Embed Size (px)
Citation preview

Immunopathologie de la polyarthrite rhumatoïde
Immunopathogenesis of rheumatoid arthritis
J. Morel (Chef de clinique-assistant) a, P. Miossec (Professeurdes Universités-praticien hospitalier) b, B. Combe (Professeurdes Universités-praticien hospitalier) a,*
a Unité Inserm U454 et service d’immuno-rhumatologie, centre hospitalier universitaire Lapeyronie,371, avenue du Doyen Gaston Giraud, 34295 Montpellier cedex 5, Franceb Unité d’immunologie clinique, service de rhumatologie, hôpital Édouard Herriot,5, place d’Arsonval,69437 Lyon cedex 03, France
MOTS CLÉSImmunopathologie ;Polyarthriterhumatoïde ;Immunité innée ;Immunité acquise ;Cytokines ;Synovite ;Destructionostéocartilagineuse
KEYWORDSImmunopathogenesis;Rheumatoid arthritis;Innate immunity;Adaptative immuneresponse;Cytokines;Synovitis;Joint destruction
Résumé La polyarthrite rhumatoïde (PR) est la maladie auto-immune dont la physiopatho-logie est la mieux connue car la plus étudiée. L’antigène responsable de la PR restetoujours inconnu, mais la très grande spécificité des anticorps dirigés contre les protéinescitrullinées donne des indications sur la structure de cet antigène probablement riche enrésidus citrullinés. La PR est une maladie multifactorielle faisant intervenir des facteursà la fois hormonaux, génétiques, environnementaux et immunologiques qui contribuentau développement d’une inflammation chronique de la membrane synoviale. Les méca-nismes immunopathologiques sont complexes et font intervenir à la fois l’immunité innée(récepteurs toll like, cytokines, complément), mais aussi l’immunité acquise avec commeprincipaux acteurs les cellules présentant l’antigène, les lymphocytes T et B. De façonschématique, l’immunopathologie de la PR peut être divisée en trois phases distinctes :une phase de déclenchement impliquant surtout l’immunité innée ; une phase d’inflam-mation de la membrane synoviale impliquant plutôt l’immunité acquise ; une phase dedestruction articulaire secondaire à l’action de cytokines (tumor necrosis factor a,interleukine 1b, métalloprotéinases, RANKL) mais aussi à la prolifération pseudotumoraledes synoviocytes par défaut d’apoptose. La meilleure connaissance de ces mécanismesimmunopathologiques a permis le développement de traitements ciblés comme les agentsmodulant le tumor necrosis factor a et l’interleukine 1, mais permet aussi d’envisagerune large gamme de nouveaux traitements dirigés contre les cytokines, les voies designalisation intracellulaires et les protéines de costimulation lymphocytaire.© 2004 Publié par Elsevier SAS.
Abstract Rheumatoid arthritis (RA) is one of the most studied auto-immune disease. Infifty years, considerable progress in the comprehension of the immunopathogenesis ofthis disease has been achieved. The antigen responsible for RA is still unknown, however,the high specificity of auto-antibodies directed against citrullinated peptides providessome information on the structure of this antigen probably rich in citrullines residues. RAis a polyfactorial disease that involves hormones, genetic and environmental factors, butalso immunological disorders which contribute to chronic synovitis. The mechanismsinvolved in the pathogenesis of the disease are complex and implicate innate immunity(toll like receptors, cytokines, complement) and adaptative immune response characte-
* Auteur correspondant.Adresse e-mail : [email protected] (B. Combe).
EMC-Rhumatologie Orthopédie 1 (2004) 218–230
www.elsevier.com/locate/emcrho
© 2004 Publié par Elsevier SAS.doi: 10.1016/j.emcrho.2004.03.003

rised by a T cell-mediated antigen specific response implicating antigen presenting cells,B and T lymphocytes. Immunopathogenesis of RA can be subdivided in three differentphases: an early phase involving innate immunity, an established phase characterised bychronic synovitis depending essentially on adaptative immune response, and a destructivephase related to cytokines effect (TNFa, IL-1b, metalloproteinases, RANKL) but also atumour-like proliferation of fibroblast such as synoviocytes related to apoptosis defect.The better understanding of RA immunopathogenesis has led to the development ofspecific therapeutic interventions such as biologic agents. Based on the pathogenicmechanisms, novel treatment targeting cytokines, intracellular signalling pathways orlymphocyte co-stimulation proteins.© 2004 Publié par Elsevier SAS.
Introduction
La polyarthrite rhumatoïde (PR) est un rhumatismeinflammatoire responsable d’une destruction del’articulation qui contribue à une impotence fonc-tionnelle parfois majeure. Même si des progrèsconsidérables ont été faits dans la compréhensionde la physiopathologie de cette maladie, son ori-gine reste toujours inconnue. Plusieurs facteursinterviennent dans le déclenchement de la mala-die : des facteurs hormonaux, le terrain génétiqueprédisposé et des facteurs environnementaux.1
Lorsque tous ces facteurs sont réunis, ils activentune réponse immunitaire innée et acquise incontrô-lée qui se traduit par une réaction inflammatoireexagérée, en particulier de la membrane syno-viale.2 Cette synoviale est une structure habituel-lement paucicellulaire avec une couche bordante,c’est-à-dire proche de la cavité articulaire qui estmince. La synoviale rhumatoïde est en revancheinfiltrée par des cellules comprenant principale-ment des macrophages, des lymphocytes B et deslymphocytes T CD4+ qui s’organisent en agrégatslymphoïdes avec parfois des centres germinauxdont la structure rappelle celle d’un ganglion. Lasynoviale rhumatoïde se caractérise également parune prolifération de la couche bordante qui estcomposée de synoviocytes et de macrophages, maisaussi par une prolifération importante de néovais-seaux. La PR est classée parmi les maladies auto-immunes en raison de nombreux signes d’autoréac-tivité, avec la présence d’autoanticorps comme lesfacteurs rhumatoïdes mais aussi les anticorps anti-fillagrine.3 Toutefois, la seule preuve qui démon-trerait de manière formelle le caractère auto-immun de la maladie serait la mise en évidence duou des auto-antigène(s) capable(s) de reproduire lamaladie par immunisation. La physiopathologie dela PR pourrait être comparée à un puzzle dontcertaines pièces sont aujourd’hui identifiées maisdont l’agencement final reste encore mal connu.De façon schématique, nous distinguons la phase dedéclenchement de la maladie, avec les différents
facteurs responsables de l’initiation de la PR, laphase d’inflammation de la membrane synovialedont la pathogénie est mieux connue et la phase dedestruction articulaire (Fig. 1).
Phase de déclenchement de la maladie
Facteurs hormonaux
La plus grande incidence de la PR chez la femme,avec un sexe-ratio de un homme pour quatre fem-mes, suggère une implication des hormones dans ledéclenchement de la PR. Les études épidémiologi-ques se sont intéressées à l’influence de la gros-sesse, de l’allaitement et des facteurs hormonauxendogènes ou exogènes comme facteurs de risquesde la PR. Pendant la grossesse, le risque de déve-lopper une PR est faible, tandis que dans l’annéequi suit le post-partum ce risque est nettement plusélevé.4 L’allaitement a été incriminé comme étantun facteur de risque, responsable de l’incidenceplus élevée dans le post-partum. En effet, uneétude portant sur 187 femmes qui avaient déve-loppé une PR après la première grossesse montreque celles qui avaient allaité leur enfant ont unrisque cinq fois supérieur d’avoir une PR.5 Cetteimplication des facteurs hormonaux endogènes estsoulignée par certaines études qui ont montré unehypoandrogénie relative chez les femmes maisaussi chez les hommes atteints de PR, avec des tauxde testostérone et de déhydroépiandrostérone plusbas. Les hormones exogènes, que ce soit la pilulecontraceptive ou le traitement hormonal substitu-tif, ne modifient pas l’incidence de la PR maissemblent retarder son début et sa sévérité6.
