Embed Size (px)
Citation preview

ORIGINAL INVESTIGATION
Hepatitis B escape mutants in Scottish blood donors
Osmany Larralde • Brian Dow • Lisa Jarvis •
Fiona Davidson • Juraj Petrik
Received: 11 September 2012 / Accepted: 1 December 2012 / Published online: 29 December 2012
� Springer-Verlag Berlin Heidelberg 2012
Abstract Hepatitis B virus (HBV) remains as the viral
infection with the highest risk of transmission by transfusion.
This risk is associated with window period donations, occult
HBV infection (OBI) and the emergence of escape mutants,
which render blood donations false negative for hepatitis B
surface antigen (HBsAg) serological testing. A retrospective
study was conducted to gain insights into the molecular epi-
demiology of HBV escape mutants in Scottish blood donors.
The criterion for selection was HBV positivity either by
serology or nucleic acid testing (NAT). HBsAg detection was
compared across several commercial immunoassays. The full
length S gene from plasma samples was PCR amplified,
cloned and expressed in HepG2 cells. Eight samples showed
HBsAg discordant results, while 5 OBI samples were found.
Four escape mutants, containing missense mutations in the S
gene, are described here. These mutations impaired HBsAg
detection both from HBV infected plasma samples and from
recombinant proteins derived from its infected donors. Phy-
logenetic analysis showed that most of the mutants were
clustered in the genotype D and were closely related to strains
from Asia and the Middle East. We report here a proline
substitution, outside the major hydrophilic region, that
impaired HBsAg detection in vivo and in vitro, warning about
the risk for the emergence of vaccine escape mutants with
mutations outside the major neutralisation site.
Keywords Occult HBV infection � HBsAg screening �HBsAg mutation � Nucleic acid testing � Phylogenetic
analysis � Immunoassay
Introduction
Despite the continuous improvement of HBsAg serological
assays since the 1970s, HBV transmission is still considered
the highest risk of viral transmission to blood components
recipients in the world. The residual risk of HBV transmission
has been reduced from 1.17 to 0.76 per million donations after
the implementation of HBV NAT in Scotland since March
2010 [1, 2]. The availability of HBV NAT has revealed cases
of OBI, defined by the presence of HBV DNA without
detectable HBsAg outside the acute phase window period, and
most of the time accompanied by the presence of anti-HBc [3,
4]. Plasma pool NAT has also considerably reduced the
residual risk due to window period donations but nil risk is not
achievable, especially because NAT still requires higher
sensitivity to detect most OBI, which generally have very low
viral load. The mechanisms of OBI are not well understood yet
and involve host immune and viral factors such as mutated
HBsAg and low level expression of HBsAg, as a consequence
of complex cis- and trans-acting viral factors [5].
Since the current risk of HBV transfusion transmission
seems to be mainly related to OBI and the emergence of
escape mutant viruses that render blood donations false
negative for HBsAg serological testing, this investigation
was aimed to analyse the frequency of these rare events in
Scottish blood donors and to use molecular techniques to
study the effect and possible origin of these mutations.
Materials and methods
Donors and sample collection
This was a retrospective study looking at a selection of
Scottish blood donors that have been found HBV positive
O. Larralde (&) � B. Dow � L. Jarvis � F. Davidson � J. Petrik
Microbiology and Components Group, RDI, Scottish National
Blood Transfusion Service/NHS, 21 Ellen’s Glen Road,
Edinburgh EH17 7QT, UK
e-mail: [email protected]
123
Med Microbiol Immunol (2013) 202:207–214
DOI 10.1007/s00430-012-0283-9

either by serology or PCR (n = 649). Samples from 1970
to 1988 were taken only from donors of west of Scotland,
whereas samples from 1989 to 2011 included other areas of
Scotland. All blood donors were voluntary and non-remu-
nerated. Six hundred and forty-nine HBV positive samples
were selected from 7,925,259 donations. Anonymised
double-coded data were used in the study protocol in order
to protect confidentiality of personal information. Plasma
samples stored at -20 �C were retrieved from archives and
analysed for several HBV markers.
