Embed Size (px)
Citation preview

Consultations in Molecular DiagnosticsIdentification of a Gene for Renal-Hepatic-PancreaticDysplasia by Microarray-Based Homozygosity Mapping
Torunn Fiskerstrand,* Gunnar Houge,*Staale Sund,† David Scheie,‡ Sabine Leh,§
Helge Boman,* and Per M. Knappskog*¶
From the Center for Medical Genetics and Molecular Medicine *
and the Department of Pathology,§ Haukeland University
Hospital, Bergen; the Department of Pathology,† Førde Central
Hospital, Førde; the Division of Pathology,‡ Rikshospitalet
University Hospital, Oslo; and the Department of Clinical
Medicine,¶ University of Bergen, Bergen, Norway
We have investigated a family where two siblings had adevelopmental disorder associated with polycystic dys-plastic kidney disease that was incompatible with post-natal survival. Additional features observed were ductalplate malformation in the liver, dysplasia of the pan-creas, and (in one individual) complete situs inversusand polymicrogyria of the cingulate gyri. The autopsyfindings were compatible with renal-hepatic-pancreaticdysplasia, a condition with unknown genetic cause atthe time of autopsy but with similarities to the Meckel-Gruber/Joubert group of recessive ciliopathies. Consan-guinity between the parents made it likely that the mu-tated gene (with known or potential function in cilia)was located within a rather large region of homozygos-ity in the affected individuals (identical by descent).Using genetic markers (50K single nucleotide polymor-phism microarrays), we found a single large homozygousregion of 21.16 Mb containing �200 genes on the long armof chromosome 3. This region contained two known cili-opathy genes: NPHP3 (adolescent nephronophthisis) andIQCB1 (NPHP5), which is associated with Senior-Lokensyndrome. In NPHP3, homozygosity for a deletion of theconserved splice acceptor dinucleotide (AG) precedingexon 20 was found. Our finding confirms the recent re-port that NPHP3-null mutations cause renal-hepatic-pan-creatic dysplasia. Also, our case illustrates that genes forrare and genetically heterogeneous recessive condi-tions may be identified by homozygosity mappingusing single nucleotide polymorphism arrays in theroutine clinical setting. (J Mol Diagn 2010, 12:125–131;
DOI: 10.2353/jmoldx.2010.090033)
The combination of cystic kidneys and hepatic fibrosis ina fetus or newborn is usually indicative of infantile auto-somal recessive polycystic kidney disease. This condi-tion is often lethal because of oligohydramnios with sub-sequent pulmonal hypoplasia. In addition to autosomalrecessive polycystic kidney disease, cystic kidneys andhepatic periportal fibrosis with or without bile-duct anom-alies (ductal plate malformation) may also be found inother neonatal lethal syndromes that belong to the rapidlyincreasing group of inherited diseases called ciliopa-thies.1 This group includes Meckel-Gruber syndrome,severe forms of autosomal dominant polycystic kidneydisease, Joubert syndrome, and Bardet-Biedl syndrome(Table 1). Because of the widespread importance of pri-mary cilia for normal organ development, ciliopathiesmay have quite variable phenotypes and degree of organinvolvement.1 Since cilia are also involved in left-rightbody asymmetry determination, ciliopathies are also acause of lateralization defects, including situs inversus.Because the recurrence risk for a couple with a child witha lethal ciliopathy is 25% in most cases, a DNA-basedprenatal diagnostic test that can be applied at an early timepoint in subsequent pregnancies is often desired. However,genetic heterogeneity and variable expression of differentmutations in the same gene complicate molecular diagnos-tics (Table 1). Thus, mutations in CEP290 may cause iso-lated retinal disease (Leber’s congenital amaurosis), Jou-bert syndrome, or Meckel-Gruber syndrome,2 and RPGRIP1Lmutations are seen in both Joubert and Meckel-Grubersyndromes.3 Antenatal lethal cases with polycystic kidneysand ductal plate malformation but without encephalocelemay be caused by mutations in the Bardet-Biedl genesBBS2, BBS4, or BBS64 or by mutations in the Meckel-Gruber genes MKS1, MKS3, and CEP2905 (Table 1).
Renal-hepatic-pancreatic dysplasia (RHPD) is anotherlethal condition reminiscent of a ciliopathy, first describedby Ivemark et al6 and sometimes referred to as Ivemark IIsyndrome (OMIM number 208540). In addition to the
Supported by a grant from Helse Vest (Western Norway Regional HealthAuthority) (911308 to P.M.K., T.F., and H.B.).
Accepted for publication July 2, 2009.
