Embed Size (px)
Citation preview

of August 18, 2018.This information is current as
Characterization of Long-Lived B CellsIn Vivo Survival of Autoreactive B Cells:
Tatyana Orekhova and Fred D. FinkelmanSuzanne C. Morris, Marta Moroldo, Edward H. Giannini,
http://www.jimmunol.org/content/164/6/3035doi: 10.4049/jimmunol.164.6.3035
2000; 164:3035-3046; ;J Immunol
Referenceshttp://www.jimmunol.org/content/164/6/3035.full#ref-list-1
, 29 of which you can access for free at: cites 54 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2000 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

In Vivo Survival of Autoreactive B Cells: Characterization ofLong-Lived B Cells1
Suzanne C. Morris,2*† Marta Moroldo, ‡ Edward H. Giannini, ‡ Tatyana Orekhova,*‡ andFred D. Finkelman*†
To determine the effects of chronic Ag stimulation on B cell survival and phenotype, we compared survival and surface markersof hen egg lysozyme (HEL)-specific B cells in Ig transgenic (Tgn) mice, which lack HEL, and in HEL-Ig transgenic mice, whichexpress soluble HEL. Serum HEL levels were maximized in HEL-Ig Tgn mice by feeding them zinc, which activates the metal-lothionein promoter that regulates HEL expression. B cell age was characterized by expression of heat-stable Ag, and B220 andB cell survival was studied by evaluating changes in B cell number when lymphopoiesis was suppressed with anti-IL-7 mAb andby identifying newly generated B cells through 5-bromo-2*-deoxyuridine incorporation. Our observations show that the mean Bcell life span is considerably reduced in HEL-Ig Tgn compared with Ig Tgn mice, but also demonstrate that some HEL-Ig Tgn Bcells survive to maturity. Some of these surviving B cells have undergone receptor editing (substitution of an endogenous Ig lightchain for the transgenic Ig light chain), so that their ability to bind HEL is decreased or absent. Surviving HEL-Ig Tgn B cells thatretain HEL specificity express decreased mIgD and little or no mIgM. mIgD expression progressively decreases with increasingHEL-Ig Tgn B cell age. These observations suggest that self Ag-specific B cells can survive in the presence of soluble self Ag bydown-regulating mIg expression, which should limit B cell signaling by Ag that might otherwise cause deletion of these cells.TheJournal of Immunology,2000, 164: 3035–3046.
T olerization of autoreactive B cells that results from aninteraction between autoantigen and B cell mIg in theabsence of T cell help is an important constraint on the
development of Ab-mediated autoimmune disease (1–3). Twoforms of Ag-induced B cell tolerance have been described: 1) ex-posure of newly developed bone marrow B cells to multivalent,cell membrane-bound Ag, in the absence of T cell help leads to Bcell apoptosis before the cells can migrate to secondary lymphoidorgans (3–7); and 2) exposure to soluble Ag, in the absence of Tcell help, leads to a more subtle type of B cell tolerance that istermed anergy (1, 8). Anergic B cells have been characterized mostthoroughly in a transgenic (Tgn)3 mouse system in which mice,whose B cells nearly all express mIgM and mIgD that binds henegg lysozyme (HEL) with high affinity (Ig Tgn mice), were bred tomice that secrete soluble HEL (HEL Tgn mice) (8, 9). HEL-spe-cific B cells and T cells in their offspring, which express bothtransgenes (HEL-Ig Tgn mice), are exposed throughout their lifespan to soluble HEL and exhibit tolerance to this Ag (9, 10).
Splenic B cells in HEL-Ig Tgn mice have been reported to havevery low levels of mIgM but near normal levels of mIgD (8, 9, 11)and to proliferate well in response to stimulation with CD40 ligandor bacterial LPS but poorly in response to mIg cross-linking (12–14). Because signals that result from mIg cross-linking allow Bcells to survive the Fas ligand stimulation that occurs during cog-nate interactions with activated T cells (15), failure of anergic Bcells to respond fully to mIg cross-linking can cause them to dierather than clonally expand during cognate B-T interactions(16–18).
Most recent studies have also concluded that B cell anergy in theHEL-Ig system is accompanied by a dramatic decrease in life spanthat results in a considerable decrease in splenic B cell number(19–21), although the serum HEL concentration (20, 21), compe-tition with B cells that are not autoreactive (22–24), and the pres-ence of CD41 T cells (24) have all been reported to influence thesurvival of the autoreactive B cells. Few of these studies, however,have examined the possibility that some autoreactive B cells inHEL-Ig Tgn mice survive for a considerable period of time, andwith the exception of a report that some B cells in HEL-Ig Tgnmice lack autoreactivity because they express an endogenous,rather than the transgenic, Ig heavy chain gene (8), none has at-tempted to characterize long-lived HEL-Ig Tgn B cells. Becauseprevious studies have not ruled out the possibility that some au-toreactive B cells survive for a long time in HEL-Ig Tgn mice, andlong term survival of autoreactive B cells would have importantimplications for the development of autoimmune disorders, wehave restudied the issue of B cell survival in HEL-Ig Tgn mice andhave looked for evidence of long-lived autoreactive B cells in asystem in which the serum HEL concentration is maintained at ahigh level.
The results of these experiments, which used three differenttechniques to evaluate B cell survival, reveal that while most Bcells in HEL-Ig Tgn mice have a decreased life span, some survive
*Division of Immunology, University of Cincinnati College of Medicine, Cincinnati,OH 45267; †Cincinnati Veterans Administration Medical Center, Cincinnati, OH45220; and‡William S. Rowe Division of Rheumatology, Department of Pediatrics,Children’s Hospital Medical Center, University of Cincinnati College of Medicine,Cincinnati, OH 45267
Received for publication April 9, 1999. Accepted for publication January 11, 2000.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby markedadvertisementin accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by a Biomedical Science Award from the National Ar-thritis Foundation, National Institutes of Health Grant P60-AR-44059-02, the Cin-cinnati Veterans Administration Medical Center, and the Children’s Hospital Re-search Foundation.2 Address correspondence and reprint requests to Dr. Suzanne C. Morris, Departmentof Veterans Affairs, Research Service 151, 3200 Vine Street, Cincinnati, OH 45220.E-mail address: [email protected] Abbreviations used in this paper: Tgn, transgenic; BrdU, 5-bromo-29-deoxyuridine;HEL, hen egg lysozyme; HSA, heat-stable Ag; m, cell membrane; S-PE, streptavidin-R-PE; S-PharRed, streptavidin-PharRed.
Copyright © 2000 by The American Association of Immunologists 0022-1767/00/$02.00
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

for a relatively long time. The survival of some of these long-livedB cells appears to be associated with receptor editing (25–27),which decreases or eliminates their affinity for HEL. A larger pop-ulation of long-lived HEL-Ig Tgn splenic B cells, however, ap-pears to retain mIg that has the ability to bind HEL, but has down-regulated its expression of both mIgM and mIgD. Thus, mIgexpression down-regulation may be a mechanism that allows au-toreactive B cell survival and creates a reservoir of long-lived au-toreactive B cells that, if activated, might induce humoral autoim-mune disease.
Materials and MethodsAnimals
C57BL/6 female mice, obtained from the Small Animals Division of theNational Cancer Institute, National Institutes of Health (Bethesda, MD),and CB20 mice, obtained from Dr. Michael Potter (National Cancer Insti-tute, National Institutes of Health) were bred in the Cincinnati VeteransAdministration Medical Center animal facility (Cincinnati, OH). C57BL/6female mice were bred to C57BL/6 male mice that were hemizygous forboth the MD4 anti-HEL Ig H and L transgene and the ML5 soluble HELtransgene (a gift from Chris Goodnow, Australian National University,Canberra, Australia) to generate mice that carry only the MD4 transgene(Ig Tgn mice) and mice that carry both the MD4 and ML5 transgenes(HEL-Ig Tgn mice). Mice were used at 8–43 wk of age. Groups of micesubjected to different treatments were age and sex matched in individualexperiments.
Typing of transgenic mice
Mice that expressed the HEL transgene and/or the anti-HEL transgenewere identified by PCR (28). DNA was isolated with QIAamp tissue kitsfor DNA isolation (Qiagen, Santa Clarita, CA). PCR reactions were per-formed as previously described (5). Goodnow (unpublished observation).Briefly, the following five oligonucleotides were used in PCR reactions:IgHF1, 59-GCGACTCCATCACCAGCGAT-39; IgHF2, 59-CTGGAGCCCTAGCCAAGGAT-39; IgHR1, 59-ACCACAGACCAGCAGGCAGA-39;HEL3F, 59-GAGCGTGAACTGCGCGAAGA-39; and HEL4R, 59-TCGGTACCCTTGCAGCGGTT-39. HEL-Ig Tgn mice have three bands, corre-sponding to the 264-bp endogenous Ig band, the 430-bp Ig transgene band,and the 160-bp lysozyme transgenic band. Ig Tgn mice have two bands, the264-bp band and the 430-bp band. Oligonucleotide primers were producedby the BIC synthesis center at the Uniformed Services University of theHealth Science (Bethesda, MD).
Experimental conditions
All mice were maintained on drinking water that contained 25 mM ZnCl2
for at least 3 days before the initiation of other treatments and for theduration of each experiment to maximize serum HEL levels in HEL-Ig Tgnmice (21). In experiments in which newly generated B cells were identifiedby 5-bromo-29-deoxyuridine (BrdU) incorporation (7, 19), 0.8 mg/ml ofBrdU (Sigma, St. Louis, MO) was also added to drinking water for adefined period of time. BrdU-containing water was shielded from light andchanged every third day.
