Embed Size (px)
Citation preview

Nyugat-magyarországi Egyetem Kémiai Intézet
KÉMIA 2
gyakorlati jegyzet
Sopron 2014

1
Nedvesség tartalom meghatározása A fában lévő nedvességtartalom a fakomponensekhez való kötődés energiájától függően több csoportra osztható. Megkülönböztetnek: 1. Szerkezeti vizet, amely kémiai kötéssel /hidrogénhíd/ kapcsolódik a fához. 2. A szénhidrátok felületén adszorpciós úton megkötött 3. Az intermicelláris térben kapillárisan kondenzált vizet. Az utóbbi a sejtfalakat itatja át, részben a cellulózlánc orientált részei közötti teret tölti ki, részben kolloidálisan kötődik a fa anyagához. A kettő között fokozatos átmenet van ezért együttesen higroszkópos nedvességnek is nevezik. 4. Szabad nedvesség, amely a fában lévő szabad üregeket tölti ki. Az a nedvességtartalom, amely a sejtfalak teljes telítettségének felel meg nevezzük higroszkópossági határnak, ez a farost telítettségi pontja. A fa nedvességtartalma a faanyag jellemzőitől, porozitásától mint belső tényezőktől, a levegő páratartalmától, a hőmérséklettől mint külső tényezőktől függ. A külső tényezők által meghatározott körülmények között a fa nedvességtartalma egy meghatározott, egyensúlyi állapotra áll be, az a fa higroszkópos pontja. A fa nedvességtartalmára jellemző következő fogalmakat szokták megadni: 1. Az abszolút száraz fa (105 oC-on súlyállandóságig szárított fa) 2. légszáraz fa (mintegy 15-20 % víztartalommal az abszolút száraz fára vonatkoztatva. Ezt a nedvességtartalmat a külső tényezők határozzák meg. 3. Frissen kivágott fa (60-100%) víztartalommal) 4. Nedves fa (vízben való leghosszabb áztatás után, 150% vagy több víz) Mint látható, a fában lévő összes vízmennyiség elsősorban a szabad nedvességtartalomtól függ, mivel a higroszkópos nedvesség értéke kevéssé változik. A nedvességtartalom meghatározása azért szükséges, mivel a feldolgozás meghatározott nedvességtartalmú fát igényel. A kémiai elemzés szempontjából pedig fontos, mert az elemzési adatokat egy összehasonlító értékre, az abszolút száraz fára kell vonatkoztatni. A meghatározások közül a legelterjedtebb a 100-105 oC-on súlyállandóságig való szárítás. A viszonylag magas hőmérsékleten azonban a vízzel együtt más illó anyagok is eltávozhatnak (éteres olajok). Másrészt a fa hosszas szárítását oxidációs folyamatok is kísérhetik, mikor a levegő oxigénje egyesül a fa anyagával, ami súlygyarapodást idézhet elő. A, Nedvességtartalom meghatározása 105 C-on súlyállandóságig történő szárítással A szárítószekrényben 105 ± 2 oC-on kiszárított és exszikkátorban lehűtött mérőedényt és a mérőedény fedelét (M) analitikai mérlegen lemérünk, majd a felaprított mintából 5-10 g-ot bemérünk és ismét lemérjük a mérőedényt fedéllel együtt (A). Ezután a mérőedényt szárítószekrénybe helyezzük, fedelét levesszük és szintén a szárítószekrénybe helyezzük a mérőedény mellé. 105 ± 2 oC-on súlyállandóságig szárítjuk. Ekkor a mérőedényre ráhelyezzük a fedelét és lemérjük a tömegét ismét (B). A súlyállandóságot akkor értük el,

2
amikor két egymásutáni szárítást követő mérés eredménye között és az eltérés 0.1%-nál kisebb. Az első szárítás időtartama 2 óra, a továbbiaké fél-fél óra. A mérőedényre minden szárítás után még a szárítószekrénybe rá kell helyezni a fedelet és így exszikkátorban lehűteni. A %-os nedvességtartalmat a következőképpen számoljuk:
Ahol M: mérőedény tömege. A: bemért minta és a mérőedény együttes tömege, B: kiszárított
minta és a bemérő edény együttes tömege. A térfogatos nedvesség meghatározási módszereknél a víznek nem elegyedő szerves oldószerekkel történő desztillációján és a desztillált víz térfogatának közvetlen mérésén alapszik. Az ilyen oldószerek a toluol, xilol. Előnye, ezeknek a módszereknek, hogy a víz eltávolítása teljesebben megy végbe idegen anyagok hozzáelegyedését kiküszöböli mérlegelést kiküszöböli megakadályozza az esetleges oxidációt. B, Nedvességtartalom meghatározása xilolos desztillációval 300 ml-es gömblombikba bemérünk 2-10 g közepesen aprított vizsgálandó famintát, 0.01 g pontossággal. A mintához 150 ml vízzel telített xilol adunk. A lombikra Marcusson feltétet és visszafolyós hűtőt szerelünk és az elegyet desztillálni kezdjük. A desztillációt addig folytatjuk, amíg a feltét beosztásos részén leolvasható, a xiloltól elkülönített víz meniszkusza nem emelkedik, tehát csak tiszta xilolt desztillál át és a két folyadékréteg éled határvonallal elválik. Ez rendszerint 1 óra múlva következik be. Ezután a rendszert hűlni hagyjuk míg a folyadék a környezet hőmérsékletére hűl és a két réteg teljesen letisztul, majd az átdesztillált víz mennyiségét a kalibrált csőben 0.05 cm3 pontossággal a beosztás segítségével leolvassuk és a bemért fa súlyára vonatkoztatva százalékosan megadjuk.
Ahol a : a: ml-ben az átdesztillált víz, G: g-ban a bemért fa tömege.
A fa oldhatósága Extrakttartalom meghatározása. A fa elemzése során az egyes alkotórészek vagy közvetlen kinyeréssel, vagy komponensek, reakciótermékek meghatározásával adják meg. Az extraktanyagokat viszont egyes vegyületcsoportok alakjában határozzuk meg. Így megkülönböztetünk: A, hideg vízben oldható részt. B, forró vízben oldható részt. C, szerves oldószerekkel extrahálható részt. Az utóbbit egyes esetekben felosztják még: 1. éterben extrahálható és 2. alkohol-benzolban extrahálható részre.

3
Egyes elemzők javasolják még az 1%-os NaOH-ban oldott rész vizsgálatát is. Az 1%-os nátronlúgoldattal való kezelés azonban a lignin és a cellulóz meghatározást teszi pontatlanná. Az ilyen összesített extrakciók arról adnak képet, hogy az adott fafajra az extraktanyagok mely típusai jellemzőek. A hideg vízben elsősorban a fa ásványi anyagai, valamint az egyes festékanyagok oldódnak, némileg oldódnak azonban egyes szerves komponensek is.
A forró vízben oldható anyagok közé tartoznak az ásványi sók, cserzőanyagok, cukrok, keményítők, pektinek egyes hemicellulózok és festékek. A forró vizes extrakciónál bizonyos fokú hidrolízis is felléphet, melyez az extrakció folyamán a hemicellulózokból képződő savak is katalizálhatnak. A forró vízben oldható rész a fa faján kívül az életkorától és a kivágás utáni tárolási időtől függ elsősorban. Mennyisége 0.3%-tól 15-%-ig terjedhet.
A szerves oldószerekkel oldató rész a gyanta- és zsírsavakat, viaszokat, éterikus olajokat, fitoszterineket tartalmazza. Jellemző rájuk, hogy a fában igen egyenlőtlenül oszlanak el. Mennyiségük az évszakkal is igen változó. Levegőn oxidálódnak és oldószerekben oldhatatlan alakba mennek át. Az extrakcióhoz leggyakrabban étert alkalmaznak, mely a fenti komponenseket mind jól oldja. Az éter ezen kívül a vízzel való bizonyos fokú elegyedése folytán jól behatol a fába, nedvesíti azt, így az oldás tökéletesebb. Az alkohol a zsíroknak és viaszoknak nem jó oldószere, a benzol illetve a toluol viszont jól oldja azokat, ellenben vízben oldhatatlan, ezért nem tud behatolni a vizet tartalmazó sejtfalakba. Ezért általában a kettő elegyét használják meghatározott arányban (Alkohol:benzol 1:1 vagy 1:3). Benzol illetve toluol helyett az egészségre egyáltalán nem veszélyes ciklohexánt is lehet alkalmazni. Az extrahálást az extraháló készülékben, pl. Soxhlet-készülékben szokták végrehajtani. A kívánt anyagok mennyiségét pedig, vagy a bemért minta súlycsökkenéséből, vagy az extraktum lepárlásával nyert száraz maradékból határozzák meg. Az extrahált anyagoknak így meghatározott értéke nem tökéletes, mert a szárítás közben extrahált anyag távozhat el, az oldószer nyomai visszamaradhatnak, valamint az extrahált anyagok szárítás közben, diszkrepálódhatnak, oxidálódhatnak. Az 1%-os lúgoldattal való kezelés a fa ún. híg lúgoldatban oldható anyagait oldja ki. Oldatba a hemicellulózok, a cellulóz lebontott része és részben a lignin megy. – Meg kell itt jegyezni, hogy a NaOH-ban való oldhatóság a NaOH koncentrációjának függvénye. Az oldhatóság maximuma 4%-os töménység mellett van. A, Hideg vízben oldható rész meghatározása. A hideg vízben oldható rész meghatározásához 2 g légszáraz fát 300 ml desztillált vízzel keverés mellett 25 oC-on kezelik 48 órán keresztül. A szűrés és 105 oC-on való szárítás után a faanyagot zárt edénybe mérik. A bekövetkezett súlycsökkenésből következtetnek a vízoldható részre. B, Forró vízzel extrahálható anyagok meghatározása. A forró vízzel extrahálható anyagok mennyiségét úgy határozzák meg, hogy 2 g fát 100 ml vízzel forrásban lévő vízfürdőn visszafolyós hűtő mellett 3 órán keresztül kezelik. A maradékot addig mossák, míg a szűrlet színtelenné nem válik. Az oldatba ment anyagok mennyiségét a leszárított faminta súlycsökkenéséből határozzák meg. Cserzőanyagok vizes kioldását némileg másképp végzik. Ez esetben a víz hőmérséklete nem éri el általában a 100 oC-ot, Bővebben lásd: Tannintartalom meghatározása