Address reprint requests to Torunn Fiskerstrand, M.D., Ph.D., Center forMedical Genetics and Molecular Medicine, Haukeland University Hospital,N-5021 Bergen, Norway. E-mail: [email protected].
Journal of Molecular Diagnostics, Vol. 12, No. 1, January 2010
Copyright © American Society for Investigative Pathology
and the Association for Molecular Pathology
DOI: 10.2353/jmoldx.2010.090033
125

signs mentioned above, cystic dysplasia of the pancreasand situs inversus or other lateralization defects can beseen. Here, we describe two siblings with severe malfor-mations compatible with RHPD, born to remotely consan-guineous parents. At the time of diagnosis, the moleculargenetic cause of RHPD was unknown. Because a searchfor a causative gene among the known ciliopathy genesis expensive, time-consuming and possibly inconclusive,we used microarrays with 50,000 single nucleotide poly-morphisms (Affymetrix 50K) to identify a chromosomallocus for such a gene, with the hypothesis that theaffected siblings had inherited the same mutation fromboth the father and the mother (ie, the mutation wasidentical by descent from the same ancestor).7 Thismethod is called homozygosity mapping.7 The affectedsiblings will also be homozygous for all of the geneticmarkers in close vicinity of the mutation, and the size ofthe homozygosity region is dependent on how closelyrelated the parents are.
Materials and Methods
Subjects
DNA was obtained from the affected deceased male andfemale fetus, three healthy siblings (one girl and twoboys), both parents and three grandparents. The parentswere double fourth cousins (Figure 1, only one inbreed-ing loop is shown). The adults gave informed consent tothe investigation and publication of the results.
Pathological Examination
A complete perinatal autopsy was performed in bothcases. In case 2, the small 16-weeks fetus was examinedin part under a dissection microscope (Olympus SZ40;Olympus Medical Systems Corp., Tokyo, Japan). Forma-lin-fixed tissue specimens from fetal viscera and placentawere processed in paraffin blocks and cut for microscopyaccording to routine laboratory procedures.
Genotyping and Homozygosity Mapping
The expected size and number of regions of homozygos-ity in the child may be estimated when the relationshipbetween the parents is known. In this case, we expectedonly one candidate region of �16 cM.8 Whole-genomescan was performed using the Affymetrix 50K Chip (Af-fymetrix, Santa Clara, CA), and the data were exportedand treated for further analysis by use of the programsGTYPE (Afymetrix) and Progeny Lab (Progeny Software,South Norfolk, UK). Regions of homozygosity were identi-fied using the PLINK software.9 Physical marker positionsare given according to NCBI Build 36.3 (http://www.ncbi.nlm.nih.gov/mapview, accessed March 1, 2008).
DNA Sequencing and Mutation Detection
PCR primers for amplification of exons and flankingintronic sequences of both NPHP3 and IQCB1 were
Table 1. Differential Diagnoses in Lethal Cystic Kidney Disease
RHPD
Autosomalrecessive
polycystic kidneydisease
Autosomaldominant
polycystic kidneydisease* Meckel-Gruber Joubert Bardet-Biedl
Multicysticdysplastickidneys,†
bilateral
Polycystic kidneys Cysticdysplasia
Small, uniformcorticomedullarycysts
Variable picture,whole nephronaffected
Cysticdysplasia
Cystic dysplasia,Nephronophthisis
Cysticdysplasiaormedullarycysts
Cysticdysplasia
Ductal platemalformation
� � (�) � � �
Pancreatic dysplasia �� (�)Situs inversus/
heterotaxia� � � �
Encephalocele,Neural tube defects
�� (�)
Polydactyly (�) � � �Molar tooth sign (�) ��Genes mutated NPHP3 PKHD1 PKD1, PKD2 MKS1
TMEM67RPGRIP1LCEP290CC2D2A
AHI1NPHP1ARL13BTMEM67RPGRIP1LCEP290CC2D2A
BBS2BBS4BBS6MKS1TMEM67CEP290
Inheritance Autosomalrecessive
Autosomalrecessive
Autosomaldominant
Autosomalrecessive
Autosomalrecessive
Autosomalrecessive
Sporadic
Typical findings in lethal forms of these conditions are listed but not symptoms known to occur in surviving individuals, like for example, retinitispigmentosa in Bardet-Biedl or Joubert syndrome.
�Rarely, autosomal dominant polycystic kidney disease may present antenatally or perinatally with severe cystic kidney disease and ductal platemalformation.
†Multicystic dysplastic kidneys: Sporadic condition, low risk of recurrence, although an autosomal dominant condition with variable penetrance havebeen described previously.
