Embed Size (px)
Citation preview

236 Biochimica et Biophysica Acta, 955 (1988) 236-242 Elsevier
BBA 33170
In terac t ion of N A D P H and tr iazine dyes
with f e r r e d o x i n - N A D P + oxidoreductase
Lois M. Levy and G r a h a m F. Butts
School of Biological Sciences, Queen Mary College, Mile End Roa~ London (U. K.)
(Received 8 January 1988)
Key words: Ferredoxin-NADP oxidoreductase; NADPH binding
The ability of NADPH to compete for binding with other |igands of known affinity has been used to provide values for the Kd of NADPH with ferredoxin-NADP + oxidoreductase (EC 1.18.1.2) (FNR). When the competing lignd is proeion red, which binds with a red-shift in spectrum, or Woodwards reagent K(N-ethyl-5-phenylisoxazulium 3'-sulfonate), which covalentiy modifies an active site earboxy| residue, the calculated Kd for the NADPH.FNR complex is greater than 8 or 0.08 raM, respectively. Because of the feeble (or non-existent) ability of NADPH to dislodge proeion red, we propose that this dye and NADPH are not binding at the same site. Procion red must, however, bind additionally at the active site (presumably without spectral perturbation) as it is a competitive inhibitor of NADPH in ferricyanide reduction assays and more crucially proves to be a novel substrate itself, being reduced to a leuco form which can be reoxidised by oxygen. Although a K d for the NADPH-FNR complex of 0.08 mM is reasonable, we point out the difficulty of interpreting this value and question its physiological significance.
introduction
We have been seeking an estimate for the K d of ferredoxin-NADP + oxidoreductase(EC 1.18.1.2) (FNR) with its nucleotide NADPH prior to stud- ies of the direct transfer ~f NADPH to the NADP-specific glyceraldehyde-3-phosphate dehy- drogenase from chloroplasts. A reliable estimate for the K d is unavailable in the literature. A value of 6.10 -e M based on flavine absorbance changes upon introduction of NADPH [1,2] is dubious as
Abbreviations: FNR, ferredoxin-NADP + oxidoreductase (EC 1.18.1.2); Woodwards reagent K, N-ethyl-5-phenyfisoxazolium 3"-sulphonate.
the observed signal is likely to be due to flavin reduction.
Difficulties in obtaining a K d for the FNR- NADPH complex also stem from the possession by FNR of low oxidase activity, so, unless oxygen is scrupulously removed, NADPH will be oxidised to NADP + over the time scales needed for several methods of studying ligand binding. Moreover, incubating FNR with NADPH causes some structural change indicated by loosening of the flavine prosthetic group and consequent loss of activity [3,4].
We have therefore sought rapid methods to determine NADPH binding.
Materials and Methods
Correspondence: G.F. Butts, School of Biological Sciences, Queen Mary College, Mile End Road, London, E1 4NS, U.K.
Ferredoxin-NADP + oxidoreductase was ob- tained from Sigma Chemical Company. ~456 ~ - - 10.7
0167-4838/88/$03.50 © 1988 Elsevier Science Publishers B.V. (Biomedical Division)
mM-1. cm-i was used to calculate the concentra- tion of the flavoprotein [5]. Procion red HE-3B was a gift from ICI plc and was purified before use by thin-layer chromatography on silica gel plates (Merck) in a solvent consisting of 2- butanol/1-propanol/ethyl acetate/water (20" 35" 10" 35 (v/v)) [6]. The major red band (RF= 0.57) was eluted with distilled water and an e540 -- 38.5 mM -1. cm -1 was used to calculate the dye's concentration [7]. Woodwards reagent K (N-ethyl- 5-phenylisoxazolium 3'-sulphonate) was obtained from Sigma Chemical Co. All other reagents were of analytical grade. Spectrophotometric work was carried out on a Varian DMS100 UV-Vis spectro- photometer.
Dye-binding studies Dye binding to FNR was measured by dif-
ference spectroscopy. The reference cuvette con- tained 1 ml of 50 mM Tris-HCl (pH 7.5) while the sample cuvette contained an equal volume of the same buffer containing 1-3 #M FNR. Equal amounts of dye were titrated into both sample and reference cuvettes and mixed by stirring. Dif- ference spectra were recorded from 450-650 nm. Absorbance readings at 568 nm were used to determine the Ko for dye. For dye displacement experiments 10 #1 of NADPH was added to both sample and reference cuvettes, to give a final concentration of 1 mM NADPH.