HBV NAT
Viral DNA was extracted from 200 ll of plasma sample
using the High Pure Viral Nucleic Acid Kit (Roche Diag-
nostics, Germany) according to the manufacturer’s
instructions. For the estimation of HBV load, a 10 ll ali-
quot of extracted HBV DNA was amplified using a nested
PCR with secondary amplification and detection by real-
time PCR using the Roche Light Cycler V3.5. BVDV is
used as an internal control; primers and probes are
described elsewhere [6]. Reaction conditions were as fol-
lows: reverse transcription (required to amplify the internal
control) at 48 �C for 45 min, immediately followed by a
single cycle of 94 �C for 2 min then 30 rounds of ampli-
fication consisting of three stages: 30 s at 94 �C, 21 s at
55 �C and 1.5 min at 72 �C. This is followed by a final
cycle of 7 min at 72 �C. Nested amplification and detection
was performed on the Light Cycler using the following
conditions, an initial denaturation for 2 min at 95 �C fol-
lowed by 35 cycles, each consisting of 2 s at 94 �C, 20 s at
55 �C and 35 s at 72 �C. After this, the samples are cooled
to 40 �C. Primers (S1, S5, S3 and S6) and probes (F4 and
P4) were designed to amplify a 157-base pair fragment of
the surface gene (Table 1). The analytical sensitivity of this
in house HBV NAT assay is 20 IU/ml (95 % level of
detection) and 4.5 IU/ml (50 % level of detection). All
samples from 2010 were routinely screened in pools of 24
for HBV DNA, HCV RNA and HIV RNA using Cobas
TaqScreen multiplex nucleic acid test, which has an ana-
lytical sensitivity of 1.9 IU/ml (95 % level of detection)
[7]. Positive samples were confirmed by the SNBTS in
house HBV NAT assay, and an estimate of the HBV DNA
load was made by comparison with samples of known virus
titre. This is a semi-quantitative assay based on crossover
values of sample versus two positive controls of 125 and
5 IU/ml.
HBV serological markers
Plasma samples were tested for HBsAg using the following
commercial assays: BIOELISA HBsAg 3.0 (Biokit, SA,
Spain), Monolisa HBsAg ultra (BIO-RAD, France),
MUREX HBsAg (Abbott Murex Biotech, Ltd, UK) and
PRISM HBsAg (Abbott Diagnostics, USA). Other sero-
logical markers of HBV infection investigated were
HBeAg, anti-HBe, anti-HBc and IgM anti-HBc (Abbott
Diagnostics, USA).
HBsAg cloning and expression
HBsAg gene was amplified by nested PCR using a high
fidelity PFU DNA polymerase system (Promega, USA).
All PCRs were done in 50 ll reaction with 19 PFU PCR
buffer with MgSO4, 0.2 mM dNTPs, 1 lM each primer,
1.25 U of PFU polymerase and 1 ll template DNA. First
round conditions consisted of initial denaturation for 2 min
at 95 �C followed by 35 cycles, each consisting of 1 min at
95 �C, 30 s at 57.7 �C (Ta) and 1 min at 72 �C. Second
round conditions were similar to first round, except Ta was
61.1 �C. The final extension was 5 min at 72 �C for both
rounds. Outer primers (O27 and O28) and inner primers
(O12 and O13) were designed to incorporate BclI and SacI
sites at each end of the amplified product for cloning
HBsAg gene into BamHI/SacI of the mammalian expres-
sion vector pTriex-5 (Table 1). Positive colonies were
identified by PCR and by KpnI/SacI digestion. Positive
clones were sequenced in both directions using TriExUp
and TriExDown primers (EMD/Merck Novagen, USA).
This cloning strategy allows expression of the whole length
HBsAg gene (681 nt) in frame with an N-terminal Strep-
tag II and a C-terminal His-tag.
Constructs were purified using EndoFree Plasmid Maxi
Kit (QIAGEN, Germany) and DNA quantified by Nano-
Drop spectrophotometer (Thermo Scientific) and gel elec-
trophoresis. Equal amounts of maxiprep plasmid DNA
were transiently transfected into a human hepatocarcinoma
cell line (HepG2), purchased from ATCC-LGC Standards.
HepG2 cells were grown in Advanced D-MEM with
GlutaMAX (Life Technologies Invitrogen, USA) and 5 %
inactivated foetal calf serum. The number of passages from
the master culture (passage 74) was minimised using a seed
lot system. Briefly, HepG2 cells were seeded at a density of
1 9 106 cells in 60 mm cell culture dishes (FALCON) and
transfected 24 h later with 5 lg of each construct and 15 ll
of GeneJuice transfection reagent (EMD/Merck Novagen,
USA). The transfection mixture was replaced by growth
medium 6 h after. Cells were incubated for 3 days at 37 �C
(5 % CO2) and the culture medium collected for analysis of
secreted HBsAg.
Sequence analysis
HBV genotype was assessed by entering DNA sequences
in the Genotyper tool of the HepSEQ-Hepatitis B resource
[8]. HBsAg serological subtype prediction was based on
208 Med Microbiol Immunol (2013) 202:207–214
123

specific amino acid (aa) positions [9]. All data processing
was performed using GENEIOUS PRO software [10]. Two
hundred and forty-three HBsAg nucleotide sequences were
selected from NCBI and aligned to obtain consensus
sequences of each HBV genotype. Analysis of HBsAg
mutations was conducted by alignment with the consensus
sequence of its genotype. The nucleotide sequence of
escape mutants was blasted in GenBank, and the closest
related strains were used for construction of a dendrogram.
