Embed Size (px)
Citation preview

1
ICH - E6 Good Clinical Practice
Cathy Liu Cathy Liu
Shanghai, Aug 30Shanghai, Aug 30thth 20102010

2
Acronyms
� ICH = International Conference on Harmonisation
� GCP = Good Clinical Practice
� FDA = Food and Drug Administration
� CFR = Code of Federal Regulations
� SOP = Standard Operating Procedure

3
Agenda
By the end of this module, you will understand:
� What, Why and How of ICH GCP?
� ICH GCP Terminology
� Sets of Responsibilities
� Understanding Data Management Role in Clinical Trials

4
Good Clinical Practice (GCP)

5
What is Good Clinical Practice?
GCP is an international ethical & scientificquality standard for designing, conducting, recording & reporting trials that involve theparticipation of human subjects .
Phase“Zero”Pre Clinical
PhaseIClinical
PhaseII / III
PhaseIII / IV

6
The Development ofGood Clinical Practice (GCP)

7
History
The history of good clinical practice (GCP) regulations reflects the growing involvement of governments in trying to ensure that drug products are safe, effective and are manufactured and marketed appropriately.

8
Key Milestones In The Development of GCP
� 1938 Food Drug & Cosmetic Act (US)� 1947 Nuremberg Code� 1961-62 Thalidomide Birth Defects� 1963 Introduction of IND (US)� 1964 Declaration of Helsinki� 1977 Sponsor / Monitor Obligations (US)� 1981 Informed Consent Regulations (US)� 1982 IRB Regulations (US) � 1986 ABPI Guidelines (UK) � 1989-92 Nordic, Japanese, EU CPMP, Australian & WHO
Guidelines� 1995 Implementation of Korea GCP� 1996 ICH Guidelines issued� 1997 ICH becomes law in some countries and becomes
incorporated in to sponsor Standard Operating Procedures (SOPs)
……

9
Dimensions of GCP
General Frameworks� WHO GCP
� ICH GCP
Regional/Applied Frameworks� EU GCP Guideline/Directive
� US CFR
National/Applied GCP Guidelines� China, India, Singapore, Malaysia, Indonesia, Russia, South
American, South Africa, Turkey

10
ICH
International Conference on Harmonisationof Technical Requirements for Registration
of Pharmaceuticals for Human Use

11
ICH - OBJECTIVES
1. To avoid useless duplication of testing and trials, thereby saving time, money and resources.

12
ICH - OBJECTIVES
2. To allow companies to prepare one set of `Core Technical Data' on Safety, Quality and Efficacy for a new drug application which will be acceptable wherever the dossier is filed (Global Dossier).
Global
Dossier

13
ICH – OBJECTIVES
3. To allow through the submission of the `Global Dossier’ the `mutual acceptance' of foreign data.

14
ICH - HARMONISATION TOPICS
� Multi-disciplinary (since Oct 94)
� Safety (S) Pre-clinical toxicity and related tests
� Efficacy (E) Clinical testing programs and safety monitoring
� Quality (Q) Pharmaceutical development and specifications

15
ICH - E6 Good Clinical Practice

16
Who is impacted by GCP
� Sponsor
� Drug manufactures
� Laboratories
� Investigator
� Study participants
� ERB
� Caregivers
� CRO
� Study site personnel
� Pharmacist
� Other specialists supporting data provision and analysis

17
Difference with Other GCPs
� Better defined distribution of responsibilities
� Text is more stringent
� Text is more user-oriented instead of topic-oriented
� Assigns all clinical trial duties to one of 3 participating parties: IRB/IEC, investigator and sponsor

18
ICH GCP CONTENT
Chapter 1: Glossary
Chapter 2: The principles of ICH GCP
Chapter 3: IRB/IEC
Chapter 4: Investigator
Chapter 5: Sponsor
Chapter 6: Clinical Trial Protocol and Protocol Amendments
Chapter 7: Investigator’s Brochure
Chapter 8: Essential Documents for the Conduct of a Clinical Trial
19
Terminology
Sponsor
InvestigatorCRO
QA/QC
IRBIEC

20
� Institutional Review Board (ICH 1.31)� An independent body constituted of medical, scientific, and non
scientific members, whose responsibility it is to ensure the protection of the rights, safety, and well-being of human subjects involved in a trial…
� Independent Ethics Committee (ICH 1.27)� An independent body constituted of medical/scientific
professionals and non-medical/nonscientific members who responsibility it is to ensure the protection of the rights, safety, and well-being of human subjects
involved in a trial and public assurance of that protection...

