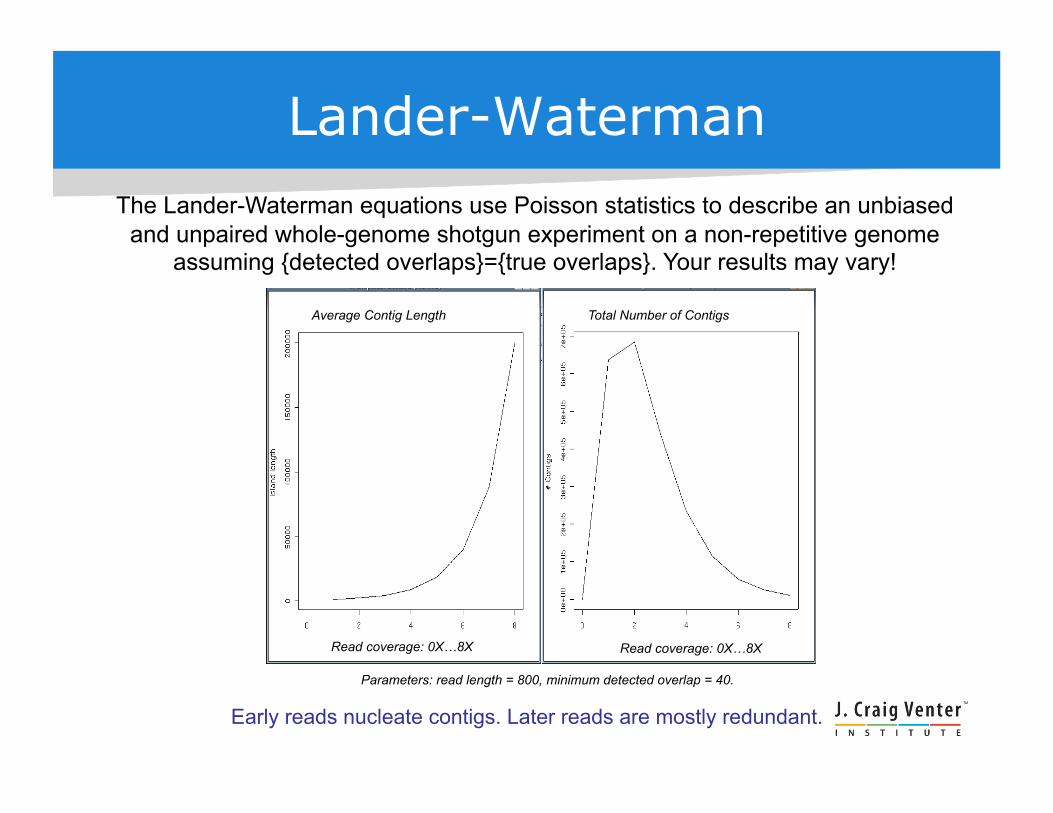
Lander-Waterman The Lander-Waterman equations use Poisson statistics to describe an unbiased
and unpaired whole-genome shotgun experiment on a non-repetitive genome assuming {detected overlaps}={true overlaps}. Your results may vary!
Parameters: read length = 800, minimum detected overlap = 40.
Average Contig Length Total Number of Contigs
Read coverage: 0X…8X Read coverage: 0X…8X
Early reads nucleate contigs. Later reads are mostly redundant.

Read-Length Effects
Assembly 1. 9 reads of length = 30bp.
Total sequenced bases = 270. Assemble with min overlap = 20bp.
Result = 7 contigs.
Assembly 2. 3 read length = 90.
Total sequenced bases = 270. Assemble with min overlap = 20bp.
Result = 1 contig.
1. Overlap Effect: For same number of sequenced bases, shorter reads require more coverage to achieve comparable N50.
2. Repeat Effect: Shorter reads resolve fewer repeats.
Repeat length = 600bp. Read length = 800bp (Sanger).
Reads span the repeats.
Repeat length = 600bp. Read length = 400bp (454). Reads bridge the repeats.
Repeat length = 600bp. Read length = 75bp (Solexa).
Repeats not resolved.
? ?
Same inputs�
Different outputs�

Mate Pair Effects
1. Variety of insert sizes will span variety of repeats.
2. Mates can resolve repeats even if not possible to tile with reads.
Inserts that span the repeat will enable scaffolds.
Larger repeats require larger insert sizes.
High coverage in mates will tile the repeat.
Mates guide multiple placement of repeat contig.

Assembly Metrics
Number of contigs, number of scaffolds Average number of contigs per scaffold Total bases in scaffolds Total span of scaffolds (including the gap lengths) Average gap length (for intra-scaffold gaps) Contig N50, Scaffold N50, Gap N50 Sum of bases in contigs>10Kbp Sum of span in scaffolds>2Kbp Min, max, and mean span of a scaffold Min, max, and mean bases in a contig Fraction of paired-end constraints satisfied in scaffolds Average read coverage in contigs

The N50 Metric
Problem: How to represent a distribution of intervals by one number?
Solution: N50 1. Measure the total span. 2. Sort the set by size. 3. Start walking from the largest interval. 4. Stop walking at the 50% mark. 5. Measure the interval on which you stopped.
10% 30% 50% 70% 90% 0 G
Intervals may represent contigs, scaffolds, gaps, alignments, etc.
Extra credit: What is N90? Why is N50 unstable?
This interval is your N50.
Start at largest.

Typical Assembly Output
• Scaffolds.fasta – Each scaffold contains one or more contigs. – Gap lengths may be represented by strings of N.
• Contigs.fasta – These are the contigs from scaffolds, without the gaps. – These may be filtered by a minimum size criterion.
• UnassembledReads.fasta – Solo reads may be called “singletons.” – Their mates may or not be assembled.
• UnassembledContigs.fasta – These are contigs not placed in scaffolds. – May be repeats, plasmids, contamination, short contigs.

Common Assembly Errors
• Chimera Assembly joins unrelated sequences, usually across a
low-copy repeat • Collapsed tandem repeat
Copy number is reduced in the assembly
• Missed join Contigs not assembled despite sequence similarity
• Assembly Forensics*
* Phillippy, Schatz, Pop (2007) Genome assembly forensics: finding the elusive mis-assembly. Genome Biology.

Consensus Problems
…GGCGT-AGCA… …GGCGT-AGCA… …GGCGTTAGCA… …GGCG-TAGCA… …GGCG-TAGCA…
…GGCGTTAGCA…
• Even sophisticated software can generate alignments that the human eye recognizes as wrong. • The root cause is that multiple sequence alignment is solved by a series of pair-wise alignments. • Celera Assembler reviews its final alignments to compress gap-rich regions. • Celera Assembler fixes this particular problem, but it misses more complex ones.
How does 1 sequencing error…
…propagate to the consensus?
These alignments were valid.
So were these.
Each consensus base has at least 3 bases supporting it.