Results
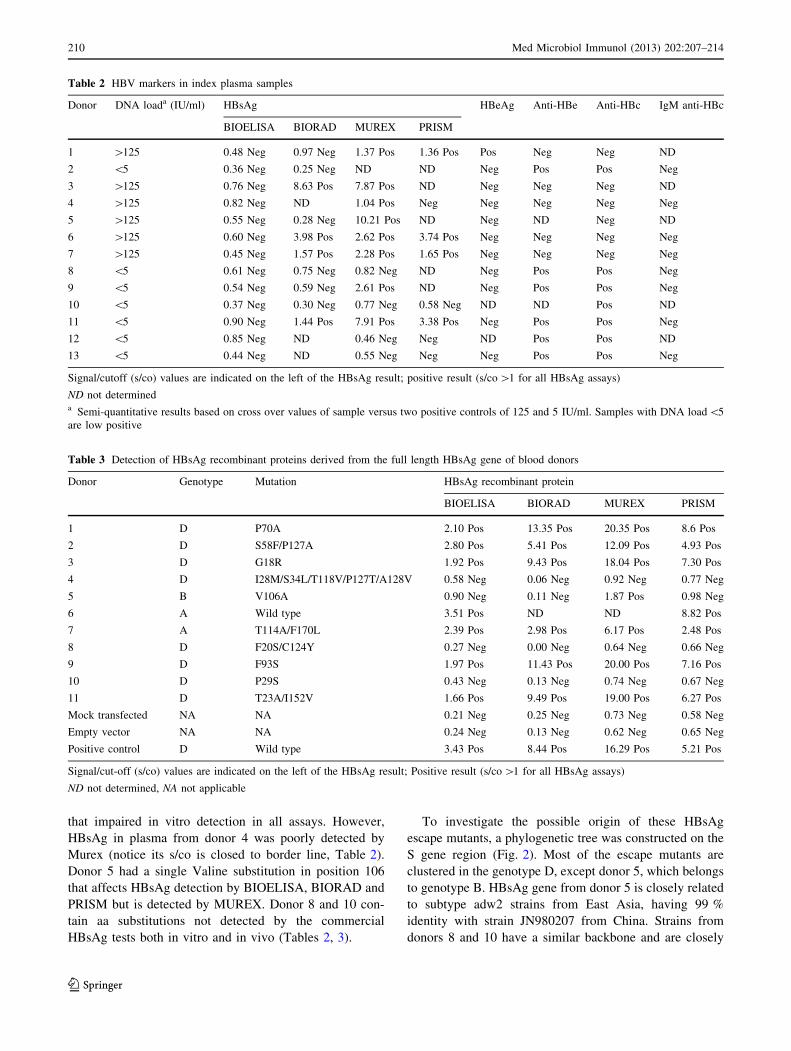
Eight of 649 HBV positive samples showed discordant
results in four commercial HBsAg serological tests, while 5
samples were HBsAg negative and HBV DNA positive
(Table 2). Donors 2, 8, 10, 12 and 13 fulfil the criteria for
OBI since in addition to being HBsAg negative in all
assays, they had a very low viral load (\5 IU/mL) and
were anti-HBc positive [4]. In contrast, the HBV DNA load
of the HBsAg discordant samples was generally above
125 IU/mL, except donors 9 and 11.
Most of the OBI donors were also anti-HBe positive,
which correlates with their low viral titre and suggests a
low degree of infectivity. Unfortunately, we could not
sequence the HBsAg gene from donors 12 and 13, which
were also positive by Cobas TaqScreen multiplex nucleic
acid test. Donor 12 was the only donor co-infected with
HIV. Donor 13 had co-circulating anti-HBs antibody
(42 IU/mL), further suggesting OBI [4].
‘‘Isolated HBsAg positivity’’ serological profile [11] was
found in index samples 3, 4, 6 and 7, which were negative
for all HBV serological markers, except HBsAg that
showed discordant results (Table 2). The HBsAg positivity
of index samples 3, 6 and 7 was confirmed by neutralisa-
tion tests.
In order to find escape mutants, the full length HBsAg
gene from index plasma samples was sequenced and
cloned in a mammalian expression vector (pTriex-5) that
allowed expression of full length HBsAg recombinant
protein in HepG2 cells (Table 3). As expected, cells
transfected with transfection reagent only (mock transfec-
ted) or transfected with pTriex-5 empty vector were neg-
ative in all assays. As a positive control, HepG2 cells were
transfected with a plasmid containing HBsAg gene, derived
from a deferred blood donor infected with HBV genotype
D wild type (its sequence coincides with the consensus
sequence of genotype D). The plasma and the recombinant
protein derived from this positive control donor had high
signal/cut-off (s/co) values in all HBsAg tests. In accor-
dance with this, recombinant protein from donor 6 (geno-
type A wild type) was also recognised by HBsAg tests.