21
� Investigator (ICH 1.34)� A person responsible for the conduct of the clinical trial at the
trial site. If a trial is conducted by a team of individuals, the investigator is the responsible leader of the team.
� Subinvestigator (ICH 1.56)� Any individual member of the clinical trial team designated and
supervised by the investigator at a trial site to perform critical trial-related procedures and/or to make important trail-related decisions (eg. associates, residents, research fellows).

22
� Sponsor (ICH 1.53)� An individual , company, institution, or organisation that takes
responsibility for the initiation, management, and/or financing of a clinical trial.
� Contract Research Organisation (ICH 1.20)� A person or an organisation contracted by the sponsor to
perform one or more of a sponsor’s trial-related duties and functions.

23
� Quality Control (QC)
� The Operational techniques and activities undertaken within the quality system to verify that the requirements for quality of the trial related activities have been fulfilled. (ICH 1.47)
• identifying a problem and implementing a solution
• continuous during the research
• carried out by the people conducting the work

24
� Quality Assurance (QA)
� All those planned and systemic actions that are established to ensure that the trial is performed and the data are generated, documented (recorded), and reported in compliance with GCP and the applicable regulatory requirement(s). (ICH 1.46)
• Quality Control
• Audit of the process (eg., site audits)
• Audit of the final product (eg., report audit)
25
PRINCIPLES OF ICH GCP

26
PRINCIPLES OF ICH GCP
1. Clinical trials should be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, and that are consistent with GCP and the applicable regulatory requirement(s).
2. Before a trial is initiated, foreseeable risks and inconveniences should be weighed against the anticipated benefit for the individual trial subject and society. A trial should be initiated and continued only if the anticipated benefits justify the risks.

27
3. The rights, safety and well-being of the trial subjects are the most important considerations and should prevail over interests of science and society.
4. The available non-clinical and clinical information on an investigational product should be adequate to support the proposed clinical trial.
PRINCIPLES OF ICH GCP

28
5. Clinical trials should be scientifically sound, and described in a clear, detailed protocol.
6. A trial should be conducted in compliance with the protocol that has received prior institutional review board (IRB)/independent ethics committee (IEC) approval/favourable opinion.
PRINCIPLES OF ICH GCP

29
7. The medical care given to, and medical decisions made on behalf of, subjects should always be the responsibility of a qualified physician or, when appropriate, of a qualified dentist.
8. Each individual involved in conducting a trial should be qualified by education, training, and experience to perform his or her respective task(s).
PRINCIPLES OF ICH GCP

30
9. Freely given informed consent should be obtained from every subject prior to clinical trial participation.
10. All clinical trial information should be recorded, handled, and stored in a way that allows its accurate reporting, interpretation and verification.
PRINCIPLES OF ICH GCP

31
11. The confidentiality of records that could identify subjects should be protected, respecting the privacy and confidentiality rules in accordance with the applicable regulatory requirement(s).
PRINCIPLES OF ICH GCP

32
12. Investigational products should be manufactured, handled, and stored in accordance with applicable Good Manufacturing Practices (GMP). They should be used in accordance with the approved protocol.
13. Systems with procedures that assure the quality of every aspect of the trial should be implemented.
PRINCIPLES OF ICH GCP

33
The Major Players
Section 4
Section 3 Section 5

34
� Safeguard the rights, safety, and well-beining of all trial subjects
� Give approval/favourable opinion on the trial conduct based on the scientific, ethic review of the trial files and qualification of investigator
� Conduct continuing review of each ongoing trial at intervals appropriate to the degree of risk to human subjects
IRB/IEC Responsibility

35
Investigators Responsibilities
1. Investigator’s Qualifications and Agreements2. Adequate Resource3. Medical Care of Trial Subjects4. Communication with IRB/IEC5. Compliance with Protocol6. Investigational Product7. Randomization Procedures and Unblinding8. Informed Consent of Trial Subjects9. Records and reports10. Progress Reports11. Safety Reporting12. Premature Termination or Suspension of a Trial13. Final Report(s) by Investigator

