Embed Size (px)
Citation preview

ORIGINAL RESEARCH COMMUNICATION
Hypoxia-Inducible Factor 1 Regulates Heatand Cold Pain Sensitivity and Persistence
Maike Kanngiesser,1 Norbert Mair,2 Hee-Young Lim,1 Katja Zschiebsch,1 Johanna Blees,3
Annett Haussler,1 Bernhard Brune,3 Nerea Ferreiros,1 Michaela Kress,2 and Irmgard Tegeder1
Abstract
Aims: The present study assessed the functions of the transcription factor hypoxia-inducible factor (HIF) insensory neurons in models of acute, inflammatory, ischemic, and neuropathic pain. The alpha subunit, HIF1a,was specifically deleted in neurons of the dorsal root ganglia by mating HIF1afl/fl mice with SNScre mice.Results: SNS-HIF1a - / - mice were more sensitive to noxious heat and cold pain stimulation than were HIF1afl/fl
control mice. They also showed heightened first-phase nociceptive responses in the formalin and capsaicin testswith increased numbers of cFos-positive neurons in the dorsal horn, and intensified hyperalgesia in early phasesafter paw inflammation and hind limb ischemia/reperfusion. The behavioral cold and heat pain hypersensi-tivity was explained by increased calcium fluxes after transient receptor potential channel activation in primarysensory neurons of SNS-HIF1a - / - mice and lowered electrical activation thresholds of sensory fibers. SNS-HIF1a - / - mice however, developed less neuropathic pain after sciatic nerve injury, which was associated withan abrogation of HIF1-mediated gene up-regulation. Innovation: The results suggest that HIF1a is protective interms of acute heat and cold pain but in case of ongoing activation in injured neurons, it may promote thedevelopment of neuropathic pain. Conclusion: The duality of HIF1 in pain regulation may have an impact on theside effects of drugs targeting HIF1, which are being developed, for example, as anticancer agents. Specifically,in patients with cancer neuropathy, however, temporary HIF1 inhibition might provide a welcome combinationof growth and pain reduction. Antioxid. Redox Signal. 00, 000–000.
Introduction
Chronic pain is a major and still poorly understoodhealth problem. Particularly, nerve injury may cause
chronic pain, which is often resistant to available therapeutics.It is still unclear why some people do not recover from neu-ropathic pain even if the injured peripheral nerve regenerates.Recent studies suggest that alterations of the redox status ininjured neurons and surrounding glia may be specificallyimportant for the development of nerve injury-evokedchronic pain (21, 22). The transcription factor, hypoxia-inducible factor 1 (HIF1) is a key regulator of redox andoxygen homeostasis, which facilitates adaptation to oxygendeficiency and other redox stresses (50), and HIF1 dysfunctions
Innovation
Therapeutic interruption of a tetrahydrobiopterin (BH4)-nitric oxide (NO)-hypoxia-inducible factor 1 (HIF1) activa-tion circle may reduce neuropathic pain. However, due tothe duality of HIF1 in nociception, targeting HIF1 for paintreatment or pain prevention likely would not selectivelyprovide the desired pain relief. The data suggest that drugstargeting HIF1, for example, for cancer treatment may causeside effects in terms of heat and cold pain sensitivity.However, in patients with cancer neuropathy, HIF1 inhibi-tors might provide a welcome combination of growth in-hibition and pain reduction.
1Pharmazentrum Frankfurt/ZAFES, Institute of Clinical Pharmacology, Goethe-University Hospital Frankfurt, Frankfurt, Germany.2Department of Physiology, University of Innsbruck, Innsbruck, Austria.3Institute for Biochemistry I, Goethe-University of Frankfurt, Frankfurt, Germany.
ANTIOXIDANTS & REDOX SIGNALINGVolume 00, Number 00, 2013ª Mary Ann Liebert, Inc.DOI: 10.1089/ars.2013.5494
1

contribute to the development of neurodegenerative diseases(37). This suggests that it may play a role in the neuronal ad-aptations to nerve injury and the development of chronic pain.
HIF1 is a heterodimer that is composed of HIF1b, whichis constitutively expressed, and HIF1a, which is stabilizedon demand. In nonhypoxic conditions, HIF1a is subject tooxygen-dependent hydroxylation and subsequent ubiquiti-nation, via the von Hippel Lindau (VHL) E3 ubiquitin ligase,and it then undergoes rapid proteasomal degradation (50). Inaddition to hypoxia, iron chelators, cobalt chloride, cytokinessuch as interleukin-1, reactive oxygen species (ROS), and ni-tric oxide (NO) can mimic a hypoxic response by stabilizationof HIF1a (35, 49).
Particularly, NO has emerged as an important signalingmolecule in nociceptive pathways (47) with the potential forboth enhancing and reducing hyperexcitability (19), depend-ing on the cellular redox status and its source. HIF1a may beone of its target molecules (7), by which it mediates its effectson gene transcription (61) and calcium fluxes (36, 58). HIF1a,in turn, likely controls NO production via regulation of theexpression of NO synthases (NOSs) (32) or GTP cyclohy-drolase 1 (GCH1), which is the rate-limiting enzyme for syn-thesis of the NOS-coenzyme, tetrahydrobiopterin (BH4). NOalso activates soluble guanylylcylase and directly nitrosylatescritical cysteine residues in target proteins, including HIF1a(31). In terms of nociception, chemical inhibitors of NOS (34)and GCH1 (54) reduce neuropathic and inflammatory pain inrodents. However, considering the potential neuroprotectivefunctions of HIF1a, this may be a double-edged sword.
It is still unclear as to what extent alterations of the redoxbalance contribute to the development of chronic pain. Re-cently, deletion of the NADPH oxidases, NOX4 and NOX2,were found to attenuate neuropathic pain after sciatic nerveinjury in mice (22, 26). Since these NOXs are target moleculesof HIF1a (13, 60), it is conceivable that HIF1a is an upstreamregulator of neuropathic pain. To address the specific func-tions of HIF1a in nociception, we deleted it specifically inperipheral nociceptive neurons of the dorsal root ganglia(DRG) and assessed nociceptive behavior, calcium fluxes,sensory fiber activation, and gene regulation in chemicallyevoked, neuropathic, inflammatory, and ischemic nociceptivemodels. HIF1a deficiency resulted in heightened acute heatand cold nociception associated with increased transient re-ceptor potential (TRP) channel sensitivity, but HIF1a defi-ciency protected the mice from nerve injury-evoked thermalhyperalgesia, likely because HIF1a-mediated up-regulation ofpro-nociceptive genes, including metalloproteinase-2(MMP2) (24) and GCH1, was abolished.
Results
Selective deletion of HIF1a in sensory neurons
HIF1 functions are mainly regulated at the protein level.However, we also observed a late increase of HIF1 mRNA inDRG after nerve injury (Fig. 1A). In situ hybridization usingHIF1a-specific riboprobes and subsequent immunostainingrevealed HIF1a mRNA in different types of neurons, includ-ing small isolectin B4-positive neurons with unmyelinatedfibers, NF200-positive large neurons with myelinated fibers,and in calcitonin gene-related peptide (CGRP)-positive pep-tidergic neurons (Fig. 1B). To assess the functions, we deletedthe alpha subunit, HIF1a in Nav1.8/SNS-positive DRG neu-
rons per cre-loxP-mediated techniques. Quantitative real-timepolymerase chain reaction (RT-PCR) and in situ hybridizationwere used for confirmation. Quantitative RT-PCR showed asignificant reduction of about 60% of HIF1a mRNA in theDRGs of SNS-HIF1a - / - mice as compared with HIF1afl/fl
littermate control mice (Fig. 1C), which is in line with the ex-pected percent reduction based on the number of Nav1.8 (SNS)-positive neurons in the DRGs. In situ mRNA hybridizationrevealed the cre-loxP-mediated HIF1a deletion in the DRG (Fig.1D) but as expected, not in the spinal cord (Fig. 1C, E).
Enhanced acute heat and cold nociceptionin SNS-HIF1a - / - mice
SNS-HIF1a - / - mice showed heat and cold pain hyper-sensitivity at baseline and in the early phases after nerve in-jury (Fig. 2A), paw inflammation (Fig. 2B), or hind limbischemia/reperfusion (I/P) injury (Fig. 2C). However, SNS-HIF1a - / - mice showed only mild increases of the baselinehypersensitivity, whereas the HIF1afl/fl control mice devel-oped strong hyperalgesia so that during the chronic phasesdifferences between genotypes were no longer evident orwere even reversed in case of the nerve injury, that is, HIF1adeficiency in the sensory neurons provided protection againstchronic neuropathic pain. This was specific for the nerve in-jury model.
Mechanical. There was no difference of mechanical no-ciception, neither at baseline nor after stimulation by nerveinjury, inflammation, or I/P. This was obvious in the dynamicAesthesiometer tests (Fig. 2, left panel) as well as in the re-sponse curves toward standard von Frey hairs (Supplemen-tary Fig. S1; Supplementary Data are available online atwww.liebertpub.com/ars).
Heat (Fig. 2 middle panel). SNS-HIF1a - / - mice showedreduced paw withdrawal latencies at baseline (Fig. 2A–C) andin the early hours after complete Freund’s adjuvant (CFA)injection (Fig. 2B) and in the early days after I/P (Fig. 2C),indicating enhanced acute heat nociception. However, in theSNS-HIF1a- / - mice, there was only a minor further decreaseof paw withdrawal latencies compared with their baselinelevels. These differences in the responses toward the injurieswere reflected by significant differences in repeated measuresanalysis of variances (rm-ANOVAs) for the between subjectfactor ‘‘genotype’’ (spared nerve injury [SNI]: F = 5.718, de-grees of freedom [df] 1, p = 0.0314; for CFA: F = 3.714, df 1,p = 0.0745, and for I/P: F = 5.086, df 1, p = 0.0406). Post hocstatistics are shown in Figure 2.
Cold (Fig. 2 right panel). SNS-HIF1a - / - mice showedcold pain hypersensitivity at baseline and in the early daysafter I/P. CFA injection evoked only mild acetone hypersen-sitivity, and no significant difference between genotypes wasobserved. However, after nerve injury, cold hypersensitivityincreased more strongly in HIF1afl/fl mice as compared withSNS-HIF1a- / - mice (rm-ANOVA for ‘‘genotype’’ SNI: F =4.673, df 1, p = 0.0485).
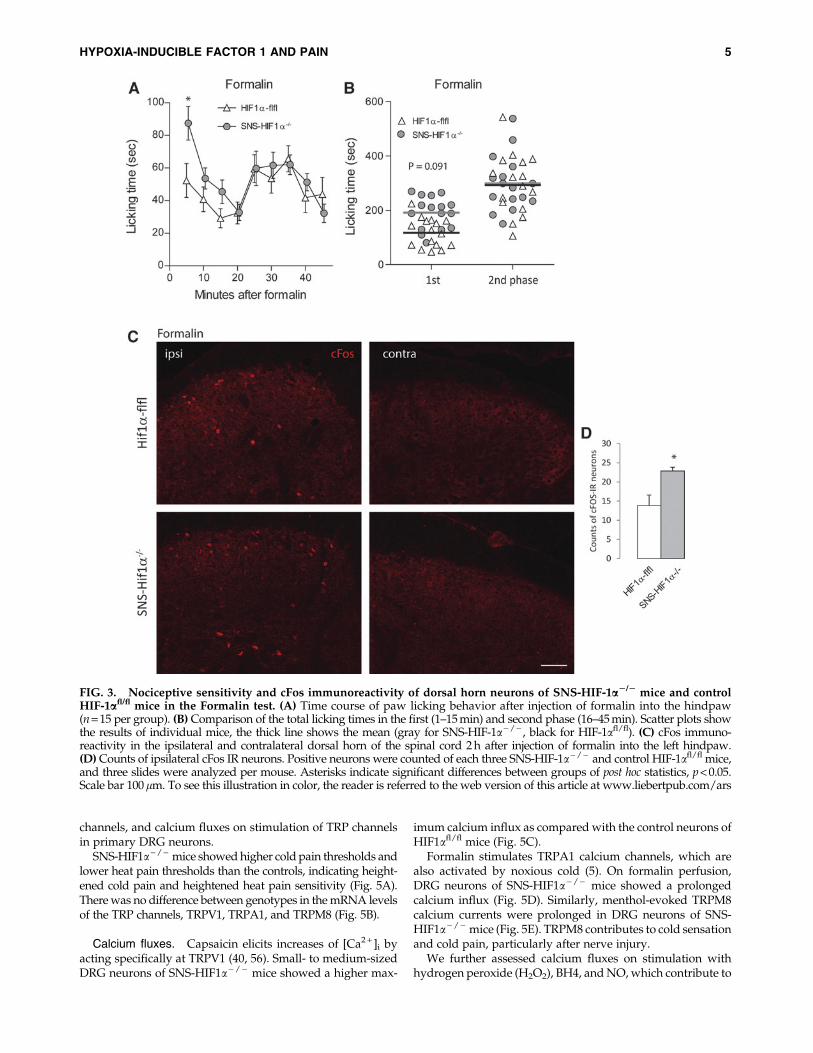
Enhanced early formalin and capsaicin-evokednociception in SNS-HIF1a - / - mice
Due to the enhanced acute heat and cold pain sensitivityin the SNS-HIF1a - / - mice, we assessed the nociceptive
2 KANNGIESSER ET AL.