Abs and immunological reagents
The following hybridomas and plasmacytomas were obtained and grown asascites in Pristane-primed athymic nude, BALB/c, or CB20 mice, andmAbs were purified from ascites by (NH4)2SO4 precipitation and DE-52(Whatman, Clifton, NJ) cation exchange column chromatography, unlessotherwise stated: RA3-6B2 (rat IgG2a anti-mouse CD45R/B220) (29),DS-1 (mouse IgG1 of theb allotype specific for mouse IgM of theaallotype) (30), Hda/1 (mouse IgG2b of theb allotype specific for mouseIgD of thea allotype) (31), AF3.33 (mouse IgG2a of thea allotype specificfor mouse IgD of theb allotype) (32), 5E4 and 2D1 (mouse IgG1 anti-HELmAbs; gift from Dennis Metzger, Toledo, OH) (33), 24G2 (rat IgG2banti-mouse FcgRII/III) (34), GK1.5 (rat IgG2b anti-mouse CD4) (35), M25(mouse IgG2b anti-human IL-7 that cross-reacts with mouse IL-7) (36),and MOPC-352 (a gift from Dr. Michael Potter, National Cancer Institute,National Institutes of Health), a mouse IgG2b that does not bind to mouseproteins and that was used as a control for M25. Some of these Abs werelabeled with FITC (37) (Calbiochem-Behring, La Jolla, CA), biotin-N-hydroxysuccinimide (38) (Calbiochem-Behring), or Cy5 reactive dye (Re-search Organics, Cleveland, OH), as suggested by the manufacturer. Bi-otin- or FITC-labeled M1/69 (anti-HSA) (39), PE-labeled 1D3 (anti-CD19)
(40), and PerCP-labeled RA3–6B2 were purchased from PharMingen (SanDiego, CA). FITC-anti-BrdU was purchased from Becton Dickinson (SanJose, CA). HEL (lysozyme from chicken egg white) was purchased fromSigma (St. Louis, MO).
Immunofluorescence staining
Spleen or bone marrow cells were depleted of erythrocytes, filtered throughnylon gauze, and suspended at 203 106 cells/ml in HBSS with 10%newborn bovine serum and 0.2% NaN3 (HNA). One hundred microliters ofcell suspension was stained for 30 min on ice with 1mg each of appro-priately labeled Abs. Cells were washed twice with HNA, then, if appro-priate, were exposed to either streptavidin-R-PE (S-PE; purchased fromLife Technologies (Gaithersburg, MD) or Becton Dickinson Immunocy-tometry Systems) or to streptavidin-PharRed (S-PharRed; purchased fromPharMingen) for 30 min on ice. All staining was performed in the presenceof 1 mg of unlabeled anti-FcgRII/III (24G2). After washing once withHNA, all samples, except those that required staining for BrdU incorpo-ration, were washed once with HBSS/0.2% sodium azide, then fixed inPBS/2% paraformaldehyde. Staining for BrdU was modified from the pro-cedure described by Allman (39). Samples that required staining for BrdUwere washed in PBS and resuspended in 0.5 ml of ice-cold 0.15 M NaCl,after which 1.2 ml of ice-cold 95% ethanol was slowly added while gentlyvortex mixing the cells. Cells were incubated on ice for 30 min, thenwashed with PBS. One milliliter of PBS/1% paraformaldehyde/0.01%Tween 20 was then added, and cells were incubated for 30 min at roomtemperature, followed by overnight storage at 4°C. The following day cellswere pelleted by centrifugation (1500 rpm for 15 min), then incubated for10 min at room temperature with 1 ml of 0.15 M saline that contained 4.2mM MgCl2 and 50 Kunitz units/ml of DNase I (Sigma). Samples were thenwashed with PBS, stained with FITC-anti-BrdU (30 min, room tempera-ture), washed with PBS, and analyzed by flow cytometry. All samples wereanalyzed with either a FACScan, FACSCalibur Analyzer equipped with ared diode laser, or with a FACS Vantage equipped with a red diode laser(Becton Dickinson, Mountain View, CA). Data analysis was performedwith LYSIS II or CellQuest software (Becton Dickinson). Light scattergates were set to exclude most nonlymphoid cells and cells that had diedbefore fixation. Cells that had been stained with a single fluorochrome-labeled Ab were used to determine compensation for overlap betweenemission spectra. The percentages of specifically stained cells and the meanfluorescence intensities of specifically stained cells were determined.
Cell counts
Nucleated cells were counted with a Coulter counter (Coulter, Miami, FL)that was set to exclude dead cells. Absolute numbers of cells that had adefined phenotype were determined by multiplying the percentage of cellsthat expressed that phenotype by the total spleen cell number.
ResultsB cell mIg is nearly saturated with HEL in ZnCl2-treated HEL-Ig Tgn mice
Preliminary studies demonstrated considerable variability in serumHEL levels and B cell numbers in HEL-Ig Tgn mice (data notshown). Because B cell survival in HEL-Ig Tgn mice may dependon the serum HEL concentration (20, 21), and HEL gene expres-sion in these mice is regulated by the metallothionein promoter,ZnCl2 was added to the drinking water of all of our mice at least3 days before initiation of each experiment and was continued forthe duration of each experiment to maintain the serum HEL con-centration at a consistently high level. In contrast to results ob-tained in another study in which HEL-Ig Tgn mice were not treatedwith zinc (18), surface mIg on splenic B cells from ZnCl2-treatedHEL-Ig Tgn mice was nearly saturated with HEL (Fig. 1). B cellsfrom HEL-Ig Tgn mice were found to bind considerably less HELthan B cells from Ig Tgn mice; thus, mIg expression is consider-ably down-regulated on HEL-specific B cells in HEL-Ig Tgn mice.
Splenic B cells that have a mature phenotype are selectivelydepleted in HEL-Ig Tgn mice
Most, but not all, previous studies of HEL-Ig Tgn mice have sug-gested that the B cell life span is considerably decreased in these
3036 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
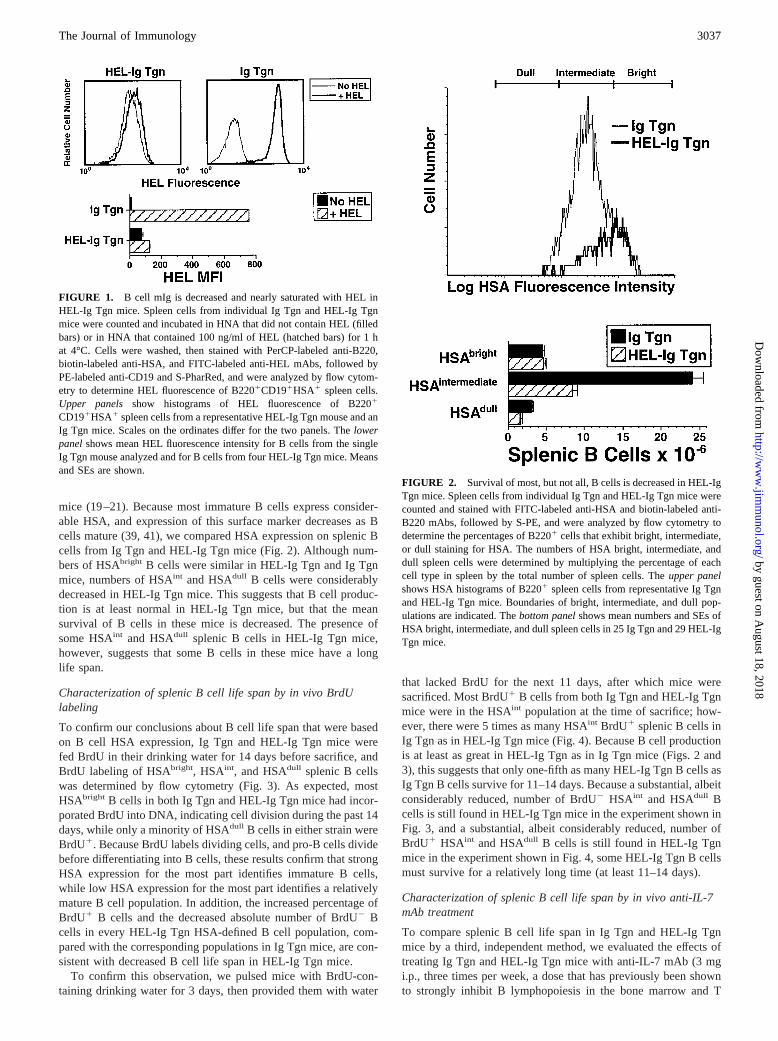
mice (19–21). Because most immature B cells express consider-able HSA, and expression of this surface marker decreases as Bcells mature (39, 41), we compared HSA expression on splenic Bcells from Ig Tgn and HEL-Ig Tgn mice (Fig. 2). Although num-bers of HSAbright B cells were similar in HEL-Ig Tgn and Ig Tgnmice, numbers of HSAint and HSAdull B cells were considerablydecreased in HEL-Ig Tgn mice. This suggests that B cell produc-tion is at least normal in HEL-Ig Tgn mice, but that the meansurvival of B cells in these mice is decreased. The presence ofsome HSAint and HSAdull splenic B cells in HEL-Ig Tgn mice,however, suggests that some B cells in these mice have a longlife span.
Characterization of splenic B cell life span by in vivo BrdUlabeling
To confirm our conclusions about B cell life span that were basedon B cell HSA expression, Ig Tgn and HEL-Ig Tgn mice werefed BrdU in their drinking water for 14 days before sacrifice, andBrdU labeling of HSAbright, HSAint, and HSAdull splenic B cellswas determined by flow cytometry (Fig. 3). As expected, mostHSAbright B cells in both Ig Tgn and HEL-Ig Tgn mice had incor-porated BrdU into DNA, indicating cell division during the past 14days, while only a minority of HSAdull B cells in either strain wereBrdU1. Because BrdU labels dividing cells, and pro-B cells dividebefore differentiating into B cells, these results confirm that strongHSA expression for the most part identifies immature B cells,while low HSA expression for the most part identifies a relativelymature B cell population. In addition, the increased percentage ofBrdU1 B cells and the decreased absolute number of BrdU2 Bcells in every HEL-Ig Tgn HSA-defined B cell population, com-pared with the corresponding populations in Ig Tgn mice, are con-sistent with decreased B cell life span in HEL-Ig Tgn mice.