36
Investigator’s Qualifications and Agreements
� Provide evidence of qualifications and experience - CV
� Select, train and keep a log of study personnel
� Allow monitoring, auditing & regulatory inspections
� Aware of and comply with GCP, regulatory requirements
� Ensure study equipment is adequate

37
Adequate Involvement in the Trial
� ACTIVE participation in the trial
� Proper delegation of tasks, NOT responsibility, to qualified individuals.
� This also includes:� Interacting with the monitor
� Keeping staff informed about all study related matters� Knowing the Protocol and any amendments� Predict recruitment accurately and keep an up to date subject
enrolment log

38
Full Compliance with the Protocol
� Acknowledged and agreed with signature on protocol signature page (original and all amendments)
� Deviations from the protocol must be discussed with the Sponsor (unless there is an immediate safety issue for a patient)
� Documentation of all protocol deviations is required

39
Initial and Ongoing Communication with IRB/IEC(1)
� Written documentation from IRB/IEC that “it is organized and operating according to GCP”
� Do not start the trial without written IRB/IEC approval
� The IRB/IEC will review and approve key documents including any patient completed materials or advertising

40
Initial and Ongoing Communication with IRB/IEC (2)
� As the trial progresses, provide IRB/IEC with:
� Any updated Investigator Brochure & safety information
� Any Protocol Amendments
� Regulatory Safety Reports (according to local requirements)
� Any other change in the study (e.g. increase in target number ofsubjects to be enrolled at your site)
� Other information that they may require for your site

41
Investigator Responsibilities:
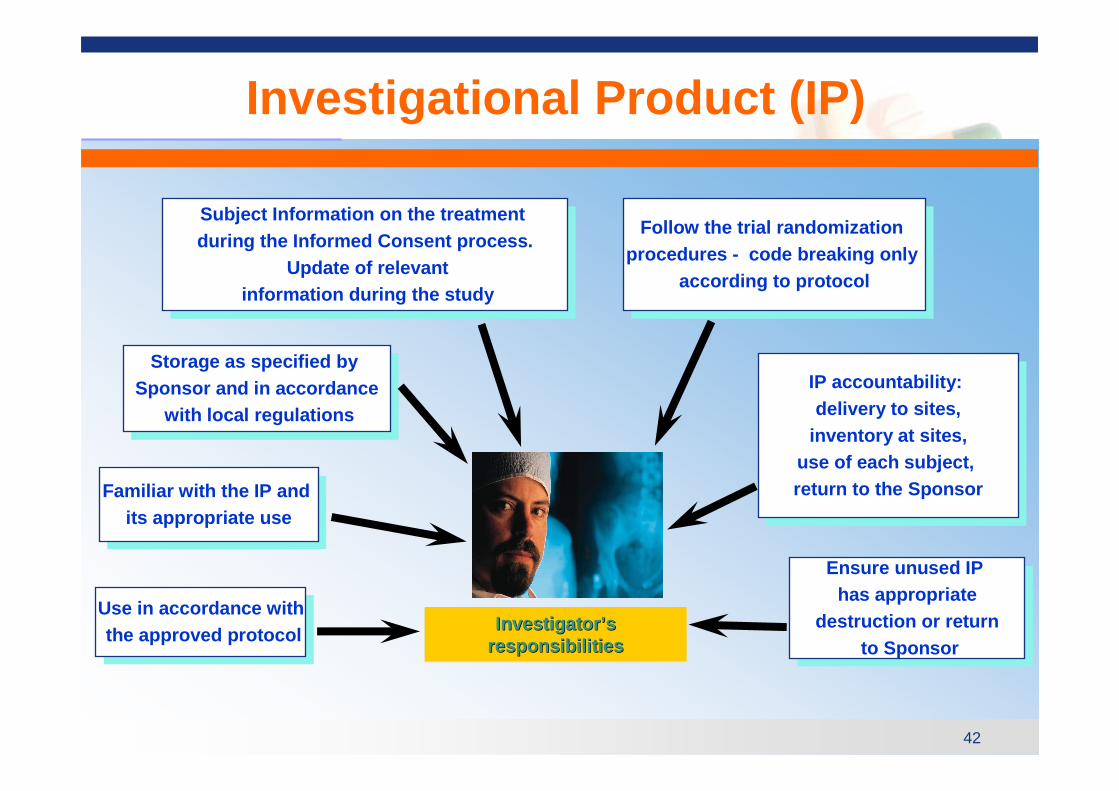
� The investigator(s) should be thoroughly familiar with the appropriate use of the investigational product(s), as described in the protocol, in the current Investigator’s Brochure, in the product information and in other information sources provided by the Sponsor. (ICH 4.1.2)
Investigational Product (IP) Accountability
42
Investigational Product (IP)
Storage as specified by Sponsor and in accordance
with local regulations
Storage as specified by Sponsor and in accordance
with local regulations
Use in accordance withthe approved protocol
Use in accordance withthe approved protocol
Familiar with the IP and its appropriate use
Familiar with the IP and its appropriate use
Follow the trial randomization procedures - code breaking only
according to protocol
Follow the trial randomization procedures - code breaking only
according to protocol
IP accountability: delivery to sites,
inventory at sites, use of each subject, return to the Sponsor
IP accountability: delivery to sites,
inventory at sites, use of each subject, return to the Sponsor
Investigator’sresponsibilitiesInvestigatorInvestigator ’’ss
responsibilitiesresponsibilities
Subject Information on the treatment during the Informed Consent process.
Update of relevantinformation during the study
Subject Information on the treatment during the Informed Consent process.
Update of relevantinformation during the study
Ensure unused IP has appropriate
destruction or returnto Sponsor
Ensure unused IP has appropriate
destruction or returnto Sponsor