responses on an injection of formalin (Fig. 3) and capsaicin(Fig. 4) into the hind paw, which stimulate transient receptorpotential channel subfamily A, member 1 (TRPA1) and tran-sient receptor potential channel subfamily V, and member 1(TRPV1) receptors, respectively. SNS-HIF1a - / - mice spentmore time licking the injected hind paw than the controls inthe early phase in both tests (Fig. 3A, B for formalin and Fig.4A, B for capsaicin). The enhanced nociceptive responses werereflected by higher numbers of cFos immunoreactive neuronsin the ipsilateral dorsal horn in SNS-HIF1a - / - mice after
formalin (Fig. 3C, quantification 3D) and after capsaicin(Fig. 4C, D).
Expression and calcium fluxes of TRP channelsin HIF1-a-deficient DRGs
The acute heat and cold nociceptive hypersensitivity and thehyperexcitability of nociceptive terminals in SNS-HIF1a- / -
mice suggested heightened responses of TRPV1 and TRPA1channels in SNS-HIF1a - / - mice. We, therefore, assessed thenociceptive temperature thresholds, expression of TRP
FIG. 1. Characterization of HIF1a mRNA expression in the DRG and spinal cord. (A) Time course of HIF1a mRNA inDRGs in naıve mice and after sciatic nerve injury. Triplicate analysis of pooled samples of three mice per sample. (B) In situhybridization of HIF1a mRNA in the DRGs of HIF-1afl/fl mice and post in situ immunostaining for isolectin B4, NF200, andCCGRP. HIF1a mRNA was expressed in different types of neurons. (C) Quantitative RT-PCR of HIF1a mRNA in DRGs andspinal cord in SNS-HIF-1a- / - and control HIF-1afl/fl mice after SNI showing the DRG specific deletion. Tissue of n = 8 mice. (D)In situ hybridization of HIF1a mRNA in the DRGs of SNS-HIF-1a- / - and HIF-1afl/fl mice showing the cre-loxP mediated deletionof HIF1a in SNS-HIF-1a- / - mice. (D) In situ hybridization of HIF1a mRNA in the dorsal and ventral horns of the spinal cord ipsi-and contralateral of the sciatic nerve injury. HIF1a mRNA was abundant in dorsal and ventral horn neurons with mild SNI-mediated ipsilateral up-regulation in injured motor neurons without differences between genotypes. In situs show representativeresults of four mice per group. Asterisks indicate statistically significant results with p < 0.05. Scale bars 50lm in (B, D) and 100 lmin (E). DRG, dorsal root ganglia; SNI, spared nerve injury; HIF1a, hypoxia inducible factor 1 alpha; RT-PCR, real-time polymerasechain reaction. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 3

FIG. 2. Nerve injury (A) inflammation (B), and I/P (C) evoked nociceptive hypersensitivity toward mechanical, heat, andcold stimuli in SNS-HIF-1a2/2 mice and control HIF-1afl/fl mice. (A) Withdrawal latencies on mechanical stimuli in dynamicAesthesiometer tests and upon heat stimulation in Hargreaves tests and cold evoked withdrawal reactions in Acetone tests ofSNS-HIF-1a - / - and control HIF-1afl/fl mice before and after injury of the sciatic nerve in the SNI model. (B) Nociceptivebehavior before and after injection of CFA into one hindpaw. (C) Nociceptive behavior before and after I/P injury of the hindlimb. Data are means – sem of n = 8 mice in each group (16 for baseline acetone). ANOVAs for repeated measurementsrevealed statistically significant differences for heat and cold evoked nociception. Asterisks indicate significant differencesbetween groups of post hoc statistics, p < 0.05. CFA, complete Freund’s adjuvant; ANOVA, analysis of variance; I/P, ischemia/reperfusion.
4
channels, and calcium fluxes on stimulation of TRP channelsin primary DRG neurons.
SNS-HIF1a- / - mice showed higher cold pain thresholds andlower heat pain thresholds than the controls, indicating height-ened cold pain and heightened heat pain sensitivity (Fig. 5A).There was no difference between genotypes in the mRNA levelsof the TRP channels, TRPV1, TRPA1, and TRPM8 (Fig. 5B).
Calcium fluxes. Capsaicin elicits increases of [Ca2 + ]i byacting specifically at TRPV1 (40, 56). Small- to medium-sizedDRG neurons of SNS-HIF1a - / - mice showed a higher max-
imum calcium influx as compared with the control neurons ofHIF1afl/fl mice (Fig. 5C).
Formalin stimulates TRPA1 calcium channels, which arealso activated by noxious cold (5). On formalin perfusion,DRG neurons of SNS-HIF1a - / - mice showed a prolongedcalcium influx (Fig. 5D). Similarly, menthol-evoked TRPM8calcium currents were prolonged in DRG neurons of SNS-HIF1a - / - mice (Fig. 5E). TRPM8 contributes to cold sensationand cold pain, particularly after nerve injury.
We further assessed calcium fluxes on stimulation withhydrogen peroxide (H2O2), BH4, and NO, which contribute to
FIG. 3. Nociceptive sensitivity and cFos immunoreactivity of dorsal horn neurons of SNS-HIF-1a2/2 mice and controlHIF-1afl/fl mice in the Formalin test. (A) Time course of paw licking behavior after injection of formalin into the hindpaw(n = 15 per group). (B) Comparison of the total licking times in the first (1–15 min) and second phase (16–45 min). Scatter plots showthe results of individual mice, the thick line shows the mean (gray for SNS-HIF-1a- / - , black for HIF-1afl/fl). (C) cFos immuno-reactivity in the ipsilateral and contralateral dorsal horn of the spinal cord 2 h after injection of formalin into the left hindpaw.(D) Counts of ipsilateral cFos IR neurons. Positive neurons were counted of each three SNS-HIF-1a- / - and control HIF-1afl/fl mice,and three slides were analyzed per mouse. Asterisks indicate significant differences between groups of post hoc statistics, p < 0.05.Scale bar 100lm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 5