4
C, Szerves oldószerekben extrahálható rész meghatározása. (1:1 ciklohexán-etanol kivonat készítése Soxhlet készülékkel) 3 g légszáraz átdesztillált fareszeléket bemérünk extrahálóhüvelybe, s ezt kevés vattacsomóval befedjük. A hüvelyt Soxhlet készülék hengeres részébe tesszük. A hüvely belső pereme a szivornyacső legmagasabb része alatt legyen. A készülék száraz lombikjába 100 ml oldószert – alkohol:ciklohexán 1:1 arányú elegyét- öntjük. A lombikot melegítjük és az extrahálást mindaddig folytatjuk, amíg a szivornyán lefolyó oldószer színtelen lesz. (Egyes előírások 6 órás extrakciót, illetve 20-24 extrakciós ciklust javasolnak) Az extrahálás befejeztével az extraktum oldószer-tartalmát rotációs bepárló segítségével eltávolítjuk. A bepárlási maradékot tartalmazó lombikot szárítószekrényben fél óráig szárítjuk exszikkátorban hűlni hagyjuk és mérjük. Az extrakttartalmat a száraz maradékból számítjuk abszolút száraz fára vonatkoztatva.
Ahol L1: lombik üres tömege, L2: extraktanyagot tartalmazó, kiszárított lombik tömege, M:
bemért fa tömege, N: fa nedvességtartalma %-ban.
A Soxhlet-berendezés vázlata.
A rotációs bepárló vázlata.
1: Forrkő (szükség esetén) 2: Oldószer illetve az extraktum 3: Gőzáramlás 4: Soxhlet-hüvely 5: Extrahált anyag 6: Szívornyacső - bemenet 7: Szívornyacső - kimenet 8: Csiszolat 9: Hűtő 10: Hűtővíz be 11: Hűtővíz ki

5
Cellulóz-tartalom meghatározás A cellulóz β-D-glükóz egységekből felépülő poliszacharid, melynek polimerizációs foka 200-nál magasabb. A fák sejtfalaiban a cellulóz porózus folytonos szövetet képet, melyben az egymással közlekedő, igen finom ultramikroszkópikus üregek találhatók. Ezekben az üregekben foglalnak helyet a hemicellulózok, melyek öt és hat szénatomot tartalmazó polimer szénhidrátok. A sejtfal anyaga – cellulóz – és a hemicellulóz között nagy a genetikai hasonlóság, ezért újabban a holocellulóz fogalmát is bevezették, mely a fa teljes szénhidrát részét tartalmazza: (cellulóz, pentozánok, hexozánok és poliuronsavak). Nyers cellulóztartalomnak nevezzük a nyersanyagoknak azt a részét, mely a ligninnek kémiai úton történő eltávolítása, valamint a kísérleti körülmények között kioldódó hemicellulózok után visszamarad. A cellulóztartalom meghatározására alapvetően kétféle módszer ismeretes. Az egyik módszer szerint a fából a lignint és a hemicellulózokat eltávolítják. Az eltávolítás a natív lignin könnyű oxidálhatóságán és a hemicellulózok viszonylag könnyű hidrolizálhatóságán alapszik. Hátránya a módszernek, hogy a legenyhébb kezelés mellett is lebomlik a cellulóz egy része. A másik módszer szerint először a holocellulózt nyerik ki, mely a lignin klórozásával és a klórozott termékek kioldásával válik lehetővé. Ha a kioldásra forró nátriumszulfit oldatot használnak, a lehidrolizált hemicellulóz is kioldódik. A, Kürschner-Hoffer féle eljárás A Kürschner-Hoffer-féle cellulóz meghatározásnál a fát salétromsav-etanol elegyével kezelik. A ligin ilyenkor nitrálódik és részben oxidálódik, így az egyidejűleg lehidrolizált hemicellulózzal együtt oldatba megy. A meghatározáshoz 1 g anyagot mérünk be, mely szükség esetén 4-5 g-ig növelhető, a salétromsav-etanol elegy arányos növelése mellett. Légszáraz fából pontosan bemérünk 1 g-ot és 200-250 ml-es lombikba és 25 ml salétromsav-etanol eleggyel öntjük le. A salétromsav-etanol feltáró elegy 96%-os etilalkohol és tömény salétromsav (sűrűség=1.513 g/ml) 4:1 arányú elegye. A lombik tartalmát visszafolyós hűtőt alkalmazva, vízfürdőn 1 óra hosszat forraljuk. (A feltáróelegy forráspontja 90 oC körül van). Az anyagot ezután ülepítjük és dekantálással előbb az oldatot, majd a rostokat lemért G2-es üvegszűrőre visszük és leszívatjuk. A szűrőre vitt rostokat 25 ml friss feltáróeleggyel visszamossuk a lombikba és további 1 óra hosszat forraljuk. A cellulózt szűrjük 10 ml etanollal, majd forró desztillált vízzel savmentesre mossuk és 105 oC-on súlyállandóságig szárítjuk. A kapott cellulózt az abszolút száraz fa mennyiségére vonatkoztatjuk.
2 1S SC 100
100 NG ( )
100
−= ⋅−⋅
Ahol:
C: cellulóztartalom (%) N: fa nedvességtartalma G: bemért nedves fa tömege S2: üvegszűrő + cellulóz tömege S1: üvegszűrő tömege

6
B, Gross-Berau eljárás A Gross-Berau cellulóz meghatározásnál a vizsgálandó fa lignintartalmát klórozással oldható vegyületekké alakítjuk és a visszamaradó anyag mennyiségét határozzuk meg. A meghatározásnál a lignin mellett az alacsonyabb polimerizációs fokú hemicellulózok is részben kioldódnak, de a magasabb polimerizációs fokú hemicellulózok nagy része visszamarad. (A meghatározás előtt célszerű extrahálni, mert a gyanta és viasznemű anyagok klór behatolását megakadályozzák). Egy előzetesen kiszárított és lemért G3-as üvegszűrőbe 1.5-2 g légszáraz anyagot mérünk, melyet először vízzel átnedvesítünk, majd leszívatva és üvegbottal nyomkodva szétoszlatjuk úgy, hogy egyenletes rétegben helyezkedjen el. A szűrőt a klór bevezetésére szolgáló csővel ellátott dugóval lezárjuk, majd egy vizes mosópalack közbeiktatásával megkezdjük a klórozást (másodpercenként legfeljebb 1-2 buborék haladjon át). Az első klórozás ideje 3-4 perc. Hosszabb klórozásnál a cellulóz is károsodást szenved, ezért a klórozás folytatása előtt a bomlástermékeket el kell távolítani az anyagból. A klórozó feltétet eltávolítva az anyagot 30 ml 3%-os kénessavval átöblítve a klórfelesleget leöntjük. Ezután a vízlégszivattyút elzárva 20-30 ml 2%-os nátriumszulfit oldatot öntünk a szűrőre, az anyagot üvegbottal felkeverjük, majd néhány percnyi állás után az oldatot leszívatjuk. A nátriumszulfitos mosást addig ismételjük, míg az oldat színtelen lesz. Ezután az anyagot 3-400 ml forró desztillált vízzel is átmossuk, majd leszívatással szárítjuk. A klórozást és a kénessaval ill. nátriumszulfitos mosást az anyag minőségétől függően többször kell ismételni. Tűlevelű fáknál, 4-7, lombos fáknál 3-5 klórozás szükséges. Az utolsó klórozás és a nátriumszulfitos mosás után az anyagot 1000 ml forró desztillált vízzel átmossuk és a szűrővel együtt szárítószekrényben 105 oC-on súlyállandóságig szárítjuk és lemérjük. C, Wise-féle eljárás 2 g anyagot 75 ml monoetanolaminnal 170 oC-on 3 órás főzéssel ligninmentesítünk. Vizes mosás után a visszamaradt ligninnyomok eltávolítására anyagra számított 40 % klórral 5 perces klóros-vizes kezelést alkalmazunk. A visszamaradt klórfelesleget kénessavas és nátrium-szulfitos mosással kötjük le. A cellulóz mennyiségét vizes mosás és kiszárítás után a szokásos módon állapítjuk meg. Egyes egynyári növények, lombos fák és tűlevelű fák vizsgálatánál a klóros kezelést 2-3-szor meg kell ismételni. D, Normann-Jenkins-féle módszer 2 g faanyagot 100 ml 3%-os nátriumszulfit oldattal melegítünk, majd leszűrve 100 ml 0.8%os hipokloritos oldattal 10 percig keverjük. A hipokloritoldat leszivatása után az anyagot 50 ml víz és 50 ml 3%-os nátriumszulfit oldat keverékével 20 percig forraljuk. A hipokloritos kezelést és monoszulfitos kifőzést megismételjük, majd a harmadik kezelés 2 ml 20 %-os kénsavval megsavanyított 100 ml 0.15%-os NaOCl oldattal történik. A klórfelesleget az előzőekkel megegyező módon végzett monoszulfitos kezeléssel távolítjuk el. A megsavanyított hipoklorit oldattal végzett kezelést többször megismételjük. A klórozás befejezésének meghatározása a G.B. cellulóz meghatározásánál leírt módon a Mäul-féle reakció alapján történik. A visszamaradt cellulóz mennyiségét forró vizes mosás után kiszárítva, a szokásos módon állapítjuk meg.