126 Fiskerstrand et alJMD January 2010, Vol. 12, No. 1

designed using the Oligo 6.3 program (National Bio-science, Plymouth, MN). Reference sequences for NPHP3and IQCB1 are NM_153240 and NM_014642 respectively.PCR products were treated with Shrimp alkaline phos-phatase/exonuclease I (Amersham Biosciences, Piscat-away, NJ) and sequenced using the PRISM BigDye Ter-minator kit (version 1.1) and an ABI 3730 GeneticAnalyzer (Applied Biosystems, Foster City, CA). Data
analysis was assisted by use of the Seqscape software(Applied Biosystems).
RNA Analyses
Blood from the parents and normal controls were col-lected in Tempus tubes (Applied Biosystems). Total RNAwas purified using the ABI 6100 system (Applied Biosys-tems), and the quality of the RNA was analyzed using theExperion system (Bio-Rad, Hercules, CA). cDNA synthe-sis was performed using the TaqMan Reverse Transcrip-tion kit (Applied Biosystems). PCR amplification of thecDNA (exons 19 to 21) was performed using the 5�-CAGATGAACTCCCGTGGC-3� and 5�-AAAGGGCAAAT-CCGTACAAGT-3� primers. The PCR products were clonedinto the pCR2.1 vector using the Invitrogen TOPO TA Clon-ing Kit (Invitrogen, Carlsbad, CA). Clones were sequencedusing the Prism BigDye terminator version 1.1 kit and the3730 Genetic Analyzer (Applied Biosystems).
Immunohistochemistry
Immunohistochemistry was performed on formalin-fixedand paraffin-embedded kidney tissue sections. Antigenretrieval was performed for 15 minutes in citraconic an-hydride in a pressure cooker. Sections were incubatedwith nephrocystin-3 antibody (N-18, 1/500; Santa CruzBiotechnology, Santa Cruz, CA). Bound antibodies weredetected by a streptavidin-biotin kit (LSAB; DakoCytoma-tion, Carpinteria, CA) or EnVision detection kit (DakoCy-tomation) in combination with a link antibody from rabbit.Diaminobenzidine was used for visualization of immuno-reactivity, followed by hematoxylin for nuclear counter-staining. In the negative controls, the primary antibodywas omitted. Age-matched normal kidney tissue servedas positive control.
Results
Clinical Findings
Case 1: Boy Delivered by Caesarian Section in Week 34
This was the couple’s first pregnancy. Caesarean sec-tion was performed in the 34th gestational week becauseof intrauterine growth retardation and oligohydramnios.Birth weight was 1775 g. The baby boy died 1 day afterdelivery due to respiratory insufficiency. An autopsy re-vealed typical Potter facies with hypertelorism, a beak-like nose, low-set ears, a short neck, and a short sternum.Lungs and kidneys were hypoplastic. The lungs weighed13.9 g (left) and 16.0 g (right) (reference weight is 19.8and 22.6 g, respectively). The kidneys weighed 3.7 g(left) and 3.6 g (right) (reference weight is 9.5 and 9.1 g,respectively10). The kidneys showed complete loss ofnormal architecture with multiple small cysts. The pan-creas seemed enlarged and fibrotic (pancreas weight,4.6 g; reference weight, 4.0 g). Microscopic examinationof the kidneys showed primitive tubules and tubular cystsin a mesenchymal stroma with rare malformed glomeruli
Figure 1. Pedigree and haplotypes in the candidate region on chromosome3q. Selected single nucleotide polymorphisms and their distance in mega-bases from the end of the short arm of chromosome 3 are shown in the leftcolumns in consecutive order. Parental haplotypes have been given differ-ent colors, with the shared (ancestral) haplotype in red. The boundaries ofthe candidate region are indicated by dashed lines. The NPHP3 gene islocated 133.88 to 133.92 Mb and the IQCB1 gene 122.97 to 123.03 Mb fromthe start of chromosome 3 (3pter).