Facteurs génétiques
Le taux de concordance pour la PR chez les ju-meaux homozygotes atteints est en moyenne de13 %. L’association génétique la plus forte est ob-servée avec les gènes codant pour les molécules
219Immunopathologie de la polyarthrite rhumatoïde
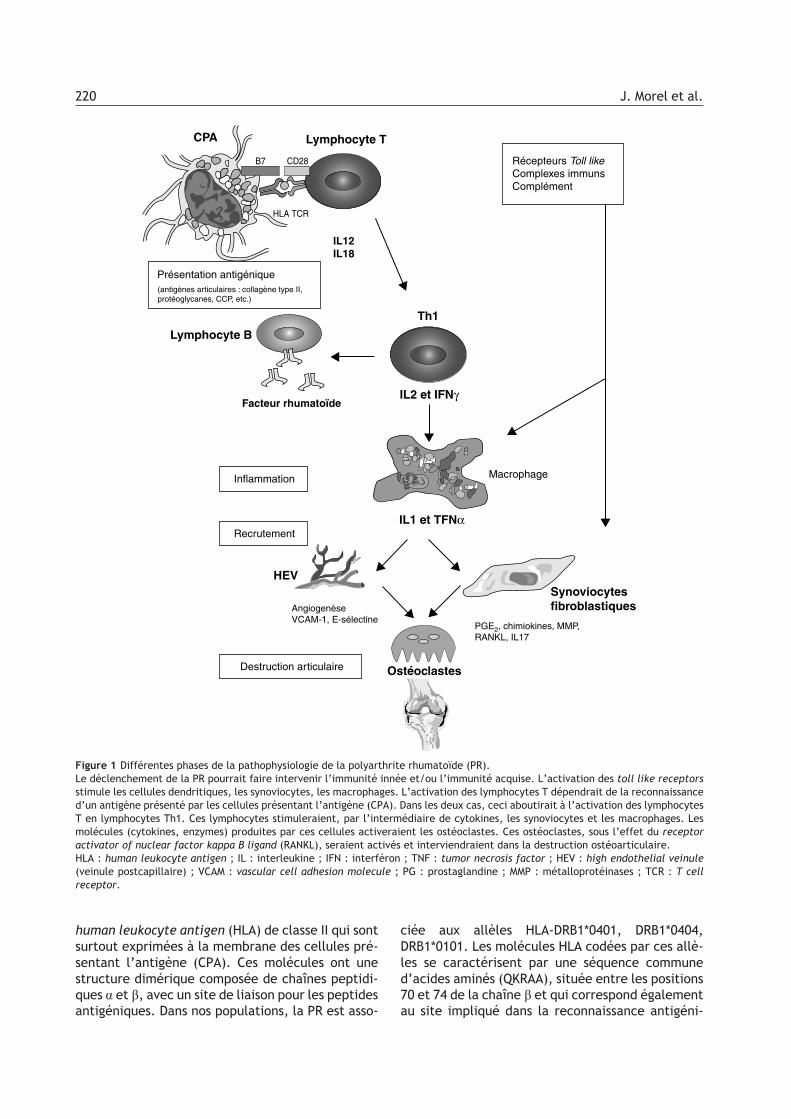
human leukocyte antigen (HLA) de classe II qui sontsurtout exprimées à la membrane des cellules pré-sentant l’antigène (CPA). Ces molécules ont unestructure dimérique composée de chaînes peptidi-ques a et b, avec un site de liaison pour les peptidesantigéniques. Dans nos populations, la PR est asso-
ciée aux allèles HLA-DRB1*0401, DRB1*0404,DRB1*0101. Les molécules HLA codées par ces allè-les se caractérisent par une séquence communed’acides aminés (QKRAA), située entre les positions70 et 74 de la chaîne b et qui correspond égalementau site impliqué dans la reconnaissance antigéni-
CPA
IL12IL18
Facteur rhumatoïde
Lymphocyte T
Lymphocyte B
B7 CD28
HLA TCR
Présentation antigénique
Récepteurs Toll likeComplexes immunsComplément
Inflammation Macrophage
Recrutement
Th1
Synoviocytesfibroblastiques
IL2 et IFNγ
IL1 et TFNα
Ostéoclastes
(antigènes articulaires : collagène type II,protéoglycanes, CCP, etc.)
Destruction articulaire
HEV
AngiogenèseVCAM-1, E-sélectine
PGE2, chimiokines, MMP,RANKL, IL17
Figure 1 Différentes phases de la pathophysiologie de la polyarthrite rhumatoïde (PR).Le déclenchement de la PR pourrait faire intervenir l’immunité innée et/ou l’immunité acquise. L’activation des toll like receptorsstimule les cellules dendritiques, les synoviocytes, les macrophages. L’activation des lymphocytes T dépendrait de la reconnaissanced’un antigène présenté par les cellules présentant l’antigène (CPA). Dans les deux cas, ceci aboutirait à l’activation des lymphocytesT en lymphocytes Th1. Ces lymphocytes stimuleraient, par l’intermédiaire de cytokines, les synoviocytes et les macrophages. Lesmolécules (cytokines, enzymes) produites par ces cellules activeraient les ostéoclastes. Ces ostéoclastes, sous l’effet du receptoractivator of nuclear factor kappa B ligand (RANKL), seraient activés et interviendraient dans la destruction ostéoarticulaire.HLA : human leukocyte antigen ; IL : interleukine ; IFN : interféron ; TNF : tumor necrosis factor ; HEV : high endothelial veinule(veinule postcapillaire) ; VCAM : vascular cell adhesion molecule ; PG : prostaglandine ; MMP : métalloprotéinases ; TCR : T cellreceptor.
220 J. Morel et al.

que.7 Cette séquence commune, appelée aussi« épitope partagé », pourrait être au cœur de laréaction auto-immune médiée par les lymphocytesT. Trois principaux modèles ont été proposés :
• l’épitope partagé pourrait reconnaître un pep-tide du soi et favoriser dans le thymus la per-sistance d’un clone de lymphocytes T auto-réactifs par sélection clonale positive ; ceclone T autoréactif pourrait dans certainesconditions être activé et déclencher une ré-ponse immunitaire spécifique contre ce pep-tide du soi ;
• l’épitope partagé se lierait spécifiquementavec l’antigène responsable de la PR ; uneétude récente a montré une affinité de l’épi-tope partagé pour les peptides citrullinés quireprésentent un groupe de peptides candidatsà l’initiation de la PR ;8
• l’épitope partagé interagirait avec un peptideantigénique exogène mais ayant une structurevoisine à un peptide du soi. Cette théorie ditedu « mimétisme moléculaire » a été observéepour une glycoprotéine du virus Epstein-Barr(la gp110), mais aussi pour la protéine ADN-Jd’Escherichia coli et les protéines de chocthermique (heat schock proteins [HSP] pour lesAnglo-Saxons).L’implication des allèles HLA-DRB1*04 etDRB1*01 dans la PR est également soulignéepar l’association étroite entre ces allèles et lasévérité de la maladie. L’allèle DRB1*04 estpratiquement constamment retrouvé dans lesPR agressives, avec des dégradations ostéoar-ticulaires plus précoces9,10 et plus importan-tes.11,12 L’allèle HLA-DRB1*01 semble égale-ment associé aux PR sévères, mais plusfaiblement. Le nombre d’allèles à risque dansle génotype du patient est corrélé avec lasévérité de la PR. La notion d’effet dose a étédéveloppée par les travaux de Weyand quimontrent que les patients homozygotes pourDRB1*04 ont un risque de développer une PRplus sévère que les sujets hétérozygotes pourcet allèle.11 D’autres polymorphismes généti-ques ont été décrits pour des gènes impliquésdans la présentation antigénique comme HLA-DM,13 ou le récepteur FCcRIII des immunoglo-bulines,14 mais aussi pour des gènes codantpour des cytokines comme le tumor necrosisfactor (TNF) a, les interleukines (IL) 1b, 4 et10.15,16,17,18 Ces polymorphismes génétiquesreprésentent des facteurs pronostiques de sé-vérité. Ainsi, le polymorphisme du gène codantpour l’IL1b est associé à des PR plus érosivestandis que le polymorphisme du gène codantpour l’IL4 est associé à des PR moins destruc-trices.17,18
Facteurs environnementaux
Les agents infectieux viraux (Epstein-Barr), bacté-riens (E. coli) et mycobactériens ont été incriminésdans le déclenchement de la PR. Une infectioncommune sur un terrain génétiquement prédisposépourrait déclencher la maladie par mimétisme mo-léculaire de certains composants de ces agentsinfectieux avec des composants de l’articulation.La protéine de choc thermique HSP65 a une struc-ture voisine avec une protéine présente dans lecytoplasme des cellules de la couche bordante.L’HSP 70 d’E. coli est reconnue par l’épitope par-tagé de la molécule HLA-DR. Les souris immuniséesavec Mycobacterium tuberculosis produisent desanticorps dirigés contre les HSP. Une réaction im-munitaire croisée pourrait donc déclencher la réac-tion inflammatoire observée dans la PR. Les agentsinfectieux peuvent induire une réponse immuni-taire innée par activation des toll like receptors(TLR).19 Ces TLR reconnaissent des molécules ex-primées par les micro-organismes : TLR4 est activéepar les composants lipopolysaccharidiques de lamembrane bactérienne et TLR9 interagit avec l’oli-gonucléotide CpG présent dans l’acide désoxyribo-nucléique (ADN) bactérien.D’autres facteurs environnementaux, comme le
statut social, la vie urbaine par rapport au mode devie rural, le régime alimentaire, ont été incriminésdans le déclenchement de la PR, mais sans que celasoit formellement prouvé. En revanche, deux étu-des longitudinales confirmées par une étude sur lesjumeaux ont montré un risque plus élevé de déve-lopper une PR chez les fumeurs de tabac.20
Phase d’inflammation de la synoviale
L’inflammation de la synoviale, ou synovite, impli-que de nombreux acteurs cellulaires, extracellulai-res et intracellulaires.