43
Informed Consent
� THIS IS ONE OF THE FUNDAMENTAL ASPECTS OF RUNNING A TRIAL
� Process for meaningful exchange of study information between the investigator and subject
� Must be personally signed and dated by all parties before any study procedures begin
� Documentation in source that the subject signed/ dated the consent prior to study participation
� Any changes to the Informed Consent form need to be signed by study participants

44
Randomisation and Unblinding
� Follow randomization procedures
� Code broken only in accordance with protocol
� Document and explain any premature unblinding

45
Safety Reporting: Adverse Events
� Report all Adverse Events (AEs)
� Report all Serious Adverse Events (SAEs) immediately (within 24 hours of being aware of event) to the sponsor with prompt follow-up information
� Report SAEs to IRB/IEC if required by local/regional regulations
� Do not use subject names (only subject numbers) and do not relay personal information such as addresses, phone numbers next of kin etc.

46
Source Documentation: Purpose
� Verify each subject’s existence
� Reported study conclusions can be reconstructed based on documentation
� Document and explain any protocol deviations

47
Recommended Contents of Source Documents
� Subject identifiers
� Medical history
� Baseline presentation
� Clear evidence of study participation
� Study treatment information

48
Recommended Contents of Source Documents (2)
� Clear evidence of study progress and ongoing clinical course/evaluation:
� Results of diagnostic tests
� Documentation of adequate care for AE/SAEs & review of lab results
� Notations for each study visit, including:
• Each visit date and any AE/SAEs occurring
• Study Medication dose changes
• Concomitant medication

49
Source Data – Vitally Important
� First place where the clinical observations were recorded
� Part of the subjects medical records or study participation notes
� Property of the investigator/hospital (and/or subject)
� Should be written in real time, not weeks or months after an event

50
Source Documentation Verification (SDV)
� Verification of CRF data entries against information in the subject’s medical records and other available source documents fulfils several purposes:� Determines completeness & accuracy of CRF data and
authenticates it
� Per ICH, a documented agreement between site and investigator on which source documents are to be accessed to verify CRF data should be in place
� SDV Agreement

51
Why is SDV important?
� “It is the responsibility of the investigator to make all
data available to the sponsor/monitor and relevant
authorities for verification and audit and inspecti on
purposes”
Ref: Applied clinical trialsSept 1994, 3;9:38-45.