To confirm this observation, we pulsed mice with BrdU-con-taining drinking water for 3 days, then provided them with water
that lacked BrdU for the next 11 days, after which mice weresacrificed. Most BrdU1 B cells from both Ig Tgn and HEL-Ig Tgnmice were in the HSAint population at the time of sacrifice; how-ever, there were 5 times as many HSAint BrdU1 splenic B cells inIg Tgn as in HEL-Ig Tgn mice (Fig. 4). Because B cell productionis at least as great in HEL-Ig Tgn as in Ig Tgn mice (Figs. 2 and3), this suggests that only one-fifth as many HEL-Ig Tgn B cells asIg Tgn B cells survive for 11–14 days. Because a substantial, albeitconsiderably reduced, number of BrdU2 HSAint and HSAdull Bcells is still found in HEL-Ig Tgn mice in the experiment shown inFig. 3, and a substantial, albeit considerably reduced, number ofBrdU1 HSAint and HSAdull B cells is still found in HEL-Ig Tgnmice in the experiment shown in Fig. 4, some HEL-Ig Tgn B cellsmust survive for a relatively long time (at least 11–14 days).
Characterization of splenic B cell life span by in vivo anti-IL-7mAb treatment
To compare splenic B cell life span in Ig Tgn and HEL-Ig Tgnmice by a third, independent method, we evaluated the effects oftreating Ig Tgn and HEL-Ig Tgn mice with anti-IL-7 mAb (3 mgi.p., three times per week, a dose that has previously been shownto strongly inhibit B lymphopoiesis in the bone marrow and T
FIGURE 1. B cell mIg is decreased and nearly saturated with HEL inHEL-Ig Tgn mice. Spleen cells from individual Ig Tgn and HEL-Ig Tgnmice were counted and incubated in HNA that did not contain HEL (filledbars) or in HNA that contained 100 ng/ml of HEL (hatched bars) for 1 hat 4°C. Cells were washed, then stained with PerCP-labeled anti-B220,biotin-labeled anti-HSA, and FITC-labeled anti-HEL mAbs, followed byPE-labeled anti-CD19 and S-PharRed, and were analyzed by flow cytom-etry to determine HEL fluorescence of B2201CD191HSA1 spleen cells.Upper panels show histograms of HEL fluorescence of B2201
CD191HSA1 spleen cells from a representative HEL-Ig Tgn mouse and anIg Tgn mice. Scales on the ordinates differ for the two panels. Thelowerpanelshows mean HEL fluorescence intensity for B cells from the singleIg Tgn mouse analyzed and for B cells from four HEL-Ig Tgn mice. Meansand SEs are shown.
FIGURE 2. Survival of most, but not all, B cells is decreased in HEL-IgTgn mice. Spleen cells from individual Ig Tgn and HEL-Ig Tgn mice werecounted and stained with FITC-labeled anti-HSA and biotin-labeled anti-B220 mAbs, followed by S-PE, and were analyzed by flow cytometry todetermine the percentages of B2201 cells that exhibit bright, intermediate,or dull staining for HSA. The numbers of HSA bright, intermediate, anddull spleen cells were determined by multiplying the percentage of eachcell type in spleen by the total number of spleen cells. Theupper panelshows HSA histograms of B2201 spleen cells from representative Ig Tgnand HEL-Ig Tgn mice. Boundaries of bright, intermediate, and dull pop-ulations are indicated. Thebottom panelshows mean numbers and SEs ofHSA bright, intermediate, and dull spleen cells in 25 Ig Tgn and 29 HEL-IgTgn mice.
3037The Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

lymphopoiesis in the thymus (36, 42, 43)) on numbers ofHSAbright, HSAint, and HSAdull splenic B cells (Fig. 5). To distin-guish B cells generated before initiation of anti-IL-7 mAb treat-ment from B cells generated after anti-IL-7 mAb treatment hadbegun, mice were fed drinking water that contained BrdU, startingon the first day of anti-IL-7 mAb treatment. Anti-IL-7 mAb treat-ment decreased the number of recently generated (BrdU1)HSAbright splenic B cells in both strains, but had a considerablygreater effect in HEL-Ig Tgn than in Ig Tgn mice (89.1 vs 56.1%,respectively). Anti-IL-7 mAb treatment also decreased HSAint
BrdU1 splenic B cell number in HEL-Ig Tgn, but not in Ig Tgnmice. It is likely that these effects resulted from the suppression ofB lymphopoiesis, rather than from reduced B cell survival, becauseanti-IL-7 had no effect on the number of splenic B cells generatedbefore the initiation of treatment with this mAb (BrdU2 B cells).Anti-IL-7 mAb treatment also had little effect on the small numberof BrdU1 HSAdull B cells in Ig Tgn or HEL-Ig Tgn mice, sug-gesting that most BrdU1 HSAdull B cells were mature, proliferat-ing cells rather than newly generated cells. Treatment of Ig Tgn orHEL-Ig Tgn mice with an isotype-matched control mAb, insteadof anti-IL-7, did not decrease the number of HSAbright or HSAint
spleen cells (data not shown). Taken together, our experimentaldata provide evidence by three independent techniques that theaverage B cell life span is shorter in HEL-Ig Tgn mice than in IgTgn mice when HEL is maintained at a high level.
Surface Ig expression and HEL binding by B cells from Ig Tgnand HEL-Ig Tgn mice
Although our studies indicate that average splenic B cell life spanis decreased in HEL-Ig Tgn mice, they also demonstrate the ex-istence of long-lived B cells in these mice. This long-lived popu-lation might represent B cells that were insensitive to the relativelyhigh concentrations of HEL in zinc-treated mice because they ex-pressed endogenous, rather than transgenic, Ig heavy chains (8) orbecause they expressed an endogenous Ig light chain as a result ofreceptor editing (25–27). Alternatively, B cells that retained theirHEL specificity might have survived by modifying their phenotypein a way that decreased sensitivity to Ag signaling, or survival mayhave been selective for a subset of B cells that is relatively insensitiveto deletion. Finally, some HEL-specific B cells may have survived bychance regardless of and without altering their properties.
To investigate these possibilities, experiments were performedto characterize mature splenic B cells in zinc-treated HEL-Ig Tgnmice. The possibility that the long-lived B cells in these mice wereunresponsive to HEL because they expressed endogenous, ratherthan transgenic, Ig heavy chains was eliminated because splenic Bcells that expressed IgM or IgD of the endogenousb allotype werenot detected in the great majority (.95%) of HEL-Ig Tgn mice inour colony (data not shown); the small number of mice that ex-hibited substantial numbers ofb allotype B cells was excludedfrom further study. To compare the extent of receptor editing insplenic B cells from Ig Tgn and HEL-Ig Tgn mice, spleen cells
FIGURE 3. HEL-Ig Tgn mice have a higher percentage of recently gen-erated splenic B cells than Ig Tgn mice. Three Ig Tgn and five HEL-Ig Tgnmice were administered BrdU-containing drinking water for 14 days beforesacrifice. Spleen cells from individual mice were counted; stained withbiotin-labeled anti-HSA mAb and Cy5-labeled anti-B220 mAb, followedby S-PE; then fixed, permeabilized, and stained with FITC-anti-BrdUmAb. Contour plots demonstrate gates drawn to identify HSA1B2201
BrdU1or2 cells that were then determined to be either HSA bright, inter-mediate, or dull by the indicated markers on fluorescence histograms. Thepercentages of BrdU2 and BrdU1 HSA bright, intermediate, and dullB2201 spleen cells were determined by flow cytometry and multiplied bytotal spleen cell numbers to determine the number of spleen cells of eachtype. Means and SEs are shown.
FIGURE 4. Characterization of B cell survival in Ig Tgn and HEL-IgTgn mice by pulsing mice with BrdU. Ig Tgn and HEL-Ig Tgn mice (fourper group) were administered BrdU-containing drinking water on days 0–2followed by regular drinking water on days 3–13 and were sacrificed onday 14. Spleen cells from all mice were individually counted and stainedwith biotin-anti-HSA mAb and Cy5-anti-B220 mAb, followed by S-PE,then fixed, permeabilized, and stained with FITC-anti-BrdU mAb. Cellswere analyzed by flow cytometry as described in Fig. 3 to determine thepercentages of BrdU1 and BrdU2B2201 cells that demonstrated bright,intermediate, or dull HSA staining; the number of cells of each phenotypewas determined by multiplying percentages by total spleen cell number.Means and SEs are shown. (Note the log scale on the abscissa.) Numbersof HSAdullBrdU1, HSAintBrdU1, and HSAbrightBrdU1 spleen cells were25, 20, and 65% as great, respectively, in HEL-Ig Tgn as in Ig Tgn mice.