7
Holocellulóz tartalom meghatározás A holocellulóz tartalom meghatározására szolgáló eljárások célja az anyag ligninmentesítése a cellulóz- és hemicellulóz tartalom károsodása nélkül. Ez a követelmény ugyan teljesen nem érhető el, azonban gyakorlatilag jól megközelíthető. Mivel a holocellulóz tartalom meghatározására szolgáló eljárásnál a lignin kioldása enyhe kémiai kezeléssel történik, a cellulóz és a hemicellulózok aránylag változatlan formában nyerhetők ki. A holocellulóz meghatározások végterméke ezért rendkívül alkalmas kiindulási anyag a poliszacharidok részletesebb vizsgálatára. A, Klórozásos módszer Előzetesen etanol-benzollal extrahált fareszelékből 2 g-ot Schott-tégelyben szívópalack tulipánjába teszünk, majd a fareszelékre annyi hideg 8 C-os vizet öntünk, hogy a nedvesítés teljes legyen, a vizet leszívatjuk, s a tégelyt pedig a klór bevezetésére szolgáló csővel ellátott dugóval bedugaszoljuk. Először 3 percig, majd a fareszeléket felkeverve 2 percig klórozzuk. A hőmérsékletet a klórozás alatt jeges vízzel alacsonyan tartjuk. A klórmentes reszeléket 2-3 percre 96%-os etanolban oldott monoetanolamin 3%-os oldatával – hőmérséklete 75-80 C legyen – felöntjük, majd az oldatot leszívatjuk és etanollal ismét mossuk. A kezelés során a fareszelék sárgás vagy vörösesbarna színű lesz. A fenti kezelést addig ismételjük, amíg a maradék a forró etanolaminos oldat hatására más nem színeződik meg. Ekkor etanollal, vízzel majd ismét etanollal semleges kémhatásig mossuk a maradékot. Befejezéskor kevés éterrel az alkoholt kiszorítjuk és 105 C-on a holocellulózt súlyállandóságig szárítjuk. B, Wise-féle eljárás A Wise-féle holocellulóztartalom meghatározásánál a lignin-tartalom kioldása ecetsavas közegben nátriumkloritos (NaClO2) oxidációval történik. A nátriumklorit előnye, hogy az oxidáció kíméletesebb, mint az egyéb oxidálószerek (Cl2, NaOCl, HNO3) esetében. A meghatározás kivitelezése: 5 g körüli anyagmennyiséget 250 ml-es Erlenmeyer-lombikba mérünk. Az anyaghoz ezután 1.5 g nátriumkloritot és 10 csepp jégecetet (kb 0.5 ml) tartalmazó 160 ml oldatot adunk és 1 órán át vízfürdőn óvatos felrázással többször átkeverjük. Egy óra múlva a lombikba 10 csepp jégecetet és kis részletekben 1.5 g szilárd nátriumkloritot adunk, majd az előbb leírt módon ismét egy óráig melegítjük. A jégecet és nátriumklorit hozzáadását és a melegítést többször megismételjük. Extrahált tűlevelű fák esetében 4, lombosfáknál 3 a főrészek száma. A nátriumkloritos kezelés befejezése után a lombikot jeges vízbe állítva lehűtjük, majd G1 jelzésű üvegszűrőn leszűrjük és jeges vízzel alaposan kimossuk. A vizes kiomosás után az anyagot acetonnal is átmossuk és alacsony hőmérsékleten (30-35 C) lehetőleg vákuumban, súlyállandóságig szárítva lemérjük. Ha a holocellulóztartalom részletesebb vizsgálatához nagyobb anyagmennyiségre van szükség, a bemérést az oldat és a vegyszerek arányos emelésével 100-200g-ig növelhetjük. C, Jayme-féle eljárás A meghatározás a Wise-féle eljáráshoz hasonlóan enyhe nátriumkloritos oxidációval történik. Eltérés az előbbi eljáráshoz képest, hogy az oxidációs idő lényegesen hosszabb, mint a Wise-féle eljárásnál és a kloritos kezelések között az anyagot kimosva a bomlástermékeket eltávolítjuk. A vizsgálandó anyagot lehetőleg mikrotómmal vágjuk 40-80 µm vastagságú szeletekre, majd

8
metilalkohol-benzol keverékkel extraháljuk és szobahőmérsékleten kiszárítjuk. Az így előkészített anyagból 2 g körüli mennyiséget lemérünk és becsiszolt dugóval és visszafolyós hűtővel ellátott 200 ml-es gömblombikban 100 ml, 0.18 g jégecetet és 1 g nátriumkloritot tartalmazó oldattal elkeverjük. A lombikot ezután vízfürdőn melegítve 12 óra hosszat 60 C-on tartjuk. A melegítés befejezése után a lombik tartalmát G2 jelzésű üvegszűrőn leszűrjük és jeges vízzel erősen kimossuk, a vizet erős leszívatással minél jobban eltávolítjuk. Az itt leírt kezelést és kimosást azonos vegyszermennyiség hozzáadásával még háromszor megismételjük. Az utolsó kezelés és kimosás urán az anyagot acetonnal is átmossuk és 25-30 oC-nál nem magasabb hőmérsékleten kiszárítjuk és lemérjük. Az eredmények kiszámítása a Wise-féle meghatározásnál leírt képlettel történik. Az alfacellulóz tartalom meghatározás Az alfacellulóz megjelölés kémiailag nem egységes összetételű anyagot jelent, hanem csupán a 17.5%-os nátrium-hidroxiddal (NaOH) a kísérleti körülmények által megszabott körülmények mellett oldhatatlan cellulóz mennyiségét fejezi ki. A meghatározás kivitelezése. A szokásos módon aprított ismert nedvességtartalmú mintából 3.5 g abszolút szárazanyag tartalomnak megfelelő anyagot 200 ml-es főzőpohárba mérünk. Az anyaghoz 50 ml 17.5%-os 20 oC hőmérsékletű nátriumhidroxid oldatot adunk és üvegbottal 5 percig keverjük, majd 20 oC-os vízfürdőbe helyezve, 4 percig állni hagyjuk. 40 perc múlva a pohárba 50 ml 20 oC-os desztillált vizet öntünk és a pohár taralmát felkeverjük, majd G2 jelzésű üvegszűrőn leszűrjük. A lúgoldat leszívatása után az anyagot 25-25 ml 9.5%-os nátrium-hidroxiddal kétszer kimossuk, majd a szűrletet egyesítve visszaöntjük és ismét átszívatjuk az anyagon. A szűrés és az első mosás összesen legfeljebb 5 percig történet. Ügyelni kell arra, hogy az anyag a szűrő teljes felületét egyenletesen lepje el és ne száradjon ki teljesen. Hemicellulóz meghatározás esetén a szűrleteket összegyűjtve félretesszük. Az anyagot ezután 600 ml desztillált vízzel átmossuk, majd 50 ml 10 %-os ecetsavat öntünk a szűrőre és 5 perces állás után leszívatjuk. A szűrő tartalmát ezután 50 ml 10%-os ecetsavval mossuk át, majd forró desztillált vízzel savmentesre mossuk. A forró vizes mosás után az anyagot erős szívatással lehetőség szerint kiszárítjuk, majd a szivattyút kikapcsolva 25 ml alkoholt öntünk az anyagra és 5 percig állni hagyjuk. Az alkoholt leszívatva, a szűrő tartalmát előzetesen kiszárított és lemért mérőedénybe visszük át és 105 oC-on súlyállandóságig szárítva lemérjük. Az alfacellulóz hamutartalmát a szokásos módon végzett kiizzítással határozzuk meg. Az eredmény kiszámítása:
( ) ( )( )
2 1G G 100 HA 100
G 100 N
− −=
−�
�
Ahol: A: alfacellulóz tartalom; G: a bemért anyag tömege (g); N: a bemért anyag nedvesség tartalma (%); G1: üres mérőedény tömege (g); G2: mérőedény + alfacellulóz tömege (g); H: alfacellulóz hamutartalma (%)

9
Lignintartalom meghatározása A lignin aromás jellegű monomerekből felépülő kolloidális alkotórésze a fának, mely a cellulózhoz és a többi szénhidráthoz valamilyen kémiai kötéssel kapcsolódik. Az egy metoxi csoportot tartalmazó koniferil alkohol a fenyőfélék, a két metoxi csoportot tartalmazó szinapil alkohol pedig a lombosfák ligninjének az építőköve. A ligin kvalitatív meghatározása A, Sósav-floroglucin próba A lignintartalmú anyagok floroglucinnal piros színeződést adnak. (A színeződést a lignin kíséretében jelenlévő, vagy a sósav hatására felszabaduló aromás aldehideknek tulajdonítják). B, Anilinszulfát-próba A lignintartalmú nyersanyagok anilinszulfát-oldattal vagy ecetsavas anilin hatására sárga színeződést adnak. C, Egyéb színreakciók Különböző fenolszármazékok a lignint kísérő számos aromás aldehidekkel (koniferilaldehid) színreakciót adnak (Fenol: kékszöld, m-krezol: kék, rezorcin: kékesibolya). Mäule-féle reakció A vizsgálandó anyaghoz néhány ml 0.1 N KMnO4 oldatot adunk és jól elkeverjük. Három perc eltelte után desztillált vízzel 10 percen át főzzük, majd a kimosott anyagot 10 % sósavoldatba tesszük. Miután a kivált barnakő feloldódott, az anyagot gyorsan kimossuk és koncentrált ammóniumhidroxidba helyezzük. Lignin jelenlétében a lombosfák mélypiros, a fenyőfák pedig barna színeződést adnak. A lignin kvantitatív meghatározása A lignin mennyiségi meghatározása közvetlen v. közvetett úton lehetséges. A lignintartalom meghatározására szolgáló közvetlen eljárások azon alapulnak, hogy erős ásványi savakkal végzett hidrolízis hatására a cellulóz és egyéb poliszacharidok vízben oldható cukrokká bomlanak le és a ligninszerű anyagok kondenzált állapotban savban oldhatatlan termékként visszamaradnak. A hidrolízishez alkalmazott sav koncentrációját tehát úgy kell megválasztani, hogy egyrészt az összes szénhidrátokat teljesen eltávolítsuk, másrészt ne képződjék humifikálódott maradék. A ligintartalom meghatározására szolgáló eljárások a szénhidrátok kioldására alkalmazott hidrolízis eltérő körülményei miatt sok esetben különböző értéket adnak, ezért különösen fontos az eredmények megadásánál minden esetben jelezni, hogy a meghatározás milyen módszer szerint történt. A ligintartalom meghatározásánál rendkívül fontos a vizsgálandó anyag megfelelő finomságra való felaprítása. Hasonlóképpen fontos a gyanták és viaszszerű anyagok eltávolítása a meghatározás előtt, ezek ugyanis a cellulóztartalmú részeket bevonva a hidrolízist gátolják és így a meghatározás eredményét többszörösére növelik. A közvetett módszerek – melyek közelítő értéket szolgáltatnak – a ligninből lehasítható metoxicsoportok, vagy a lignin által addícionált klór mennyiségének a meghatározásán alapszanak.

