Embed Size (px)
Citation preview

1
Amyotrophic Lateral Sclerosis Mutant VAPB Causes a Nuclear Envelope Defect 1
2
Duvinh Tran, Antonious Chalhoub, Allana Schooley, Wendy Zhang and Johnny K. Ngsee 3
4
Neuroscience, Ottawa Hospital Research Institute, Cellular and Molecular Medicine, 5
University of Ottawa, 451 Smyth Road, Ottawa, Ontario, K1H 8M5, Canada 6
7
Corresponding Author: Johnny K. Ngsee 8
TEL (613) 562-5800 x 2251 9
E-MAIL [email protected] 10
11
12
13
Key words: Amyotrophic Lateral Sclerosis, VAPB, Nuclear envelope, gp210, Nup214, 14
Emerin, ERGIC 15
Running title: VAPB Nuclear Envelope Defect 16
17
Abbreviations List 18
ALS Amyotrophic Lateral Sclerosis 19
EMD Emerin 20
ER endoplasmic reticulum 21
ERGIC ER-Golgi intermediate compartment 22
FFAT two phenylalanines on an acidic track 23
INM inner nuclear membrane 24
NE nuclear envelope 25
NPC nuclear pore complex 26
Nup nucleoporin 27
ONM outer nuclear membrane 28
29
30
© 2012. Published by The Company of Biologists Ltd.Jo
urna
l of C
ell S
cien
ceA
ccep
ted
man
uscr
ipt
JCS online publication date 27 March 2012

2
Summary 31
A proline to serine substitution (P56S) in VAPB causes an autosomal dominant form of 32
Amyotrophic Lateral Sclerosis (ALS). We show that the mutation also causes a nuclear envelope 33
(NE) defect. Transport of Nucleoporins (Nups) and Emerin (EMD) to the NE is blocked, 34
resulting in their sequestration in dilated cytoplasmic membranes. Simultaneous overexpression 35
of the FFAT motif (two phenylalanines on an acidic track) antagonizes this mutant VAPB effect 36
and restores transport to the NE. VAPB function is required for transport to the NE with 37
knockdown of endogenous VAPB recapitulating this phenotype. Moreover, we identified this 38
compartment as ER-Golgi intermediate compartment (ERGIC) with NE membrane proteins 39
transiting to ERGIC before VAPB-dependent retrograde transport to the NE. 40
41
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

3
Introduction 42
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease caused 43
by death of motor neurons. Familial ALS8 is caused by a mutation in the VAPB gene 44
(Nishimura et al., 2004; Chen et al., 2010). Overexpresion of the P56S mutation leads to 45
formation of large ER-derived membranes (Nishimura et al., 2004; Kanekura et al., 2006; 46
Teuling et al., 2007; Prosser et al., 2008; Suzuki et al., 2009). VAPB is an integral membrane 47
protein with an N-terminal Major Sperm Protein (MSP) domain and a C-terminal transmembrane 48
domain. The cytoplasmic MSP domain interacts with the FFAT motif (two phenylalanine in an 49
acidic track) (Loewen et al., 2003) found in oxysterol-binding protein (OSBP or ORP) family 50
(Wyles et al., 2002), NIR phospholipid transfer proteins (Lev, 2004), and ceramide transport 51
protein (Kawano et al., 2006). Coordinated membrane recruitment of these proteins by VAPs is 52
thought to regulate lipid composition at membrane contact sites that in turn affects organelle 53
morphology (Amarilio et al., 2005; Peretti et al., 2008). Substitution of Pro56 with Ser exposes a 54
hydrophobic patch (Furuita et al., 2010; Kim et al., 2010) that renders the protein highly prone to 55
aggregation (Kanekura et al., 2006). We have previously shown that co-overexpression of a 56
FFAT-containing fragment but not when the two Phe were substituted with Ala (named AAAT) 57
resolved the abnormal ER morphology induced by mutant VAPB (Prosser et al., 2008). Here we 58
report that the mutation also causes a NE defect characterized by separation of the outer (ONM) 59
and inner nuclear membrane (INM). This defect is caused by disruption of transport of NE 60
proteins as loss of VAPB led to their accumulation in dilated cytoplasmic foci containing 61
ERGIC-53. This suggests that NE proteins transit through ERGIC with VAPB function required 62
for final transit to the NE. 63
64 Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