Renal-Hepatic-Pancreatic Dysplasia 127JMD January 2010, Vol. 12, No. 1
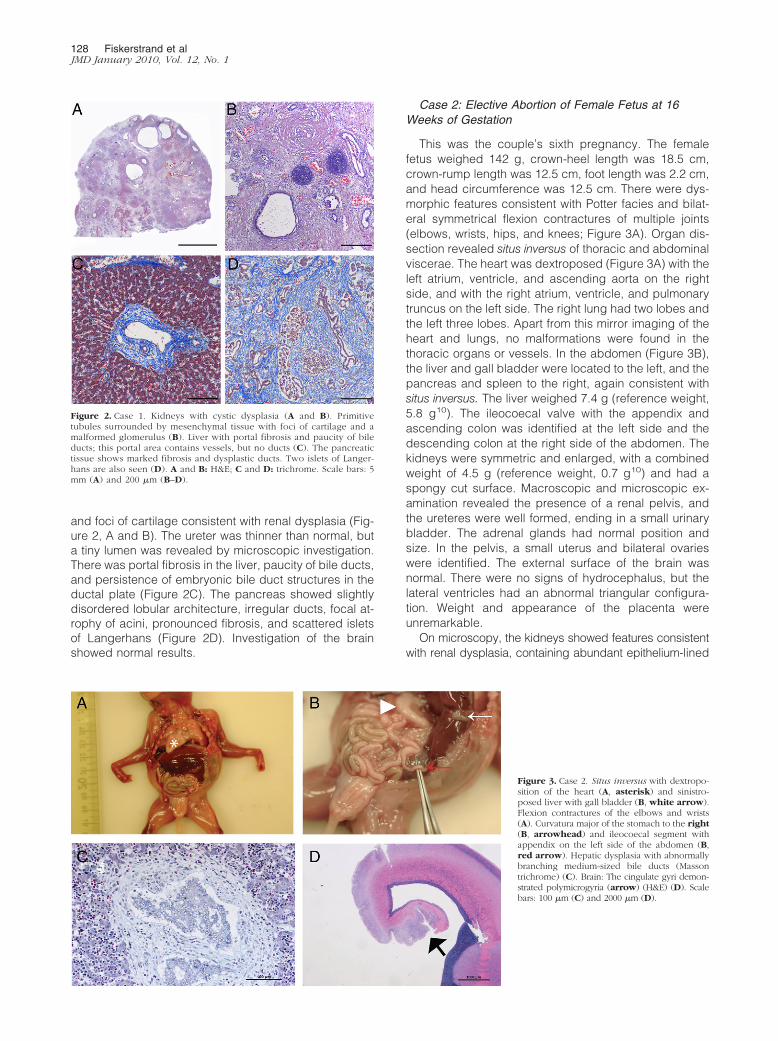
and foci of cartilage consistent with renal dysplasia (Fig-ure 2, A and B). The ureter was thinner than normal, buta tiny lumen was revealed by microscopic investigation.There was portal fibrosis in the liver, paucity of bile ducts,and persistence of embryonic bile duct structures in theductal plate (Figure 2C). The pancreas showed slightlydisordered lobular architecture, irregular ducts, focal at-rophy of acini, pronounced fibrosis, and scattered isletsof Langerhans (Figure 2D). Investigation of the brainshowed normal results.
Case 2: Elective Abortion of Female Fetus at 16Weeks of Gestation
This was the couple’s sixth pregnancy. The femalefetus weighed 142 g, crown-heel length was 18.5 cm,crown-rump length was 12.5 cm, foot length was 2.2 cm,and head circumference was 12.5 cm. There were dys-morphic features consistent with Potter facies and bilat-eral symmetrical flexion contractures of multiple joints(elbows, wrists, hips, and knees; Figure 3A). Organ dis-section revealed situs inversus of thoracic and abdominalviscerae. The heart was dextroposed (Figure 3A) with theleft atrium, ventricle, and ascending aorta on the rightside, and with the right atrium, ventricle, and pulmonarytruncus on the left side. The right lung had two lobes andthe left three lobes. Apart from this mirror imaging of theheart and lungs, no malformations were found in thethoracic organs or vessels. In the abdomen (Figure 3B),the liver and gall bladder were located to the left, and thepancreas and spleen to the right, again consistent withsitus inversus. The liver weighed 7.4 g (reference weight,5.8 g10). The ileocoecal valve with the appendix andascending colon was identified at the left side and thedescending colon at the right side of the abdomen. Thekidneys were symmetric and enlarged, with a combinedweight of 4.5 g (reference weight, 0.7 g10) and had aspongy cut surface. Macroscopic and microscopic ex-amination revealed the presence of a renal pelvis, andthe ureteres were well formed, ending in a small urinarybladder. The adrenal glands had normal position andsize. In the pelvis, a small uterus and bilateral ovarieswere identified. The external surface of the brain wasnormal. There were no signs of hydrocephalus, but thelateral ventricles had an abnormal triangular configura-tion. Weight and appearance of the placenta wereunremarkable.