Single Cell Assembly Challenges
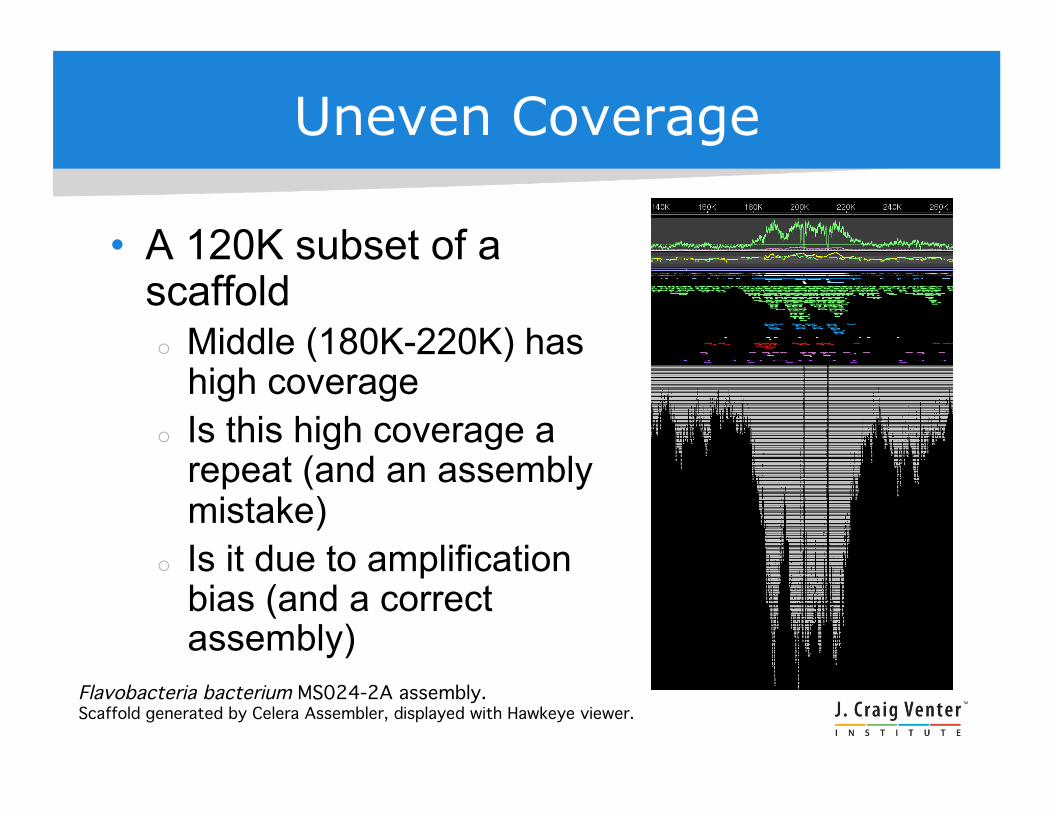
Uneven Coverage
• A 120K subset of a scaffold Middle (180K-220K) has
high coverage Is this high coverage a
repeat (and an assembly mistake)
Is it due to amplification bias (and a correct assembly)
Flavobacteria bacterium MS024-2A assembly. !Scaffold generated by Celera Assembler, displayed with Hawkeye viewer.!
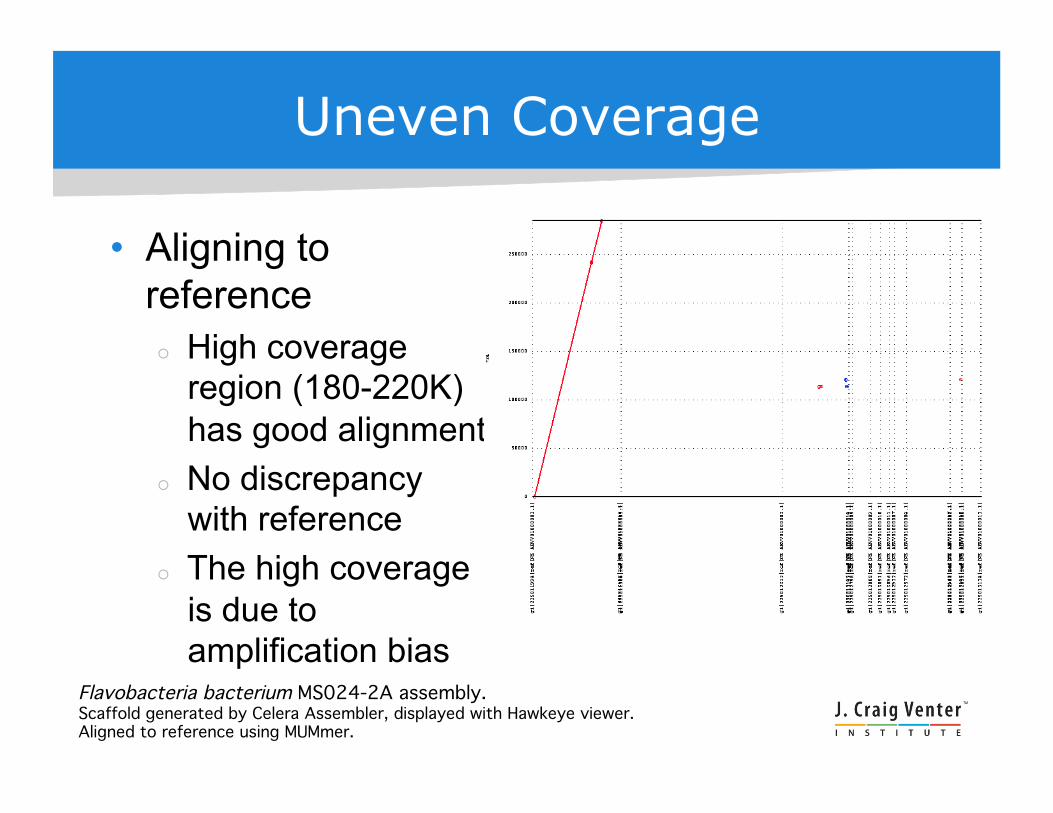
Uneven Coverage
• Aligning to reference High coverage
region (180-220K) has good alignment
No discrepancy with reference
The high coverage is due to amplification bias
Flavobacteria bacterium MS024-2A assembly. !Scaffold generated by Celera Assembler, displayed with Hawkeye viewer.!Aligned to reference using MUMmer.!

Coverage in scaffolds including gaps and surrogates. Bar at 0 is truncated; value is 152.9M. Count at each position reflected sum of spanning reads, regardless of alignment gaps. (Bonobo i7 Assembly, Bonobo Consortium)

Uneven Coverage (cont)
Flavobacteria bacterium MS024-2A assembly. !Scaffold generated by Celera Assembler!

Uneven Coverage (cont)
Flavobacteria bacterium MS024-2A assembly. !Scaffold generated by Celera Assembler!

Chimera
• 85% of chimeras correspond to a sequence inversion (A, C). 15% of chimeras resulted from the joining of two segments in direct orientation (B, D).
• The order of the two segments could be reversed. The first segment in the chimera (black arrows) can join to a segment that is downstream (A, B) or upstream (C, D).

Bioinformatics Challenges

Assembly Tools
• Bioinformatics tools are often not user-friendly • They must be compiled from code and give cryptic
error messages • Require Unix knowledge/IT support

Bioinformatics Availability
• A collection of bioinformatics tools
• Can be run on the Amazon EC2
• Designed to skip the complexity of finding/downloading/building tools
http://www.cloudbiolinux.com/

Preparing Inputs to Assembly

Hands On: Preparing Data

Assembling with Celera Assembler

Hands On: Assembling with CA

Assembly.scf.fasta >scf138 CTTAATTTTGGAATGAGCAAATCAAACGGCTCTCTTTCTTCTCTCTTCCGACGATCATTTCTCTCCTTCA TCCTTTTTCTCTATTGAATTTATGGTTGTGAGACACTTGCCTATGCCGTTCAAAATTTTACATTAGAGTG GATCTAAACCAAATTGGACACTGTGTTATCTGGTTTTAGCTTGAAGTAATTTCTAACAAGAATGGTTGCT TCAAAAAGACAAATATAAAGTGAAATTGATTGCCTCATTCTCTTCACCCTCATTCAAACTAAAATTGAAG CTTAAAGAAAGAAGTAAAGGAATCAAAAATGGCAGCCTGCACGTATGAGATGACGAATTCTAAAATTTCA AGCTCTGCGTTATATTTATTTTGAGTTATACGTTATCTTAATTAATTGGGTTGCTATAATTATAACTTAT TTCGCTCCACCTAGATTTTTAACGGCCAAAGTGAGTTTAGTTGGTGGTATTAAAATGACTTTTTTATTTA AAAATCGAAGGTTTAGTTTTTACTATGCAATTAGTGTACCAAAAGAAATTTTTATACCTATTTAAATCAA
Celera Assembler Output
Assembly.asm {MDI ref:(20KB,1) mea:20608.635 std:3273.333 min:1539 max:31474 buc:28 his: 8 0 1 52 69 59 104 146 85 66
Assembly.qc [Scaffolds] TotalScaffolds=4127 TotalContigsInScaffolds=8653 MeanContigsPerScaffold=2.10 MinContigsPerScaffold=1 MaxContigsPerScaffold=73 TotalBasesInScaffolds=200660111 MeanBasesInScaffolds=48621 MinBasesInScaffolds=446 MaxBasesInScaffolds=4058172 N25ScaffoldBases=1609364 N50ScaffoldBases=780164 N75ScaffoldBases=385348 ScaffoldAt100000000=780347 ScaffoldAt200000000=1027
Assembly.posmap.frgscf FPQMBNN02SL55F 138 331191 331696 f FPMLNGL01BTUZ1 138 331112 331621 f FO77AEH01COVYY 138 331152 331619 f FPKRC2D02RHQ3C 138 331238 331546 r FPDPMZT01DTV0D 138 331089 331532 f FPBG22E02Q51Y0 138 331209 331532 r FPMX3IK01CS2XJ 138 331140 331521 f FPMLNGL02QFBKX 138 331297 331502 f FO6AT3P02QPVK0 138 331018 331488 r FO6AT3P02TCI7F 138 331095 331488 r FO77AEH01A5XKF 138 330986 331485 r FO77AEH01CKY2Z 138 331037 331485 r FO77AEH01BD0T1 138 331096 331485 r FO76IZV02R25OI 138 330986 331481 f FO76IZV02P6FZH 138 330986 331481 f FPDPEIT01BA86D 138 330972 331473 r