4
Results and Discussions 65
VAPB-P56S Causes Swelling of the NE 66
To gain insights to the consequences of the mutant protein, we performed an electron 67
microscopic analysis on CHO overexpressing VAPB-P56S or simultaneously overexpressing 68
mutant VAPB and FFAT- or AAAT-containing fragment. Convoluted ER tubules have been 69
observed in HeLa overexpressing mutant VAPB (Teuling et al., 2007; Fasana et al., 2009), and 70
these were also evident in some cells. However, the most prominent features in cells 71
overexpressing mutant VAPB alone or together with AAAT were large cytoplasmic vacuole-like 72
structures and a dilated or “herniated” NE (Fig. 1A-C). The vacuole-like structures likely 73
correspond to dilated mutant VAPB membranes viewed under light microscopy (Kanekura et al., 74
2006; Teuling et al., 2007; Prosser et al., 2008). Interestingly, ~75% of these cells also showed a 75
prominent NE defect characterized by separation of the INM and ONM at discrete regions (Fig. 76
1A, arrows). The gap between the two membranes can be as much as 500 nm apart with a mean 77
of 160 ± 32 nm, which is over two-fold greater than the 70 ± 5 nm gap in control cells (Fig. 1D). 78
Connecting ER tubules when detected also appeared dilated (Fig. 1C, #). These ER and NE 79
defects were not observed in cells co-overexpressing mutant VAPB and the FFAT fragment, 80
consistent with the ability of this motif to neutralize the adverse effects of the mutant protein. 81
Thus, overexpression of mutant VAPB not only causes formation of aberrant cytoplasmic 82
membranes but also results in separation of the two nuclear membranes. 83
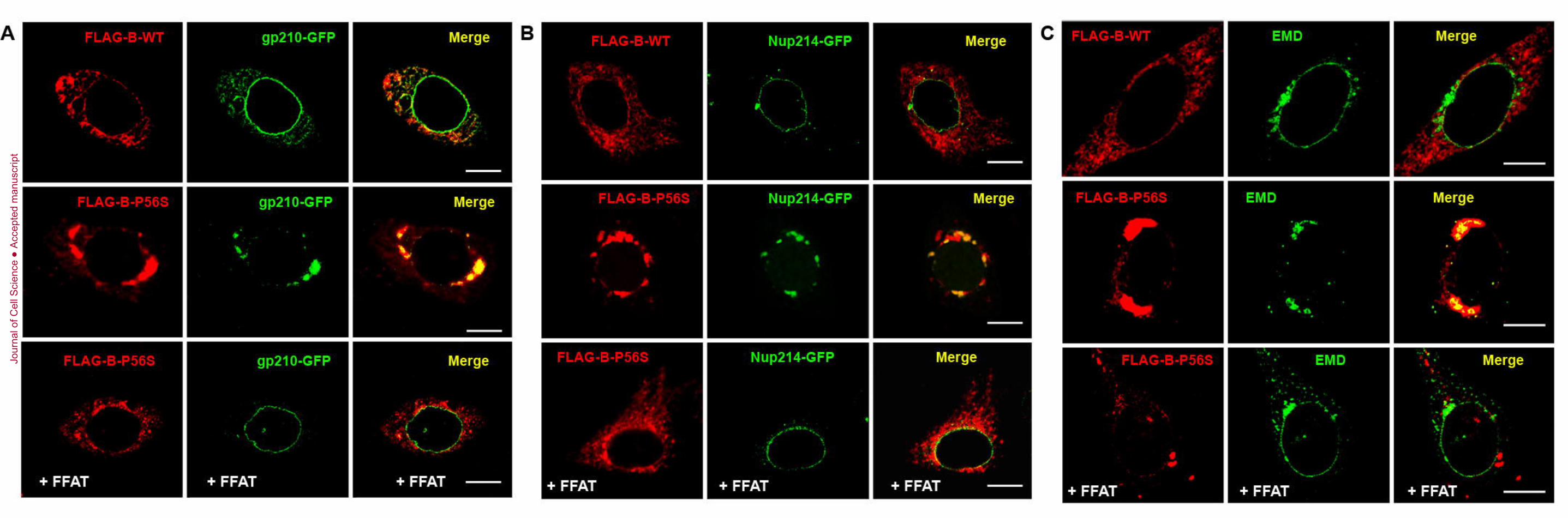
Retention of Nup and Emerin (EMD) in Mutant VAPB-Induced Dilated Membranes 84
We next examined mislocalization of Nups as a possible cause since nuclear pores 85
spanning the two nuclear membranes help maintain their close apposition. We examined two 86
Nups with vastly different residence times at the NE and membrane topology: gp210 is a highly 87
dynamic integral membrane protein of the nuclear pore complex (NPC), while Nup214 which 88
forms the structural scaffold has a NE residence time an order of magnitude higher (Daigle et al., 89
2001; Rabut et al., 2004). In empty vector and VAPB-WT transfected cells, gp210-GFP formed a 90
ring-like pattern encompassing the rim of the NE as well as localized to scattered cytoplasmic 91
puncta (Fig. 2A), consistent with previous studies. Cytoplasmic gp210-GFP co-localized 92
extensively with VAPB in tubular membranes. Its NE localization was unaffected by up to 3-fold 93
overexpression of the transgene based on fluorescence intensity measurements relative to 94
endogenous VAPB. In contrast, gp210-GFP was excluded from the NE and retained in mutant 95
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