Recombinant proteins derived from index samples of
donors 1 and 7 contained aa substitutions in the HBsAg;
however, these mutations did not seem to affect its anti-
genicity (Table 3). Follow-up samples from donors 1, 6
and 7 seroconverted to IgM anti-HBc, total anti-HBc and
anti-HBe, confirming that the index samples were taken
during acute infection. We also found evidence of sero-
conversion to anti-HBc and anti-HBs in a subsequent
sample of donor 4, proving that the ‘‘isolated HBsAg
positivity’’ serological profile of index samples 4, 6 and 7
was not due to sample contamination. There were no fol-
low-up samples available from donor 3 to discard sample
contamination; however, the rigorous algorithms in place
for sample collection and testing make contamination of
this sample very unlikely. Interestingly, recombinant pro-
teins from chronic infected donors 2, 9 and 11, also con-
tained aa substitutions without apparent effect on
recombinant HBsAg detection by commercial assays.
Four escape mutants were found in this study, defined as
mutations in the HBsAg gene that impaired HBsAg
detection in at least one commercial assay using HBV
infected plasma samples (in vivo) and confirmed with
recombinant proteins derived from its infected donors
(in vitro). Positions of aa substitutions of these escape
mutants are represented by shadow circles with arrows in
Fig. 1. Donor 4 contains T118V/P127T/A128V triple
mutation in the major hydrophilic region and I28M/S34L
double aa substitution in the downstream cytosolic loop
Table 1 Primers and probes for
HBsAg gene detection and
cloning
Oligonucleotide Sequence (50 ? 30)
S1 outer primer CATCAGGAYTCCTAGGACCCCT
S5 outer primer GAGGCATAGCAGCAGGATGMAGAGG
S3 inner primer CGTGTTACAGGCGGKGTKTTTCTTGT
S6 inner primer ATGATAAAACGCCGCAGACACATC
F4 probe AATTCGCAGTCCCMAAYCTCCA—FL
P4 probe LC705—TCACTCACCAACYTSYTGTCCT—PH
O27 outer primer CCTGCTGGTGGCTCCAGTTC
O28 outer primer AGTTGGCGAGAAAGTGAAAGCCTG
O12 inner primer CTAGTGATCACATGGAGAACATCACATCAGG
O13 inner primer CTAGGAGCTCGAATGTATACCCAAAGACAAA
Med Microbiol Immunol (2013) 202:207–214 209
123
that impaired in vitro detection in all assays. However,
HBsAg in plasma from donor 4 was poorly detected by
Murex (notice its s/co is closed to border line, Table 2).
Donor 5 had a single Valine substitution in position 106
that affects HBsAg detection by BIOELISA, BIORAD and
PRISM but is detected by MUREX. Donor 8 and 10 con-
tain aa substitutions not detected by the commercial
HBsAg tests both in vitro and in vivo (Tables 2, 3).
To investigate the possible origin of these HBsAg
escape mutants, a phylogenetic tree was constructed on the
S gene region (Fig. 2). Most of the escape mutants are
clustered in the genotype D, except donor 5, which belongs
to genotype B. HBsAg gene from donor 5 is closely related
to subtype adw2 strains from East Asia, having 99 %
identity with strain JN980207 from China. Strains from
donors 8 and 10 have a similar backbone and are closely
Table 2 HBV markers in index plasma samples
Donor DNA loada (IU/ml) HBsAg HBeAg Anti-HBe Anti-HBc IgM anti-HBc
BIOELISA BIORAD MUREX PRISM
1 [125 0.48 Neg 0.97 Neg 1.37 Pos 1.36 Pos Pos Neg Neg ND
2 \5 0.36 Neg 0.25 Neg ND ND Neg Pos Pos Neg
3 [125 0.76 Neg 8.63 Pos 7.87 Pos ND Neg Neg Neg ND
4 [125 0.82 Neg ND 1.04 Pos Neg Neg Neg Neg Neg
5 [125 0.55 Neg 0.28 Neg 10.21 Pos ND Neg ND Neg ND
6 [125 0.60 Neg 3.98 Pos 2.62 Pos 3.74 Pos Neg Neg Neg Neg
7 [125 0.45 Neg 1.57 Pos 2.28 Pos 1.65 Pos Neg Neg Neg Neg
8 \5 0.61 Neg 0.75 Neg 0.82 Neg ND Neg Pos Pos Neg
9 \5 0.54 Neg 0.59 Neg 2.61 Pos ND Neg Pos Pos Neg
10 \5 0.37 Neg 0.30 Neg 0.77 Neg 0.58 Neg ND ND Pos ND
11 \5 0.90 Neg 1.44 Pos 7.91 Pos 3.38 Pos Neg Pos Pos Neg
12 \5 0.85 Neg ND 0.46 Neg Neg ND Pos Pos ND
13 \5 0.44 Neg ND 0.55 Neg Neg Neg Pos Pos Neg
Signal/cutoff (s/co) values are indicated on the left of the HBsAg result; positive result (s/co [1 for all HBsAg assays)
ND not determineda Semi-quantitative results based on cross over values of sample versus two positive controls of 125 and 5 IU/ml. Samples with DNA load\5
are low positive
Table 3 Detection of HBsAg recombinant proteins derived from the full length HBsAg gene of blood donors
Donor Genotype Mutation HBsAg recombinant protein