52
Study Records
4 simple rules:
� Accuracy
� Completeness
� Legibility
� Timelines

53
Essential Documents for the Conductof the Clinical Trial
ICH Section 8

54
Essential Documents
� Permit evaluation of trial conduct and data quality
� Demonstrate compliance of the investigator and the sponsor
� Assist in trial management
� Confirm validity of trial conduct and integrity of data during audit / inspection
� Important to have these documents during the trial and as an archive after the trial

55
Sponsor Responsibilities
•QA and QC (ICH-GCP 5.1)•Duties and Functions (ICH-GCP 5.7, 5.2)•Qualified Individuals (ICH-GCP 5.3-5.5)
•Data Handling (ICH-GCP 5.5)•Record Keeping (ICH-GCP 5.5)•Investigator Selection
•Compensation and Financing (ICH GCP 5.8-5.9)
•Regulatory Authority, IRB (ICH-GCP 5.10-5.11)
•Investigational Product (ICH GCP 5.12-5.14)•Record Access ( ICH GCP 5.15)
•Safety Information (ICH-GCP 5.16)
•Adverse Drug Reaction (ADR) Reporting (ICH-GCP 5.17)•Monitoring (ICH-GCP 5.18)
•Audit ( ICH GCP 5.19)
•Noncompliance (ICH GCP 5.20)
•Premature Termination of a Trial•Clinical Trial/Study Reports (ICH-GCP 5.22)

56
QA and QC (ICH-GCP 5.1)
�Implementing and maintaining QA and QC systems with written Standard Operating Procedures (SOPs)
�Written agreement with all involved parties to ensure direct access to sites, source docs & reports
� For monitoring, auditing, IEC/ERB review, regulatory inspection
� As part of protocol or separate agreement
�Verification that each subject consented in writing to direct access to their medical records
� QC for each stage of data handling
� For reliability and correctness of data

57
Duties and Functions (ICH-GCP 5.7, 5.2)
� May transfer sponsor’s duties and functions to Contract Research Organizations (CRO)
� Sponsor keeps responsibility for quality and integrity of trial data� Transfer to be specified in writing
� CRO should implement QA and QC
� Define, establish and allocate all trial-related duties and functions prior to initiating a trial

58
Qualified Individuals (ICH-GCP 5.3-5.5)
� Qualified Medical Personnel� Advise on trial related medical questions or problems
� Qualified individuals throughout all stages of Trial Process:� Design
� Trial Management
� Data Handling
� Record Keeping
59
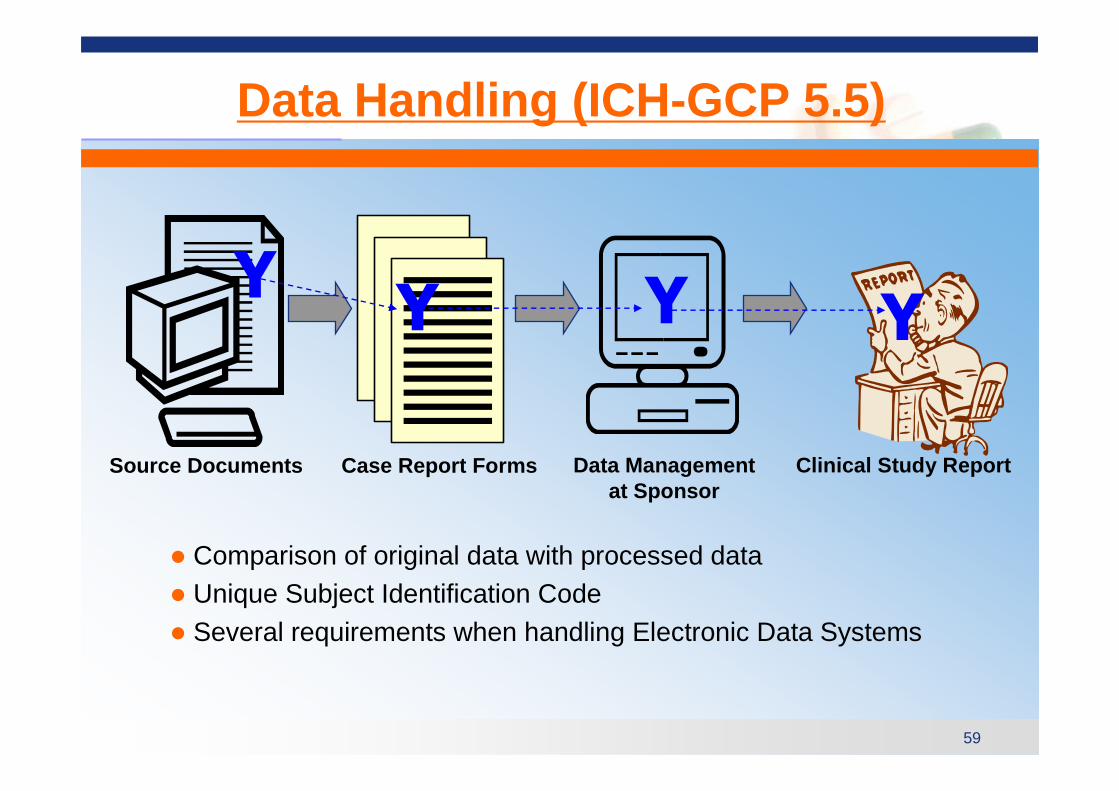
Data Handling (ICH-GCP 5.5)
� Comparison of original data with processed data� Unique Subject Identification Code� Several requirements when handling Electronic Data Systems
Source Documents Clinical Study ReportData Managementat Sponsor
Case Report Forms
YY Y Y

