Embed Size (px)
Citation preview

— —< <
Geometry Optimization of Polymersby the Elongation Method
MASAKI MITANI AND YURIKO AOKIDepartment of Chemistry, Faculty of Science, Hiroshima University, Kagamiyama 1-3-1,Higashi-Hiroshima 739, Japan
AKIRA IMAMURADepartment of Chemistry, Faculty of Science, Hiroshima University, Kagamiyama 1-3-1, Higashi-Hiroshima 739, Japan; Group, PRESTO, Research Development Corporation of Japan, ResearchConsortium, Tsukuba 300-26, Japan
Received April 9, 1996; accepted April 25, 1996
ABSTRACT
Theoretical studies on the electronic and the geometrical structures for various moleculesby the molecular orbital or the density functional theory have recently been developedand applied widely under the progress of computer technologies. At present, it ispossible to carry out a theoretical investigation on electronic properties for small moleculesat the Hartree]Fock and the post-Hartree]Fock levels by the improvement of advancedprogram packages. However, it is difficult to perform the theoretical calculations onelectronic structures for large polymers with the aperiodic sequence of molecularsegments, because the theoretical treatment of random systems has not yet beenestablished. We recently proposed the elongation method as a useful theoretical approachto obtain the electronic states of any polymers without the periodic geometry of molecularfragments. In the previous works, the reliability of our treatment has been shown by theapplication to many polymers under single-point calculations with fixed moleculargeometry. Thus, as the next step of our study, an attempt for the geometry optimizationof large polymers by the elongation method was made in this work. As the first samplesof geometry optimization, the periodic polymers of polyethylene, polyacetylene, andpolyglycine were examined. Also, as the second samples, the locally aperiodic polymersof polyacetylene with local defects of positively and negatively charged solitons weretested. Total energies, optimized geometries, and electron densities were checked bythose obtained from the conventional molecular orbital method. Q 1997 John Wiley &Sons, Inc.
( )International Journal of Quantum Chemistry, Vol. 64, 301]323 1997Q 1997 John Wiley & Sons, Inc. CCC 0020-7608 / 97 / 030301-23

MITANI, AOKI, AND IMAMURA
Introduction
p to now, under the remarkable progress ofU computer technologies such as a work stationwith fast CPU and large memory or an easy to usegraphic user interface, computational study by thequantum chemical approach has extensively beendeveloped in the field of computational and exper-imental chemistries. The molecular orbital calcula-tion has already become a common and powerfultool to investigate the electronic properties of vari-ous molecules. The methodology of molecular or-bital theory for both the ground and excited statesof molecules is very sophisticated, and right now,the electronic and geometrical structures of smallmolecules can be obtained theoretically at theHartree]Fock and the post-Hartree]Fock levels of
w xapproximations 1 . Furthermore, many populargeneral program packages which can be used eas-ily have been advanced in the field of moleculecalculation. Also, it is possible to treat the periodicsystems such as polymers, surfaces, and crystalstheoretically by the crystal orbital method underthe periodic boundary condition. The characteristicfeatures of the periodic systems in the band struc-ture or the density of states can be estimatedcomputationally without the end effects according
w xto the periodic Hartree]Fock scheme 2, 3 , and,recently, ways to treat the electron correlation ef-fects in the periodic systems have been in progressw x4]6 . On the contrary, compared with molecularcalculations, the improvement of program pack-ages for the crystal calculation has been delayed.However, the program packages which enable usto obtain the bulk electronic properties for one- tothree-dimensional crystals by ab initio calculations
wwere released recently as CRYSTAL88 and 92 7,x8 . The studies of crystalline systems by these
w xprograms were reported 9]14 . As mentionedabove, the theoretical treatment of small moleculesand periodic crystals has already been well estab-lished.
However, it is very difficult to apply these con-ventional quantum chemical approaches to thelarge aperiodic systems of random polymers, ad-sorbed surfaces, and defective crystals, because thesize of these systems extending in three dimen-sions is too large to calculate the whole system bythe cluster model based on the molecular orbitalmethod and the periodicity of these systems is sobroken that we cannot apply the periodic supercell
model based on the crystal orbital method. In theusual way, part of these large systems is approxi-mated as a cluster of a supercell, but it should benoted that this approximation is not necessarilysatisfactory enough. In particular, the perturbedperiodic system with disordered sites includes thefollowing two states, i.e., the periodic-extendedstate which is suitable to be described by thecrystal orbital as a crystal with the extended bulknature, and the aperiodic-localized state which canbe represented appropriately by the molecular or-bital as a cluster with the localized molecular na-ture. In the molecular approach for the clustermodel, the periodic condition is estimated byembedding a molecule in the any approximatedcrystal field represented by point charges. In thecrystal approach for the supercell model, the non-periodic nature is evaluated by adapting a largecell including disordered sites as a unit cell for thetranslation symmetry of the periodic system. But,the molecular calculation cannot reproduce com-pletely the bulk electron density distribution in acluster, because of the cluster edges on whichneighboring molecules do not exist. For the crystalapproach, the interactions among neighboring dis-ordered sites affect the electronic structure of acentral supercell, because of the periodicity inwhich surrounding supercells exist around thesupercell.
The effective theoretical treatment in large, ex-tended, nonperiodic systems has not yet been es-tablished. Trials for the quantum chemical calcula-
w xtions for the extended molecular systems 15]23w xand the perturbed periodic systems 24]31 are
now in progress. In the treatment of these systems,the fragmented cluster for a large molecule or theembedded cluster for a perturbed crystal is oftenadapted. However, in these approaches, the parti-tion of the systems into the interaction and thenoninteraction regions must be presumed, and,also, the former is evaluated exactly while thelatter is estimated approximately in calculating theelectronic structures of the entire systems. Thus,the determination of the perturbed and the unper-turbed spaces is arbitrary, and, then, the obtainedresults depend sensitively on the manner of divi-sion in the systems. Also, attempts for the recon-
w xstruction of the band structures 32, 33 or thew xextrapolation of the bulk properties 34]36 of pe-
riodic polymers from the finite cluster calculationsare now under way.
The serious problem in the application of themolecular orbital and the crystal orbital calcula-
VOL. 64, NO. 3302

GEOMETRY OPTIMIZATION OF POLYMERS
tions to the extended system without the periodicgeometry is that the larger the size of systembecomes, the more enormous the computationalefforts become for the calculations of integrals andthe diagonalization of matrices in the iterations of
Ž .the self-consistent-field SCF procedure. Espe-cially, the above-mentioned steps in the computa-tional procedure consume much CPU time in theab initio calculation. In the large extended system,the latter step may become more serious in com-parison with the former step, because the many-centered integrals with the basis functions sepa-rated far from each other can be neglected orapproximated in any way while the dimension ofthe matrices for the eigenvalue problem cannot bereduced in any way by the large size of the sys-tem. Moreover, it is impossible to determine apriori the optimal size of a cluster or a supercellwhich can describe the electronic structure of thelarge system reasonably. This fact requires us torepeat the calculations for the models with varioussizes to find the most effective model by checkingthe convergence in the electronic property.
To overcome the above-mentioned problems, itshould be pointed out that the general interactionspace among the partitioned subsystems for theextended system must be determined uniquely bya theoretical formulation. For that purpose, werecently proposed an approach as the elongationmethod. This method includes the following con-tinuous three processes: First, we calculate theelectronic structure of a cluster or a supercell withsuitable sizes as a starting system by the molecularorbital or the crystal orbital method. Second, toextend the starting system by the elongation pro-cedure, the interaction orbitals are extracted fromall the orbitals included in the starting systemunder the perturbation caused by the connectionwith other fragments. Third, the eigenvalue prob-lem is solved only within the interaction space toobtain the electronic structure of the whole ex-tended system. By repeating these procedures suc-cessively, various fragments can be combined witha cluster, or disordered parts can be created into asupercell. In these processes, we must divide thetotal orbital space of a system into two subspacesof the interaction and the noninteraction spacesunder a perturbation. For that purpose, we re-cently proposed and developed the elongation
wmethod by the uniform localization procedure 37,x38 at various levels of approximation and applied
w xit to many molecular systems 39]43 . Moreover,
the stationary conditions of the electronic structureagainst the extension of molecular system has beenintroduced into the elongation calculation as acluster-series model and adapted to various poly-
w xmer systems 44]47 . The utility of our approachin the crystal orbital calculation has already been
w xsuggested 48]50 .In the cluster-series calculation with the station-
ary conditions by the elongation procedure, a clus-ter molecule is extended by connecting a molecu-lar fragment with a cluster terminal one by one toobtain the polymer including any species of seg-ments with the desired length. The stationary or-bitals in the series of clusters can be extracted byfinding the orbitals which satisfy both the or-thonormality and the variational conditions in theextended cluster. Then, the repeated extensions oflarge clusters can be performed successively byfreezing the stationary orbitals, i.e., the stationaryorbitals can be identified as the unaltered orbitalsin the original cluster before and after the exten-sion of the cluster. These stationary orbitals can bedetermined uniquely by a simple method usingthe unitary transformation of orbitals in the occu-pied and the virtual spaces of clusters as shown in
w xthe previous articles 44]47 which can be re-garded as an essentially similar procedure to the
w xcorresponding orbital 51 or the interaction fron-w xtier orbital 52, 53 . By freezing the stationary or-
bitals in a series of cluster extending calculations,we can reduce effectively the dimensions of theeigenvalue problems for extended system in theSCF procedures.
In the previous work, we reported the reliabilityof the elongation approach by applying to varioussystems. The single-point calculations with fixedgeometrical parameters of molecules were carriedout in these studies. Therefore, as the next stage inour study, we tried to perform the geometry opti-mization of polymers by the elongation method.
In the present article, we show the results of testapplications of the elongation approach to periodicpolymers of polyethylene, polyacetylene, andpolyglycine and also aperiodic polymers of defec-tive polyacetylene with positively and negativelycharged solitons. The calculations were done byintroducing the elongation method into the pro-gram package for various semiempirical molecular
w xorbital methods of MOPAC Ver. 6.0 54 and byw xusing the AM1 Hamiltonian 55 on IBM RISC
Systemr6000 in our laboratory.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 303

MITANI, AOKI, AND IMAMURA
Method
In the elongation calculation for a continuousseries of any cluster, a starting cluster with anappropriate size is extended successively by con-necting any adding fragments with any clusterends. The extraction of the interaction space in anextended system plays an important role for theefficient treatment of a large molecule. By ourelongation procedure, we can select the specificorbitals from the total orbital space of the systemas the stationary orbitals which can be removedmore effectively from the basis orbitals of theeigenvalue problem.
