Embed Size (px)
Citation preview

Ciba Foundation Symposium 147 -
IgE, MAST CELLS AND THE
ALLERGIC RESPONSE
A Wiley-lnterscience Publication
1989
JOHN WILEY & SONS -~ ~ ~
Chichester New York . Brisbane . Toronto Singapore


IgE, MAST CELLS AND THE
ALLERGIC RESPONSE

The Ciba Foundation is an international scientific and educational charity. It was established in 1947 by the Swiss chemical and pharmaceutical company of ClBA Limited-now CIBA-GEIGY Limited. The Foundation operates independently in London under English trust law.
The Ciba Foundation exists to promote international cooperation in biological, medical and chemical research. It organizes about eight international multidisciplinary symposia each year on topics that seem ready for discussion by a small group of research workers. The papers and discussions are published in the Ciba Foundation symposium series. The Foundation also holds many shorter meetings (not published), organized by the Foundation itself or by outside scientific organizations. The staff always welcome suggestions for future meetings.
The Foundation's house at 41 Portland Place, London W1N 48N, provides facilities for meetings of all kinds. Its Media Resource Service supplies information to journalists on all scientific and technological topics. The library, open seven days a week to any graduate in science or medicine, also provides information on scientific meetings throughout the world and answers general enquiries on biomedical and chemical subjects. Scientists from any part of the world may stay in the house during working visits to London.

Ciba Foundation Symposium 147 -
IgE, MAST CELLS AND THE
ALLERGIC RESPONSE
A Wiley-lnterscience Publication
1989
JOHN WILEY & SONS -~ ~ ~
Chichester New York . Brisbane . Toronto Singapore

OCiba Foundation 1989
Published in 1989 by John Wiley & Sons Ltd, Chichester, UK.
Suggested series entry for library catalogues: Ciba Foundation Symposia
Ciba Foundation Symposium 147 ix + 282 pages, 40 figures, 23 tables
Library of Congress Cataloging-in-Publication Data
IgE, mast cells, and the allergic response. p. cm.-(Ciba Foundation symposium ; 147)
“Symposium on IgE, Mast Cells, and the Allergic Response, held at
Edited and organized by Derek Chadwick and others. “A Wiley-Interscience publication.” Includes bibliographical references. ISBN 0 471 92309 5 1. Immunoglobulin E-Congresses.
the Ciba Foundation, London, 11-13 April 1989”-Contents p.
2. Mast cells-Immunology- Congresses. 3. Allergy-Pathogenesis-Congresses. 1. Chadwick, Derek. (1989 : Ciba Foundation) I l l . Series.
[ DNLM: 1 . Hypersensitivity-congresses. 2. IgE-congresses. 3. Mast Cells-congresses. W3 C161F v. 147 / W D 300 I24 19891 QR186.8.E2I44 1989 6 16.07 ’g-dc2O DNLM/DLC
11. Symposium on IgE, Mast Cells and the Allergic Response
for Library of Congress 89-24895 CIP
British Library Cataloguing in Publication Data
IgE, mast cells and the allergic response.-(Ciba Foundation Symposium; 147) 1 . Mammals. Immunoglobulins, IgE 1. Chadwick, Derek 11. Evered, David 111. Whelan, Julie 1111. Series 599.02 ‘ 93
ISBN 0 471 92309 5
Phototypeset by Dobbie Typesetting Limited, Devon. Printed and bound in Great Britain by Bath Press, Bath, Avon

Contents
Symposium on IgE, Mast Cells and the Allergic Response, held at the Ciba
The topic for this symposium was proposed by Professor Heiner Frost
Editors: Derek Chadwick, David Evered (Organizers) and Julie Whelan
Foundation, London, 11-13 April 1989
H. Metzger Introduction I
F. D. Finkelman, I. M. Katona, J. F. Urban Jr and W. E. Paul Control of in vivo IgE production in the mouse by interleukin 4 3 Discussion 17
H. Kikutani, A. Yokota, N. Uchibayashi, K. Yukawa, T. Tanaka, K. Sugiyama, E. L. Barsumian, M. Suemura and T. Kishimoto Structure and function of Fc, receptor I1 (Fc,RII/CD23): a point of contact between the effector phase of allergy and B cell differentiation 23 Discussion 3 1
M. F. Gurish and K. F. Austen Different mast cell mediators produced by different mast cell phenotypes 36 Discussion 45
S. J. Galli, B. K. Wershil, J. R. Gordon and T. R. Martin Mast cells: immunologically specific effectors and potential sources of multiple cytokines during IgE-dependent responses 53 Discussion 64
F. L. Pearce Non-IgE-mediated mast cell stimulation 74 Discussion 87
H. Metzger, J.-P. Kinet, H. Blank, L. Miller and C. Ra The receptor with high affinity for IgE 93 Discussion 10 1
C. Fewtrell, F. C. Mohr, T. A. Ryan and P. J. Millard Calcium: an important second messenger in mast cells Discussion 128
114
V

vi Contents
J. Yodoi, M. Hosoda, Y. Maeda, S. Sato, M. Takami and T. Kawabe Low affinity IgE receptors: regulation and functional roles in cell activation 133 Discussion 148
A. Capron, M. Capron, C. Grangette and J. P. Dessaint IgE and inflammatory cells 153 Discussion 160
D. G. Marsh, P. Zwollo, S. K. Huang and A. A. Ansari Molecular genetics of human responsiveness to allergens Discussion 183
171
S. Takafuji, S. Suzuki, M. Muranaka and T. Miyamato Influence of environmental factors on IgE production Discussion 20 1
188
K. J. Turner Epidemiology of the allergic response 205 Discussion 222
J. A. Warner, D. W. MacGlashan Jr and L. M. Lichtenstein Heterogeneity of human Fc,RI-bearing cells 230 Discussion 24 1
A. L. de Weck Conventional and new approaches to hyposensiti- zation 254 Discussion 259
Final general discussion 264
H. Metzger Closing statement 270
Index of contributors 272
Subject index 274

Participants
P. W. Askenase Department of Internal Medicine, Section of Allergy & Clinical Immunology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06510, USA
K. F. Austen Department of Rheumatology & Immunology, Harvard Medical School, The Seeley G Mudd Building, Room 604, 250 Longwood Avenue, Boston, MA 021 15, USA
J. Brostoff Department of Immunology, The Middlesex Hospital Medical School, London W1, UK
A. Capron Centre d’Immunologie et de Biologie Parasitaire, Institut Pasteur, 1 rue du Professeur A. Calmette, BP 245, F-59019 Lille Cedex, France
M. Capron Centre d’Immunologie et de Biologie Parasitaire, Institut Pasteur, 1 rue du Professeur A. Calmette, BP 245, F-59019 Lille Cedex, France
A. de Weck Institute for Clinical Immunology, Inselspital, CH-3010 Bern, Switzerland
G. Delespesse Notre-Dame Hospital, Allergy Research Laboratory, 1560 Sherbrooke Street East, Montreal, Quebec, Canada H2L 4M1
C. Fewtrell Department of Pharmacology, NY State College of Veterinary Medicine, Cornell University, Ithaca, NY 14853-6401, USA
F. Finkelman Department of Internal Medicine, Uniformed Services University of Health Sciences, Bethesda, MD 208 14-4799, USA
H. Frost Pharma Research, CIBA-GEIGY AG, CH-4002 Basle, Switzerland
S. J. Galli Departments of Pathology, Beth Israel Hospital and Harvard Medical School, 330 Brookline Avenue, Boston, MA 02215, USA
vii

