Embed Size (px)
Citation preview

© 2019 The Pharmaceutical Society of Japan
Vol. 67, No. 12 1301Chem. Pharm. Bull. 67, 1301–1313 (2019)
Regular Article
Comparative Study of Pharmacopoeias in Japan, Europe, and the United States: Toward the Further Convergence of International Pharmacopoeial Standards
Yujiro Kameyama,*,a Maki Matsuhama,a Chie Mizumaru,a Rieko Saito,a Tsuyoshi Ando,a and Seiko Miyazaki*,b
a Division of Pharmacopoeia and Standards for Drugs, Office of Review Management, Pharmaceuticals and Medical Devices Agency; Shin-Kasumigaseki Building, 3–3–2 Kasumigaseki, Chiyoda-ku, Tokyo 100–0013, Japan: and b Division of Social Pharmacy and Public Health, Showa Pharmaceutical University; 3–3165 Higashi-Tamagawagakuen, Machida, Tokyo 194–8543, Japan.Received July 25, 2019; accepted September 25, 2019
A pharmacopoeia’s core mission is to protect public health by creating and making available public standards to help ensure the quality of drugs. In recent years, pharmacopoeias around the world have har-monized their standards in the present context of globalized drug supply chains and markets. For example, the Pharmacopoeial Discussion Group has worked to harmonize excipient monographs and general chapters. In addition, the International Meeting of World Pharmacopoeias has been held by the WHO to discuss infor-mation exchange and international collaboration, among other topics. To contribute further to the protection of public health in the globalized drug market, we conducted a comparative study of the pharmacopoeias in Japan, Europe, and the United States. We aimed to examine current differences among the Japanese Phar-macopoeia, the European Pharmacopoeia, and the United States Pharmacopeia–National Formulary and to identify areas that require further collaboration among the three pharmacopoeias. In this study, we analyzed monographs and general chapters listed in the three pharmacopoeias. We identified the features of the mono-graphs and general chapters listed in each pharmacopoeia, as well as differences across the pharmacopoeias. Moreover, on the basis of our findings, we suggest standards that require further collaboration among the pharmacopoeias in certain preferred areas. The comparison data produced by this study are expected to be used to develop strategies for future revisions of pharmacopoeias around the world.
Key words Pharmacopoeia; Pharmacopoeial Standards; Japanese Pharmacopoeia; European Pharmacopoeia; United States Pharmacopeia–National Formulary; Pharmacopoeial Discussion Group
IntroductionThe WHO “Good Pharmacopoeial Practices” defines a
pharmacopoeia’s core mission as protecting public health by creating and making available public standards to help ensure the quality of drugs.1) Pharmacopoeias exist to serve this role in 56 countries and three regions.2) For example, the Japanese Pharmacopoeia (JP) is an official document that defines the specifications, criteria, and standard testing methods neces-sary to properly assure the quality of drugs in Japan, based on Paragraph 1, Article 41 of the Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical Devices (PMD Act)—the most fundamental law on health care products in Japan.3) Similarly, the United States Pharmacopeia–National Formulary (USP–NF) is used to control the quality of drugs distributed in the United States, as prescribed in Sections 201(g) and 501(b) of the Federal Food, Drug, and Cosmetic Act.4,5) In Europe, the European Pharmacopoeia (Ph. Eur.) has been published in addition to national-level pharmacopoeia such as the British Pharmaco-poeia. The Ph. Eur. is mandatory when requesting marketing authorization in the member states of European Pharmaco-poeia Commission, as prescribed in European Union Direc-tives 2001/83/EC and 2001/82/EC.6,7) Each pharmacopoeia is embedded in its own national or regional regulatory envi-ronment.1) Although these pharmacopoeias have contributed to the quality assurance of drug substances and products by
providing common standards for users in each region/country, with the globalization of drug markets, the independent stan-dards in each pharmacopoeia increase users’ burden of having to perform analytical procedures in different ways and using different acceptance criteria to satisfy pharmacopoeia require-ments that vary across regions.
The harmonization of standards among pharmacopoeias is one way to reduce this burden. In 1989, the Pharmacopoeial Discussion Group (PDG) was formed, with representatives from the organizations that developed the JP, Ph. Eur., and USP–NF, to work toward the harmonization of pharmaco-poeial standards such as excipient monographs and general chapters.8) Furthermore, the International Meeting of World Pharmacopoeias has been held by the WHO to facilitate in-formation exchange and international collaboration, among other aims.9) The harmonization of pharmacopoeial standards conducted by groups such as the PDG has contributed to re-ducing users’ burden caused by different requirements among pharmacopoeias.
In recent years, innovative and complex drugs such as monoclonal antibodies have been marketed, and drug prices have risen rapidly. In addition, the further globalization of drug supply chains often diversifies the manufacturing process and material grade, even for the same drug, because of dif-fering pharmacopoeial and regulatory requirements depend-ing on the country/region. This results in cost increases and
* To whom correspondence should be addressed. e-mail: [email protected]; [email protected]
Highlighted Paper selected by Editor-in-Chief

1302 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
delayed access to drugs. These trends threaten patient access to drugs. Therefore, further harmonization of pharmacopoeial standards is needed by many stakeholders.
However, the areas that can be discussed for harmonization across pharmacopoeias are restricted by the limited resources of each pharmacopoeia, and these areas for discussion are not sufficiently prioritized. For efficient collaboration among the pharmacopoeias, it is essential to understand the differences across individual pharmacopoeias and to identify preferred areas for collaboration among the pharmacopoeias. Although multiple studies have been carried out on pharmacopoeias around the world,10–13) overall pharmacopoeial standards have not yet been clarified because of the large number of standards in each pharmacopoeia. To facilitate the further harmoniza-tion of pharmacopoeial standards, for the JP, Ph. Eur., and USP–NF, we analyzed the overall monographs and general chapters. These pharmacopoeias are used in many countries and are the member pharmacopoeias of the PDG. Reflecting on the study results, we propose areas that should be prefer-entially selected for collaboration among the three pharmaco-poeias to help assure the quality of drugs and improve patient access to drugs in the globalized market.
MethodsThe JP 17th Edition (JP17),14) Ph. Eur. Edition 9.0 (Ph. Eur.
9.0),15) and USP 39–NF 3416)—the latest versions of each phar-macopoeia as of January 1, 2017—were analyzed and com-pared in this study. The pharmacopoeial standards are gener-ally composed of two types of standards: monographs and general chapters. A pharmacopoeial monograph is a standard that describes the properties and minimum quality require-ments for raw materials or drug products. A general chapter is a standard that provides general requirements and information for a test method or technical guidance on drugs. We analyzed and compared the standards among the JP, Ph. Eur., and USP–NF, dividing our investigation into monographs and general chapters.
Analysis and Comparison of Monographs In the com-parison of monographs, we analyzed the constitution of the monographs listed in each pharmacopoeia, examining the percentages of chemical products, biotechnological/biologi-cal products, herbal products, excipients, radiopharmaceuti-cals, homeopathic products, and the other types of products listed in each pharmacopoeia. Additionally, we identified the monographs listed in common among the JP, Ph. Eur., and USP–NF. Because some of the monograph titles differ across pharmacopoeias although the target of the monograph is same substance, we used the Chemical Abstracts Service (CAS) registry number (a numeric identifier containing up to 10 digits, divided into three parts by hyphens) to identify monographs on the same substance.17) According to the prepa-ration guidelines for monograph drafts in the JP, Ph. Eur., and USP–NF, each pharmacopoeia requires a CAS registry num-ber to identify a substance if it is known.18–20) We extracted the titles and CAS registry numbers of the monographs and compared the monographs across the JP17, Ph. Eur. 9.0, and USP39–NF34 using the CAS registry numbers. As is the case for monograph titles, policies on the development of mono-graphs differ across the pharmacopoeias. For example, differ-ent hydrates/polymorphs can be included in the same USP–NF monograph under a flexible monograph approach, as described
in the “USP Guideline for Submitting Requests for Revision to USP–NF.”16) In contrast, the policy on hydrate forms in the Ph. Eur. specified in the “Technical Guide for the Elaboration of Monographs” states that, when a substance exists both in water-free form and in the form of (a) hydrate(s) with different water contents, if all of these forms are used, they are normal-ly treated as individual substances requiring separate mono-graphs. The same rule applies for solvates. Because of these differences in policies on monograph construction across the pharmacopoeias, it is difficult to compare the number of monographs listed in each pharmacopoeia. In this study, we compared the number of monographs among the three pharmacopoeias using the monograph construction of the JP. For comparisons between the Ph. Eur. and the USP–NF, we compared the number of monographs using the monograph construction of the Ph. Eur. because its policy on monograph construction is similar to that of the JP.