On microscopy, the kidneys showed features consistentwith renal dysplasia, containing abundant epithelium-lined
Figure 2. Case 1. Kidneys with cystic dysplasia (A and B). Primitivetubules surrounded by mesenchymal tissue with foci of cartilage and amalformed glomerulus (B). Liver with portal fibrosis and paucity of bileducts; this portal area contains vessels, but no ducts (C). The pancreatictissue shows marked fibrosis and dysplastic ducts. Two islets of Langer-hans are also seen (D). A and B: H&E; C and D: trichrome. Scale bars: 5mm (A) and 200 �m (B–D).
Figure 3. Case 2. Situs inversus with dextropo-sition of the heart (A, asterisk) and sinistro-posed liver with gall bladder (B, white arrow).Flexion contractures of the elbows and wrists(A). Curvatura major of the stomach to the right(B, arrowhead) and ileocoecal segment withappendix on the left side of the abdomen (B,red arrow). Hepatic dysplasia with abnormallybranching medium-sized bile ducts (Massontrichrome) (C). Brain: The cingulate gyri demon-strated polymicrogyria (arrow) (H&E) (D). Scalebars: 100 �m (C) and 2000 �m (D).
128 Fiskerstrand et alJMD January 2010, Vol. 12, No. 1
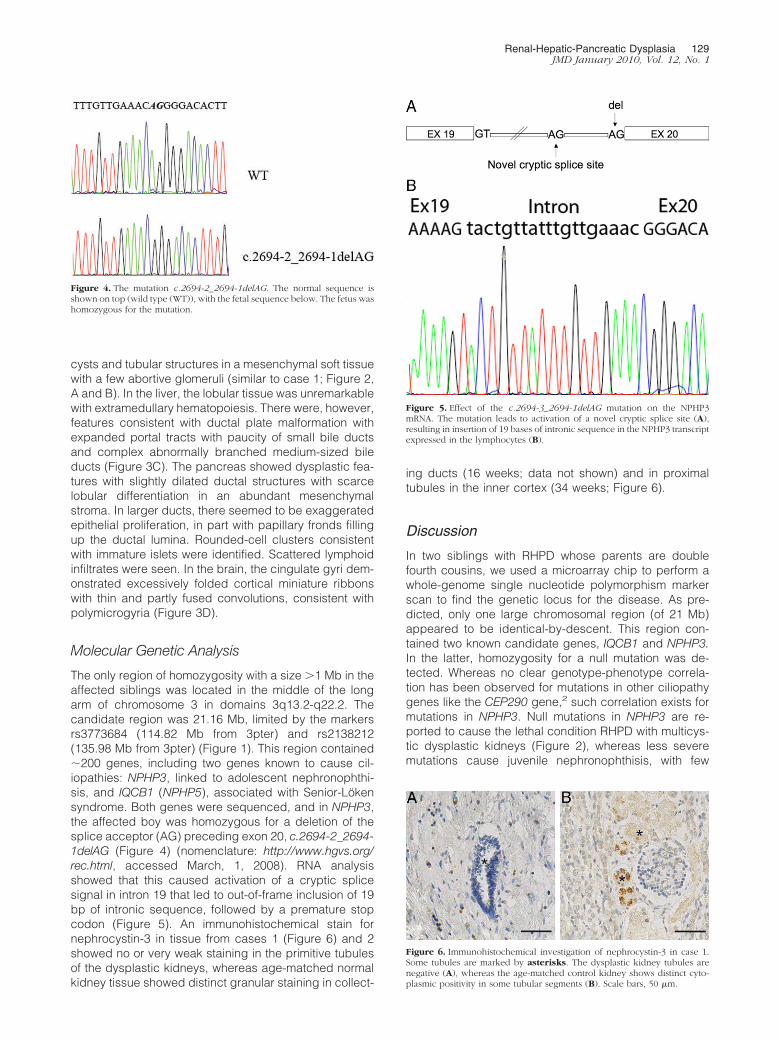
cysts and tubular structures in a mesenchymal soft tissuewith a few abortive glomeruli (similar to case 1; Figure 2,A and B). In the liver, the lobular tissue was unremarkablewith extramedullary hematopoiesis. There were, however,features consistent with ductal plate malformation withexpanded portal tracts with paucity of small bile ductsand complex abnormally branched medium-sized bileducts (Figure 3C). The pancreas showed dysplastic fea-tures with slightly dilated ductal structures with scarcelobular differentiation in an abundant mesenchymalstroma. In larger ducts, there seemed to be exaggeratedepithelial proliferation, in part with papillary fronds fillingup the ductal lumina. Rounded-cell clusters consistentwith immature islets were identified. Scattered lymphoidinfiltrates were seen. In the brain, the cingulate gyri dem-onstrated excessively folded cortical miniature ribbonswith thin and partly fused convolutions, consistent withpolymicrogyria (Figure 3D).