viii Participants
H. J. Could Department of Biophysics, King’s College London, 26-29 Drury Lane, London WC2B 5RL, UK
C. H. Heusser Immunology (R1056-409), CIBA-GEIGY AG, CH 4002 Basle, Switzerland
M. Jaju (Ciba Foundation Bursar) RRL (H), B.M. Medical Research Centre, Mahavir Hospital, A.C. Guards, Hyderabad 500004 (AP), India
J. P. Kinet Arthritis & Rheumatism Branch, Chemical Immunology Section, NIAMSD, Bethesda, MD 20892, USA
T. Kishimoto Division of Immunology, Institute for Molecular & Cellular Biology, Osaka University, 1-3 Yamada-oka, Suita, Osaka 565, Japan
L. M. Lichtenstein* Clinical Immunology Division, Johns Hopkins University School of Medicine, The Good Samaritan Hospital, 5601 Loch Raven Boulevard, Baltimore, MD 21239, USA
D. W. MacGlashan Department of Medicine, Johns Hopkins University School of Medicine, The Good Samaritan Hospital, 5601 Loch Raven Boulevard, Baltimore, MD 21239, USA
G . Marone Department of Clinical Immunology, Faculty of Medicine & Surgery 11, University of Naples, Via S Pansini 5, 1-80131 Naples, Italy
D. G. Marsh Clinical Immunology Division, Johns Hopkins University School of Medicine, The Good Samaritan Hospital, 5601 Loch Raven Boulevard, Baltimore, MD 21239, USA
H. Metzger (Chairman) Department of Health & Human Sciences, Building 10 Room 9N228, National Institutes of Health, Bethesda, MD 20892, USA
F. L. Pearce Department of Chemistry, University College London, 20 Gordon Street, London WClH OAJ, UK
J. Ring Dermatologische Klinik und Poliklinik der Universitat, Frauenlobstrasse 9- 1 1 , D-8000 Munich 2, Federal Republic of Germany
*Present uddress: Johns Hopkins Asthma & Allergy Center, 301 Bayview Boulevard, Baltimore, MD 21224, USA.

Participants ix
D. R. Stanworth Rheumatology & Allergy Research Unit, Department of Immunology, The Medical School, University of Birmingham, Vincent Drive, Birmingham B15 2JT, UK
S. Takafuji Department of Medicine & Physical Therapy, Faculty of Medicine, University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113, Japan
K. J. Turner Department of Microbiology, University of Western Australia, Queen Elizabeth I1 Medical Centre, Nedlands, Western Australia 6009
J. Yodoi Institute for Immunology, Faculty of Medicine, Kyoto University, Yoshida, Sakyo, Kyoto 606, Japan


Introduction H. Metzger
Building 10 Room 9N 228, National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Institutes of Health, Bethesda, MD 20892, USA
Allergy like autoimmunity is an aberration of the immune response, and the two disorders present us with similar dilemmas. The most fundamental question is why certain individuals produce antibodies to harmless stimuli which elicit minor or no responses in normals. In the autoimmune individual the dilemma is compounded because the reaction breaks the fundamental canon of the immune response: tolerance to self. In the allergic individual the puzzle is deepened by the unique isotype specificity of the abnormal response: the abnormally produced antibodies are chiefly of the IgE class.
To solve these riddles we need to understand what initiates and regulates normal immune responses; uncertainties in this fundamental aspect of immunity constrain our ability to explain its lapses. Nevertheless, though our knowledge is imperfect, we can try to assess which defects in the normal response engender the abnormal one. In the case of the excessive IgE responses of allergy, are they due to abnormal mechanisms of antigen presentation: to errors in isotype ‘switching’ (and, if so, by what mechanism): or to excessive or deficient activities of isotype-specific Fc receptors (or binding factors)?
Animal studies suggest that inbred strains differ in their tendency to mount IgE responses, but even within a single strain these responses can be substantially modified depending on what ‘adjuvant’ accompanies the particular stimulus. Thus genetically determined constitutive differences are amplified by extrinsic, i.e. environmental, factors. Although the precise mechanisms that are affected still elude us, considerable progress has been made in defining the soluble and cell-bound molecules that can affect the immune response in general and the IgE response in particular.
Similarly, considerable progress has been made in defining the molecules whose activities ultimately lead to the signs and symptoms that afflict the allergic individual. Although less progress has been made in unravelling the biochemistry of the cellular responses initiated by these molecules, sophisticated methods by which these reactions can be examined are becoming available and there is no reason to doubt that answers will be forthcoming.
1989 IgE, Mast Cells and the Allergic Response. Wiley, Chichester (Ciba Foundation Symposium 147) p 1-2
1

2 Introduction
Much of the emphasis in research on allergic phenomena has focused on the rapid responses of mast cell (or basophils) mediated by interaction of the cell- bound IgE with antigen. It is apparent, however, that this is not the whole story; other cells and other mechanisms for triggering mast cells may be important, particularly in the later phases of allergic reactions, and some of these will be dealt with in this symposium.
Increased knowledge about these matters allows one to envision and test new therapeutic approaches. Caution is called for because we lack knowledge about the possible physiological functions of mast cells and basophils and because we do know that IgE responses play an important if not critical role in protecting us against parasites. But in its most general aspect this kind of therapeutic dilemma is faced whenever one intervenes pharmacologically, and caution should not lead to pessimism.
One final note before we begin our discussions. As physician-scientists we are often beguiled by the fascinating mechanisms which challenge our intellects to an extent that we may fail to see the diseases we are grappling with in their larger context. That is, we may lose sight of the socio-environmental factors, changes in which might profoundly affect the very incidence of such diseases. Thus the answer to the dreadful toll that automobile accidents incur lies not only in more sophisticated methods of triage, trauma surgery, and burn management, but also-even preferably-in better seat-restraining devices, driver education and drunken-driving legislation. As physicians we cannot absolve ourselves from playing active roles in identifying the epidemiological factors that affect the diseases we are dealing with, and in persuading society to confront them. As we shall hear during the latter parts of this symposium, such factors may well play a role in the incidence of allergic diseases, and we need to pay continuing attention to this area.

Control of in vivo in the mouse by Fred D. Finkelman*, lldy M. Katonat*
IgE production nterleukin 4 Joseph F. Urban JrS and William E. Paulo
Depaflments of Medicine* and Pediatricst, Uniformed Services University of the Health Sciences, Bethesda. MD 208 14; Helminthic Diseases Laboratory$, Agricultural Research Service, US Department of Agriculture, Beltsville, MD 20705: and Laboratory of Immunology", National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD 20892, USA
Abstract. In vitro studies have demonstrated that the cytokine IL-4 can, with the proper co-stimuli, induce IgE secretion. We have demonstrated in in vivo studies with a monoclonal anti-IL-4 antibody that this cytokine is required for the generation of the polyclonal primary IgE responses induced by injecting mice with GaM6 antibody or inoculating them with larvae of the nematode parasite Nippostrongylus brasiliensis (Nb), as well as for the secondary TNP-specific IgE response induced by immunizing mice with TNP-KLH on alum. We now report studies that demonstrate that: (1) the secondary polyclonal IgE response induced by repeated Nb inoculation, while mostly inhibitable by anti-IL-4 antibody, has an IL-4-independent component; (2) whereas treatment with anti-IL-4 antibody during a primary Nb inoculation does not prevent the rapid generation of a large IgE response during a second inoculation, treatment with anti-IL-4 antibody during both primary and secondary inoculation inhibits the development of a secondary IgE response by greater than 99%; (3) an established ongoing chronic IgE response, induced by inoculation of mice with larvae of the nematode parasite Heligmosomoides polygyrus (Hp), can be reduced by greater than 95% by administration of anti-IL-4 antibody; and (4) an anti-IL-4 receptor antibody effectively, efficiently and selectively blocks the GaM6 antibody-induced IgE response. These observations suggest that approaches aimed at blocking IL-4 effects may be useful for treating IgE-mediated diseases.
1989 IgE, Mast Cells and the Allergic Response. Wiley, Chichester (Ciba Foundation Symposium 147) p 3-22
During the last few years considerable evidence has accumulated that the IgE response in mouse and man is, to a large extent, regulated by two cytokines, interleukin 4 (IL-4) and y-interferon (IFN-y). The observations that led to this concept had their origin in studies which demonstrated that a culture supernatant of an activated T cell tumour induced lipopolysaccharide (LPS)-activated B lymphocytes to secrete less IgG3 and more IgGl (Isakson et a1 1982). The substance in the culture supernatant that was responsible for this activity was
3