Acteurs cellulaires
Le mécanisme physiopathologique de la PR est basésur le complexe tricellulaire : CPA/lymphocytesT/synoviocytes.
Cellules présentant l’antigèneLes macrophages, les lymphocytes B et les cellulesdendritiques sont capables de présenter un anti-gène aux lymphocytes T. Ces cellules expriment eneffet à la surface de leur membrane des moléculesHLA de classe II qui sont indispensables au déclen-chement d’une réponse immunitaire médiée par leslymphocytes T. Les CPA ne sont pas toutes douées
221Immunopathologie de la polyarthrite rhumatoïde

du pouvoir de phagocytose, mais elles ont un pointcommun qui est leur aptitude à l’endocytose desmolécules extracellulaires et à la protéolyse de cesmolécules à l’intérieur des lysosomes. Les cellulesdendritiques (CD) sont les cellules présentatricesprofessionnelles du système immunitaire et sontsupposées être les cellules qui présentent initiale-ment l’antigène aux lymphocytes T dans la PR.21
Dans la synoviale rhumatoïde, les CD sont trouvéesprincipalement dans les agrégats lymphocytaires eten périphérie des vaisseaux, suggérant que les CDproviennent du sang périphérique. Les chimiokinescapables d’attirer les cellules dendritiques, MIP-3aet b, les chimiokines CC, sont trouvées en abon-dance dans la synoviale. Les chimiokines CCL19 etCCL21 et leur récepteur CCR7 interviennent dansl’adressage des CD dans la synoviale et plus préci-sément dans l’infiltrat lymphocytaire.22 Les CD pré-sentes dans la synoviale rhumatoïde expriment desmarqueurs de différenciation qui témoignent d’uncontact préalable avec les lymphocytes T. Ces ob-servations suggèrent que les CD seraient activéesdans les organes lymphoïdes et migreraient ensuitedans la synoviale. Les CD pourraient participer à lapérennisation de l’inflammation par un défaut derégulation de la présentation antigénique. Ainsi, undéfaut d’apoptose des CD favorisé par des facteursantiapoptotiques présents dans l’environnementsynovial pourrait prolonger anormalement leur du-rée de vie. Le ou les antigènes arthritogènes se-raient ainsi présentés de manière anormalementprolongée et pourraient de ce fait favoriser la pé-rennisation de l’inflammation. Les CD ont un rôled’éducation des lymphocytes T régulateurs CD4+ etCD25+ pour envoyer des messages de régulation.
Lymphocytes TLes lymphocytes T autoréactifs sont capables deréagir avec des peptides du soi. Chez les patientsatteints de PR, la proportion de ces lymphocytes Tautoréactifs serait plus élevée que chez les sujetsnormaux et serait due à une anomalie de la sélec-tion thymique. Dans le thymus, les lymphocytes Tqui reconnaîtraient les autopeptides associés auxmolécules HLA de classe II présentées par les CPAsubiraient une délétion clonale par apoptose. Cettesélection dite négative serait déficiente chez lesmalades atteints de PR et contribuerait à la survied’un plus grand nombre de lymphocytes T autoréac-tifs. Cette hypothèse est supportée par les diffé-rences observées entre le répertoire des lymphocy-tes T des malades atteints de PR et celui des sujetsnormaux,11 mais aussi par l’association génétiqueavec certains gènes HLA de classe II. Les lymphocy-tes naïfs, après reconnaissance d’un antigène, vontse différencier en lymphocytes T producteurs d’in-
terféron c, d’IL2 ou encore d’IL17. Cette réponseest dite de type Th1 par opposition à une réponsede type Th2 qui se traduit plutôt par une productiond’IL4. Dans la synoviale rhumatoïde, les lymphocy-tes Th1 sont particulièrement abondants.23,24 Ceslymphocytes T sont recrutés à partir du sang péri-phérique et s’organisent en agrégats qui ressem-blent par leur morphologie à l’architecture follicu-laire des ganglions lymphoïdes, avec également laprésence de veinules postcapillaires (high endothe-lial venules). La plupart des lymphocytes T syno-viaux expriment à la fois les marqueurs CD4 etCD45RO, et sont donc des lymphocytes T auxiliairesmémoires. Ces lymphocytes T peuvent être à nou-veau activés par les CPA par engagement des molé-cules du T cell receptor, des molécules HLA-DR,mais aussi de molécules de costimulation commeCD28 et B7. L’activation des lymphocytes T est sousle contrôle des lymphocytes T régulateurs CD4+ etCD25+. Ces lymphocytes T régulateurs sont capa-bles d’inhiber l’expansion clonale des lymphocytesT CD4+. La molécule CTLA4 exprimée sur les lym-phocytes T régulateurs 1 interagit avec la protéineCD28 exprimée sur les lymphocytes T CD4+ et induitun message inhibiteur. Ces lymphocytes T régula-teurs pourraient intervenir dans la physiopatholo-gie de la PR.25 Les lymphocytes T activés interagis-sent avec les cellules endothéliales qui composentl’endothélium des veinules postcapillaires.26 Cescellules endothéliales sont activées par des cytoki-nes produites par les monocytes ou les lymphocytesT activés. Ces cellules endothéliales et les lympho-cytes T activés expriment alors des molécules d’ad-hésion, d’abord des sélectines (E-sélectine,L-sélectine), puis des intégrines (aEb7, a4b7,a4b1), qui interagissent entre elles.27 L’interactionlymphocyte T/ cellules endothéliales permet ladiapédèse des lymphocytes T circulants qui passentalors dans la membrane synoviale. Les lymphocytesT migrent ensuite dans la synoviale en exprimant àleur surface membranaire des récepteurs aux chi-miokines comme CCR5 qui reconnaissent des chi-miokines telles que RANTES (regulated upon activa-tion normal T-cell expressed and secreted),produites dans la synoviale.28 Ces lymphocytes T,nouvellement arrivés dans la synoviale, produisentdes cytokines de type Th1 qui activent les cellulesrésidentes : lymphocytes B, macrophages rési-dents, cellules endothéliales et fibroblastes.Celles-ci libèrent à leur tour des chimiokines et desmolécules d’adhésion qui favorisent le recrutementde monocytes et de polynucléaires neutrophilescirculants.29 L’ensemble du processus de recrute-ment des cellules circulantes du compartiment san-guin vers le compartiment synovial est appelél’adressage ou homing pour les Anglo-Saxons