nociceptive signaling by activation of redox-sensitive TRPchannels and N and P/Q type voltage-gated calcium chan-nels (36, 54, 58) and by regulation of intracellular calciumrelease (11).
H2O2 evoked an increase of [Ca2 + ]i in small- to medium-sized DRG neurons, which was increased in SNS-HIF1a - / -
mice in both maximum and duration (Fig. 5E). Similarly, BH4evoked stronger and longer lasting increases in [Ca2 + ]i inDRG neurons of SNS-HIF1a - / - mice compared with controlneurons (Fig. 5F) and NO evoked a double peak of [Ca2 + ]i.The second raise, likely showing release from intracellularstores, was stronger in DRG neurons of SNS-HIF1a - / - mice(Fig. 5G). Statistical results are presented in Figure 5.
Excitability of nociceptive nerve fibersin SNS-HIF1a - / - mice
To assess whether the enhanced sensitivity to acute noci-ceptive stimuli was associated with a hypersensitivity ofperipheral nerve terminals and fibers, we performed electro-physiological recordings on peripheral fibers of DRG neu-rons in saphenous nerve-skin preparations isolated fromSNS-HIF1a- / - mice and HIF1afl/fl mice (Fig. 6). Electricalthresholds ranged from 1 to 70 V. The proportion of fiberswith low activation thresholds (0.1–5 and 5–10 V) was higherin SNS-HIF1a - / - mice compared with HIF1afl/fl (Fig. 6A, n.s.p = 0.176). The cumulative electrical stimulation curves were
FIG. 4. Nociceptive sensitivity and cFos immunoreactivity of dorsal horn neurons of SNS-HIF-1a2/2 mice and controlHIF-1afl/fl mice in the Capsaicin test. (A) Time course of paw licking behavior after injection of capsaicin into the hindpaw(n = 18 per group). (B) Comparison of the total licking times in the first (1–15 min) and second phase (16–45 min). Scatter plots showthe results of individual mice, the thick line shows the mean (gray for SNS-HIF-1a- / - , black for HIF-1afl/fl). (C) cFos immuno-reactivity in the ipsilateral and contralateral dorsal horn of the spinal cord 2 h after injection of capsaicin into the left hindpaw.(D) Counts of ipsilateral cFos IR neurons. Positive neurons were counted of each three SNS-HIF-1a- / - and control HIF-1afl/fl mice,and three slides were analyzed per mouse. Asterisks indicate significant differences between groups of post hoc statistics, p < 0.05.Scale bar 100 lm. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
6 KANNGIESSER ET AL.

FIG. 5. TRP channel expression and function in DRG neurons of SNS-HIF-1a2/2 and control HIF-1afl/fl mice. (A)Temperature thresholds for cold and heat pain. SNS-HIF-1a - / - mice showed enhanced sensitivity for both heat (loweredthreshold) and cold (elevated threshold, i.e., reaction already at higher temperatures). (B) Fold mRNA expression ofTRPV1, TRPA1, and TRPM8 in DRGs of SNS-HIF-1a - / - and control HIF-1afl/fl mice relative to the mean of TRPV1 of allsamples set to 1 (n = 5 mice per group). (C–H) Calcium imaging in primary DRG neurons of adult SNS-HIF-1a - / - (red)and HIF-1afl/fl (blue) mice stimulated by perfusion with 500 nM capsaicin (C), 0.01% formalin (D), 250 lM menthol (E),1 mM H2O2 (F), 100 lM BH4 (G), and 500 lM NO donor, DEA-NO (H). [Ca2 + ]i was measured fluorometrically in neuronsloaded with fura-2 as absorbance ratio at 340–380 nm (DF 340/380). The figures show the fold changes of DF as comparedwith the baseline. The thick line is the mean, the shaded light blue (HIF-1afl/fl) and light red (SNS-HIF-1a - / - ) areas showthe standard deviation. Neurons (numbers in the figures) of four to six mice were analyzed in each group. t-Test results forthe maximum (Max) and AUCs are shown in the figures, p < 0.05 for all tests. AUCs, area under the curves; BH4,tetrahydrobiopterin; NO, nitric oxide. To see this illustration in color, the reader is referred to the web version of this articleat www.liebertpub.com/ars
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 7

significantly shifted to lower thresholds. The EC50 was 7.72 Vfor HIF1afl/fl (95% confidence interval [CI]: 7.28–8.18) and6.54 V for SNS-HIF1a- / - (95% CI: 6.01–7.05). Correspond-ingly, a significantly higher proportion of fibers from SNS-HIF1a - / - mice had high conduction velocities ( > 0.5 m/s)(Fig. 6B, p = 0.021). In addition, more fibers were activated bycold (n.s.), whereas response rates toward heat did not differbetween genotypes (Fig. 6C).
Expression of HIF1a regulated genesin the DRGs after nerve injury
The enhanced TRP channel sensitivity provided an expla-nation for the observed acute thermal pain hypersensitivity inSNS-HIF1a - / - mice. However, the relative protection againstthe nerve injury evoked nociceptive hypersensitivity duringthe chronic phase that likely depended on different long-lasting adaptations. We, therefore, analyzed HIF1a-mediatedgene regulation in the DRGs using a commercial HIF1a-cDNAarray (Fig. 7A). At baseline, there were only minor differencesbetween genotypes. In addition to HIF1a itself, mRNA levelsof the glucose transporter Glut3, the uncoupling protein(UCP3), and platelet-derived growth factor alpha were re-duced in the DRGs of naıve SNS-HIF1a - / - mice. However,after SNI, there was a strong up-regulation of several genes inthe control mice, which did not occur in SNS-HIF1a - / - mice.The strongest increases applied to HIF1a itself, erythropoie-tin (EPO), vascular endothelial growth factor (VEGF), pro-survival factors including B-cell lymphoma 2 (Bcl2) and ringfinger protein 7 (alias sensitive to apoptosis, SAG), the histoneacetyltransferase EP300, which acts as a coactivator of HIF1a,and for genes, which play a role in myelination and Schwanncell or satellite cell function, including N-myc downstream-regulated gene 1 (NDRG1), MMP2, and cathepsin D (CTSD).After CFA injection, fewer genes were differentially regulatedbetween SNS-HIF1a - / - and control mice (SupplementaryFig. S2). Only EPO, EP300, and CTSD showed a stronger in-crease in HIF1afl/fl mice.
The number of neurons with up-regulation and nucleartranslocation of the injury marker activating transcriptionfactor 3 (ATF-3) did not differ between genotypes (Fig. 7B),and the percentages were within the expected ranges for theSNI model, confirming that the axonal injury was equallyperformed. However, we observed differences in the nerveinjury-evoked regulation of GCH1 (Fig. 7C). GCH1 mRNAand protein were up-regulated in the DRGs after sciatic nerveinjury in HIF1afl/fl mice (Fig. 7C, D). This up-regulation wassignificantly weaker in SNS-HIF1a - / - mice (Fig. 7C, D), andit was associated with reduced biopterin levels after nerveinjury (Fig. 7E). Biopterin did not increase in the DRGs in theCFA model, and there was no difference between genotypes.Neopterin concentrations in the DRGs were below quantifi-cation limit (0.1 ng/ml) in SNS-HIF1a - / - mice and rangedfrom 0.219 to 0.278 ng/ml in the control mice.
Discussion
In the present study, we show that mice deficient in HIF1ain primary nociceptive neurons of the DRG have heightenedheat and cold pain on acute noxious, inflammatory, and is-chemic stimuli but reduced nerve injury-evoked chronicneuropathic pain. The acute hypersensitivity was associatedwith enhanced calcium fluxes in DRG neurons on stimulationof heat, cold, and redox-sensitive TRP channels and on acti-vation of BH4/NO signaling. In addition, electrical stimula-tion of the saphenous nerve revealed an increase of A-fiberresponses with low activation thresholds and high conduc-tion velocities. On the other hand, the relative protectionagainst neuropathic pain was associated with a reduced up-regulation of a number of typical HIF1 target genes. In re-verse, this would mean that HIF1 protects against early phaseheat and cold pain but promotes chronic neuropathic pain ifits activation is persistent and results in regulations of HIF1-responsive genes. Such dual functions of HIF1 are known invarious systems and may arise from differential activationpathways and differential selections of target genes. The
FIG. 6. Electrophysiological recordings from sensory fibers of the saphenous nerve in an ex vivo skin-nerve preparationderived from SNS-HIF-1a2/2 and control HIF-1afl/fl mice (n510 mice in each group). Fibers were identified by electricalthresholds and conduction velocities. (A) Percentages of fibers responding at the specified electrical stimulation range. Theproportion of fibers responding at low activation voltages was increased in SNS-HIF-1a - / - . The EC50 levels of cumulativeresponse curves were 7.72 V for HIF-1afl/fl and 6.54 V for SNS-HIF-1a- / - , p < 0.05. (B) Percentages of fibers responding atthe specified range of conduction velocities. Fisher’s exact tests showed a significant shift toward a high velocity in SNS-HIF-1a - / - . (C) Percentages of fibers responding on cold or heat stimulation (Fisher’s exact tests, n.s.). The number of analyzedfibers was n = 55 for HIF-1afl/fl and 43 for SNS-HIF-1a - / - . p < 0.05 for all tests.
8 KANNGIESSER ET AL.

