Embed Size (px)
Citation preview

Improving Clinical Significance of PCR: Use of Propidium Monoazideto Distinguish Viable from Dead Staphylococcus aureus andStaphylococcus epidermidis
Hideo Kobayashi,1,2 Margret Oethinger,1 Marion J. Tuohy,1 Gerri S. Hall,1 Thomas W. Bauer1,2
1Institute of Pathology and Laboratory Medicine, The Cleveland Clinic, Cleveland, Ohio, 2Orthopaedic and Rheumatologic Institute, The ClevelandClinic Foundation, 9500 Euclid Avenue, Cleveland, Ohio 44195
Received 18 September 2008; accepted 29 January 2009
Published online 25 March 2009 in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/jor.20872
ABSTRACT: Molecular techniques, such as the polymerase chain reaction (PCR) have high sensitivity when used to diagnose infection, butmay detect DNA, RNA, and proteins from dead, as well as viable, bacteria. Propidium monoazide (PMA) is a DNA binding agent, that has theability to penetrate only dead cells with compromised membranes and has been used in conjunction with real-time PCR to distinguish intactfrom dead bacterial cells. In this study, intact, heat-inactivated (dead), and intact/dead admixed Staphylococcus aureus (S. aureus) andStaphylococcus epidermidis (S. epidermidis) were treated with PMA or left untreated before DNA extraction. We quantified levels of 16SrDNA and tuf gene by real-time quantitative PCR (qPCR), to test the ability of PMA to distinguish intact from dead bacteria. Our resultsindicated that PMA inhibited detection of dead bacteria, and the qPCR results reflected the number of intact bacteria without being impactedby the presence of the dead bacteria. This approach of combining qPCR with and without PMA treatment has promise to limit false-positivePCR results when used to diagnose infections, but needs to be further validated in clinical samples. � 2009 Orthopaedic Research Society.
Published by Wiley Periodicals, Inc. J Orthop Res 27:1243–1247, 2009
Keywords: propidium monoazide (PMA); bacterial viability; Staphylococcus aureus; Staphylococcus epidermidis; real-time polymerase
chain reaction
Polymerase chain reaction (PCR) is recognized as asensitive tool for detecting bacteria associated withorthopedic infections, and is being used for rapiddiagnosis and detection of non-cultivable pathogens.1,2
However, a major limitation is the lack of differentiationbetween viable and dead bacteria.3,4 Several previousstudies5,6 that have evaluated synovial fluid or ortho-pedic implants after total hip or knee arthroplasty havereported evidence of bacteria in a high proportion ofcases. For example, Tunney et al.5 tested the detectionrates of culture versus PCR in 120 patients who hadundergone total hip revision surgery. They reportedthat 22% of cases were culture positive, but 72% werePCR positive. Similarly, Mariani et al.6 analyzedsynovial fluid aspirates from 50 patients with symptomsafter total knee arthroplasty and reported that 6 caseswere standard culture positive, but 32 cases were PCRpositive. It is not yet known if the high prevalence ofbacteria identified in those studies represents the causeof patients’ infection or false-positive results. Our recentresults1 in which PCR was used to identify evidence ofbacteria in apparently aseptically loose implantsshowed bacterial DNA in 12%, which is a smallerproportion to that of Tunney et al.5 or Mariani et al.6
However, in vitro studies have also shown that PCR canidentify DNA from dead bacteria.3 While the detectionof dead bacteria is interesting, it carries less clinicalimportance than detecting viable bacteria. If PCR isused to diagnose persistent infection at the second stagerevision for a previously infected prosthesis despiteantibiotic therapy, it is especially important to deter-
mine if PCR-positive results are derived from viable ornonviable bacteria since dead bacteria could cause‘‘false-positive’’ PCR results (i.e., a true positive fromthe assay perspective, but a false-positive with respectto patient care).
Propidium monoazide (PMA) has recently been usedas a DNA binding agent to differentiate intact frommembrane-compromised bacterial cells.7–10 PMA hasthe ability to penetrate dead cells with compromisedmembranes, and to intercalate into the DNA uponexposure to intense visible light. This results in covalentcross-linkage of the two DNA strands and blockssubsequent amplification of target DNA sequences byPCR. Unbound PMA is decomposed by photolysis anddoes not interfere with PCR. In contrast, PMA is unableto penetrate through the membrane of intact bacterialcells. Thus, when bacterial cells are extracted andtreated with PMA before DNA extraction, only DNAfrom the intact cells is amplified by the PCR.