222 J. Morel et al.
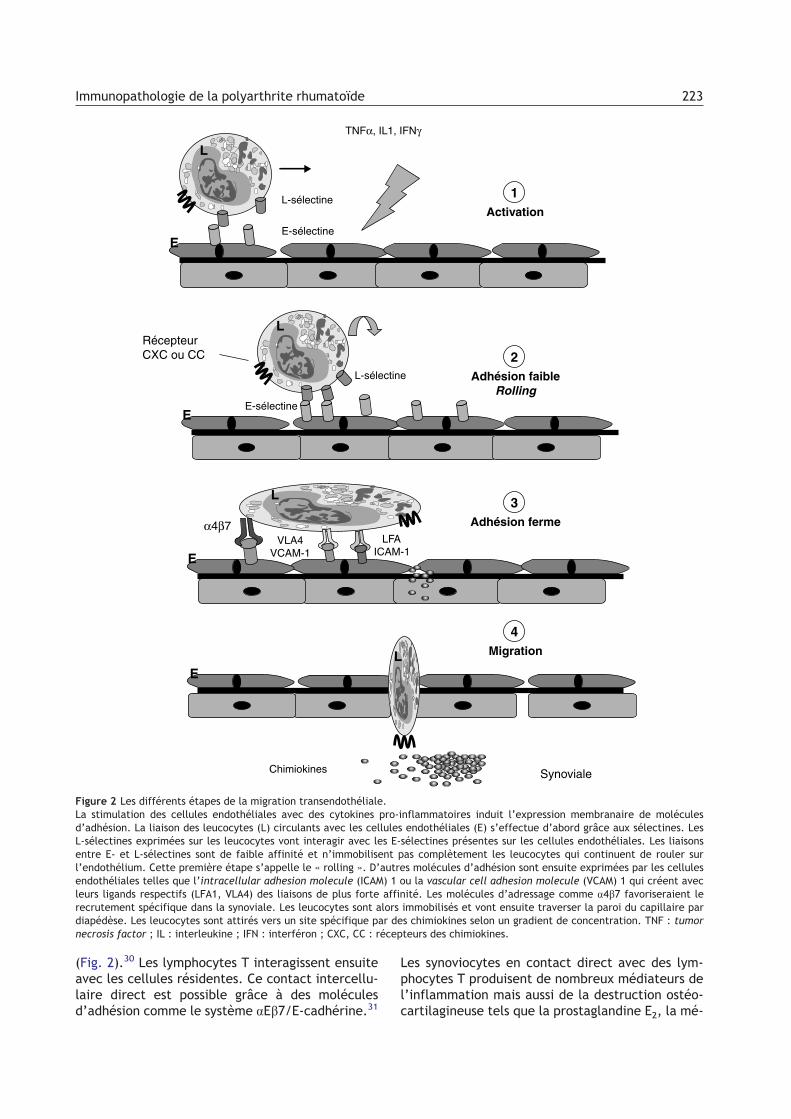
(Fig. 2).30 Les lymphocytes T interagissent ensuiteavec les cellules résidentes. Ce contact intercellu-laire direct est possible grâce à des moléculesd’adhésion comme le système aEb7/E-cadhérine.31
Les synoviocytes en contact direct avec des lym-phocytes T produisent de nombreux médiateurs del’inflammation mais aussi de la destruction ostéo-cartilagineuse tels que la prostaglandine E2, la mé-
TNFα, IL1, IFNγ
L-sélectine
RécepteurCXC ou CC
L-sélectine
VLA4VCAM-1
LFAICAM-1
Chimiokines
α4β7
E-sélectine
E-sélectine
Activation
L
L
L
E
E
E
1
Adhésion faibleRolling
2
Adhésion ferme
3
Migration
Synoviale
4
LE
Figure 2 Les différents étapes de la migration transendothéliale.La stimulation des cellules endothéliales avec des cytokines pro-inflammatoires induit l’expression membranaire de moléculesd’adhésion. La liaison des leucocytes (L) circulants avec les cellules endothéliales (E) s’effectue d’abord grâce aux sélectines. LesL-sélectines exprimées sur les leucocytes vont interagir avec les E-sélectines présentes sur les cellules endothéliales. Les liaisonsentre E- et L-sélectines sont de faible affinité et n’immobilisent pas complètement les leucocytes qui continuent de rouler surl’endothélium. Cette première étape s’appelle le « rolling ». D’autres molécules d’adhésion sont ensuite exprimées par les cellulesendothéliales telles que l’intracellular adhesion molecule (ICAM) 1 ou la vascular cell adhesion molecule (VCAM) 1 qui créent avecleurs ligands respectifs (LFA1, VLA4) des liaisons de plus forte affinité. Les molécules d’adressage comme a4b7 favoriseraient lerecrutement spécifique dans la synoviale. Les leucocytes sont alors immobilisés et vont ensuite traverser la paroi du capillaire pardiapédèse. Les leucocytes sont attirés vers un site spécifique par des chimiokines selon un gradient de concentration. TNF : tumornecrosis factor ; IL : interleukine ; IFN : interféron ; CXC, CC : récepteurs des chimiokines.
223Immunopathologie de la polyarthrite rhumatoïde

talloprotéinase MMP-1, l’IL6.32,33 Les lymphocytesT interagissent aussi avec les macrophages et lescellules présentatrices de l’antigène, amplifiantainsi la réaction inflammatoire.34 Les lymphocytesT ont pendant longtemps tenu le rôle vedette dansla physiopathologie de la PR.35 In vitro, l’utilisationde tests de prolifération employant des lymphocy-tes T a permis d’identifier plusieurs autoantigènescomme des candidats potentiels pouvant être res-ponsables de la PR. Le collagène de type II, lesprotéoglycanes, les aggrecanes, certaines protéi-nes liées au cartilage, les protéines HSP et beau-coup d’autres antigènes présents dans l’articula-tion ont été incriminés dans la pathogénie de la PRpar leur capacité à activer les lymphocytes T, maisaussi à induire la production d’anticorps spécifi-ques, voire d’arthrite chez la souris. Les modèlesd’arthrite expérimentale comme l’arthrite au col-lagène et l’arthrite à adjuvant sont clairementdépendants des lymphocytes T. Si l’importance deslymphocytes T est indiscutable dans la maladie,leur présence n’est pas indispensable à l’entretiende l’inflammation locale de l’articulation qui dé-pend plus des synoviocytes.
SynoviocytesIls constituent le principal composant cellulaire dela couche bordante de la membrane synoviale avecdeux types de synoviocytes : les macrophages et lessynoviocytes fibroblastiques. Les premiers appar-tiennent comme les cellules dendritiques à la li-gnée monocytaire, tandis que les seconds ont plu-tôt une origine mésenchymateuse. Lesmacrophages se distinguent des synoviocytes fibro-blastiques par l’expression de marqueurs de diffé-renciation tels que CD68 et CD14, des moléculesHLA de classe II et des récepteurs Fc des immuno-globulines.36 Les synoviocytes macrophagiques ac-tivés seraient les véritables moteurs de la réactioninflammatoire en produisant deux types de média-teurs : des médiateurs « primaires » ne nécessitantpas de synthèse protéique tels que les prostaglan-dines, les leucotriènes, les radicaux libres et lesenzymes contenues dans les granules et qui partici-pent de façon importante à la destruction tissu-laire ; des médiateurs secondaires requérant unesynthèse protéique constituée principalement parles cytokines pro-inflammatoires IL1 et TNFa.37 Cescytokines jouent un rôle clef dans le développe-ment de la réaction inflammatoire locale et systé-mique car elles sont capables, grâce à des systèmesautocrines et paracrines, d’induire à leur tour lasynthèse de la plupart des protéines de l’inflamma-tion. Ces cytokines induisent la synthèse de média-teurs primaires de l’inflammation (prostaglandineE2, monoxyde d’azote, radicaux libres) par une
action autocrine mais aussi par une action para-crine sur les cellules avoisinantes comme les chon-drocytes, les cellules endothéliales et les synovio-cytes. Ces derniers, stimulés par l’IL1 et le TNF a,produisent des facteurs de croissance et des cyto-kines pro-inflammatoires. Les synoviocytes ont unecapacité de prolifération qui ressemble par certainsaspects à celle des cellules cancéreuses. Ils possè-dent des caractéristiques qui se rapprochent descellules tumorales par leur morphologie, la perted’inhibition de contact, mais aussi l’activation deplusieurs oncogènes.38 La prolifération anormaledes synoviocytes de PR pourrait s’expliquer commepour les tumeurs cancéreuses par un défaut d’apop-tose.39 Les protéines p53, FAS ligand ainsi que lesvoies de signalisation NFjB et PI3 kinase sont parti-culièrement impliquées dans le phénomène de ré-sistance des synoviocytes à l’apoptose.