Dealing with Coverage Bias
• Shaded node is a repeat • Links together distinct regions of a genome • Most assemblers start by disconnecting repetitive nodes

Coverage in CA
• Celera Assembler uses the A-Statistic P(read starting at base) = arrival rate = α = P(k starts in ρ bases) is Binomial
– We approximate via Poisson with giving us For a 2-copy repeat, the arrival rate is doubled, giving us A-Statistic = log P(unique)/P(2-copy) = When A-Statistic is negative, consider node a repeat
• Read coverage is not perfect Sequencing bias due to GC-content Borderline cases Incorrect coverage expectation
– Polyploid genomes – Metagenomes

Fixing Repeat Detection: Toggling
overlap unitig scaffold
Celera Assembler pipeline: default
reads scaffolds
overlap unitig scaffold
Celera Assembler pipeline: toggling
reads
Find large “repeat” unitigs placed once in scaffolds. Mark them “unique.”
scaffold scaffolds
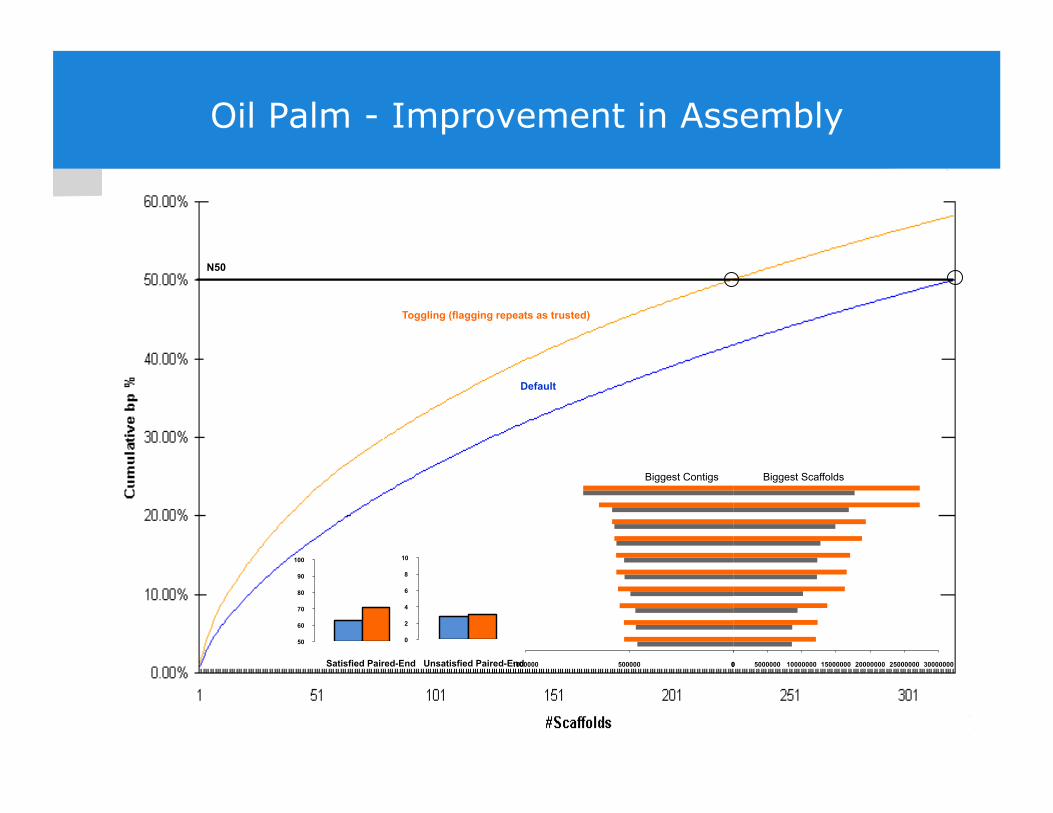
Oil Palm - Improvement in Assembly
0 500000 1000000 0 5000000 10000000 15000000 20000000 25000000 30000000
50
60
70
80
90
100
0
2
4
6
8
10
Biggest Contigs Biggest Scaffolds
Toggling (flagging repeats as trusted)
Satisfied Paired-End Unsatisfied Paired-End
Default
N50

Hands on: How To Toggle
• doToggle At the end of a successful assembly, search for placed surrogates and toggle them to
be unique unitigs. Re-run the assembly starting from scaffolder Either 0 or 1, default is 0
• Toggling Useful Options toggleUnitigLength
– Minimum length for a surrogate to be toggled. – Any number >= 1, default is 2000
toggleNumInstances – Number of instances for a surrogate to be toggled. If 0 is specified, all non-singleton unitigs
are toggled to unique status. – Any number >=0, default is 1. – In future version of CA, setting it to 0 will toggle all non-singleton unitigs >= toggleUnitigLength
• Output Pre-toggling results: asm/9-teminator Post-toggling results: asm/10-toggled/9-terminator Same contents as from a regular assembly

Incorporating Finishing Reads
• Goals of finishing Close sequencing gaps by generating new reads Get higher coverage in low-coverage sections Generate reads to resolve repeat instances
• Manual finishing is expensive • Finishing reads generated from PCR product
or clone Constrain possible assembly: between PCR
product ends or clone ends • Formulate as a constraint the scaffolder
should satisfy