As far as we know, there is only one report on theeffectiveness of the PMA assay for Staphylococcusaureus (S. aureus).7 The purpose of this study wasto use in vitro assays with mixtures of intact anddead bacteria to investigate the effectiveness of PMAin conjunction with real-time quantitative PCR(qPCR) to distinguish intact from dead S. aureus andStaphylococcus epidermidis (S. epidermidis), the organ-isms most commonly involved with periprostheticinfections.11
METHODSBacteria and CulturesBiofilm-formative S. aureus (ATCC 12600) and biofilm-formative S. epidermidis (289F22) were used in the in vitrostudy. Each bacterial strain was grown to mid-exponential
JOURNAL OF ORTHOPAEDIC RESEARCH SEPTEMBER 2009 1243
Correspondence to: Thomas W. Bauer (T: þ1-216-444-6830; F: þ1-216-445-6967; E-mail: [email protected])
� 2009 Orthopaedic Research Society. Published by Wiley Periodicals, Inc.

phase in brain heart infusion (BHI) broth at 378C and split intotwo cultures. One culture was heated in a boiling water bathfor 10 min, the other was left untreated. As described below,500 ml aliquots of each suspension were harvested for DNAextraction after incubation with (or without) PMA (Biotium,Inc., Hayward, CA). DNA extraction was performed intriplicate. At the same time, cell suspensions were plated forcolony counts. For quantification of bacteria by conventionalmicrobiological culture, an aliquot of the suspension wasserially diluted and spread in duplicate on blood agar plates(TSA II 5% SB, Becton, Dickinson and Company, Sparks, MD).After incubation for 18–24 h, colony-forming units (CFU) werecounted for each plate, averaged, and expressed as CFU/ml.The heat-treated tubes were cooled to room temperature, andthe absence of viable CFU was determined.
Mixture of Intact with Dead BacteriaA constant quantity of heat-inactivated cultures (250 ml, nodilution) was mixed with the unheated (intact) cultures of fivedifferent concentrations (250 ml, 1:1–1:105 dilutions). Controlgroups of unheated cultures of five different concentrations(250 ml, 1:1–1:105 dilutions) without PMA treatment were alsoprepared. Conversely, unheated (intact) cultures of constantconcentration (250 ml, no dilution) were mixed with the heat-inactivated cultures at five different concentrations (250 ml,1:1–1:105 dilutions). All these dilutions were made with BHIbroth. The intact-dead cell mixtures (500 ml) were then treatedwith PMA as described below.
PMA Cross-LinkingThe methods and the optimal concentration of the PMA assayhave been described previously.7 Briefly, 1.25 ml of 20 mMPMA was added to 500 ml culture aliquots to final concen-trations of 50 mM. Light-transparent 1.7-ml microcentrifugetubes (Fisher Scientific, Pittsburgh, PA) were used. Followingan incubation period of 5 min in the dark with occasionalflipping, samples were light exposed for 2 min using a 650-Whalogen light source (sealed beam lamp, FCW 120 V; GELighting, General Electric Co., Cleveland, OH). The sampletubes were placed at 20 cm from the light source and were laidhorizontally on ice with continuous tilting. After photo-induced cross-linking, bacterial suspensions were centrifugedat 7,600 rpm for 10 min for DNA extraction.
DNA Extraction and Real-Time PCRDNA extraction was performed from 500 ml of each suspensionusing DNeasy Blood & Tissue kit (Qiagen Inc., Valencia, CA)according to the manufacturer’s instructions. One hundredmicroliters of DNA extract was obtained for each sample.Quantitative PCR assays targeted 16S rDNA1 and tuf using aRotorgene 30001 (Corbett Research, Sydney, Australia). Tufprimer sequences (BioChem, Salt Lake City, UT) were:forward primer, 50-ATGCCACAAACTCGTGA-30, reverse pri-mer, 50-ACCACGACCAGTGATTGAGAA-30.