4 Finkelman et al
subsequently identified as IL-4 (Vitetta et a1 1985). It was soon afterwards demonstrated that B cells activated by LPS can also be induced to secrete IgE by a substance that is secreted by some activated T cells; this substance also proved to be IL-4 (Coffman & Carty 1986, Coffman et a1 1986). The induction of an IgE response was found, however, to require the presence of a higher concentration of IL-4 for a longer period of time than does the induction of an IgGl response (Snapper et a1 1988). In vitro studies in mice have also established that IL-4 is required for the induction of an IgE response by B cells that are activated by antigen-specific helper T cells (Coffman et a1 1988), and that IL-4 probably stimulates isotype switching at the DNA level, since it induces the production of germ-line CY1 and C, mRNA transcripts (Stavnezer et a1 1988, Rothman et a1 1988).
The cytokine IFN-7 has been shown to antagonize many in v i m effects of 1L-4 on mouse cells, including the induction of IgGl and IgE secretion (Coffman & Carty 1986). These inhibitory effects are not due to a general inhibition of Ig secretion, since IFN-y stimulates increased IgG2a secretion by LPS- and helper T cell-activated B cells (Snapper & Paul 1987). Because many activated murine T cells produce IL-4 but not IFN-y, or IFN-7 but not IL-4, the selective activation of IL-4-secreting T cells (Th2 cells) might stimulate an IgE response, whereas the selective activation of IFN-7 secreting T cells (Thl cells) might block such a response (Mosmann et a1 1986).
These in vitro studies in mice have, to a large extent, been reproduced in human cell culture systems. IL-4 has been shown to selectively induce IgE secretion when added to cultures of otherwise unstimulated human peripheral blood mononuclear cells (PBMC) (Pene et a1 1988, Sarfati & Delespesse 1988). IgE secretion in this system is dependent upon the presence of T lymphocytes and monocytes and is blocked by IFN-y, as well as by other IFNs (Pene et a1 1988). While most human T cells clones, unlike many mouse T cell clones, secrete both IL-4 and IFN-y, they differ greatly in the ratios of these cytokines that they produce (Del Prete et al 1988). The ability of these human T cell clones to induce an IgE response by purified human B cells has been shown to vary directly with their secretion of IL-4 and inversely with their secretion of IFN--, (Del Prete et a1 1988). Although human PBMC can, in some culture systems, be induced to secrete IgE by interleukin 2 in the absence of added IL-4, the presence of T lymphocytes in these systems raises the possibility that endogenous IL-4 production is stimulating IgE secretion. It is also possible that the precursors of IgE-secreting cells, in one of these systems, are B lymphocytes that had already been induced by in vivo exposure to IL-4 to switch to the expression of membrane IgE (Splawski et a1 1989). It is particularly pertinent that highly purified human B cells, when stimulated in vilro with Epstein-Barr virus, are induced to secrete IgE by IL-4, but not by IL-1, 1L-2, IL-5 or IL-6, and that IgE secretion in this system is selectively suppressed by low concentrations of IFN-y (Thyphronitis et a1 1989). It is not clear from studies in any of these in v i m human systems

IL-4 control of in vivo IgE production 5
that the selection of IgG isotypes is regulated by IL-4 and/or IFN-y, as was found in the mouse.
Initial in vivo studies
While these in vitro murine and human studies all support the view that IL-4 and IFN-y have important and opposite effects on the control of the IgE production, they cannot define the importance of these cytokines in the control of IgE production in vivo, since they leave open the possibilities that: (1) these cytokines may not be produced in vivo in the proper quantity, at the proper place, and at the proper time to have the same effects as those demonstrated in vitro; (2) other, less well-defined stimuli might be more potent or plentiful inducers or suppressors of IgE production and might supplant IL-4 and IFN-y in vivo; and (3 ) in vivo conditions might suppress the effects of IL-4 and IFN--, that have been demonstrated in vitro. For these reasons, systems were developed or adapted in which to study the roles of these cytokines in the development of in vivo IgE responses.
We have previously reported studies in which we investigated the effects of recombinant IFN-7 or monoclonal anti-IL-4 or anti-IFN-y antibodies (1 1B11 and XMG-1, respectively) on the development of Ig responses to three different stimuli. First, we demonstrated that injection of mice with an affinity-purified goat antibody to mouse IgD (GaM6) stimulates the production of large, T cell- dependent, IgGl and IgE responses (Finkelman et a1 1987). While the IgGl response generated in this system is approximately 1000-fold greater than the IgE response, both serum isotypes usually increase by factors of 100 or more above baseline levels. The same mice usually develop serum IgG2a levels that are approximately 4- to 6-fold above baseline levels, but little or no increases in serum IgG3, IgG2b or IgA levels. T cells from the spleens of these mice produce small, but detectable increased quantitites of IL-4, starting three days after GaM6 injection (Finkelman et a1 1986a). IL-4 is essential for the development of an IgE response in this system, since injection of an anti-IL-4 antibody suppresses IgE production by greater than 99% (Finkelman et a1 1986b, 1988a). Surprisingly, anti-IL-4 antibody has no reproducible effects on IgGl or IgG2a production in this system. Kinetic experiments performed with anti- IL-4 and anti-CD4 antibodies in this system suggest that IL-4 has its stimulatory effect on IgE production 3-5 days after GaM6 injection, even though IgE production is not detectable until Day 6 and does not peak until Days 7-8 (Finkelman et a1 1989). These same experiments also indicate that some form of T cell help other than IL-4 is required late in the immune response to stimulate IgE, but not IgGl production. Experiments with IFN-y and a monoclonal anti- IFN--/ antibody have provided results that are completely consistent with the in vitro studies. Anti-IFN-y antibody enhanced polyclonal IgGl and IgE responses and suppressed the polyclonal IgG2a response, while injection of