Now, we consider the extension process inwhich a molecule A is connected with a fragmentB to produce an extended cluster A q B. In ausual way, A or A q B is calculated as onemolecule individually, and the basis orbitals of
Ž .eigenvalues problems are atomic orbitals AOs inA or A q B. In the calculation of A q B, we can
Ž .employ molecular orbitals MOs of A and B, i.e.,the same result for A q B can be obtained byusing MOs instead of AOs as the basis orbitals ofthe eigenvalue problem. But, the dimensions of theAO-based and the MO-based matrices diagonal-ized in the SCF procedure are the same. To per-form a more effective computation of A q B, thesuitable interaction space between A and B inevaluating the electronic structure of A q B shouldbe determined in any manner. In other words, if itis possible to extract the MOs in A which arealtered and unaltered by the effect of bonded frag-ment B, we may include only the former MOs inthe eigenvalue problem and we may be able toremove the latter MOs from the problem, i.e., theeigenvalue problem can be solved within the spaceof a part of MOs in A and all of AOs in B toobtain the electronic structure of A q B. This pro-cedure may be more effective for the system inwhich the size of A is much larger than that of Bsuch as the continuous extensions of clusters. So,by the iterations of this procedure by any desiredtimes, we can synthesize theoretically the polymerwith any various segments as A q B q ??? qNmore efficiently than can the conventional calcula-tion using all AOs in the system.
As mentioned above, the selection of stationaryMOs in extended systems is an important prob-lem. In the previous work, we introduced the
stationary conditions of the electronic structureagainst the extension of clusters and the simplemethod to extract effectively the stationary MOs inextended clusters which can be removed from theeigenvalue problem in continuous extensions. Bythis treatment, we can calculate the extended poly-mers without increasing the dimensions of matri-ces which should be diagonalized in the eigen-
w xvalue problems 44]47 .The stationary conditions are defined as fol-
lows: the stationary MOs must satisfy both theorthonormality and the variational conditions, i.e.,the MOs in an original cluster A which are alsowell-defined MOs in an extended cluster A q Bcan be selected by finding the MOs which satisfythe conditions. Both conditions are written as fol-lows:
( )i The orthonormality condition:
a bŽ . Ž .c A c A s d d ,² :i j i j ab
a bŽ . Ž .² :c A f A q B s 0,i r
( )ii The variational condition:
o vˆŽ . Ž . Ž .c A F A q B c A s 0,¦ ;i j
o vˆŽ . Ž . Ž .c A F A q B f A q B² :i s
v oˆŽ . Ž . Ž .s c A F A q B f A q B s 0,¦ ;j t
where the symbols a, b s o or v, the superscripts oand v indicate occupied and virtual MOs, respec-
Ž̂ .tively, and F A q B is the Fock operator of the� oŽ .4 � vŽextended cluster A q B. f A q B and f At s
.4q B represent the remaining nonstationary MOsof the cluster A q B. Therefore, the MOs in the
� oŽ .4 � vŽ .4cluster A of f A and c A can be calledi jstationary MOs against the extension from A toA q B. Thus, if the electronic structures of both
Ž̂ .clusters A and A q B are determined if F A q Bis known, it is possible to define the stationaryspace uniquely based on these orthonormality andvariational conditions.
The way to extract the stationary MOs has al-ready been proposed and applied to elongationcalculations for various polymers. Two-step proce-dures are introduced according to two conditionsw x44]47 .
The first step is to specify the MOs which aresatisfied and unsatisfied with the orthonormality
VOL. 64, NO. 3304

GEOMETRY OPTIMIZATION OF POLYMERS
condition in the extended system. To select suchMOs, we form two rectangular overlap matricesbetween original MOs in A and AOs in B asfollows:
o o oŽ .² :S s c x i s 1, . . . , N ; r s 1, . . . , M ,i r i r
Ž .1a
v v vŽ .S s c x j s 1, . . . , N ; r s 1, . . . , M .² :jr j r
Ž .1b
� o4 � v4 o vwhere c and c are N and N original MOsi jin the occupied and the virtual spaces in A, re-
� 4spectively, and x indicates adding AOs in Brwith the number of M. Then, the products SoSoH
and SvSvH are diagonalized in order to evaluatethe eigenvalues of these matrices, respectively.From the magnitude of the obtained eigenvalues,
� 4we divide c , which is the original occupied and� 4virtual MOs, in C9 , which is the set of MOs with
� 4the zero eigenvalue, and x 9 , which consists of� 4MOs with the nonzero eigenvalue w9 and of
� 4adding AOs x . Thus, it can be considered thatthe former space satisfies and the latter spacebreaks the orthonormality condition in the ex-tended system.
The second step is to extract the MOs which aresatisfies and unsatisfied with the variational condi-tion in the total orbital space. For that purpose, thefollowing rectangular Fock matrices are defined:
X o X vo Ž̂ .F s c F A q B c¦ ;i j i j
Ž X o X v . Ž .i s 1, . . . , N ; j s 1, . . . , N , 2aX o Xo Ž̂ .F s c F A q B x² :i s i s
Ž o v v . Ž .i s 1, . . . , N9 ; s s N9 q 1, . . . , N9 q M9 , 2bX v X ov Ž̂ .F s c F A q B c¦ ;ji j i
Ž v o . Ž .j s 1, . . . , N9 ; i s 1, . . . , N9 , 2cX v Xv Ž̂ .F s c F A q B x¦ ;jt j t
Ž v o o . Ž .j s 1, . . . , N9 ; t s N9 q 1, . . . , N9 q M9 , 2d
where N9o and N X v are the numbers of occupied� 4and virtual MOs in the orbital space of c 9 , and
M9 denotes the dimension of MOs and AOs in the� 4orbital space of x 9 , which are obtained in the
first step. To find the stationary and the interaction� 4 o oHMOs included in c 9 , then the matrices F F
and F v F vH are diagonalized, and, next,the MOswhose eigenvalues are less or greater than anappropriate threshold value are selected as the
� 4 � 4stationary MOs c 0 or the interaction MOs w0� 4from occupied and virtual spaces in c 9 , respec-
tively.� 4Thus, finally, the original MOs c can be classi-
� 4fied into the stationary MOs c 0 which satisfy theorthonormality and the variational conditions, the
� 4interaction MOs including the MOs w9 which donot satisfy the orthonormality condition, and the
FIGURE 1. Polymer systems and initial values for their( )geometrical parameters optimized in this study: a
˚ ˚( ) ( )polyethylene with r C } C = 1.54 A, r C } H = 1.09 A,( ) ( ) ( )/ CCC = / CCH = 109.58, and / CCCH = "60.08;
˚( ) ( ) ( )b polyacetylene with r C } C = 1.46 A, r C C =˚ ˚( ) ( ) ( )1.35 A, r C } H = 1.09 A, and / CCC = / CCH =
˚( ) ( ) ( )120.08; c polyglycine with r N } C = 1.47 A, r C } C˚ ˚ ˚( ) ( ) (= 1.53 A, r C } N = 1.32 A, r C O = 1.24 A, r N }
˚) ( ) ( ) ( )H = r C } H = 1.00 A, / NCC = / NCH = 109.58,( ) ( ) ( ) ( )/ CCN = 114.08, / CNC = / CNH = 123.08 / CCO
( ) ( )= 121.08, and / CNCH = "60.08, and d positive and˚( ) ( ) ( )e negative solitons with r C } C = 1.46 A, r C C =
˚ ˚( ) ( ) ( )1.35 A, r C } H = 1.09 A, and / CCC = / CCH =( ) ( ) ( )120.08. Periodic polymers of a , b , and c or aperiodic
( ) ( )polymers of d and e were extended in one or twodirections by replacing the one or two hydrogen atomsat cluster terminals by adding fragments, respectively.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 305
MITANI, AOKI, AND IMAMURA
� 4MOs w0 which do not satisfy the variationalcondition. By these simple ways using the unitarytransformation within the original cluster, we cancreate the stationary and the interaction MOsuniquely. Then, the stationary MOs can be frozenin the next extension of cluster by removing thosefrom the total MOs.
The stationary conditions include all terms inA q B which cause the orbital mixing among Aand B in the rectangular matrix F as the polariza-tion effect within A in F 0 and F v and the chargei j jitransfer and exchange interactions between A andB in F 0 and F v. Therefore, the stationary MOs cano s jtbe recognized as the unaltered MOs in a after theextension of A q B by B, and this orbital extrac-tion is expected to be reasonable.
To apply this treatment exactly, the Fock opera-tor A q B must be known as mentioned in the
w xoriginal formulation 44]46 , i.e., the stationaryMOs are determined exactly after the calculationsof both clusters A and A q B. Also, an approxi-mated treatment was developed in which the sta-tionary MOs are obtained in the extension calcula-
w xtion from A to A q B 47]50 . In other words, thestationary MOs and the Fock operator in the ex-tended cluster are obtained simultaneously by thestepwise procedure in which the eigenvalue prob-lem of A q B is solved repeatedly within the
� 4 � 4 � 4interaction space x [ w9 [ w0 by a series ofthe efficient SCF steps starting with the Fock oper-ator of A and the core Hamiltonian between Aand B. The detailed explanations of both formula-tions is included in these previous articles.
Finally, it is noted that since the semiempiricalmolecular orbital calculation was adapted in thepresent study, the orthonormality condition wasautomatically satisfied, and the first step can beskipped in extracting the stationary MOs.
Results and Discussion
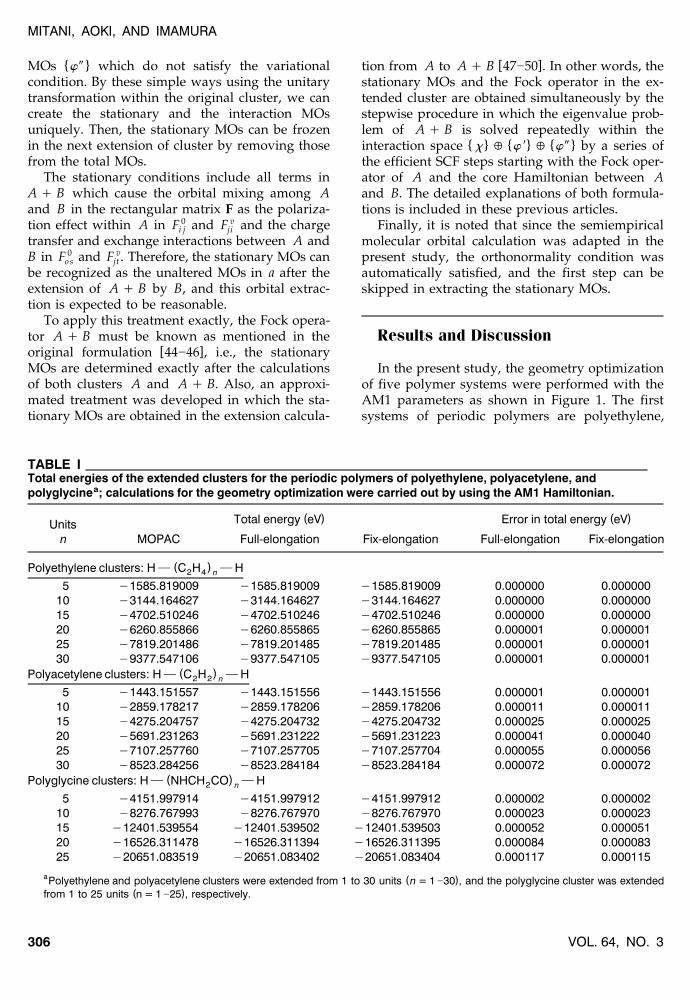
In the present study, the geometry optimizationof five polymer systems were performed with theAM1 parameters as shown in Figure 1. The firstsystems of periodic polymers are polyethylene,
TABLE ITotal energies of the extended clusters for the periodic polymers of polyethylene, polyacetylene, and
apolyglycine ; calculations for the geometry optimization were carried out by using the AM1 Hamiltonian.