The PCR master mix consisted of 4.0 or 4.5 mmol/L MgCl2,0.2 mmol/L of each primer, 12.5 mL of 2�SensiMix (Quantace,Norwood, MA) and 0.5 mL of 50�SYBR green I (Quantace) for avolume of 23 mL master mix. Two microliters of DNA extractwere added to the reaction mixture, for a final reaction volumeof 25 mL for each tube. The cycling conditions were: (1) 16SrRNA–958C for 10 min, followed by 45 cycles of denaturationat 958C for 5 s, annealing at 578C for 20 s, and 728C for 15 s; and(2) tuf–958C for 10 min, followed by 45 cycles of denaturation at958C for 10 s, annealing at 508C for 30 s, and 728C for 15 s. A
DNA standard curve was generated by serial dilutions of asample with known bacterial concentration and plotted asqPCR cell number versus the threshold cycle (Ct) at whichthe amplified DNA in the sample exceeded the detection thres-hold during the PCR. The difference between the detectionthreshold cycles of untreated and PMA-treated bacteriawas calculated for each sample pair as follows: DCt¼Ct with PMA�Ct without PMA. In addition, the qPCR cell numberof individual samples was calculated from the standardcurves.
Statistical AnalysisResults of intact and dead bacteria, expressed in Ct dif-ferences (DCt), were compared using the unpaired Student’st-tests. p-Values less than 0.01 were considered statisticallysignificant.
RESULTSEffect of PMATreatment of intact cells with PMA was associated witha small DCt, i.e., the Ct value of bacteria treated withPMA was only slightly higher than the Ct value ofuntreated cells (Fig. 1A, B left, C left). When resultswere expressed in qPCR-derived cell counts and bothPMA-treated and PMA-untreated samples were readfrom the standard curve that was generated from PMA-untreated cells, PMA treatment caused intact cells todrop by less than one log, whereas PMA-treated deadcells dropped 3–4 log (Fig. 1D, E). The qPCR cell countsobtained from bacterial cultures without PMA treat-ment were positive, irrespective of bacterial viability(Fig. 1A, D, E), confirming that PCR identifies DNAfrom dead as well as intact bacteria.
PMA treatment resulted in a significant increase of Ctvalues of target DNA derived from dead bacteria (Fig. 1B,C right) and the Ct values were almost as high as a Ctvalue of the negative control (Ultra Clean PCR water; MoBio Laboratories, Inc., Carlsbad, CA) (Fig. 1A), i.e., PCRturned positive almost as late as the negative control.The qPCR results emphasized that PMA inhibited thePCR detection of dead bacteria, but only slightlyinfluenced detection of intact bacteria (Fig. 1D, E). TheqPCR results for 16S rDNA and tuf gene in S. aureus andS. epidermidis were almost identical (Table 1).
Mixture of Intact with Dead BacteriaThe qPCR bacterial number from intact/dead mixturesof heat-inactivated cultures (constant concentration)and unheated cultures (five different concentrations)with PMA treatment (Fig. 2, black bars) were similar tounheated cultures without PMA (control group; Fig. 2,gray bars); i.e., increasing proportions of intact bacterialed to proportional increases in the bacterial numberdetected by qPCR. The results suggest that theamplification of target DNA from the dead bacteriawas effectively inhibited by PMA, and that the qPCRnumber reflected the intact bacteria, irrespective of thepresence of the dead bacteria.
When the unheated cultures (constant concentration)were mixed with the heat-inactivated cultures (five