3038 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
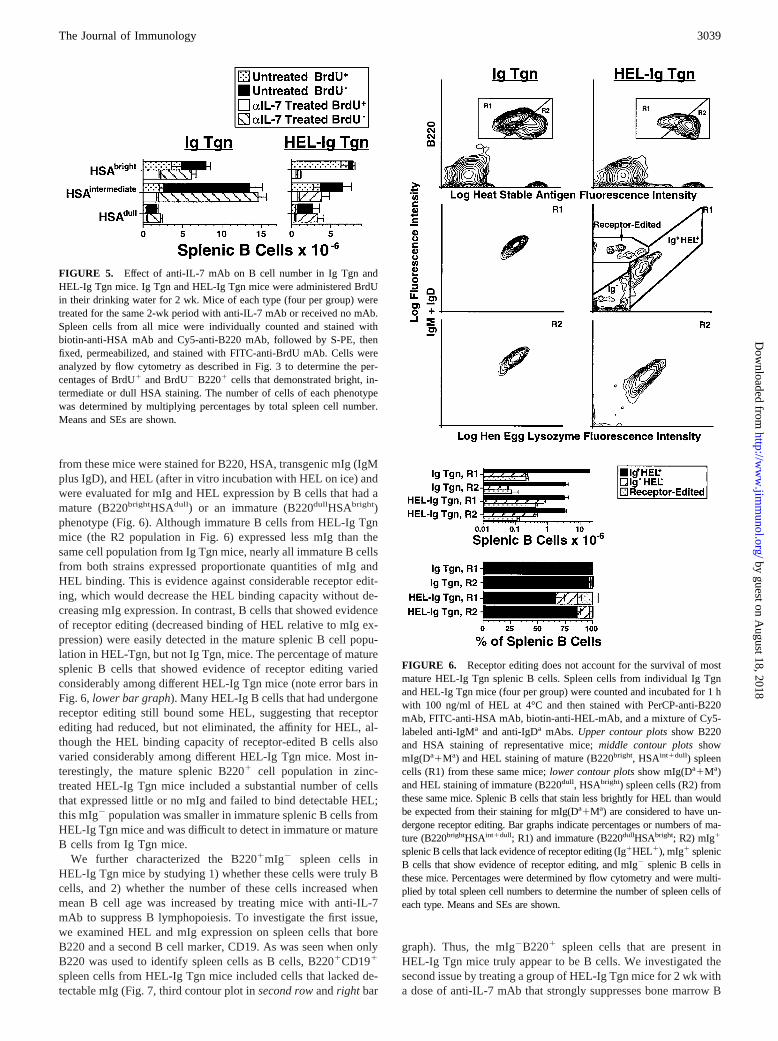
from these mice were stained for B220, HSA, transgenic mIg (IgMplus IgD), and HEL (after in vitro incubation with HEL on ice) andwere evaluated for mIg and HEL expression by B cells that had amature (B220brightHSAdull) or an immature (B220dullHSAbright)phenotype (Fig. 6). Although immature B cells from HEL-Ig Tgnmice (the R2 population in Fig. 6) expressed less mIg than thesame cell population from Ig Tgn mice, nearly all immature B cellsfrom both strains expressed proportionate quantities of mIg andHEL binding. This is evidence against considerable receptor edit-ing, which would decrease the HEL binding capacity without de-creasing mIg expression. In contrast, B cells that showed evidenceof receptor editing (decreased binding of HEL relative to mIg ex-pression) were easily detected in the mature splenic B cell popu-lation in HEL-Tgn, but not Ig Tgn, mice. The percentage of maturesplenic B cells that showed evidence of receptor editing variedconsiderably among different HEL-Ig Tgn mice (note error bars inFig. 6, lower bar graph). Many HEL-Ig B cells that had undergonereceptor editing still bound some HEL, suggesting that receptorediting had reduced, but not eliminated, the affinity for HEL, al-though the HEL binding capacity of receptor-edited B cells alsovaried considerably among different HEL-Ig Tgn mice. Most in-terestingly, the mature splenic B2201 cell population in zinc-treated HEL-Ig Tgn mice included a substantial number of cellsthat expressed little or no mIg and failed to bind detectable HEL;this mIg2 population was smaller in immature splenic B cells fromHEL-Ig Tgn mice and was difficult to detect in immature or matureB cells from Ig Tgn mice.
We further characterized the B2201mIg2 spleen cells inHEL-Ig Tgn mice by studying 1) whether these cells were truly Bcells, and 2) whether the number of these cells increased whenmean B cell age was increased by treating mice with anti-IL-7mAb to suppress B lymphopoiesis. To investigate the first issue,we examined HEL and mIg expression on spleen cells that boreB220 and a second B cell marker, CD19. As was seen when onlyB220 was used to identify spleen cells as B cells, B2201CD191
spleen cells from HEL-Ig Tgn mice included cells that lacked de-tectable mIg (Fig. 7, third contour plot insecond rowandright bar
graph). Thus, the mIg2B2201 spleen cells that are present inHEL-Ig Tgn mice truly appear to be B cells. We investigated thesecond issue by treating a group of HEL-Ig Tgn mice for 2 wk witha dose of anti-IL-7 mAb that strongly suppresses bone marrow B
FIGURE 5. Effect of anti-IL-7 mAb on B cell number in Ig Tgn andHEL-Ig Tgn mice. Ig Tgn and HEL-Ig Tgn mice were administered BrdUin their drinking water for 2 wk. Mice of each type (four per group) weretreated for the same 2-wk period with anti-IL-7 mAb or received no mAb.Spleen cells from all mice were individually counted and stained withbiotin-anti-HSA mAb and Cy5-anti-B220 mAb, followed by S-PE, thenfixed, permeabilized, and stained with FITC-anti-BrdU mAb. Cells wereanalyzed by flow cytometry as described in Fig. 3 to determine the per-centages of BrdU1 and BrdU2 B2201 cells that demonstrated bright, in-termediate or dull HSA staining. The number of cells of each phenotypewas determined by multiplying percentages by total spleen cell number.Means and SEs are shown.
FIGURE 6. Receptor editing does not account for the survival of mostmature HEL-Ig Tgn splenic B cells. Spleen cells from individual Ig Tgnand HEL-Ig Tgn mice (four per group) were counted and incubated for 1 hwith 100 ng/ml of HEL at 4°C and then stained with PerCP-anti-B220mAb, FITC-anti-HSA mAb, biotin-anti-HEL-mAb, and a mixture of Cy5-labeled anti-IgMa and anti-IgDa mAbs. Upper contour plotsshow B220and HSA staining of representative mice;middle contour plotsshowmIg(Da1Ma) and HEL staining of mature (B220bright, HSAint1dull) spleencells (R1) from these same mice;lower contour plotsshow mIg(Da1Ma)and HEL staining of immature (B220dull, HSAbright) spleen cells (R2) fromthese same mice. Splenic B cells that stain less brightly for HEL than wouldbe expected from their staining for mIg(Da1Ma) are considered to have un-dergone receptor editing. Bar graphs indicate percentages or numbers of ma-ture (B220brightHSAint1dull; R1) and immature (B220dullHSAbright; R2) mIg1
splenic B cells that lack evidence of receptor editing (Ig1HEL1), mIg1 splenicB cells that show evidence of receptor editing, and mIg2 splenic B cells inthese mice. Percentages were determined by flow cytometry and were multi-plied by total spleen cell numbers to determine the number of spleen cells ofeach type. Means and SEs are shown.
3039The Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

lymphopoiesis (8, 36, 42). Anti-IL-7 mAb treatment selectivelydecreased the number of immature splenic B cells (Fig. 7, secondvs third contour plot inthird row and bar graphon left), decreasedthe number of mIg1 B cells, and increased the number of mIg2 Bcells (Fig. 7, third vs fourth contour plot insecond rowand bargraph onright). These effects most likely resulted from inhibitionof B lymphopoiesis rather than from a nonspecific effect of injec-tion of a large quantity of mouse IgG or from inhibition of IL-7effects on mature T cells, which may have decreased T cell help.Injection of an isotype-matched control mAb at the same dose asanti-IL-7 had no significant effect on any of these populations (Fig.7, third vs fifth contour plots insecond row; second vs fourth
contour plots inthird row and bar graphs), and injection of ananti-CD4 mAb at a dose that effectively suppresses T cell help (35,42) selectively decreased the number of mature B cells and failedto increase the percentage of splenic B cells that were mIg2 (Fig.7, second vs fifth contour plots inthird row, third vs sixth contourplot in second row,and bar graphs). Thus, the effects of anti-IL-7mAb on HEL-Ig Tgn splenic B cells are specific and are not relatedto inhibition of T cell help.
Membrane IgM and IgD expression by HEL-Ig Tgn B cells
The presence of a B220bright HSAdull mIgdull-negativespleen cellpopulation in HEL-Ig Tgn mice raised the question of which Ig
FIGURE 7. Anti-IL-7 treatment is associated with loss of mIg by splenic HEL-Ig Tgn B cells. HEL-Ig Tgn mice (four per group) were left untreatedor were injected i.p with 3 mg of either anti-IL-7 mAb (to suppress new B cell production) or a control isotype-matched mAb three times per week for2 wk or were injected i.v. with 1 mg of anti-CD4 mAb once a week (to block T cell help). Mice were sacrificed on day 14, and their spleen cells werecounted and incubated at 4°C for 1 h with either 100 ng/ml of HEL or buffer only and then stained with PerCP-labeled anti-B220, Cy5-labeled anti-IgMa
and/or anti-IgDa, and FITC-labeled anti-HSA or FITC-labeled anti-HEL, followed by PE-labeled anti-CD19. Spleen cells from an Ig Tgn mouse and a CB20mouse (Igb allotype) were similarly treated. Cells were then analyzed by flow cytometry.Upper contour plotsshow spleen cells from a representativemouse, gated for scatter and for the CD191B2201 (B cell) population. Thesecond rowof contour plots shows HEL and mIg fluorescence for theCD191B2201 population. Gates define the receptor-edited (R-E), non-receptor-edited mIg1HEL1 (mIg1), and mIg2 splenic B cell populations. Thethirdrow of contour plots show B220 and HSA fluorescence for CD191B2201 spleen cells. Numbers of mature, immature, mIg1, mIg2, and receptor-editedspleen cells were determined by multiplying the appropriate percentages by the total spleen cell number. Bar graphs show means and SEs. By the criteriashown, a single Ig Tgn mouse that was simultaneously studied had 20.63 106 mIg1 cells, 0.123 106 mIg2 cells, and 0.113 106 receptor-edited cellsin its spleen.