FIG. 7. Expression of HIF1-regulated genes. (A) Relative intensity of mRNA expression of HIF1-regulated genes in theipsilateral L4/5 DRGs of SNS-HIF-1a - / - and control HIF-1afl/fl mice at baseline and 21 days after SNI. Samples of pooledDRG tissue of each four animals were analyzed in duplicate in a HIF1 cDNA plate array. Intensities were normalized tob-actin, which was set to 100%. Respective results 7 days after CFA injection are shown in Supplementary Figure S2. (B)Immunofluorescence analysis of ATF-3 in L5 DRGs of naıve mice and 3 days after SNI in SNS-HIF-1a- / - and HIF-1afl/fl mice.Nuclear ATF3 is a marker for neurons with injured axons. The fraction did not differ between genotypes. (C) mRNA analysisand (D) Western Blot analysis of GCH1 expression in the DRGs of naıve and SNI treated SNS-HIF-1a- / - and HIF-1afl/fl mice.HSP90 was used as a loading control. Example and quantitative result of n = 4 mice per group and time point. (E) Biopterinconcentrations in DRGs of naıve (N) and nerve-injured SNS-HIF-1a- / - and control HIF-1afl/fl mice analyzed with liquidchromatography coupled with tandem mass spectrometry, n = 4 mice per group. Asterisks indicate significant differencesbetween genotypes with p < 0.05. GCH1, GTP cyclohydrolase 1; ATF-3, activating transcription factor 3; HSP90, heat shockprotein 90. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 9

duality poses problems to therapeutic approaches targetingHIF1 (30, 59). Experiments in cells have shown that only thesequential HIF1 sensitization and then desensitization areable to rescue cells from cell death (16), suggesting thatswitching it off may be as important as switching it on. Po-tential signaling pathways are depicted in Figure 8.
The enhanced TRP channel sensitivity of the primary af-ferent neurons in SNS-HIF1a - / - mice agrees with previousreports showing that hypoxia increases TRPV1 sensitivity (27,44) and activates TRPA1 (52). The latter functions as an oxy-gen sensor in that the fine tuning of its activity is mediatedthrough prolylhydroxylases, which either inhibit or activateTRPA1 depending on the oxygen status (52). Besides thesetypical ‘‘pain-TRPs,’’ hypoxia or ROS, including H2O2, maystimulate TRPC channels, which are localized on the nerveterminals, and, recently, TRPC5 has been recognized as a‘‘cold pain channel’’ (62). In agreement, we observed intensi-fied calcium fluxes in HIF1a-deficient neurons on stimulationwith H2O2, which activates TRPC channels (42) and redox-sensitive TRPM2 (23, 51), and elicits nociceptive withdrawalreactions after an injection into the hindpaw (4, 45). HIF1amay counteract H2O2 through transcriptional regulations of
sestrin and other redoxins (18, 39). Presumably, HIF1a defi-ciency may increase the effect of ROS on TRP channels be-cause of the lack of counterbalancing regulations. However,we do not know exactly how HIF1a affects TRP channels at amolecular level. Considering its major function as a tran-scription factor, one might expect alterations of TRP mRNAlevels. This was, however, not the case, suggesting that HIF1may rather impact one of the many regulatory mechanismswhich determine TRP channel membrane insertion or channelgating. Both mechanisms are redox sensitive (46).
The effects of SNS-HIF1a - / - deficiency on acute nocicep-tion were mild but concisely pointed to an increase of acuteheat and cold nociception and early-phase nociceptive hy-persensitivity in various pro-inflammatory models, includingpaw inflammation, I/P, capsaicin, and formalin. The ob-served sensitization of the TRP channels readily explains theacute heat pain hypersensitivity and the enhanced capsaicinand formalin-evoked nociceptive responses in SNS-HIF1a - / -
mice. However, this does not explain the attenuation ofneuropathic pain in the chronic stage after nerve injury. Sincethe benefit of HIF1a deletion in terms of the behavioral re-sponses only occurred after nerve injury and manifested
FIG. 8. Illustration of the potential interactions of HIF1a, ROS, and TRP channels in nociception. Activation of transientreceptor potential channels, TRPV1 and TRPA1, by noxious heat or cold, or other stimuli, including capsaicin, formalin andreactive oxygen species, elicits an influx of calcium through the channel. Calcium then activates various calcium-sensitiveproteins directly or by binding to CaM, which acts as a calcium sensor for CamKII and nNOS. The nociceptive stimulusthereby increases the production of NO and NOx and ROS. A major source of NOx and ROS is an uncoupled NOS, whichresults from an imbalance between NOS-dimers and the NOS coenzyme, BH4. BH4 is produced by GCH1. NO, NOx, andROS can directly modify transient receptor potential, TRP channels through S-nitrosylation or oxidation of crucial cysteineresidues. Particularly, TRPA1 is sensitive to redox modification, whereas TRPV1 likely is more indirectly NO-regulatedthrough S-nitrosylation of various kinases, which phosphorylate the channel, including CamKII and PKC or by redoxmodification of the ATPase N-ethyl-maleimide-sensitive protein (NSF), which disassembles the SNARE complex and drivesmembrane fusion. PKC is activated on nociceptive stimulation and acts as a second messenger of prostaglandins, BK,chemokines, or NGF. These pro-inflammatory mediators are released by inflammatory cells, which are also a major source ofNO and ROS such as H2O2. The latter is an activator of redox sensitive TRPC channels and TRPA1. An inflammatorynociceptive stimulus additionally causes ROS generation through NOX, and COX. In case of a nerve injury, damage tomitochondria substantially contributes to generation of ROS in neurons and Schwann cells, likely overwhelming the detox-ifying capacity of a complicated network of redoxins, SOD, katalases, GSH, and other ROS scavengers. These ROS guardiansusually allow for ROS signaling without causing damage. One of the ROS-sensitive proteins is the HIF1a. During normoxiaand low ROS, HIF1a is hydroxylated at proline residues through PHDs, resulting in ubiquitin binding and degradation ofHIF1a in the proteasome. On hypoxia, PHDs are inactivated and HIF1a is stabilized. NO and ROS also stabilize HIF1a innormoxic conditions directly. HIF1a then translocates to the nucleus, complexes with HIF1b/Arnt and coactivators, includingEP300/CBP and then binds to promoter regions of its target genes to elicit transcription of genes involved in survival, redoxcontrol, apoptosis, inflammation, and angiogenesis. Under physiological conditions, HIF1a likely mainly regulates expressionof anti-oxidative enzymes to counterbalance ROS generated during everyday metabolism. Deficiency thereof may subtlyincrease the ROS load, leading to a stronger or longer-lasting oxidative modification of TRPs and an increase of theirsensitivity. This may constitute a pre-conditioning stimulus in that it forces neurons to adapt to the mild redox stress so thatthey are better prepared to deal with the rush of ROS elicited by injury of the neuron or its axon. A nerve injury evokedoverflow of damaging ROS is caused by mitochondrial damage, uneven up-regulation of GCH1 and nNOS, upregulation ofNOXs, and enhanced lysosomal production and by extracellular ROS, which are released by infiltrating inflammatory cells.Such extracellular ROSs may also contribute to the sensitization of nociceptive nerve terminals during inflammatory condi-tions. However, in contrast to nerve injury, stimulation of nociceptive nerve terminals will not elicit a rush of damaging ROSswithin the neuron but rather a subtle increase of signaling ROS, which are produced, for example, by activated neuronal NOS.Hence, axonal injury has an exceptional pathophysiology with exceptional functions of HIF1 of injured neurons. PersistentHIF1 activation will then stimulate not only transcription of pro-survival genes but also pro-apoptotic and pro-inflammatorygenes, so that the initial benefit of HIF1a activation may turn into a disadvantage. As a result, HIF1a deficient mice developonly mild neuropathic pain after nerve injury despite of the baseline hypersensitivity. Ubi, ubiquitin; GSH, glutathione; GSSG,glutathione disulfide; GSS, glutathione synthase; GSR, glutathione reductase, Trx, thioredoxin; TrxR, thioredoxin reductase;Prdx, peroxiredoxin; VHL, von Hippel Lindau protein; PLC phospholipase C; PIP2, phosphatidylinositol 4,5-bisphosphate;CaM, calmodulin; CamKII, calmodulin kinase II; nNOS, neuronal nitric oxide synthase; NOS, NO synthase; NOx, reactivenitrogen species; ROS, reactive oxygen species; PKC, protein kinase C; BK, bradykinin; NGF, nerve growth factor; H2O2,hydrogen peroxide; NOX, NADPH-oxidases; COX, cyclooxygenases; SOD, superoxide dismutase; PHDs, prolylhydroxylases.To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
‰
10 KANNGIESSER ET AL.

days up to several weeks after the injury, it may be due toHIF1a-mediated gene regulation in the DRGs. In support, weobserved only mild differences in HIF1a target genes atbaseline but multiple after nerve injury and, importantly, theup-regulation was abrogated in SNS-HIF1a - / - mice.
The pain protection provided by HIF1a deficiency wasexclusively observed in the nerve injury model, which is theonly model in which the axon is directly injured and wheredamaging ROS are generated within the injured neuron,likely mainly by mitochondrial damage (33, 43). In the in-flammatory and I/P models, ROS are rather mainly generatedin the tissue surrounding the nerve terminals, for example, byimmune cells or are signaling ROS produced by NOS (55).Transient challenges with tissue-derived ROS may well con-tribute to a sensitization of the nerve terminals, but likely donot cause axonal damage, structural synaptic adaptations, orsubstantial glial activation. The same holds true for signalingROS produced by neuronal NOS.
The nerve injury model, therefore, differs from all othermodels in that the injured neurons lose their peripheral inputand trophic support, generate pathological spontaneous elec-trical activity, and, eventually, die as a consequence of the ax-onal injury (53). The latter is followed by loss and restructuringof synapses, which permanently alters signaling pathways. Theexceptional pathophysiology is reflected by transcriptionalchanges of thousands of genes in the DRGs after sciatic nerveinjury and concomitant glia activation alongside the nocicep-tive signaling pathway (48). The present data suggest that
HIF1a- / - may be a part of these adaptations and may promotethe persistence, specifically of neuropathic pain.
Due to the multitude of HIF1-regulated genes, it is notpossible to ascribe the development of allodynia to a specificone. Likely, the effect results from the sum of transcriptionaldifferences. HIF1a mostly up-regulates genes, which helpsurvive hypoxia or oxidative stress. However, the preventionof cell death also requires its timely deactivation after initialactivation (30). Indeed, we observed an up-regulation of pro-survival genes in HIF1aflfl mice, which did not occur in SNS-HIF1a - / - mice, including Bcl2, ring finger protein 7 (RNF7),EPO, and VEGF. It has been shown that EPO up-regulation inDRG neurons depends on NO-HIF1a signaling (25) and pro-motes regeneration after axonal injury (10, 14). TherapeuticEPO also reduced hyperalgesia in models of diabetic andcisplatin-induced neuropathy (10). However, such neuro-protective effects would only be advantageous for stressed orpartially injured neurons but not for those that are beyondrepair. HIF1a-mediated up-regulation of anti-apoptotic genesmight result in a protraction of the Wallerian degenerationand dying of injured neurons, which amount to about 30% inmouse DRGs after sciatic nerve injury (53). Consequently,pathological firing may also be protracted, but still irre-versible. This idea would agree with a previous studyshowing that HIF1a-deficiency in the brain unexpectedly re-duced ischemia-evoked damage (3).
HIF1a-deficiency may also constitute to some kind of pre-conditioning stimulus, because its deficiency may require the
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 11