1244 KOBAYASHI ET AL.
JOURNAL OF ORTHOPAEDIC RESEARCH SEPTEMBER 2009
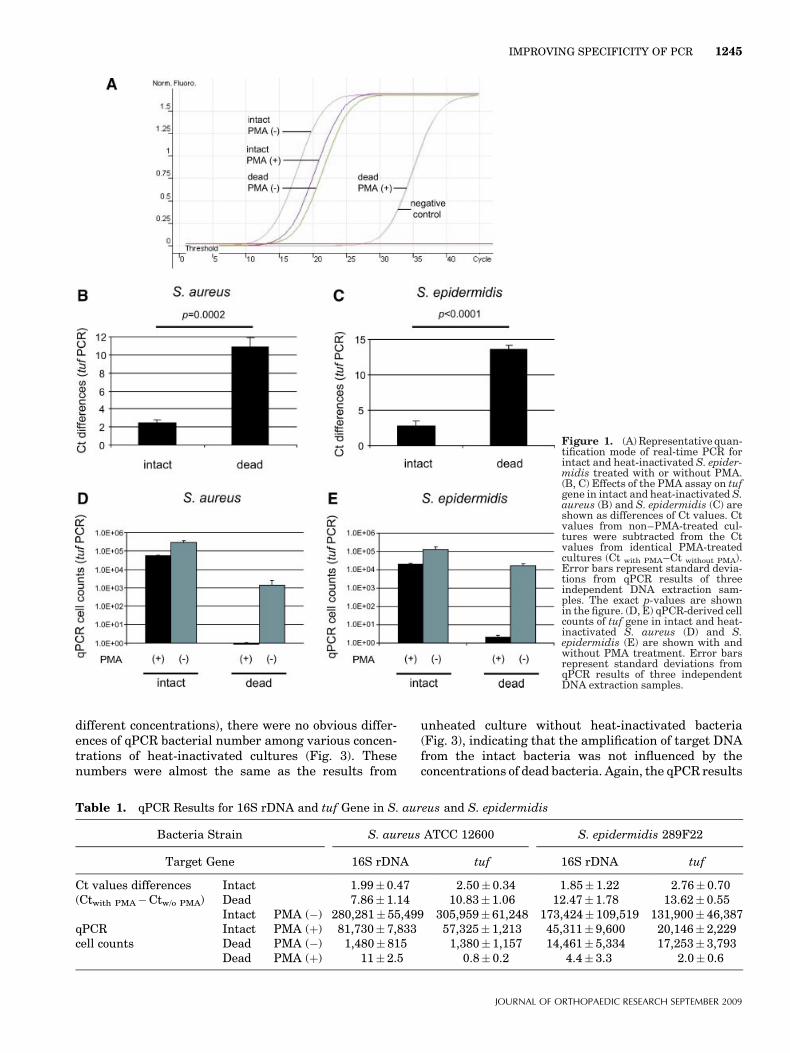
different concentrations), there were no obvious differ-ences of qPCR bacterial number among various concen-trations of heat-inactivated cultures (Fig. 3). Thesenumbers were almost the same as the results from
unheated culture without heat-inactivated bacteria(Fig. 3), indicating that the amplification of target DNAfrom the intact bacteria was not influenced by theconcentrations of dead bacteria. Again, the qPCR results
Figure 1. (A) Representative quan-tification mode of real-time PCR forintact and heat-inactivated S. epider-midis treated with or without PMA.(B, C) Effects of the PMA assay on tufgene in intact and heat-inactivated S.aureus (B) and S. epidermidis (C) areshown as differences of Ct values. Ctvalues from non–PMA-treated cul-tures were subtracted from the Ctvalues from identical PMA-treatedcultures (Ct with PMA–Ct without PMA).Error bars represent standard devia-tions from qPCR results of threeindependent DNA extraction sam-ples. The exact p-values are shownin the figure. (D, E) qPCR-derived cellcounts of tuf gene in intact and heat-inactivated S. aureus (D) and S.epidermidis (E) are shown with andwithout PMA treatment. Error barsrepresent standard deviations fromqPCR results of three independentDNA extraction samples.