BIOELISA BIORAD MUREX PRISM
1 D P70A 2.10 Pos 13.35 Pos 20.35 Pos 8.6 Pos
2 D S58F/P127A 2.80 Pos 5.41 Pos 12.09 Pos 4.93 Pos
3 D G18R 1.92 Pos 9.43 Pos 18.04 Pos 7.30 Pos
4 D I28M/S34L/T118V/P127T/A128V 0.58 Neg 0.06 Neg 0.92 Neg 0.77 Neg
5 B V106A 0.90 Neg 0.11 Neg 1.87 Pos 0.98 Neg
6 A Wild type 3.51 Pos ND ND 8.82 Pos
7 A T114A/F170L 2.39 Pos 2.98 Pos 6.17 Pos 2.48 Pos
8 D F20S/C124Y 0.27 Neg 0.00 Neg 0.64 Neg 0.66 Neg
9 D F93S 1.97 Pos 11.43 Pos 20.00 Pos 7.16 Pos
10 D P29S 0.43 Neg 0.13 Neg 0.74 Neg 0.67 Neg
11 D T23A/I152V 1.66 Pos 9.49 Pos 19.00 Pos 6.27 Pos
Mock transfected NA NA 0.21 Neg 0.25 Neg 0.73 Neg 0.58 Neg
Empty vector NA NA 0.24 Neg 0.13 Neg 0.62 Neg 0.65 Neg
Positive control D Wild type 3.43 Pos 8.44 Pos 16.29 Pos 5.21 Pos
Signal/cut-off (s/co) values are indicated on the left of the HBsAg result; Positive result (s/co [1 for all HBsAg assays)
ND not determined, NA not applicable
210 Med Microbiol Immunol (2013) 202:207–214
123

related to subtype ayw2 Middle East strains. Donor 4
shares the T118V/P127T/A128V triple mutation with
Japanese and Indian strains of the subtype ayw3 but has
some minor differences in the backbone. Donor 4 backbone
is also closely related to FN295546 strain, isolated in
England.
Discussion
HBsAg is an integral membrane protein, glycosylated at
asparagine 146 (Fig. 1). Current models predict that it is
anchored in the endoplasmic reticulum (ER) through four
transmembrane domains [12, 13]. It also comprises two
cytosolic loops (aa 24–80 and aa 194–201) and the major
hydrophilic region (MHR; aa 99–160), exposed on the
virion surface [14]. In particular, the immunodominant
‘‘A’’ determinant (aa 124–147) is the site towards which
the neutralising antibodies are directed and the target of the
reagents used in many HBsAg diagnostic assays [15, 16].
The conformation of the MHR is stabilised by conserved
cysteine disulphide bonds [17].
Escape mutant isolated from OBI donor 8 contains two
aa substitutions: F20S on the transmembrane region and
C124Y on the ‘‘A’’ determinant. The lack of HBsAg
detection may be due to the elimination of disulphide
bonds caused by the substitution of cysteine by tyrosine.
This is supported by in vitro experiments showing that
HBsAg detection in the C121A/C124A double mutant was
disturbed by the elimination of disulphide bonds [17]. Our
results also confirmed a very recent study showing that
HBsAg with C124Y mutation, isolated from OBI donors,
impaired HBsAg detection in seven commercial assays
[18].
Diagnostic failure of aa substitutions in threonine 118
and proline 127 has been previously suggested [19–22].
Clinical effects of these mutations are not clear in the lit-
erature, since they were generally detected with other aa
substitutions and no attempt was made to show in vitro
effect of these mutations on HBsAg antigenicity. Here, we
show in vivo and in vitro effect of these mutations on
HBsAg antigenicity. However, in order to assess individual
aa contributions, further molecular studies are needed.
The P29S and V106A single mutations, located in the
downstream cytosolic loop and MHR respectively, have
not been previously described to impair HBsAg detection
in vivo. They are both located in highly conserved regions
within the S gene product: residues 25–43 and 69–109 [23].
A similar proline substitution (P29A) has been studied
using an in vitro infection assay, showing that it prevented
the assembly of HBV virions. P29A significantly reduced
HBsAg secretion measured by ELISA kit from Diasorin
[14]. The same group has shown that hepatitis D virus
mutants containing V106A mutation had a decreased
in vitro infectivity but its antigenicity was not affected
when it was measured using HBsAg kits from BIORAD
and Diasorin [24]. This seems to be in contradiction with
our measurements from BIORAD. However, we detected
V106A recombinant protein by MUREX and the s/co
values of PRISM were closed to border line. Discrepancies
could be attributed to the use of different in vitro experi-
mental systems. Therefore, we speculate that the V106A
escape mutant phenotype might be due to a decrease in the
amount of secreted HBsAg, similar to G145R, the most
Fig. 1 Schematic
representation of HBsAg
folding. Transmembrane
domains are depicted as boxes.