Lymphocytes BLa théorie du complexe trimoléculaire minimise lerôle tenu par les lymphocytes B. Cependant, l’effi-cacité du rituximab, un anticorps dirigé contre lemarqueur CD20 des lymphocytes B, responsable dela déplétion des lymphocytes B chez les patientsatteints de PR, souligne leur importance dans laphysiopathologie de la PR.40 Leur contribution dansla pathogénie de la PR se situe à plusieurs niveaux.Les lymphocytes B peuvent se comporter comme devéritables CPA car ils sont capables de présenterdes antigènes aux lymphocytes TCD4+. En effet,grâce aux facteurs rhumatoïdes membranaires, ilscaptent très efficacement des complexes im-muns.41 Les antigènes présents dans ces complexesimmuns pourraient subir un « apprêtement » avantd’être présentés par les molécules HLA-DR. L’impli-cation des lymphocytes B dans la présentation anti-génique est supportée par l’augmentation de l’ex-pression des molécules HLA-DR observée à lasurface des lymphocytes B présents dans la syno-viale rhumatoïde.42 L’analyse du répertoire deslymphocytes B synoviaux a révélé qu’ils étaientactivés dans la membrane synoviale.43 Les lympho-cytes B produisent certains autoanticorps détectésdans la PR tels que les facteurs rhumatoïdes et lesanticorps anticollagène. D’autres autoanticorpspourraient intervenir dans la pathogénie de la PRcomme BIP (immunoglobulin heavy gene bindingprotein), human RNP-33 (RA33). Ces anticorps nesont pas spécifiques de la maladie. En revanche, lesanticorps dirigés contre des protéines citrullinéesproduites par déimination de résidu arginine parune peptidylarginine déiminase sont plus spécifi-ques de la PR.44 Les anticorps anti-citrullinatedcyclic peptide reconnaissent également les résiduscitrullinés de protéines comme la fillagrine, le col-
224 J. Morel et al.

lagène ou la fibrine. Le rôle de ces peptides citrul-linés reste maintenant à démontrer. Le rôle deslymphocytes B a été souligné par le modèle K/BxN.Ces souris générées fortuitement développentspontanément une arthrite directement liée à desanticorps dirigés contre un antigène ubiquitaire, laglucose-6 phospho-isomérase. Le transfert du sé-rum de ces souris induit en effet une arthrite.45 Cesautoanticorps forment des complexes immuns quise déposent dans la paroi des vaisseaux et provo-quent des vascularites. Ces complexes immuns fa-voriseraient la production de cytokines pro-inflammatoires par les monocytes et joueraientégalement un rôle dans la formation du pannussynovial.29 Cependant, ces anticorps sont rarementretrouvés dans la PR et ne sont pas spécifiques àcette maladie.
Acteurs intercellulaires : les cytokines
Les cellules communiquent entre elles par contactde cellule à cellule ou en utilisant des messagersintercellulaires appelés cytokines. Il existe quatregrandes familles de cytokines : les interleukines,les interférons, les facteurs de croissance et leschimiokines. Ces cytokines jouent un rôle majeurdans la réponse immunitaire innée et acquise. Dansla PR, il existe un déséquilibre entre les cytokinespro- et anti-inflammatoires (Fig. 3). Paradoxale-ment, les cytokines produites par les lymphocytesde type Th1 (IL2 et interféron c) ne sont trouvées
qu’à de faibles concentrations dans les articula-tions de PR. Seule l’IL17, qui est une cytokineproduite par les lymphocytes T CD4+ Th1 et Th0, estplus exprimée dans le liquide articulaire des pa-tients atteints de PR que de ceux atteints d’arth-rose.46 L’IL15 est un puissant stimulant de la pro-duction d’IL17 par les lymphocytes T. Les cytokinesproduites par les synoviocytes, telles que le TNFa,l’IL1, l’IL15, l’IL18, l’IL6, mais aussi les facteurs decroissance et les chimiokines, sont présentes à desconcentrations élevées dans le liquide synovialmais aussi dans le sérum des patients atteints dePR.36,47 L’IL1b et le TNFa sont des médiateurs clefsde l’inflammation. Ils contrôlent la production denombreuses cytokines comme le fibroblast growthfactor, le vascular endothelial growth factor et leschimiokines, mais aussi des molécules d’adhésionqui interviennent également dans la réaction in-flammatoire en favorisant l’angiogenèse et le re-crutement des cellules dans la synoviale. Le rôlecentral de ces deux cytokines dans la pathogénie dela PR a été démontré ces dernières années avecl’utilisation des traitements capables de les neutra-liser. Ces traitements anti-TNFa et anti-IL1 ontmontré une efficacité anti-inflammatoire remar-quable en réduisant l’angiogenèse et l’infiltrat in-flammatoire dans la synoviale.48,49,50 L’IL17 etl’IL18 induisent l’expression de nombreux média-teurs de l’inflammation par activation du facteurde transcription NFjB.46,51 Parmi les autres cytoki-nes pouvant participer à la physiopathologie de laPR, l’IL6 semble particulièrement intéressante.L’IL6 est une cytokine qui cumule des propriétés àla fois pro- et anti-inflammatoires.52 En effet,d’une part l’IL6 induit les protéines de la phaseaiguë de l’inflammation, et d’autre part elle estcapable de freiner la production de l’IL1, du TNF aet des chimiokines. Son rôle dans la pathogénie dela PR est encore mal connu mais indiscutable. Laneutralisation de l’IL6 par un anticorps monoclonaldirigé contre le récepteur de l’IL6 (MRA) donne eneffet des résultats très encourageants dans la PR.53
Acteurs intracellulaires : les voiesde signalisation
Lorsqu’une cytokine se fixe sur un récepteur mem-branaire, elle provoque une modification deconformation du récepteur qui aboutit à la phos-phorylation du récepteur lui-même ou d’une en-zyme associée à ce récepteur. Cette premièrephosphorylation entraîne l’activation en cascaded’autres enzymes appelées les protéines kinasesqui activent à leur tour les facteurs de transcription(Fig. 4). Ces facteurs de transcription régulent lasynthèse de protéines en agissant directement sur
Lymphocyte T
CD28
IL15GM-CSFTNFα
IL17INFγ
IL4IL10
IL10IL1RaS TNF-R
IL6IL8GM-CSF
IL1IL18TNFα
Synoviocytesfibroblastiques
Macrophage
-
-
+
+
+
+
TCR
Figure 3 Le réseau cytokinique dans la polyarthrite rhumatoïde.Les lymphocytes T activent les cellules synoviales résidentes parles cytokines Interféron (IFN) c et interleukine (IL)17. Les syno-viocytes, les macrophages et les cellules dendritiques produi-sent des cytokines capables de stimuler ou d’inhiber les cellulesrésidentes présentes dans la synoviale. Les cytokines pro-inflammatoires et anti-inflammatoires sont respectivement in-diquées par les signes + et -. GM-CSF : granulocyte-macrophagecolony-stimulating factor ; TNF : tumor necrosis factor ; IL1 Ra :antagoniste du récepteur de l’IL-1 ; sTNF-R= récepteur solubledu TNFa; Syno. F : synoviocytes fibroblastiques ; TCR : T cellreceptor.
225Immunopathologie de la polyarthrite rhumatoïde

le promoteur des gènes. L’activation des facteursde transcription est induite par des protéines kina-ses qui ont une activité phosphorylante. Cettephosphorylation du facteur de transcription permetsa translocation du cytoplasme vers le noyau ouencore augmente son affinité pour l’ADN par chan-gement conformationnel. Les principales voies designalisation impliquées dans l’inflammation sontTRAF/IjBK/NFjB, la voie des mitogen activatedprotein kinases (MAPK)/AP-1, la voie de la phospho-inositide-3 kinase et la voie JAK/STAT. NFjB estactivé lorsqu’il est dissocié de IKB. La dissociationde ces deux molécules est favorisée par la phospho-rylation de IKB par la kinase IKB (IKK). Pour lesMAPK, il existe trois familles : p38, Erk1/2 et JNK.Toutes les trois sont exprimées dans le tissu syno-vial des patients atteints de PR. L’activation de p38induit la synthèse des cytokines pro-inflammatoirescomme le TNF a, l’IL1, l’IL6 et l’IL8, soit paractivation directe de la transcription des gènes,soit par stabilisation de l’acide ribonucléique mes-sager.54,55,56,57 La p38 MAPK contrôle également la
synthèse d’autres molécules impliquées dans l’in-flammation comme les chimiokines et les molécu-les d’adhésion, mais aussi dans la synthèse desmétalloprotéinases responsables de la destructioncartilagineuse ou la synthèse des prostaglandi-nes.58,59 La phospho-inositide-3 kinase est une en-zyme induisant la phosphorylation de lipides mem-branaires, les phospho-inositides. La phospho-inositide-3 kinase est impliquée dans laprolifération et l’adhésion cellulaire, mais aussidans l’apoptose et l’angiogenèse.60 Les facteurs detranscription AP-1 et NFjB semblent particulière-ment impliqués pour deux raisons. Premièrement,ils sont surexprimés dans les synoviocytes extraitsde synoviale rhumatoïde par rapport à la synovialed’arthrose et cette expression est corrélée à l’acti-vité de la maladie.61 Deuxièmement, AP-1 et NFjBinterviennent dans des voies de signalisation quicontrôlent la synthèse de protéines participant àl’inflammation de la synoviale et à la destructionarticulaire. NFjB et AP-1 régulent l’activation desgènes codant pour les métalloprotéinases, maisaussi pour des cytokines pro-inflammatoires,comme l’IL6 et le TNF a,62 mais aussi des média-teurs de l’inflammation (prostaglandines), de l’an-giogenèse (vascular endothelial growth factor, fi-broblasts growth factor, chimiokines) et durecrutement cellulaire dans la synoviale (moléculesd’adhésion et chimiokines).63,64 Ces molécules designalisation représentent de nouvelles cibles thé-rapeutiques dans le traitement de la PR. Des inhi-biteurs de p38 MAPK sont actuellement en essaithérapeutique chez l’homme.