Analysis and Comparison of General Chapters In the comparison of general chapters, we analyzed the kinds of general chapters that are listed in common among the JP, Ph. Eur., and USP–NF. The construction of general chapters dif-fers across the Ph. Eur., USP–NF, and JP. In the JP, the gener-al chapters are composed of general test methods and general information chapters that provide conceptual descriptions of test methods or quality control/assurance. Similarly, the gener-al chapters in the Ph. Eur. are composed of methods of analy-sis chapters and general texts, which correspond, respectively, to the general test methods and general information chapters in the JP. In the USP–NF, the general chapters are composed of general product quality chapters that describe the basic requirements for drug products, general test methods, and in-formational chapters numbered above ‹999›, which correspond to the general information chapters in the JP. The details of the contents of general chapters vary across the three phar-macopoeias. Therefore, chapters describing the same principle or the same type of apparatus across pharmacopoeias were considered equal, regardless of the details these chapters’ con-tents. In this study, because the classification constructions of general chapters differ among the three pharmacopoeias, the general chapters on testing methods listed in the JP were clas-sified using the classifications of the responsible expert com-mittees/groups working to develop pharmacopoeial standards for this pharmacopoeia. Testing methods that are not listed in the JP were classified on the basis of the classifications of general chapters and of the responsible expert committees/groups for the Ph. Eur. and the USP–NF. The testing methods were classified as physicochemical testing methods, physical testing methods, microbiological testing methods, and testing methods for drug formulations. The classification of general chapters is shown in Tables S1, S2, S3, and S4.
ResultsAnalysis and Comparison of Monographs Because
some of the monograph titles differ across pharmacopoeias although the target of the monographs is the same substance, we extracted both the titles and the CAS registry numbers of the monographs and compared the monographs among the JP17, Ph. Eur. 9.0, and USP39–NF34 using the CAS registry numbers. We summarized the numbers of monographs listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 and the numbers of monographs listed in common in two pharmacopoeias or in
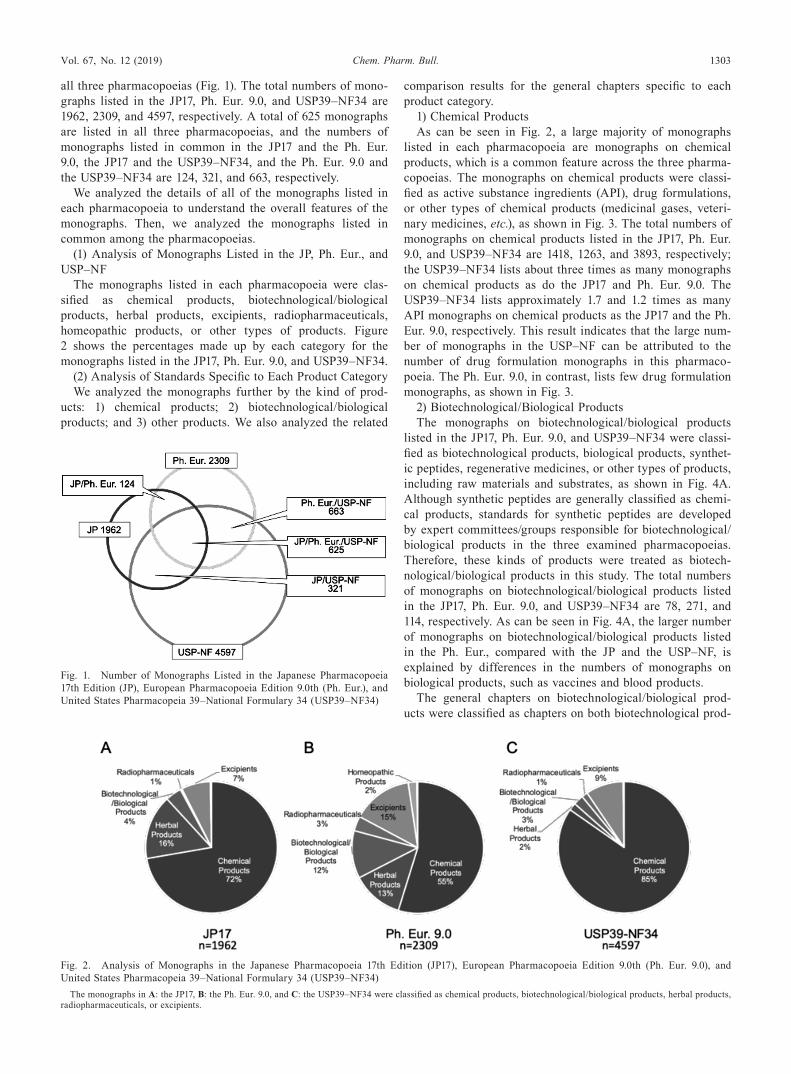
Vol. 67, No. 12 (2019) 1303Chem. Pharm. Bull.
all three pharmacopoeias (Fig. 1). The total numbers of mono-graphs listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 are 1962, 2309, and 4597, respectively. A total of 625 monographs are listed in all three pharmacopoeias, and the numbers of monographs listed in common in the JP17 and the Ph. Eur. 9.0, the JP17 and the USP39–NF34, and the Ph. Eur. 9.0 and the USP39–NF34 are 124, 321, and 663, respectively.
We analyzed the details of all of the monographs listed in each pharmacopoeia to understand the overall features of the monographs. Then, we analyzed the monographs listed in common among the pharmacopoeias.
(1) Analysis of Monographs Listed in the JP, Ph. Eur., and USP–NF
The monographs listed in each pharmacopoeia were clas-sified as chemical products, biotechnological/biological products, herbal products, excipients, radiopharmaceuticals, homeopathic products, or other types of products. Figure 2 shows the percentages made up by each category for the monographs listed in the JP17, Ph. Eur. 9.0, and USP39–NF34.
(2) Analysis of Standards Specific to Each Product CategoryWe analyzed the monographs further by the kind of prod-
ucts: 1) chemical products; 2) biotechnological/biological products; and 3) other products. We also analyzed the related
comparison results for the general chapters specific to each product category.
1) Chemical ProductsAs can be seen in Fig. 2, a large majority of monographs
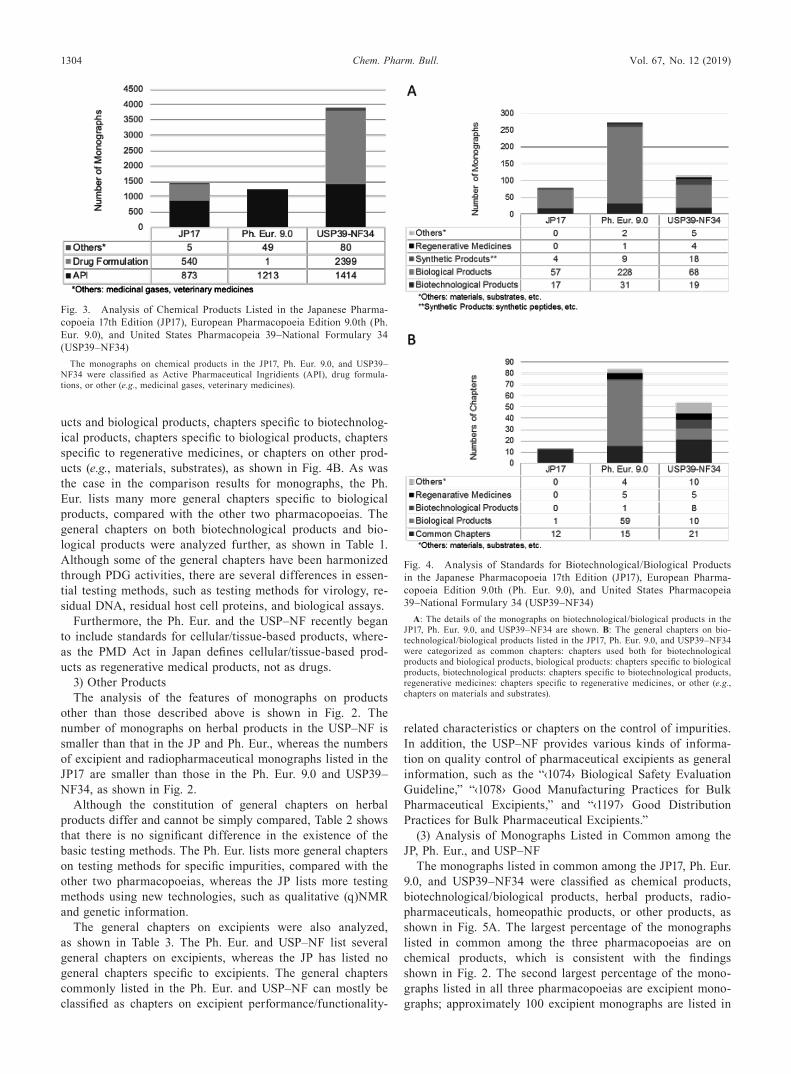
listed in each pharmacopoeia are monographs on chemical products, which is a common feature across the three pharma-copoeias. The monographs on chemical products were classi-fied as active substance ingredients (API), drug formulations, or other types of chemical products (medicinal gases, veteri-nary medicines, etc.), as shown in Fig. 3. The total numbers of monographs on chemical products listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 are 1418, 1263, and 3893, respectively; the USP39–NF34 lists about three times as many monographs on chemical products as do the JP17 and Ph. Eur. 9.0. The USP39–NF34 lists approximately 1.7 and 1.2 times as many API monographs on chemical products as the JP17 and the Ph. Eur. 9.0, respectively. This result indicates that the large num-ber of monographs in the USP–NF can be attributed to the number of drug formulation monographs in this pharmaco-poeia. The Ph. Eur. 9.0, in contrast, lists few drug formulation monographs, as shown in Fig. 3.