Finishing Reads in CA
• Given the set of bounded finishing reads F, and WGS reads W
• Bounded algorithm Incorporate unitigs into gaps Place reads in repeat instances
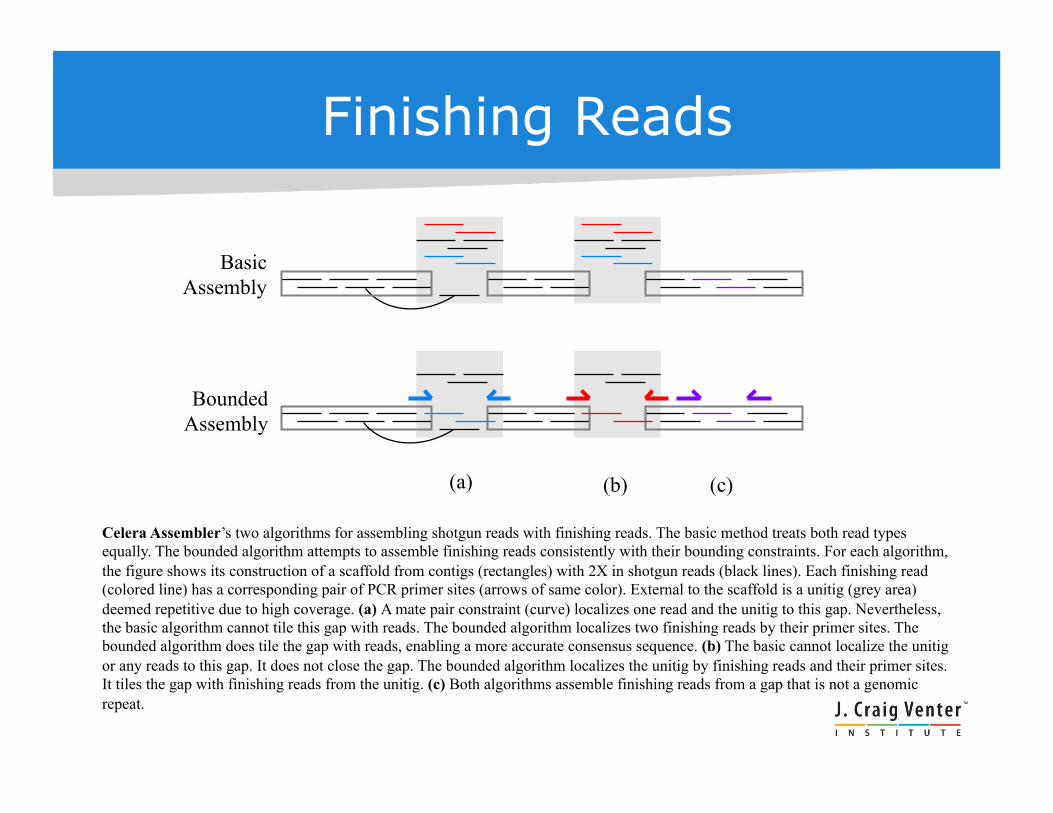
Basic Assembly
Bounded Assembly
Celera Assembler’s two algorithms for assembling shotgun reads with finishing reads. The basic method treats both read types equally. The bounded algorithm attempts to assemble finishing reads consistently with their bounding constraints. For each algorithm, the figure shows its construction of a scaffold from contigs (rectangles) with 2X in shotgun reads (black lines). Each finishing read (colored line) has a corresponding pair of PCR primer sites (arrows of same color). External to the scaffold is a unitig (grey area) deemed repetitive due to high coverage. (a) A mate pair constraint (curve) localizes one read and the unitig to this gap. Nevertheless, the basic algorithm cannot tile this gap with reads. The bounded algorithm localizes two finishing reads by their primer sites. The bounded algorithm does tile the gap with reads, enabling a more accurate consensus sequence. (b) The basic cannot localize the unitig or any reads to this gap. It does not close the gap. The bounded algorithm localizes the unitig by finishing reads and their primer sites. It tiles the gap with finishing reads from the unitig. (c) Both algorithms assemble finishing reads from a gap that is not a genomic repeat.
Finishing Reads
(a) (b) (c)
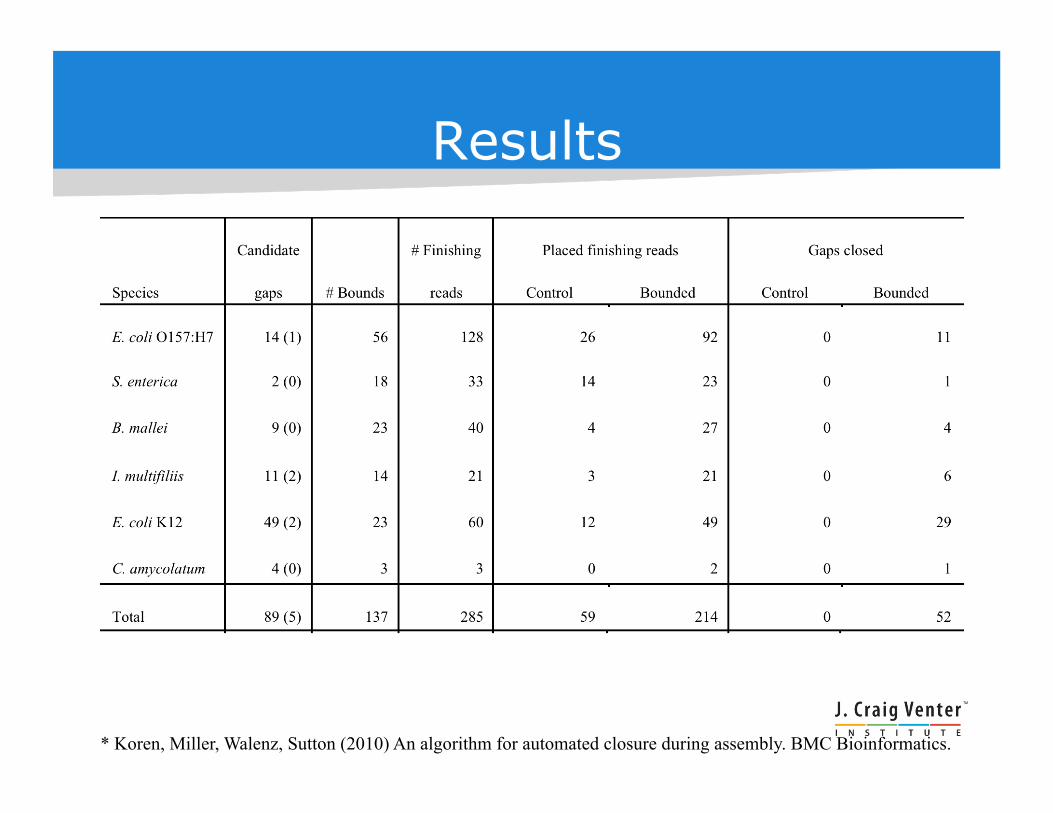
Results
* Koren, Miller, Walenz, Sutton (2010) An algorithm for automated closure during assembly. BMC Bioinformatics.

Specifying Finishing Reads
• Special record in FRG file Only supported in FRG v2 Example of a PLC:
{PLC act:A frg:X frg:Y frg:Z }
Finishing read ID
Boundary #1 ID
Boundary #2 ID
• Reads X, Y, and Z must be defined in an {FRG • X, Y and Z can appear in an {LKG • Y (and Z) can be either the left or right bound

Hands On: Finishing Reads in CA

Assembling with Newbler

Hands On: Assembling with Newbler

Hands On: Finishing Reads in Newbler

454Scaffolds.fna >scaffold00001 length=48466 tttgtctaaagtaaatcttagcttaaggcagtgatatcgatggattagaaaGGATTTGGA TTGGGGCATTGGTGTAAAAGAAAAACCCTCCGCTTGGGAGGGCTAAATGGCTTtATTTTG TTGAACCTATAaTTTCCCAAATTTCAATAGCTTTAAAACCGTTTGGCATCAAATCGCCCA TCTTGGTTTTATTCGCAATAGAAGCAGGtAATGGAAATTCTCCTGCTAGATACTTCATAA CGTTAGTCATTGAGCCAAAAGCGTCCCAAGTCAGTATTGTTGATGAGTCCTCGTAATATG GAGCAATTcTTTAAAACCCGTCCTCTCTCAGTGtATTTtCAATGTTACAATCTTAGAAAA TCACATAGACGCGAGTTTTTAGTTATGAGTACTATTCTATTGAATTTCTATATTCTGAAG GGGTAACTGATTTtATTTTTTtAAatACTCTATTAAAATTTTCTTGGGAATTAAACCCTG ATTTTTCTACAAGTGCCACAATTGTATATTTATCTAAAAATTTATTTTGGATTAAATCAC
Newbler Output
454Contigs.ace AS 389 201659
CO contig00001 9348 676 210 U tttgtctaaagtaaatcttagcttaaggcagtgatatcgatggattagaa aGGATTTGGATTGGGGCATTGGTGTAAAAGAAAAACCCTCCGCTTGGGAG GGCTAAATGGCTTt*ATTTTGTTGAACCTATAa*TTTCCCAAATTTCAAT AGCTTTAAAACCGTTTGGCATCAAATCGCCCATCTTGGTTTTATTCGCAA TAGAAGCAGGt*AATGG*AAATTCT*CCTGCTAGATACTTCATAACGTTA GT*CATTGAGCCAAAAGCGTCCCAAGTCAGTATTGTTGATGAGTCCTCGT AATATGGAGCAATT*c*TTTAAAACCCGTCCTCTCTCAGTGt*ATTTtCA ATGTTACAATCTTAGAAAATCACATAGACGCG*AGTTTTTAGTTATGAGT ACTATTCTATTGAATTTCTATATTCTGAAGGGGTAACTGATTTtATTTTT TtAAa*t*ACTCTATTAAAATTTTCTTGGGAATTAAACCCTG*ATTTTTC TACAAGTGCCACAATTGTATATTTAT*CTAAAAATTTATTTTGGATTAAA TCACAAGCGAAATCTATTTTCAAAGAATTGATGTAAAATGGTGTAGTGAC ATTTAACTTTTGTTTGATAGAATCACGTATATCATTTTGAGTTATTCCTG TATCTACTGACAAATTTACTATAGAATAATTATTTTGAAGGAATGCTTTT GTTTCTTTAAGGTAATGGTTAAaGTTTtGAATTAATTCCTT
454NewblerMetrics.txt pairedReadStatus numberWithBothMapped = 2124; numberWithOneUnmapped = 69; numberMultiplyMapped = 1; numberWithBothUnmapped = 23; library { libraryName = "sanger3KB_fios"; pairDistanceAvg = 2933.3; pairDistanceDev = 2933.3; } scaffoldMetrics { numberOfScaffolds = 86; numberOfBases = 1566754; avgScaffoldSize = 18218; N50ScaffoldSize = 149992; largestScaffoldSize = 282146;
454ReadStatus.txt EM9Q5SV01A4E99 Assembled contig00227 89333 - contig00227 89174 + EM9Q5SV01ECMLE Assembled contig00383 42212 + contig00383 42263 - EM9Q5SV01A835M Assembled contig00384 25320 - contig00384 25146 + EM9Q5SV01APR37 Assembled contig00383 19524 - contig00383 19375 + EM9Q5SV01CUR52 Assembled contig00227 104203 - contig00227 104109 + EM9Q5SV01CMCSJ Assembled contig00227 228655 - contig00227 228548 + EM9Q5SV01CCIN0 Assembled contig00229 64432 + contig00229 64490 - EM9Q5SV01BXHGV Assembled contig00229 44737 + contig00229 44848 - EM9Q5SV01B8QV8 Assembled contig00294 51080 - contig00294 50993 + EM9Q5SV01B5IA1 Assembled contig00383 21777 + contig00383 21877 - EM9Q5SV01CMFK1 Assembled contig00287 190037 + contig00287 190168 - EM9Q5SV01EORKA Assembled contig00287 11293 + contig00287 11432 - EM9Q5SV01A44JD Assembled contig00022 23173 + contig00022 23294 - EM9Q5SV01BCVA3 Singleton EM9Q5SV01CS1HJ Assembled contig00384 120543 - contig00384 120467 + EM9Q5SV01A8ZAK Assembled contig00294 9939 - contig00294 9863 + EM9Q5SV01DGAKL Assembled contig00227 72188 - contig00227 72027 + EM9Q5SV01AVH7L PartiallyAssembled contig00131 1215 + contig00131 1267 - EM9Q5SV01AX7FC Assembled contig00287 144376 - contig00287 144298 + EM9Q5SV01EDHFV Assembled contig00384 35590 + contig00384 35669 - EM9Q5SV01ANK9N Assembled contig00384 143759 + contig00384 144032 - EM9Q5SV01AW150 Assembled contig00383 71921 - contig00383 71836 + EM9Q5SV01CKSJH Singleton EM9Q5SV01AZYCP Assembled contig00383 92675 + contig00383 92904 - EM9Q5SV01AWB3A Assembled contig00130 11303 + contig00130 11422 - EM9Q5SV01EABU1 Assembled contig00229 15683 - contig00229 15631 + EM9Q5SV01AVZ12 Assembled contig00265 2758 - contig00265 2700 + EM9Q5SV01EVB54 Assembled contig00384 114869 + contig00384 114977 - EM9Q5SV01CFUI9 Assembled contig00002 16971 + contig00002 17038 - EM9Q5SV01BH1OF Assembled contig00384 51892 + contig00384 52043 - EM9Q5SV01CI83R Assembled contig00154 5324 - contig00154 5263 + EM9Q5SV01CQC2E Assembled contig00227 43908 + contig00227 44062 -