3040 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
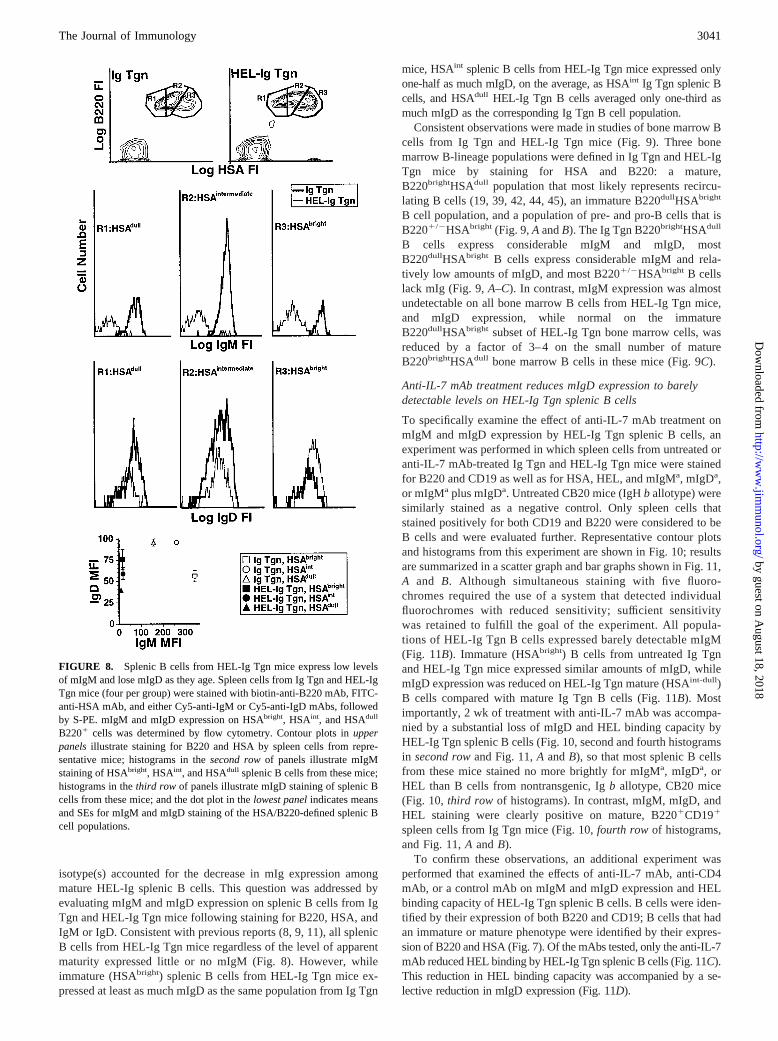
isotype(s) accounted for the decrease in mIg expression amongmature HEL-Ig splenic B cells. This question was addressed byevaluating mIgM and mIgD expression on splenic B cells from IgTgn and HEL-Ig Tgn mice following staining for B220, HSA, andIgM or IgD. Consistent with previous reports (8, 9, 11), all splenicB cells from HEL-Ig Tgn mice regardless of the level of apparentmaturity expressed little or no mIgM (Fig. 8). However, whileimmature (HSAbright) splenic B cells from HEL-Ig Tgn mice ex-pressed at least as much mIgD as the same population from Ig Tgn
mice, HSAint splenic B cells from HEL-Ig Tgn mice expressed onlyone-half as much mIgD, on the average, as HSAint Ig Tgn splenic Bcells, and HSAdull HEL-Ig Tgn B cells averaged only one-third asmuch mIgD as the corresponding Ig Tgn B cell population.
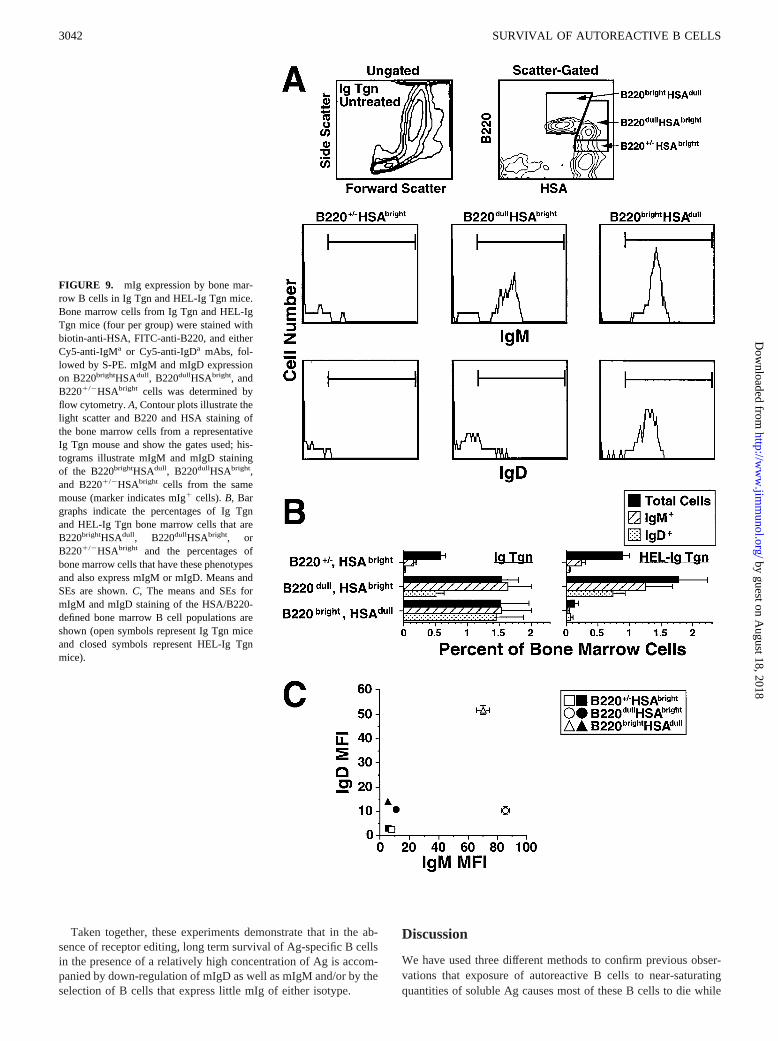
Consistent observations were made in studies of bone marrow Bcells from Ig Tgn and HEL-Ig Tgn mice (Fig. 9). Three bonemarrow B-lineage populations were defined in Ig Tgn and HEL-IgTgn mice by staining for HSA and B220: a mature,B220brightHSAdull population that most likely represents recircu-lating B cells (19, 39, 42, 44, 45), an immature B220dullHSAbright
B cell population, and a population of pre- and pro-B cells that isB2201/2HSAbright (Fig. 9,A andB). The Ig Tgn B220brightHSAdull
B cells express considerable mIgM and mIgD, mostB220dullHSAbright B cells express considerable mIgM and rela-tively low amounts of mIgD, and most B2201/2HSAbright B cellslack mIg (Fig. 9,A–C). In contrast, mIgM expression was almostundetectable on all bone marrow B cells from HEL-Ig Tgn mice,and mIgD expression, while normal on the immatureB220dullHSAbright subset of HEL-Ig Tgn bone marrow cells, wasreduced by a factor of 3–4 on the small number of matureB220brightHSAdull bone marrow B cells in these mice (Fig. 9C).
Anti-IL-7 mAb treatment reduces mIgD expression to barelydetectable levels on HEL-Ig Tgn splenic B cells
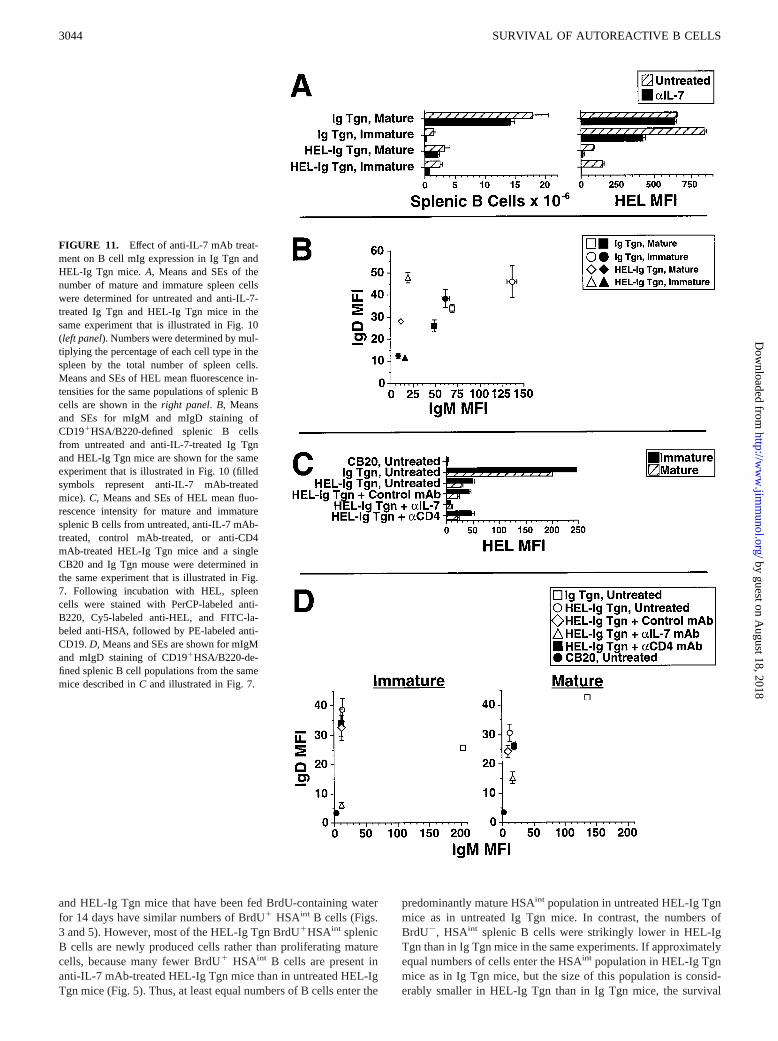
To specifically examine the effect of anti-IL-7 mAb treatment onmIgM and mIgD expression by HEL-Ig Tgn splenic B cells, anexperiment was performed in which spleen cells from untreated oranti-IL-7 mAb-treated Ig Tgn and HEL-Ig Tgn mice were stainedfor B220 and CD19 as well as for HSA, HEL, and mIgMa, mIgDa,or mIgMa plus mIgDa. Untreated CB20 mice (IgHb allotype) weresimilarly stained as a negative control. Only spleen cells thatstained positively for both CD19 and B220 were considered to beB cells and were evaluated further. Representative contour plotsand histograms from this experiment are shown in Fig. 10; resultsare summarized in a scatter graph and bar graphs shown in Fig. 11,A and B. Although simultaneous staining with five fluoro-chromes required the use of a system that detected individualfluorochromes with reduced sensitivity; sufficient sensitivitywas retained to fulfill the goal of the experiment. All popula-tions of HEL-Ig Tgn B cells expressed barely detectable mIgM(Fig. 11B). Immature (HSAbright) B cells from untreated Ig Tgnand HEL-Ig Tgn mice expressed similar amounts of mIgD, whilemIgD expression was reduced on HEL-Ig Tgn mature (HSAint-dull)B cells compared with mature Ig Tgn B cells (Fig. 11B). Mostimportantly, 2 wk of treatment with anti-IL-7 mAb was accompa-nied by a substantial loss of mIgD and HEL binding capacity byHEL-Ig Tgn splenic B cells (Fig. 10, second and fourth histogramsin second rowand Fig. 11,A andB), so that most splenic B cellsfrom these mice stained no more brightly for mIgMa, mIgDa, orHEL than B cells from nontransgenic, Igb allotype, CB20 mice(Fig. 10, third row of histograms). In contrast, mIgM, mIgD, andHEL staining were clearly positive on mature, B2201CD191
spleen cells from Ig Tgn mice (Fig. 10,fourth rowof histograms,and Fig. 11,A andB).