2) Biotechnological/Biological ProductsThe monographs on biotechnological/biological products
listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 were classi-fied as biotechnological products, biological products, synthet-ic peptides, regenerative medicines, or other types of products, including raw materials and substrates, as shown in Fig. 4A. Although synthetic peptides are generally classified as chemi-cal products, standards for synthetic peptides are developed by expert committees/groups responsible for biotechnological/biological products in the three examined pharmacopoeias. Therefore, these kinds of products were treated as biotech-nological/biological products in this study. The total numbers of monographs on biotechnological/biological products listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 are 78, 271, and 114, respectively. As can be seen in Fig. 4A, the larger number of monographs on biotechnological/biological products listed in the Ph. Eur., compared with the JP and the USP–NF, is explained by differences in the numbers of monographs on biological products, such as vaccines and blood products.
The general chapters on biotechnological/biological prod-ucts were classified as chapters on both biotechnological prod-
Fig. 1. Number of Monographs Listed in the Japanese Pharmacopoeia 17th Edition (JP), European Pharmacopoeia Edition 9.0th (Ph. Eur.), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
Fig. 2. Analysis of Monographs in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
The monographs in A: the JP17, B: the Ph. Eur. 9.0, and C: the USP39–NF34 were classified as chemical products, biotechnological/biological products, herbal products, radiopharmaceuticals, or excipients.
1304 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
ucts and biological products, chapters specific to biotechnolog-ical products, chapters specific to biological products, chapters specific to regenerative medicines, or chapters on other prod-ucts (e.g., materials, substrates), as shown in Fig. 4B. As was the case in the comparison results for monographs, the Ph. Eur. lists many more general chapters specific to biological products, compared with the other two pharmacopoeias. The general chapters on both biotechnological products and bio-logical products were analyzed further, as shown in Table 1. Although some of the general chapters have been harmonized through PDG activities, there are several differences in essen-tial testing methods, such as testing methods for virology, re-sidual DNA, residual host cell proteins, and biological assays.
Furthermore, the Ph. Eur. and the USP–NF recently began to include standards for cellular/tissue-based products, where-as the PMD Act in Japan defines cellular/tissue-based prod-ucts as regenerative medical products, not as drugs.
3) Other ProductsThe analysis of the features of monographs on products
other than those described above is shown in Fig. 2. The number of monographs on herbal products in the USP–NF is smaller than that in the JP and Ph. Eur., whereas the numbers of excipient and radiopharmaceutical monographs listed in the JP17 are smaller than those in the Ph. Eur. 9.0 and USP39–NF34, as shown in Fig. 2.
Although the constitution of general chapters on herbal products differ and cannot be simply compared, Table 2 shows that there is no significant difference in the existence of the basic testing methods. The Ph. Eur. lists more general chapters on testing methods for specific impurities, compared with the other two pharmacopoeias, whereas the JP lists more testing methods using new technologies, such as qualitative (q)NMR and genetic information.
The general chapters on excipients were also analyzed, as shown in Table 3. The Ph. Eur. and USP–NF list several general chapters on excipients, whereas the JP has listed no general chapters specific to excipients. The general chapters commonly listed in the Ph. Eur. and USP–NF can mostly be classified as chapters on excipient performance/functionality-
related characteristics or chapters on the control of impurities. In addition, the USP–NF provides various kinds of informa-tion on quality control of pharmaceutical excipients as general information, such as the “‹1074› Biological Safety Evaluation Guideline,” “‹1078› Good Manufacturing Practices for Bulk Pharmaceutical Excipients,” and “‹1197› Good Distribution Practices for Bulk Pharmaceutical Excipients.”
(3) Analysis of Monographs Listed in Common among the JP, Ph. Eur., and USP–NF
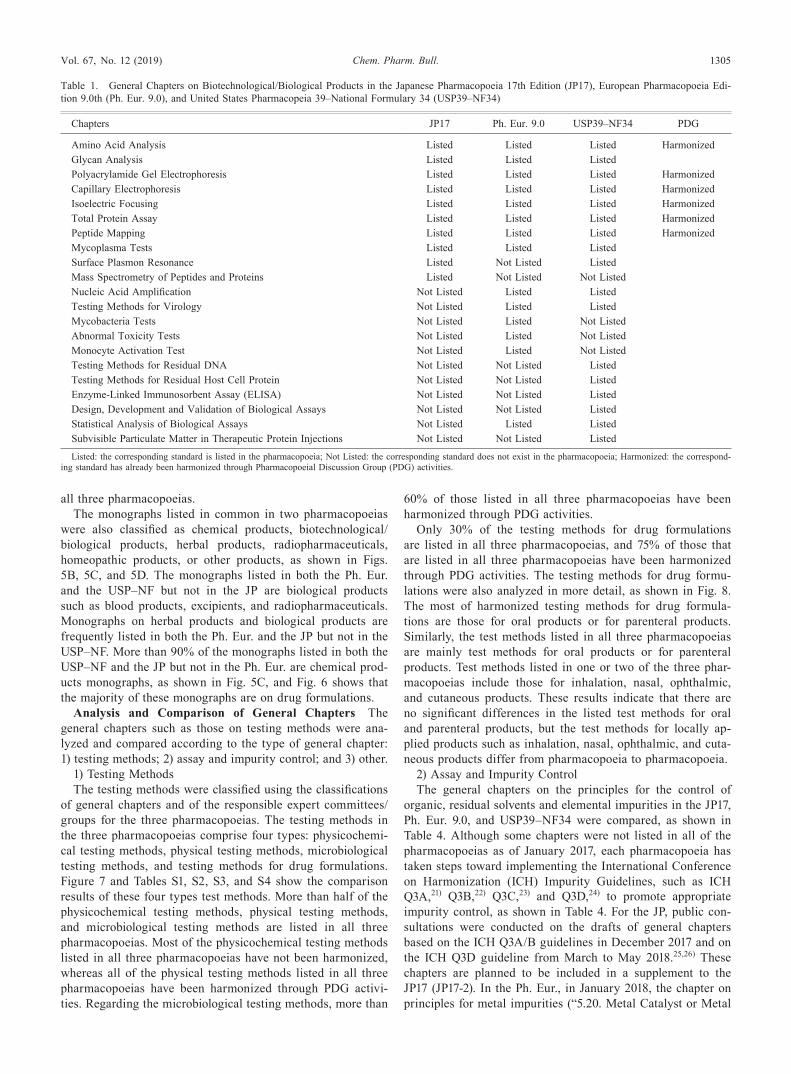
The monographs listed in common among the JP17, Ph. Eur. 9.0, and USP39–NF34 were classified as chemical products, biotechnological/biological products, herbal products, radio-pharmaceuticals, homeopathic products, or other products, as shown in Fig. 5A. The largest percentage of the monographs listed in common among the three pharmacopoeias are on chemical products, which is consistent with the findings shown in Fig. 2. The second largest percentage of the mono-graphs listed in all three pharmacopoeias are excipient mono-graphs; approximately 100 excipient monographs are listed in
Fig. 4. Analysis of Standards for Biotechnological/Biological Products in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharma-copoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
A: The details of the monographs on biotechnological/biological products in the JP17, Ph. Eur. 9.0, and USP39–NF34 are shown. B: The general chapters on bio-technological/biological products listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 were categorized as common chapters: chapters used both for biotechnological products and biological products, biological products: chapters specific to biological products, biotechnological products: chapters specific to biotechnological products, regenerative medicines: chapters specific to regenerative medicines, or other (e.g., chapters on materials and substrates).
Fig. 3. Analysis of Chemical Products Listed in the Japanese Pharma-copoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
The monographs on chemical products in the JP17, Ph. Eur. 9.0, and USP39–NF34 were classified as Active Pharmaceutical Ingridients (API), drug formula-tions, or other (e.g., medicinal gases, veterinary medicines).
Vol. 67, No. 12 (2019) 1305Chem. Pharm. Bull.
all three pharmacopoeias.The monographs listed in common in two pharmacopoeias
were also classified as chemical products, biotechnological/biological products, herbal products, radiopharmaceuticals, homeopathic products, or other products, as shown in Figs. 5B, 5C, and 5D. The monographs listed in both the Ph. Eur. and the USP–NF but not in the JP are biological products such as blood products, excipients, and radiopharmaceuticals. Monographs on herbal products and biological products are frequently listed in both the Ph. Eur. and the JP but not in the USP–NF. More than 90% of the monographs listed in both the USP–NF and the JP but not in the Ph. Eur. are chemical prod-ucts monographs, as shown in Fig. 5C, and Fig. 6 shows that the majority of these monographs are on drug formulations.