( ) ( )Total energy eV Error in total energy eVUnitsn MOPAC Full-elongation Fix-elongation Full-elongation Fix-elongation
( )Polyethylene clusters: H } C H } H2 4 n
5 y1585.819009 y1585.819009 y1585.819009 0.000000 0.00000010 y3144.164627 y3144.164627 y3144.164627 0.000000 0.00000015 y4702.510246 y4702.510246 y4702.510246 0.000000 0.00000020 y6260.855866 y6260.855865 y6260.855865 0.000001 0.00000125 y7819.201486 y7819.201485 y7819.201485 0.000001 0.00000130 y9377.547106 y9377.547105 y9377.547105 0.000001 0.000001
( )Polyacetylene clusters: H } C H } H2 2 n
5 y1443.151557 y1443.151556 y1443.151556 0.000001 0.00000110 y2859.178217 y2859.178206 y2859.178206 0.000011 0.00001115 y4275.204757 y4275.204732 y4275.204732 0.000025 0.00002520 y5691.231263 y5691.231222 y5691.231223 0.000041 0.00004025 y7107.257760 y7107.257705 y7107.257704 0.000055 0.00005630 y8523.284256 y8523.284184 y8523.284184 0.000072 0.000072
( )Polyglycine clusters: H } NHCH CO } H2 n
5 y4151.997914 y4151.997912 y4151.997912 0.000002 0.00000210 y8276.767993 y8276.767970 y8276.767970 0.000023 0.00002315 y12401.539554 y12401.539502 y12401.539503 0.000052 0.00005120 y16526.311478 y16526.311394 y16526.311395 0.000084 0.00008325 y20651.083519 y20651.083402 y20651.083404 0.000117 0.000115
a ( )Polyethylene and polyacetylene clusters were extended from 1 to 30 units n = 1 ]30 , and the polyglycine cluster was extended( )from 1 to 25 units n = 1 ]25 , respectively.
VOL. 64, NO. 3306
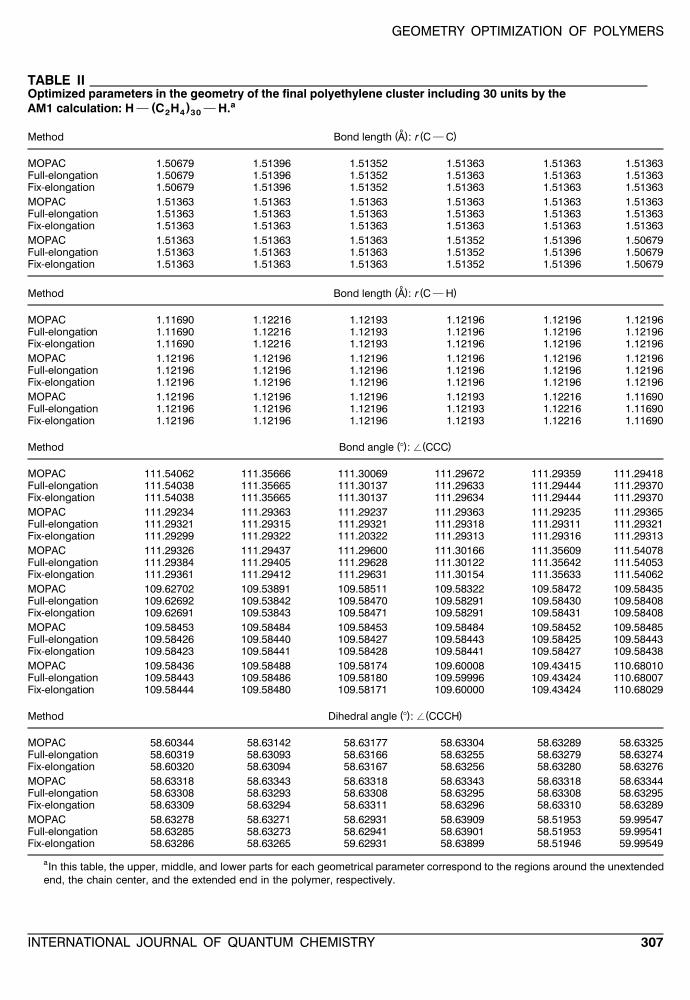
GEOMETRY OPTIMIZATION OF POLYMERS
TABLE IIOptimized parameters in the geometry of the final polyethylene cluster including 30 units by the
a( )AM1 calculation: H } C H } H.2 4 30
˚( ) ( )Method Bond length A : r C } C
MOPAC 1.50679 1.51396 1.51352 1.51363 1.51363 1.51363Full-elongation 1.50679 1.51396 1.51352 1.51363 1.51363 1.51363Fix-elongation 1.50679 1.51396 1.51352 1.51363 1.51363 1.51363MOPAC 1.51363 1.51363 1.51363 1.51363 1.51363 1.51363Full-elongation 1.51363 1.51363 1.51363 1.51363 1.51363 1.51363Fix-elongation 1.51363 1.51363 1.51363 1.51363 1.51363 1.51363MOPAC 1.51363 1.51363 1.51363 1.51352 1.51396 1.50679Full-elongation 1.51363 1.51363 1.51363 1.51352 1.51396 1.50679Fix-elongation 1.51363 1.51363 1.51363 1.51352 1.51396 1.50679
˚( ) ( )Method Bond length A : r C } H
MOPAC 1.11690 1.12216 1.12193 1.12196 1.12196 1.12196Full-elongation 1.11690 1.12216 1.12193 1.12196 1.12196 1.12196Fix-elongation 1.11690 1.12216 1.12193 1.12196 1.12196 1.12196MOPAC 1.12196 1.12196 1.12196 1.12196 1.12196 1.12196Full-elongation 1.12196 1.12196 1.12196 1.12196 1.12196 1.12196Fix-elongation 1.12196 1.12196 1.12196 1.12196 1.12196 1.12196MOPAC 1.12196 1.12196 1.12196 1.12193 1.12216 1.11690Full-elongation 1.12196 1.12196 1.12196 1.12193 1.12216 1.11690Fix-elongation 1.12196 1.12196 1.12196 1.12193 1.12216 1.11690
( ) ( )Method Bond angle 8 : / CCC
MOPAC 111.54062 111.35666 111.30069 111.29672 111.29359 111.29418Full-elongation 111.54038 111.35665 111.30137 111.29633 111.29444 111.29370Fix-elongation 111.54038 111.35665 111.30137 111.29634 111.29444 111.29370MOPAC 111.29234 111.29363 111.29237 111.29363 111.29235 111.29365Full-elongation 111.29321 111.29315 111.29321 111.29318 111.29311 111.29321Fix-elongation 111.29299 111.29322 111.20322 111.29313 111.29316 111.29313MOPAC 111.29326 111.29437 111.29600 111.30166 111.35609 111.54078Full-elongation 111.29384 111.29405 111.29628 111.30122 111.35642 111.54053Fix-elongation 111.29361 111.29412 111.29631 111.30154 111.35633 111.54062MOPAC 109.62702 109.53891 109.58511 109.58322 109.58472 109.58435Full-elongation 109.62692 109.53842 109.58470 109.58291 109.58430 109.58408Fix-elongation 109.62691 109.53843 109.58471 109.58291 109.58431 109.58408MOPAC 109.58453 109.58484 109.58453 109.58484 109.58452 109.58485Full-elongation 109.58426 109.58440 109.58427 109.58443 109.58425 109.58443Fix-elongation 109.58423 109.58441 109.58428 109.58441 109.58427 109.58438MOPAC 109.58436 109.58488 109.58174 109.60008 109.43415 110.68010Full-elongation 109.58443 109.58486 109.58180 109.59996 109.43424 110.68007Fix-elongation 109.58444 109.58480 109.58171 109.60000 109.43424 110.68029
( ) ( )Method Dihedral angle 8 : / CCCH
MOPAC 58.60344 58.63142 58.63177 58.63304 58.63289 58.63325Full-elongation 58.60319 58.63093 58.63166 58.63255 58.63279 58.63274Fix-elongation 58.60320 58.63094 58.63167 58.63256 58.63280 58.63276MOPAC 58.63318 58.63343 58.63318 58.63343 58.63318 58.63344Full-elongation 58.63308 58.63293 58.63308 58.63295 58.63308 58.63295Fix-elongation 58.63309 58.63294 58.63311 58.63296 58.63310 58.63289MOPAC 58.63278 58.63271 58.62931 58.63909 58.51953 59.99547Full-elongation 58.63285 58.63273 58.62941 58.63901 58.51953 59.99541Fix-elongation 58.63286 58.63265 59.62931 58.63899 58.51946 59.99549
aIn this table, the upper, middle, and lower parts for each geometrical parameter correspond to the regions around the unextendedend, the chain center, and the extended end in the polymer, respectively.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 307
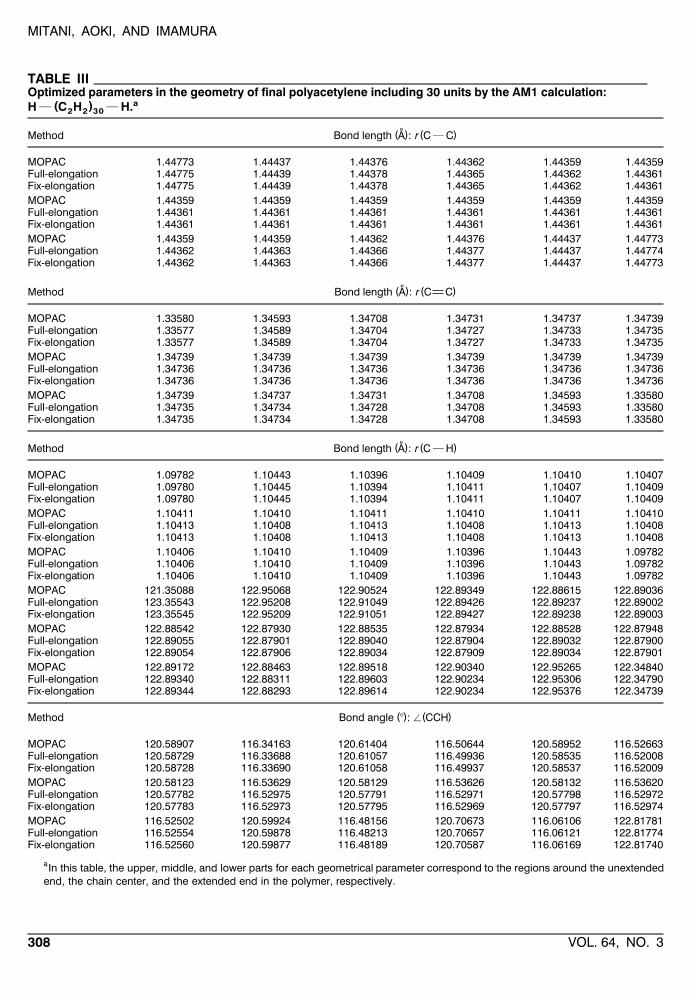
MITANI, AOKI, AND IMAMURA
TABLE IIIOptimized parameters in the geometry of final polyacetylene including 30 units by the AM1 calculation:
a( )H } C H } H.2 2 30
˚( ) ( )Method Bond length A : r C } C
MOPAC 1.44773 1.44437 1.44376 1.44362 1.44359 1.44359Full-elongation 1.44775 1.44439 1.44378 1.44365 1.44362 1.44361Fix-elongation 1.44775 1.44439 1.44378 1.44365 1.44362 1.44361MOPAC 1.44359 1.44359 1.44359 1.44359 1.44359 1.44359Full-elongation 1.44361 1.44361 1.44361 1.44361 1.44361 1.44361Fix-elongation 1.44361 1.44361 1.44361 1.44361 1.44361 1.44361MOPAC 1.44359 1.44359 1.44362 1.44376 1.44437 1.44773Full-elongation 1.44362 1.44363 1.44366 1.44377 1.44437 1.44774Fix-elongation 1.44362 1.44363 1.44366 1.44377 1.44437 1.44773
˚( ) ( )Method Bond length A : r C C
MOPAC 1.33580 1.34593 1.34708 1.34731 1.34737 1.34739Full-elongation 1.33577 1.34589 1.34704 1.34727 1.34733 1.34735Fix-elongation 1.33577 1.34589 1.34704 1.34727 1.34733 1.34735MOPAC 1.34739 1.34739 1.34739 1.34739 1.34739 1.34739Full-elongation 1.34736 1.34736 1.34736 1.34736 1.34736 1.34736Fix-elongation 1.34736 1.34736 1.34736 1.34736 1.34736 1.34736MOPAC 1.34739 1.34737 1.34731 1.34708 1.34593 1.33580Full-elongation 1.34735 1.34734 1.34728 1.34708 1.34593 1.33580Fix-elongation 1.34735 1.34734 1.34728 1.34708 1.34593 1.33580
˚( ) ( )Method Bond length A : r C } H