10
A, A ligin kvantitatív meghatározása módosított Klason eljárással. (König-Komarov módszere) 1 g légszáraz fát 15 ml 72%-os kénsavoldattal 2.5 órán át 24-25 C-on kezeljük. A lombik tartalmát időről-időre felkeverjük, hogy annak csomósodását elkerüljük. Az elegyet ezután 200 ml desztillált vízzel hígítjuk és egy órán át visszafolyós hűtővel ellátott lombikban forraljuk. A lignint le hagyjuk ülepedni, majd előzetesen kiszárított és lemért G4-es szűrőn szűrjük. A kapott anyagot semleges kémhatásig mossuk és 105 oC-on tömegállandóságig szárítjuk. A kapott lignin mennyiségét az extrahálatlan abszolút száraz fára vonatkoztatva számíjuk. B, A lignin meghatározása Halse szerint 1 g légszáraz fát analitikai mérlegen 250 ml-es jódszámlombikba mérünk +/- 0.2 mg pontossággal. Az anyagra 50 ml 38%-os sósavoldatot öntünk úgy, hogy azzal a pohár falára tapadó rostokat lemossuk. Az anyagot üvegbottal átkeverjük majd 5 perc múlva 5 ml tömény kénsavat öntünk a keverékhez. A lombikot ledugaszoljuk jól összerázzuk és 24 óráig állni hagyjuk. Ez idő alatt a cellulóztartalom elcukrosodik. Ezután a lombik tartalmát forró desztillált vízzel kvantitatíve 800 ml-es főzőpohárba mossuk és a folyadék térfogatát 500 ml-re egészítjük ki desztillált vízzel. Az oldatot 2 percig forraljuk, majd a lignint ülepítjük, ezután analitikai üvegszűrőn (G4) szűrjük és desztillált vízzel savmentesre mossuk. Mosás után a szárazra szívatott szűrőt 105 +/- 2 oC-on tömegállandóságig szárítjuk és az exszikkátorban hűtött mintát mérjük. A lignin tartalmat a bemért száraz fa tömegére vonatkoztatva százalékosan számítjuk. Feltárási fok meghatározása A feltárási fok megállapodás szerinti mérőszám, mely a fehérítetlen cellulóz jellemzésére szolgál. A feltárási fok meghatározására különböző módszereket dolgoztak ki. Az eljárások alapja az, hogy a cellulózban lévő lignint és más szerves kísérő anyagokat oxidációs eljárással lebontjuk. A lignin, hemicellulóz és egyéb szerves anyagok oxidációs lebontásának mértéke elsősorban az oxidálószer mennyiségétől, minőségétől, koncentrációjától valamint a reakció idejétől és hőmérsékletétől függ. A vizsgálat kivitelezésénél tehát mindig be kell tartani az előírt körülményeket. A feltárási fok meghatározására szolgáló eljárások közül kiemelkedik a Kappa-szám meghatározás. Kappa-szám meghatározás Kappa-számon azt a 0.02 M KMnO4 oldat mennyiséget értjük ml-ben kifejezve, amelyet 1 g abszolút száraz cellulóz a vizsgálat körülményei között elfogyaszt. A vizsgálat akkor tekinthető szabványosnak, ha a bemért cellulóz a kálium-permanganát mennyiségének 30-70%-át fogyasztja el. A meghatározás kivitelezése Ha a Kürschner-Hoffer féle módszerrel előállított cellulózt vizsgáljuk, 0.8-1.2 g körüli mintamennyiségből kell kiindulni. Az analítikai mérlegen bemért vizsgálandó anyagot dörzsmozsárban 250 ml desztillált víz kis részletéve csomómentesre foszlatjuk, majd egy bőnyakú 1000 ml-es Erlenmeyer-lombikba öntjük és a maradék vízzel átmossuk. Ezzel egyidejűleg egy 250 ml-es főzőpohárba 50 ml 0.02 M KMnO4 oldatot pipettázunk és mérőhengerrel 50 ml 4 M H2SO4 oldatot öntünk hozzá. A vizsgálandó anyag és a reagens hőmérsékletét termosztátban 25 oC-ra állítjuk be. (Célszerű a desztillált vizet és a reagens oldatokat előzőleg termosztátba helyezni.) 100 ml össztérfogatú reagens oldatot gyors mozdulattal a cellulóztartalmú oldathoz öntjük a

11
stopper óra egyidejű megnyomásával. A reagens oldatot 150 ml desztillált vízzel három részletben gyorsan utána öblítjük. Így a reakcióelegy összes térfogata mintegy 500 ml lesz. A stopper indításától az Erlenmeyer lombikot körkörös mozdulattal egyenletesen rázzuk, majd pontosan 10 perc múlva a reakciót leállítjuk úgy, hogy 10 ml 0.1 M KI oldatot öntünk hozzá mérőhengerből. A kivált jódot a rostok leszűrése nélkül 0.1 M Na2S2O3 oldattal keményítő indikátor (4-5 ml) alkalmazása mellett megtitráljuk. A lejátszódó kémiai folyamatok:
Lignin + 2 KMnO4 + H2SO4 = oxidált-lignin + K2SO4 + MnSO4 + H2O
2 KMnO4 + 10 KI + 8 H2SO4 = 6 K2SO4 + 2 MnSO4 + 5 I2 + 8 H2O
I2 + 2Na2S2O3 = 2 NaI + Na2S4O6 Az eredmény kiszámolása
1 2
0.02 0.1
a c b cp
⋅ ⋅= −
a: KMnO4 oldat térfogata ml-ben (50 ml) c1: KMnO4 oldat pontos koncentrációja mol/dm3-ben b: Na2S2O3 oldat fogyása ml-ben c2: Na2S2O3 oldat pontos koncentrációja mol/dm3-ben p: az oxidálódásra fogyott KMnO4 oldat ml-ben
pK
G=
K: Kappa-szám G: a bemért cellulózminta minta absz. száraz tömege (g). Megjegyzés: A meghatározás a szabvány vizsgálat fele mennyiségeit tartalmazza. Ha p nem pontosan 50%-a a 0.1 M-os KMnO4 oldat térfogatának, a következő korrekciót lehet elvégezni.
( )plog K lg 0.00093 2 p 25
G= + ⋅ −
Hőmérsékleti korrekció: Ha termosztát nem áll rendelkezése a Kappa-számot a következőképpen kell kiszámítani:
[ ]'K K 1 0.013 (25 t)= ⋅ + ⋅ −
Ahol t a reakció hőmérséklete oC-ban.

12
Égésmeleg és fűtőérték meghatározása A fának mint tüzelőanyagnak a felhasználása még mindig nagyon jelentős. Mint tüzelőanyagnak, de mint szerves vegyületnek is igen fontos jellemzője az égésmeleg és a fűtőérték. Az égésmeleg az a hőmennyiség kalóriában kifejezve, amely 1 kg anyag elégetésénél felszabadul, ha: A, A vizsgált anyag hőmérséklete az égés előtt és az égéstermék hőmérséklete az égés után egyaránt 20 oC. B, A szén és kéntartalom az égés után CO2 ill. SO2 alakban van jelen az égéstermékben. C, Az anyagban eredetileg jelenlévő nedvesség és az égés folyamán keletkező víz cseppfolyós állapotban van jelen. Az égésmeleg jele E, egysége KJ/kg, ill. J/g. A fűtőérték értéke annyiban különbözik az égésmelegtől, hogy az égésmeleg értékéből az égéstermékben lévő víz elpárologtatásához szükséges hőmennyiséget le kell vonnunk, vagyis ez esetben az anyagban eredetileg jelen lévő és keletkezett víz gőz alakban van jelen: A fűtőérték jele F, egysége, ugyancsak kJ/kg. A kalorimétereket működésük alapján izotermikus, anizoterm és adiabatikus kaloriméterekre osztjuk. Az izotermikus kaloriméterek működési elve a következő: A mérendő hőmennyiséggel halmazállapot változást hozunk létre, miközben a hőmérsékletet a halmazállapot változásnak megfelelő hőmérsékleten tartjuk. Az átalakult anyag mennyiségének és az átalakulási hőnek ismeretében a bevitt hőmennyiség kiszámítható. Az adiabatikus kalorimétereknek a környezettől hőátadás tekintetében tökéletesen el vannak szigetelve. Általában lassú folyamatoknál alkalmazzák, a hőátadás megakadályozása végett a termikus köpenyt hasonló sebességgel fűtik. Az anizoterm kaloriméterek ismert hőkapacitású edények, melyek a környezettől lehetőleg el vannak szigetelve. A mérendő hőmennyiséget a kaloriméter köpenyében lévő folyadékkal közölve, annak hőmérséklet változásából a felvett hő kiszámítható.
Q c t= ⋅∆ Q: a közölt hőmennyiség (kJ) c: a folyadék és a kaloriméter melegedő részeinek hőkapacitása (kJ/C) ∆t: hőmérséklet változás Mivel a környezettel való hőcsere nem küszöbölhető ki, meg kell határozni az ebből adódó hibát. E célból meg kell mérni a kaloriméter járását, vagyis azt, hogy mennyire változik a kaloriméter hőmérséklete a környezet hatására, miközben a hőmennyiséget leadó folyamat lejátszódik benne.