5
VAPB-containing dilated cytoplasmic membranes (Fig. 2A). Similarly, Nup214-GFP that 96
normally localizes to the NE was sequestered in mutant VAPB-containing cytoplasmic 97
membranes, albeit not all mutant VAPB-containing membranes were Nup214-GFP positive (Fig. 98
2B). This effect was not cell line specific as HeLa showed similar Nup214 relocation defect 99
(Supplemental Fig. S1). The normal NE localization of both gp210-GFP and Nup214-GFP were 100
restored upon co-overexpression of the FFAT fragment (Fig. 2A,B), consistent with the ability of 101
this motif to resolve mutant VAPB effects. Thus, cytoplasmic retention of these Nups along with 102
aggregated mutant VAPB inhibits their transport to the NE. 103
To determine whether trafficking of other NE proteins is also affected, we examined the 104
distribution of EMD, an integral membrane protein that shuttles between the ER and INM 105
(Zuleger et al., 2011). Endogenous EMD formed a ring-like pattern along the rim of the NE in 106
both control and VAPB-WT transfected cells (Fig. 2C). It partially co-localized with VAPB-WT 107
in cytoplasmic puncta throughout the cell as well as in discreet regions adjacent to or at the NE. 108
In contrast, EMD was retained in dilated cytoplasmic membranes and excluded from the NE in 109
mutant VAPB overexpressing cells (Fig. 2C). These EMD-containing membranes were often 110
positioned adjacent to but not part of the NE. Co-overexpression of the FFAT fragment with 111
VAPB-WT had no effect on EMD distribution, but restored EMD localization to the NE in 112
mutant VAPB cells (Fig. 2C). Since EMD is retained in the INM in part by binding to A-type 113
lamins in the nuclear lamina (Vaughan et al., 2001; Ostlund et al., 2006), we examined the 114
distribution of Lamin A/C and found no change in its distribution pattern or evidence of nuclear 115
deformation (J.K.N., unpublished data). Thus, loss of EMD from the INM is likely due to 116
disruption of transport to the INM rather than loss of retention at the INM. 117
Cytoplasmic Retention is due to Loss of VAPB Function 118
Mutant VAPB is aggregation-prone and recruitment of endogenous VAPB to insoluble 119
aggregates is thought to result in a dominant negative effect (Teuling et al., 2007; Suzuki et al., 120
2009; Kim et al., 2010). To determine whether the defect is due to loss of VAPB function and to 121
exclude non-specific sequestration of NE proteins with aggregated mutant VAPB, we examined 122
their distribution upon siRNA knockdown of endogenous VAPB. Co-transfection with the empty 123
pLKO.1 vector had no effect on the distribution pattern of Nup214-GFP, whereas siVAPB 124
resulted in relocation of Nup214-GFP to cytoplasmic foci and loss of NE localization (Fig. 3A). 125
The cytoplasmic foci likely represent sites of NPC assembly since Nup214 is a soluble Nup 126
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