Dye inhibition of ferricyanide reduction Ferricyanide reduction was followed at 420 nm
in a reaction mixture containing 50 mM Tris-HCl (pH 7.5), I mM potassium ferricyanide, 0.02-0.1 mM NADPH, 0-12 #M procion red and FNR.
Inactivation protection by NADPH The rate of inactivation of FNR by Woodwards
reagent K was followed in a medium containing 0.7 #M FNR, 50 mM Tris-HCl (pH 7.5), 0.5 mM Woodwards reagent K 0-0.3 mM NADPH. (For the determination of the K i for Woodwards re- agent K, its concentration was varied from 0-1 mM.) The modification reaction was stopped at given times by removal of aliquots into sodium acetate, to give a final sodium acetate concentra- tion of 0.1 M. Residual enzyme activity was mea- sured by following NADPH-dependent fer- ricyanide reduction.
237
Dye reduction Procion red reduction and NADFH oxidation
were followed at 540 and 345 rim, respectively, in a reaction mixture containing 50 mM Tris-HCl (pH 7.5), 2.2 i/LM FNR, 7.14 #M procion red and
' l r ' ~
4.2.10 -4 M NAD:H. In experiments where the oxygen concentration was to be substantially re- duced, all reaction mixture components were bub- bled with nitrogen gas. A value of 6.103 M - ] . cm-] was r/.sed for e34s nm of NADPH.
Results
Dye-binding studies The spectrum of triazine dyes is red-shifted on
binding to the nucleotide binding domain of several proteins, this binding being competitive with the nucleotide [8]. Such a phenomena has been zeported for procion red and NADP + bind- ing to FNR [7]. We have sought to use this tech- nique to obtain a K d for the FNR-NADPH corn-
0"lS I
0.10 :
Absorbance
O'OS
o4 / L l , , ~ _ ~ d o4 / o4 9 / ,.2 ,~ x2o / f ~
:;::[ .7 /a\ -o.otj/ /A\\
-O'OSso 490 S30 S'?O 610 Wavelength trim)
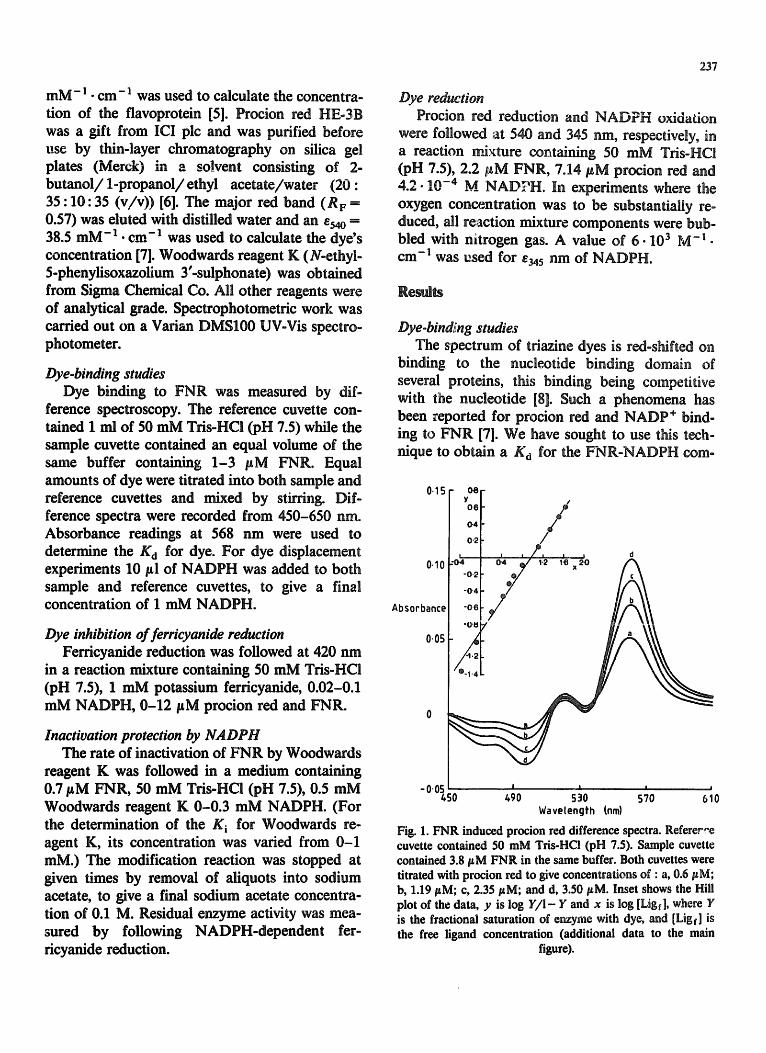
Fig. 1. FNR induced procion red difference spectra. Referer,'e cuvette contained 50 mM Tris-HCl (pH 7.5). Sample cuvette contained 3.8 #M FNR in the same buffer. Both cuvettes were titrated with procion red to give concentrations of : a, 0.6 #M; b, 1.19/tM; c, 2.35/~M; and d, 3.50 #M. Inset shows the Hill plot of the data, y is log ¥ / I - Y and x is log [Ligf], where Y is the fractional saturation of enzyme with dye, and [Ligt] is the free ligand concentration (additional data to the main
figure).