development of tolerance toward higher baseline levels ofeveryday metabolic ROS. The adaptation to mild redox stressmay increase the ability to deal with the rush of ROS causedby injury of the neuron or its axon, similar to mild hypoxiathat fortifies against strong hypoxia. Subtly heightened TRPchannel sensitivity may contribute to such a preadaptation,explaining why HIF1a–deficient mice developed only mildneuropathic pain inspite of, or precisely because of, thebaseline hypersensitivity.
More than the typical HIF1a-regulated genes, we werespecifically interested in potential BH4/NOS–HIF1a–BH4/NOS activation circles, because NOS is a major source ofsignaling ROS and a major contributor to nociception. Poly-morphisms in GCH1, the enzyme controlling BH4 andthereby NO synthesis, determine pain sensitivity and persis-tence in humans (9, 54), and both BH4 and NO cause hyper-algesia on local, spinal, and systemic injection (17, 41, 54).They are redox molecules that activate voltage-gated and TRPcalcium channels and increase the intracellular release ofcalcium (20, 58). These effects are likely caused by direct redoxmodification of target proteins (8, 46, 55, 58), which may bemodified via HIF1a-dependent activity or expression of re-doxins (18, 39). The present data show that HIF1a deficiencyprevents nerve injury-evoked up-regulation of GCH1 andreduces BH4 production in the DRGs. Since GCH1 inhibitionattenuates neuropathic pain, it is likely that prevention of itsup-regulation contributed to the antinociceptive benefit ofHIF1a-deletion in the nerve injury model.
Interestingly, we observed some up-regulations of genesthat are mainly expressed in satellite or Schwann cells, in-cluding NDRG1, which plays a major role in myelination (28,38) and MMP2, which contributes to neuropathic pain (24, 29)although the deletion of HIF1a was specific for the DRGneurons. There is no leaky cre-recombinase expression in glialcells in SNScre mice (1); so, the deletion of HIF1a in the neu-rons apparently secondarily altered functions of surroundingglial cells or infiltrating immune cells in the DRGs, possibly viarelease of pro-inflammatory HIF1 target gene products. Ac-tivated glia contributes to the development of neuropathicpain (48) and HIF1a is a crucial regulator or macrophagephysiology (57). Beyond the functions in sensory neurons,HIF1a activation in glial and immune cells may impact neu-ropathic pain.
In summary, our results show that HIF1a protects againstacute heat and cold pain via regulation of TRP channel sen-sitivity. However, its ongoing activation after nerve injurycontributed to the maintenance of neuropathic pain, likelydue to up-regulation of HIF1 target genes and up-regulationof GCH1 and its product, BH4. This would further drive NOproduction and, hence, further HIF1 activation. Loss of con-trol of this positive activation circle may be detrimental.
Materials and Methods
Animals
We generated mice deficient in the alpha subunit ofHIF1 (HIF1a) in peripheral primary sensory neurons (SNS-HIF1a) via cre-loxP-mediated recombination. Mice carryingthe HIF1a flox allele (HIF1afl/fl) were mated with mice ex-pressing cre-recombinase under control of the promoter of thesensory neuron-specific tetrodotoxin-resistant sodium chan-nel SNS/Nav1.8 (SNS-cre) (1, 6). Genotyping was done for the
HIF1a floxed allele (Primer 5¢-3¢: ggagctatctctctagacc, gcagt-taagagcactagttg) and for cre-recombinase as described (1)(Primer 5¢-3¢: gaaagcagccatgtccaatttactgaccgtac; gcgcgcctgaa-gatatagaaga).
Male and female 8–16-week-old SNS-HIF1afl/fl and theirHIF1afl/fl littermates were used for experiments with ‡ 8 miceper group. Mice had free access to food and water and werehoused in climate- and light-controlled quiet rooms with a12-h light–dark cycle. The experiments were approved by thelocal ethics committee for animal research (Darmstadt, Ger-many), adhered to the guidelines for pain research in con-scious animals of the International Association for the Studyof PAIN (IASP), and were in line with the European andGerman regulations for animal research.
Nerve injury and inflammatory models of chronic pain
Surgery was performed under 1.5%–2% isoflurane anes-thesia. For the SNI model of neuropathic pain, two of the threeperipheral branches of the sciatic nerve, the common peronealand the tibial nerves, were ligated with silk (6-0) and distallytransected, leaving the sural nerve intact (12). Mechanicaland heat withdrawal latencies as well as acetone tests weredone before and after SNI 2–3 times weekly for approximately4 weeks after SNI.
For the chronic inflammatory model, 20 ll of CFA wereinjected into the plantar side of the left hindpaw. Mechanicaland heat withdrawal latencies were recorded before andseveral times after CFA injection for approximately 1 week.
To induce the I/P injury, the blood circulation of the lefthind limb was blocked with a rubber band that was tightlyfixed around the upper hind limb for 1 h and then removed.Isoflurane anesthesia was maintained during the constrictionand during the early reperfusion phase for 30 min. Animalswere then allowed to recover in their home cages.
Behavioral experiments
All tests were performed by an investigator who wasblinded to the mouse genotype. After habituation, we deter-mined the latency for paw withdrawal using a DynamicPlantar Aesthesiometer (Ugo Basile) to assess sensitivity tomechanical stimulation. The steel rod was pushed against theplantar paw with ascending force (0–5 g over a 10-s period,0.2 g/s) and then maintained at 5 g until the paw was with-drawn. The paw withdrawal latency (PWL) was the mean ofthree consecutive trials with at least 30 s intervals. Mechanicalnociception was also assessed with graded strength von Freyhairs (Stoelting). With each hair, 10 stimuli were applied to thehindpaw and the number of responses was counted. The re-sponse percentages were then plotted versus the mechanicalforce.
Cold allodynia was analyzed in the acetone test. A drop ofacetone was applied to the mouse plantar paw and with-drawal responses, including licking, lifting, and shaking thepaw, were recorded with a stop watch for 90 s starting im-mediately after acetone application.
Sensitivity to painful heat was assessed by means of aHargreaves Test (Plantar Test 390G; IITC Life Science). Aheating lamp was placed with a mirror system underneath therespective hindpaw and by pressing the start button, the lampstarted emitting a laser beam until the paw was withdrawn,which automatically stopped the lamp. The mean PWL of
12 KANNGIESSER ET AL.