6 Finkelman et al
recombinant IFN-y had the opposite effects (Finkelman et a1 1988b). Thus, IFN- y can be produced in sufficient quantity to influence Ig isotype selection during a physiological humoral immune response. It is noteworthy, however, that mice became obviously ill when injected with quantities of IFN-7 that were required to inhibit IgE production.
To supplement the GaMd studies we also did experiments with anti-IL-4 antibody in which mice were inoculated with the third-stage larvae of the nematode parasite Nippostrongylus brasiliensis (Nb). Adult Nb are expelled from the mouse gut approximately 10 days after inoculation in a CD4+ T cell- dependent process (Katona et a1 1988). An IgE response equivalent to that made by GaMd-injected mice is detectable 1-3 days later, along with an IgGl response that is less than one-twentieth that made by GaMd-injected mice (Finkelman et a1 1986b). Unlike GaMd-injected mice, no detectable IgG2a response is made by BALB/c mice inoculated with Nb; indeed, serum IgG2a levels in these mice decrease (Lebrun & Spiegelberg 1987). Injection of monoclonal anti-IL-4 antibody, however, has the same effect in these mice as it has in GaMd-injected mice; the IgE response is inhibited by approximately 99% while the IgGl response is not affected (Finkelman et a1 1988a). The differences in the effects of anti-IL-4 antibody on IgE and IgG1 responses in this system are not explainable simply by the lower IL-4 requirement for the induction of an IgGl response than an IgE response, since the dose of anti-IL-4 antibody used in these studies blocked the usually observed increase in B cell membrane class I 1 MHC expression in Nb-infected mice, a phenomenon that requires even lower concentrations of IL-4 in vitro than does the induction of an IgGl response ( C . Snapper & F. D. Finkelman, unpublished observation). While serum IgE levels in Nb-inoculated mice start to decline 13 or 14 days after inoculation, the rate of decline is slow compared to that observed in GaMd-injected mice. This allowed us to study whether established IgE responses are sustained by the continued production of IL-4. This proved to be true, since injection of anti-IL-4 antibody at the time of the peak IgE response hastened the decline in serum IgE levels (Finkelman et al 1988a).
The third in vivo system that we have studied is one in which the priming and boosting of mice with a low dose of TNP-KLH on alum stimulates the production of IgGl, IgG2a and IgE anti-TNP antibody responses. This system allowed us to test whether IL-4 is required for the generation of a secondary antigen-specific IgE response. We found that anti-IL-4 antibody, injected at the time of boosting with TNP-KLH on alum, completely blocked the generation of the secondary IgE anti-TNP response but had no specific effect on the generation of the secondary IgG1 or IgG2a anti-TNP antibody responses (Finkelman et a1 1988a). Furthermore, as in the Nb system, injection of anti-IL-4 antibody at the time of the peak secondary IgE anti-TNP response made it decline more rapidly. Thus, as is true for a primary, polyclonal IgE response, IL-4 is required for the generation and maintenance of a secondary, antigen-specific IgE response.