13
i1-i0: előperiódus i0-in: főperiódus in-i2: utóperiódus
A görbe alapján a grafikus megoldással a ∆t a következőképpen állapítható meg. Átlagoljuk a főperiódus első és utolsó hőmérsékletét:
o nk
t tt
2
+=
A tk és a görbe metszéspontjából merőlegest állítunk az x-tegelyre (idő-tengely). Meghosszabbítjuk az elő- és utóperiódus egyeneseit, mely a merőlegest két pontban (a,b) metszi. A két metszéspont különbsége a ∆t. A kaloriméter hőmérsékletét egyperces időközökben 5 percen át leolvassuk (előperiódus). A reakció időtartama alatt (főperiódus) 10-15 percig a leolvasást folytatjuk, majd miután a környezettel való hőcsere stacionáriussá vált, 5 percig az utóperiódust észleljük. A ∆t megállapítását a következőképpen végezzük el: Az elő- és utószakasz alapján kiszámítjuk a kaloriméternek azt a hőmérséklet-változását, mely a főszakasz alatt a környezettel való hőkicserélődése folytán következik be. Ezzel az értékkel a főszakasz alatt mért hőmérsékletváltozást korrigálni kell.
n o
s o
t t U ex n U
t t 2
− ∆ − ∆= − ⋅∆ + ⋅−
(1)
o 1t te
k
−∆ = (2)
Az ideális idő-hőmérsékleti görbe az, amelyet akkor kapnánk, ha végtelenül gyors reakció végtelenül kis hőtehetetlenségű rendszerben játszódik le.
A reális idő-hőmérséklet görbe ettől eltérő, exoterm reakció esetén a következő alakot veheti fel

14
2 nt tU
r
−∆ = (3)
Ahol n: a főperiódusban percenként leolvasott hőmérsékletek közeinek a száma x: korrekciós tag ∆e: az előkísérlet két hőmérsékletleolvasása közötti időre eső közepes hőmérsékletváltozás. ∆U: az utókísérlet két hőmérséklet leolvasása közötti időre eső közepes hőmérséklet változás to: a főkísérlet első, az előkísérlet utolsó hőmérséklete t1: az előkísérlet első hőmérséklete ts: a főkísérlet második hőmérséklete tn: a főkísérlet utolsó hőmérséklete t2: az utókísérlet utolsó hőmérséklete k: az előperiódusban a hőmérsékleti leolvasási közök száma r: az utóperiódusban a hőmérsékleti leolvasási közök száma Az x korrekciós tag előjelének megfelelően értékét a főperiódusban mért hőmérsékletkülönbséghez hozzáadjuk vagy levonjuk.
n ot t t x∆ = − + (4)
A hőmennyiség meghatározásához ismernünk kell a kaloriméter hőkapacitását, mely a kaloriméteredény, egyéb vízbe merülő részek és a víz hőkapacitásából tevődik össze. Ezen hőkapacitások összege a vízérték (V), mely alatt az a grammokban kifejezett vízmennyiséget értjük, melynek 1 oC-al való felmelegítéséhez ugyanakkora hőmennyiség szükséges, mint a szóban forgó testek és a víz 1 oC-al való felmelegítéséhez (mivel a víz fajhője szobahőmérsékleten 1.0 cal/g). A vízérték meghatározás céljából ismert (Qo) hőmennyiséget közlünk a kaloriméterrel. Meghatározva a hőmérsékletváltozást, a vízérték kiszámítható. Ismert hőmennyiséget pl. tiszta benzoesav vagy nádcukor ismert hőmennyiségének az elégetésével lehet a kaloriméterbe bevinni. Pl. benzoesav égéshője Qo = 6325.4 cal/g. 1 cal = 4.184 J, (Qo= 26465.4 J/g).
( )n o
o
V t t xQ b
G
⋅ − += − Σ (5)
ahol V: a kaloriméter vízértéke G: a vizsgálathoz bemért anyag tömege (g) x: korrekciós tag Σb: a drótszál, pamut, stb., égés melegéből tevődik össze, kb. 20 cal (83.68 J) Ebből a vízértéket kifejezve, a következő összefüggést kapjuk:
o
n o
Q G bV
t t x
⋅ + Σ=− +
(6)

15
A bombakaloriméter és tartozékok lényeges részeinek ismertetése A kaloriméterbomba a kaloriméteredényben lévő meghatározott tömegű, ill. térfogatú vízben áll. A mennyiséget úgy kell megválasztani, hogy a bomba fedelét ellepje. Az edény nikkelezett henger az elektromosan meghajtott keverő számára kiképzett hellyel. Az edényt hőszigetelő köpenybe helyezzük és rossz hővezető anyagból készített fedővel fedjük. A fémkeverőt szabályozható fordulatszámú elektromotor hajtja. A keverő súrlódását el kell kerülni. Függőleges keverő esetén a célszerű löketszám 70-80/perc. A hőmérsékletmérést ún. Beckman-féle hőmérővel hajtjuk végre. Ez nagy higanygömbbel és kis keresztmetszetű kapillárissal elkészített hőmérő. Skálája erősen nyújtott, így mérési köze 5-6 oC, osztása viszont 0.01 oC. A hőmérő felső részén lévő tartalékedény segítségével a higanytartalom változtatható, így tetszés szerinti hőmérsékleten használható. A Bertelot-Mahler féle kaloriméterbomba V2A acélból készült kb. 300 ml űrtartalmú edény, mely azonos anyagból készült fedővel légmentesen elzárható. A fedő gázbeeresztő és gázát kibocsátó csavaros szeleppel és szigetelt elektróddal van ellátva. A beeresztő szelep furatához egy csaknem a bomba fenekéig érő vékony acélcső csatlakozik, az erre forrasztott gyűrűbe kerül a platina v. kvarctégely. Mérés elvégzése A finomra aprított faanyagból 1.0 g-ot kéziprésen pasztillázunk, majd a pasztilla tömegét analitikai mérlegen lemérjük. A bomba pólusait virágdróttal összekötjük és pamutszállal hozzáerősítjük a pasztillát. A bombát összeszereljük. Redukálószelep segítségével 30 bar nyomásig töltjük oxigéngázzal a bombát. Ezután az elektródákat a gyújtószerkezettel összekapcsoljuk és a kaloriméter edénybe helyezzük, melybe előzőleg annyi vizet tettünk, hogy a bomba fedelét is befedje. A kaloriméter edényben lévő víz kezdeti hőmérséklete 1.0-1.5 oC-al alacsonyabb legyen a köpenyvíz hőmérsékleténél. Ha a kaloriméter hőmérsékletemelkedése állandósult, elkezdjük a mérést. Öt percen keresztül, percenként leolvassuk a hőmérsékletértékeket. A hatodik leolvasásnál (t0), mely egyben a főkísérlet kezdeti hőmérséklete is, a gyújtás áramkörét zárjuk. A robbanásszerűen lezajló égést a gyútjóáramkörbe iktatott égő felvillanása jelzi. A továbbiakban a hőmérséklet leolvasásokat percenként kell elvégezni mindaddig, amíg a hőmérséklet emelkedése meg nem szűnt. Az utolsó leolvasás (tn) egyúttal az utókísérlet első leolvasása. Itt ugyancsak 5 percen keresztül végezzük a hőmérsékletleolvasást, s a mérés elvégzése után a bombát szétszereljük és meggyőződünk a minta tökéletes elégéséről. A kalorimetrálást ismert égésmelegű benzoesavval megismételjük és az (5) valamint (6) képletek segítségével meghatározzuk a kaloriméter vízértékét (a G helyére a bemért benzoesav tömegét helyettesítve be, valamint a megfelelő hőmérséklet adatokkal számolva). A kaloriméter vízértékének ismeretében a famintára kapott mérési eredményekből az (1), (2), (3) és (4) képletek segítségével a már ismert összefüggésből kiszámoljuk az égésmeleget (a G helyére a bemért faminta tömegét helyettesítve be, valamint a megfelelő hőmérséklet adatokkal számolva):
( )n oV t t xE b
G
⋅ − += − Σ

16
A fűtőértéket az alábbi összefüggés alapján számoljuk:
F = E – 24.51*(9*H + H2O) E: égésmeleg (KJ/kg vagy J/g) F: fűtőérték ( KJ/kg vagy J/g) H: fa %-os hidrogéntartalma, értéke: 4.48% H2O: a fa %-os nedvességtartalma (a korábbi mérésekben N-el jelölve!) 24.51 a tömegegységnyi víz 100 °C-ra való melegítéséhez és elpárologtatásához szükséges hőmennyiség. A faanyag savtartalmának meghatározása A fában lévő savas karakterű anyagokon elsősorban a poliózokhoz kapcsolódó acetil csoportokat értjük, de a savas jelleg kialakításához más karbonsav származékok (formil, aromás karbonsav, stb.) is hozzájárulnak, jóllehet ez utóbbiak mennyisége elhanyagolható az ecetsavhoz képest. Az acetil csoportok jelentős része kötött állapotban fordul elő a fában, jellemzően éter kötésen keresztül kapcsolódik a szénhidrátokhoz. A faanyag különböző feldolgozása során a hő-, illetve a hidrotermikus hatás következményeként az éterkötés elhidrolizál, így ecetsav szabadul fel. A keletkezett ecetsav mennyisége erősen fafaj függő (pl. tölgyfélékben, akácban magas, bükkben alacsonyabb), de befolyásolja az életkor, illetve a döntés körülményei (évszak, stb.). A faanyag savtartalmának ismerete rendkívül fontos a fafeldolgozás során, mivel meghatározza a - Cementkötésű forgácslap mechanikai tulajdonságait, - A karbamid alapú ragasztóanyagok kötési idejét, - Fafeltárási folyamatokat, - Ecetsav lehasadás feldolgozás (pl. gőzölés, termikus kezelés, stb.) során. A faanyag savtartalmát felosztjuk szabad- és összes savtartalomra, illetve a kettő különbségét jelentő kötött savtartalomra. A szabad savtartalmat vízoldható savtartalomnak is nevezik, ami a meghatározás módjára utal. A savtartalom meghatározás a savasságot okozó gyenge savak analitikai meghatározásán alapul. Ennek megismeréséhez tekintsük át néhány fogalmat: Ekvivalencia pont: Azt a pH értéket, amely az oldatban ekvivalens mennyiségű sav és lúg egymásra hatásakor beáll ekvivalencia pontnak nevezzük. Erős savak és erős bázisok esetében ezt akkor érjük el, amikor a H+ koncentrációja megegyezik a OH- koncentrációjával, vagyis semleges kémhatásnál, pH=7-nél. Ha gyenge savat erős bázissal titrálunk a gyenge sav sója keletkezik, amely hidrolizálva lúgos kémhatású lesz. Ebből következik, ha pl. ecetsavat NaOH-dal titrálunk az ekvivalencia pont nem pH=7-nél lesz, hanem a keletkező oldat azon pH értéke lesz, amely megegyezik az ugyanilyen koncentrációjú Na-acetát pH-jával. Az ekvivalencia pont meghatározása történhet kísérleti úton vagy számítással. A számítás elve Gyenge savak erős bázissal alkotott sói hidrolizálva lúgos kémhatásúak. Az oldat pH-ja függ a gyenge sav disszociációs egyensúlyi állandójától és az oldat koncentrációjától.