MOPAC 1.09782 1.10443 1.10396 1.10409 1.10410 1.10407Full-elongation 1.09780 1.10445 1.10394 1.10411 1.10407 1.10409Fix-elongation 1.09780 1.10445 1.10394 1.10411 1.10407 1.10409MOPAC 1.10411 1.10410 1.10411 1.10410 1.10411 1.10410Full-elongation 1.10413 1.10408 1.10413 1.10408 1.10413 1.10408Fix-elongation 1.10413 1.10408 1.10413 1.10408 1.10413 1.10408MOPAC 1.10406 1.10410 1.10409 1.10396 1.10443 1.09782Full-elongation 1.10406 1.10410 1.10409 1.10396 1.10443 1.09782Fix-elongation 1.10406 1.10410 1.10409 1.10396 1.10443 1.09782MOPAC 121.35088 122.95068 122.90524 122.89349 122.88615 122.89036Full-elongation 123.35543 122.95208 122.91049 122.89426 122.89237 122.89002Fix-elongation 123.35545 122.95209 122.91051 122.89427 122.89238 122.89003MOPAC 122.88542 122.87930 122.88535 122.87934 122.88528 122.87948Full-elongation 122.89055 122.87901 122.89040 122.87904 122.89032 122.87900Fix-elongation 122.89054 122.87906 122.89034 122.87909 122.89034 122.87901MOPAC 122.89172 122.88463 122.89518 122.90340 122.95265 122.34840Full-elongation 122.89340 122.88311 122.89603 122.90234 122.95306 122.34790Fix-elongation 122.89344 122.88293 122.89614 122.90234 122.95376 122.34739
( ) ( )Method Bond angle 8 : / CCH
MOPAC 120.58907 116.34163 120.61404 116.50644 120.58952 116.52663Full-elongation 120.58729 116.33688 120.61057 116.49936 120.58535 116.52008Fix-elongation 120.58728 116.33690 120.61058 116.49937 120.58537 116.52009MOPAC 120.58123 116.53629 120.58129 116.53626 120.58132 116.53620Full-elongation 120.57782 116.52975 120.57791 116.52971 120.57798 116.52972Fix-elongation 120.57783 116.52973 120.57795 116.52969 120.57797 116.52974MOPAC 116.52502 120.59924 116.48156 120.70673 116.06106 122.81781Full-elongation 116.52554 120.59878 116.48213 120.70657 116.06121 122.81774Fix-elongation 116.52560 120.59877 116.48189 120.70587 116.06169 122.81740
aIn this table, the upper, middle, and lower parts for each geometrical parameter correspond to the regions around the unextendedend, the chain center, and the extended end in the polymer, respectively.
VOL. 64, NO. 3308
GEOMETRY OPTIMIZATION OF POLYMERS
TABLE IVOptimized parameters in the geometry of final polyglycine including 25 units by the AM1 calculation:
a( )H } NHCH CO } H.2 25
˚( ) ( )Method Bond length A : r N } C
MOPAC 1.41275 1.43366 1.43479 1.43511 1.43523 1.43529Full-elongation 1.41272 1.43363 1.43475 1.43507 1.43519 1.43524Fix-elongation 1.41272 1.43363 1.43475 1.43507 1.43519 1.43524
MOPAC 1.43535 1.43535 1.43535 1.43536 1.43536 1.43536Full-elongation 1.43531 1.43531 1.43532 1.43532 1.43532 1.43532Fix-elongation 1.43531 1.43531 1.43532 1.43532 1.43532 1.43532
MOPAC 1.43535 1.43534 1.43533 1.43531 1.43521 1.43508Full-elongation 1.43532 1.43533 1.43532 1.43531 1.43521 1.43508Fix-elongation 1.43532 1.43533 1.43532 1.43531 1.43521 1.43508
˚( ) ( )Method Bond length A : r C } C
MOPAC 1.53642 1.53337 1.53371 1.53387 1.53394 1.53398Full-elongation 1.53634 1.53325 1.53360 1.53374 1.53381 1.53386Fix-elongation 1.53634 1.53325 1.53360 1.53374 1.53381 1.53386
MOPAC 1.53403 1.53403 1.53403 1.53403 1.53403 1.53403Full-elongation 1.53391 1.53391 1.53391 1.53392 1.53392 1.53392Fix-elongation 1.53391 1.53392 1.53391 1.53392 1.53392 1.53392
MOPAC 1.53400 1.53399 1.53395 1.53389 1.53392 1.51064Full-elongation 1.53393 1.53392 1.53390 1.53388 1.53392 1.51064Fix-elongation 1.53393 1.53392 1.53390 1.53388 1.53392 1.51064
˚( ) ( )Method Bond length A : r C } N
MOPAC 1.37920 1.37587 1.37529 1.37494 1.37483 1.37474Full-elongation 1.37935 1.37604 1.37544 1.37512 1.37501 1.37490Fix-elongation 1.37935 1.37604 1.37544 1.37512 1.37501 1.37490
MOPAC 1.37467 1.37366 1.37466 1.37465 1.37465 1.37465Full-elongation 1.37483 1.37482 1.37481 1.37481 1.37480 1.37480Fix-elongation 1.37482 1.37482 1.37480 1.37481 1.37480 1.37480
MOPAC 1.37469 1.37471 1.37475 1.37481 1.37488 1.37514Full-elongation 1.37479 1.37479 1.37480 1.37483 1.37489 1.37515Fix-elongation 1.37479 1.37479 1.37480 1.37483 1.37489 1.37515
˚( ) ( )Method Bond length A : r C O
MOPAC 1.24679 1.24825 1.24803 1.24815 1.24813 1.24817Full-elongation 1.24676 1.24821 1.24801 1.24811 1.24810 1.24813Fix-elongation 1.24676 1.24821 1.24801 1.24811 1.24810 1.24813
MOPAC 1.24818 1.24818 1.24818 1.24818 1.24818 1.24818Full-elongation 1.24815 1.24814 1.24815 1.24815 1.24815 1.24815Fix-elongation 1.24814 1.24814 1.24815 1.24815 1.24815 1.24815
MOPAC 1.24816 1.24815 1.24813 1.24814 1.24797 1.23003Full-elongation 1.24815 1.24814 1.24814 1.24813 1.24797 1.23003Fix-elongation 1.24815 1.24814 1.24814 1.24813 1.24797 1.23003
( )Continued
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 309
MITANI, AOKI, AND IMAMURA
TABLE IV( )Continued
˚( ) ( )Method Bond length A : r N } H
MOPAC 0.98375 0.99372 0.99472 0.99482 0.99492 0.99494Full-elongation 0.98376 0.99366 0.99469 0.99477 0.99487 0.99489Fix-elongation 0.98376 0.99366 0.99469 0.99477 0.99487 0.99489
MOPAC 0.99497 0.99498 0.99498 0.99498 0.99498 0.99498Full-elongation 0.99493 0.99493 0.99493 0.99494 0.99494 0.99494Fix-elongation 0.99493 0.99494 0.99493 0.99494 0.99494 0.99494
MOPAC 0.99497 0.99497 0.99495 0.99494 0.99492 0.99329Full-elongation 0.99494 0.99494 0.99494 0.99494 0.99492 0.99329Fix-elongation 0.99494 0.99494 0.99494 0.99494 0.99492 0.99329
˚( ) ( )Method Bond length A : r C } H
MOPAC 1.13267 1.13007 1.12997 1.12995 1.12993 1.12992Full-elongation 1.13268 1.13010 1.12999 1.12997 1.12995 1.12995Fix-elongation 1.13268 1.13010 1.12999 1.12997 1.12995 1.12995
MOPAC 1.12991 1.12991 1.12991 1.12991 1.12991 1.12991Full-elongation 1.12993 1.12993 1.12993 1.12993 1.12993 1.12993Fix-elongation 1.12993 1.12993 1.12993 1.12993 1.12993 1.12993
MOPAC 1.12991 1.12991 1.12992 1.12993 1.12992 1.13142Full-elongation 1.12993 1.12993 1.12993 1.12993 1.12992 1.13142Fix-elongation 1.12993 1.12993 1.12993 1.12993 1.12992 1.13142
( ) ( )Method Bond angle 8 : / NCC
MOPAC 113.20565 112.62505 112.54960 112.49716 112.48728 112.47429Full-elongation 113.20562 112.64203 112.55815 112.51415 112.49788 112.49026Fix-elongation 113.20565 112.64205 112.55815 112.51415 112.49788 112.49026
MOPAC 112.46695 112.46657 112.46703 112.46493 112.46764 112.46399Full-elongation 112.48057 112.47996 112.47931 112.47932 112.47854 112.47904Fix-elongation 112.48136 112.47950 112.48015 112.47886 112.47882 112.47838
MOPAC 112.47014 112.46744 112.47284 112.47498 112.51409 112.81717Full-elongation 112.47831 112.47748 112.47837 112.47646 112.51411 112.81740Fix-elongation 112.47849 112.47731 112.47867 112.47676 112.51371 112.81766
( ) ( )Method Bond angle 8 : / CCN
MOPAC 115.99434 116.05768 116.02800 116.04485 116.03882 116.04553Full-elongation 116.00732 116.05426 116.03542 116.04400 116.04453 116.04468Fix-elongation 116.00736 116.05430 116.03545 116.04403 116.04456 116.04475
MOPAC 116.04308 116.04428 116.04453 116.04307 116.04586 116.04207Full-elongation 116.04602 116.04600 116.04590 116.04629 116.04581 116.04659Fix-elongation 116.04620 116.04549 116.04603 116.04550 116.04630 116.04601
MOPAC 116.04747 116.04250 116.04741 116.04477 116.05412 116.11713Full-elongation 116.04660 116.04651 116.04574 116.04640 116.05368 116.11727Fix-elongation 116.04671 116.04628 116.04617 116.04631 116.05330 116.11771
( )Continued
VOL. 64, NO. 3310
GEOMETRY OPTIMIZATION OF POLYMERS
TABLE IV( )Continued
( ) ( )Method Bond angle 8 : / CNC
MOPAC 120.80427 120.72326 120.73087 120.72935 120.74060 120.73790Full-elongation 120.77027 120.70690 120.69997 120.70885 120.71093 120.71759Fix-elongation 120.77019 120.70682 120.69989 120.70878 120.71087 120.71756