Phase de destruction articulaire
Des progrès considérables ont été réalisés ces der-nières années dans la compréhension des mécanis-mes impliqués dans la destruction ostéoarticulaire.La destruction ostéoarticulaire est la conséquencede la prolifération pseudotumorale de la synovialeet de l’action des cytokines. La nette réduction desdestructions articulaires chez les patients traitéspour leur PR par des antagonistes de l’IL1 et du TNFa démontrent clairement le rôle structural de cescytokines dans cette maladie.49,65,66 L’IL1 et le TNFa participent à cette destruction articulaire eninduisant non seulement la synthèse de facteurs decroissance nécessaires à la prolifération de la syno-viale, mais aussi la production par les synoviocytesde métalloprotéinases, de cathepsines et de colla-génases responsables de la dégradation des princi-paux composants du cartilage.67,68,69,70 Des don-nées récentes suggèrent l’implication du systèmeRANK/RANKL dans la résorption osseuse sous-
Figure 4 L’activation en cascade des protéines kinases active lesfacteurs de transcription (FT). Lorsque qu’une cytokine se fixesur un récepteur membranaire, elle provoque une modificationde conformation du récepteur qui aboutit à la phosphorylationdu récepteur lui-même ou d’une enzyme associée à ce récep-teur. Cette première phosphorylation entraîne l’activation encascade d’autres enzymes appelées les protéines kinases quiactivent à leur tour les facteurs de transcription. MAPK=mitogenactivated protein kinase ; PI 3 K : phospho-inositides 3 kinase ;IKK : I kappa B kinase ; NFjB : nuclear factor kappa B ; AP-1 :activating protein 1 ; STAT : signal transducer and activator oftranscription ; RTK : récepteur des protéines tyrosines kinases ;PK : protéines kinases ; ATP : adénosine triphosphate ; ADP :adénosine diphosphate ; P : phosphore.
226 J. Morel et al.

chondrale des patients atteints de PR. Le receptoractivator of NFjB ligand (RANKL) est une nouvellecytokine exprimée à la surface des cellules de lalignée ostéoblastique, mais aussi des lymphocytesactivés et des cellules endothéliales.71,72,73 La pro-duction de RANKL est régulée par les cytokinespro-inflammatoires telles que l’IL1 et le TNF a,74
mais aussi l’IL17. RANK est le récepteur membra-naire de RANKL et l’ostéoprotégérine la forme solu-ble du récepteur. La liaison de RANKL à son récep-teur membranaire RANK, présent sur lespréostéoclastes, favorise la différenciation et l’ac-tivation des ostéoclastes.75 RANKL est trouvé à desconcentrations élevées dans le sérum et le liquidesynovial des patients atteints de PR. Le rôle deRANKL dans la destruction articulaire est fortementsuggéré par les observations faites dans les modèlesanimaux. Dans le modèle de polyarthrite induitepar le transfert de sérum provenant de souris K/BxNappliqué aux souris déficientes en RANKL (RANKL-/-), les souris développent une arthrite mais pasd’érosions osseuses.76 Dans différents modèlesd’arthrites expérimentales, l’administration d’os-téoprotégérine réduit les érosions osseuses et aug-mente la densité minérale osseuse.77 Les synovio-cytes et les cellules endothéliales pourraientparticiper à la destruction articulaire par l’inter-médiaire de ce système par leur capacité à expri-mer du RANKL lorsqu’elles sont stimulées.72,78 Lessynoviocytes de PR ont des caractéristiques qui serapprochent des cellules tumorales par leur mor-phologie, la perte d’inhibition de contact, maisaussi l’augmentation de plusieurs proto-oncogènestels que Bcl2, Myc, Ras, fos ou encore la phospho-inositide-3 kinase.38 L’intérêt s’est donc naturelle-ment porté sur certains gènes suppresseurs destumeurs, comme le gène codant pour la protéinep53. Une mutation dans la séquence aminoacide dela protéine p53, qui est observée dans certainestumeurs, est également trouvée dans les synoviocy-tes de PR.79 Cette mutation de la protéine p53contribuerait au prolongement de la durée de viedes cellules. La prolifération anormale des synovio-cytes de PR pourrait donc être expliquée, commepour les tumeurs cancéreuses, par un défaut detransmission d’un signal de mort pour la cellule,appelée aussi apoptose.39 Le phosphatase and ten-sin homologue (PTEN) est un autre suppresseurtumoral qui a été récemment impliqué dans laprolifération incontrôlée et invasive des synoviocy-tes de PR.38 PTEN, qui est un inhibiteur naturel dela phospho-inositide-3 kinase, est sous-exprimédans les synoviocytes. Les connaissances actuellesne permettent cependant pas de déterminer sil’hyperplasie synoviale observée dans la PR résultevéritablement d’une prolifération anormale ou
d’un défaut d’apoptose des synoviocytes. L’ab-sence de différence en termes de vitesse de proli-fération entre les synoviocytes extraits de PR etceux obtenus à partir de sujets arthrosiques estplutôt un argument contre une dysrégulation de laprolifération des synoviocytes rhumatoïdes. Lesdonnées récentes privilégient plutôt l’hypothèsed’une dérégulation de l’apoptose dans les synovio-cytes. Les phénomènes d’apoptose sont en effetrares dans la synoviale et pourraient résulter d’unesurexpression de facteurs antiapoptotiques.L’apoptose induite par le système Fas/FasL neconcerne qu’un pourcentage limité de synoviocytes(20 %) et la majorité des cellules sont résistantes àl’apoptose. Cette résistance des synoviocytes pour-rait donc résulter d’une surexpression de facteursantiapoptotiques qui restent à identifier. Les syno-viocytes obtenus à partir de patients atteints de PRse distinguent de ceux des sujets normaux ou arth-rosiques par leur capacité à adhérer fortement aucartilage, à l’envahir et enfin à le détruire.69
Conclusion
La physiopathologie de la PR reste complexe et faitintervenir de nombreux acteurs cellulaires, maisaussi inter- et intracellulaires. Les progrès réalisésces dernières années concernent surtout la com-préhension des mécanismes de l’inflammation de lasynoviale rhumatoïde. Cette meilleure compréhen-sion de la pathogénie de la synovite rhumatoïde apermis de développer des cibles thérapeutiques,mais permet aussi d’envisager dans l’avenir procheune large gamme de nouveaux traitements dirigéscontre les cytokines (anti-IL6, -IL17, -IL18, -RANKL,-CCR5), contre les voies de signalisation (MAPK,phospho-inositide-3 kinase) ou encore l’activationdes cellules (CTLA4 immunoglobuline, rituximab).
Références
1. Sany J, Combe B, Jorgensen C. Polyarthrite rhumatoïde del’adulte (I). Aspects cliniques. Encycl Méd Chir (ElsevierSAS, Paris). Appareil locomoteur 1997 14-220-A-10.
2. Firestein GS. Evolving concepts of rheumatoid arthritis.Nature 2003;423:356–361.
3. Sany J. La polyarthrite rhumatoïde de l’adulte : concep-tion actuelle. Paris: John Libbey Eurotext; 2003.
4. Silman A, Kay A, Brennan P. Timing of pregnancy in rela-tion to the onset of rheumatoid arthritis. Arthritis Rheum1992;35:152–155.
5. Brennan P, Silman A. Breast-feeding and the onset ofrheumatoid arthritis. Arthritis Rheum 1994;37:808–813.
6. Cutolo M, Seriolo B, Villaggio B, Pizzorni C, Craviotto C,Sulli A. Androgens and estrogens modulate the immune andinflammatory responses in rheumatoid arthritis. Ann N YAcad Sci 2002;966:131–142.
227Immunopathologie de la polyarthrite rhumatoïde

7. Gregersen PK, Silver J, Winchester RJ. The shared epitopehypothesis: an approach to understanding the moleculargenetics of susceptibility to rheumatoid arthritis. ArthritisRheum 1987;30:1205–1213.
8. Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA,Cairns E. The conversion of arginine to citrulline allows fora high-affinity peptide interaction with the rheumatoidarthritis-associated HLA-DRB1*0401 MHC class II molecule.J Immunol 2003;171:538–541.