17
2
lg7 spKc
pH+
+= , psav = -lgKsav
Az ecetsav disszociációs egyensúlyi állandója 20 °C-on : Ksav = 1,86 • 10-5 psav = 4,73 A Kísérleti meghatározás A szabad savtartalmat az aprított faanyag vizes áztatásból származó szűrletből, az összes savtartalmat Na-acetátos szűrletből határozzuk meg. Szűrletek készítése - vizes kivonat (szabad savtartalom meghatározás): 2,50 g facsiszolatot 30 ml desztillált vízben 24 órán át áztatunk, majd átszűrjük szűrőpapíron egy 100 ml-es mérőlombikba. A mintát négyszer 17,5 ml desztillált vízzel átmossuk és hagyjuk lecsepegni. Ezután a lombikot jelig töltjük desztillált vízzel. -Na-acetátos kivonat (összes savtartalom meghatározás): 2,50 g facsiszolatot 30 ml 0,1 molos Na-acetátban 24 órán át áztatunk, majd átszűrjük szűrőpapíron egy 100 ml-es rázólombikba. A mintát egyszer 17,5 ml 0,14 molos Na-acetáttal, majd háromszor 17,5 ml desztillált vízzel átmossuk és hagyjuk lecsepegni. Ezután a lombikot jelig töltjük desztillált vízzel. Mérés kivitelezés A kapott szűrletek teljes mennyiségét 150 ml-es főzőpohárba öntjük, majd pH mérő segítségével (potenciometrikus végpont meghatározás) 50 ml-es bürettából 0,005-0,01 M NaOH oldattal megtitráljuk. Mivel a savtartalom értéke tág határok között változhat, ezért a felhasználandó lúg koncentrációját a gyakorlatvezető közli. Mérési eredmények feldolgozása Táblázatot készítünk, amelyben a lúg fogyást és az ahhoz tartozó pH értékét tüntetjük fel, valamint az első, illetve a második derivált értékét határozzuk meg. Ez utóbbi lépésre azért van szükség, mert a titrálások során gyakran zavarják a meghatározást a szűrletben lévő egyéb anyagok, ami eredményeképpen a kapott adatsorra illesztett görbe grafikus kiértékelésének megbízhatósága csökken. (Az eredmény kiszámolásához célszerű az Excel program használata. Ennek segítségével könnyen tudjuk képezni a pH változások első és második deriváltját, ami majd az inflexiós pont pontos meghatározását teszi lehetővé.) ml lúg oldat pH dpH d2pH 0 5,43 - - 0,5 6,21 6,21-
5,43=0,78 -
1 6,85 6,85-6,21=0,64
0,64-0,78
1,5 7,12 7,12-6,85=0,27
0,27-0,64
2 7,37 7,37-7,12=0,25
0,25-0,27
… … … …
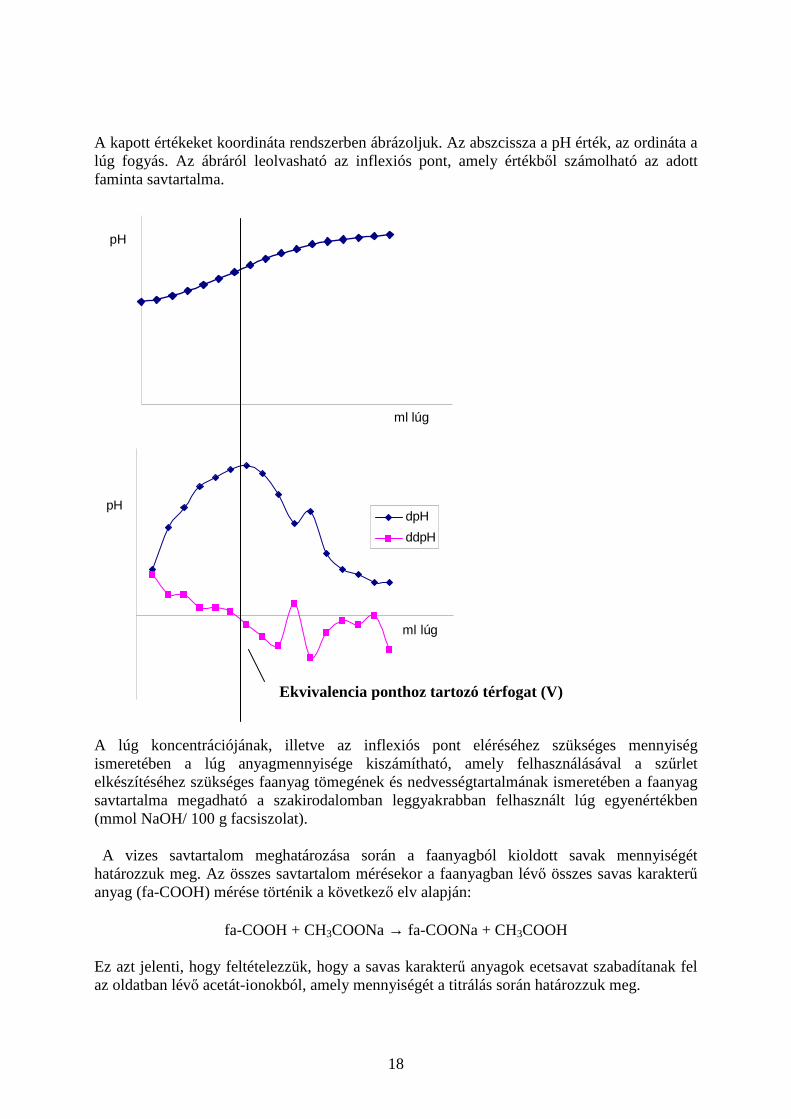
18
A kapott értékeket koordináta rendszerben ábrázoljuk. Az abszcissza a pH érték, az ordináta a lúg fogyás. Az ábráról leolvasható az inflexiós pont, amely értékből számolható az adott faminta savtartalma.
ml lúg
pH
dpH
ddpH
ml lúg
pH
A lúg koncentrációjának, illetve az inflexiós pont eléréséhez szükséges mennyiség ismeretében a lúg anyagmennyisége kiszámítható, amely felhasználásával a szűrlet elkészítéséhez szükséges faanyag tömegének és nedvességtartalmának ismeretében a faanyag savtartalma megadható a szakirodalomban leggyakrabban felhasznált lúg egyenértékben (mmol NaOH/ 100 g facsiszolat). A vizes savtartalom meghatározása során a faanyagból kioldott savak mennyiségét határozzuk meg. Az összes savtartalom mérésekor a faanyagban lévő összes savas karakterű anyag (fa-COOH) mérése történik a következő elv alapján:
fa-COOH + CH3COONa → fa-COONa + CH3COOH Ez azt jelenti, hogy feltételezzük, hogy a savas karakterű anyagok ecetsavat szabadítanak fel az oldatban lévő acetát-ionokból, amely mennyiségét a titrálás során határozzuk meg.
Ekvivalencia ponthoz tartozó térfogat (V)

19
A két savtartalom különbsége a kötött savtartalmat jellemzi. A szabad savtartalom számítása
1000
Vn c= ⋅
1000100
100100
nSZ
NG
⋅= ⋅−
Az összes savtartalom számítása
1000
Vn c= ⋅
1000100
100100
nO
NG
⋅= ⋅−
A kötött savtartalom számítása K = O – SZ K – A faanyag kötött savtartalma (mmol/100 g sz. fa) Faanyag totálfenol tartalmának meghatározása A növényi fenolok (elsősorban flavonoidok, tanninok, lignánok, stb.) jelentős szerepet töltenek be a növények életében, a szövetek élettani funkcióiban. A mechanikailag, vagy fertőzés által megsértett szövetben védelmi funkciót fejtenek ki. A reakcióban keletkező sötét kinon vegyületek és polimerjeik kémiailag gátat emelnek a sérülés nyomán bekövetkező fertőzésnek, illetve a már megtörtént fertőzés továbbterjedésének, emellett a fa színének kialakításához is hozzájárulnak. Az igen jelentős élettani funkciók mellett a növényi fenolok kis koncentrációjuk ellenére szignifikánsan meghatározzák a faanyag színét, színstabilitását, a faanyag tartósságát, ellenállóságát, sőt mechanikai, fizikai tulajdonságait is. A geszt magasabb fenol tartalommal rendelkezik mint a szíjács ez magyarázza leginkább a geszt kedvező tulajdonságait is.
A faanyag feldolgozása (szárítás, hőkezelés, modifikáció, stb.) és használata (fotodegradáció, kilúgozódás, környezeti hatások stb.) során a fa eredeti fenol vegyületei jelentősen átalakulhatnak.
V – A NaOH mérőoldat fogyása az ekvivalencia pontig (cm3) c – A NaOH mérőoldat pontos koncentrációja (mol/dm3) n – A meghatározás során elreagált NaOH anyagmennyisége (mol) G – A bemért faanyag tömege (g) N – A bemért faanyag nedvességtartalma (%) SZ – A faanyag szabad savtartalma (mmol/100 g sz. fa)
V – A NaOH mérőoldat fogyása az ekvivalencia pontig (cm3) c – A NaOH mérőoldat pontos koncentrációja (mol/dm3) n – A meghatározás során elreagált NaOH anyagmennyisége (mol) G – A bemért faanyag tömege (g) N – A bemért faanyag nedvességtartalma (%) O – A faanyag összes savtartalma (mmol/100 g sz. fa)