Table 1. qPCR Results for 16S rDNA and tuf Gene in S. aureus and S. epidermidis
Bacteria Strain S. aureus ATCC 12600 S. epidermidis 289F22
Target Gene 16S rDNA tuf 16S rDNA tuf
Ct values differences Intact 1.99� 0.47 2.50� 0.34 1.85� 1.22 2.76� 0.70(Ctwith PMA�Ctw/o PMA) Dead 7.86� 1.14 10.83� 1.06 12.47� 1.78 13.62� 0.55
Intact PMA (�) 280,281� 55,499 305,959� 61,248 173,424� 109,519 131,900� 46,387qPCR Intact PMA (þ) 81,730� 7,833 57,325� 1,213 45,311� 9,600 20,146� 2,229cell counts Dead PMA (�) 1,480� 815 1,380� 1,157 14,461� 5,334 17,253� 3,793
Dead PMA (þ) 11� 2.5 0.8� 0.2 4.4� 3.3 2.0� 0.6
IMPROVING SPECIFICITY OF PCR 1245
JOURNAL OF ORTHOPAEDIC RESEARCH SEPTEMBER 2009
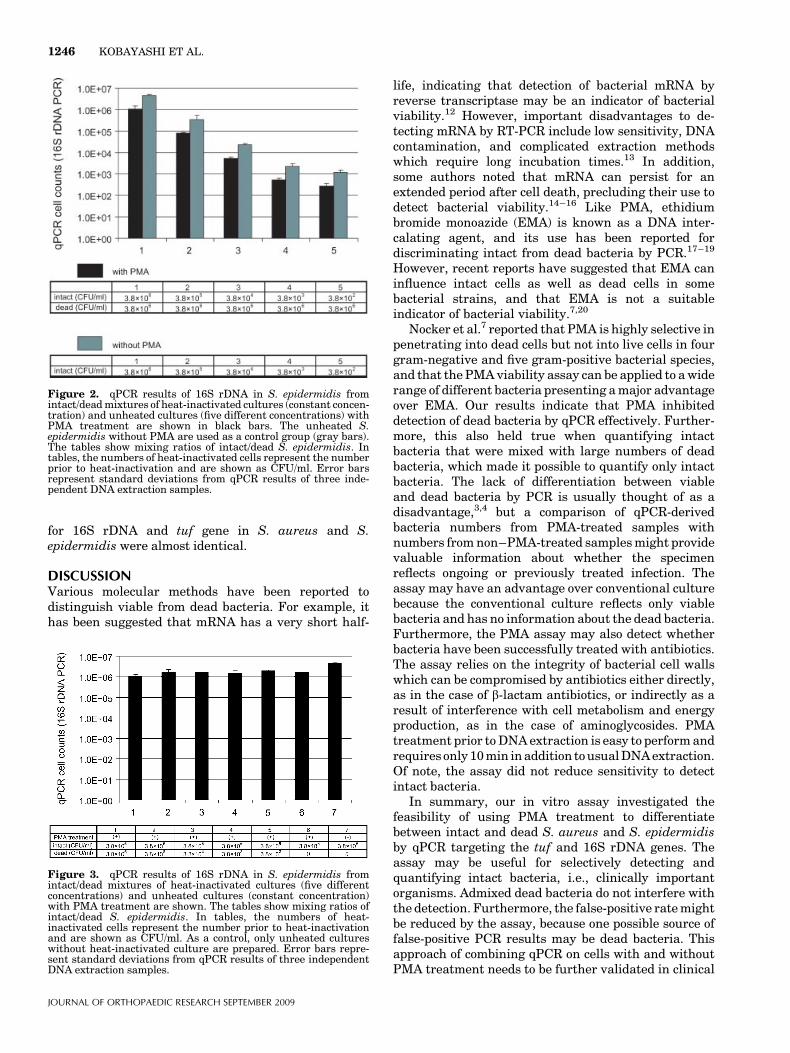
for 16S rDNA and tuf gene in S. aureus and S.epidermidis were almost identical.