9. Emery P, Salmon M, Bradley H, Wordsworth P, Tunn E,Bacon PA, et al. Genetically determined factors as predic-tors of radiological change in patients with early symetri-cal arthritis. Br Med J 1992;305:1387–1389.
10. Gough AK, Faint J, Salmon M, Hassel A, Wordsworth P,Pilling D, et al. Genetic typing of patients with inflamma-tory arthritis at presentation can be used to predict out-come. Arthritis Rheum 1994;37:1166–1170.
11. Weyand CM, MacCarthy TG, Goronzy JJ. Correlationbetween disease phenotype and genetic heterogeneity inrheumatoid arthritis. J Clin Invest 1995;95:2120–2126.
12. Combe B, Eliaou JF, Meyer O, Clot J, Sany J. Prognosticfactors in rheumatoid arthritis. Comparative study of twosubsets of patients according to severity of articular dam-age. Br J Rheumatol 1995;34:529–534.
13. Pinet V, Combe B, Avinens O, Caillat-Zucman S, Sany J,Clot J, et al. Polymorphism of the HLA-DMA and DMB genesin rheumatoid arthritis. Arthritis Rheum 1997;40:854–858.
14. Nieto A, Caliz R, Pascual M, Mataran L, Garcia S, Martin J.Involvement of Fcgamma receptor IIIA genotypes in sus-ceptibility to rheumatoid arthritis. Arthritis Rheum 2000;43:735–739.
15. Lard LR, van Gaalen FA, Schonkeren JJ, Pieterman EJ,Stoeken G, Vos K, et al. Association of the -2849interleukin-10 promoter polymorphism with autoantibodyproduction and joint destruction in rheumatoid arthritis.Arthritis Rheum 2003;48:1841–1848.
16. Udalova IA, Richardson A, Ackerman H, Wordsworth P,Kwiatkowski D. Association of accelerated erosive rheuma-toid arthritis with a polymorphism that alters NF-kappaBbinding to the TNF promoter region. Rheumatology 2002;41:830–831.
17. Buchs N, Silvestri T, di Giovine FS, Chabaud M, Vannier E,Duff GW, et al. IL-4 VNTR gene polymorphism in chronicpolyarthritis. The rare allele is associated with protectionagainst destruction. Rheumatology 2000;39:1126–1131.
18. Buchs N, di Giovine FS, Silvestri T, Vannier E, Duff GW,Miossec P. IL-1B and IL-1Ra gene polymorphisms and dis-ease severity in rheumatoid arthritis: interaction withtheir plasma levels. Genes Immun 2001;2:222–228.
19. Klinman D. Does activation of the innate immune systemcontribute to the development of rheumatoid arthritis?Arthritis Rheum 2003;48:590–593.
20. Silman AJ, Newman J, MacGregor AJ. Cigarette smokingincreases the risk of rheumatoid arthritis. Results from anationwide study of disease-discordant twins. ArthritisRheum 1996;39:732–735.
21. Pettit AR, Thomas R. Dendritic cells: the driving forcebehind autoimmunity in rheumatoid arthritis? ImmunolCell Biol 1999;77:420–427.
22. Page G, Lebecque S, Miossec P. Anatomic localization ofimmature and mature dendritic cells in an ectopic lym-phoid organ: correlation with selective chemokine expres-sion in rheumatoid synovium. J Immunol 2002;168:5333–5341.
23. Berek C, Schroder AE. A germinal center-like reaction inthe nonlymphoid tissue of the synovial membrane. Ann N YAcad Sci 1997;815:211–217.
24. Panayi GS, Corrigall VM, Pitzalis C. Pathogenesis of rheu-matoid arthritis. The role of T cells and other beasts.Rheum Dis Clin North Am 2001;27:317–334.
25. Cao D, Malmstrom V, Baecher-Allan C, Hafler D,Klareskog L, Trollmo C. Isolation and functional character-ization of regulatory CD25brightCD4+ T cells from thetarget organ of patients with rheumatoid arthritis. Eur JImmunol 2003;33:215–223.
26. Shimizu Y, Newman W, Tanaka Y, Shaw S. Lymphocyteinteractions with endothelial cells. Immunol Today 1992;13:106–112.
27. Damle NK, Eberhardt C, Van der Vieren M. Direct interac-tion with primed CD4+ CD45R0+ memory T lymphocytesinduces expression of endothelial leukocyte adhesionmolecule-1 and vascular cell adhesion molecule-1 on thesurface of vascular endothelial cells. Eur J Immunol 1991;21:2915–2923.
28. Oppenheimer-Marks N, Davis LS, Bogue DT, Ramberg J,Lipsky PE. Differential utilization of ICAM-1 and VCAM-1during the adhesion and transendothelial migration ofhuman T lymphocytes. J Immunol 1991;147:2913–2921.
29. Korganow AS, Fournier C, Pasquali JL, Martin T. Place dusystème immunitaire dans la physiopathologie de la poly-arthrite rhumatoïde. Méd Thér 1996;2:267–274.
30. Jalkanen S, Steere AC, Fox RI, Butcher EC. A distinctendothelial cell recognition system that controls lympho-cyte traffic into inflamed synovium. Science 1986;233:556–558.
31. Trollmo C, Nilsson IM, Sollerman C, Tarkowski A. Expres-sion of the mucosal lymphocyte integrin alpha E beta 7 andits ligand E-cadherin in the synovium of patients withrheumatoid arthritis. Scand J Immunol 1996;44:293–298.
32. Burger D, Rezzonico R, Li JM, Modoux C, Pierce RA, Wel-gus HG, et al. Imbalance between interstitial collagenaseand tissue inhibitor of metalloproteinases 1 in synoviocytesand fibroblasts upon direct contact with stimulated Tlymphocytes: involvement of membrane-associated cytok-ines. Arthritis Rheum 1998;41:1748–1759.
33. Chabaud M, Aarvak T, Garnero P, Natvig JB, Miossec P.Potential contribution of IL-17-producing Th(1)cells todefective repair activity in joint inflammation: partialcorrection with Th(2)-promoting conditions. Cytokine2001;13:113–118.
34. Aarvak T, Natvig JB. Cell-cell interactions in synovitis:antigen presenting cells and T cell interaction in rheuma-toid arthritis. Arthritis Res 2001;3:13–17.
35. Firestein GS, Zvaifler NJ. How important are T cells inchronic rheumatoid synovitis? Arthritis Rheum 1990;33:768–773.
36. Firestein GS, Alvaro-Gracia JM, Maki R, Alvaro-Garcia JM.Quantitative analysis of cytokine gene expression in rheu-matoid arthritis. J Immunol 1990;144:3347–3353.
37. Russo-Marie F. La réaction inflammatoire dans la polyarth-rite rhumatoïde. Méd Thér 1996;2:261–266.
38. Pap T, Franz JK, Hummel KM, Jeisy E, Gay R, Gay S.Activation of synovial fibroblasts in rheumatoid arthritis:lack of expression of the tumour suppressor PTEN at sitesof invasive growth and destruction. Arthritis Res 2000;2:59–64.
39. Pope RM. Apoptosis as a therapeutic tool in rheumatoidarthritis. Nat Rev Immunol 2002;2:527–535.
40. De Vita S, Zaja F, Sacco S, De Candia A, Fanin R, Ferrac-cioli G. Efficacy of selective B cell blockade in the treat-ment of rheumatoid arthritis: evidence for a pathogeneticrole of B cells. Arthritis Rheum 2002;46:2029–2033.
228 J. Morel et al.

41. Tighe H, Chen PP, Tucker R, Kipps TJ, Roudier J,Jirik FR, et al. Function of B cells expressing a humanimmunoglobulin M rheumatoid factor autoantibody intransgenic mice. J Exp Med 1993;177:109–118.
42. Eliaou JF, Andary M, Favier F, Carayon P, Poncelet P,Sany J, et al. Increase of class II HLA molecules on themembrane of B lymphocytes from patients with rheuma-toid arthritis. Autoimmunity 1988;1:217–222.
43. Kim HJ, Berek C. B cells in rheumatoid arthritis. ArthritisRes 2000;2:126–131.
44. van Boekel MA, Vossenaar ER, van den Hoogen FH, vanVenrooij WJ. Autoantibody systems in rheumatoidarthritis: specificity, sensitivity and diagnostic value.Arthritis Res 2002;4:87–93.
45. Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis pro-voked by linked T and B cell recognition of a glycolyticenzyme. Science 1999;286:1732–1735.
46. Miossec P. Interleukin-17 in rheumatoid arthritis if T cellscontribute to inflammation and destruction through syn-ergy. Arthritis Rheum 2003;48:594–601.