Molecular Genetic Analysis
The only region of homozygosity with a size �1 Mb in theaffected siblings was located in the middle of the longarm of chromosome 3 in domains 3q13.2-q22.2. Thecandidate region was 21.16 Mb, limited by the markersrs3773684 (114.82 Mb from 3pter) and rs2138212(135.98 Mb from 3pter) (Figure 1). This region contained�200 genes, including two genes known to cause cil-iopathies: NPHP3, linked to adolescent nephronophthi-sis, and IQCB1 (NPHP5), associated with Senior-Lokensyndrome. Both genes were sequenced, and in NPHP3,the affected boy was homozygous for a deletion of thesplice acceptor (AG) preceding exon 20, c.2694-2_2694-1delAG (Figure 4) (nomenclature: http://www.hgvs.org/rec.html, accessed March, 1, 2008). RNA analysisshowed that this caused activation of a cryptic splicesignal in intron 19 that led to out-of-frame inclusion of 19bp of intronic sequence, followed by a premature stopcodon (Figure 5). An immunohistochemical stain fornephrocystin-3 in tissue from cases 1 (Figure 6) and 2showed no or very weak staining in the primitive tubulesof the dysplastic kidneys, whereas age-matched normalkidney tissue showed distinct granular staining in collect-
ing ducts (16 weeks; data not shown) and in proximaltubules in the inner cortex (34 weeks; Figure 6).
Discussion
In two siblings with RHPD whose parents are doublefourth cousins, we used a microarray chip to perform awhole-genome single nucleotide polymorphism markerscan to find the genetic locus for the disease. As pre-dicted, only one large chromosomal region (of 21 Mb)appeared to be identical-by-descent. This region con-tained two known candidate genes, IQCB1 and NPHP3.In the latter, homozygosity for a null mutation was de-tected. Whereas no clear genotype-phenotype correla-tion has been observed for mutations in other ciliopathygenes like the CEP290 gene,2 such correlation exists formutations in NPHP3. Null mutations in NPHP3 are re-ported to cause the lethal condition RHPD with multicys-tic dysplastic kidneys (Figure 2), whereas less severemutations cause juvenile nephronophthisis, with few
Figure 4. The mutation c.2694-2_2694-1delAG. The normal sequence isshown on top (wild type (WT)), with the fetal sequence below. The fetus washomozygous for the mutation.
Figure 5. Effect of the c.2694-3_2694-1delAG mutation on the NPHP3mRNA. The mutation leads to activation of a novel cryptic splice site (A),resulting in insertion of 19 bases of intronic sequence in the NPHP3 transcriptexpressed in the lymphocytes (B).
Figure 6. Immunohistochemical investigation of nephrocystin-3 in case 1.Some tubules are marked by asterisks. The dysplastic kidney tubules arenegative (A), whereas the age-matched control kidney shows distinct cyto-plasmic positivity in some tubular segments (B). Scale bars, 50 �m.
Renal-Hepatic-Pancreatic Dysplasia 129JMD January 2010, Vol. 12, No. 1

cysts, tubular atrophy, and interstitial fibrosis.11 However,two cases with a juvenile nephronopthisis-like phenotypedespite presumed null mutations in NPHP3 have beendescribed, indicating that this correlation may not alwaysbe clear-cut. The first case was homozygous for an oblig-atory splice acceptor-site mutation (IVS26 � 1 G�A), andthis child had no extrarenal manifestations of disease.12
The second case was combined heterozygous for aframeshift mutation (435_438delAAGT) and a splice mu-tation (IVS24-1G�C) in NPHP313 and was also heterozy-gous for a frameshift mutation in the NPHP4 gene. Thischild had liver fibrosis and reached end-stage renal dis-ease at age 3 years. RNA analysis was not reported inthese cases. It is possible that at least in the case ho-mozygous for IVS26 � 1 G�A, a partly functional tran-script could exist due to alternative splicing and alsobecause the mutation affected the penultimate exon ofthe gene, possibly resulting in a truncated but somewhatfunctional protein. The finding of instances with likelytriallelic inheritance also adds to the phenotypic hetero-geneity in ciliopathies.13,14 Thus, it is possible that allelicvariation or mutations in other genes involved in ciliadevelopment and function may affect the phenotype inNPHP3-related disease.