DISCUSSIONVarious molecular methods have been reported todistinguish viable from dead bacteria. For example, ithas been suggested that mRNA has a very short half-
life, indicating that detection of bacterial mRNA byreverse transcriptase may be an indicator of bacterialviability.12 However, important disadvantages to de-tecting mRNA by RT-PCR include low sensitivity, DNAcontamination, and complicated extraction methodswhich require long incubation times.13 In addition,some authors noted that mRNA can persist for anextended period after cell death, precluding their use todetect bacterial viability.14–16 Like PMA, ethidiumbromide monoazide (EMA) is known as a DNA inter-calating agent, and its use has been reported fordiscriminating intact from dead bacteria by PCR.17–19
However, recent reports have suggested that EMA caninfluence intact cells as well as dead cells in somebacterial strains, and that EMA is not a suitableindicator of bacterial viability.7,20
Nocker et al.7 reported that PMA is highly selective inpenetrating into dead cells but not into live cells in fourgram-negative and five gram-positive bacterial species,and that the PMA viability assay can be applied to a widerange of different bacteria presenting a major advantageover EMA. Our results indicate that PMA inhibiteddetection of dead bacteria by qPCR effectively. Further-more, this also held true when quantifying intactbacteria that were mixed with large numbers of deadbacteria, which made it possible to quantify only intactbacteria. The lack of differentiation between viableand dead bacteria by PCR is usually thought of as adisadvantage,3,4 but a comparison of qPCR-derivedbacteria numbers from PMA-treated samples withnumbers from non–PMA-treated samples might providevaluable information about whether the specimenreflects ongoing or previously treated infection. Theassay may have an advantage over conventional culturebecause the conventional culture reflects only viablebacteria and has no information about the dead bacteria.Furthermore, the PMA assay may also detect whetherbacteria have been successfully treated with antibiotics.The assay relies on the integrity of bacterial cell wallswhich can be compromised by antibiotics either directly,as in the case of b-lactam antibiotics, or indirectly as aresult of interference with cell metabolism and energyproduction, as in the case of aminoglycosides. PMAtreatment prior to DNA extraction is easy to perform andrequires only 10 min in addition to usual DNA extraction.Of note, the assay did not reduce sensitivity to detectintact bacteria.
In summary, our in vitro assay investigated thefeasibility of using PMA treatment to differentiatebetween intact and dead S. aureus and S. epidermidisby qPCR targeting the tuf and 16S rDNA genes. Theassay may be useful for selectively detecting andquantifying intact bacteria, i.e., clinically importantorganisms. Admixed dead bacteria do not interfere withthe detection. Furthermore, the false-positive rate mightbe reduced by the assay, because one possible source offalse-positive PCR results may be dead bacteria. Thisapproach of combining qPCR on cells with and withoutPMA treatment needs to be further validated in clinical
Figure 2. qPCR results of 16S rDNA in S. epidermidis fromintact/dead mixtures of heat-inactivated cultures (constant concen-tration) and unheated cultures (five different concentrations) withPMA treatment are shown in black bars. The unheated S.epidermidis without PMA are used as a control group (gray bars).The tables show mixing ratios of intact/dead S. epidermidis. Intables, the numbers of heat-inactivated cells represent the numberprior to heat-inactivation and are shown as CFU/ml. Error barsrepresent standard deviations from qPCR results of three inde-pendent DNA extraction samples.
Figure 3. qPCR results of 16S rDNA in S. epidermidis fromintact/dead mixtures of heat-inactivated cultures (five differentconcentrations) and unheated cultures (constant concentration)with PMA treatment are shown. The tables show mixing ratios ofintact/dead S. epidermidis. In tables, the numbers of heat-inactivated cells represent the number prior to heat-inactivationand are shown as CFU/ml. As a control, only unheated cultureswithout heat-inactivated culture are prepared. Error bars repre-sent standard deviations from qPCR results of three independentDNA extraction samples.
1246 KOBAYASHI ET AL.
JOURNAL OF ORTHOPAEDIC RESEARCH SEPTEMBER 2009

samples, but it is anticipated that the test may beespecially helpful to identify persistent infection atthe time of second stage re-implantation for a previouslytreated infected arthroplasty.
ACKNOWLEDGMENTSThis work was supported in part by test development fundsfrom The Cleveland Clinic and by an educational gift fromNippon Stryker KK.
REFERENCES1. Kobayashi N, Bauer TW, Tuohy MJ, et al. 2006. The
comparison of pyrosequencing molecular gram stain, culture,and conventional gram stain for diagnosing orthopaedicinfections. J Orthop Res 24:1641–1649.
2. Yang S, Ramachandran P, Hardick A, et al. 2008. Rapid PCR-based diagnosis of septic arthritis by early gram-typeclassification and pathogen identification. J Clin Microbiol46:1386–1390.
3. Josephson KL, Gerba CP, Pepper IL. 1993. Polymerase chainreaction detection of nonintact bacterial pathogens. ApplEnviron Microbiol 59:3513–3515.
4. Young G, Turner S, Davies JK, et al. 2007. Bacterial DNApersists for extended periods after cell death. J Endod 33:1417–1420.