MOPAC 120.74524 120.74508 120.74440 120.74731 120.74298 120.74859Full-elongation 120.72045 120.72137 120.72187 120.72190 120.72273 120.72202Fix-elongation 120.72033 120.72171 120.72204 120.72281 120.7222220.72290
MOPAC 120.73726 120.74146 120.73074 120.72770 120.71083 120.93960Full-elongation 120.72322 120.72306 120.72119 120.72158 120.71134 120.93914Fix-elongation 120.72288 120.72326 120.72065 120.72141 120.71181 120.93860
( ) ( )Method Bond angle 8 : / CCO
MOPAC 123.39053 122.90626 122.79702 122.74121 122.72171 122.70823Full-elongation 123.40751 122.93375 122.82256 122.76989 122.74880 122.73680Fix-elongation 123.40765 122.93383 122.82266 122.76997 122.74888 122.73691
MOPAC 122.69549 122.69482 122.69450 122.69354 122.69457 122.69332Full-elongation 122.72272 122.72167 122.72081 122.72041 122.71969 122.71973Fix-elongation 122.72301 122.72077 122.72108 122.72005 122.71994 122.71942
MOPAC 122.70213 122.70498 122.71416 122.72610 122.69465 124.37739Full-elongation 122.71877 122.71963 122.72249 122.72694 122.69488 122.37764Fix-elongation 122.71885 122.71965 122.72274 122.72705 122.69480 122.37782
( ) ( )Method Bond angle 8 : / NCH
MOPAC 110.48342 109.33649 109.29887 109.29586 109.29195 109.29274Full-elongation 110.48839 109.33603 109.30159 109.29619 109.29500 109.29396Fix-elongation 110.48824 109.33583 109.30142 109.29602 109.29485 109.29383
MOPAC 109.29169 109.29152 109.29132 109.29180 109.29102 109.29206Full-elongation 109.29248 109.29244 109.29244 109.29230 109.29241 109.29215Fix-elongation 109.29225 109.29235 109.29217 109.29237 109.29229 109.29229
MOPAC 109.29093 109.29228 109.29135 109.29257 109.27720 109.37664Full-elongation 109.29141 109.29081 109.29044 109.29210 109.27734 109.37660Fix-elongation 109.29133 109.29081 109.29032 109.29205 109.27747 109.37660
( ) ( )Method Bond angle 8 : / CNH
MOPAC 121.24435 121.29569 121.32029 121.33020 121.32883 121.33280Full-elongation 121.26343 121.30279 121.33598 121.33931 121.34289 121.34152Fix-elongation 121.26301 121.30239 121.33558 121.33892 121.34253 121.34117
MOPAC 121.33147 121.33195 121.33250 121.33101 121.33347 121.33029Full-elongation 121.34293 121.34265 121.34269 121.34275 121.34242 121.34273Fix-elongation 121.34350 121.34244 121.34308 121.34224 121.34259 121.34237
MOPAC 121.33485 121.33122 121.33529 121.33375 121.34801 121.04513Full-elongation 121.34118 121.33987 121.34018 121.33824 121.34743 121.04542Fix-elongation 121.34124 121.33974 121.34043 121.33821 121.34715 121.04552
( )Continued
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 311
MITANI, AOKI, AND IMAMURA
TABLE IV( )Continued
( ) ( )Method Dihedral angle 8 : / CNCH
MOPAC 58.68962 58.70633 58.71515 58.71773 58.72034 58.72045Full-elongation 58.68663 58.70573 58.71190 58.71626 58.71718 58.71889Fix-elongation 58.68650 58.70561 58.71179 58.71615 58.71707 58.71878
MOPAC 58.72199 58.72204 58.72193 58.72246 58.72175 58.72273Full-elongation 58.71969 58.71990 58.71996 58.71998 58.72011 58.72000Fix-elongation 58.71944 58.71986 58.71970 58.72007 58.71999 58.72016
MOPAC 58.72113 58.72216 58.62066 58.72071 58.70921 58.31431Full-elongation 58.71961 58.71938 58.71882 58.72000 58.70926 58.31423Fix-elongation 58.71955 58.71941 58.71870 58.71993 58.70942 58.31410
aIn this table, the upper, middle, and lower parts for each geometrical parameter correspond to the regions around the unextendedend, the chain center, and the extended end in the polymer, respectively.
polyacetylene, and polyglycine, and the secondsystems of aperiodic polymers are defective poly-acetylene with positively and negatively chargedsolitons. The periodic and the aperiodic clusterswere extended in one and two directions by re-
placing one and two hydrogen atoms at one andboth sides of the cluster ends by one and twoadding fragments, respectively. The main chains ofclusters for these systems were fixed to be planerand other geometrical parameters for which we list
TABLE VMaximum errors in optimized parameters of final polyethylene, polyacetylene, and polyglycine clusters
aby the two types of elongation calculations.
Polyethylene
( ) ( ) ( ) ( ) ( )Method r C } C r C } H / CCC / CCH / CCCH
Full-elongation 0.00000 0.00000 0.00125 y0.00049 y0.00059Fix-elongation 0.00000 0.00000 0.00126 y0.00051 y0.00058
Polyacetylene
( ) ( ) ( ) ( ) ( )Method r C } C r C C r C } H / CCC / CCH
Full-elongation 0.00004 y0.00004 0.00003 0.00634 y0.00708Fix-elongation 0.00004 y0.00004 0.00003 0.00635 y0.00707
Polyglycine
( ) ( ) ( ) ( ) ( ) ( )Method r N } C r C } C r C } N r C } O r N } H r C } H
Full-elongation y0.00005 y0.00013 0.00018 y0.00004 y0.00006 0.00003Fix-elongation y0.00005 y0.00013 0.00018 y0.00004 y0.00006 0.00003
( ) ( ) ( ) ( )Method / NCC / CCN / CNC / CCO
Full-elongation 0.01699 0.01298 y0.03400 0.02868Fix-elongation 0.01700 0.01302 y0.03408 0.02876
( ) ( ) ( )Method / NCH / CNH / CNCH
Full-elongation 0.00497 0.01908 y0.00325Fix-elongation 0.00482 0.01866 y0.00336
aThe values of lengths and angles are shown in angstroms and degrees, respectively.
VOL. 64, NO. 3312
GEOMETRY OPTIMIZATION OF POLYMERS
the initial values in the caption of Figure 1 wereoptimized in extending the clusters. In the elonga-tion calculations, the parameters related to theatoms on which electron densities were frozengreater than 99.99999% were fixed. The percentageof frozen density is determined as the ratio ofcontribution from the stationary orbitals on theatomic density. In other words, the condition canbe considered to be the weight of unaltered den-sity on the atom before and after the cluster exten-sion, i.e., the bond length between two atoms whichsatisfied the condition was fixed in the next exten-sion, and the bond angle and the dihedral anglewere also fixed in a similar way. Then, full opti-mization calculation of all the parameters in thegeometry was tested by the elongation method.All optimization calculations by the originalMOPAC and the elongation method were per-formed with ‘‘EF’’ and ‘‘PRECISE’’ options in theprogram package. The value of 10y6 was used asthe threshold value to extract the stationary or-bitals by the elongation procedure in this work.
The obtained results with regard to total ener-gies, optimized parameters, atomic charges, andcomputational times by applying the usual molec-ular orbital and the two-type elongation methodswere compared with each other to check our ap-proach. The total energies of periodic polymers arelisted in Table I. The agreements of results forthese three systems between the usual and theelongation calculations are excellent. The energiesfor both types of elongations are almost consistentwith each other. The errors for the largest clustersare as follows: y0.000001 eV in polyethylene with30 units, y0.000072 eV in polyacetylene with 30
Ž .units, and y0.000117 eV full and y0.000115 eVŽ .fix in polyglycine with 25 units. These excellentagreements in total energies suggest that the opti-mized geometries are also in excellent agreementbetween both of the conventional and the elonga-tion methods.