Assembly Analysis
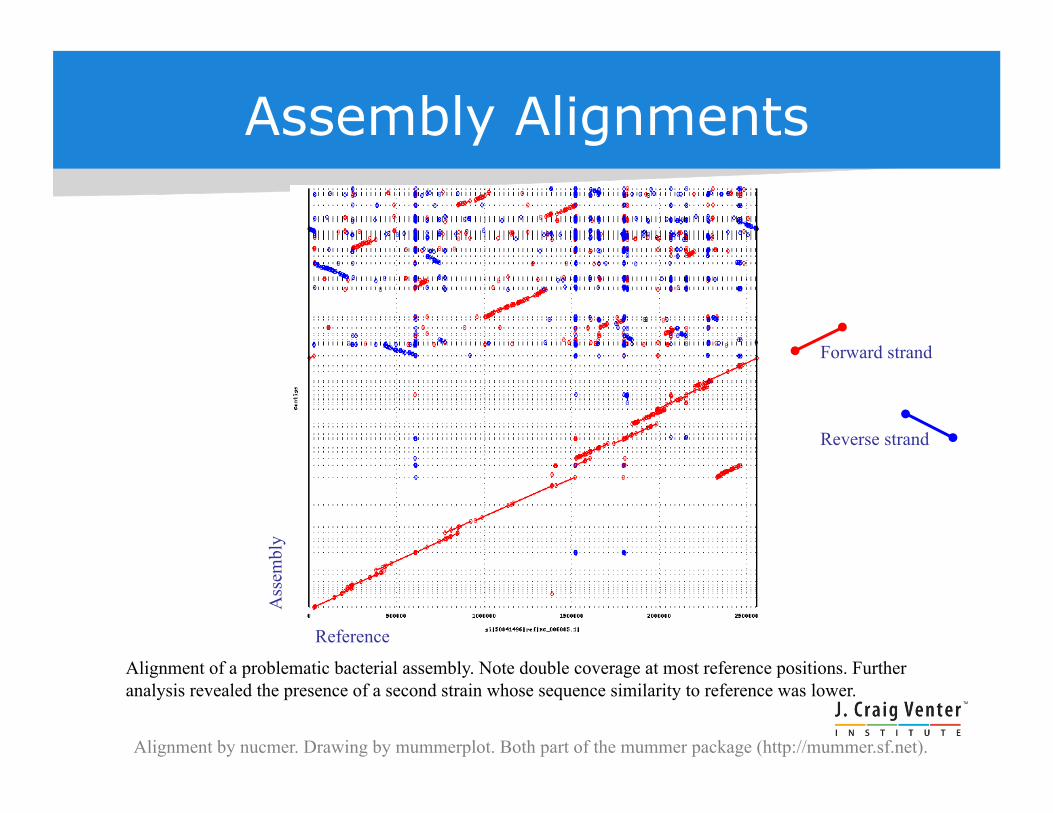
Assembly Alignments
Alignment of a problematic bacterial assembly. Note double coverage at most reference positions. Further analysis revealed the presence of a second strain whose sequence similarity to reference was lower.
Ass
embl
y
Reference
Forward strand
Reverse strand
Alignment by nucmer. Drawing by mummerplot. Both part of the mummer package (http://mummer.sf.net).