238
6
1 A c t i v i t y
3 (AA mi n "t)
[Procion Red] IpH)
~, 12.03 ll,z, 2
~, 6'02
I" 2,~
o &
-30 -20 - I 0 I I I I I t
0 10 2 0 3 0 6 0 SO 1
[N A DPH ] (mR -1)
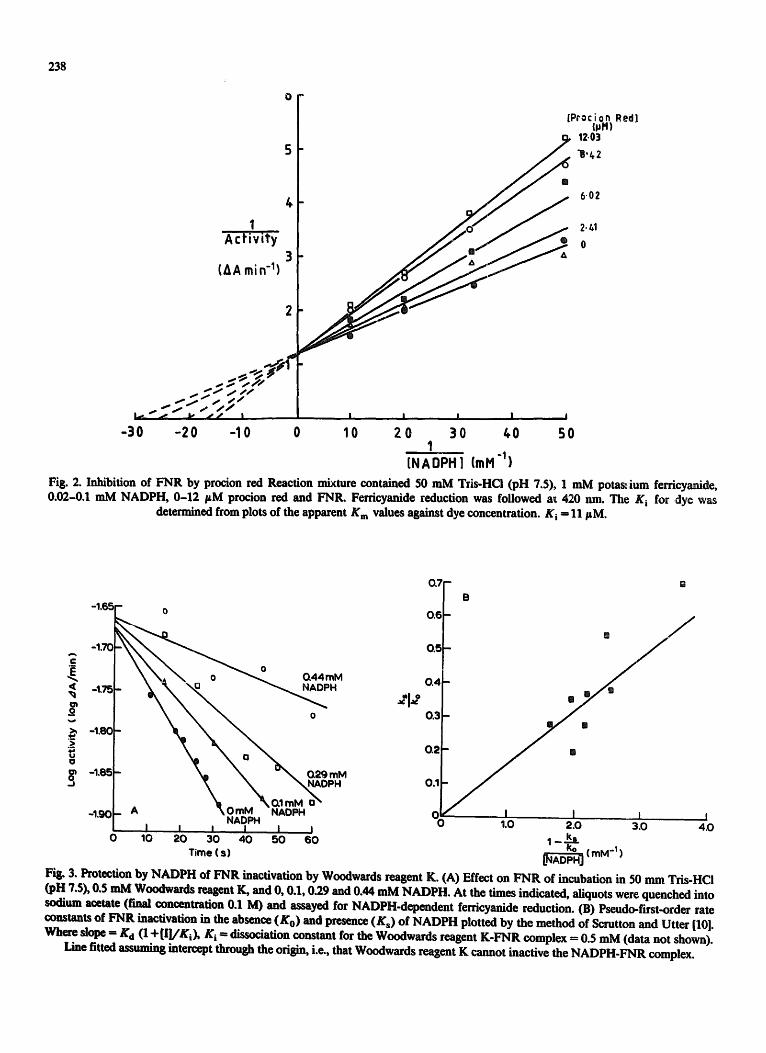
Fig. 2. Inhibition of FNR by procion red Reaction mixture contained 50 mM Tzis-HCi (pH 7.5), 1 mM potas~,ium ferricyanide, 0.02-0.1 mM NADPH, 0-12 FM procion red and FNR. Ferricyanide reduction was followed a~ 420 nm. The K i for dye was
determined from plots of the apparent K m values against dye concentration. K i -- 11 FM.