The consensus amino acid
sequence of genotype D is
shown in circles (MHR,
exposed on the virion surface,
and a portion of downstream
transmembrane domain and
cytosolic loop I). CHO
represents a facultative N-linked
glycosylation site at N146.
Cysteine disulphide bonds are
depicted by a discontinuousline. Amino acid substitutions
contributing to HBsAg
detection’s failure are shown by
shadow circles with arrowspointing towards a square
Med Microbiol Immunol (2013) 202:207–214 211
123

common escape mutant on vaccinated individuals [16, 25,
26].
Most of the work on escape mutants has been focused in
identifying mutations in the virion exposed MHR [16, 18,
22, 27]. Only a few papers describe mutations outside the
‘‘A’’ determinant that affect HBsAg antigenicity and/or
secretion [28–30]. We have identified a P29S mutation,
outside the MHR, that impaired HBsAg detection in vivo
and in vitro. Since the neutralising antibodies raised by
current HBV vaccines target the ‘‘A’’ determinant region,
there is still a risk for the emergence of drug associated
vaccine escape mutants with mutations outside the major
neutralisation site. In theory, this new vaccine escape
mutants could infect both naive and hepatitis B immunized
individuals [31].
Discrepancy of HBsAg results in some of the plasma
donor samples may be explained by differences in sensi-
tivity of HBsAg commercial tests. All the kits used in this
study have a high analytical sensitivity (\0.12 IU/ml).
However, PRISM is the commercial assay with the highest
analytical sensitivity, followed by MUREX, BIORAD and
BIOELISA. The last one also seems to have a reduced
sensitivity to genotypes C/adr and D/ayw3 [32]. The lower
sensitivity of BIOELISA may explain its seronegative
results with all the discordant samples (Table 2). With
regard to escape mutant detection, MUREX was the only
kit that detected V106A mutation (Table 3). In addition,
we have not considered here other trans-acting factors like
HBV regulatory elements, polymerase and HBcAg muta-
tions that could decrease HBV replication and HBsAg
expression in vivo [33].
The strains comprising genotype B originated mainly
from China, Japan and Southeast Asia. [9]. Most strains
specifying ayw3 subtype are found in subgenotypes D2 and
D3. Indian strains specifying ayw3 belong to subgenotype
D2 and usually contain T118V mutation [23]. P127T
mutation is an additional aa substitution that determines
ayw3 and adw3 serotypes [9]. This mutation is commonly
found in subgenotype D3, where it seems to be derived
independently in a drug addict population [23]. A recent
study has shown that Indian strains specifying ayw3 sero-
type, specifically subgenotype D3, are associated with OBI
[34]. On the other hand, we report here two donors with
OBI that belong to genotype D serotype ayw2. Strains
Fig. 2 Dendrogram based on
243 S gene sequences obtained
from GenBank, displaying a
selection of the HBV strains
with the highest homology to
sequences from escape mutants
(donors 4, 5, 8 and 10).
Accession number, country of
origin, genotype and serotype
are given at the nodes
212 Med Microbiol Immunol (2013) 202:207–214
123

specifying ayw2 subtype are found mostly in subgenotypes
D1 and D4. Moreover, the strains from Middle East mainly
belong to subgenotype D1 [9, 23].
The high homology of these escape mutants with worldwide
HBV strains corresponds with current estimates that 58 % of
HBV infected population living in Scotland are of non-British
origin (30 % from East Asia, 10 % from South Asia and 18 %
from other countries) (Wallace L.A., Sexual Health and Blood
Borne Virus Conference, Glasgow, June 2012).
OBI is a main risk factor for transfusion-transmitted
infection that requires further research. Difficulties with
virological characterisation of some HBV infected donors
are mostly related to limited availability to look-back and
follow-up samples, limited volume of archive samples,
very low viral load and lack of clinical data.
The SNBTS currently have a strategy of double testing
based on both NAT and serology. The first line routine
screening tests are commercial tests of the highest sensi-
tivity in the market: HBsAg PRISM (0.021 IU/ml) and
Cobas TaqScreen multiplex nucleic acid test (1.9 IU/ml).
Despite the implementation of HBV NAT, the emergence
of escape mutants is still a risk factor to blood safety since
NAT still requires higher sensitivity to detect most OBI,
which generally has very low viral load. Also mutations
may occur in the primer binding regions, rendering escape
mutants undetected by both NAT and serology. NAT
sensitivity could be improved by individual donation
screening [5] but is not cost effective. In our current blood
testing algorithm, we confirm individual donations after
initial screening by an independent HBV NAT test and a
series of serological tests. No confirmed HBV transfusion-
transmitted infection has been reported since 2005. To
maintain this high standard of blood safety, we need to be
in a position of predicting and monitoring the emergence of
new escape mutants.