Two Assemblies from 454
• Cucumber 27M Titanium reads 3K, 8K, 20K inserts 199M in scaffolds 797K scaffold N50 87K contig N50 Read fates:
– 65% contig – 31% degen – 1% singleton
• Bonobo 250M Titanium reads 3K, 8K, 20K inserts 2.9G in scaffolds 9.6M scaff N50 67K contig N50 Read fates:
– 88% contig – 7% degen – 3% singleton
Bonobo assembly by CA 5.4. Consortium led by Max Planck Institute. Cucumber assembly by CA 5.3. Consortium led by 454 Life Sciences.
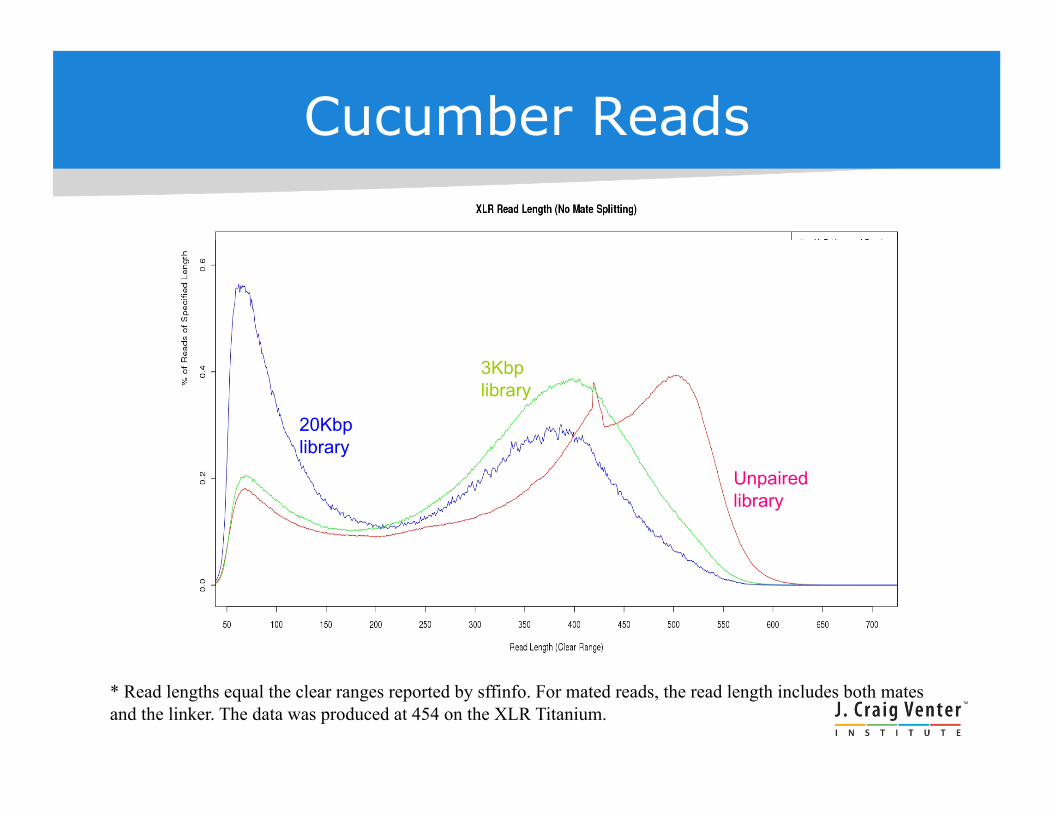
Cucumber Reads
20Kbp library
3Kbp library
Unpaired library
* Read lengths equal the clear ranges reported by sffinfo. For mated reads, the read length includes both mates and the linker. The data was produced at 454 on the XLR Titanium.
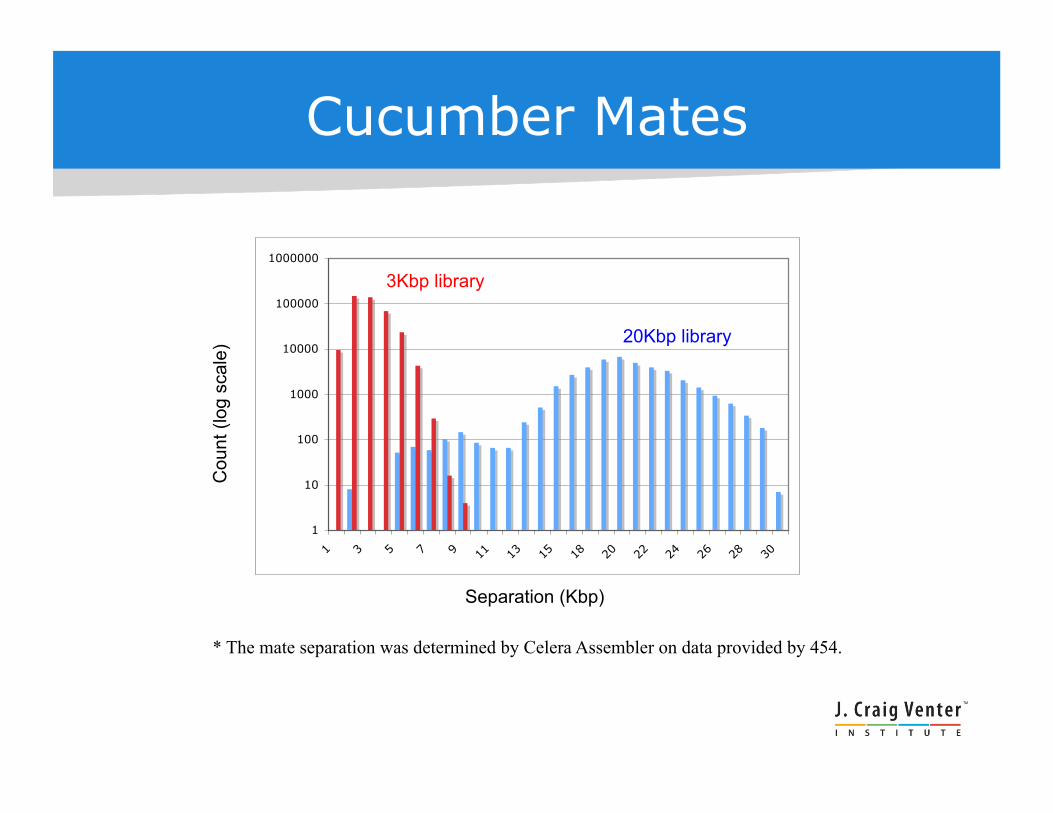
Cucumber Mates
Separation (Kbp)
1
10
100
1000
10000
100000
1000000
1 3 5 7 911
13
15
18
20
22
24
26
28
30
Cou
nt (l
og s
cale
)
3Kbp library
20Kbp library
* The mate separation was determined by Celera Assembler on data provided by 454.

Library Effect
Large-insert libraries are over-represented in degenerates.
Large-insert reads were more often rejected for being too short or perfect prefix.
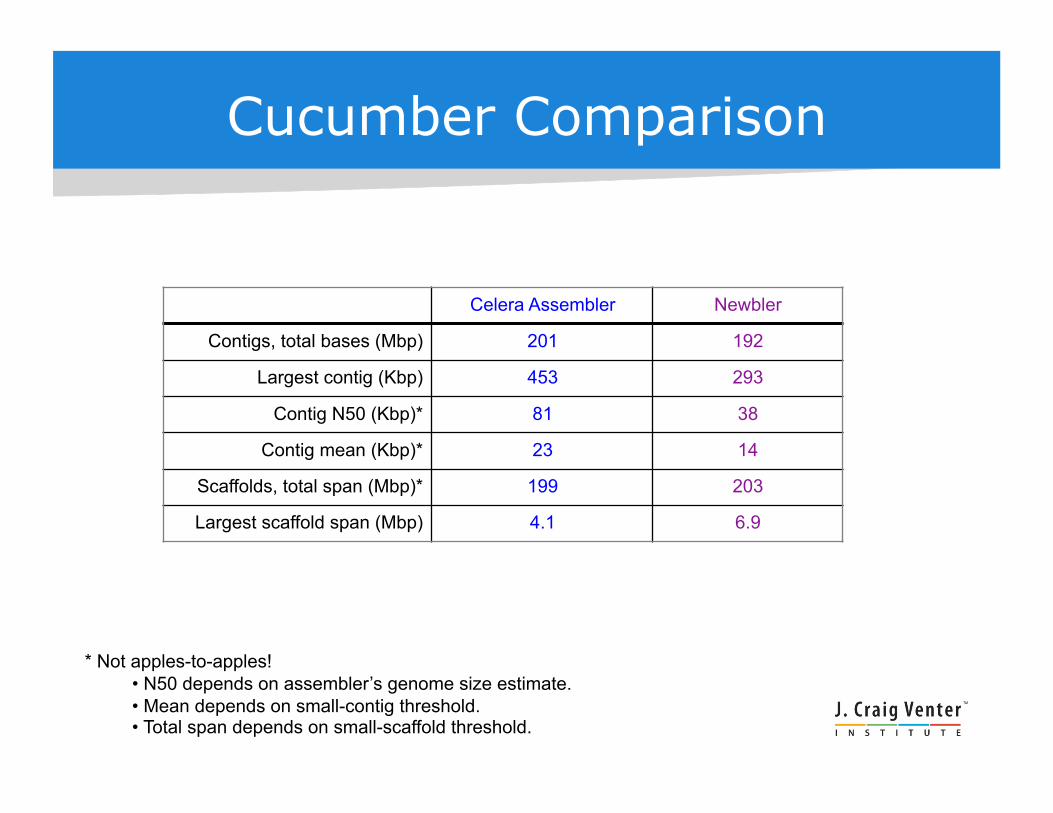
Cucumber Comparison
Celera Assembler Newbler
Contigs, total bases (Mbp) 201 192
Largest contig (Kbp) 453 293
Contig N50 (Kbp)* 81 38
Contig mean (Kbp)* 23 14
Scaffolds, total span (Mbp)* 199 203
Largest scaffold span (Mbp) 4.1 6.9
* Not apples-to-apples! • N50 depends on assembler’s genome size estimate. • Mean depends on small-contig threshold. • Total span depends on small-scaffold threshold.

Cucumber assembly
Max N10 N10 N25 N25 N50 N50 N95 N95 Total Combinedlength count length count length count length count length count length
Scaffolds 4,649,969 6 2,717,851 22 1,506,488 68 815,116 363 73,997 3,610 202,566,885Alignments 3,813,521 7 2,292,931 28 1,098,439 88 642,517 458 63,463 3,501 198,183,263Contigs 522,294 70 219,018 242 146,370 690 87,421 3,121 10,379 7,901 200,010,660
Alignments of CA assembly to the Newbler assembly approach the scaffold size, which is the maximum.