6
recruited to the membrane to form the core scaffold during NPC assembly (Bodoor et al., 1999). 127
This accumulation of Nup214-GFP suggests that VAPB is required for transport of pre-128
assembled NPC to the NE. It is consistent with the view that mutant VAPB acts in a dominant 129
negative manner. 130
Transport of EMD was similarly inhibited by knockdown of VAPB. Endogenous EMD 131
was relocated from the NE to large cytoplasmic puncta throughout the cell upon siVAPB (Fig. 132
3B). Thus, VAPB function is essential for transport of Nup214, EMD and possibly other 133
membrane proteins to the NE. Loss of NE localization is not simply due to inadvertent 134
sequestration of Nups with mutant VAPB aggregates but a consequence of loss of VAPB 135
function. 136
VAPB Affects ERGIC Trafficking 137
To identify this VAPB-containing intracellular compartment, we examined its co-138
localization with known organelle markers. Only cells with low level of expression of transfected 139
VAPB-WT were examined to avoid potential overexpression artifacts. We found VAPB-WT co-140
localized extensively with GFP-ERGIC-53 cytoplasmic puncta (Fig. 4A). The perinuclear Golgi 141
ribbons were devoid of VAPB, suggesting VAPB resides primarily at ERGIC. When subjected 142
to intensity correlation analysis, excluding the Golgi ribbons, VAPB and ERGIC-53 showed a 143
mean Pearson’s correlation and Mander’s overlap coefficients of 0.73 ± 0.01 and 0.79 ± 0.02 144
(from 3 replicates of 10-17 cells), respectively. These high coefficients suggest co-localization 145
with recycling ERGIC-53. To determine whether VAPB affects trafficking through ERGIC, we 146
examined the distribution of endogenous ERGIC-53 upon siVAPB knockdown. ERGIC-53 147
relocated from the Golgi to expanded membranes in 45.6% ± 0.9% (n=3) of siVAPB cells (Fig. 148
4B) compared to 4.9% ± 1.9% in pLKO.1 control, indicating that relocation was not an artifact 149
of ectopic ERGIC-53 expression. Quantitation of individual peripheral cytoplasmic puncta 150
indicated a shift towards larger ERGIC-53 puncta in the knockdown cells (Fig. 4C). This 151
indicates that VAPB is essential in maintaining ERGIC morphology and retrograde trafficking of 152
ERGIC-53. To verify that Nups are retained in ERGIC in a VAPB-dependent manner, we stained 153
the knockdown cells with mAb414, a monoclonal anybody that recognizes several FG repeat 154
Nups (Davis and Blobel, 1986). These Nups were normally localized to the rim of the NE and in 155
small cytoplasmic puncta in control cells, but were retained in expanded GFP-ERGIC-53-156
containing membranes upon siVAPB (Fig. 4C). Distribution of EMD was similarly affected, 157
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

7
relocating to the expanded ERGIC along with GFP-ERGIC-53 in siVAPB cells (Fig. 4D). 158
Quantitation of mAb414 and EMD cytoplasmic puncta excluding the NE also indicated a shift 159
towards larger puncta in the knockdown cells (Fig. 4C). Together, this indicates that NE and 160
transmembrane Nups transit through ERGIC, and VAPB is required for final transport to the NE. 161
Our study shows that overexpression of mutant VAPB not only causes ER abnormalities 162
but also separation of the ONM from the INM at discreet regions of the NE. Interestingly, this 163
abnormality is similar to that caused by disruption of gp210 (Drummond and Wilson, 2002), 164
which leads to formation of nuclear pore intermediates that fail to dilate into functional pores. 165
This suggests that positioning of fully assembled pores at the junction of the two nuclear 166
membranes structurally contributes to their close apposition. Cytoplasmic retention of certain 167
Nups and membrane proteins upon siRNA knockdown clearly indicates that VAPB plays an 168
essential role in their transport to the NE. This disruption in transport will likely affect the 169
overall composition of NPC and compromise the structural integrity as well as functional 170
properties of the pores. Interestingly, bulk anterograde ER-to-Golgi transport of VSVG remains 171
largely unaffected upon siVAPB knockdown (Fig. S2), suggesting that VAPB is primarily 172
involved in regulating retrograde transport through ERGIC. 173
Co-overexpression of the FFAT fragment clearly counteracts the adverse effects of 174
mutant VAPB. We propose that interaction with the FFAT fragment might interfere with mutant 175
VAPB aggregate formation. The P56S mutation does not directly affect FFAT binding nor does 176
it result in complete loss of function since the mutant protein is able to rescue the Drosophila 177
deletion phenotype (Chai et al., 2008). Its susceptibility to form insoluble aggregates may recruit 178
endogenous VAPB (Suzuki et al., 2009), and block access to the FFAT binding site as the 179
aggregates increase in size (Kim et al., 2010). This not only generates a dominant negative effect 180
but possibly additional toxic properties. Binding to the FFAT fragment may impart 181
conformational changes that reduce aggregate formation, allowing partial restoration of wild-182
type functions and averting aggregate toxicity. 183
Cytoplasmic retention of Nups and EMD in mutant VAPB-containing membranes is not 184
due to inadvertent sequestration with insoluble aggregates since siRNA knockdown of 185
endogenous VAPB also results in their cytoplasmic retention. Given that wild-type VAPB co-186
localizes with recycling ERGIC-53, and siVAPB knockdown results in expansion and retention 187
of Nups and EMD with ERGIC-53, it suggests that these NE membrane proteins do not reach the 188
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