We give the optimized geometrical parametersof the largest clusters for three polymers in TablesII]IV. In these tables, three parts of the geometries
TABLE VINet charges on carbon atoms in the largest polyethylene and polyacetylene clusters and on nitrogen
aatoms in the largest polyglycine cluster obtained from AM1 calculations.
Net charges on carbon or nitrogen atoms
Atom site MOPAC Full-elongation Fix-elongation
( )Polyethylene clusters: H } C H } H2 4 30
10 y0.157577 y0.157554 y0.15755420 y0.157578 y0.157556 y0.15755530 y0.157578 y0.157556 y0.15755640 y0.157578 y0.157556 y0.15755650 y0.157578 y0.157557 y0.15755760 y0.210431 y0.210433 y0.210433
( )Polyacetylene clusters: H } C H } H2 2 30
10 y0.126258 y0.125468 y0.12546820 y0.125204 y0.124434 y0.12443430 y0.124993 y0.124248 y0.12424840 y0.124829 y0.124202 y0.12420050 y0.124188 y0.123919 y0.12391760 y0.204348 y0.204317 y0.204317
( )Polyglycine clusters: H } NHCH CO } H2 25
5 y0.375050 y0.375399 y0.37539910 y0.374484 y0.374691 y0.37473015 y0.374438 y0.374620 y0.37462320 y0.374503 y0.374607 y0.37460625 y0.375874 y0.375879 y0.375878
aThe numbering of atoms is defined as follows: The first and the last atoms correspond to the atoms in the both end units at theunextended and the extended terminals, respectively.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 313
MITANI, AOKI, AND IMAMURA
which correspond to the unextended terminal atthe starting cluster, the central part in the ex-tended chain, and the extended terminal at theadding fragment are shown. The good results wereobtained as for three types of parameters of bondlengths, bond angles, and dihedral angles all overthe chain. The maximum errors in all the opti-mized parameters are summarized in Table V, andit is clear that the elongation method has an ade-quate ability also for the geometry optimizations.
The net charges on carbon atoms in polyeth-ylene and polyacetylene with 30 units and on
nitrogen atoms in polyglycine with 25 units areshown in Table VI. From these results, we see thatthe elongation calculations give reasonable valuesfor atomic charges in comparison with the conven-tional calculations, i.e., the elongation approachcan sufficiently reproduce the electron density dis-tributions on atoms in large polymers by the opti-mization procedure.
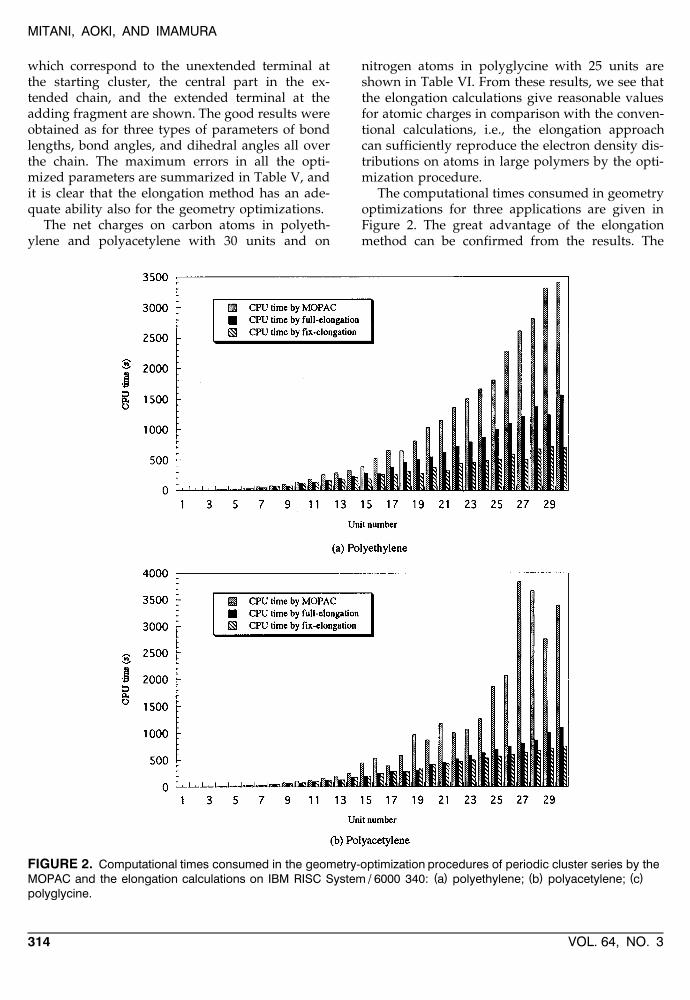
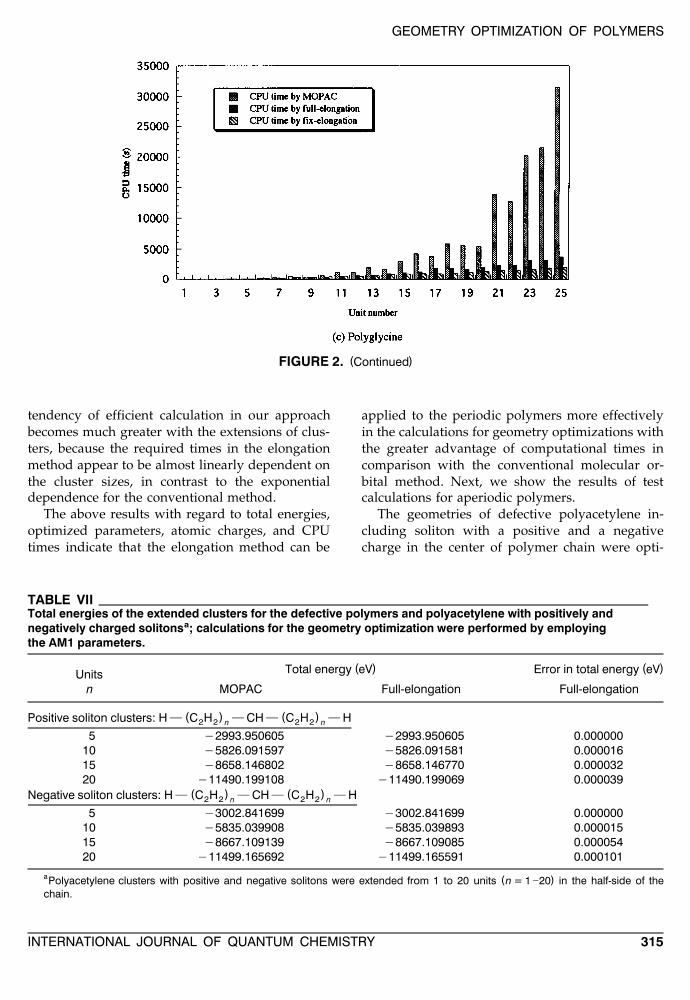
The computational times consumed in geometryoptimizations for three applications are given inFigure 2. The great advantage of the elongationmethod can be confirmed from the results. The
FIGURE 2. Computational times consumed in the geometry-optimization procedures of periodic cluster series by the( ) ( ) ( )MOPAC and the elongation calculations on IBM RISC System / 6000 340: a polyethylene; b polyacetylene; c
polyglycine.
VOL. 64, NO. 3314
GEOMETRY OPTIMIZATION OF POLYMERS
( )FIGURE 2. Continued
tendency of efficient calculation in our approachbecomes much greater with the extensions of clus-ters, because the required times in the elongationmethod appear to be almost linearly dependent onthe cluster sizes, in contrast to the exponentialdependence for the conventional method.
The above results with regard to total energies,optimized parameters, atomic charges, and CPUtimes indicate that the elongation method can be
applied to the periodic polymers more effectivelyin the calculations for geometry optimizations withthe greater advantage of computational times incomparison with the conventional molecular or-bital method. Next, we show the results of testcalculations for aperiodic polymers.
The geometries of defective polyacetylene in-cluding soliton with a positive and a negativecharge in the center of polymer chain were opti-
TABLE VIITotal energies of the extended clusters for the defective polymers and polyacetylene with positively and
anegatively charged solitons ; calculations for the geometry optimization were performed by employingthe AM1 parameters.
( ) ( )Total energy eV Error in total energy eVUnitsn MOPAC Full-elongation Full-elongation
( ) ( )Positive soliton clusters: H } C H } CH } C H } H2 2 2 2n n
5 y2993.950605 y2993.950605 0.00000010 y5826.091597 y5826.091581 0.00001615 y8658.146802 y8658.146770 0.00003220 y11490.199108 y11490.199069 0.000039
( ) ( )Negative soliton clusters: H } C H } CH } C H } H2 2 2 2n n
5 y3002.841699 y3002.841699 0.00000010 y5835.039908 y5835.039893 0.00001515 y8667.109139 y8667.109085 0.00005420 y11499.165692 y11499.165591 0.000101
a ( )Polyacetylene clusters with positive and negative solitons were extended from 1 to 20 units n = 1 ]20 in the half-side of thechain.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 315
MITANI, AOKI, AND IMAMURA
TABLE VIIIOptimized parameters in the geometry of final polyacetylene for positive soliton including 40 units
a( ) ( )by using the AM1 Hamiltonian: H } C H } CH } C H } H.2 2 20 2 2 20
˚( ) ( )Method Bond length A : r C } C
MOPAC 1.39653 1.41024 1.42206 1.43083 1.43650 1.43980Full-elongation 1.39657 1.41038 1.42225 1.43101 1.43664 1.43991
MOPAC 1.44254 1.44303 1.44328 1.4442 1.44349 1.44353Full-elongation 1.44260 1.44308 1.44333 1.44347 1.44354 1.44358
MOPAC 1.44357 1.44358 1.44362 1.44376 1.44437 1.44776Full-elongation 1.44360 1.44363 1.44366 1.44376 1.44437 1.44776
˚( ) ( )Method Bond length A : r C C
MOPAC 1.38218 1.37087 1.36179 1.35569 1.35200 1.34992Mull-elongation 1.38274 1.37076 1.36166 1.35559 1.35192 1.34986
MOPAC 1.34819 1.34786 1.34768 1.34757 1.34751 1.34747Full-elongation 1.34815 1.34782 1.34764 1.34754 1.34747 1.34744
MOPAC 1.34743 1.34741 1.34734 1.34710 1.34595 1.33581Full-elongation 1.34738 1.34737 1.34731 1.34710 1.34595 1.33581
˚( ) ( )Method Bond length A : r C } H
MOPAC 1.10842 1.10492 1.10820 1.10465 1.10762 1.10427Full-elongation 1.10840 1.10494 1.10817 1.10466 1.10758 1.10428
MOPAC 1.10460 1.10387 1.10449 1.10391 1.10441 1.10394Full-elongation 1.10460 1.10387 1.10449 1.10391 1.10441 1.10394
MOPAC 1.10401 1.10418 1.10405 1.10403 1.10441 1.09787Full-elongation 1.10401 1.10418 1.10405 1.10403 1.10441 1.09787
( ) ( )Method Bond angle 8 : / CCC
MOPAC 122.43447 122.94694 122.44073 122.93961 122.46210 122.93266Full-elongation 122.44657 122.92676 122.44689 122.92129 122.46813 122.91700
MOPAC 122.75405 122.91187 122.78225 122.90771 122.80340 122.90478Full-elongation 122.75621 122.91176 122.78301 122.90940 122.80281 122.90747
MOPAC 122.90169 122.86124 122.90228 122.88330 122.95552 123.33124Full-elongation 122.90140 122.86190 122.90137 122.88390 123.95509 123.33152
( ) ( )Method Bond angle 8 : / CCH
MOPAC 118.49757 119.12459 117.95686 119.64386 117.50053 120.03044Full-elongation 118.48903 119.14008 117.94515 119.65840 117.48769 120.04786
MOPAC 120.56657 116.59349 120.57207 116.57907 120.57485 116.56925Full-elongation 120.57009 116.58874 120.57467 116.57411 120.57608 116.56441
MOPAC 116.53689 120.59801 116.49162 120.70818 116.06838 122.82421Full-elongation 116.53630 120.59863 116.49121 120.70859 116.06822 122.82463
aIn this table, the upper middle, and lower parts for each geometrical parameter correspond to the regions around the unextendedcenter in charged defect, the chain center in the half-side, and the extended terminal in the polymer, respectively.