The low prevalence of escape mutants found (0.05 per
100,000 donations) reflects the low rate of hepatitis B
infection among Scottish blood donors but it could be
masked by undetected OBI cases that may have occurred
prior to HBV NAT introduction. Nevertheless, the epide-
miological situation is changing since there is a notorious
increase in the incidence of hepatitis B infection in Scot-
land in recent years [35, 36]. Whether this is due to
immigration from countries with high HBV prevalence
and/or the lack of universal HBV vaccination is still a
matter of debate. Indeed, it seems clear that immigration
from high prevalence countries and the use of new antiviral
drugs may contribute to the emergence of new escape
mutants and more cases of OBI.
Acknowledgments We are very thankful to Helen Munro and Tony
Jordan for providing serology results, Jacqui Doran for HBV NAT
and all SNBTS teams involved in sample collection and storage.
Conflict of interest The authors declare that they have no conflict
of interest.
References
1. Brant L, Cawley C, Reynolds C, Byrne L (2008) Infection sur-
veillance annual report 2007. The NHS Blood and Transplant and
Health Protection Agency. http://www.hpa.org.uk/webc/HPAweb
File/HPAweb_C/1227255714122. Accessed 22 Nov 2012
2. Brailsford S, Davison K, Reynolds C, Homer K, Vasconcelos M
(2011) Safe supplies: new horizons. Annual Review from the
NHS Blood and Transplant/Health Protection Agency. http://
www.hpa.org.uk/webc/HPAwebFile/HPAweb_C/1317136707628.
Accessed 22 Nov 2012
3. Allain JP (2004) Occult hepatitis B virus infection: implications
in transfusion. Vox Sang 86:83–91
4. Allain JP, Candotti D (2009) Diagnostic algorithm for HBV safe
transfusion. Blood Transfus 7:174–182
5. Allain JP, Cox L (2011) Challenges in hepatitis B detection
among blood donors. Curr Opin Hematol 18:461–466
6. Cleland A, Nettleton P, Jarvis L, Simmonds P (1999) Use of
bovine viral diarrhoea virus as an internal control for amplifica-
tion of hepatitis C virus. Vox Sang 76:170–174
7. Jarvis L, Becker J, Tender A, Cleland A, Queiros L, Aquiar A,
Azevedo J, Aprili G, Bressan F, Torres P, Nieto S, Ursitti A,
Montoro J, Vila E, Ramada C, Saldanha J (2008) Evaluation of
the Roche cobas s 201 system and cobas TaqScreen multiplex test
for blood screening: a European multicenter study. Transfusion
48:1853–1861
8. Gnaneshan S, Ijaz S, Moran J, Ramsay M, Green J (2007)
HepSEQ: international public health repository for hepatitis B.
Nucleic Acids Res 35:D367–D370
9. Kay A, Zoulim F (2007) Hepatitis B virus genetic variability and
evolution. Virus Res 127:164–176
10. Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M,
Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer
T, Ashton B, Meintjes P, Drummond A (2012) Geneious basic: an
integrated and extendable desktop software platform for the
organization and analysis of sequence data. Bioinformatics
28:1647–1649
11. Ponde RA (2011) The underlying mechanisms for the ‘‘isolated
positivity for the hepatitis B surface antigen (HBsAg)’’ serolog-
ical profile. Med Microbiol Immunol 200:13–22
12. Persson B, Argos P (1994) Prediction of transmembrane seg-
ments in proteins utilising multiple sequence alignments. J Mol
Biol 237:182–192
13. Eble BE, MacRae DR, Lingappa VR, Ganem D (1987) Multiple
topogenic sequences determine the transmembrane orientation of
the hepatitis B surface antigen. Mol Cell Biol 7:3591–3601
14. Blanchet M, Sureau C (2006) Analysis of the cytosolic domains
of the hepatitis B virus envelope proteins for their function in
viral particle assembly and infectivity. J Virol 80:11935–11945
15. Zuckerman JN, Zuckerman AJ (2003) Mutations of the surface
protein of hepatitis B virus. Antiviral Res 60:75–78
16. Ma Q, Wang Y (2012) Comprehensive analysis of the prevalence
of hepatitis B virus escape mutations in the major hydrophilic
region of surface antigen. J Med Virol 84:198–206
17. Mangold CM, Streeck RE (1993) Mutational analysis of the
cysteine residues in the hepatitis B virus small envelope protein.