each of the three tests with at least 10 min intervals was usedfor statistical analysis.
In addition, we determined cold and heat pain temperaturethresholds with a Cold-Hot Plate that gradually decreased(20�C–4�C) or increased (20�C–52�C) the temperature ofthe metal plate by 10�C/min. The temperature at which themouse shows first withdrawal reactions is recorded as therespective threshold.
For the formalin test, 20 ll of 5% formalin were injected intothe left hind paw and the licking time was recorded for 45 minin 5 min intervals, starting directly after an injection of theirritant into the paw. The time the mouse spent licking thepaw during the first and second phases of the test was usedfor statistical comparisons.
To assess nociception on TRPV1 channel activation, 20 ll0.1% capsaicin dissolved in 0.9% sodium chloride were in-jected into the left hind paw and the licking time was recordedas described earlier for the formalin test.
Sensory neurons activation thresholdsand nerve conduction velocity
We employed extracellular recordings of action poten-tials in single fibers isolated from the saphenous nerve ofSNS-HIF1a - / - mice and control HIF1afl/fl littermates (2, 6).Briefly, the saphenous nerve was dissected, the proximal endwas mounted into an organ bath chamber and superfusedwith an oxygen-saturated modified synthetic interstitial fluidsolution containing (in mM) 108 NaCl, 3.48 KCl, 3.5 MgSO4,26 NaHCO3, 1.7 NaH2PO4, 2.0 CaCl2, 9.6 sodium gluconate,5.5 glucose, and 7.6 sucrose at a temperature of 31�C – 1�C andpH 7.4 – 0.05. The distal end of the saphenous nerve waspulled into a separate chamber, and fine filaments wereplaced on a gold wire recording electrode. Action potentials insingle sensory neurons were recorded extracellularly, ampli-fied (5000-fold), filtered (low pass 1 KHz, high-pass 100 Hz),visualized on an oscilloscope, and stored on a PC-type com-puter. For offline analysis, the Spike/Spidi software packagewas used (15). The nerve trunk was stimulated with square-wave pulses of increasing magnitude (up to 100 V), and theactivation threshold and conduction velocity of the nerve fi-bers were computed. Fibers conducting with conduction ve-locity < 2 m/s were considered unmyelinated C-fibers. Thereceptive field was identified by mechanical probing of theskin with a glass rod. The standard heat stimulus linearly risesthe intracutaneous temperature from 31�C to 50�C within 20 s.The standard cold stimulus decreases the temperature from31�C to 3�C within 4 s and holds this temperature for 20 s.Fibers were considered be sensitive if three or more actionpotentials were evoked during the stimulus.
Quantitative RT-PCR
Total RNA was extracted from homogenized tissue ac-cording to the protocol provided in the RNAeasy tissue MiniKit (Qiagen, Hilden, Germany), and reverse transcribed usingthe Verso� cDNA Kit (Thermo Scientific). Quantitative RT-PCR was performed for HIF1a, TRPV1, TRPA1, and TRPM8using an ABI prism 7700 TaqMan thermal cycler (AppliedBiosystems) using the Sybrgreen detection system with 18Sribosomal RNA as control and primer sets recommended bythe manufacturer as follows: HIF1a: Forward: 5¢-ggcgaag-caaagagtctgaag-3¢ and Reverse: 5¢-ccatctgtgccttcatctcat-3¢;
TRPV1: Forward: 5¢-aaggctctatgatcgcagga-3¢ and Reverse: 5¢-cagattgagcatggctttga-3¢; TRPA1: Forward: 5¢-aggagaccctgct-tcacaga-3¢ and Reverse: 5¢-tggagagcgtccttcagaat-3¢; TRPM8:Forward: 5¢-tgcctgctgtttcagaaatg-3¢ and Reverse: 5¢-taaaccga-tgcctcatctcc-3¢. Specific PCR product amplification was con-firmed with gel electrophoresis. Transcript regulation wasdetermined using the relative standard curve method ac-cording to the manufacturer’s instructions (Applied Biosystems).
cDNA array
Total RNA was extracted from DRGs L4–6, and biotin-dUTP labeled cDNA was generated by using the cDNAArray-Kit (Mouse HIF-Regulated cDNA Plate Array; Signo-sis) according to the manufacturer’s instructions. The cDNAwas hybridized with the target genes at 45�C overnight. Afterwashing and blocking free sites, a streptavidin-HRP substratemix was added and incubated for 45 min at room tempera-ture. After washing, the luminescent substrates were addedand light signals were captured after 1 min with a lumin-ometer (Mithras LB 940; Berthold Technologies). Analysiswas done with the MikroWin 2000 software.
Biopterin and neopterin analysis
After acidic oxidation of homogenized tissue with iodine,pteridines were obtained by solid phase extraction usingOasis MCX extraction cartridges (Waters GmbH). Con-centrations of total biopterin, neopterin, and the internalstandard rhamnopterin were determined by liquid chroma-tography coupled to tandem mass spectrometry as previouslydescribed (54).
Immunofluorescence studies
Mice were terminally anesthetized and perfused transcar-dially with 0.9% saline followed by 4% paraformaldehyde(PFA) in 0.1 M phosphate-buffered saline (PBS, pH 7.4). TheL4 and L5 spinal cord segments, DRGs, and brains were dis-sected and postfixed for 2 h and then transferred into 20%sucrose in PBS for overnight cryoprotection at 4�C. The tissuewas embedded in Tissue-Tek� O.C.T. Compound (ScienceServices) and cut at 14 lm in transverse sections on a cryo-tome. Sections were permeabilized for 5 min in phosphate-buffered saline with Tween 20 (PBST) (0.1% Tween in PBS),blocked for 1 h with 1% bovine serum albumin (BSA) in PBST,and incubated overnight at 4�C with primary antibodiesagainst c-Fos or ATF-3 (both 1:1000; Santa Cruz) dissolved in1% BSA in PBST. After washing in PBS, sections were incu-bated for 2 h at room temperature with a species-specificsecondary antibody conjugated with Alexa 488 or 594 (In-vitrogen). Slides were rinsed in PBS and embedded in Fluor-omount (Southern Biotech). Microscopy was done on aninverted fluorescent microscope (Axiovert 200; Zeiss) andanalyzed with AxioVisison 4.0 software (Zeiss).
In situ hybridization and pot in situ immunostaining
Sense and antisense riboprobes for mouse HIF1a wereobtained by cloning PCR products into the pCR4 TOPO se-quencing vector (Invitrogen) and subsequent in vitro tran-scription and labeling with digoxigenin (Dig-labeling kit;Roche Diagnostics). The riboprobe encompassed 532 bp, nu-cleotide 1–532 of mouse HIF1a cDNA, accession number
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 13

u59496, where the A of the start ATG codon is referred to asnucleotide 1. The forward primer was 5¢-atggagggcgccg-gcggcgag-3¢, reverse 5¢-ggcttgttagggtgcacttc-3¢. Fresh frozenDRGs, spinal cord, and brain were cut at 14 lm on a cryotome.Air-dried slides were fixed for 20 min in 4% PFA in 0.1 M PBSand acetylated. Sections were prehybridized for 2 h at 65�Cand hybridized overnight at 65�C with sense and antisenseprobes diluted 1:200 in the prehybridization mix (50% form-amide, 5 · saline-sodium citrate buffer (SSC), 5 · Denhardt’ssolution, 500 lg/ml herring sperm DNA, and 250 lg/ml yeasttransfer RNA), washed in 0.2% SSC at 65�C and incubatedwith anti-Dig-AP (1:1000; Roche Diagnostics) in 0.12 M maleicacid buffer with 0.15 M NaCl, pH 7.5 and 1% blocking reagent(Roche Diagnostics), washed in PBS, equilibrated in alkalinebuffer (0.1 M Tris-HCl, 0.1 M NaCl, 0.05 M MgCl2, pH 9.5, and2 mM levamisole), developed with BM Purple AP substrate(Roche Diagnostics), and embedded in Fluoromount or sub-jected to post in situ immunostaining with neuronal markers.Isolectin B4 labels small glutamatergic neurons with unmy-elinated fibers. NF200 labels large neurons with myelinatedfibers and CGRP is a marker for peptidergic neurons. Imageswere obtained using an inverse microscope (Axiovert 200;Zeiss) that was equipped with an AxioCam MRm andAxioVision 4.0 software (Zeiss).
Western blot analysis
Tissue samples were homogenized in PhosphoSafe Buffer(Sigma), and protease inhibitor mix (Pefabloc; Roche Diag-nostics). Proteins were separated by SDS-PAGE (30 lg/lane),transferred onto nitrocellulose membranes (AmershamPharmacia Biotech) by wet blotting, and incubated with pri-mary antibodies overnight at 4�C. Heat shock protein (HSP90)was used as a loading control. For GCH1, a rabbit antiserumwas used (54). After incubation with the secondary antibody,conjugated with IRDye 680 (1:10,000; LI-COR Biosciences),blots were visualized and analyzed on the Odyssey InfraredImaging System (LI-COR Biosciences). The ratio of the re-spective protein band to the control band was used for semi-quantitative analysis.
Primary neuronal cultures
Primary neuron-enriched cultures of DRG neurons wereprepared by dissecting DRG of adult mice into Hanks’Balanced Salt Solution (Dulbecco) and 10 mM hydro-xyethylpiperazin ethane sulfonic acid (HEPES) buffer, fol-lowed by digestion with 5 mg/ml collagenase A and 1 mg/mldispase II (Roche Diagnostics). Triturated cells were centri-fuged through a 10% BSA solution, plated, and cultivated onpoly-l-lysine and laminin-coated cover slips in serum-freeNeurobasal medium (GibcoBRL) containing 2% (v/v) B27supplement (GibcoBRL), 50 lg/ml Pen-Strep (Sigma), 10 lMarabinosylcytosine (Ara-C; Sigma), 100 ng/ml nerve growthfactor (NGF; GibcoBRL), and 200 mM l-glutamine (Gib-coBRL) at 5% CO2. Calcium imaging experiments were per-formed 24–36 h after plating.
Calcium imaging
Cultured adult DRG neurons were loaded for 1 h with10 lM fura-2 (Invitrogen) in Neurobasal medium with 0.02%pluronic (w/v). The temperature was maintained at 37�Cthroughout the measurements. The neurons were continu-
ously perfused with Ringer solution (in mM: 136 NaCl,5.4 KCl, 1.8 CaCl2, 1 MgCl2, 0.33 NaH2PO4, 10 Glucose, and10 HEPES) at a speed of 2 ml/min. Images were captured at arate of 1 frame per 2 s with a high-speed camera. Intracellular[Ca2 + ]i was assessed fluorimetrically as absorbance ratio at340 and 380 nm excitation (F340/380) (510 nm for emission).Baseline ratios were recorded for 100 s before bath applicationof capsaicin (500 nM), formalin (0.01%), menthol (250 lM),H2O2 (1 mM), DEA-NO (500 lM), or BH4 (100 lM). Thestimulation time was 40 and 100 s depending on the stimulus.After a wash-out period, cells were perfused with high potas-sium (50 mM KCl in Ringer) to check the viability of the neu-rons. Data are presented as changes in the fluorescence ratios(F340/F380) normalized to baseline ratios. The peak foldchange of [Ca2 + ]i and the area under the ‘‘fold change versustime curves’’ (AUCs) were used for statistical comparisons.
Statistics
SPSS 21.0 was used for statistical evaluation. Data arepresented as means – SD or SEM (for behavior). Behavioraldata were analyzed using analysis of variance (ANOVA) forrepeated measurements for time courses and one-way AN-OVA or t-tests to compare areas under the curve. The latterwere calculated according to the linear trapezoidal rule.Counts of neurons, quantitative RT-PCR, biopterin, andwestern blot results were analyzed with Student’s t-tests (fortwo groups) or one-way ANOVA. Ordinal data were com-pared with the Mann–Whitney U-test (two groups) or theKruskal–Wallis test. Fisher’s exact tests were used to comparethe fractions of C-fibers responding to electrical, cold, andheat stimuli. The cumulative response curves on electricalstimulation were fitted to a standard sigmoidal Emax modelwith variable Hill slope. The goodness of fit was assessed by‘‘sum of squares’’ and residual plots. p Was set at 0.05 for allstatistical comparisons. In case of multiple comparisons,we used a correction of alpha according to Bonferroni. Thenumber of animals used for the experiments are given in therespective figure legends.
Acknowledgments
The authors thank Sandra Labocha for technical assistanceand acknowledge the financial support of the Deutsche For-schungsgemeinschaft (SFB815 A12 to I.T., A8 to B.B.).
Authors’ Contributions
M.K. and N.M. performed experiments and analyzed data,H.-Y.L. and J.B. provided advice and experimental support,A.H. and K.Z. performed experiments, B.B. and M.K. providedadvice, supervised experiments, and revised the article, N.F.analyzed data, and I.T. designed the study, performed exper-iments, analyzed data, made the figures, and wrote the article.
Author Disclosure Statement
No competing financial interests exist.
References
1. Agarwal N, Offermanns S, and Kuner R. Conditional genedeletion in primary nociceptive neurons of trigeminal gan-glia and dorsal root ganglia. Genesis 38: 122–129, 2004.
2. Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE,Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory
14 KANNGIESSER ET AL.