- I , 6 5
-1,70 c
< -I,75 t~ o
~ -1.8o 2 o ~ -~'~:
-1,9(
- 0
0 4 4 m M -
_ o
\ .I~ 0 mM NADP I - "" .% 0.1 mM,_, a" A m
N A D P H I I I I I l
0 10 20 30 40 50 60 Time (s)
0.7
0.6
O.~
B
o 4 - •
0 .3 -
0 2 -
0.1
o~ ~ i 1.0 2.0
~ (mlVt -I )
I I 3.0 4.0
Fig. 3. Protection by HADPH of ~qR inactivation by Woodwards reagent K. (A) Effect on ~ q R of incubation in 50 nun Tris-HCl (pH Z$), 0.5 mM Woodwards reagent K, and 0, 0.1, 0.29 and 0.44 mM NADPH. At the times indicated, aliquots were quenched into sodium acetate (final concentration 0.1 M) and assayed for HADPH-dependent ferricyanide reduction. (B) Pseudo-first-order rate constants of FNR inactivation in the absence (Ko) and presence (Ks) of NADPH plotted by the method of Scrutton and Utter [10]. ~Vha'¢ SlOl~ = K d (1 +[[]/Ki) , K i = dissociation constant for the Woodwards reagent K-FNR complex = 0.5 mM (data not shown).
Line fitted assuming intercept through the origin, i.e., that Woodwards reagent K cannot inactive the HADPH-FNR complex.
plex. Fig. 1 shows the difference spectra when procion red is titrated into both sample and refer- ence cuvettes, with FNR present in the sample cuvette only. Spectral perturbation was complete within the time required to mix and run the spec- trum and no time-dependent changes were seen when a single wavelength was observed at 568 nm from 10 s after dye addition. The inset of Fig. 1 shows a Hill plot of absorbance difference and free dye concentration, data from which a slope of 0.93 and a Kd for dye of 8 /tM is calculated. When I mM NADPH is introduced into a sample cuvette containing 11 pM dye and 3 #M FNR, there is no observed decrease in the absorbance at 568 nm. A 5% decrease in signal would be easily noticed. Assuming that NADPH is a competitive inhibitor of dye binding, then this failure to dis- lodge the dye means that the K d for NADPH must be greater than 8 raM. We also failed to dislodge dye with 1 mM NADP +. Moreover, titrating dye into a cuvette already containing 1 mM NADP + does not alter the data for Fig, I and insert.
239
Dye inhibition of ferricyanide reduction If dye and NADPH are binding at the same
site with Kd values of 8 #M and greater than 8 mM respectively then dye should be a good com- petitive inhibitor of FNR catalysed oxidation of NADPH. Fig. 2 shows proeion red inh/bitlon of FNR catalysed ferricyanide reduction. The kinet- ics are consistent with dye acting as a reversible inhibitor, competitive with NADPH with a K i ffi 11 #M. The apparent K m for NADPH - 33 #M.
The results so far can be interpreted in terms of dye binding strongly (K a = 8 #M) and NADPH binding weakly ( K d > 8 raM) at the same site with the only surprising feature being the large difference between the K d ar id K m for NADPH. The next section, however, casts doubts on the high value for the NADPH K d.
Inactivation protection by NADPH Woodwards reagent K has been shown to inac-
tive FNR by modifying a particular carbo×yl group; inactivation being prevented by NADP + binding [9]. Fig. 3A shows that NADPH binding
45C-
i
w
T Q <
, z
250
- 7.5
- A I I I I I 5 10 15 20 25
Time (min)
~5
5.5 L
4.5
3.5
41~
41~
( 1 . Q < z
o
02
j . 13 - ~ -
4 6 12 16 20 24 Time (mln)
6~
4T 2 ~
t .