20
A faanyag minősége szempontjából nélkülözhetetlen paraméter az ún. „totálfenol” vagy összfenol tartalom mérése, mely információt ad az össze fenolos vegyület mennyiségéről, ezáltal a faanyag minőségéről is. Ez a paraméter egy adott fafajon belül, az életkorral, magassággal vagy a szöveti szerkezettel is jelentősen változhat, pontos mérése ezért is indokolt. Totálfenol tartalom mérése Folin-Ciocâltău módszerével A mérés során termikusan modifikált faanyagok totálfenol tartalmát határozzuk meg. A termikus kezelés hatására a lignin részben depolimerizálódik és jelentős mennyiségű egyszerű fenol vegyület (C8 - C12) keletkezik. A keletkező anyagok meghatározzák és javítják a faanyag színét, tartósságát, méretstabilitását. Emellett azonban a lignin átalakulása kedvezőtlenül befolyásolja a faanyag mechanikai (MOE, hajlítószilárdság, sűrűség) tulajdonságait. A falignin átalakulásának mértéke a totálfenol tartalommal jól számszerűsíthető, mely egyben információt ad a termikus kezelés intenzitásáról is. Extrakció A vizsgálandó faanyagot felaprítjuk (1), daráljuk (2) és szitáljuk (3). A 0.2 – 0.63 mm szitafrakciót használjuk az extrakcióhoz: 0.250 g faanyagot bemérünk (4) és 6 szakaszban extraháljuk 80%-os metanollal ultrahangos fürdőben (szakaszonként 8 ml extrahálószerrel 30 percig (5)). Az egyes szakaszokban kapott extraktumokat 50 ml-es mérőlombikba gyűjtjük Whatman GF/A üvegszálas szűrőn keresztül (6). A hatodik szakasz után jelre töltjük a mérőlombikot (7).
2 1
3 4
5 6
7

21
Mérés Pontosan kimért 0.5 ml extraktumhoz 2.5 ml 10x-es hígítású Folin-Ciocâltău reagenst adunk, majd 1 perc elteltével az elegyhez 2.0 ml 0.7M-os Na2CO3 oldatot mérünk. A reakcióelegyet pontosan 5 percig 50 °C-os vízfürdőben melegítjük, ezalatt kifejlődik és stabilizálódik az oldat kék színe. Ezután az elegyet hideg vízfürdőben visszahűtjük és a totál fenol tartalmat spektrofotometriásan meghatározzuk 760 nm hullámhosszon, mindegyik reakcióelegyből három párhuzamos mérést végzünk. Standardként kvercetint alkalmazunk. A mérési eredményeket kiértékelő egyenes segítségével értékeljük ki. A mérési eredményeket súlyállandóságig szárított faanyagra vonatkoztatjuk.
A mérés kivitelezése előtt azonban tekintsünk át néhány fogalmat illetve a módszer elvi hátterét… A spektrofotometria elve
A vegyi anyagok (atomok és molekulák) és az elektromágneses sugárzás kölcsönhatásának vizsgálata jelentős szerepet játszik ezen anyagok mind minőségi, mind mennyiségi elemzésében. Az elektromágneses sugárzás azon hullámhossztartománya, amely optikai eszközökkel vizsgálható, az optikai sugárzás, amely felöleli az ultraibolya (UV), a látható (VIS) és az infravörös (IR) fény tartományát. Azokat az analitikai módszereket, amelyek során az anyagoknak különböző hullámhosszúságú fénnyel történő kölcsönhatását a hullámhossz függvényében vizsgáljuk, spektroszkópiai módszereknek nevezzük. A spektroszkópiai módszerek között külön területet jelent az atomok ill. a molekulák vizsgálata, azaz az atom- és a molekula-spektroszkópia. Mind az atomok, mind a molekulák esetében vizsgálhatjuk az anyag által kibocsátott fényt (emissziós spektroszkópia) ill. az elnyelt fényt (abszorpciós spektroszkópia). A gyakorlatokon a fény ultraibolya tartományában vizsgáljuk különböző vegyületek fényelnyelését.
--IR-><---------------------------VIS---------------------------><-UV-
A fény kettős természete Hullámtermészetét bizonyítja a visszaverődés, interferencia, polarizáció jelensége. A fény hullámhossza [ λ (m) ], rezgésszáma [ µ (s-1) ], a közeg törésmutatója [ n ] és a fény vákuumban mérhető sebessége [ c = 3·108 m/s ] között a következő összefüggés áll fenn:
λ · µ = c / n A fény anyagi természetét bizonyítja, hogy a fénysugár diszkrét fotonokból áll. A fotonok energiája [ E (J) ] arányos a sugárzás frekvenciájával. Az arányossági tényező a Planck-állandó [ h = 6,62·10-34 J·s ].

22
E = h · µ A fény energiája tehát annál nagyobb, minél nagyobb a frekvenciája ill. minél kisebb a hullámhossza. A spektroszkópiai módszerek során a minőségi és mennyiségi elemzéshez a fény hullámhosszát valamint intenzitását mérjük. Spektrofotometriás mérési módszer Elektromágneses sugárzás hatására az anyagok a különböző hullámhosszú sugárkomponensekből különböző mennyiséget nyelnek el. Valamely, a fényelnyeléssel arányos mennyiséget a hullámhossz függvényében ábrázolva abszorpciós spektrumot kapunk. Minőségi elemzésre az elnyelési sávok hullámhosszának meghatározása, mennyiségi elemzésre pedig az elnyelés nagyságának mérése nyújt módot. A spektrofotometriás mérés során UV, látható vagy IR fénnyel történő besugárzással energiát közlünk a mintával. Ezen közölt energia hatására megváltozik a molekulák energiaállapota, amelyet a következő összefüggéssel írhatunk le:
Eössz = Eelektron + Erezgési + Eforgási Vagyis a molekulák összes energiája a kötést létrehozó elektronok energiájának, valamint az atomok rezgési és forgási energiájának összegeként írható le. (A molekulákon belül az atomokat, atomcsoportokat nem egymáshoz meghatározott távolságra rögzítetten, hanem egymáshoz közeledve ill. egymástól távolodva, rugó mentén rezegve ill. tengely körül forogva kell elképzelni.) Energiaelnyelés (fényabszorpció) akkor jön létre, ha a besugárzó fény energiája (h • µ) megegyezik a molekula két lehetséges energianívója közötti különbséggel. A molekula elektron-, rezgési és forgási energiája a sugárzás abszorpciója közben külön-külön vagy egyszerre is megváltozhat. A lehetséges változások láthatók a következő ábrán:

23
A forgási állapotok között a legkisebb a különbség (az ábrán C-vel jelzett átmenet). A forgási (C), valamint a rezgési és a forgási átmenetek (B) gerjesztéséhez elegendő a kis energiájú infravörös sugárzás. Legnagyobb a különbség az elektronállapotok energiái között (A), gerjesztésükre az ultraibolya és a látható tartomány alkalmas. Ha a gerjesztési energia az elektronállapotot megváltoztatja, akkor azzal sokféle rezgési és forgási állapotváltozás is együtt jár. Ez, valamint a minta és az oldószer molekuláinak kölcsönhatása magyarázza, hogy a molekulák abszorpciós spektrumában nem diszkrét vonalak, hanem folytonos görbék mutatják a fényelnyelést. Mennyiségi elemzés és lépései A mintaoldatot tartalmazó küvettát egy Io intenzitású fénnyalábbal megvilágítva és az I áthaladt sugárzást detektálva azt tapasztaljuk, hogy a fény intenzitása csökken. A csökkenés oka a minta fényelnyelésén kívül a fény visszaverődése és szóródása:
Io = Ivisszavert + Iszórt + Iabszorbeált + Iáthaladt Ahhoz, hogy a visszaverődés és a szóródás okozta fényintenzitás-csökkenést kiküszöböljük, a fénysugarat egy másik fényúton átvezetjük egy ún. „vak” oldaton, amely a vizsgálandó anyag kivételével mindent tartalmaz, mint ami a minta küvettában van. A fényvisszaverődés és a szóródás mértéke a „vak” oldalon azonos lesz a minta oldalival, mivel a küvetták ugyanolyan méretűek és kialakításúak. Ha a vak oldalon kilépő fénysugár intenzitását (mely egyben megegyezik a mérendő mintánkba belépő fénysugár intenzitásával!) Io-nak tekintjük, akkor ehhez képest a mintánkból kilépő I fényintenzitás ennél alacsonyabb értékű lesz. A csökkenést kizárólag a mintában lévő vizsgálandó anyag fényelnyelése okozza, ezáltal a fényintenzitás-csökkenés mértéke lehetőséget nyújt mennyiségi elemzésre. A fényelnyelés mennyiségi analitikai alkalmazása a Lambert-Beer törvényen alapul, amely szerint monokromatikus sugárzás esetén a mintába belépő fénysugár Io intenzitása arányos az oldat c koncentrációjával, az l rétegvastagsággal, és függ az anyagi minőségtől. A = lg (Io / I) = e · l · c
Io: a vakoldatból kilépő fénysugár intenzitása I: a mintaoldatból kilépő fénysugár intenzitása c: az oldat koncentrációja e: a vizsgálandó anyag fényelnyelési együtthatója (abszorpciós koefficiense). I / Io hányados: fényáteresztő képesség vagy transzmittancia (T), −−−−lg (I / Io): abszorbancia. [A ≡ lg (Io / I )]
Ismert anyag koncentrációjának meghatározásánál az elmondottak alapján a következőképpen járunk el:
1. Felvesszük egy tetszőleges minta abszorpciós spektrumát. 2. Kiválasztjuk az egyik abszorpciós maximumhoz tartozó hullámhosszt a spektrumból. 3. A standard vegyületből koncentráció-sorozatot készítünk. 4. A kiválasztott hullámhosszon megmérjük a különböző koncentrációhoz tartozó