To confirm these observations, an additional experiment wasperformed that examined the effects of anti-IL-7 mAb, anti-CD4mAb, or a control mAb on mIgM and mIgD expression and HELbinding capacity of HEL-Ig Tgn splenic B cells. B cells were iden-tified by their expression of both B220 and CD19; B cells that hadan immature or mature phenotype were identified by their expres-sion of B220 and HSA (Fig. 7). Of the mAbs tested, only the anti-IL-7mAb reduced HEL binding by HEL-Ig Tgn splenic B cells (Fig. 11C).This reduction in HEL binding capacity was accompanied by a se-lective reduction in mIgD expression (Fig. 11D).
FIGURE 8. Splenic B cells from HEL-Ig Tgn mice express low levelsof mIgM and lose mIgD as they age. Spleen cells from Ig Tgn and HEL-IgTgn mice (four per group) were stained with biotin-anti-B220 mAb, FITC-anti-HSA mAb, and either Cy5-anti-IgM or Cy5-anti-IgD mAbs, followedby S-PE. mIgM and mIgD expression on HSAbright, HSAint, and HSAdull
B2201 cells was determined by flow cytometry. Contour plots inupperpanelsillustrate staining for B220 and HSA by spleen cells from repre-sentative mice; histograms in thesecond rowof panels illustrate mIgMstaining of HSAbright, HSAint, and HSAdull splenic B cells from these mice;histograms in thethird row of panels illustrate mIgD staining of splenic Bcells from these mice; and the dot plot in thelowest panelindicates meansand SEs for mIgM and mIgD staining of the HSA/B220-defined splenic Bcell populations.
3041The Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
Taken together, these experiments demonstrate that in the ab-sence of receptor editing, long term survival of Ag-specific B cellsin the presence of a relatively high concentration of Ag is accom-panied by down-regulation of mIgD as well as mIgM and/or by theselection of B cells that express little mIg of either isotype.
Discussion
We have used three different methods to confirm previous obser-vations that exposure of autoreactive B cells to near-saturatingquantities of soluble Ag causes most of these B cells to die while
FIGURE 9. mIg expression by bone mar-row B cells in Ig Tgn and HEL-Ig Tgn mice.Bone marrow cells from Ig Tgn and HEL-IgTgn mice (four per group) were stained withbiotin-anti-HSA, FITC-anti-B220, and eitherCy5-anti-IgMa or Cy5-anti-IgDa mAbs, fol-lowed by S-PE. mIgM and mIgD expressionon B220brightHSAdull, B220dullHSAbright, andB2201/2HSAbright cells was determined byflow cytometry.A, Contour plots illustrate thelight scatter and B220 and HSA staining ofthe bone marrow cells from a representativeIg Tgn mouse and show the gates used; his-tograms illustrate mIgM and mIgD stainingof the B220brightHSAdull, B220dullHSAbright,and B2201/2HSAbright cells from the samemouse (marker indicates mIg1 cells).B, Bargraphs indicate the percentages of Ig Tgnand HEL-Ig Tgn bone marrow cells that areB220brightHSAdull, B220dullHSAbright, orB2201/2HSAbright and the percentages ofbone marrow cells that have these phenotypesand also express mIgM or mIgD. Means andSEs are shown.C, The means and SEs formIgM and mIgD staining of the HSA/B220-defined bone marrow B cell populations areshown (open symbols represent Ig Tgn miceand closed symbols represent HEL-Ig Tgnmice).
3042 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

still immature (19, 46–49). 1) The decreased number of splenic Bcells in HEL-Ig Tgn mice, as opposed to Ig Tgn mice, was shownto result from a selective deficiency in B cells that express surfacemarkers characteristic of mature B cells (relatively high quantitiesof B220 and relatively low quantities of HSA) (Figs. 2 and 6). 2)Labeling of dividing B cells, including newly generated B cells,with BrdU demonstrated that similar numbers of splenic B cellswere labeled in Ig Tgn and HEL-Ig Tgn mice when the mice weretreated with BrdU for 14 days (Fig. 3), but fewer labeled splenic Bcells were present in HEL-Ig Tgn than in Ig Tgn mice 11 days aftera 3-day labeling period (Fig. 4). Furthermore, B cells that hadsurvived for.14 days were particularly deficient in HEL-Ig Tgnmice regardless of their apparent maturity, as characterized by sur-face expression of B220 and HSA (Fig. 3). 3) Treatment withanti-IL-7 mAb, which suppresses B lymphopoiesis, decreasedsplenic B cell numbers more in HEL-Ig Tgn than in Ig Tgn mice(Fig. 5), while treatment with an isotype-matched control mAb didnot decrease splenic B cell numbers (data not shown). The effect ofanti-IL-7 mAb on splenic B cell number most likely resulted frominhibition of B lymphopoiesis rather than from nonspecific effectsof injecting mice with a large quantity of mouse IgG2b or blockingIL-7 stimulation of T cells or other cells that could provide help toB cells, because anti-IL-7 mAb treatment had no effect on thesurvival of B cells that had been produced before the initiation ofanti-IL-7 treatment (i.e., the BrdU2 B cells in Fig. 5), and theeffects of anti-IL-7 mAb treatment were not reproduced by inject-ing mice with an isotype-matched control Ab or an anti-CD4 mAb(Fig. 7).
In view of previous debate about the survival of autoreactive Bcells in HEL-Ig Tgn mice (8, 13, 19) and the potential problemswith each of the methods that have been used to evaluate survival,we believe that investigation of B cell life span by these threemethods was necessary to securely demonstrate decreased survivalof most HEL-Ig Tgn B cells. 1) Although B220 and HSA expres-sion can be used to characterize B cells as mature or immature (19,39, 42), this characterization is not foolproof. Previous studieshave shown, for example, that mature marginal zone B cells can beHSAbright (50), and we found considerable numbers of HSAbright Bcells in Ig Tgn mice that failed to label after mice were treated for14 days with BrdU (Fig. 3). 2) B cells that label with BrdU may beproliferating mature B cells rather than newly generated B cells. 3)Suppression of B lymphopoiesis by anti-IL-7 mAb may be incom-plete, and decreased B cell production in the bone marrow appearsto be partially compensated for by an increased likelihood thatnewly produced bone marrow B cells will migrate to the spleen(Fig. 5 and data not shown). Furthermore, neutralization of IL-7might affect B cells indirectly by inhibiting the activating effects ofIL-7 on T cells (51–54). Despite the potential problems with eachof these three methods, our observation that all the methods lead tosimilar conclusions about B cell survival reinforces the likelihoodthat these conclusions are correct.
In addition to confirming that the binding of soluble Ag to Ag-specific B cells decreases the average life span of these cells, ourobservations demonstrate that this decrease in B cell survival re-sults not only from decreased survival of newly generated B cells,but also from decreased survival of more mature B cells. Ig Tgn
FIGURE 10. Effect of anti-IL-7 mAbtreatment on B cell mIg expression in Ig Tgnand HEL-Ig Tgn mice. Ig Tgn and HEL-IgTgn mice (three or four per group) were leftuntreated or were injected i.p. with 3 mg ofanti-IL-7 mAb three times per week for 2 wkto suppress new B cell production. Spleencells from individual Ig Tgn and HEL-Ig Tgnmice as well as from a CB20 mouse (Igballotype) were counted and incubated for 1 hwith 100 ng/ml of HEL at 4°C and stainedwith PerCP-anti-B220, biotin-anti-HSA,FITC-anti-HEL, and Cy5-labeled anti-IgMa
and/or anti-IgDa mAb, followed by PE-la-beled anti-CD19 and S-PharRed, then ana-lyzed by flow cytometry.Upper panelsshowcontour plots from representative animals ofthe type labeled. Gates used for analysis areshown. Histograms in thesecond rowshowthe intensity of fluorescence staining ofB2201CD191 spleen cells from an untreatedmouse (thin line) or an anti-IL-7 mAb-treated mouse (bold line) for IgMa, IgDa,IgMa and IgDa, or HEL. In histograms in thethird and fourth rows spleen cells from aCB20 mouse (Igb allotype) are used as anegative control for staining with the anti-Iga allotype reagents and for staining withHEL. The scale on the ordinate differsamong panels.