Analysis and Comparison of General Chapters The general chapters such as those on testing methods were ana-lyzed and compared according to the type of general chapter: 1) testing methods; 2) assay and impurity control; and 3) other.
1) Testing MethodsThe testing methods were classified using the classifications
of general chapters and of the responsible expert committees/groups for the three pharmacopoeias. The testing methods in the three pharmacopoeias comprise four types: physicochemi-cal testing methods, physical testing methods, microbiological testing methods, and testing methods for drug formulations. Figure 7 and Tables S1, S2, S3, and S4 show the comparison results of these four types test methods. More than half of the physicochemical testing methods, physical testing methods, and microbiological testing methods are listed in all three pharmacopoeias. Most of the physicochemical testing methods listed in all three pharmacopoeias have not been harmonized, whereas all of the physical testing methods listed in all three pharmacopoeias have been harmonized through PDG activi-ties. Regarding the microbiological testing methods, more than
60% of those listed in all three pharmacopoeias have been harmonized through PDG activities.
Only 30% of the testing methods for drug formulations are listed in all three pharmacopoeias, and 75% of those that are listed in all three pharmacopoeias have been harmonized through PDG activities. The testing methods for drug formu-lations were also analyzed in more detail, as shown in Fig. 8. The most of harmonized testing methods for drug formula-tions are those for oral products or for parenteral products. Similarly, the test methods listed in all three pharmacopoeias are mainly test methods for oral products or for parenteral products. Test methods listed in one or two of the three phar-macopoeias include those for inhalation, nasal, ophthalmic, and cutaneous products. These results indicate that there are no significant differences in the listed test methods for oral and parenteral products, but the test methods for locally ap-plied products such as inhalation, nasal, ophthalmic, and cuta-neous products differ from pharmacopoeia to pharmacopoeia.
2) Assay and Impurity ControlThe general chapters on the principles for the control of
organic, residual solvents and elemental impurities in the JP17, Ph. Eur. 9.0, and USP39–NF34 were compared, as shown in Table 4. Although some chapters were not listed in all of the pharmacopoeias as of January 2017, each pharmacopoeia has taken steps toward implementing the International Conference on Harmonization (ICH) Impurity Guidelines, such as ICH Q3A,21) Q3B,22) Q3C,23) and Q3D,24) to promote appropriate impurity control, as shown in Table 4. For the JP, public con-sultations were conducted on the drafts of general chapters based on the ICH Q3A/B guidelines in December 2017 and on the ICH Q3D guideline from March to May 2018.25,26) These chapters are planned to be included in a supplement to the JP17 (JP17-2). In the Ph. Eur., in January 2018, the chapter on principles for metal impurities (“5.20. Metal Catalyst or Metal
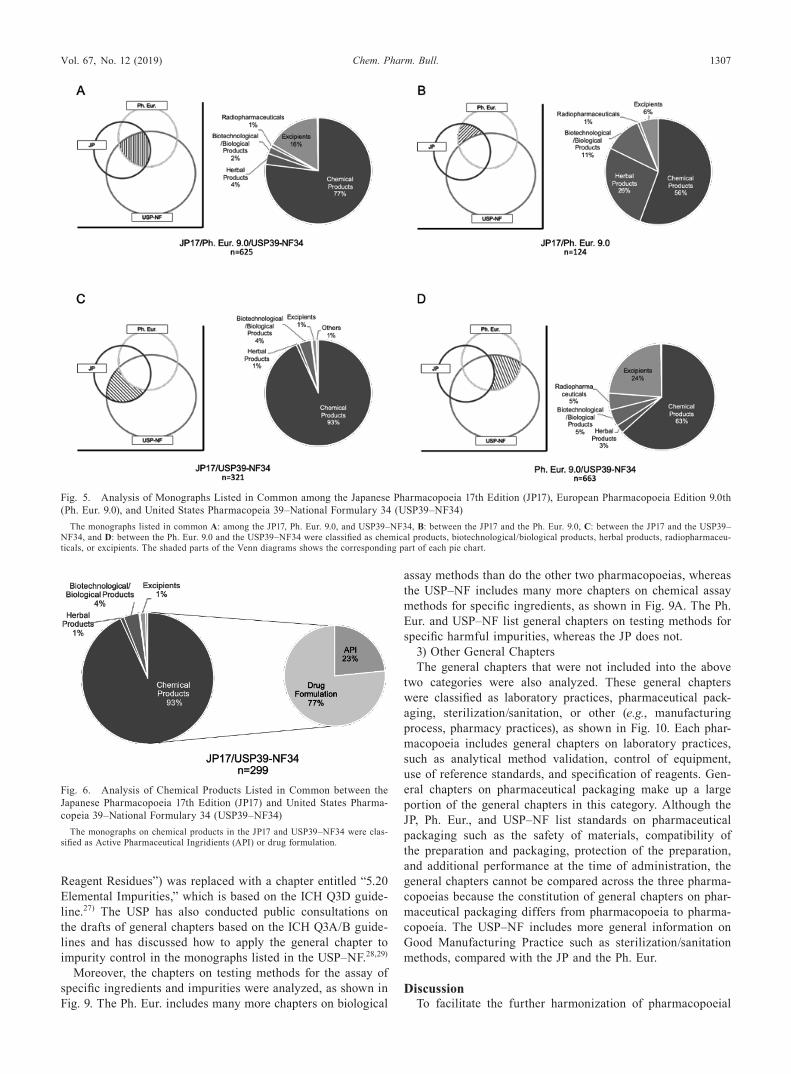
Table 1. General Chapters on Biotechnological/Biological Products in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edi-tion 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
Chapters JP17 Ph. Eur. 9.0 USP39–NF34 PDG
Amino Acid Analysis Listed Listed Listed HarmonizedGlycan Analysis Listed Listed ListedPolyacrylamide Gel Electrophoresis Listed Listed Listed HarmonizedCapillary Electrophoresis Listed Listed Listed HarmonizedIsoelectric Focusing Listed Listed Listed HarmonizedTotal Protein Assay Listed Listed Listed HarmonizedPeptide Mapping Listed Listed Listed HarmonizedMycoplasma Tests Listed Listed ListedSurface Plasmon Resonance Listed Not Listed ListedMass Spectrometry of Peptides and Proteins Listed Not Listed Not ListedNucleic Acid Amplification Not Listed Listed ListedTesting Methods for Virology Not Listed Listed ListedMycobacteria Tests Not Listed Listed Not ListedAbnormal Toxicity Tests Not Listed Listed Not ListedMonocyte Activation Test Not Listed Listed Not ListedTesting Methods for Residual DNA Not Listed Not Listed ListedTesting Methods for Residual Host Cell Protein Not Listed Not Listed ListedEnzyme-Linked Immunosorbent Assay (ELISA) Not Listed Not Listed ListedDesign, Development and Validation of Biological Assays Not Listed Not Listed ListedStatistical Analysis of Biological Assays Not Listed Listed ListedSubvisible Particulate Matter in Therapeutic Protein Injections Not Listed Not Listed Listed
Listed: the corresponding standard is listed in the pharmacopoeia; Not Listed: the corresponding standard does not exist in the pharmacopoeia; Harmonized: the correspond-ing standard has already been harmonized through Pharmacopoeial Discussion Group (PDG) activities.

1306 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
Table 2. General Chapters on Herbal Products in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
Chapter JP17 Ph. Eur. 9.0 USP39–NF34
Sampling Listed Listed ListedSample Preparation Listed Listed ListedMicroscopic Examination Listed Listed ListedStomatal Density Not Listed Listed ListedChemical Identification Test Not Listed Not Listed ListedDNA-Based Identification Test Not Listed Not Listed ListedHeavy Metals Listed Not Listed ListedForeign Matter Listed Listed ListedPesticide Residues Listed Listed ListedLoss on Drying Listed Listed ListedSwelling Index Not Listed Listed Not ListedTotal Ash Listed Listed ListedAcid-insoluble Ash Listed Listed ListedWater-soluble Ash Not Listed Not Listed ListedEthanol-soluble Extract Listed Not Listed ListedWater-soluble Extract Listed Not Listed ListedDiethyl Ether-soluble Extract Listed Not Listed ListedEssential Oil Content Listed Listed ListedWater in Essential Oils Not Listed Listed Not ListedForeign Esters in Essential Oils Not Listed Listed Not ListedFatty Oils and Resinified Essential Oils in Essential Oils Not Listed Listed Not ListedOdour and Taste of Essential Oils Not Listed Listed Not ListedResidue on Evaporation of Essential Oils Not Listed Listed Not ListedSolubility in Alcohols of Essential Oils Not Listed Listed Not ListedAssay of 1 8-Cineole in Essential Oils Not Listed Listed Not ListedStarch Content Not Listed Not Listed ListedAristolochic Acids Listed Listed Not ListedAflatoxins Listed Listed ListedOchratoxins Not Listed Listed Not ListedTannins Not Listed Listed Not ListedBitterness Not Listed Listed Not ListedMicrobial Limit Listed Not Listed Not ListedqNMR for Assay of Herbal Products Listed Not Listed Not ListedGenetic Information for Purity Test Listed Not Listed Not ListedTLC for Herbal Products Listed Listed ListedQuantitative Marker Listed Not Listed Not ListedNames of Herbal Products Listed Listed Not ListedHerbal Drug Extracts Not Listed Listed Not Listed
Listed: the corresponding standard is listed in the pharmacopoeia; Not Listed: the corresponding standard does not exist in the pharmacopoeia.