J Virol 67:4588–4597
18. Huang CH, Yuan Q, Chen PJ, Zhang YL, Chen CR, Zheng QB,
Yeh SH, Yu H, Xue Y, Chen YX, Liu PG, Ge SX, Zhang J, Xia
NS (2012) Influence of mutations in hepatitis B virus surface
Med Microbiol Immunol (2013) 202:207–214 213
123

protein on viral antigenicity and phenotype in occult HBV strains
from blood donors. J Hepatol 57:720–729
19. Brojer E, Grabarczyk P, Liszewski G, Mikulska M, Allain JP,
Letowska M (2006) Characterization of HBV DNA?/HBsAg-
blood donors in Poland identified by triplex NAT. Hepatology
44:1666–1674
20. Carman WF, Van Deursen FJ, Mimms LT, Hardie D, Coppola R,
Decker R, Sanders R (1997) The prevalence of surface antigen
variants of hepatitis B virus in Papua New Guinea, South Africa,
and Sardinia. Hepatology 26:1658–1666
21. Sayiner AA, Agca H, Sengonul A, Celik A, Akarsu M (2007) A
new hepatitis B virus vaccine escape mutation in a renal trans-
plant recipient. J Clin Virol 38:157–160
22. Hsu HY, Chang MH, Ni YH, Chiang CL, Chen HL, Wu JF, Chen
PJ (2010) No increase in prevalence of hepatitis B surface antigen
mutant in a population of children and adolescents who were
fully covered by universal infant immunization. J Infect Dis
201:1192–1200
23. Norder H, Courouce AM, Coursaget P, Echevarria JM, Lee SD,
Mushahwar IK, Robertson BH, Locarnini S, Magnius LO (2004)
Genetic diversity of hepatitis B virus strains derived worldwide:
genotypes, subgenotypes, and HBsAg subtypes. Intervirology
47:289–309
24. Salisse J, Sureau C (2009) A function essential to viral entry
underlies the hepatitis B virus ‘‘a’’ determinant. J Virol
83:9321–9328
25. Carman WF, Zanetti AR, Karayiannis P, Waters J, Manzillo G,
Tanzi E, Zuckerman AJ, Thomas HC (1990) Vaccine-induced
escape mutant of hepatitis B virus. Lancet 336:325–329
26. Kalinina T, Iwanski A, Will H, Sterneck M (2003) Deficiency in
virion secretion and decreased stability of the hepatitis B virus
immune escape mutant G145R. Hepatology 38:1274–1281
27. Avellon A, Echevarria JM (2006) Frequency of hepatitis B virus
‘a’ determinant variants in unselected Spanish chronic carriers.
J Med Virol 78:24–36
28. Chua PK, Wang RY, Lin MH, Masuda T, Suk FM, Shih C (2005)
Reduced secretion of virions and hepatitis B virus (HBV) surface
antigen of a naturally occurring HBV variant correlates with the
accumulation of the small S envelope protein in the endoplasmic
reticulum and Golgi apparatus. J Virol 79:13483–13496
29. Ito K, Qin Y, Guarnieri M, Garcia T, Kwei K, Mizokami M,
Zhang J, Li J, Wands JR, Tong S (2010) Impairment of hepatitis
B virus virion secretion by single-amino-acid substitutions in the
small envelope protein and rescue by a novel glycosylation site.
J Virol 84:12850–12861
30. Warner N, Locarnini S (2008) The antiviral drug selected hepa-
titis B virus rtA181T/sW172* mutant has a dominant negative
secretion defect and alters the typical profile of viral rebound.
Hepatology 48:88–98
31. Clements CJ, Coghlan B, Creati M, Locarnini S, Tedder RS,
Torresi J (2010) Global control of hepatitis B virus: does treat-
ment-induced antigenic change affect immunization? Bull World
Health Organ 88:66–73
32. Scheiblauer H, El-Nageh M, Diaz S, Nick S, Zeichhardt H,
Grunert HP, Prince A (2010) Performance evaluation of 70
hepatitis B virus (HBV) surface antigen (HBsAg) assays from
around the world by a geographically diverse panel with an array
of HBV genotypes and HBsAg subtypes. Vox Sang 98:403–414
33. Bar-Yishay I, Shaul Y, Shlomai A (2011) Hepatocyte metabolic
signalling pathways and regulation of hepatitis B virus expres-
sion. Liver Int 31:282–290
34. Chandra PK, Biswas A, Datta S, Banerjee A, Panigrahi R,
Chakrabarti S, De BK, Chakravarty R (2009) Subgenotypes of
hepatitis B virus genotype D (D1, D2, D3 and D5) in India:
differential pattern of mutations, liver injury and occult HBV
infection. J Viral Hepat 16:749–756
35. Barclay ST, Cameron S, Mills PR, Priest M, Ross F, Fox R,
Goulding C, Forrest EH, Morris AJ, Neilson M, Stanley AJ
(2010) The changing face of hepatitis B in greater Glasgow:
epidemiological trends 1993–2007. Scott Med J 55:4–7
36. Mackenzie AR, Molyneaux PJ, Cadwgan AM, Laing RB,
Douglas JG, Smith CC (2003) Increasing incidence of acute
hepatitis B virus infection referrals to the Aberdeen Infection
Unit: a matter for concern. Scott Med J 48:73–75
214 Med Microbiol Immunol (2013) 202:207–214
123





