47. Alvaro-Gracia JM, Zvaifler NJ, Brown CB, Kaushansky K,Firestein GS. Cytokines in chronic inflammatory arthritis.VI. Analysis of the synovial cells involved in granulocyte-macrophage colony-stimulating factor production andgene expression in rheumatoid arthritis and its regulationby IL-1 and tumor necrosis factor-alpha. J Immunol 1991;146:3365–11371.
48. Bresnihan B, Alvaro-Gracia JM, Cobby M, Doherty M, Dom-ljan Z, Emery P, et al. Treatment of rheumatoid arthritiswith recombinant human interleukin-1 receptor antago-nist. Arthritis Rheum 1998;41:2196–2204.
49. Jiang Y, Genant HK, Watt I, Cobby M, Bresnihan B, Aitchi-son R, et al. A multicenter, double-blind, dose-ranging,randomized, placebo-controlled study of recombinanthuman interleukin-1 receptor antagonist in patients withrheumatoid arthritis: radiologic progression and correla-tion of Genant and Larsen scores. Arthritis Rheum 2000;43:1001–1009.
50. Smeets TJ, Kraan MC, van Loon ME, Tak PP. Tumor necrosisfactor alpha blockade reduces the synovial cell infiltrateearly after initiation of treatment, but apparently not byinduction of apoptosis in synovial tissue. Arthritis Rheum2003;48:2155–2162.
51. Morel JC, Park CC, Woods JM, Koch AE. A novel role forinterleukin-18 in adhesion molecule induction through NFkappa B and phosphatidylinositol (PI) 3-kinase-dependentsignal transduction pathways. J Biol Chem 2001;276:37069–37075.
52. Wong PK, Campbell IK, Egan PJ, Ernst M, Wicks IP. The roleof the interleukin-6 family of cytokines in inflammatoryarthritis and bone turnover. Arthritis Rheum 2003;48:1177–1189.
53. Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y,Bird H, et al. Therapeutic benefit of blocking interleukin-6activity with an anti-interleukin-6 receptor monoclonalantibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. ArthritisRheum 2002;46:3143–3150.
54. Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S,Green D, et al. A protein kinase involved in the regulationof inflammatory cytokine biosynthesis. Nature 1994;372:739–746.
55. Beyaert R, Cuenda A, Vanden Berghe W, Plaisance S,Lee JC, Haegeman G, et al. The p38/RK mitogen-activatedprotein kinase pathway regulates interleukin-6 synthesisresponse to tumor necrosis factor. Embo J 1996;15:1914–1923.
56. Suzuki M, Tetsuka T, Yoshida S, Watanabe N, Kobayashi M,Matsui N, et al. The role of p38 mitogen-activated proteinkinase in IL-6 and IL-8 production from the TNF-alpha- orIL-1beta-stimulated rheumatoid synovial fibroblasts. FEBSLett 2000;465:23–27.
57. Miyazawa K, Mori A, Miyata H, Akahane M, Ajisawa Y,Okudaira H. Regulation of interleukin-1beta-inducedinterleukin-6 geneexpression in human fibroblast-like syno-viocytes by p38 mitogen-activated protein kinase. J BiolChem 1998;273:24832–24838.
58. Herlaar E, Brown Z. p38 MAPK signalling cascades in inflam-matory disease. Mol Med Today 1999;5:439–447.
59. Berenbaum F, Humbert L, Bereziat G, Thirion S. Concomi-tant recruitment of ERK1/2 and p38 MAPK signalling path-way is required for activation of cytoplasmic phospholipaseA2 via ATP in articular chondrocytes. J Biol Chem 2003;278:13680–13687.
60. Roymans D, Slegers H. Phosphatidylinositol 3-kinases intumor progression. Eur J Biochem 2001;268:487–498.
61. Firestein GS, Manning AM. Signal transduction and tran-scription factors in rheumatic disease. Arthritis Rheum1999;42:609–621.
62. May MJ, Ghosh S. Signal transduction through NF-KB.Immunol Today 1998;19:80–88.
63. Dayer JM, de Rochemonteix B, Burrus B, Demczuk S,Dinarello CA. Human recombinant interleukin 1 stimulatescollagenase and prostaglandin E2 production by humansynovial cells. J Clin Invest 1986;77:645–648.
64. Alvaro-Gracia JM, Zvaifler NJ, Firestein GS. Cytokines inchronic inflammatory arthritis. V. Mutual antagonismbetween interferon-gamma and tumor necrosis factor-alpha on HLA-DR expression, proliferation, collagenaseproduction, and granulocyte macrophage colony-stimulating factor production by rheumatoid arthritis syn-oviocytes. J Clin Invest 1990;86:1790–1798.
65. Lipsky PE, van der Heijde DM, St Clair EW, Furst DE,Breedveld FC, Kalden JR, et al. Infliximab and methotrex-ate in the treatment of rheumatoid arthritis. N Engl J Med2000;343:1594–1602 Anti-tumor necrosis factor trial inrheumatoid arthritis with concomitant therapy studygroup.
66. Genovese MC, Bathon JM, Martin RW, Fleischmann RM,Tesser JR, Schiff MH, et al. Etanercept versus methotrex-ate in patients with early rheumatoid arthritis: two-yearradiographic and clinical outcomes. Arthritis Rheum 2002;46:1443–1450.
67. Okada Y, Morodomi T, Enghild JJ, Suzuki K, Yasui A,Nakanishi I, et al. Matrix metalloproteinase 2 from humanrheumatoid synovial fibroblasts. Purification and activa-tion of the precursor and enzymic properties. Eur J Bio-chem 1990;194:721–730.
68. Case JP, Lafyatis R, Remmers EF, Kumkumian GK,Wilder RL. Transin/stromelysin expression in rheumatoidsynovium. A transformation-associated metalloproteinasesecreted by phenotypically invasive synoviocytes. Am JPathol 1989;135:1055–1064.
69. Edwards JC. Fibroblast biology. Development and differen-tiation of synovial fibroblasts in arthritis. Arthritis Res2000;2:344–347.
70. Feldmann M, Maini RN. Anti-TNF alpha therapy of rheuma-toid arthritis: what have we learned? Annu Rev Immunol2001;19:163–196.
71. Hofbauer LC, Khosla S, Dunstan CR, Lacey DL, Boyle WJ,Riggs BL. The roles of osteoprotegerin and osteoprotegerinligand in the paracrine regulation of bone resorption. JBone Miner Res 2000;15:2–12.
229Immunopathologie de la polyarthrite rhumatoïde

72. Collin-Osdoby P, Rothe L, Anderson F, Nelson M, Mal-oney W, Osdoby P. Receptor activator of NF-kappa B andosteoprotegerin expression by human microvascularendothelial cells, regulation by inflammatory cytokines,and role in human osteoclastogenesis. J Biol Chem 2001;276:20659–20672.
73. Roux S, Orcel P. Bone loss. Factors that regulate osteoclastdifferentiation: an update. Arthritis Res 2000;2:451–456.
74. Hofbauer LC, Lacey DL, Dunstan CR, Spelsberg TC,Riggs BL, Khosla S. Interleukin-1beta and tumor necrosisfactor-alpha, but not interleukin-6, stimulate osteoprote-gerin ligand gene expression in human osteoblastic cells.Bone 1999;25:255–259.
75. Hofbauer LC, Heufelder AE. Role of receptor activator ofnuclear factor-kappaB ligand and osteoprotegerin in bonecell biology. J Mol Med 2001;79:243–253.
76. Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR,Choi Y, et al. TRANCE/RANKL knockout mice are protectedfrom bone erosion in a serum transfer model of arthritis.Am J Pathol 2001;159:1689–1699.
77. Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M,Lang S, et al. Tumor necrosis factor alpha-mediated jointdestruction is inhibited by targeting osteoclasts withosteoprotegerin. Arthritis Rheum 2002;46:785–792.
78. Takayanagi H, Iizuka H, Juji T, Nakagawa T, Yamamoto A,Miyazaki T, et al. Involvement of receptor activator ofnuclear factor kappaB ligand/ osteoclast differentiationfactor in osteoclastogenesis from synoviocytes in rheuma-toid arthritis. Arthriris Rheum 2000;43:259–269.
79. Firestein GS, Echeverri F, Yeo M, Zvaifler NJ, Green DR.Somatic mutations in the p53 tumor suppressor gene inrheumatoid arthritis synovium. Proc Natl Acad Sci USA1997;94:10895–10900.
230 J. Morel et al.





