Table 3. General Chapters on Excipients in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
Chapters JP17 Ph. Eur. 9.0 USP39–NF34
Functionality-Related Characteristics Not Listed Listed ListedEthylene Oxide/Dioxan Not Listed Listed ListedEthylene Glycol and Diethylene Glycol in Ethoxylated Substances Not Listed Listed ListedDimethylaniline Not Listed Listed ListedMethoxy Determination Not Listed Not Listed ListedSulfur Dioxide Not Listed Listed ListedExcipient Biological Safety Evaluation Not Listed Not Listed ListedGood Manufacturing Practices Not Listed Not Listed ListedGood Distribution Practices Not Listed Not Listed ListedCertificate of Analysis Not Listed Not Listed ListedLabeling Not Listed Not Listed ListedChange Control Not Listed Not Listed Listed
Listed: the corresponding standard is listed in the pharmacopoeia; Not Listed; the corresponding standard does not exist in the pharmacopoeia.
Vol. 67, No. 12 (2019) 1307Chem. Pharm. Bull.
Reagent Residues”) was replaced with a chapter entitled “5.20 Elemental Impurities,” which is based on the ICH Q3D guide-line.27) The USP has also conducted public consultations on the drafts of general chapters based on the ICH Q3A/B guide-lines and has discussed how to apply the general chapter to impurity control in the monographs listed in the USP–NF.28,29)
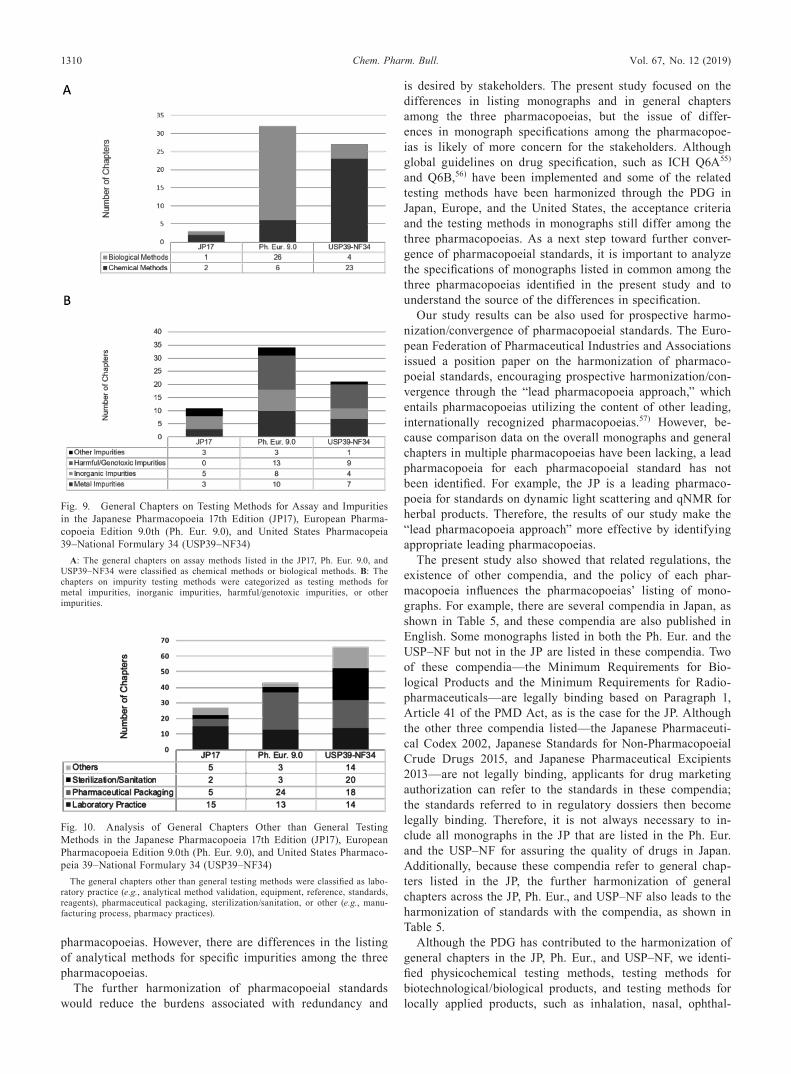
Moreover, the chapters on testing methods for the assay of specific ingredients and impurities were analyzed, as shown in Fig. 9. The Ph. Eur. includes many more chapters on biological
assay methods than do the other two pharmacopoeias, whereas the USP–NF includes many more chapters on chemical assay methods for specific ingredients, as shown in Fig. 9A. The Ph. Eur. and USP–NF list general chapters on testing methods for specific harmful impurities, whereas the JP does not.
3) Other General ChaptersThe general chapters that were not included into the above
two categories were also analyzed. These general chapters were classified as laboratory practices, pharmaceutical pack-aging, sterilization/sanitation, or other (e.g., manufacturing process, pharmacy practices), as shown in Fig. 10. Each phar-macopoeia includes general chapters on laboratory practices, such as analytical method validation, control of equipment, use of reference standards, and specification of reagents. Gen-eral chapters on pharmaceutical packaging make up a large portion of the general chapters in this category. Although the JP, Ph. Eur., and USP–NF list standards on pharmaceutical packaging such as the safety of materials, compatibility of the preparation and packaging, protection of the preparation, and additional performance at the time of administration, the general chapters cannot be compared across the three pharma-copoeias because the constitution of general chapters on phar-maceutical packaging differs from pharmacopoeia to pharma-copoeia. The USP–NF includes more general information on Good Manufacturing Practice such as sterilization/sanitation methods, compared with the JP and the Ph. Eur.
DiscussionTo facilitate the further harmonization of pharmacopoeial
Fig. 5. Analysis of Monographs Listed in Common among the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
The monographs listed in common A: among the JP17, Ph. Eur. 9.0, and USP39–NF34, B: between the JP17 and the Ph. Eur. 9.0, C: between the JP17 and the USP39–NF34, and D: between the Ph. Eur. 9.0 and the USP39–NF34 were classified as chemical products, biotechnological/biological products, herbal products, radiopharmaceu-ticals, or excipients. The shaded parts of the Venn diagrams shows the corresponding part of each pie chart.
Fig. 6. Analysis of Chemical Products Listed in Common between the Japanese Pharmacopoeia 17th Edition (JP17) and United States Pharma-copeia 39–National Formulary 34 (USP39–NF34)
The monographs on chemical products in the JP17 and USP39–NF34 were clas-sified as Active Pharmaceutical Ingridients (API) or drug formulation.

1308 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
standards, for the JP, Ph. Eur., and USP–NF, we compared and analyzed the overall monographs and general chapters listed in the three pharmacopoeias. As a common feature of the JP, Ph. Eur., and USP–NF, monographs on chemical products account for the largest portion of monographs listed in each pharmacopoeia. The USP–NF lists about three times as many chemical product monographs, compared with the JP and the Ph. Eur. We found that this large number of chemical product monographs in the USP–NF is explained by the number of drug formulation monographs, as shown in Fig. 3. Further-more, Fig. 5C shows that more than 90% of the monographs listed in both the USP–NF and the JP but not in the Ph. Eur. are chemical product monographs. Most of these monographs are drug formulation monographs.