o
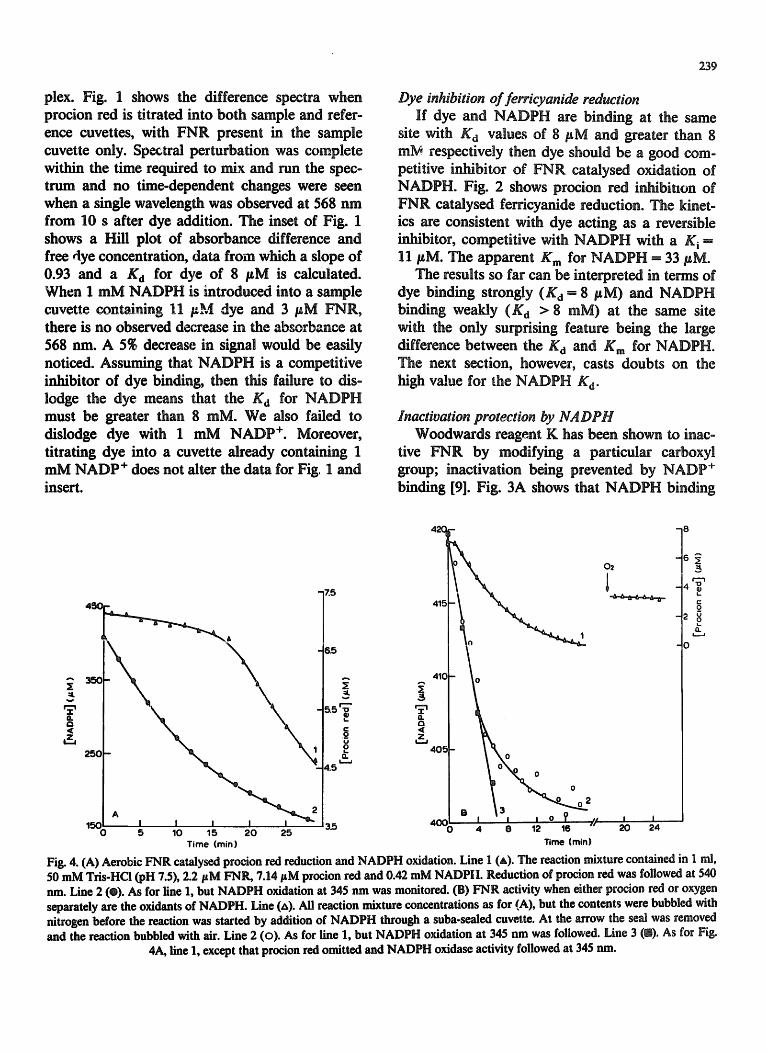
Fig. 4. (A) Aerobic FNR catalysed procion red reduction and NADPH oxidation. Line 1 (A). The reaction mixture contained in 1 nil, 50 mM Tris-HCl (pH 7.5), 2.2/zM FNIL 7.14 pM procion red and 0.42 mM NADP|I. Reduction of procion red was followed at 540 nm. Line 2 (e). As for line 1, but NADPH oxidation at 345 run was monitored. (B) FNR activity when either procion red or oxygen separately are the oxidants of NADPH. Line (zx). All reaction mixture concentrations as for (A), but the contents were bubbled with nitrogen before the reaction was started by addition of NADPH through a suba-sealed cuvette. At the arrow the seal was removed and the reaction bubbled with air. Line 2 (o). As for line 1, but NADPH oxidation at 345 nm was followed. Line 3 (D). As for Fig.
4A, line 1, except that procion red omitted and NADPH oxidase activity followed at 345 nm.

240
also protects against inactivation. Fig. 3B is a plot of inactivation data which allows a Kd for NADPH to be calculated from the slope of the plot [10]. A value of 8- 10 -5 M is obtained which is at least two orders of magnitude less than that obtained by the dye displacement method.
Dye reduction Although we have no spectral evidence of dye
displacement from FNR by NADPH, Figs. 4A and B, lines 1, show that spectral changes do occur over extended periods in mixtures contain- ing FNR, procion red and NADPH. Bleaching of the dye spectrum is also achieved by mixing dye and sodium dithionite alone [11]. It is not achieved by NADPH in the absence of FNR nor by super- oxide or hydrogen peroxide (products of the auto- xidation of reduced FNR). We therefore propose that procion red is a substrate for FNR-catalysed reduction by NADPH. The rate of NADPH oxidation (Fig. 4A, line 2) is proportional to en- zyme concentration at least up to 2.2/~M FNR.
The biphasic nature of the rate of dye reduction under aerobic conditions (Fig. 4A, line 1), we take to be due to back oxidation of enzymically reduced dye by dissolved oxygen until much of the oxygen is removed by reduction. When oxygen is substan- tiany depleted by bubbling with nitrogen before starting the reaction with NADPH (Fig. 4B, line 1), there is no apparent lag in the rate of dye reduction. NADPH is oxidised substantially faster in the presence of both oxygen and dye (Fig. 4A, line 2) than in the presence of either dye alone (Fig. 413, line 2) or oxygen alone (Fig. 4B, line 3).