The mutation c.2694-2_2694-1delAG in our ethnic Nor-wegian family was also reported in a Turkish family in afemale fetus and a female sibling who died perinatally(both were homozygous).11 Thus, this may represent arecurrent mutation. Both siblings had enlarged multicys-tic kidneys, oligohydramnios, and ductal plate malforma-tion. The fetus had normal findings in the pancreas. Noinformation was given about the pancreas in the baby girlthat died perinatally. None of the siblings had lateraliza-tion defects or polymicrogyria, as seen in our case 2,further demonstrating variability of phenotype in individ-uals with the same mutation. Polymicrogyria of the cingu-late gyrus, as we found in case 2, has not been reportedbefore in RHPD and is probably a rare event in ciliopa-thies, although it may be underreported. Dixon-Salazar etal15 found frontal polymicrogyria and thin corpus callo-sum in three cases with Joubert syndrome with mutationsin the AHI1 gene, and they argued that this finding ofsupratentorial brain affection pointed to a role for thisgene also in cortical development. Han et al16 showedthat intact primary cilia and cilia-associated sonic hedge-hog signaling are essential for development of adult neu-ral stem cells. Nephrocystin-3 is expressed in neuraltissues and interacts with inversin11 and nephrocystin-1,12 but whether it also interacts with jouberin (the proteinencoded by AHI1) is not known. Jouberin, however, in-teracts with nephrocystin-1.17
Diagnostic stratification of cystic kidney disease infetuses and stillborns is challenging because many of theconditions have overlapping clinical pictures. After athorough clinical and pathological characterization, theremay be many candidate genes that could harbor muta-tion(s). A strategy to find the single major disease-caus-ing gene among many candidates would be useful. Incases where parents are consanguineous and the dis-ease is presumed to be recessive, homozygosity map-ping is the method of choice to find the locus (position) of
the mutated gene. Most often, such a locus contains oneor a few genes known to be involved in a ciliopathy, inother cases, new ciliopathy genes may be searchedfor. Many ciliopathy genes remain to be discovered, asillustrated by the fact that about half of, for example,Joubert syndrome cases18 and even more of theMeckel-Gruber syndrome cases,19 remain geneticallyunexplained, after all relevant ciliopathy genes havebeen sequenced. The new high-throughput single nu-cleotide polymorphism arrays have made homozygos-ity mapping7 feasible also as a routine screening pro-cedure, with an analytic turnaround time of about aweek after DNA samples from a sufficient number offamily members have been collected.
Acknowledgments
The technical assistance of Jorunn Skeie Bringsli, GuriMatre (Center for Medical Genetics and Molecular Med-icine), and Kalairasy Kugarajh (The Norwegian KidneyBiopsy Registry) was highly appreciated.
References
1. Badano JL, Mitsuma N, Beales PL, Katsanis N: The ciliopathies: anemerging class of human genetic disorders. Annu Rev GenomicsHum Genet 2006, 7:125–148
2. Frank V, den Hollander AI, Bruchle NO, Zonneveld MN, Nurnberg G,Becker C, Du Bois G, Kendziorra H, Roosing S, Senderek J, NurnbergP, Cremers FP, Zerres K, Bergmann C: Mutations of the CEP290 geneencoding a centrosomal protein cause Meckel-Gruber syndrome.Hum Mutat 2008, 29:45–52
3. Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, GolzioC, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I,Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, GublerMC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, MacherMA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F,Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U,Schneider-Maunoury S, Attie-Bitach T, Saunier S: The ciliary geneRPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubertsyndrome type B) and Meckel syndrome. Nat Genet 2007,39:875–881
4. Karmous-Benailly H, Martinovic J, Gubler MC, Sirot Y, Clech L, OzilouC, Auge J, Brahimi N, Etchevers H, Detrait E, Esculpavit C, AudollentS, Goudefroye G, Gonzales M, Tantau J, Loget P, Joubert M, GaillardD, Jeanne-Pasquier C, Delezoide AL, Peter MO, Plessis G, Simon-Bouy B, Dollfus H, Le Merrer M, Munnich A, Encha-Razavi F,Vekemans M, Attie-Bitach T: Antenatal presentation of Bardet-Biedlsyndrome may mimic Meckel syndrome. Am J Hum Genet 2005,76:493–504