60
Record Keeping (ICH-GCP 5.5)
� Retain all of the sponsor-specific essential documents
� Inform the investigators/institutions in writing of the need for record retention
� Notify in writing when trial related records are no longer needed

61
Compensation and Financing (ICH GCP 5.8-5.9)
� Insurance of investigator against any claims arising from the trial (except malpractice and/or negligence)
� Compensation to subject(s) for trial-related injuries
� Compensation to subjects according to regulatory requirements
� Financial aspects of the trial documented in agreement between sponsor and investigator/ institution

62
Regulatory Authority, IRB (ICH-GCP 5.10-5.11)
� Submission to regulatory authority before study start for review, acceptance & permission
� Dated and allow identification of the protocol
� Sponsor should obtain from the investigator
� Name and address of IEC/IRB
� Statement that IEC/IRB is constituted and operates according to GCP and applicable laws
� Documented IEC/IRB approval
� Copy of the modification(s) made, if required by IEC/IRB, and approval by the IRB
� Copy of any reapprovals, if applicable

63
Investigational Product (ICH GCP 1.33)
A pharmaceutical form of an active ingredient or placebo being tested or used as a reference in a clinical trial
� including a product with a marketing authorization� when used or assembled (formulated or packaged) in a way
different from the approved form, OR
� when used for an unapproved indication, OR
� when used to gain further information about an approved use
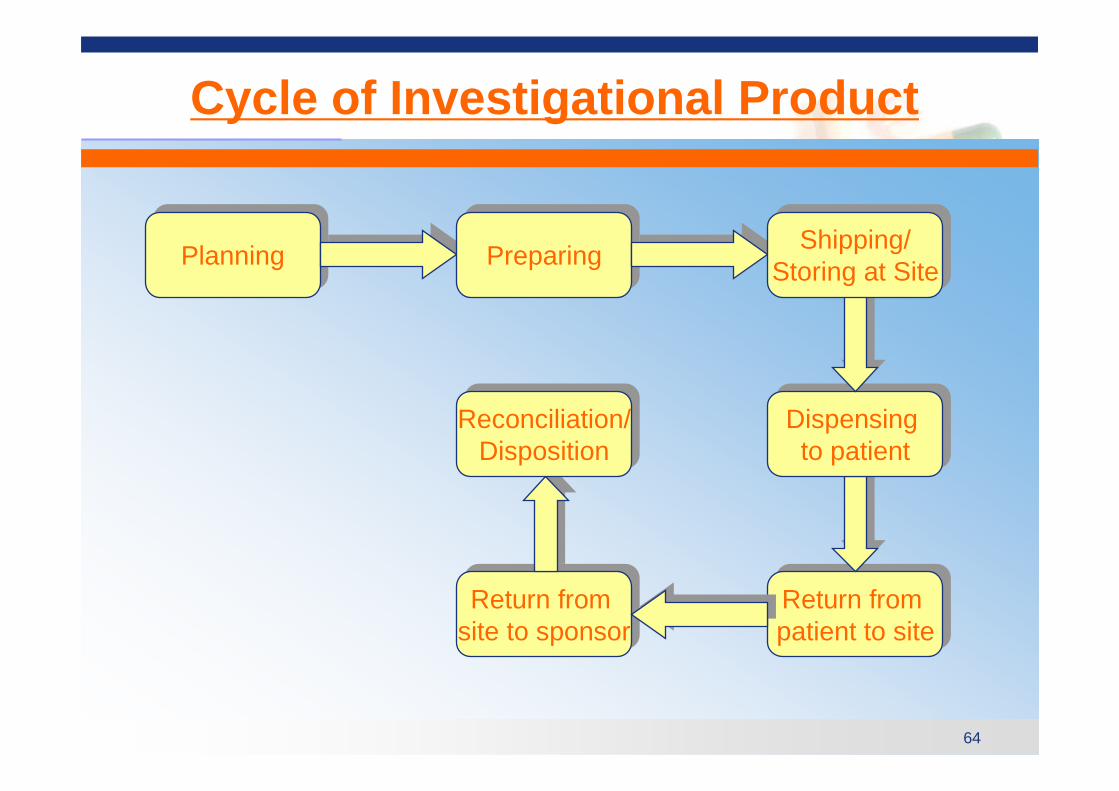
64
Cycle of Investigational Product
Return from patient to siteReturn from patient to site
PlanningPlanning PreparingPreparing
Return from site to sponsorReturn from
site to sponsor
Dispensing to patient
Dispensing to patient
Shipping/Storing at Site
Shipping/Storing at Site
Reconciliation/Disposition
Reconciliation/Disposition