These differences in listing monographs can be explained by variations in policies and related regulations across the pharmacopoeias. The USP–NF policy is to include standards for all drugs, including drug formulations that are legally marketed in the United States,30) whereas the JP focuses on drugs that are essential for health care and medicinal treat-ment rather than all drugs marketed in Japan.31) The Ph. Eur. has historically focused on the development of API and ex-cipient monographs. European Union Directive 2003/63/EC, which details the requirements for marketing authorization ap-plications, does not describe the relationship between dossier specification and drug formulation monographs of the Ph. Eur. clearly,6) but the “Specifications and Control Tests on the Fin-ished Products” guideline states that applicants for marketing
authorization should determine the appropriate specifications of drug formulations by referring to the general monographs in Ph. Eur., which prescribe basic requirements for drug for-mulations.32) Recently, the Ph. Eur. commission has discussed the inclusion of drug formulation monographs in the Ph. Eur and has developed one pilot monograph, “Sitagliptin tablets,” as shown in Fig. 3.33,34)
We also found that the Ph. Eur. lists more monographs on biological products such as vaccines and blood products, com-pared with the JP and the USP–NF. In Japan, the Minimum Requirements for Biological Products, issued according to Ar-ticle 42(1) of the PMD Act, provide the standards for biologi-cal products.35) Although some monographs on vaccines and blood products are listed in the JP, these monographs refer to the monographs in the Minimum Requirements for Biological Products. In the United States, the Code of Federal Regula-tions, Title 21, Part 600–660 and related guidelines issued by the Food and Drug Administration cover quality control and production for biological products.36) Although past edi-tions of the USP–NF listed more monographs on biological products, some of these monographs were deleted around 2010 because of one or more of the following issues: lack of quality information, the product was no longer marketed in the United States, tests were mentioned by name but no procedures were provided, licenses were revoked, the product was no longer manufactured in the same manner, or the production of the ar-ticle was covered in the Food and Drug Administration regu-lation concerning biologics.37–52) Some of biological product
Fig. 7. Testing Methods Listed in Common among the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
The testing methods were compared, divided into A: physicochemical testing methods, B: physical testing methods, C: microbiological testing methods, and D: testing methods for drug formulations. The pie chart on the left for each testing method shows the numbers of pharmacopoeias listing the method (3 Ph.: 3 pharmacopoeias, 2 Ph.: 2 pharmacopoeias, 1 Ph.: 1 pharmacopoeia). The pie chart on the right for each testing method shows the percentages of testing methods that were harmonized and non-harmonized through Pharmacopoeial Discussion Group (PDG) activities.

Vol. 67, No. 12 (2019) 1309Chem. Pharm. Bull.
monographs listed in the Ph. Eur. are not included in the JP or the USP–NF, but corresponding standards do exist in regula-tions in Japan and the United States.
Some excipient and radiopharmaceutical monographs are listed in both the Ph. Eur. and the USP–NF but not in the JP, as shown Fig. 5D. Similarly to biological products, the corresponding standards are listed in other compendia (the “Japanese Pharmaceutical Excipients”53) and the “Minimum Requirements for Radiopharmaceuticals”54)). Likewise, the herbal product monographs that are listed in both the JP and the Ph. Eur. but not in the USP–NF (Fig. 5B) are listed in the “Herbal Medicine Compendium,” which is a compendium of herbal products in the United States.
Furthermore, we identified the types of general chapters for further collaboration among the three pharmacopoeias. Table 1 shows that some of the general chapters on biotechnologi-cal/biological products have been harmonized through PDG activities, but there are multiple differences among the three
pharmacopoeias in the listing of essential testing methods such as testing methods for virology, residual DNA, residual host cell proteins, and biological assays. Additionally, the JP does not list any general chapters for excipients, whereas some of the general chapters for the functionality of excipients and for impurity testing methods are listed both the Ph. Eur. and the USP–NF, as shown in Table 3. Figure 7A shows that most physicochemical testing methods are not subject to harmoni-zation through PDG activities, although about 60% of the test-ing methods are listed in all three pharmacopoeias. There are differences in the listing of testing methods for locally applied drug formulations among the three pharmacopoeias, whereas most testing methods for tablets and injections are listed in all three and have been harmonized through PDG activities, as shown in Figs. 7D and 8. Furthermore, we found that each pharmacopoeia is implementing the ICH Q3A, Q3B, Q3C, and Q3D guidelines, which has contributed to the convergence of requirements for the control of impurities among the three
Fig. 8. Testing Methods for Drug Formulations Listed in Common among the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmaco-poeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
A: The testing methods for drug formulations that have been harmonized through Pharmacopoeial Discussion Group (PDG) activities were classified according to the type of drug formulation. The testing methods for drug formulations listed in B: three, C: two, and D: one of the three pharmacopoeias were categorized in the same way.
Table 4. General Chapters on Impurity Control in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
Type of Impurities JP17 Ph. Eur. 9.0 USP39–NF34
Organic Impurities: ICH Q3A/B guidelines
Not Listed (Planned to be Listed) Listed (5.10. Control of Impurities in Substances for pharmaceutical use)
Not Listed (Planned to be Listed)
Residual Solvents: ICH Q3C guideline
Listed (2.46 Residual Solvents) Listed (5.4. Residual Solvents) Listed (‹467› Residual Solvents)
Elemental Impurities: ICH Q3D guideline
Not Listed (Planned to be Listed) Not Listed (Planned to be Listed) Listed (‹232› Elemental Impurities-Limits)
Listed: the corresponding standard is listed in the pharmacopoeia; Not Listed: the corresponding standard does not exist in the pharmacopoeia. Chapter names are listed in parentheses for each pharmacopoeia.
1310 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
pharmacopoeias. However, there are differences in the listing of analytical methods for specific impurities among the three pharmacopoeias.
The further harmonization of pharmacopoeial standards would reduce the burdens associated with redundancy and
is desired by stakeholders. The present study focused on the differences in listing monographs and in general chapters among the three pharmacopoeias, but the issue of differ-ences in monograph specifications among the pharmacopoe-ias is likely of more concern for the stakeholders. Although global guidelines on drug specification, such as ICH Q6A55) and Q6B,56) have been implemented and some of the related testing methods have been harmonized through the PDG in Japan, Europe, and the United States, the acceptance criteria and the testing methods in monographs still differ among the three pharmacopoeias. As a next step toward further conver-gence of pharmacopoeial standards, it is important to analyze the specifications of monographs listed in common among the three pharmacopoeias identified in the present study and to understand the source of the differences in specification.
Our study results can be also used for prospective harmo-nization/convergence of pharmacopoeial standards. The Euro-pean Federation of Pharmaceutical Industries and Associations issued a position paper on the harmonization of pharmaco-poeial standards, encouraging prospective harmonization/con-vergence through the “lead pharmacopoeia approach,” which entails pharmacopoeias utilizing the content of other leading, internationally recognized pharmacopoeias.57) However, be-cause comparison data on the overall monographs and general chapters in multiple pharmacopoeias have been lacking, a lead pharmacopoeia for each pharmacopoeial standard has not been identified. For example, the JP is a leading pharmaco-poeia for standards on dynamic light scattering and qNMR for herbal products. Therefore, the results of our study make the “lead pharmacopoeia approach” more effective by identifying appropriate leading pharmacopoeias.
The present study also showed that related regulations, the existence of other compendia, and the policy of each phar-macopoeia influences the pharmacopoeias’ listing of mono-graphs. For example, there are several compendia in Japan, as shown in Table 5, and these compendia are also published in English. Some monographs listed in both the Ph. Eur. and the USP–NF but not in the JP are listed in these compendia. Two of these compendia—the Minimum Requirements for Bio-logical Products and the Minimum Requirements for Radio-pharmaceuticals—are legally binding based on Paragraph 1, Article 41 of the PMD Act, as is the case for the JP. Although the other three compendia listed—the Japanese Pharmaceuti-cal Codex 2002, Japanese Standards for Non-Pharmacopoeial Crude Drugs 2015, and Japanese Pharmaceutical Excipients 2013—are not legally binding, applicants for drug marketing authorization can refer to the standards in these compendia; the standards referred to in regulatory dossiers then become legally binding. Therefore, it is not always necessary to in-clude all monographs in the JP that are listed in the Ph. Eur. and the USP–NF for assuring the quality of drugs in Japan. Additionally, because these compendia refer to general chap-ters listed in the JP, the further harmonization of general chapters across the JP, Ph. Eur., and USP–NF also leads to the harmonization of standards with the compendia, as shown in Table 5.
Although the PDG has contributed to the harmonization of general chapters in the JP, Ph. Eur., and USP–NF, we identi-fied physicochemical testing methods, testing methods for biotechnological/biological products, and testing methods for locally applied products, such as inhalation, nasal, ophthal-
Fig. 9. General Chapters on Testing Methods for Assay and Impurities in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharma-copoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmacopeia 39–National Formulary 34 (USP39–NF34)
A: The general chapters on assay methods listed in the JP17, Ph. Eur. 9.0, and USP39–NF34 were classified as chemical methods or biological methods. B: The chapters on impurity testing methods were categorized as testing methods for metal impurities, inorganic impurities, harmful/genotoxic impurities, or other impurities.
Fig. 10. Analysis of General Chapters Other than General Testing Methods in the Japanese Pharmacopoeia 17th Edition (JP17), European Pharmacopoeia Edition 9.0th (Ph. Eur. 9.0), and United States Pharmaco-peia 39–National Formulary 34 (USP39–NF34)
The general chapters other than general testing methods were classified as labo-ratory practice (e.g., analytical method validation, equipment, reference, standards, reagents), pharmaceutical packaging, sterilization/sanitation, or other (e.g., manu-facturing process, pharmacy practices).