IL-4 control of in vwo IgE production 7
These observations suggested that inhibition of the production or the effects of IL-4, or treatment with IFN-y, might, by lowering IgE antibody production, provide a useful method for treating allergic disorders. Since injection of IFN-y has made mice ill, while anti-IL-4 antibody injection has had no apparent detrimental effects, we have concentrated our studies on the latter approach. Experiments have been performed to answer the following questions: (1) Can all secondary IgE responses be prevented by anti-IL-4 antibody? (2) To what extent does administration of anti-IL-4 antibody during a primary response affect the generation of an IgE response to a subsequent antigenic challenge? (3) To what extent can an ongoing, established IgE response be inhibited by anti-IL-4 antibody? (4) Will reagents that inhibit IL-4 receptor (IL-4R) function block IgE production as completely and effectively as does anti-IL-4 antibody?
Inhibition of a secondary, Nb-induced, polyclonal IgE response by anti-IL-4 antibody
Whereas the inoculation of previously untreated BALB/c mice with third-stage larvae of Nb induces an IgE response of 20-40 pg/ml that appears 1 1 - 13 days after inoculation, a repeat inoculation with third-stage larvae of the same parasite stimulates a response that typically peaks at 100 to 200 pg/ml, seven days after inoculation. To determine whether this response is IL-4-dependent, previously untreated mice or mice that had initially been inoculated with Nb 4-5 weeks earlier were inoculated with third-stage Nb larvae and injected with saline, anti- IL-4 antibody, or a control rat IgGl antibody.
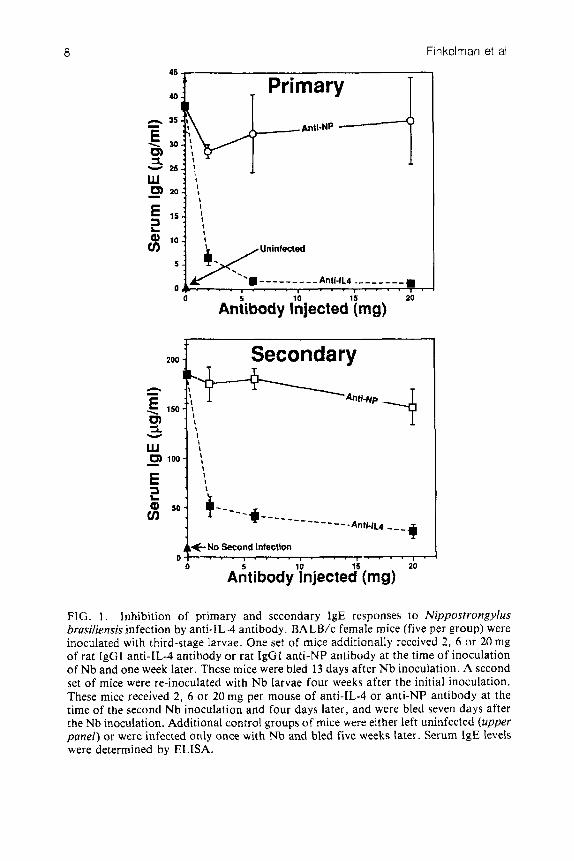
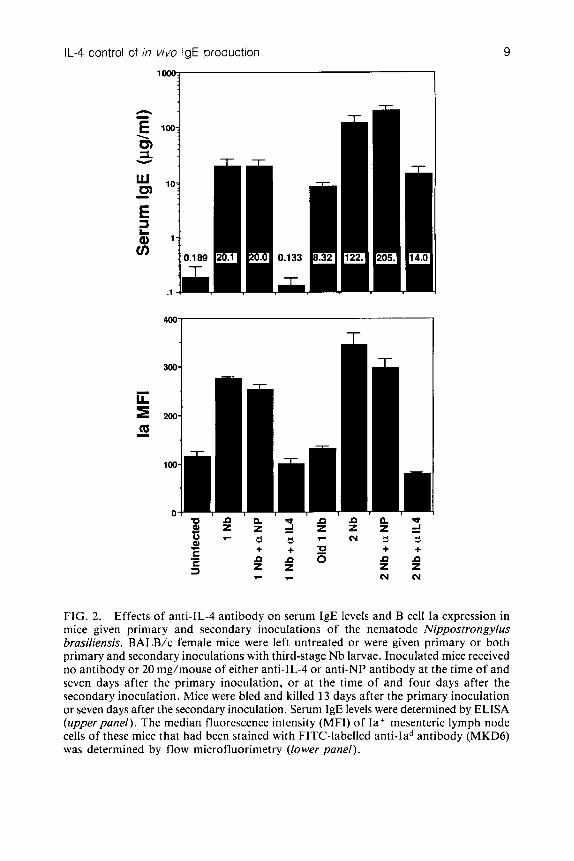
Although anti-IL-4 antibody greatly reduced the size of the polyclonal IgE response that was generated by the Nb re-inoculation, it never totally eliminated this response. Dose-response studies (Fig. 1) establish that most of the secondary IgE response is blocked by a relatively low dose of anti-IL-4 antibody (2 mg per mouse, given at the time of Nb inoculation and four days later), while a 10-fold larger dose of anti-IL-4 antibody still fails to inhibit this response completely. It is unlikely that the incomplete inhibition of the secondary IgE response is a result of incomplete neutralization of IL-4, since the large increase in B cell membrane Ia expression that accompanies the repeat Nb infection, and which should be a very sensitive indicator of the IL-4 effect on B lymphocytes, is completely blocked by anti-IL-4 antibody administration (Fig. 2). These results suggest that at least a small percentage of a secondary IgE response may be IL-4 independent. This would be expected if the stimulatory effect of IL-4 on IgE production were restricted to the induction of an isotype switch, and some of the IgE that is produced in mice given a second Nb inoculation arises from memory cells that had undergone such a switch during the primary inoculation. These results also suggest that therapy aimed at blocking the production of the effects of IL-4 will be unable to totally eliminate the development of a secondary IgE response in individuals that have already
a Finkelman et al
w 5 . . . . . . . . . . . . . .
0 20
Aniibody &jected'img)
200 Secondary
0 1
c'
Y
No Second Infection . . . . . . . . . . . . . . . . . . . . - i
FIG. 1. lnhibition of primary and secondary IgE responses to Nippostrongylus brasiliensis infection by anti-IL-4 antibody. BA LB/c female mice (five per group) were inoculated with third-stage larvae. One set of mice additionally received 2 , 6 or 20 mg of rat IgGl anti-IL-4 antibody or rat IgGI anti-NP antibody at the time of inoculation of Nb and one week later. These mice were bled 13 days after N b inoculation. A second set of mice were re-inoculated with Nb larvae four weeks after the initial inoculation. These mice received 2, 6 o r 20 mg per mouse of anti-IL-4 or anti-NP antibody at the time of the second Nb inoculation and four days later, and were bled seven days after the Nb inoculation. Additional control groups of mice were either left uninfected (upper panel) or were infected only once with Nb and bled five weeks later. Serum IgE levels were determined by ELISA.
IL-4 control of in vivo IgE production 9
FIG. 2. Effects of anti-IL-4 antibody on serum IgE levels and B cell Ia expression in mice given primary and secondary inoculations of the nematode Nippostrongylus brasiliensis. BALB/c female mice were left untreated or were given primary or both primary and secondary inoculations with third-stage Nb larvae. Inoculated mice received no antibody or 20 mg/mouse of either anti-IL-4 or anti-NP antibody at the time of and seven days after the primary inoculation, or at the time of and four days after the secondary inoculation. Mice were bled and killed 13 days after the primary inoculation or seven days after the secondary inoculation. Serum IgE levels were determined by ELISA (upper panel). The median fluorescence intensity (MFI) of Ia+ mesenteric lymph node cells of these mice that had been stained with FITC-labelled anti-Iad antibody (MKD6) was determined by flow microfluorimetry (lower panel).
10 Finkelman et al
generated IgE memory l3 cells for a specific antigen, but will greatly reduce the magnitude of the IgE response that is generated.
Effect of anti-IL-4 antibody administered during a primary IgE response on the development of a secondary IgE response
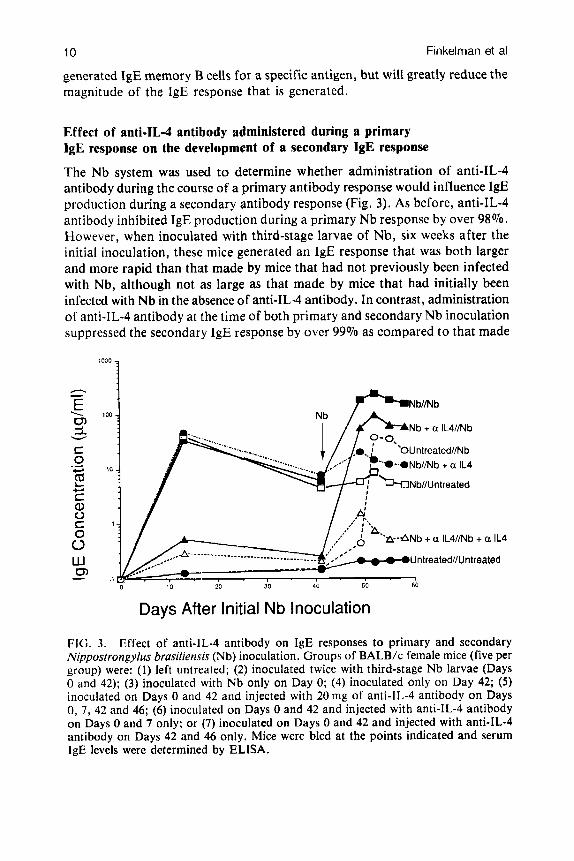
The Nb system was used to determine whether administration of anti-IL-4 antibody during the course of a primary antibody response would influence 1gE production during a secondary antibody response (Fig. 3). As before, anti-IL-4 antibody inhibited IgE production during a primary Nb response by over 98%. However, when inoculated with third-stage larvae of Nb, six weeks after the initial inoculation, these mice generated an IgE response that was both larger and more rapid than that made by mice that had not previously been infected with Nb, although not as large as that made by mice that had initially been infected with Nb in the absence of anti-IL-4 antibody. In contrast, administration of anti-IL-4 antibody at the time of both primary and secondary Nb inoculation suppressed the secondary IgE response by over 99% as compared to that made
I00 1 Nb
/ A NbIlNb
Nb + a IL4//Nb
*-,ONb//Nb + a IL4
+ a IL4
UntreatedNUntreated -
I 0 40 50 60
Y . , - I , I . I ' 8
10 20 30
Days After Initial Nb Inoculation
FIG. 3. Effect of anti-IL-4 antibody on IgE responses to primary and secondary Nippostrongylus brusiliensis (Nb) inoculation. Groups of BALB/c female mice (five per group) were: (1) left untrcated; (2) inoculated twice with third-stage Nb larvae (Days 0 and 42); (3) inoculated with Nb only on Day 0; (4) inoculated only on Day 42; ( 5 ) inoculated on Days 0 and 42 and injected with 20 mg of anti-lL-4 antibody on Days 0, 7, 42 and 46; (6) inoculated on Days 0 and 42 and injected with anti-IL-4 antibody on Days 0 and 7 only; or (7) inoculated on Days 0 and 42 and injected with anti-lL-4 antibody on Days 42 and 46 only. Mice were bled at the points indicated and serum IgE levels were determined by ELISA.