3043The Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from
and HEL-Ig Tgn mice that have been fed BrdU-containing waterfor 14 days have similar numbers of BrdU1 HSAint B cells (Figs.3 and 5). However, most of the HEL-Ig Tgn BrdU1HSAint splenicB cells are newly produced cells rather than proliferating maturecells, because many fewer BrdU1 HSAint B cells are present inanti-IL-7 mAb-treated HEL-Ig Tgn mice than in untreated HEL-IgTgn mice (Fig. 5). Thus, at least equal numbers of B cells enter the
predominantly mature HSAint population in untreated HEL-Ig Tgnmice as in untreated Ig Tgn mice. In contrast, the numbers ofBrdU2, HSAint splenic B cells were strikingly lower in HEL-IgTgn than in Ig Tgn mice in the same experiments. If approximatelyequal numbers of cells enter the HSAint population in HEL-Ig Tgnmice as in Ig Tgn mice, but the size of this population is consid-erably smaller in HEL-Ig Tgn than in Ig Tgn mice, the survival
FIGURE 11. Effect of anti-IL-7 mAb treat-ment on B cell mIg expression in Ig Tgn andHEL-Ig Tgn mice.A, Means and SEs of thenumber of mature and immature spleen cellswere determined for untreated and anti-IL-7-treated Ig Tgn and HEL-Ig Tgn mice in thesame experiment that is illustrated in Fig. 10(left panel). Numbers were determined by mul-tiplying the percentage of each cell type in thespleen by the total number of spleen cells.Means and SEs of HEL mean fluorescence in-tensities for the same populations of splenic Bcells are shown in theright panel. B, Meansand SEs for mIgM and mIgD staining ofCD191HSA/B220-defined splenic B cellsfrom untreated and anti-IL-7-treated Ig Tgnand HEL-Ig Tgn mice are shown for the sameexperiment that is illustrated in Fig. 10 (filledsymbols represent anti-IL-7 mAb-treatedmice). C, Means and SEs of HEL mean fluo-rescence intensity for mature and immaturesplenic B cells from untreated, anti-IL-7 mAb-treated, control mAb-treated, or anti-CD4mAb-treated HEL-Ig Tgn mice and a singleCB20 and Ig Tgn mouse were determined inthe same experiment that is illustrated in Fig.7. Following incubation with HEL, spleencells were stained with PerCP-labeled anti-B220, Cy5-labeled anti-HEL, and FITC-la-beled anti-HSA, followed by PE-labeled anti-CD19.D, Means and SEs are shown for mIgMand mIgD staining of CD191HSA/B220-de-fined splenic B cell populations from the samemice described inC and illustrated in Fig. 7.
3044 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

time of cells that enter this population must be shorter in HEL-IgTgn than in Ig Tgn mice. These observations contradict previoussuggestions that B cells experience a single period of susceptibilityto Ag-induced deletion (47), but support studies performed in nor-mal mice demonstrating that mIgD cross-linking shortens the lifespan of most mature B cells (42).
The most novel observation of our studies concerns the minorityof B cells that survive to maturity in HEL-Ig Tgn mice. The pres-ence of a mature B cell population in these mice is demonstratedby the existence of HSAint and HSAdull splenic B cells in thesemice that fail to label when the mice are fed BrdU for 2 wk andpersist when B lymphopoiesis is suppressed for 2 wk with anti-IL-7 mAb. Few of these B cells have lost the ability to bind HELby expressing endogenous Ig heavy chains, because B cells thatexpress Ig heavy chains of the endogenousb allotype are unde-tectable in most HEL-Ig mice in our colony, even when we selec-tively examine HSAint-dull splenic B cells (data not shown). Incontrast, a considerable, but variable, percentage of HEL-Igsplenic B cells has undergone receptor editing. Many of these re-ceptor-edited B cells still bind HEL, but bind it with lower affinitythan do B cells that express the transgenic Ig light chain, as dem-onstrated by a reduction in the ratio of HEL binding to mIg ex-pression (Fig. 6). Most HSAint-dull splenic B cells in HEL-Ig Tgnmice, however, appear to remain HEL specific, but have not un-dergone receptor editing; rather, their reduced expression of mIgDas well as mIgM may promote survival in the presence of HEL bylimiting their ability to interact with this Ag. Recently producedbone marrow or splenic B cells in HEL-Ig Tgn mice express nor-mal quantities of mIgD, but appear to progressively lose mIgD asthey age. This loss of mIgD becomes particularly apparent whenthe average age of B cells in HEL-Ig Tgn mice is increased bytreating the mice for 2 wk with anti-IL-7 mAb; most splenic Bcells in these mice expressed barely detectable mIgM or mIgD andbound little HEL (Figs. 10 and 11,A and B). It is not certainwhether the low mIgD expression by mature HEL-Ig Tgn B cellsresults from selective survival of B cells that express low levels ofmIgD or is induced by prolonged mIg-mediated signaling. It islikely, however, that prolonged mIg-mediated signaling, ratherthan selection, causes at least some of the decrease in mIgD ex-pression that follows treatment with anti-IL-7 mAb, because anti-IL-7 mAb treatment of HEL-Ig Tgn mice appears to cause anincrease in the absolute number of splenic Ig2 B cells (Fig. 7).
B cells that have escaped Ag-induced deletion by decreasingmIg expression might provide a reservoir of autoreactive cells thatcould promote protective immunity by allowing Ab responses tobe generated to a pathogen-associated Ag that cross-reacts with anautoantigen. Alternatively, activation of these B cells by autoim-mune T cells or by pathogen-induced inflammation might lead toproduction of disease-causing autoantibodies. Identification of thestimuli that can activate these mIg-deficient autoreactive B cells toincrease mIg expression, proliferate, and/or differentiate into Ab-secreting cells and determination of whether autoreactive B cellscan survive as mIglow or mIg2 cells in normal, nontransgenic an-imals should facilitate determination of whether these cells may beimportant in protective immunity or autoimmune disorders.
References1. Basten, A., R. Brink, P. Peake, E. Adams, J. Crosbie, S. Hartley, and
C. C. Goodnow. 1991. Self tolerance in the B-cell repertoire.Immunol. Rev.122:5.
2. Metcalf, E. S., and N. R. Klinman. 1976. In vitro tolerance induction of neonatalmurine B cells.J. Exp. Med. 143:1327.
3. Nemazee, D. A., and K. Burki. 1989. Clonal deletion of B lymphocytes in atransgenic mouse bearing anti-MHC class I antibody genes.Nature 337:562.
4. Nemazee, D., and K. Burki. 1989. Clonal deletion of autoreactive B lymphocytesin bone marrow chimeras.Proc. Natl. Acad. Sci. USA 20:8039.
5. Hartley, S. B., J. Crosbie, R. Brink, A. B. Kantor, A. Basten, and C. C. Goodnow.1991. Elimination from peripheral lymphoid tissues of self-reactive B lympho-cytes recognizing membrane-bound antigens.Nature 353:765.
6. Russell, D. M., Z. Dembic, G. Morahan, J. F. Miller, K. Burki, and D. Nemazee.1991. Peripheral deletion of self-reactive B cells.Nature 354:308.
7. Hartley, S. B., M. P. Cooke, D. A. Fulcher, A. W. Harris, S. Cory, A. Basten, andC. C. Goodnow. 1993. Elimination of self-reactive B lymphocytes proceeds intwo stages: arrested development and cell death.Cell 72:325.
8. Goodnow, C. C., J. Crosbie, S. Adelstein, T. B. Lavoie, S. J. Smith-Gill,R. A. Brink, H. Pritchard-Briscoe, J. S. Wotherspoon, R. H. Loblay, K. Raphael,et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice.Nature 334:676.
9. Goodnow, C. C., J. Crosbie, H. Jorgensen, R. A. Brink, and A. Basten. 1989.Induction of self-tolerance in mature peripheral B lymphocytes.Nature 342:385.
10. Adelstein, S., H. Pritchard-Briscoe, T. A. Anderson, J. Crosbie, G. Gammon,R. H. Loblay, A. Basten, and C. C. Goodnow. 1991. Induction of self-tolerancein T cells but not B cells of transgenic mice expressing little self antigen.Science251:1223.
11. Bell, S. E., and C. C. Goodnow. 1994. A selective defect in IgM antigen receptorsynthesis and transport causes loss of cell surface IgM expression on tolerant Blymphocytes.EMBO J. 13:816.
12. Cooke, M. P., A. W. Heath, K. M. Shokat, Y. Zeng, F. D. Finkelman,P. S. Linsley, M. Howard, and C. C. Goodnow. 1994. Immunoglobulin signaltransduction guides the specificity of B cell-T cell interactions and is blocked intolerant self-reactive B cells.J. Exp. Med. 179:425.
13. Goodnow, C. C., R. Brink, and E. Adams. 1991. Breakdown of self-tolerance inanergic B lymphocytes.Nature 352:532.
14. Eris, J. M., A. Basten, R. A. Brink, K. Doherty, M. R. Kehry, and P. D. Hodgkin.1994. Anergic self-reactive B cells present self antigen and respond normally toCD40-dependent T cell signals but are defective in antigen-receptor mediatedfunctions.Proc. Natl. Acad. Sci. USA 91:4392.