8
NE by lateral diffusion through the interconnecting ER and ONM network, but transit through 189
ERGIC. Progression from these ERGIC foci is clearly dependent on VAPB with its loss of 190
function either through siRNA knockdown or dominant negative effect of mutant VAPB 191
overexpression inhibiting their exit and consequently expansion of ERGIC. While the 192
mechanism by which VAPB facilitates this retrograde transport step remains to be determined, 193
progressive deterioration of the NE is a predictable consequence of disrupting this transport step 194
and could contribute to age-dependent onset of the disease. 195
196
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

9
Materials and Methods 197
Expression Plasmids, Cell Culture and Transfection 198
FLAG-tagged VAPB and Myc-tagged FFAT constructs were described previously 199
(Prosser et al., 2008). Gp210-GFP and Nup214-GFP were from EUROSCARF. Chinese Hamster 200
Ovary (CHO-K1) and HeLa were maintained at 37°C in MEMα and DMEM (Invitrogen, 201
Carlsbad, CA, USA), respectively, and supplemented with 100 units/ml penicillin, 100 µg/ml 202
streptomycin and 10% fetal bovine serum (FBS). Cells were transfected with LipofectAMINE 203
(Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. 204
Immunofluorescence and Electron Microscopy 205
Transfected cells were fixed with 4% paraformaldehyde in phosphate-buffered saline 206
(PBS) for 30 min at room temperature. After neutralization with PBS-glycine (PBS with 100 207
mM glycine), cells were permeabilized in blocking buffer (PBS, 1% bovine serum albumin, 2% 208
normal goat serum, 0.4% saponin and 0.1% Triton X-100). Primary and secondary antibodies 209
were diluted in blocking buffer and incubated for at least 1h. Cover slips were mounted with 210
SlowFade Gold with or without DAPI (Invitrogen Carlsbad, CA, USA). Primary antibodies used 211
included anti-FLAG (Applied Biological Materials, Richmond, BC, Canada), mAb414 212
(Covance, Princeton, NJ, USA), ERGIC-53 (Sigma-Aldrich, St. Louis, MO, USA), and EMD 213
(MANEM1, Developmental Studies Hybridoma Bank, Iowa City, IA, USA). Secondary 214
antibodies were conjugated with Alexa 488 or 594 (Invitrogen, Carlsbad, CA, USA). 215
Images were captured on a LSM 510confocal microscope with a 1.4 numerical aperture 216
63X oil-immersion objective and processed with Image J (NIH, Bethesda, MD, USA). ERGIC-217
53, mAb414 and EMD cytoplasmic particles sizes excluding the perinuclear Golgi ribbons and 218
NE were measured with particle analysis plug-in. Over 10 cells were chosen from each group 219
and from 3 replicates. Maximal Feret’s diameter of individual peripheral puncta (>200 per cell) 220
was grouped into 100 nm bins. Unpaired two-tailed Student’s t-test was used to determine 221
statistical significance. 222
For electron microscopy, cells were fixed with 1.6% glutaraldehyde in 0.1 M sodium 223
cacodylate buffer pH 7.2 for 2-6 h. After contrasting with 1% osmium tetroxide and dehydration 224
in increasing concentrations of ethanol, cells were embedded in Spurr’s low viscosity epoxy 225
(Polysciences, Warrington, PA, USA). Resulting ultra-thin sections were stained with 5% Uranyl 226
Acetate for 15 min and Reynold’s Lead Citrate Solution for 5 min. Digital images were taken 227
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