Cucumber Degens
• A degenerate is… Two or more reads in a unitig (high-confidence contig) That could not be promoted to contig
– Negative A-stat due to short length or high coverage That could not be incorporated into scaffolds
– Would need 2 consistent mates at least • Lots of small degenerates
Number of degenerates= 665,311 Combined length= 332 Mbp
– Equivalent to 165% of scaffold bases! Largest= 21 Kbp. Average length= 499 bp
• Lots of reads in degenerates Reads in degenerate= 40% of all reads Reads in degenerate and so is mate= 2.9% of reads Non-degen reads with degen mate= 0.3% of reads
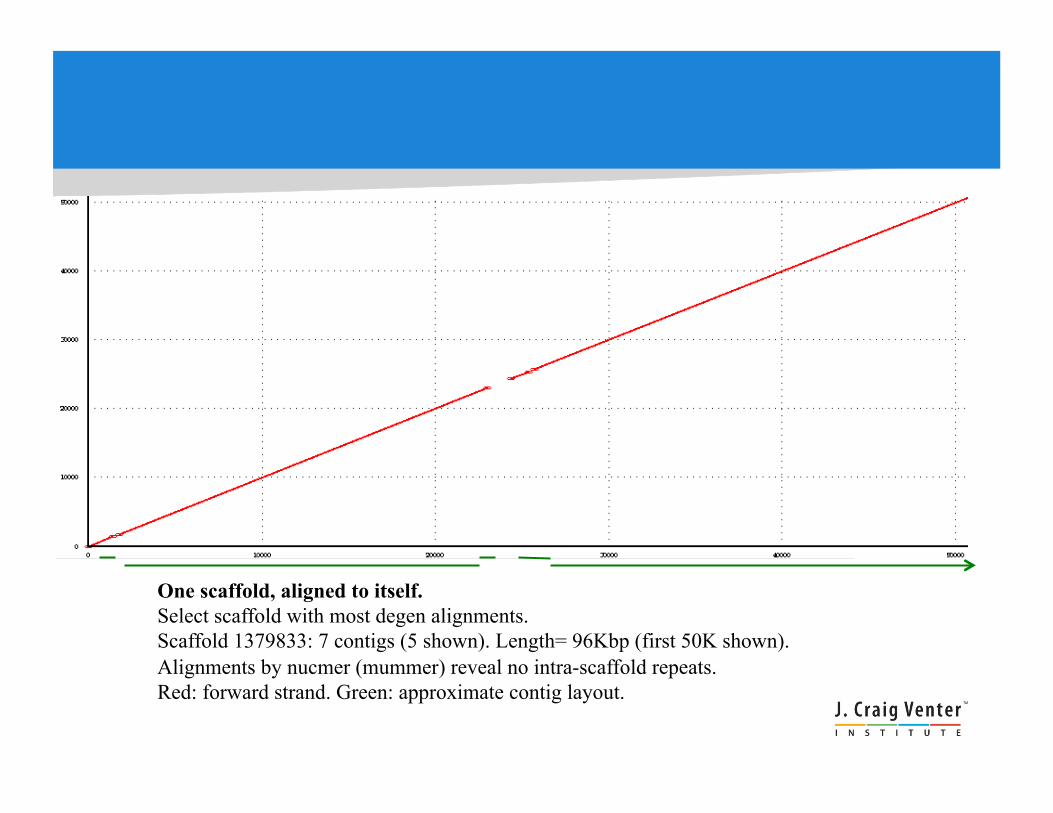
One scaffold, aligned to itself. Select scaffold with most degen alignments. Scaffold 1379833: 7 contigs (5 shown). Length= 96Kbp (first 50K shown). Alignments by nucmer (mummer) reveal no intra-scaffold repeats. Red: forward strand. Green: approximate contig layout.
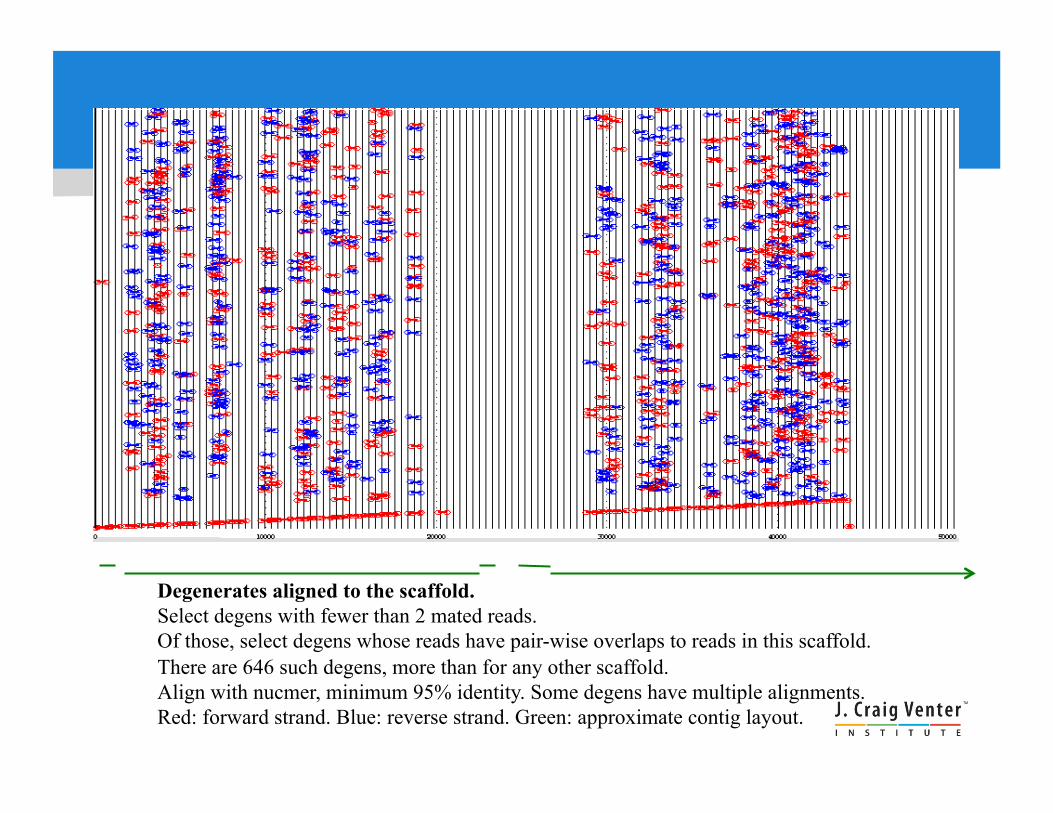
Degenerates aligned to the scaffold. Select degens with fewer than 2 mated reads. Of those, select degens whose reads have pair-wise overlaps to reads in this scaffold. There are 646 such degens, more than for any other scaffold. Align with nucmer, minimum 95% identity. Some degens have multiple alignments. Red: forward strand. Blue: reverse strand. Green: approximate contig layout.
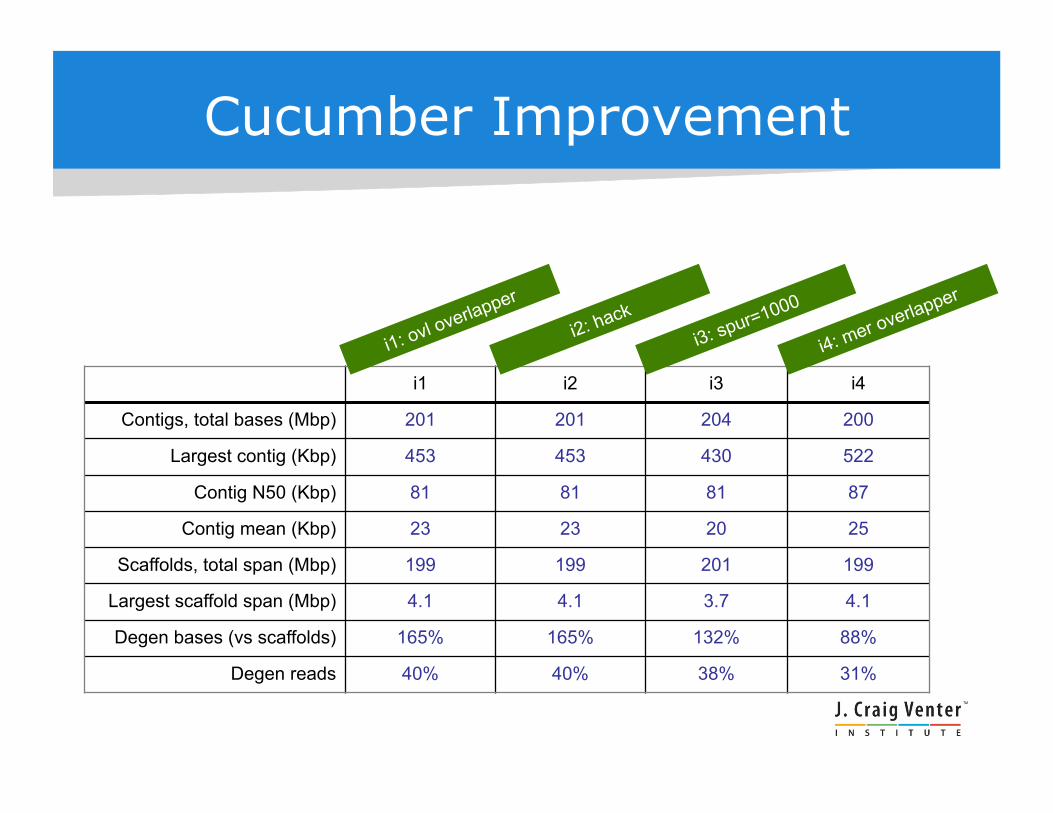
Cucumber Improvement
i1 i2 i3 i4
Contigs, total bases (Mbp) 201 201 204 200
Largest contig (Kbp) 453 453 430 522
Contig N50 (Kbp) 81 81 81 87
Contig mean (Kbp) 23 23 20 25
Scaffolds, total span (Mbp) 199 199 201 199
Largest scaffold span (Mbp) 4.1 4.1 3.7 4.1
Degen bases (vs scaffolds) 165% 165% 132% 88%
Degen reads 40% 40% 38% 31%