65
� Ongoing safety evaluation of the Investigational Product
� Promptly notify all Investigators and Regulatory Authorities of findings that
� Could affect adversely the safety of subject
� Impact the conduct of the trial
� Alter the IEC/IRB approval to continue the trial
Safety Information (ICH-GCP 5.16)

66
Adverse Drug Reaction (ADR) Reporting (ICH-GCP 5.17)
� Expedited reporting of all serious, unexpected adverse drug reactions to
� Investigators
� IEC/IRB (where required) and
� Regulatory Authorities
� Safety updates and periodic reports to regulatory authorities (if required)

67
Monitoring – Purpose (ICH-GCP 5.18.1)
� Purposes of trial monitoring is to verify that:
� Rights and well-being of human subject protected
� Reported data are accurate, complete and verifiable
from source documents
� Trial conducted in compliance with protocol, with GCP
and local law

68
Monitors (ICH-GCP 5.18.2)
� Monitors appointed by sponsor
� Appropriately trained, scientific and/or clinical
knowledge
� needed
� Qualification should be documented
� Thoroughly familiar with� Investigational product
� Protocol
� Written information provided to subjects
� Sponsor’s SOPs
� GCP and applicable regulatory requirements

69
Clinical Trial/Study Reports (ICH-GCP 5.22)
� Clinical study reported prepared and submitted to the regulatory agencies (as required) for completed and terminated studies.
� CT reports in marketing applications should meet standards of the ICH Guideline for Structure and Content
of CT study reports.

70
Case Report Form (CRF) and Data Handling

71
Case Report Forms (CRFs or eCRFs)
� A printed, optical, or electronic document designed to record all of the protocol required information to be reported to the sponsor on each trial subject

72
Case Report Forms...
� Part of the clinical study
� Property of the sponsor
� Source data should not be a replica of the content of the CRF!

73
CRF as Source Document
� The CRF may suffice as the source document for data needed solely for trial purposes (i.e. not essential to the clinical care of subjects and not routinely recorded in clinical practice)
� patient self-rating assessments
� visual analogue scales
� repeated vital signs, etc.

74
ICH Expectations of CRFs
� Accurate, complete, legible, and timely
� Data consistent with the source documents or discrepancies explained
� Changes are signed, dated, and explained, without obscuring the original entry (i.e. maintain an audit trail)
ICH 4.9.1
ICH 4.9.2
ICH 4.9.3

75
Tips for Completing a CRF
� Print neatly and legibly
� Make all entries with black ink and press firmly so all copies are legible
� Avoid use of abbreviations and acronyms whenever possible Use only abbreviations that are clear and in standard medical use
� Comments must be clear and concise
� Write only in the designated areas of the CRF

76
Tips for Correcting a CRF
� Draw a single line through the incorrect entry
� Do not "write over" or erase an incorrect entry or re-copy the original page
� Do not use correction materials (e.g. correction fluid or tape)
� Write the correct data nearby
� Date and initial the correction