10
with a JEOL 1230 transmission electron microscope at 60kV adapted with a 2000 x 2000 pixel 228
bottom mount Hamamatsu CCD digital camera. 229
Lentiviral pLKO.1 based plasmids were from Open Biosystems (Hunstville, AL, USA). 230
TRCN0000153862 and TRCN0000152888 matched both human and mouse VAPB sequences. 231
Empty pLKO.1 vector or eGFP shRNA were used as controls. In some cases, co-transfection 232
with monomeric RFP (mRFP) was used to identify the transfected cells. Cells were processed 233
48h post-transfection. 234
235
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

11
Acknowledgements 236
This work was supported by an operating grant from the Canadian Institutes of Health Research. 237
A.C. was supported by NSERC of Canada. We thank Ms. Kalina Abrol for her technical support. 238
239
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

12
Figure Legends 240
Figure 1. Overexpression of VAPB-P56S causes NE defect. (A) Large vacuole-like membrane 241
structures (*) and swelling of nuclear envelope (arrows) in mutant VAPB transfected CHO cells 242
after 48h. (B) Magnified image of the NE defect with some containing membrane vesicles within 243
the perinuclear space (arrowhead). (C) Magnified image of a dilated interconnecting ER tubule 244
(#). Scale bar, 500 nm. (D) Measurement of the gap between ONM and INM in control and 245
mutant VAPB transfected cells. 246
Figure 2. NE proteins are retained in mutant VAPB-containing membranes. CHO cells were co-247
transfected with FLAG-tagged VAPB (WT or P56S) and gp210-GFP (A) or Nup214-GFP (B). 248
FFAT was co-transfected with mutant VAPB in the bottom panels. (C) Cells were transfected 249
with FLAG-VAPB-WT or P56S and stained with anti-FLAG (red) and anti-EMD (green). FFAT 250
was co-transfected with mutant VAPB in the bottom panel. Scale bar, 10 µm. All images are 251
representative of at least 90% of transfected cells from three replicates. 252
Figure 3. Cytoplasmic retention of Nup214-GFP and EMD upon siVAPB knockdown. (A) HeLa 253
was co-transfected with Nup214-GFP and Lentiviral constructs (empty pLKO.1 or siVAPB 254
152888) and mounted with DAPI. (B) Endogenous EMD distribution in cells co-transfected with 255
mRFP and empty pLKO.1 or siVAPB. Scale bar, 10 µm. 256
Figure 4. VAPB is localized to the ERGIC. (A) HeLa was transfected with VAPB-WT and GFP-257
ERGIC-53. Only low expressing cells were analyzed. (B) Distribution of endogenous ERGIC-53 258
in cells co-transfected with empty pLKO.1 or siVAPB and mRFP. (C) Distribution histograms of 259
maximal Feret’s diameter of the cytoplasmic puncta in 100 nm bins for ERGIC-53, mAb414 and 260
EMD. Asterisks (*) indicate p<0.05. (D) Cells were transfected as in (B), but further stained with 261
mAb414 antibodies or with anti-EMD antibodies (E). Scale bar, 10 µm. 262
Supplemental Figures 263
Figure S1. Cellular distribution of Nup214 in HeLa. HeLa cells were co-transfected with FLAG-264
VAPB (WT or P56S), and Nup214-GFP. Cells were fixed after 48h and stained with anti-FLAG 265
antibodies. Scale bar, 10 µm. 266
Figure S2. Transport of anterograde VSVGts042-GFP in siVAPB knockdown cells. (A) HeLa cells 267
were co-transfected with pLKO.1 empty vector or siVAPB and VSVGts042-GFP. VSVGts042-GFP 268
was trapped in the ER at 42ºC for 5h, and shifted to 32ºC for the time indicated before fixation. 269
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