5. Leitch CC, Zaghloul NA, Davis EE, Stoetzel C, Diaz-Font A, Rix S,Alfadhel M, Lewis RA, Eyaid W, Banin E, Dollfus H, Beales PL,Badano JL, Katsanis N: Hypomorphic mutations in syndromic en-cephalocele genes are associated with Bardet-Biedl syndrome. NatGenet 2008, 40:443–448
6. Ivemark BI, Oldfelt V, Zetterstrom R: Familial dysplasia of kidneys,liver and pancreas: a probably genetically determined syndrome.Acta Paediatr 1959, 48:1–11
7. Lander ES, Botstein D: Homozygosity mapping: a way to map humanrecessive traits with the DNA of inbred children. Science 1987,236:1567–1570
8. Genin E, Todorov AA, Clerget-Darpoux F: Optimization of genomesearch strategies for homozygosity mapping: influence of markerspacing on power and threshold criteria for identification of candidateregions. Ann Hum Genet 1998, 62:419–429
9. Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D,Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC: PLINK: a tool set
130 Fiskerstrand et alJMD January 2010, Vol. 12, No. 1

for whole-genome association and population-based linkage analy-ses. Am J Hum Genet 2007, 81:559–575
10. Guihard-Costa AM, Menez F, Delezoide AL: Organ weights in humanfetuses after formalin fixation: standards by gestational age and bodyweight. Pediatr Dev Pathol 2002, 5:559–578
11. Bergmann C, Fliegauf M, Bruchle NO, Frank V, Olbrich H, KirschnerJ, Schermer B, Schmedding I, Kispert A, Kranzlin B, Nurnberg G,Becker C, Grimm T, Girschick G, Lynch SA, Kelehan P, Senderek J,Neuhaus TJ, Stallmach T, Zentgraf H, Nurnberg P, Gretz N, Lo C,Lienkamp S, Schafer T, Walz G, Benzing T, Zerres K, Omran H: Lossof nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreaticdysplasia. Am J Hum Genet 2008, 82:959–970
12. Olbrich H, Fliegauf M, Hoefele J, Kispert A, Otto E, Volz A, Wolf MT,Sasmaz G, Trauer U, Reinhardt R, Sudbrak R, Antignac C, Gretz N,Walz G, Schermer B, Benzing T, Hildebrandt F, Omran H: Mutationsin a novel gene, NPHP3, cause adolescent nephronophthisis, tapeto-retinal degeneration and hepatic fibrosis. Nat Genet 2003, 34:455–459
13. Hoefele J, Wolf MT, O’Toole JF, Otto EA, Schultheiss U, DeschenesG, Attanasio M, Utsch B, Antignac C, Hildebrandt F: Evidence ofoligogenic inheritance in nephronophthisis. J Am Soc Nephrol 2007,18:2789–2795
14. Katsanis N: The oligogenic properties of Bardet-Biedl syndrome.Hum Mol Genet 2004, 13(Spec No 1):R65–R71
15. Dixon-Salazar T, Silhavy JL, Marsh SE, Louie CM, Scott LC, Gururaj A,
Al-Gazali L, Al-Tawari AA, Kayserili H, Sztriha L, Gleeson JG: Muta-tions in the AHI1 gene, encoding jouberin, cause Joubert syndromewith cortical polymicrogyria. Am J Hum Genet 2004, 75:979–987
16. Han YG, Spassky N, Romaguera-Ros M, Garcia-Verdugo JM, AguilarA, Schneider-Maunoury S, Alvarez-Buylla A: Hedgehog signaling andprimary cilia are required for the formation of adult neural stem cells.Nat Neurosci 2008, 11:277–284
17. Eley L, Gabrielides C, Adams M, Johnson CA, Hildebrandt F, SayerJA: Jouberin localizes to collecting ducts and interacts with nephro-cystin-1. Kidney Int 2008, 74:1139–1149
18. Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY,Audollent S, Attie-Bitach T, Holden KR, Dobyns WB, Traver D,Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F,Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM,Woods CG, Gleeson JG: Mutations in the cilia gene ARL13B lead tothe classical form of Joubert syndrome. Am J Hum Genet 2008,83:170–179
19. Baala L, Audollent S, Martinovic J, Ozilou C, Babron MC,Sivanandamoorthy S, Saunier S, Salomon R, Gonzales M, RattenberryE, Esculpavit C, Toutain A, Moraine C, Parent P, Marcorelles P, DaugeMC, Roume J, Le Merrer M, Meiner V, Meir K, Menez F, Beaufrere AM,Francannet C, Tantau J, Sinico M, Dumez Y, MacDonald F, MunnichA, Lyonnet S, Gubler MC, Genin E, Johnson CA, Vekemans M,Encha-Razavi F, Attie-Bitach T: Pleiotropic effects of CEP290(NPHP6) mutations extend to Meckel syndrome. Am J Hum Genet2007, 81:170–179
Renal-Hepatic-Pancreatic Dysplasia 131JMD January 2010, Vol. 12, No. 1





