IL-4 control of in vivo IgE production 11
by mice infected twice in the absence of anti-IL-4 antibody, and by over 80% as compared to that made by mice given anti-IL-4 antibody only at the time of the secondary infection. These observations indicate that: (1) ‘priming’ for a secondary IgE response can occur even when the primary response is largely blocked by neutralization of IL-4; and (2) when an IgE response to an initial immunization is blocked by neutralization of IL-4, continued IL-4 neutralization is both able and necessary to block the development of an IgE response to a second immunization.
Inhibition of an ongoing, established IgE response by anti-IL-4 antibody
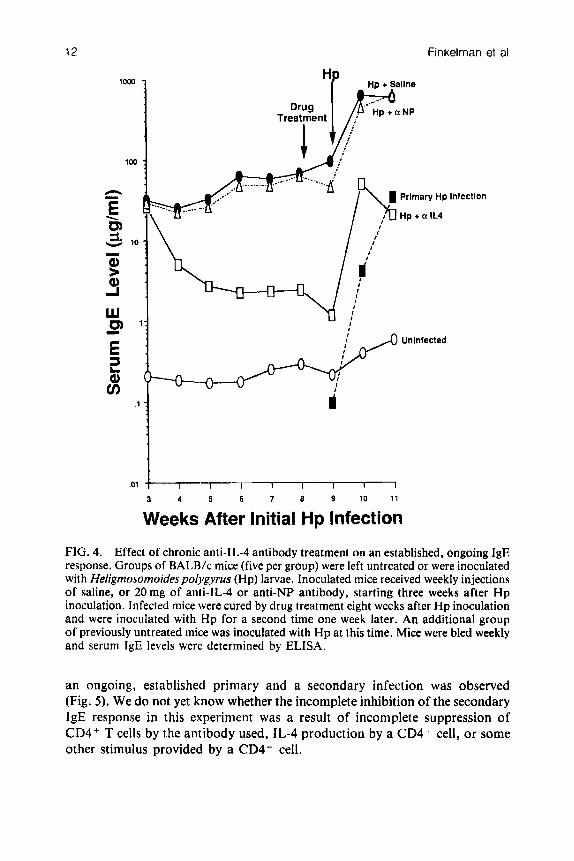
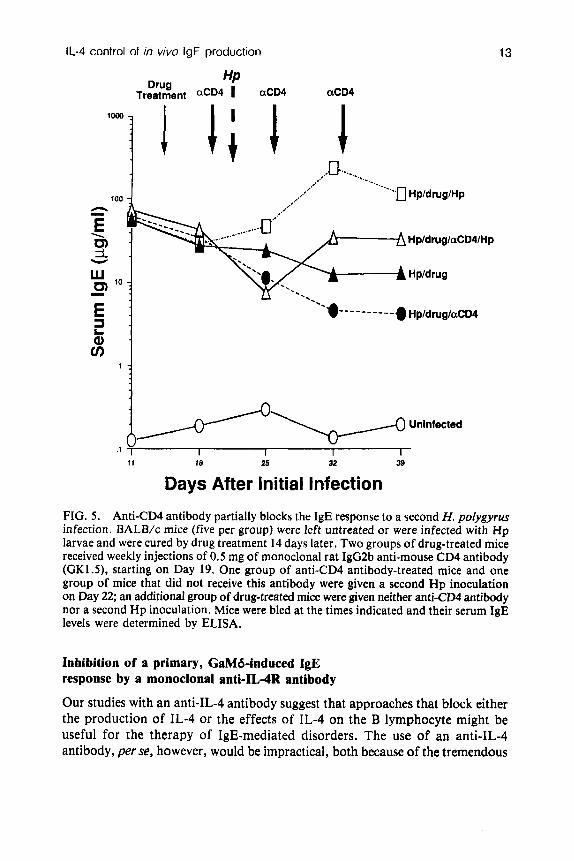
Our studies with Nb have established that anti-IL-4 antibody causes serum IgE levels to decline rapidly when given at the time of a peak IgE response (Finkelman et al 1988a), and also considerably inhibits the generation of an IgE response by a second Nb inoculation. Both situations differ, however, from that experienced by an allergic individual, who may undergo continuous immunogenic stimulation. Since such an individual would most likely present for treatment only after an IgE response had become established, an anti- IL-4-based therapeutic regimen would have to be capable of inhibiting an established, ongoing IgE response in order to be of value. To test whether anti-IL-4 antibody treatment might be able to inhibit such a response, we used a model in which mice are infected with larvae of the nematode Heligmosomoides polygyrus (Hp, also known as Nematospiroides dubius). Adult Hp, unlike Nb, survive in the gut of infected mice for months, during which time they stimulate a stable, large serum IgE response (Urban et a1 1989). Weekly injections of anti-IL-4 antibody were started three weeks after Hp inoculation, at which time a large IgE response had already been established. This treatment caused serum IgE levels to decrease by a factor of 20 over a six-week period, despite the continued presence of Hp; most of this decrease occurred during the first 1-2 weeks of treatment (Fig. 4). In contrast, serum IgE levels continued to rise in Hp-infected mice that were treated with saline or with a control antibody. Treatment of Hp-infected mice with an anti-helminthic drug terminates Hp infections and allows mice subsequently to be reinfected with this same parasite. This reinfection stimulates a large secondary serum IgE response, which can exceed 500pg/ml. As was observed with secondary Nb infections, the IgE response to a second Hp inoculation was not eliminated by chronic anti-IL-4 antibody treatment, but was inhibited by 90-95% (Fig. 4). These observations indicate that anti-IL-4 antibody is a strong, although incomplete, inhibitor even of established, ongoing IgE responses. In experiments with Hp-infected mice in which treatment with a cytotoxic anti-CD4 antibody was substituted for anti-IL-4 antibody treatment, similar considerable but incomplete inhibition of the IgE responses to both
12 Finkelman et al
Infection
.01
3 4 5 6 7 8 9 10 11
Weeks After Initial Hp Infection
FIG. 4. Effect of chronic anti-lL-4 antibody treatment on an established, ongoing IgE response. Groups of BALB/c mice (five per group) were left untreated or were inoculated with Heligmosomoidespolygyrus (Hp) larvae. Inoculated mice received weekly injections of saline, or 20mg of anti-lL-4 or anti-NP antibody, starting three weeks after H p inoculation. Infected mice were cured by drug treatment eight weeks after Hp inoculation and were inoculated with H p for a second time one week later. An additional group of previously untreated mice was inoculated with H p at this time. Mice were bled weekly and serum IgE levels were determined by ELISA.
an ongoing, established primary and a secondary infection was observed (Fig. 5) . We do not yet know whether the incomplete inhibition of the secondary IgE response in this experiment was a result of incomplete suppression of CD4+ T cells by the antibody used, IL-4 production by a CD4- cell, or some other stimulus provided by a CD4- cell.
IL-4 control of in vivo IgE production
3 Q) v)
L
1 :
13
HP Drug Treatment aCD4 I aCD4 aCD4
Uninfected
,1
1 1 18 25 32 39
Days After Initial Infection
FIG. 5 . Anti-CD4 antibody partially blocks the IgE response to a second H. polygyrus infection. BALB/c mice (five per group) were left untreated or were infected with Hp larvae and were cured by drug treatment 14 days later. Two groups of drug-treated mice received weekly injections of 0.5 mg of monoclonal rat IgG2b anti-mouse CD4 antibody (GK1.5), starting on Day 19. One group of anti-CD4 antibody-treated mice and one group of mice that did not receive this antibody were given a second Hp inoculation on Day 22; an additional group of drug-treated mice were given neither anti-CD4 antibody nor a second Hp inoculation. Mice were bled at the times indicated and their serum IgE levels were determined by ELISA.
Inhibition of a primary, GaMd-induced IgE response by a monoclonal anti-IL4R antibody
Our studies with an anti-IL-4 antibody suggest that approaches that block either the production of IL-4 or the effects of IL-4 on the B lymphocyte might be useful for the therapy of IgE-mediated disorders. The use of an anti-IL-4 antibody, per se, however, would be impractical, both because of the tremendous
14 Finkelman et al
quantities that would be required to achieve a clinically significant effect in a human and because of the problems that would be associated with repeated administration of a foreign protein. A more reasonable approach would be the use of an inhibitor of IL4R function. As an initial test of this approach it would be useful to determine whether an antibody that blocked IL-4R function could be used in vivo to selectively inhibit an IgE antibody response. The recent cloning of the murine IL-4R gene and production of a rat IgG2a monoclonal antibody that binds with high affinity to the mouse IL-4R and blocks IL-4 binding at the Immunex Corporation has allowed us t o test this approach. BALB/c mice were left untreated, or injected with 800vg of GaMd with no additional antibody, or with 1 mg of anti-IL-4R antibody, or with 1 mg of a control rat IgG2a anti-TNP antibody. Sera were obtained eight days later, after which the mice were killed and their spleens were weighed (Table 1). As compared to untreated animals, mice that were injected with GaMd alone demonstrated an approximately 6.5-fold increase in spleen weight, a 132-fold increase in serum IgGI level, a 17-fold increase in serum IgG2a level, and a 554-fold increase in serum IgE level. The injection of anti-IL-4R antibody along with GaMd did not affect the GaMd-induced increase in spleen weight but inhibited by greater than 95 To the GaMd-induced increase in serum IgE level. Relatively modest inhibition of the GaMb-induced increase in serum IgGl and enhancement of the GaMd-induced increase in serum IgG2a were also seen in anti-IL-4R antibody-treated mice. 'These last two effects were quite interesting, since, while IL-4 has been shown to stimulate IgG1 production and suppress IgG2a production in some in iiitro systems (Vitetta et a1 1985, Snapper & Paul 1987). anti-IL-4 antibody has not had these effects in vivo, even when used at doses that inhibit IgE production by more than 99% (Finkelman et a1 1988a, 1989). Further studies with anti-IL-4R antibody are required before conclusions can be drawn about its ability to inhibit IgE production under different circumstances and about its effects on IgG isotype selection. The results of this initial
TABLE 1 Effects of anti-IL-4 receptor antibody on the CaMS-induced immune response
Serum immunoglobulin levels
Treatment Spleen weight IgGl IgC2a IgE
Untreated 81.6 (1.05)a 0.29 (1.07) 0.11 (1.12) 0.14 (1.52) GaMd 527 (1.02) 38.3 (1.08) 1.92 (1.12) 77.5 (1.17)
G a M 6 + aTNP 929 (1.03) 42.4 (1.04) 1.79 (1.14) 44.6 (1.17) GaMd + aIL-4R 540 (1.02) 29.9 (1.04) 4.06 (1.09) 1.95 (1.13)
BALB/c female mice (five per group) were injected intravenously with the antibodies shown and bled and killed eight days later. Doses of antibodies used were 800 pg of GaMd and 1 mg of anti- TNP or anti-IL-4R. Serum lgGl and Ig02a levels were determined by radial immunodiffusion; 1gE levels by ELISA. Spleens weights are in mg, lgGl and IgG2a levels in mg/ml, IgE levels in pg/ml. 'Data are shown as geometric means x / + (geometric standard errors).