13
(B) Fraction of cells with Golgi-localized VSVGts042-GFP after release from 42ºC. Error bars 270
represent s.e.m. (n=3). 271
272
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

14
References 273
Amarilio, R., Ramachandran, S., Sabanay, H. and Lev, S. (2005). Differential regulation of 274 endoplasmic reticulum structure through VAP-Nir protein interaction. J. Biol. Chem. 280, 5934-275 5944. 276 Bodoor, K., Shaikh, S., Salina, D., Raharjo, W. H., Bastos, R., Lohka, M. and Burke, B. 277 (1999). Sequential recruitment of NPC proteins to the nuclear periphery at the end of mitosis. J. 278 Cell Sci. 112, 2253-2264. 279 Chai, A., Withers, J., Koh, Y. H., Parry, K., Bao, H., Zhang, B., Budnik, V. and Pennetta, 280 G. (2008). hVAPB, the causative gene of a heterogeneous group of motor neuron diseases in 281 humans, is functionally interchangeable with its Drosophila homologue DVAP-33A at the 282 neuromuscular junction. Hum. Mol. Genet. 17, 266-280. 283 Chen, H.-J., Anagnostou, G., Chai, A., Withers, J., Morris, A., Adhikaree, J., Pennetta, G. 284 and de Belleroche, J. S. (2010). Characterisation of the properties of a novel mutation in VAPB 285 in familial ALS. J. Biol. Chem. 285, 40266-40281. 286 Daigle, N., Beaudouin, J., Hartnell, L., Imreh, G., Hallberg, E., Lippincott-Schwartz, J. and 287 Ellenberg, J. (2001). Nuclear pore complexes form immobile networks and have a very low 288 turnover in live mammalian cells. J. Cell Biol. 154, 71-84. 289 Davis, L. I. and Blobel, G. (1986). Identification and characterization of a nuclear pore complex 290 protein. Cell 45, 699-709. 291 Drummond, S. P. and Wilson, K. L. (2002). Interference with the cytoplasmic tail of gp210 292 disrupts "close apposition" of nuclear membranes and blocks nuclear pore dilation. J. Cell Biol. 293 158, 53-62. 294 Fasana, E., Fossati, M., Ruggiano, A., Brambillasca, S., Hoogenraad, C. C., Navone, F., 295 Francolini, M. and Borgese, N. (2009). A VAPB mutant linked to amyotrophic lateral sclerosis 296 generates a novel form of organized smooth endoplasmic reticulum. Faseb J. 24, 1419-1430. 297 Furuita, K., Jee, J., Fukada, H., Mishima, M. and Kojima, C. (2010). Electrostatic interaction 298 between oxysterol-binding protein and VAMP-associated protein A revealed by NMR and 299 mutagenesis studies. J. Biol. Chem. 285, 12961-12970. 300 Kanekura, K., Nishimoto, I., Aiso, S. and Matsuoka, M. (2006). Characterization of 301 Amyotrophic Lateral Sclerosis-linked P56S Mutation of Vesicle-associated Membrane Protein-302 associated Protein B (VAPB/ALS8). J. Biol. Chem. 281, 30223-30233. 303 Kawano, M., Kumagai, K., Nishijima, M. and Hanada, K. (2006). Efficient Trafficking of 304 Ceramide from the Endoplasmic Reticulum to the Golgi Apparatus Requires a VAMP-associated 305 Protein-interacting FFAT Motif of CERT. J. Biol. Chem. 281, 30279-30288. 306 Kim, S., Leal, S. S., Ben Halevy, D., Gomes, C. M. and Lev, S. (2010). Structural 307 requirements for VAP-B oligomerization and their implication in amyotrophic lateral sclerosis-308 associated VAP-B(P56S) neurotoxicity. J. Biol. Chem. 285, 13839-13849. 309 Lev, S. (2004). The role of the Nir/rdgB protein family in membrane trafficking and 310 cytoskeleton remodeling. Exp. Cell Res. 297, 1-10. 311 Loewen, C. J., Roy, A. and Levine, T. P. (2003). A conserved ER targeting motif in three 312 families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 22, 2025-2035. 313 Nishimura, A. L., Mitne-Neto, M., Silva, H. C., Richieri-Costa, A., Middleton, S., Cascio, 314 D., Kok, F., Oliveira, J. R., Gillingwater, T., Webb, J. et al. (2004). A mutation in the vesicle-315 trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral 316 sclerosis. Am. J. Hum. Genet. 75, 822-831. 317
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