K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ,Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, and Kuner R.Cannabinoids mediate analgesia largely via peripheral type1 cannabinoid receptors in nociceptors. Nat Neurosci 10: 870–879, 2007.
3. Baranova O, Miranda LF, Pichiule P, Dragatsis I, JohnsonRS, and Chavez JC. Neuron-specific inactivation of thehypoxia inducible factor 1 alpha increases brain injury in amouse model of transient focal cerebral ischemia. J Neurosci27: 6320–6332, 2007.
4. Barriere DA, Rieusset J, Chanteranne D, Busserolles J,Chauvin MA, Chapuis L, Salles J, Dubray C, and Morio B.Paclitaxel therapy potentiates cold hyperalgesia in strepto-zotocin-induced diabetic rats through enhanced mitochon-drial reactive oxygen species production and TRPA1sensitization. Pain 153: 553–561, 2012.
5. Bautista DM, Movahed P, Hinman A, Axelsson HE, SternerO, Hogestatt ED, Julius D, Jordt SE, and Zygmunt PM.Pungent products from garlic activate the sensory ionchannel TRPA1. Proc Natl Acad Sci USA 102: 12248–12252,2005.
6. Bockhart V, Constantin CE, Haussler A, Wijnvoord N,Kanngiesser M, Myrczek T, Pickert G, Popp L, Sobotzik JM,Pasparakis M, Kuner R, Geisslinger G, Schultz C, Kress M,and Tegeder I. Inhibitor kappaB Kinase beta deficiency inprimary nociceptive neurons increases TRP channel sensi-tivity. J Neurosci 29: 12919–12929, 2009.
7. Brune B and Zhou J. The role of nitric oxide (NO) in stabilityregulation of hypoxia inducible factor-1alpha (HIF-1alpha).Curr Med Chem 10: 845–855, 2003.
8. Bull R, Finkelstein JP, Galvez J, Sanchez G, Donoso P,Behrens MI, and Hidalgo C. Ischemia enhances activation byCa2 + and redox modification of ryanodine receptor chan-nels from rat brain cortex. J Neurosci 28: 9463–9472, 2008.
9. Campbell CM, Edwards RR, Carmona C, Uhart M, Wand G,Carteret A, Kim YK, Frost J, and Campbell JN. Polymorph-isms in the GTP cyclohydrolase gene (GCH1) are associatedwith ratings of capsaicin pain. Pain 141: 114–118, 2009.
10. Chattopadhyay M, Walter C, Mata M, and Fink DJ.Neuroprotective effect of herpes simplex virus-mediatedgene transfer of erythropoietin in hyperglycemic dorsal rootganglion neurons. Brain 132: 879–888, 2009.
11. Cheng LZ, Lu N, Zhang YQ, and Zhao ZQ. Ryanodine re-ceptors contribute to the induction of nociceptive input-evoked long-term potentiation in the rat spinal cord slice.Mol Pain 6: 1, 2010.
12. Decosterd I and Woolf CJ. Spared nerve injury: an animalmodel of persistent peripheral neuropathic pain. Pain 87:149–158, 2000.
13. Diebold I, Petry A, Hess J, and Gorlach A. The NADPHoxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol Biol Cell 21: 2087–2096, 2010.
14. Digicaylioglu M and Lipton SA. Erythropoietin-mediatedneuroprotection involves cross-talk between Jak2 and NF-kappaB signalling cascades. Nature 412: 641–647, 2001.
15. Forster C and Handwerker HO. Automatic classification andanalysis of microneurographic spike data using a PC/AT.J Neurosci Methods 31: 109–118, 1990.
16. Ginouves A, Ilc K, Macias N, Pouyssegur J, and Berra E.PHDs overactivation during chronic hypoxia ‘‘desensitizes’’HIFalpha and protects cells from necrosis. Proc Natl Acad SciUSA 105: 4745–4750, 2008.
17. Guhring H, Tegeder I, Lotsch J, Pahl A, Werner U, Reeh PW,Rehse K, Brune K, and Geisslinger G. Role of nitric oxide in
zymosan induced paw inflammation and thermal hyper-algesia. Inflamm Res 50: 83–88, 2001.
18. Hawkes HJ, Karlenius TC, and Tonissen KF. Regulation ofthe human thioredoxin gene promoter and its key sub-strates: a study of functional and putative regulatory ele-ments. Biochim Biophys Acta 1840: 303–314, 2013.
19. Jin XG, Chen SR, Cao XH, Li L, and Pan HL. Nitric oxideinhibits nociceptive transmission by differentially regulatingglutamate and glycine release to spinal dorsal horn neurons.J Biol Chem 286: 33190–33202, 2011.
20. Kakizawa S, Yamazawa T, Chen Y, Ito A, Murayama T,Oyamada H, Kurebayashi N, Sato O, Watanabe M, Mori N,Oguchi K, Sakurai T, Takeshima H, Saito N, and Iino M.Nitric oxide-induced calcium release via ryanodine receptorsregulates neuronal function. Embo J 31: 417–428, 2012.
21. Kallenborn-Gerhardt W, Lu R, Syhr KM, Heidler J, vonMelchner H, Geisslinger G, Bangsow T, and Schmidtko A.Antioxidant Activity of Sestrin 2 Controls Neuropathic PainAfter Peripheral Nerve Injury. Antioxid Redox Signal 2013[Epub ahead of print]; DOI: 10.1089/ars.2012.4958
22. Kallenborn-Gerhardt W, Schroder K, Del Turco D, Lu R,Kynast K, Kosowski J, Niederberger E, Shah AM, BrandesRP, Geisslinger G, and Schmidtko A. NADPH oxidase-4maintains neuropathic pain after peripheral nerve injury.J Neurosci 32: 10136–10145, 2012.
23. Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E,Minami T, Takada Y, Kume T, Katsuki H, Mori Y, andAkaike A. A critical role of TRPM2 in neuronal cell death byhydrogen peroxide. J Pharmacol Sci 101: 66–76, 2006.
24. Kawasaki Y, Xu ZZ, Wang X, Park JY, Zhuang ZY, Tan PH,Gao YJ, Roy K, Corfas G, Lo EH, and Ji RR. Distinct roles ofmatrix metalloproteases in the early- and late-phase devel-opment of neuropathic pain. Nat Med 14: 331–336, 2008.
25. Keswani SC, Bosch-Marce M, Reed N, Fischer A, SemenzaGL, and Hoke A. Nitric oxide prevents axonal degenerationby inducing HIF-1-dependent expression of erythropoietin.Proc Natl Acad Sci USA 108: 4986–4990, 2011.
26. Kim D, You B, Jo EK, Han SK, Simon MI, and Lee SJ.NADPH oxidase 2-derived reactive oxygen species in spinalcord microglia contribute to peripheral nerve injury-inducedneuropathic pain. Proc Natl Acad Sci USA 107: 14851–14856,2010.
27. Kim KS, Yoo HY, Park KS, Kim JK, Zhang YH, and Kim SJ.Differential effects of acute hypoxia on the activation of TRPV1by capsaicin and acidic pH. J Physiol Sci 62: 93–103, 2012.
28. King RH, Chandler D, Lopaticki S, Huang D, Blake J,Muddle JR, Kilpatrick T, Nourallah M, Miyata T, Okuda T,Carter KW, Hunter M, Angelicheva D, Morahan G, andKalaydjieva L. Ndrg1 in development and maintenance ofthe myelin sheath. Neurobiol Dis 42: 368–380, 2011.
29. Kobayashi H, Chattopadhyay S, Kato K, Dolkas J, Kikuchi S,Myers RR, and Shubayev VI. MMPs initiate Schwann cell-mediated MBP degradation and mechanical nociception af-ter nerve damage. Mol Cell Neurosci 39: 619–627, 2008.
30. Koshiji M and Huang LE. Dynamic balancing of the dual na-ture of HIF-1alpha for cell survival. Cell Cycle 3: 853–854, 2004.
31. Li F, Sonveaux P, Rabbani ZN, Liu S, Yan B, Huang Q,Vujaskovic Z, Dewhirst MW, and Li CY. Regulation of HIF-1alpha stability through S-nitrosylation. Mol Cell 26: 63–74,2007.
32. Lu DY, Liou HC, Tang CH, and Fu WM. Hypoxia-inducediNOS expression in microglia is regulated by the PI3-kinase/Akt/mTOR signaling pathway and activation of hypoxia in-ducible factor-1alpha. Biochem Pharmacol 72: 992–1000, 2006.
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 15