IL-4 control of in vivo IgE production 15
experiment, however, support the view that pharmacological approaches that are aimed at inhibiting IL-4R function may be useful means of selectively inhibiting IgE production.
Conclusions
Although IgE plays a central role in the pathogenesis of many allergic reactions, and IgE responses have been known for several years to be regulated differently from other antibody responses, therapy of allergic reactions has generally been aimed at inhibition of mediator release and mediator receptor function, rather than inhibition of IgE production. The in vivo studies that we have described reinforce the results of a series of in vitro investigations with murine and human lymphoid cells and lead to the conclusion that the lymphokines IL-4 and IFN-7 have a central role in the control of IgE antibody production. Our studies in a variety of systems indicate that IgE responses, whether polyclonal or antigen- specific, primary or secondary, initial or established and ongoing, can all be considerably inhibited by an anti-IL-4 antibody. Our studies also indicate, in contrast to what might have been expected from the results of in vitro investigations, that this inhibition of IgE secretion can be achieved without interfering with other, possibly protective, immune responses. Furthermore, our initial experiment with an anti-IL4R antibody strongly suggests that IL-4R inhibition will have similar effects. It thus seems reasonable to believe that approaches aimed at limiting the production or effects of IL-4 may yield a new and useful approach to the treatment of IgE-mediated disease.
A ckn o wledgemen ts We thank Ms Joanne Holmes and Mr Steven Kang for their expert technical contributions to the studies described here, and Drs Kenneth Grabstein, Robert Coffman, Timothy Mosmann, Clifford Snapper and James Mond for their helpful discussions and advice. The great generosity of the Immunex Corporation in providing us with their anti-IL-4R antibody even before this antibody was described in the literature is particularly appreciated. This research was supported in part by Uniformed Services University of the Health Sciences research protocols RO 8308 and CO 8634, National Institute of Health grants R01-A121328 and lR29AI2615001Al and US Department of Agriculture grant CRIS 1205-34000-009. Opinions and assertions contained herein are the private ones of the authors and are not to be construed as official or reflecting the views of the Department of Defense or the Uniformed Services University of the Health Sciences.
References
Coffman RL, Carty J 1986 A T cell activity that enhances polyclonal IgE production
Coffman RL, Ohara J, Bond MW, Carty J, Zlotnik A, Paul WE 1986 B cell stimulatory and its inhibition by interferon-y. J Immunol 136:949-954

16 Finkelman et al
factor-I enhances the IgE response of lipopolysaccharide-activated B cells. J Immunol
Coffman RL, Seymour BWP, Lebman DA et a1 1988 The role of helper T cell products in B cell differentiation and isotype regulation. Immunol Rev 102:5-28
Del Prete G, Maggi E, Parronchi P et a1 1988 IL-4 is an essential factor for the IgE synthesis induced in v i m by human T cell clones and their supernatants. J Immunol
Finkelman FD, Ohara J , Goroff DK et al 1986a Production of BSF-1 during an in vivo, T-dependent immune response. J Immunol 137:2878-2885
Finkelman FD, Katona IM, Urban JF Jr, Snapper CM, Ohara J , Paul WE 1986b Suppression of in vivo polyclonal IgE responses by monoclonal antibody to the lymphokine B-cell stimulatory factor-]. Proc Natl Acad Sci USA 83: 9675-9678
Finkelman FD, Snapper CM, Mountz JD, Katona IM 1987 Polyclonal activation of the murine immune system by a goat antibody to mouse IgD. IX. Induction of a polyclonal IgE response. J Immunol 138:2826-2830
Finkelman FD, Katona IM, Urban JF Jr et a1 1988a IL-4 is required to generate and sustain in vivo IgE responses. J lmmunol 141:2335-2341
Finkelman FD, Katona IM, Mosmann TR, Coffman RL 1988b IFN-y regulates the isotypes of Ig secreted during in vivo humoral immune responses. J Immunol
Finkelman FD, Holmes J, Urban JF Jr, Paul WE, Katona IM 1989 T help requirements for the generation of an in vivo IgE response: a late acting form of T cell help other than IL-4 is required for IgE but not for IgGl production. J Immunol
Isakson PC, Pure E, Vitetta ES, Krammer PH 1982 T cell-derived B cell differentiation factor(s). Effect on the isotype switch of murine B cells. J Exp Med 155:734-748
Katona IM, Urban JF Jr, Finkelman FD 1988 The role of L3T4' and Lyt-2+ T cells in the IgE response and immunity to Nippostrongylus brusiliensis. J Immunol
Lebrun P, Spiegelberg HL 1987 Concomitant immunoglobulin E and immunoglobulin GI formation in Nippostrongyius brus;iiensis-infected mice. J lmmunol 139:1459-1465
Mosmann TR, Cherwinski H, Bond MW. Giedlin MA, Coffman RL 1986 Two types of murine helper T cell clones. I. Definition according to profiles of lymphocyte activities and secreted proteins. J Immunol 136:2348-2357
Pene J. Rousset F, Briere F et al 1988 IgE production by normal human lymphocytes is induced by interleukin 4 and suppressed by interferons y and CY and prostaglandin EZ. Proc Natl Acad Sci USA 85:6880-6884
Rothman P, Lutzker S, Cook W, Coffman R, Alt FW 1988 Mitogen plus interleukin-4 induction of CE transcripts in B lymphoid cells. J Exp Med 168:2385-2389
Sarfati M, Delespesse G 1988 Possible role of human lymphocyte receptor for IgE (CD23) or its soluble fragments in the in vitro synthesis of human IgE. J Immunol
Snapper CM, Paul WE 1987 Interferon-y and B cell stimulatory factor-1 reciprocally regulate Ig isotype production. Science (Wash DC) 236:944-947
Snapper CM, Finkelman FD, Paul WE 1988 Differential regulation of IgGl and IgE synthesis by interleukin 4. J Exp Med 167:183-196
Splawski JB, Jelinek DF, Lipsky PE 1989 Immunomodulatory role of IL-4 on the secretion of Ig by human B cells. J Immunol 142:1569-1575
Stavnerer J, Radcliffe G, Lin Y-C et a1 1988 Immunoglobulin heavy-chain switching may be directed by prior induction of transcripts from constant-region genes. Proc Natl Acad Sci USA 85:7704-7708
136:4538-4541
140:4193-4198
140: 1022- 1027
142:403-408
140:3206-3211
141:2195-2199