Vol. 67, No. 12 (2019) 1311Chem. Pharm. Bull.
mic, and cutaneous products, as areas where there should be further collaboration, as shown in Fig. 8. In recent years, the numbers of biotechnological products, such as monoclonal antibodies, and locally applied drug formulations, such as inhalational products, approved by the regulatory authorities in each region/country has increased rapidly. Because the specification of biotechnological/biological products and lo-cally applied products varies across products and manufactur-ing processes, it is necessary for each pharmacopoeia to set testing methods with specifications that are flexible rather than unified to assure the quality of these complex products. For example, the Ph. Eur. lists a monoclonal antibody monograph on infliximab concentrated solution, as of January 2019.58) In this monograph, depending on the product and manufacturing process, some specifications are not prescribed to provide flex-ibility, but the testing methods are provided, along with the criteria for verifying their performance.59) Biotechnological products, such as a monoclonal antibody, can target a disease that cannot be targeted using chemical compounds and have rapidly expanded in the world’s drug market. The number of biosimilars is also expected to further increase in the future. Likewise, the number of locally applied products will increase because some regulatory agencies promote the development generics for locally applied products to suppress high drug prices through drug price competition.60) Using established and common testing methods listed in the monographs, indus-tries are able to reduce developments time and regulators are able to easily evaluate the submitted data. Thus, the develop-ment and harmonization of the testing methods identified in this study become increasingly important.
The comparison data in this study are also expected to be used to develop a strategy for future revisions to pharmaco-poeias worldwide. For example, we found that some physico-chemical testing methods (e.g., size-exclusion chromatography, X-ray fluorescence spectrometry, and circular dichroism) are listed in both the Ph. Eur. and the USP–NF but not in the JP, as shown in Tables S1, S2, S3, and S4. We also found that general chapters on the characteristics of excipients and the testing methods for specific harmful/genotoxic impurities are not listed in the JP. As for chapters on the characteris-tics of excipients, the Ph. Eur. and the USP–NF include the excipients’ properties related to the formulation performance and the manufacturing process in “5.15. Functionality-Related Characteristics Excipients” and “‹1059› Excipient Perfor-mance,” respectively. The principles on properties in each pharmacopoeia have influenced the constitution of the excipi-ent monographs. The JP divides some excipient monographs by properties related to formulation performance and the man-ufacturing process, but other pharmacopoeias have integrated these excipients into one monograph by providing the proper-ties as general information. Therefore, developing a general
chapter on the properties related to formulation performance and the manufacturing process for the JP might be useful to further harmonize excipient monographs through the PDG.
The globalization of drug markets diversifies the supply chain and manufacturing methods, causing pharmacopoeias to prepare new regulatory frameworks to assure the quality of drugs while maintaining flexibility in quality control. For example, harmful/genotoxic impurities result from a lack of material control, manufacturing process control, and/or sup-ply chain control; therefore, monographs, in some cases, are required to include purity tests for harmful/genotoxic impuri-ties. However, such tests would increase users’ burden because of redundancy in cases where the materials, manufacturing process, and supply chain are appropriately controlled. In such cases, the JP addresses this diversification of the environment around drugs through General Notices 12 (“Manufacture”) and 35 (“Potential Adulteration”). The development of testing methods for harmful/genotoxic impurities as a general chapter in the JP will make these General Notices effective and will help users to prevent contamination with harmful/genotoxic impurities.
In the present study, we identified the standards listed in common in the JP, Ph. Eur., and USP–NF, as well as the stan-dards specific to each pharmacopoeia. Furthermore, we ana-lyzed the features of standards listed in each pharmacopoeia depending on the type of product and standards. Moreover, we identified standards that should be included in the JP and those that should be selected for further coordination among the pharmacopoeias, based on our study results. We anticipate that our findings will help pharmacopoeias worldwide to de-velop strategies for harmonization and/or the convergence of international pharmacopoeial standards, which will improve patient access to drugs in a globalized market.
ConclusionIn this study, we identified the standards listed in common
among the JP, Ph. Eur., and USP–NF, as well as the standards specific to each pharmacopoeia. This study also showed that related regulations, the existence of other compendia, and the policy of each pharmacopoeia influences the listing of monographs in each pharmacopoeia, but harmonization of the pharmacopoeial general chapters that are referred to in the related compendia or regulations will contribute to the fur-ther harmonization of compendial standards and regulations across Japan, Europe, and the United States. We also identi-fied physicochemical testing methods, testing methods for biotechnological/biological products, and testing methods for locally applied products as areas for further collaboration. The comparison data in this study are also expected to be used to develop strategies for future revisions to pharmacopoeias worldwide.
Table 5. Types of Monographs in Official Compendia in Japan
Type of Monographs Compendium Number of Monographs
Drug Substances Japanese Pharmaceutical Codex 2002 Part I 715Drug Products Japanese Pharmaceutical Codex 2002 Part II 171Biological Products Minimum Requirements for Biological Products (Revision Date December 25, 2017) 109Radiopharmaceuticals Minimum Requirements for Radiopharmaceuticals (Revision Date March 29, 2013) 47Herbal Products Japanese Standards for Non-Pharmacopoeial Crude Drugs 2015 75Pharmaceutical Excipients Japanese Pharmaceutical Excipients 2013 489

1312 Vol. 67, No. 12 (2019)Chem. Pharm. Bull.
Acknowledgments We sincerely appreciate the advice and support of past and present co-workers at the Division of Pharmacopoeia and Standards for Drugs of the Pharmaceu-ticals and Medical Devices Agency (PMDA). We would like to express our sincere gratitude to Takao Yamori, Ph.D., of PMDA for giving the opportunity to write this article. We are grateful to Naoyuki Yabana, Ph.D., of PMDA for the initial planning of this study. The authors would also like to ac-knowledge the valuable advice of Haruhiro Okuda, Ph.D. We also thank Jennifer Barrett, Ph.D., for editing a draft of this manuscript.
The views expressed in this manuscript are those of the authors and do not necessarily reflect the views of the Phar-maceuticals and Medical Devices Agency or the Japanese Pharmacopoeia.
Conflict of Interest The authors declare no conflict of interest.
Supplementary Materials The online version of this ar-ticle contains supplementary materials.
References 1) World Health Organization, Annex 1 of WHO Technical Report Se-
ries, 996, 81–129 (2016). 2) World Health Organization, “Index of World Pharmacopoeias
and Pharmacopoeial Authorities.”: ‹http://www.who.int/medicines/ publications/pharmacopoeia/index-of-pharmacopoeias_17012018.pdf›, cited 10 January, 2018.
3) The Law on Securing Quality, Efficacy and Safety of Products including Pharmaceuticals and Medical devices, The Japanese Phar-macopoeia, etc., Article 41 (2015).
4) Federal Food, Drug, and Cosmetic Act. Public Law 115–176; Defini-tions, Section 201(g) (2018).
5) Federal Food, Drug, and Cosmetic Act. Public Law 115–176; Adul-terated Drugs and Devices, Section 501(b) (2018).
6) Directive 2003/63/EC of the European Parliament and of the Coun-cil of 25 June 2001 on the Community code relating to medicinal products for human use. Official Journal of the European Union L-159, 46–94 (2003).
7) Directive 2001/82/EC of the European Parliament and of the Coun-cil of 6 November 2001 on the Community code relating to vet-erinary medicinal products. Official Journal of the European Union L-311, 1 (2009).
8) Pharmacopoeial Discussion Group, “Statement of Harmonization Policy.”: ‹https://www.pmda.go.jp/files/000158075.pdf›, cited June 10, 2019.
9) World Health Organization, “Meeting Reports, Internation-al Meeting of World Pharmacopoeias, Geneva, 29 Febru-ary 2 - March, 2012.”: ‹http://www.who.int/medicines/areas/quality_safety/quality_assurance/resources/iint-meeting-world-pharmacopoeias_final_qas12-467_06032012.pdf?ua=1›, cited March 6, 2012.
10) Matsuhama M., Mizumaru C., Miyazaki S., RSMP, 8, 55–68 (2018).11) The Sub-Committee I of the Western Pacific Regional Forum for
the Harmonization of Herbal Medicines (FHH), “Comparative Studies on Pharmacopoeial Definitions, Requirements and Informa-tion for Crude Drugs among FHH Member Countires in 2007.”: ‹http://www.nihs.go.jp/dpp/FHH/pdf/00_content.pdf›, cited April 14, 2011.
12) Roth L., Adler M., Jain T., Bempong D., Bull. World Health Organ., 96, 378–385 (2018).
13) European Industrial Gases Assosiation AISBL, “Comparison of European, US & Japanese Pharmacopoeia Monographs For Me-
dicinal Gases.”: ‹https://www.eiga.eu/index.php?eID=dumpFile&t=f&f=3295&token=1d87bcc82d5da58bc1235ad0815312237ea22acd›, cited June 10, 2019.
14) “The Japanese Pharmacopoeia,” 17th ed. (JP17), Ministry of Health, Labour and Welfare, Tokyo, 2016.
15) “The European Pharmacopoeia,” 9 ed. (Ph. Eur. 9.0), European Di-rectorate for the Quality of Medicines, Strasbourg, 2016.