77
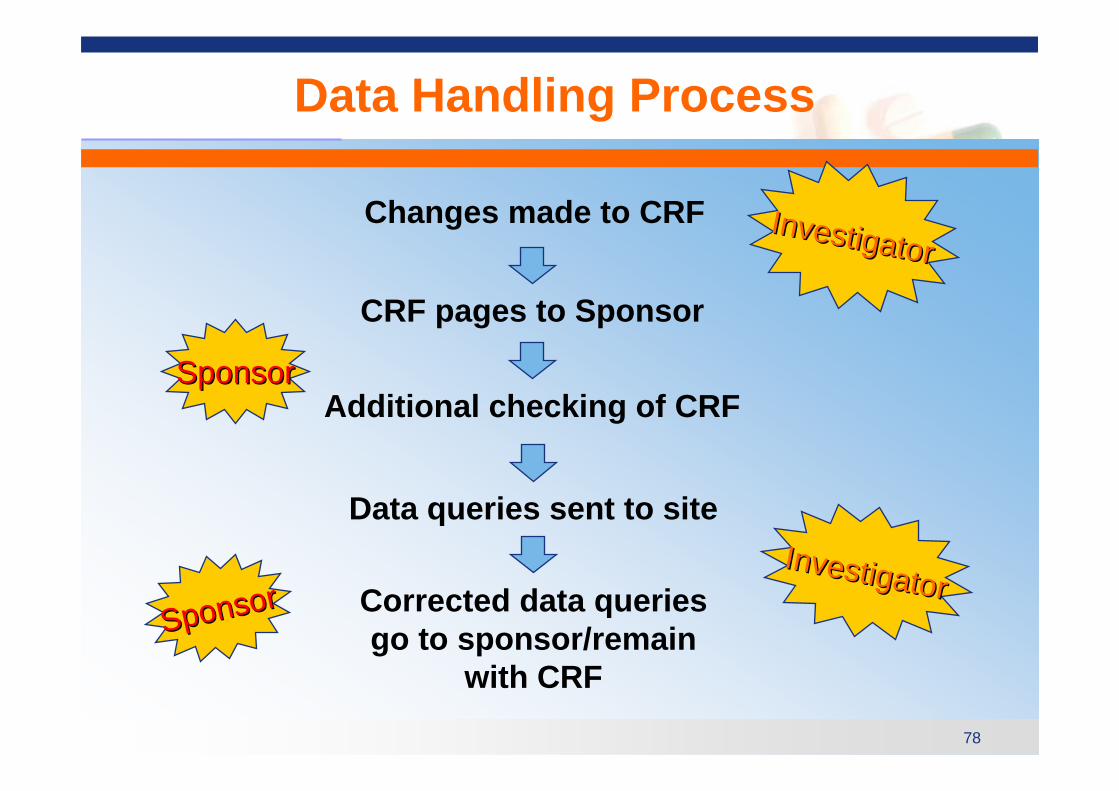
Data Handling Process
Patients enter studyPatients enter study
Review CRFReview CRF
CRFs completedCRFs completed
Any changes/clarifications requestedAny changes/clarifications requested
InvestigatorInvestigator
MonitorMonitor
MonitorMonitor
78
Data Handling Process
Changes made to CRF
Additional checking of CRF
CRF pages to Sponsor
Data queries sent to site
Corrected data queries go to sponsor/remain
with CRF
SponsorSponsor
InvestigatorInvestigator
InvestigatorInvestigator
SponsorSponsor

79
Good Source Documentation...
� Data that are:
� Attributable
� Legible
� ‘Written at that moment’
� Original
� Accurate

80
Electronic Subject Records
� Data must be retrievable and reproducible in a readable form
� Direct access to the relevant data must be available
� Printouts (signed & dated) are required of the electronic data if;
� monitor cannot view subject data directly, or
� reliability of computer system is in question

81
Understanding GCP

82
Good Clinical Practice_ Summary
A set of responsibilities
� Shared responsibilities
� Individual responsibilities
“ A process that make all parties to a study
responsible for patient safety and study quality.”

83
Module Objectives re-cap
You should now understand:
� What, Why and How of ICH GCP?
� ICH GCP Terminology
� Sets of Responsibilities
� Understanding Data Management Role in Clinical Trials

84





