VOL. 64, NO. 3316
GEOMETRY OPTIMIZATION OF POLYMERS
TABLE IXOptimized parameters in the geometry of final polyacetylene for negative soliton including 40 units
a( ) ( )by using the AM1 Hamiltonian: H } C H } CH } C H } H.2 2 20 2 2 20
˚( ) ( )Method Bond length A : r C } C
MOPAC 1.39376 1.40741 1.41950 1.42874 1.43491 1.43864Full-elongation 1.39379 1.40757 1.41971 1.42896 1.43509 1.43875
MOPAC 1.44194 1.44260 1.44297 1.44319 1.44332 1.44341Full-elongation 1.44200 1.44264 1.44300 1.44322 1.44334 1.44343
MOPAC 1.44350 1.44353 1.44358 1.44373 1.44435 1.44770Full-elongation 1.44352 1.44355 1.44361 1.44374 1.44435 1.44770
˚( ) ( )Method Bond length A : r C C
MOPAC 1.38045 1.36912 1.36064 1.3550 1.35160 1.34969Full-elongation 1.38034 1.36895 1.36046 1.35484 1.35149 1.34961
MOPAC 1.34810 1.34780 1.34764 1.34754 1.34748 1.34745Full-elongation 1.34806 1.34776 1.34761 1.34751 1.34746 1.34742
MOPAC 1.34741 1.34738 1.34732 1.34707 1.34592 1.33579Full-elongation 1.34739 1.34736 1.34729 1.34707 1.34592 1.33579
˚( ) ( )Method Bond length A : r C } H
MOPAC 1.09987 1.10778 1.10005 1.10736 1.10055 1.10671Full-elongation 1.09984 1.10782 1.10003 1.10738 1.10054 1.10669
MOPAC 1.10360 1.10440 1.10372 1.10434 1.10380 1.10429Full-elongation 1.10363 1.10437 1.10374 1.10431 1.10383 1.10426
MOPAC 1010412 1.10402 1.10412 1.10390 1.10445 1.09777Full-elongation 1.10412 1.10402 1.10412 1.10390 1.10445 1.09777
( ) ( )Method Bond angle * : / CCC
MOPAC 124.68754 122.68453 124.48956 122.70216 124.17260 122.72965Full-elongation 124.71845 122.67041 124.50524 122.70243 124.17053 122.74133
MOPAC 123.05402 122.84516 123.00933 122.85267 122.97793 122.85886Full-elongation 123.04405 122.85742 123.00204 122.86097 122.97239 122.86727
MOPAC 122.88630 122.90458 122.89213 122.92042 122.95271 123.36406Full-elongation 122.88590 122.90514 122.89121 122.92075 122.95224 123.36398
( ) ( )Method Bond angle 8 : / CCH
MOPAC 117.39943 199.21245 117.00876 119.68459 116.74717 120.03477Full-elongation 117.38036 119.22740 116.99182 119.69704 117.73623 120.03932
MOPAC 120.56513 116.49631 120.57267 116.50308 120.57676 116.50890Full-elongation 120.55799 116.49751 120.56799 116.50236 120.57320 116.50845
MOPAC 116.51607 120.59742 116.47382 120.70321 116.05493 122.81043Full-elongation 116.51544 120.59803 116.47357 120.70350 116.05487 122.81052
aIn this table, the upper, middle, and lower parts for each geometrical parameter correspond to the regions around the unextendedcenter in charged defect, the chain center in the half-side, and the extended terminal in the polymer, respectively.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 317
MITANI, AOKI, AND IMAMURA
TABLE XMaximum errors in optimized parameters of final solitonic polyacetylene clusters by the
aelongation calculations.
Positive soliton
( ) ( ) ( ) ( ) ( )Method r C C r C C r C H / CCC / CCH
Full-elongation 0.00019 y0.00013 y0.00004 y0.02018 0.01742
Negative soliton
( ) ( ) ( ) ( ) ( )Method r C C r C C r C H / CCC / CCH
Full-elongation 0.00022 y0.00016 y0.00005 y0.02191 y0.01907aThe values of lengths and angles are shown in angstroms and degrees, respectively.
mized in cluster extending calculations. Even afterthe elongations for 1 to 20 units of these systems,the geometrical parameters of clusters were notable to be fixed; thus, full optimizations of geome-tries were carried out by the elongation method.Because the convergencies of geometry optimiza-tion procedures for these systems were very poor,all calculations for solitonic polyacetylene wereperformed by employing the ‘‘EF’’ optimizationoptions with ‘‘DMAX s 0.0921’’ and ‘‘DMAX s0.022’’ for positive and negative solitons, respec-tively.
In Table VII, we list total energies of polyacet-ylene clusters including two types of defects forpositively and negatively charged solitons. The
calculated energy values by both of the conven-tional and our methods are in good agreement,and the errors of the elongation method appeareven in the largest clusters extended by 20 units tobe only 0.000039 and 0.000101 eV for positive andnegative defective structures, respectively.
The optimized parameters in the polymer ge-ometries of positive and negative solitons are givenin Table VIII and IX, and the maximum errors inthese parameters are summarized in Table X. Thereliability of optimization calculations by the elon-gation procedure all over the whole range ofcharged chains can be confirmed from the resultsof bond lengths and bond angles indicated in thesetables.
TABLE XINet charges on carbon atoms in the largest solitonic polyacetylene clusters obtained from
aAM1 calculations.
Net charges on carbon atoms
Atom site MOPAC Full-elongation
( ) ( )Positive soliton clusters: H } C H } CH } C H } H2 2 2 220 20
1 0.092352 0.09237711 y0.038778 y0.03929221 y0.102468 y0.10245431 y0.116217 y0.11609341 y0.201882 y0.201873
( ) ( )Negative soliton clusters: H } C H } CH } C H } H2 2 2 220 20
1 y0.351432 y0.35174211 y0.212609 y0.21170021 y0.147232 y0.14635031 y0.132168 y0.13184741 y0.206819 y0.206787
aThe numbering of atoms is defined as follows: The 1st and the 41th atoms correspond to the central and the end atoms at thecharged center and the extended terminal in the half-side of the chain, respectively.
VOL. 64, NO. 3318
GEOMETRY OPTIMIZATION OF POLYMERS
The net charges on carbon atoms in defectivepolyacetylene chains with 40 units are shown inTable XI. The reasonable charge distribution onpolymers can be obtained by using our approachfor both of the positive and negative solitons. So, itcan be considered that the elongation method canbe applied to the positively and negatively chargedsystems.
In Figure 3, the computational times consumedby the conventional molecular orbital and theelongation calculations for both solitons are given.Much faster computations for these polymers could
be done by the elongation method. The conver-gence of geometry optimization by the conven-tional MOPAC calculations appear to be muchdifferent for the clusters with various sizes.
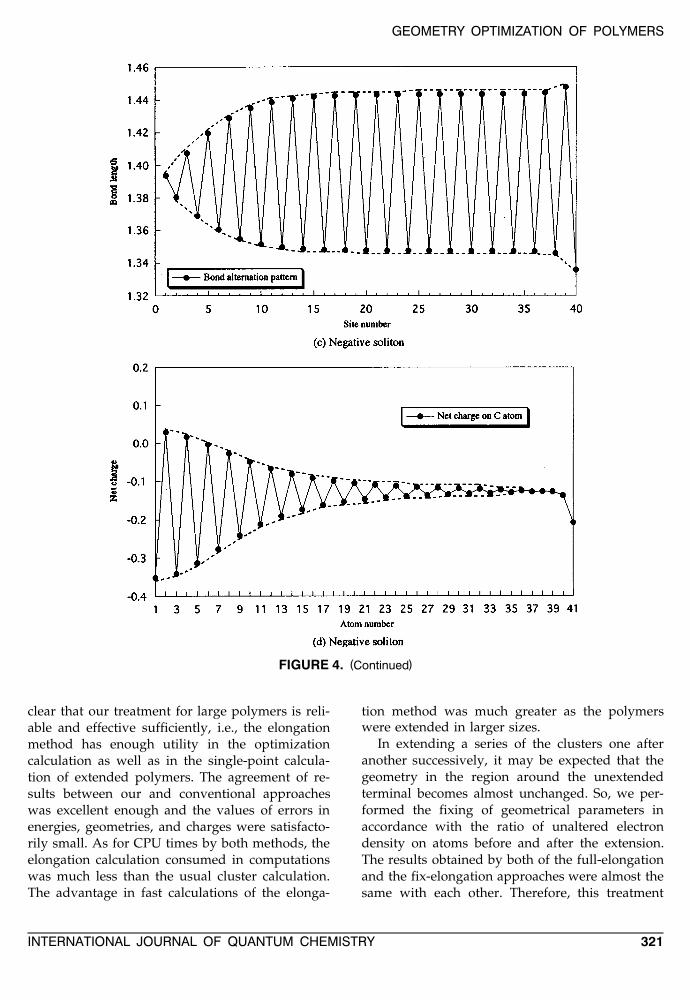
The feature of soliton defects in polyacetylenecan be characterized by the bond-alternation pat-terns between neighboring atoms and the charge-density distributions on the chain. We show thesecharacters obtained by the elongation calculationsin Figure 4 for positive and negative solitons in-cluding 81 carbon atoms in the polymer chains.These figures suggest that the defective structures
FIGURE 3. Computational times consumed in the geometry optimization procedures of aperiodic cluster series by the( ) ( )MOPAC and the elongation calculations on IBM RISC System / 6000 580: a positive soliton; b negative soliton.