15
Ostlund, C., Sullivan, T., Stewart, C. L. and Worman, H. J. (2006). Dependence of 318 diffusional mobility of integral inner nuclear membrane proteins on A-type lamins. Biochemistry 319 45, 1374-1382. 320 Peretti, D., Dahan, N., Shimoni, E., Hirschberg, K. and Lev, S. (2008). Coordinated lipid 321 transfer between the endoplasmic reticulum and the Golgi complex requires the VAP proteins 322 and is essential for Golgi-mediated transport. Mol. Biol. Cell 19, 3871-3884. 323 Prosser, D. C., Tran, D., Gougeon, P. Y., Verly, C. and Ngsee, J. K. (2008). FFAT rescues 324 VAPA-mediated inhibition of ER-to-Golgi transport and VAPB-mediated ER aggregation. J. 325 Cell Sci. 121, 3052-3061. 326 Rabut, G., Doye, V. and Ellenberg, J. (2004). Mapping the dynamic organization of the 327 nuclear pore complex inside single living cells. Nat. Cell Biol. 6, 1114-1121. 328 Suzuki, H., Kanekura, K., Levine, T. P., Kohno, K., Olkkonen, V. M., Aiso, S. and 329 Matsuoka, M. (2009). ALS-linked P56S-VAPB, an aggregated loss-of-function mutant of 330 VAPB, predisposes motor neurons to ER stress-related death by inducing aggregation of co-331 expressed wild-type VAPB. J. Neurochem. 108, 973-985. 332 Teuling, E., Ahmed, S., Haasdijk, E., Demmers, J., Steinmetz, M. O., Akhmanova, A., 333 Jaarsma, D. and Hoogenraad, C. C. (2007). Motor neuron disease-associated mutant vesicle-334 associated membrane protein-associated protein (VAP) B recruits wild-type VAPs into 335 endoplasmic reticulum-derived tubular aggregates. J. Neurosci. 27, 9801-9815. 336 Vaughan, A., Alvarez-Reyes, M., Bridger, J. M., Broers, J. L., Ramaekers, F. C., Wehnert, 337 M., Morris, G. E., Whitfield, W. G. F. and Hutchison, C. J. (2001). Both emerin and lamin C 338 depend on lamin A for localization at the nuclear envelope. J. Cell Sci. 114, 2577-2590. 339 Wyles, J. P., McMaster, C. R. and Ridgway, N. D. (2002). Vesicle-associated membrane 340 protein-associated protein-A (VAP-A) interacts with the oxysterol-binding protein to modify 341 export from the endoplasmic reticulum. J. Biol. Chem. 277, 29908-29918. 342 Zuleger, N., Kelly, D. A., Richardson, A. C., Kerr, A. R. W., Goldberg, M. W., Goryachev, 343 A. B. and Schirmer, E. C. (2011). System analysis shows distinct mechanisms and common 344 principles of nuclear envelope protein dynamics. J. Cell Biol. 193, 109-123. 345 346 347 348
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t

Jour
nal o
f Cel
l Sci
ence
Acc
epte
d m
anus
crip
t
















![LONO1 Encoding a Nucleoporin Is Required for ......LONO1 Encoding a Nucleoporin Is Required for Embryogenesis and Seed Viability in Arabidopsis1[C][W][OA] Christopher Braud2, Wenguang](https://img.pdfslide.net/doc/110x75/60f86cb4c550140b5d3d1bda/lono1-encoding-a-nucleoporin-is-required-for-lono1-encoding-a-nucleoporin.jpg)



![LONO1 Encoding a Nucleoporin Is Required for Embryogenesis … · LONO1 Encoding a Nucleoporin Is Required for Embryogenesis and Seed Viability in Arabidopsis1[C][W][OA] Christopher](https://img.pdfslide.net/doc/110x75/5f33c74a6e74b45879570c2c/lono1-encoding-a-nucleoporin-is-required-for-embryogenesis-lono1-encoding-a-nucleoporin.jpg)