IL-4 control of in vivo IgE production 17
Thyphronitis G , Tsokos GC, June CH, Levine AD, Finkelman FD 1989 IgE secretion by Epstein-Barr virus-infected purified human B lymphocytes is stimulated by interleukin-4 and suppressed by interferon-?. Proc Natl Acad Sci USA 865580-5584
Urban JF Jr, Katona IM, Finkelman FD 1989 The role of CD4+ T cells in the IgE response and immunity to Heligmosomoides polygyrus. FASEB (Fed Am SOC Exp Biol) J 3:A1357
Vitetta ES, Ohara J , Myers CD, Layton JE, Krammer PH, Paul WE 1985 Serological, biochemical, and functional identity of B cell-stimulatory factor 1 and B cell differentiation factor for IgGl. J Exp Med 162:1726-1731
DISCUSSION
Metzger: When you were attempting to block the IgE response to the parasitic infection with Nippostrongylus brasiliensis with antibody to IL-4, did you ever give the antibody continuously?
Finkelman: It was never given continuously, although these are IgG antibodies which have a half-life of three or four days, and even after six weeks some of the antibodies may have been in the circulation.
Austen: Is there an antibody available in the mouse to the low affinity IgE receptor, and, if so, do you have data on the effect of blocking that receptor on the IgE responses?
Finkelman: An antibody has been produced by Dan Conrad called B3B4 (Rae et a1 1987). This is a rat IgG2a antibody which binds to and blocks with high affinity the mouse version of CD23, the low affinity IgE receptor. Ildy Katona has done experiments in which mice either injected with anti-IgD or inoculated with N. brasiliensis also received that antibody. We have not been able to demonstrate any decrease in IgE production in either system, although we can show that we are blocking by approximately 95% the IgE-binding capacity of B cells that carry that receptor (Katona et a1 1989).
Austen: Have you used that B3B4 antibody in a hapten-specific system? Finkelman: We have not yet investigated its effects in a hapten-specific system
or on a secondary response. Monique Capron: Is it possible to use the anti-mouse IL-4 antibody in the
rat? Have you tried this? Finkelman: No, unfortunately, because the anti-mouse IL-4 is made in the
rat, and it does not work as an autoantibody. Monique Capron: Have you tried to raise anti-IL-4 antibodies in another
animal species? Finkelman: No. There are however polyclonal anti-human IL-4 antibodies,
produced in rabbits. Genzyme has such an antibody for sale. Banchereau’s group at Unicet in Lyons has also made a rabbit antibody that blocks human IL-4 function (Jacques Banchereau, personal communication).
Heusser: We have produced anti-human IL-4 monoclonal antibodies and have selected one which efficiently neutralizes human IL-4 activity in vitro. It inhibits

18 Discussion
IL-4-dependent IgE production efficiently and CD23 expression as well as IL-Cdependent T cell proliferation.
Lichtenstein: I have always been dubious about achieving anything clinically by these means; you would need to reduce IgE production by more than 99.9%, because the mast cell requires so few molecules to be triggered-although you are getting much closer to this than people used to get. You are working in vivo; have you done any biological tests, such as skin testing the animal, to see whether, when you reduce the IgE level by a certain amount, you are having an effect on the biological response?
Finkelman: Steve Galli has started experiments with us to look at some of those questions.
Kishimoto: I f you could establish an IL-4 transgenic mouse, you could make allergic mice.
Finkelman: That is a good point. I gather that Dr A. Abbas at Harvard is making such transgenic mice now. A number of people have injected large quantities of IL-4 into mice and have seen surprisingly little in the way of positive effects on the production of IgE, yet the amount of IL-4 being made in our anti-IgD-treated mice, for example, is tiny, and we see a substantial increase in serum IgE. The difference may be that we need to get IL-4 to the right place at the right time, along with the correct additional stimulatory signals, to get an IgE response, and it may be difficult to achieve this simply by having large quantities of IL-4 produced in an unregulated fashion. Do you have any knowledge of these experiments?
Kishimoto: No. With regard to 1L-6 and plasmacytoma, however, we could make IL-6 transgenic mice which generate plasmacytomas.
You showed that the anti-IL-4 receptor antibody inhibited the IgE response, but there was no inhibition of the growth of spleen cells. All IL-4 receptor- positive cells should be inhibited by this antibody. Why couldn’t you show any effect on lymphocyte growth?
Finkelman: My bias would be that lymphokines acting in vivo are much more specific in their effects than one would guess from the in vitro experiments. IL-4 is believed to be an autocrine growth factor for Th2 CD4+ cells, yet we are seeing CD4+ Th2 effects in the presence of a large dose of the anti-IL-4 receptor antibody. I suspect that there is another, more important autocrine factor which hasn’t been identified yet. An alternative possibility is that there is much more 1L-4 receptor on a T cell than it needs to respond to IL-4 and that even blocking that receptor by 98%, say, allows the remaining 2% to function sufficiently to give the results that we see.
de Weck: What dose of anti-IL-4 receptor antibody was used in this experiment, and do you have any idea of the kinetics of action of this antibody?
Finkelman: The dose was 1 mg per mouse. It was given at the time of the injection of the anti-IgD. We have not been able to look sequentially at the effects in these mice, because we wanted to kill them to weigh the spleen. We




![[PPT]PowerPoint Presentation - mulyanipharmaco | Learning ... · Web viewsyok. Obat penyebab (Penisilin, streptomisin, ) ALLERGEN IgE SYNTHESIS MAST CELL DEGRANUL Mediator LOCAL ANAPHYLAXIS](https://img.pdfslide.net/doc/110x75/5af43a957f8b9ae9488bc4e7/pptpowerpoint-presentation-mulyanipharmaco-learning-viewsyok-obat-penyebab.jpg)
