15. Rothstein, T., J. Wang, D. Panka, L. Foote, Z. Wang, B. Stanger, H. Cui, S. Ju,and A. Marshak-Rothstein. 1995. Protection against Fas-dependent Th1-mediatedapoptosis by antigen receptor engagement in B cells.Nature 374:163.
16. Rathmell, J. C., M. P. Cooke, W. Y. Ho, J. Grein, S. E. Townsend, M. M. Davis,and C. C. Goodnow. 1995. CD95 (Fas)-dependent elimination of self-reactive Bcells upon interaction with CD41 T cells.Nature 376:181.
17. Rathmell, J. C., S. E. Townsend, J. C. Xu, R. A. Flavell, and C. C. Goodnow.1996. Expansion or elimination of B cells in vivo: dual roles for CD40- and Fas(CD95)-ligands modulated by the B cell antigen receptor.Cell 87:319.
18. Foote, L.C., A. Marshak-Rothstein, and T. L. Rothstein. 1998. Tolerant B lym-phocytes acquire resistance to Fas-mediated apoptosis after treatment with inter-leukin 4 but not after treatment with specific antigen unless a surface immuno-globulin threshold is exceeded.J. Exp. Med. 187:847.
19. Fulcher, D. A., and A. Basten. 1994. Reduced life span of anergic self-reactive Bcells in a double-transgenic model.J. Exp. Med. 179:125.
20. Fulcher, D. A., A. B. Lyons, S. L. Korn, M. C. Cook, C. Koleda, C. Parish,B. Fazekas de St. Groth, and A. Basten. 1996. The fate of self-reactive B cellsdepends primarily on the degree of antigen receptor engagement and the avail-ability of T cell help. J. Exp. Med. 183:2313.
21. Cook, M. C., A. Basten, and B. Fazekas de St. Groth. 1997. Outer periarteriolarlymphoid sheath arrest and subsequent differentiation of both naive and tolerantimmunoglobulin transgenic B cells is determined by B cell receptor occupancy.J. Exp. Med. 186:631.
22. Cyster, J. G., and C. C. Goodnow. 1995. Antigen-induced exclusion from folli-cles and anergy are separate and complementary processes that influence periph-eral B cell fate.Immunity 6:691.
23. Cyster, J. G., S. B. Hartley, and C. C. Goodnow. 1994. Competition for follicularniches excludes self-reactive cells from the recirculating B-cell repertoire.Nature371:389.
24. Schmidt, K. N., and J. G. Cyster. 1999. Follicular exclusion and rapid eliminationof hen egg lysozyme autoantigen-binding B cells are dependent on competitor Bcells, but not on T cells.J. Immunol. 162:284.
25. Gay, D., T. Saunders, S. Camper, and M. Weigert. 1993. Receptor editing: anapproach by autoreactive B cells to escape tolerance.J. Exp. Med. 177:999.
26. Tiegs, S. L., D. M. Russell, and D. Nemazee. 1993. Receptor editing in self-reactive bone marrow B cells.J. Exp. Med. 177:1009.
27. Radic, M. Z., J. Erikson, S. Litwin, and M. Weigert. 1993. B lymphocytes mayescape tolerance by revising their antigen receptors.J. Exp. Med. 177:1165.
28. Chen, S., and G. A. Evans. 1990. A simple screening method for transgenic miceusing the polymerase chain reaction.BioTechniques 8:32.
29. Coffman, R. L., and I. L. Weissman. 1981. B220: a B cell-specific member of theT200 glycoprotein family.Nature 289:681.
30. Sieckmann, D., A. M. Stall, and B. Subbarao. 1991. A mouse monoclonal anti-body specific for an allotypic determinant of the IgHa allele of murine IgM:genetic and functional analysis of Igh-6a epitopes using anti-IgM monoclonalantibodies.Hybridoma 10:121.
31. Zitron, I. M., and B. L. Clevinger. 1980. Regulation of murine B cells throughsurface immunoglobulin. I. Monoclonal anti-d antibody that induces allotype-specific proliferation.J. Exp. Med. 152:1135.
32. Stall, A. M., and M. R. Loken. 1984. Allotypic specificities of murine IgD andIgM recognized by monoclonal antibodies.J. Immunol. 132:787.
33. Metzger, D.W., L.-K. Ch’ng, A. Miller, and E. E. Sercarz. 1984. The expressedlysozyme-specific B cell repertoire I. Heterogeneity in the monoclonal anti-henegg white lysozyme specificity repertoire, and its difference from the in siturepertoire.Eur. J. Immunol. 14:87.
3045The Journal of Immunology
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

34. Unkeless, J. C. 1979. Characterization of a monoclonal antibody directed againstmouse macrophage and lymphocyte Fc receptors.J. Exp. Med. 150:580.
35. Wilde, D. B., P. Marrack, J. Kappler, D. P. Dialynas, and F. W. Fitch. 1983.Evidence implicating L3T4 in class II MHC antigen reactivity: monoclonal an-tibody GK1. 5 (anti-L3T4a) blocks class II MHC antigen-specific proliferation,release of lymphokines, and binding by cloned murine helper T lymphocyte lines.J. Immunol. 131:2178.
36. Grabstein, K. H., T. Waldschmidt, F. D. Finkelman, B. Hess, A. Alpert,N. Boiani, A. Namen, and P. Morrissey. 1993. Inhibition of murine B lympho-poiesis in vivo by an anti-interleukin-7 monoclonal antibody.J. Exp. Med. 178:257.
37. Finkelman, F. D., and I. Scher. 1979. Rhesus monkey B lymphocyte surfaceimmunoglobulin: analysis with a fluorescence activated cell sorter.J. Immunol.122:1757.
38. Sher, I., J. A. Titus, and F. D. Finkelman. 1983. The ontogeny and distribution ofB cells in normal and mutant immune-defective CBA/N mice: two parameteranalysis of surface IgM and IgD.J. Immunol. 130:619.
39. Allman, D. M., S. E. Ferguson, V. M. Lentz, and M. P. Cancro. 1993. PeripheralB cell maturation. II. Heat-stable antigenhi splenic B cells are an immature de-velopmental intermediate in the production of long-lived marrow-derived B cells.J. Immunol. 151:4431.
40. Krop, L., A. R. deFougerolles, R. R. Hardy, M. Allison, M. S. Schlissel, andD. T. Fearon. 1996. Antibody to CD19 suppresses self-renewal of B-1 lympho-cytes.Eur. J. Immunol. 26:238.
41. Hardy, R. R., C. E. Carmack, S. A. Shinton, J. D. Kemp, and K. Hayakawa. 1991.Resolution and characterization of pro-B and pre-pro-B cell stages in normalmouse bone marrow.J. Exp. Med. 173:1213.
42. Finkelman, F. D., J. M. Holmes, O. I. Dukhanina, and S. C. Morris. 1995.Crosslinking of membrane IgD, in the absence of T cell help, kills mature B cellsin vivo. J. Exp. Med. 181:515.
43. Bhatia, S. K., L. T. Tygrett, K. H. Grabstein, and T. J. Waldschmidt. 1995. Theeffect of in vivo IL-7 deprivation on T cell maturation.J. Exp. Med. 181:1399.
44. Nieuwenhuis, P., and W. L. Ford. 1976. Comparative migration of B- and T-lymphocytes in the rat spleen and lymph nodes.Cell. Immunol. 23:254.
45. Sprent, J. 1973. Circulating T and B lymphocytes of the mouse.I. Migratory properties.Cell. Immunol. 7:10.
46. Klinman, N. R., A. F. Schrater, and D. H. Katz. 1981. Immature B cells as thetarget for in vivo tolerance induction.J. Immunol. 126:1970.
47. Carsetti, R., G. Kohler, and M. C. Lamers. 1995. Transitional B cells are thetarget of negative selection in the B cell compartment.J. Exp Med. 181:2129.
48. Rilery, R. L., and N. R. Klinman. 1986. The affinity threshold for antigenictriggering differs for tolerance susceptible immature precursors vs. mature pri-mary B cells.J. Immunol. 136:3147.
49. Nossal, G. J. V. 1983. Cellular mechanisms of immunologic tolerance.Annu.Rev. Immunol. 1:33.
50. Erickson, L. D., L. T. Tygrett, S. K. Bhatia, K. H. Grabstein, andT. J. Waldschmidt. 1996. Differential expression of CD22 (Lyb8) on murine Bcells. Int. Immunol. 8:1121.
51. Murray, R., T. Suda, N. Wrighton, F. Lee, and A. Zlotnik. 1989. IL-7 is a growthand maintenance factor for mature and immature thymocyte subsets.Int. Immu-nol. 1:526.
52. Peschon, J., P. J. Morrissey, K. H. Grabstein, F. J. Ramsdell, E. Maraskovsky,B. C. Gliniak, L. S. Park, S. F. Ziegler, D. E. Williams, C. B. Ware, et al. 1994.Early lymphocyte expansion is severely impaired in interleukin 7 receptor-defi-cient mice.J. Exp. Med. 180:1955.
53. Chazen, G. D., G. M. B. Pereira, G. LeGros, S. Gillis, and E. M. Shevach. 1989.Interleukin 7 is a T-cell growth factor.Proc. Natl. Acad. Sci. USA 86:5923.
54. Morrissey, P. J., R. G. Goodwin, R. P. Nordan, D. Anderson, K. H. Grabstein,D. Cosman, J. Sims, S. Lupton, B. Acres, and S. G. Reed. 1989. Recombinantinterleukin 7, pre-B cell growth factor, has costimulatory activity on purifiedmature T cells.J. Exp. Med. 169:707.
3046 SURVIVAL OF AUTOREACTIVE B CELLS
by guest on August 18, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from





