33. Martin LJ, Adams NA, Pan Y, Price A, and Wong M. Themitochondrial permeability transition pore regulates nitricoxide-mediated apoptosis of neurons induced by targetdeprivation. J Neurosci 31: 359–370, 2011.
34. Meller ST, Lewis SJ, Bates JN, Brody MJ, and Gebhart GF. Isthere a role for an endothelium-derived relaxing factor innociception? Brain Res 531: 342–345, 1990.
35. Metzen E, Zhou J, Jelkmann W, Fandrey J, and Brune B.Nitric oxide impairs normoxic degradation of HIF-1alpha byinhibition of prolyl hydroxylases. Mol Biol Cell 14: 3470–3481, 2003.
36. Miyamoto T, Dubin AE, Petrus MJ, and Patapoutian A.TRPV1 and TRPA1 mediate peripheral nitric oxide-inducednociception in mice. PLoS One 4: e7596, 2009.
37. Ogunshola OO and Antoniou X. Contribution of hypoxia toAlzheimer’s disease: is HIF-1alpha a mediator of neurode-generation? Cell Mol Life Sci 66: 3555–3563, 2009.
38. Okuda T, Higashi Y, Kokame K, Tanaka C, Kondoh H, andMiyata T. Ndrg1-deficient mice exhibit a progressive de-myelinating disorder of peripheral nerves. Mol Cell Biol 24:3949–3956, 2004.
39. Olson N, Hristova M, Heintz NH, Lounsbury KM, and vander Vliet A. Activation of hypoxia-inducible factor-1 protectsairway epithelium against oxidant-induced barrier dysfunc-tion. Am J Physiol Lung Cell Mol Physiol 301: L993–L1002, 2011.
40. Petruska JC, Napaporn J, Johnson RD, Gu JG, and CooperBY. Subclassified acutely dissociated cells of rat DRG: his-tochemistry and patterns of capsaicin-, proton-, and ATP-activated currents. J Neurophysiol 84: 2365–2379, 2000.
41. Pickert G, Myrczek T, Ruckert S, Weigert A, Haussler A,Ferreiros N, Brune B, Lotsch J, and Tegeder I. Inhibition ofGTP cyclohydrolase reduces cancer pain in mice and en-hances analgesic effects of morphine. J Mol Med (Berl) 90:1473–1486, 2012.
42. Rice ME. H2O2: a dynamic neuromodulator. Neuroscientist17: 389–406, 2011.
43. Rigaud M, Gemes G, Weyker PD, Cruikshank JM, KawanoT, Wu HE, and Hogan QH. Axotomy depletes intracellularcalcium stores in primary sensory neurons. Anesthesiology111: 381–392, 2009.
44. Ristoiu V, Shibasaki K, Uchida K, Zhou Y, Ton BH, FlontaML, and Tominaga M. Hypoxia-induced sensitization oftransient receptor potential vanilloid 1 involves activation ofhypoxia-inducible factor-1 alpha and PKC. Pain 152: 936–945, 2011.
45. Sawada Y, Hosokawa H, Matsumura K, and Kobayashi S.Activation of transient receptor potential ankyrin 1 by hy-drogen peroxide. Eur J Neurosci 27: 1131–1142, 2008.
46. Scheving R, Wittig I, Heide H, Albuquerque B, Steger M,Brandt U, and Tegeder I. Protein S-nitrosylation and deni-trosylation in the mouse spinal cord upon injury of the sci-atic nerve. J Proteomics 75: 3987–4004, 2012.
47. Schmidtko A, Tegeder I, and Geisslinger G. No NO, nopain? The role of nitric oxide and cGMP in spinal painprocessing. Trends Neurosci 32: 339–346, 2009.
48. Scholz J and Woolf CJ. The neuropathic pain triad: neurons,immune cells and glia. Nat Neurosci 10: 1361–1368, 2007.
49. Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasisand disease pathophysiology. Trends Mol Med 7: 345–350, 2001.
50. Sharp FR and Bernaudin M. HIF1 and oxygen sensing in thebrain. Nat Rev Neurosci 5: 437–448, 2004.
51. Takahashi N, Kozai D, Kobayashi R, Ebert M, and Mori Y. Rolesof TRPM2 in oxidative stress. Cell Calcium 50: 279–287, 2011.
52. Takahashi N, Kuwaki T, Kiyonaka S, Numata T, Kozai D,Mizuno Y, Yamamoto S, Naito S, Knevels E, Carmeliet P,Oga T, Kaneko S, Suga S, Nokami T, Yoshida J, and Mori Y.TRPA1 underlies a sensing mechanism for O2. Nat Chem Biol7: 701–711, 2011.
53. Tandrup T, Woolf CJ, and Coggeshall RE. Delayed loss ofsmall dorsal root ganglion cells after transection of the ratsciatic nerve. J Comp Neurol 422: 172–180, 2000.
54. Tegeder I, Costigan M, Griffin RS, Abele A, Belfer I,Schmidt H, Ehnert C, Nejim J, Marian C, Scholz J, Wu T,Allchorne A, Diatchenko L, Binshtok AM, Goldman D,Adolph J, Sama S, Atlas SJ, Carlezon WA, Parsegian A,Lotsch J, Fillingim RB, Maixner W, Geisslinger G, Max MB,and Woolf CJ. GTP cyclohydrolase and tetrahydrobio-pterin regulate pain sensitivity and persistence. Nat Med 12:1269–1277, 2006.
55. Tegeder I, Scheving R, Wittig I, and Geisslinger G. SNO-ingat the Nociceptive Synapse? Pharmacol Rev 63: 366–389, 2011.
56. Ueno S, Tsuda M, Iwanaga T, and Inoue K. Cell type-specificATP-activated responses in rat dorsal root ganglion neurons.Br J Pharmacol 126: 429–436, 1999.
57. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S,Schledzewski K, Kzhyshkowska J, and Brune B. Knockout ofHIF-1alpha in tumor-associated macrophages enhances M2polarization and attenuates their pro-angiogenic responses.Carcinogenesis 31: 1863–1872, 2010.
58. Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S,Hara Y, Tominaga M, Shimizu S, Sato Y, and Mori Y. Nitricoxide activates TRP channels by cysteine S-nitrosylation. NatChem Biol 2: 596–607, 2006.
59. Yu X, Fang Y, Liu H, Zhu J, Zou J, Xu X, Jiang S, and Ding X.The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor inrat remnant kidney depends on the timing of administration.Nephrol Dial Transplant 27: 3110–3119, 2012.
60. Yuan G, Khan SA, Luo W, Nanduri J, Semenza GL, andPrabhakar NR. Hypoxia-inducible factor 1 mediates in-creased expression of NADPH oxidase-2 in response to in-termittent hypoxia. J Cell Physiol 226: 2925–2933, 2011.
61. Zhou J and Brune B. NO and transcriptional regulation:from signaling to death. Toxicology 208: 223–233, 2005.
62. Zimmermann K, Lennerz JK, Hein A, Link AS, KaczmarekJS, Delling M, Uysal S, Pfeifer JD, Riccio A, and ClaphamDE. Transient receptor potential cation channel, subfamilyC, member 5 (TRPC5) is a cold-transducer in the peripheralnervous system. Proc Natl Acad Sci USA 108: 18114–18119,2011.
Address correspondence to:Prof. Irmgard Tegeder
Institute of Clinical PharmacologyGoethe-University Hospital Frankfurt
Theodor Stern Kai 7, Bd. 74Frankfurt 60590
Germany
E-mail: [email protected]
Date of first submission to ARS Central, June 25, 2013; date offinal revised submission, September 27, 2013; date of accep-tance, October 21, 2013.
16 KANNGIESSER ET AL.

Abbreviations Used
ANOVA¼ analysis of varianceATF-3¼ activating transcription factor 3
AUC¼ area under the curveBcl2¼B-cell lymphoma 2BH4¼ tetrahydrobiopterin
BK¼ bradykininBSA¼ bovine serum albuminCa2+¼ calciumCaM¼ calmodulin
CamKII¼ calmodulin kinase IICFA¼ complete Freund’s adjuvant
cFOS¼ transcription factorCGRP¼ calcitonin gene related peptide
CI¼ confidence intervalCTSD¼ cathepsin D
df¼degrees of freedomDIG¼digoxigenin
DRG¼dorsal root gangliaEP300¼E1A binding protein p300,
HIF1 coactivatorEPO¼ erythropoietin
GCH1¼GTP cyclohydrolaseGlut3¼ glucose transporter 3 (gene SLC2A3)GSH¼ glutathioneGSR¼ glutathione reductaseGSS¼ glutathione synthase
GSSG¼ glutathione disulfide (oxidized GSH)H2O2¼hydrogen peroxide
HEPES¼hydroxyethylpiperazin ethanesulfonic acid
HIF1¼hypoxia-inducible factor 1HIF1a¼hypoxia-inducible factor 1 alphaHIF1b¼hypoxia-inducible factor 1 betaHSP90¼heat shock protein 90
I/P¼ ischemia/reperfusionMMP2¼metalloproteinase 2
NDRG1¼N-myc downstream regulated gene 1NGF¼nerve growth factor
nNOS¼neuronal nitric oxide synthaseNO¼nitric oxide
NOS¼NO synthaseNOx¼ reactive nitrogen species
NOX2¼NADPH oxidases 2NOX4¼NADPH oxidases 4
PBS¼phosphate-buffered salinePBST¼phosphate-buffered saline
with Tween 20PFA¼paraformaldehyde
PHDs¼prolylhydroxylasesPIP2¼phosphatidylinositol 4,5-bisphosphatePKC¼protein kinase CPrdx¼peroxiredoxinPWL¼paw withdrawal latency
rm-ANOVA¼ repeated-measures analysis of varianceRNF7/SAG¼ ring finger protein 7/sensitive
to apoptosisROS¼ reactive oxygen species
RT-PCR¼ real-time polymerase chain reactionSDS-PAGE¼ sodium dodecyl sulfate
polyacrylamide gel electrophoresisSNARE¼ complex of syntaxin 1,
SNAP-25 and VAMPSNI¼ spared nerve injury
SNS/Nav1.8¼ sensory neuron tetrodotoxin-resistantsodium channel (gene SCN10A)
SOD¼ superoxide dismutaseSSC¼ sodium citrate bufferTRP¼ transient receptor potential
TRPA1¼ transient receptor potential,subfamily A, member 1
TRPC¼ transient receptor potential, subfamily CTRPM8¼ transient receptor potential,
subfamily M, member 8TRPV1¼ transient receptor potential
vanilloid subfamily, member 1Trx¼ thioredoxin
TrxR¼ thioredoxin reductaseUbi¼ubiquitin
UCP3¼uncoupling proteinVEGF¼vascular endothelial growth factorVHL¼von Hippel Lindau
HYPOXIA-INDUCIBLE FACTOR 1 AND PAIN 17





