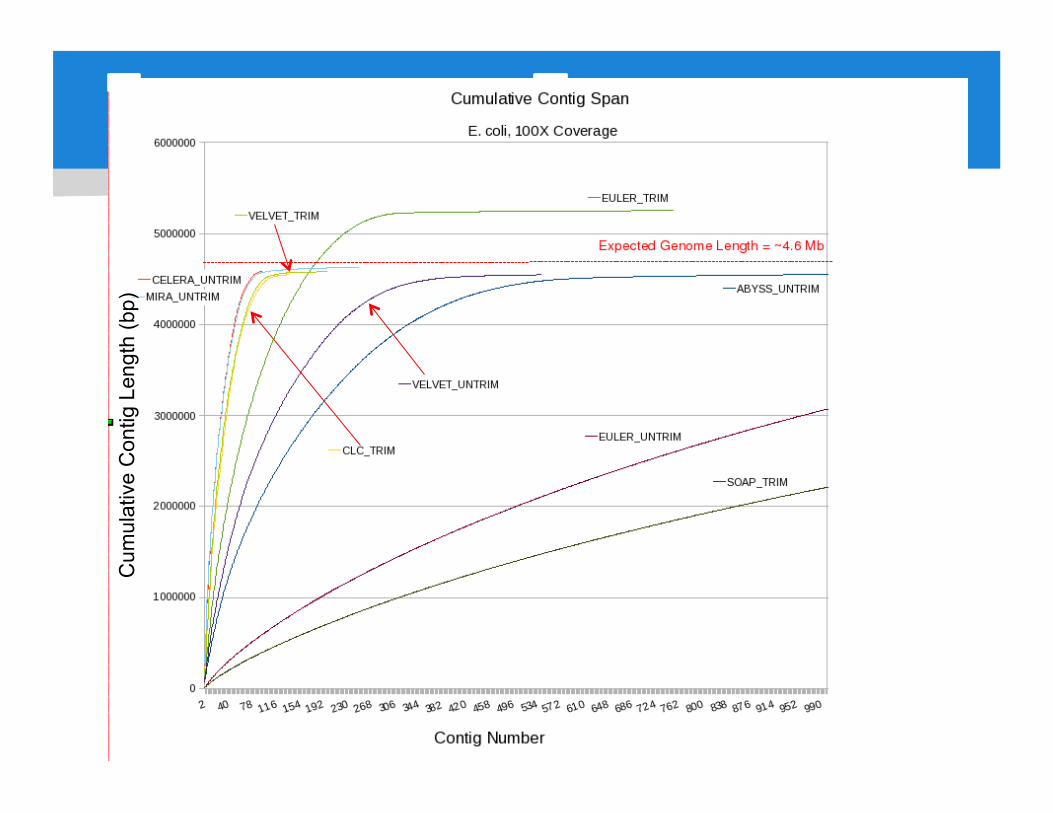
Illumina Assemblies
• The NAT experiment at JCVI Bacterial DNA from E. coli and Y. pestis Test Illumina 100bp PE at various coverage Test addition of 454 fragment or PE Test all assemblers
Cum
ulat
ive
Con
tig L
engt
h (b
p)

Results: Performance Metrics For E. coli Assemblies
0
200
400
600
800
1000
1200
1400
1600
25x trim 50x trim 100x trim
150x trim
%C
PU U
sed
Readset
CLC illumina
Velvet illumina
SOAP illumina
MIRA illumina
EULER illumina 150000
1500000
15000000
25x trim 50x trim 100x trim
150x trim
Virt
ual M
emor
y (k
b) –
Log
sc
ale
Readset
CLC illumina
Velvet illumina
SOAP illumina
MIRA illumina
EULER illumina
100000
1000000
10000000
100000000
25x trim 50x trim 100x trim 150x trim
RA
M (k
b) –
log
scal
e
CLC illumina
Velvet illumina
SOAP illumina
MIRA illumina
EULER illumina
Hands On: Assembly Comparison

Assembly Viewers

Assembly Error?
Prochlorococcus from GOS III metagenomic assembly. !Scaffold generated by Celera Assembler, displayed with Hawkeye viewer.!Investigation by Doug Rusch, JCVI.!
Green: mate pairs satisfied by the assembly.!
Coverage discontinuity.!

Another Viewpoint
Prochlorococcus fragment recruitment to scaffold. !GOS reads aligned to GOS III assembly, displayed with JCVI Fragment Recruitment Viewer.!Investigation by Doug Rusch, JCVI.!
Color: collection site.!
Recruitment discontinuity !corresponds to 16s-23S rRNA operon!
100% identity!
50% identity!
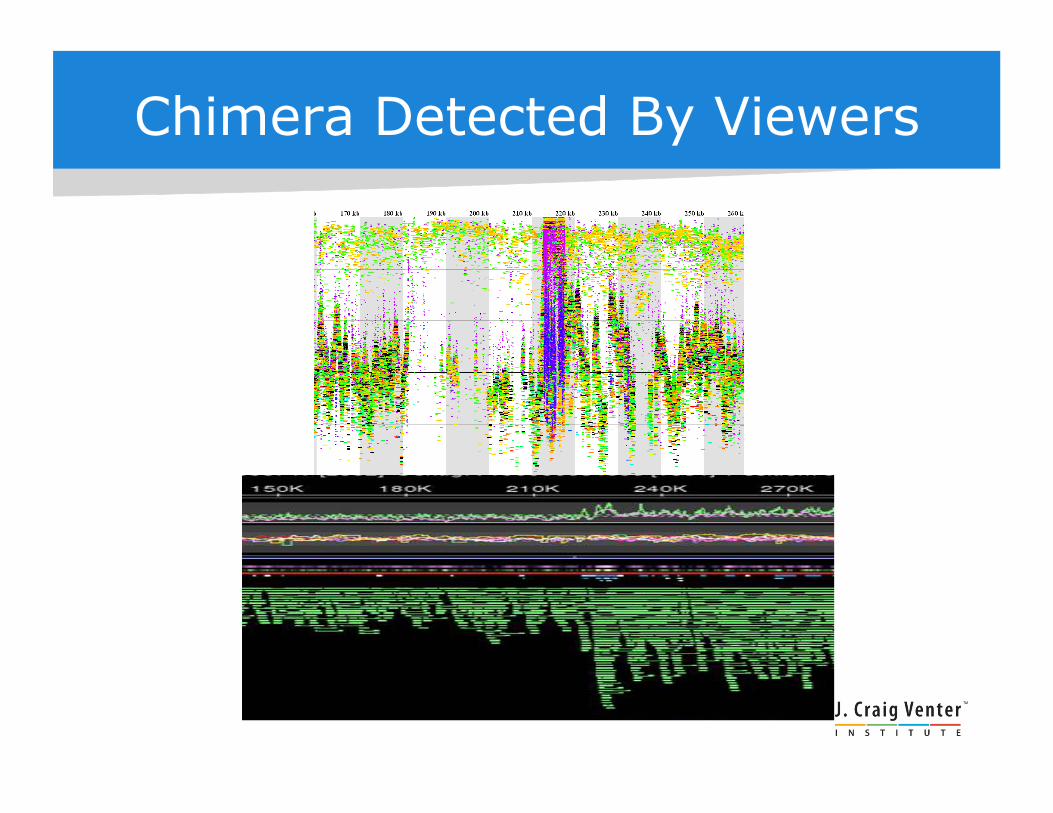
Chimera Detected By Viewers

Introduction to Viewers: Consed

Introduction to Viewers: Hawkeye

Hands On: Viewing Using Hawkeye

Introduction to Viewers: Gap4
Introduction to Viewers: Gap5

Hands On: Viewing Using Gap4/5

Questions





