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 319
MITANI, AOKI, AND IMAMURA
extend up to almost 15 carbon atoms from thecharged center in half side of the chain, and, also,the end effects can be recognized in the region ofterminal two carbon atoms at the cluster end.
Conclusion
In this work, we tried to apply the elongationmethod at the semiempirical AM1 level to thegeometry optimization of five polymer systems.
The periodic clusters of polyethylene, polyacety-lene, and polyglycine were tested as the first appli-cation, and, also, the aperiodic clusters of de-fective polyacetylene including positively andnegatively charged solitons were checked as thesecond application.
From the comparisons of results for total ener-gies, optimized parameters, atomic charges, andcomputational times by the elongation calculationof a cluster-series model with those by the conven-tional calculation for a cluster model, it became
FIGURE 4. Bond-alternation patterns and net charge distributions on carbon atoms in main chains by the elongation( ) ( )method: a, b positive soliton, c, d negative soliton.
VOL. 64, NO. 3320
GEOMETRY OPTIMIZATION OF POLYMERS
( )FIGURE 4. Continued
clear that our treatment for large polymers is reli-able and effective sufficiently, i.e., the elongationmethod has enough utility in the optimizationcalculation as well as in the single-point calcula-tion of extended polymers. The agreement of re-sults between our and conventional approacheswas excellent enough and the values of errors inenergies, geometries, and charges were satisfacto-rily small. As for CPU times by both methods, theelongation calculation consumed in computationswas much less than the usual cluster calculation.The advantage in fast calculations of the elonga-
tion method was much greater as the polymerswere extended in larger sizes.
In extending a series of the clusters one afteranother successively, it may be expected that thegeometry in the region around the unextendedterminal becomes almost unchanged. So, we per-formed the fixing of geometrical parameters inaccordance with the ratio of unaltered electrondensity on atoms before and after the extension.The results obtained by both of the full-elongationand the fix-elongation approaches were almost thesame with each other. Therefore, this treatment
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 321

MITANI, AOKI, AND IMAMURA
should be useful to compute the large polymersefficiently for the systems with almost linear shapeof main chains.
In this work, we concluded that the elongationmethod can permit an effective treatment for boththe electronic and geometrical structures. But, weassumed that the polymers have planar structures.Of course, the geometries of real polymers havemuch flexibility with various conformations by thelarge degrees of freedom such as those of polypep-tides or biopolymers. So, we will carry out thecalculation including the optimization for the con-formation of main chain in a polymer by the elon-gation method in the near future and it may bereasonably expected that we will obtain good re-sults for flexible polymers as shown in the presentarticle.
ACKNOWLEDGMENT
This work was supported by a Grant-in-Aid forScientific Research from the Ministry of Educationof Japan.
References
1. W. J. Hehre, L. Radom, J. A. Pople, and P.v.R. Schleyer, AbŽ .Initio Molecular Orbital Theory Wiley, New York, 1986 .
Ž2. J. Ladik, Quantum Theory of Polymers as Solids Plenum,.New York, London, 1989 .
3. C. Pisani, R. Dovesi, and C. Roetti, Hartree]Fock Ab InitioTreatment of Crystalline Systems, Lecture Notes in Chemistry
Ž .Vol. 48 Springer-Verlag, Heidelberg, 1988 .
Ž .4. S. Suhai, Int. J. Quantum Chem. 40, 559 1991 .
Ž .5. S. Suhai, Int. J. Quantum Chem. 42, 193 1992 .
Ž .6. W. Forner, Int. J. Quantum Chem. 43, 221 1992 .¨7. R. Dovesi, C. Pisani, C. Roetti, M. Causa, and V. R.`
ŽSaunders, CRYSTAL88, QCPE No. 577 Quantum Chem-.istry Program Exchange .
8. R. Dovesi, V. R. Saunders, and C. Roetti, CRYSTAL92Žmanual available upon request from V. R. Saunders,
.Daresbury Laboratory, Daresbury, England .
9. A. C. Hess and V. R. Saunders, J. Phys. Chem. 96, 4367Ž .1992 .
10. R. D. Poshusta, D. C. Tseng, A. C. Hess, and M. I.Ž .McCarthy, J. Phys. Chem. 97, 7295 1993 .
11. M. I. McCarthy, A. C. Hess, N. M. Harrison, and V. R.Ž .Saunders, J. Chem. Phys. 98, 6387 1993 .
12. C. A. Scamehorn, A. C. Hess, and M. I. McCarthy, J. Chem.Ž .Phys. 99, 2786 1993 .
13. L. Ojamae, K. Hermansson, R. Dovesi, C. Roetti, and V. R.¨Ž .Saunders, J. Chem. Phys. 100, 2128 1994 .
14. C. A. Scamehorn, N. M. Harrison, and M. I. McCarthy,Ž .J. Chem. Phys. 101, 1547 1994 .
15. B. Kirtman and C. P. DeMelo, J. Chem. Phys. 86, 1624Ž .1987 .
Ž .16. K. A. Robins and B. Kirtman, J. Chem. Phys. 99, 6777 1993 .
17. S. Roszak, P. C. Hariharan, and J. J. Kaufman, J. Comp.Ž .Chem. 11, 1072 1990 .
18. S. Roszak and J. J. Kaufman, Int. J. Quantum Chem. 42, 917Ž .1992 .
19. G. G. Ferenczy, J.-L. Rivail, P. R. Surjan, And G. Naray-´ ´Ž .Szabo, J. Comp. Chem. 13, 830 1992 .´
Ž .20. J. M. Cullen, J. Comp. Chem. 13, 901 1992 .
Ž . Ž .21. W. Yang, J. Mol. Struct. Theochem 255, 461 1992 .
22. B. K. Novosadov, O. Y. Nikitin, and L. A. Gribov, J. Mol.Ž .Struct. 268, 223 1992 .
23. O. Y. Nikitin, B. K. Novosadov, and L. A. Gribov, J. Mol.Ž .Struct. 268, 237 1992 .
Ž .24. M. Seel, Int. J. Quantum Chem. 26, 753 1984 .
Ž . Ž .25. J. Ladik, J. Mol. Struct. Theochem 230, 127 1991 .
26. H. Nakatsuji, Int. J. Quantum Chem., Quantum Chem.Ž .Symp. 26, 725 1992 .
27. Y. Fukunishi and H. Nakatsuji, J. Chem. Phys. 907, 6535Ž .1992 .
28. C. Pisani, R. Dovesi, R. Nada, and S. Tamiro, Surf. Sci. 216,Ž .489 1989 .
29. C. Pisani, R. Dovesi, R. Nada, and L. N. Kantorovich,Ž .J. Chem. Phys. 92, 7448 1990 .
30. C. Pisani, R. Orlando, and F. Cora, J. Chem. Phys. 97, 4195`Ž .1992 .
Ž .31. P. J. Feibelman, Phys. Rev. B 46, 15416 1992 .
32. C. X. Cui, M, Kertesz, and Y. Jiang, J. Phys. Chem. 94, 5172Ž .1990 .
33. J. Chandrasekhar and P. K. Das, J. Phys. Chem. 96, 679Ž .1992 .
34. E. J. Weniger and C. M. Liegener, Int. J. Quantum Chem. 38,Ž .55 1990 .
Ž .35. J. Cioslowski, J. Chem. Phys. 92, 1236 1990 .
36. J. Cioslowski and M. B. Lepetit, J. Chem. Phys. 95, 3536Ž .1991 .
Ž .37. V. Magnasco and A. Perico, J. Chem. Phys. 47, 971 1967 .
Ž .38. V. Magnasco and A. Perico, J. Chem. Phys. 48, 800 1968 .
39. A. Imamura, Y. Aoki, and K. Maekawa, J. Chem. Phys. 95,Ž .5419 1991 .
40. A. Imamura, Y. Aoki, K. Nishimoto, Y. Kurihara, and A.Ž .Nagao, Int. J. Quantum Chem. 52, 309 1994 .
Ž .41. Y. Aoki and A. Imamura, J. Chem. Phys. 97, 8432 1992 .
42. Y. Aoki, S. Suhai, and A. Imamura, Int. J. Quantum Chem.Ž .52, 267 1994 .
43. Y. Aoki, S. Suhai, and A. Imamura, J. Chem. Phys. 101,Ž .10808 1994 .
Ž .44. K. Maekawa and A. Imamura, J. Chem. Phys. 98, 534 1993 .
45. K. Maekawa and A. Imamura, J. Chem. Phys. 98, 7086Ž .1993 .
VOL. 64, NO. 3322

GEOMETRY OPTIMIZATION OF POLYMERS
46. K. Maekawa and A. Imamura, Int. J. Quantum Chem. 47,Ž .449 1993 .
Ž .47. M. Mitani and A. Imamura, J. Chem. Phys. 101, 7712 1994 .48. M. Mitani, Y. Aoki, and A. Imamura, J. Chem. Phys. 100,
Ž .2346 1994 .49. M. Mitani, Y. Aoki, and A. Imamura, Int. J. Quantum
Ž .Chem. 54, 167 1995 .Ž .50. M. Mitani and A. Imamura, J. Chem. Phys. 103, 663 1995 .
51. A. T. Amos and G. G. Hall, Proc. R. Soc. Lond. Ser. A 263,Ž .483 1961 .
52. K. Fukui, N. Koga, and H. Fujimoto, J. Am. Chem. Soc. 103,Ž .196 1981 .
53. H. Fujimoto, N. Koga, and K. Fukui, J. Am. Chem. Soc. 103,Ž .7452 1981 .
Ž .54. J. J. P. Stewart, MOPAC Ver. 6.00 VAX version , QCPE No.Ž .455 Quantum Chemistry Program Exchange and Ver. 6.01
Ž . ŽAIX version , JCPE No. 016 Japanese Chemistry Program.Exchange .
55. M. J. S. Dewar, E. G. Zoebisch, E. F. Healy, and J. J. P.Ž .Stewart, J. Am. Chem. Soc. 107, 3902 1985 .
INTERNATIONAL JOURNAL OF QUANTUM CHEMISTRY 323