Discussion
The data for the perturbation of procion red spectrum on binding to FNR (Fig. 1) are con- sistent with a reversible binding of dye rather than stoichiometric irreversible binding. Moreover, re- versibility of binding is evidenced by a decrease in the absorbance difference spectrum on dilution by a factor greater than the dilution factor.
That procion red is reduced by NADPH and FNR, means that absorbance decreases seen when NADPB is added to a mixture of FNR and dye [7] might easily be mistaken for the blue-shift expected on displacement of dye by NADPH. The
initially slow nature of the time-dependent changes in absorbance (due to the reoxidation of reduced dye by oxygen (Fig. 4A, line 1)), might explain the failure to notice dye reduction.
Although in our hands NADP(H) fails to affect that dye which is spectraUy perturbed on binding, our results are consistent with those of Carrillo and Vallejos [7] in that the dye displacement method fails to reveal a nucleotide-binding site approaching the affinity of one revealed by other techniques. Thus, the data of Carrillo and Vallejos [7] may be used to calculate a Kd for NADP + of 2 raM, yet values around 3 .10 -5 M have been obtained from spectral changes of NADP + on binding to FNR [12-14]. Similarly data presented here from dye displacement or inactivation protec- tion by NADPH provide K d values for NADPH of greater than 8 .10 - 3 o r 8 . 1 0 - 5 , respectively.
That a 100-fold excess of NADPH, binding with a Kd of 80 /~M, fails to displace any red- shifted dye, binding with a Kd of 8 /~M would argue that dye and NADPH do not bind to the same site. However, dye is a competitive inhibitor of NADPH oxidation and is itself enzymicaUy reduced, presumably on binding to the NADPH site. Taken together these data indicate two bind- ing sites for dye, one at the nucleotide site at which dye binds without spectral shift and another elsewhere where the dye spectrum is red-shifted on binding. With evidence of rather non-specific bi- nding of triazine dyes to proteins, some of which do not even contain a nucleotide binding site [15], it is no surprise to find dye binding at places additional to the active site.
The reduction rate of procion red is slow even in the absence of oxygen. Assuming that the K m
for dye is equivalent to its Ki in the ferricyanide reduction assay (11 IgM), then the apparent turnover number in the fixed NADPH concentra- tion of 0.42 mM is 0.84 rain-1 compared to 2900 rain-1 for ferricyanide reduction.
Because dye is not reduced by non-enzymically generated superoxide and H202 in concentrations greater than can be generated by the oxidase activity of FNR and because enzymically reduced dye can be back oxidised by oxygen (Fig. 4B, line 1), we assume that in reactions containing both dye and oxygen, it is dye which mediates oxygen reduction rather than vice versa.

It is noticeable that the NADPH oxidation rate in the presence of both dye and oxygen (Fig. 4A, line 2) is greater than the sum of rates when dye or oxygen are present separately (Fig. 4B, lines 2 and 3). This synergism would be explained if the re- lease of reduced dye was rate limiting and oxygen could back-oxidise reduced dye without the necessity of dye coming free from FNR.
Fig. 4B, line 1, shows a rate of dye reduction (0.74 #M. min -1) which is only 31~ of that of NADPH oxidation (2.4 lgM. min -1. Fig. 4B, line 2). This could be explained by residual oxygen providing a sink for some of the electrons from NADPH. Thus after 12 rain incubation the dif- ference in the amount of NADPH oxidised and dye reduced is 10 #M which is only 4~; of the oxygen which would be in solutions in equilibrium with air. Moreover, by this time (when trace oxygen would have been removed by reduction), dye is being reduced at 65~ of the rate of NADPH oxidation. We need not expect absolute stoichio- metry at this point even if oxygen is completely eliminated. This is because by this time a substan- tial portion of the dye has been reduced to its hydrazino form with a loss of absorption at 540 nm. A further reduction can now follow to the cleaved products with free amino groups, a pro- cess which will use NADPH but can result in no further changes in absorbance at 540 nm.