5. Tunney MM, Patrick S, Curran MD, et al. 1999. Detection ofprosthetic hip infection at revision arthroplasty by immuno-fluorescence microscopy and PCR amplification of the bacte-rial 16S rRNA gene. J Clin Microbiol 37:3281–3290.
6. Mariani BD, Martin DS, Levine MJ, et al. 1996. The CoventryAward. Polymerase chain reaction detection of bacterialinfection in total knee arthroplasty. Clin Orthop Relat Res331:11–22.
7. Nocker A, Cheung CY, Camper AK. 2006. Comparison ofpropidium monoazide with ethidium monoazide for differ-entiation of live vs. dead bacteria by selective removal of DNAfrom dead cells. J Microbiol Methods 67:310–320.
8. Nocker A, Sossa KE, Camper AK. 2007. Molecular monitoringof disinfection efficacy using propidium monoazide in combi-nation with quantitative PCR. J Microbiol Methods 70:252–260.
9. Nocker A, Sossa-Fernandez P, Burr MD, et al. 2007. Use ofpropidium monoazide for live/dead distinction in microbialecology. Appl Environ Microbiol 73:5111–5117.
10. Vesper S, McKinstry C, Hartmann C, et al. 2008. Quantifyingfungal viability in air and water samples using quantitativePCR after treatment with propidium monoazide (PMA). JMicrobiol Methods 72:180–184.
11. Peersman G, Laskin R, Davis J, et al. 2001. Infection in totalknee replacement: a retrospective review of 6489 total kneereplacements. Clin Orthop Relat Res 392:15–23.
12. Birmingham P, Helm JM, Manner PA, et al. 2008. Simulatedjoint infection assessment by rapid detection of live bacteriawith real-time reverse transcription polymerase chain reac-tion. J Bone Joint Surg [Am] 90:602–608.
13. McKillip JL, Jaykus LA, Drake M. 1999. Nucleic acidpersistence in heat-killed Escherichia coli. O157:H7 fromcontaminated skim milk. J Food Prot 62:839–844.
14. Sheridan GE, Masters CI, Shallcross JA, et al. 1998. Detectionof mRNA by reverse transcription-PCR as an indicator ofviability in Escherichia coli cells. Appl Environ Microbiol64:1313–1318.
15. Birch L, Dawson CE, Cornett JH, et al. 2001. A comparison ofnucleic acid amplification techniques for the assessment ofbacterial viability. Lett Appl Microbiol 33:296–301.
16. Sung K, Hiett KL, Stern NJ. 2005. Heat-treated Campylo-bacter spp. and mRNA stability as determined by reversetranscriptase-polymerase chain reaction. Foodborne PathogDis 2:130–137.
17. Soejima T, Iida K, Qin T, et al. 2007. Photoactivated ethidiummonoazide directly cleaves bacterial DNA and is applied toPCR for discrimination of live and dead bacteria. MicrobiolImmunol 51:763–775.
18. Wang S, Levin RE. 2006. Discrimination of intact Vibriovulnificus cells from dead cells in real-time PCR. J MicrobiolMethods 64:1–8.
19. Lee JL, Levin RE. 2007. Quantification of total intact bacteriaon fish fillets by using ethidium bromide monoazide real-timepolymerase chain reaction. Int J Food Microbiol 118:312–317.
20. Flekna G, Stefanic P, Wagner M, et al. 2007. Insufficient dif-ferentiation of live and dead Campylobacter jejuni and Listeriamonocytogenes cells by ethidium monoazide (EMA) com-promises EMA/real-time PCR. Res Microbiol 158:405–412.
IMPROVING SPECIFICITY OF PCR 1247
JOURNAL OF ORTHOPAEDIC RESEARCH SEPTEMBER 2009










![FirstCaseofPleuralEmpyemaCausedby Staphylococcus simulans ... · Staphylococcus saprophyticus, and Staphylococcus lugdu-nensis[1]. S.simulanscommonly affects cows, sheep, goats,](https://img.pdfslide.net/doc/110x75/60a9850bbd5f8210840e7181/firstcaseofpleuralempyemacausedby-staphylococcus-simulans-staphylococcus-saprophyticus.jpg)


