16) “The United States Pharmacopeia and The National Formulary,” 39th ed. (USP39-NF34), United States Pharmacopeial Convention, Rockville, MD, 2016.
17) American Chemical Society, “CAS REGISTRY and CAS Regis-try Number FAQs.”: ‹https://www.cas.org/support/documentation/chemical-substances/faqs›, cited June 10, 2019.
18) United States Pharmacopeial Convention, “USP Guidelines for Sub-mitting Requests for Revision to USP-NF: Submission Guideline for Chemical Medicines.”: ‹http://www.usp.org/sites/default/files/usp/document/get-involved/submission-guidelines/chemical_medicines_rfr_guideline_-28apr16.pdf›, cited April 29, 2016.
19) European Directorate for the Quality of Medicines, “Technical Guide for Elaboration of Monographs 7th Edition.”: ‹https://www.edqm.eu/sites/default/files/technical_guide_for_the_elaboration_of_ monographs_7th_edition_2015.pdf›, cited October 1, 2015.
20) Pharmaceuticals and Medical Devices Agency, “Guideline for draft-ing The Japanese Pharmacopoeia 18th Edition (Engl. Transl.).”: ‹http://www.pmda.go.jp/rs-std-jp/standards-development/jp/0003.html›, cited January 18, 2017.
21) International Council for Harmonisation of Technical Require-ments for Pharmaceuticals for Human Use, “ICH HARMONISED TRIPARTITE GUIDELINE, IMPURITIES IN NEW DRUG SUBSTANCES – Q3A(R2).”: ‹http://www.ich.org/fileadmin/Public_ Web_Site/ICH_Products/Guidelines/Quality/Q3A_R2/Step4/Q3A_ R2__Guideline.pdf›, cited October 25, 2006.
22) International Council for Harmonisation of Technical Require-ments for Pharmaceuticals for Human Use, “IMPURITIES IN NEW DRUG PRODUCTS – Q3B(R2).”: ‹http://www.ich.org/filead-min/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3B_R2/Step4/Q3B_R2__Guideline.pdf›, cited June 2, 2006.
23) International Council for Harmonisation of Technical Require-ments for Pharmaceuticals for Human Use, “ICH HARMONISED GUIDELINE, IMPURITIES FOR RESIDUAL SOLVENTS – Q3C(R6).”: ‹http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3C/Q3C__R6___Step_4.pdf›, cited October 20, 2016.
24) International Council for Harmonisation of Technical Require-ments for Pharmaceuticals for Human Use, “ICH HARMONISED GUIDELINE, GUIDELINE FOR ELEMENTAL IMPURITIES – Q3D.”: ‹http://www.ich.org/fileadmin/Public_Web_Site/ICH_ Products/Guidelines/Quality/Q3D/Q3D_Step_4.pdf›, cited Decem-ber 16, 2014.
25) Pharmaceuticals and Medical Devices Agency, “JP drafts for public comments, March 1 – May 31, 2018: 2.66 Elemental Impurities - Procedures.”: ‹https://www.pmda.go.jp/files/000223171.pdf›, cited June 10, 2019.
26) Pharmaceuticals and Medical Devices Agency, “JP drafts for public comments, March 1 – May 31, 2018: General Information Elemental Impurities in Drug Products (Engl. Transl.).”: ‹https://www.pmda.go.jp/files/000222966.pdf›, cited June 10, 2019.
27) European Directorate for the Quality of Medicines, “UPDATE ON THE PH. EUR. POLICY ON ELEMENTAL IMPURITIES.”: ‹https://www.edqm.eu/sites/default/f iles/press_release_pheur_ policy_on_elemental_impurities_update_ january_2017.pdf›, cited January 11, 2017.
28) United States Pharmacopeial Convention, Pharmacop. Forum, 43 (2017).
29) United States Pharmacopeial Convention, “Overview of USP Gen-eral Chapters ‹476› and ‹1086›.”: ‹http://www.usp.org/sites/default/

Vol. 67, No. 12 (2019) 1313Chem. Pharm. Bull.
files/usp/document/events-training/workshops/usp-476-1086.pdf›, cited June 10, 2019.
30) United States Pharmacopeial Convention, “USP Guideline for Sub-mitting Requests for Revision to USP-NF.”: ‹http://www.usp.org/sites/default/files/usp/document/get-involved/submission-guidelines/general-information-for-all-submissions.pdf›, cited June 10, 2019.
31) Ministry of Health, Labour and Welfare, “The Basic Principles for Preparation of JP 18th Edition (Engl. Transl.).”:‹https://www.mhlw.go.jp/file/06-Seisakujouhou-11120000-Iyakushokuhinkyoku/jp18kihonsousin.pdf ›, cited October 19, 2016.
32) European Medicines Agency, “Specifications and Control Tests on the Finished Products.”: ‹https://www.ema.europa.eu/en/documents/scientific-guideline/specifications-control-tests-finished-product_en.pdf›, cited June 10, 2019.
33) European Directorate for the Quality of Medicines, “Gen-eral principle for Monographs on Finished Products (FPs) con-taining chemically defined active substances.”: ‹https://www.edqm.eu/sites/default/files/general_principles_for_monographs_ on_finished_products_ june_2017_e.pdf›, cited June 1, 2017.
34) Keitel S., “Finished Product Monographs in the European Pharma-copoeia.”: ‹http://www.biopharminternational.com/finished-product-monographs-european-pharmacopoeia?pageID=1›, cited June 1, 2014.
35) “The Minimum Requirements for Biological Products,” Ministry of Health, Labour and Welfare, Tokyo, 2018.
36) “General Biological Products Standards. Code of Federal Regula-tions,” Part 610, Title 21, 2018.
37) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1164 (2010).
38) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1168 (2010).
39) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1168 (2010).
40) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1169 (2010).
41) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1170 (2010).
42) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1170 (2010).
43) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1184 (2010).
44) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1185 (2010).
45) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1187 (2010).
46) United States Pharmacopeial Convention, Pharmacop. Forum, 36, 1215 (2010).
47) United States Pharmacopeial Convention, Pharmacop. Forum, 36,
1218 (2010).48) United States Pharmacopeial Convention, Pharmacop. Forum, 36,
1501 (2010).49) United States Pharmacopeial Convention, Pharmacop. Forum, 36,
1503 (2010).50) United States Pharmacopeial Convention, Pharmacop. Forum, 37
(2011).51) United States Pharmacopeial Convention, Pharmacop. Forum, 37
(2011).52) United States Pharmacopeial Convention, “Herbal Medicines Com-
pendium.”: ‹https://hmc.usp.org›, cited June 10, 201953) “Japanese Pharmaceutical Excipients,” Ministry of Health, Labour
and Welfare, Tokyo, 2018.54) “The Minimum Requirements for Radiopharmaceuticals (Engl.
Transl.),” Ministry of Health, Labour and Welfare, Tokyo, 2013.55) International Council for Harmonisation of Technical Re-
quirements for Pharmaceuticals for Human Use, “ICH HARMONISED TRIPARTITE GUIDELINE, SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR NEW DRUG SUBSTANCES AND NEW DRUG PRODUCTS: CHEMICAL SUBSTANCES– Q6A.”: ‹https://www.ich.org/filead min/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6A/Step4/Q6Astep4.pdf›, cited October 6, 1999.
56) International Council for Harmonisation of Technical Re-quirements for Pharmaceuticals for Human Use, “ICH HARMONISED TRIPARTITE GUIDELINE, SPECIFICATIONS: TEST PROCEDURES AND ACCEPTANCE CRITERIA FOR BIOTECHNOLOGICAL/BIOLOGICAl PRODUCTS– Q6B.”: ‹https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q6B/Step4/Q6B_Guideline.pdf›, cited March 10, 1999.
57) European Federation of Pharmaceutical Industries and Asso-ciations, “EFPIA Position Paper on the Impact of Non-harmonised Requirements in Local Pharmacopoeias and Opportunities to Promote Alignments of Public Standards.”: ‹https://www.efpia.eu/media/412590/efpia-position-paper-on-non-harmonised-require-ments-in-local-pharmacopoeias.pdf›, cited June 10, 2019.
58) European Directorate for the Quality of Medicines, “New mono-graph for Infliximab concentrated solution: the first monograph on monoclonal antibody in the Ph. Eur.”: ‹https://www.edqm.eu/en/news/new-monograph-infliximab-concentrated-solution-first-mono-graph-monoclonal-antibody-ph-eur›, cited 14 December, 2017.
59) European Directorate for the Quality of Medicines, Infliximab con-centrated solution (2928), Pharmeuropa, 28 (2016).
60) Food and Drug Administration, “Formal Meetings Between FDA and ANDA Applicants of Complex Products Under GDUFA Guid-ance for Industry —Draft Guidance—”: ‹https://www.fda.gov/media/107626/download›, cited June 10, 2019.





