R I N H N
~ S020-
Phenylsulphonate group of of procion red HE-3B
N ~ C O N H 2
I R
Nicotinamide group of NADP +
One would imagine that while oxygen will oxidise the dihydrazino derivative back to the diazo com- F'mnd, it has no chance to reoxidise the doubly reduced and cleaved products. Tiffs agrees well with the fact that when oxygen is introduced into an anaerobic system which is reducing dye less than the total amount of dye is recovered (Fig. 4B, line 1). The fractional recovery of the original dye absorbance decreases with the anaerobic reduction
241
period. It is noticeable that there have been no reports
of enzymatic reduction of triazine dyes by NAD(P)H. Procion red reduction in this instance is in fact made possible by the possession by FNR of a flavine prosthetic group. Thus NADPH will reduce this and depart (as NADP +), thereby pro- viding access for dye binding at the same site. In any dehydrogenase without a prosthetic group, a ternary complex will be required (Enz.dye. NAD(P)H), in which case no dye reduction will be expected unless the dye mimics substrate rather than nucleotide. It is to be noted that with the natural substrate NADP + and reduced ferredoxin, FNR has a ternary complex (sequential) mecha- nism [16], so conceivably procion red may bind at the ferredoxin site. Oxidised ferredoxin does not, however, dislodge red-shifted dye.
It is interesting to note that if one assumes that the phenylsulphonate ring of the dye is an ana- logue of the nicotinamide ring of NADPH, then the electron transfer from the flavine to the diazo group is somewhat misaligned from the C-4 nico- tinamide from which the flavine receives its elec- trons.
This study has highlighted some of the difficul- ties in obtaining binding constants for the NADPH-FNR complex. Although we now have a value for the K d useful for solTle practical pur- poses, we are not convinced of the physiological relevance of this. For instance, we are not sure of the status of the flavine in the presence of NADPH, whether it is oxidised, fully reduced, or semiquinone (due to single electron transfer from reduced flavine to oxygen) or a combination of all three. Which of these in complex with NADPH is kinetically significant is unknown. Indeed, with the report of a ternary complex mechanism [16] and the possibility of an ordered release of prod- uct it is quite conceivable that no FNR-NADPH binary complex is kinetically competent. We are therefore using other methods to study the overall passage of reducing power from reduced ferredox- in to glyceraldehyde 3-phosphate.
R e f e r e n c e s
1 Batie, C.J. and Kamin, H. (1982) in Flavins and Flavoproteins (Massey, V. and Williams, C.H., Jr., eds.), Vol. 7, pp. 679-686, Elsevier.

242
2 Batie, C.J. and Kamin, H. (1986) J. Biol. Chem. 261, 11214-11223.
3 Zanetti, G. and Forti, G. (1966) J. Biol. Chem. 241, 279-285. 4 Forti, G. and Sturani, E. (1968) Eur. J. Biochem. 3, 461-472. 5 Forti, G. (1966) Brookhaven Symp. Biol. 19, 195-201. 6 Low, C.R. and Pearson, J.C. (1984) Methods Enzymol. 104,
97-113. 7 Carrglo, N. and Vallejos, R.H. (1983) Biochirn. Biophys.
Acta 742, 285-294. 8 Stellwagen, E. (1977) Acc. Chem. Res. 10, 92-98. 9 Carrillo, N., Arana, J.L. and Vallejos, R.H. (1981) J. Biol.
Chem, 256, 6823-6828.
10 Scrutton, M. and Utter, M. (1965) J. Biol. Chem. 240, 3714-3723.
11 Clonis, Y.D. (1982) J. Chromatogr. 236, 69-80. 12 Foust, G.P., Mayhew, S.O. and Massey, V. (1969) J. Biol.
Chem. 244, 964-970. 13 Zanetfi, O. (1976) Biochim. Biophys. Acta 445, 14-24. 14 Batie, C.J. and Kamin, H. (1984) J. Biol. Chem. 259,
8832-8839. 15 Dean, P.D.G. and Watson, D.H. (1973) J. Chromatogr.
165, 301-319. 16 Masald, R., Yoshikawa, S. and Matsubara, H. (1982) Bio-
chim. Biophys. Acta 700, 101-109.





![Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase ...Pyrethrin Biosynthesis: The Cytochrome P450 Oxidoreductase CYP82Q3 Converts Jasmolone To Pyrethrolone1[OPEN] Wei Li,a](https://img.pdfslide.net/doc/110x75/5e2d08c0200c602a86070292/pyrethrin-biosynthesis-the-cytochrome-p450-oxidoreductase-pyrethrin-biosynthesis.jpg)