24
abszorbancia-értékeket. 5. A mért abszorbanciákat a koncentráció függvényében ábrázolva egyenest kapunk (kalibrációs egyenes), amelyről leolvasható az ismeretlen koncentrációjú minta mért abszorbanciájához tartozó koncentráció. 6. A kalibrációs egyenes inverz függvénye az ún. „kiértékelő egyenes” amely az abszorbancia függvényében ábrázolja az oldat koncentrációját. A továbbiakban ezt használjuk.
Spektrofotométer és elvi felépítése
Számolás – mennyiségi kiértékelés
k = 0.2445 * A + 0.0045R2 = 0.9985
0
0.05
0.1
0.15
0.2
0.25
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
Totálfenol - kiértékel ő görbe
Abszorbancia
mm
ol k
verc
etin
/ lit
er
Kiértékelő egyenes és az egyenes egyenlete totálfenol tartalom meghatározásához (példa!). R2 : az egyenes határozottsági foka. A kiértékelő egyenes a meghatározásnak megfelelően 760 nm hullámhosszon került felvételre.
A kiértékelő egyenes egyenlete: k = a*A + b
7. A minta kiértékelését úgy végezzük, hogy megmérjük az abszorbancia értéket és a
kiértékelő egyenes alapján az alábbi képlet alapján számítjuk a totálfenol tartalmat k: totálfenol koncentráció a reakcióelegyben (mmol kvercetin/liter) a: kalibrációs egyenes meredeksége

25
A: egy adott minta 760 nm-en mért abszorbanciája b: tengelymetszet Ctf: totálfenol tartalom (mmol kvercetin/ 100 g száraz fa) G: az extrakcióhoz bemért fa tömege (g) H: a minta hígítása N: bemért fa nedvességtartalma (%)
( ) 50100
(100 )1000 ( )
100
tf
a A b HC
G N⋅ + ⋅ ⋅= ⋅ ⋅ −⋅
Egy adott mintára három párhuzamos mérés történt (Ctf1, Ctf2, Ctf3). Végeredményként a totálfenol tartalmak átlagát (Ctf_átlag) és a tapasztalati szórást (s) kell megadni mintánként. A szórás kiszámolása a következő képlettel történik:
2 2 21 2 3( ) ( ) ( )
1tfátlag tf tfátlag tf tfátlag tfC C C C C C
sn
− + − + −=
−
s: szórás n: párhuzamos mérések száma (3) Ctfátlag: totálfenol tartalom átlaga (mmol kvercetin/100 g száraz fa) Ctf1, Ctf2, Ctf3: párhuzamos mérések Faanyag összes kioldható szénhidrát (cukor) tartalmának meghatározása Dubois módszerével Az „összes kioldható szénhidrát” fogalom a vizes metil-alkohollal kioldható szénhidrát komponenseket jelöli. Ezek túlnyomórészt mono- és oligoszacharidok (cukrok). A fa szerkezeti szénhidrát komponensei az alkalmazott körülmények közt nem oldódnak ki. A kioldható cukrok a szövetek élettani folyamataiban vesznek részt, koncentrációjuk a vegetációs időszakban és szövetenként is jelentősen változhat. A faanyag kezelése (termikus, hidrolítikus, enzimes, stb.) valamint degradációja során a szerkezeti szénhidrátok bomlása is bekövetkezhet. Ennek termékei a kioldható mono- és oligoszacharidok. Ezek a vegyületek további reakciókban illékony anyagokká (aldehidek, pl. furfurol, metil-furfurol) alakulnak. Az összes kioldható szénhidrát tartalom a fás szövet biológiai állapotára, aktivitására, faanyag esetében az a szerkezeti szénhidrátok (cellulóz, hemicellulóz) átalakulására, degradációjára vonatkozóan ad gyors, megbízható mennyiségi képet. Extrakció A vizsgálandó faanyagot felaprítjuk, daráljuk majd szitáljuk. A 0.2 – 0.63 mm szitafrakciót használjuk az extrakcióhoz: 0.050 g faanyagot bemérünk és 6 szakaszban extraháljuk 80%-os metanollal ultrahangos fürdőben (szakaszonként 8 ml extrahálószerrel 30 percig). Az egyes szakaszokban kapott extraktumokat 50 ml-es mérőlombikba gyűjtjük Whatman GF/A üvegszálas szűrőn keresztül. A hatodik szakasz után jelre töltjük a mérőlombikot. Mérés

26
Dugós kémcsőbe pontosan kimért 1 ml extraktumhoz 1 ml 5%-os vizes fenol oldatot adunk, majd homogenizálás után az elegyhez óvatosan 5 ml koncentrált kénsav oldatot mérünk. A kémcsövet ledugózzuk, a reakcióelegyet pontosan 10 percig szobahőmérsékleten állni hagyjuk, majd 20 percig 25 oC-os vízfürdőben hűtjük le. Az összes kioldható szénhidrát tartalmat spektrofotometriásan határozzuk meg 490 nm-es hullámhosszon. Standardként glükózt alkalmazunk. A mérési eredményeket kiértékelő egyenes segítségével értékeljük ki. A mérési eredményeket súlyállandóságig szárított faanyagra vonatkoztatjuk.
Számolás – mennyiségi kiértékelés
k = 119.9 * A - 1.2115R2 = 0.9995
0
20
40
60
80
100
120
140
160
180
0 0.2 0.4 0.6 0.8 1 1.2 1.4 1.6
Összes kioldható szénhidrát - kiértékel ő görbe
Abszorbancia
mg
glük
óz /
liter
Kiértékelő egyenes és az egyenes egyenlete kioldható szénhidrát tartalom meghatározásához (példa!). A kiértékelő egyenes a meghatározásnak megfelelően 490 nm hullámhosszon került felvételre. R2 : az egyenes határozottsági foka
A kiértékelő egyenes egyenlete: k = a*A + b k: összcukor koncentráció a reakcióelegyben (mg glükóz/liter) a: kalibrációs egyenes meredeksége A: egy adott minta 490 nm-en mért abszorbanciája b: tengelymetszet Cc: összcukor tartalom (mg glükóz/ g száraz fa) G: az extrakcióhoz bemért fa tömege (g) H: a minta hígítása N: bemért fa nedvességtartalma (%)
( ) 50(100 )
1000 ( )100
c
a A b HC
G N⋅ + ⋅ ⋅= ⋅ −⋅
Egy adott mintára három párhuzamos mérés történt (Cc1, Cc2, Cc3). Végeredményként az összcukor tartalmak átlagát (Ctc_átlag) és a tapasztalati szórást (s) kell megadni mintánként. A

27
tapasztalati szórás kiszámolása a következő képlettel történik:
2 2 21 2 3( ) ( ) ( )
1cátlag c cátlag c cátlag cC C C C C C
sn
− + − + −=
−
s: szórás n: párhuzamos mérések száma (3) Cc: összcukor tartalom átlaga (mg glükóz/g száraz fa) Cc1, Cc2, Cc3: párhuzamos mérések
Kromatográfiás vizsgálatok A kromatográfia keverékek egyes komponenseinek elválasztására szolgáló eljárás. Azon alapszik, hogy a különböző anyagok különböző fázisokhoz való eltérő affinitásuk és diffúziós tulajdonságaik miatt megoszlást mutatnak a két fázis (egy ún. „állófázis” és egy ún. „mozgófázis”) között. A két fázis közt beálló dinamikus egyensúlyi állapotok azt eredményezik, hogy az egyes komponensek gyorsabban, vagy lassabban vándorolnak az állófázison. Az elválasztás jelenségét eredményező jelenségek alapján a kromatográfiás eljárásokat a következő csoportokba soroljuk be: - adszorpciós kromatográfia - megoszlásos kromatográfia - ioncserés kromatográfia - affinitás kromatográfia - gélkromatográfia Vékonyréteg kromatográfia A vékonyréteg kromatográfia elve A vékonyréteg-kromatográfia (VRK) olyan elválasztási technika, amelyhez egy üveg-, fém- vagy műanyaghordozón vékony rétegben (0.1-0.2 mm) egyenletesen szétterített, megfelelő állófázist alkalmazunk (szilikagél, alumínium-oxid, stb.). A vizsgálandó anyagok oldatait a kifejlesztés előtt felvisszük az állófázisra. Ezután a réteglapot kifejlesztő kamrába helyezzük, melybe előzőleg a kifejlesztés céljára leginkább alkalmas oldószer elegyet (mozgófázis) juttatunk. Az elválasztás (amely adszorpción, megoszláson, ioncserén vagy ezen folyamatok kombinációján alapul) úgy történik, hogy a mozgófázis a kapilláris erők következtében az állófázison előre halad, eközben az egyes komponensek a fázisokhoz való eltérő affinitásuk következtében ugyanabba az irányba haladnak, azonban egymástól eltérő sebességgel, ezáltal adott kifejlesztési idő alatt eltérő utat tesznek meg. A VRK analízis lépései 1. Minta felvitele A vizsgálandó folyadékelegyet kapilláris vagy fecskendő segítségével felvisszük a lemezre. A mintafelvitel helye a réteglap aljától 5-6 mm-re, szélétől kb 1.5-2 cm-re történjen. A felvitt pontok/sávok közt 0.5-1.5 cm távolságot érdemes tartani nehogy a sávok összefolyjanak a kifejlesztés során.

28
pontszerű mintafelvitel sávszerű mintafelvitel 2. Kifejlesztés A felvitt mintákat tartalmazó lemezt egy jól zárható üvegedénybe (kifejlesztő kád) helyezzük, mely tartalmazza az előzetesen kiválasztott mozgófázist. Fontos, hogy a felcseppentés helyének a folyadék felszíne fölött kell lennie. Az oldószer a kapilláris hatás következtében vándorol előre a rétegen. Finomszemcsés (HPTLC minőségű) réteglap és felszálló kifejlesztés esetén a lemezt addig hagyjuk a kifejlesztő kádban, amíg a mozgófázis frontja a réteglap aljától számított 6 cm-t el nem érte. Ezután vegyük ki a lapot a kádból és szárítsuk meg, lehetőleg hideg, vagy langyos levegőáramban (pl. hajszárítóval).
Felszálló kifejlesztő kád Horizontális kifejlesztő kád (nyíl: kifejlesztés iránya) (nyíl: kifejlesztés iránya) 3. Előhívás, megjelenítés A megszárított rétegen ezután láthatóvá kell tenni az elválasztott komponenseket a további értékeléshez. Szerencsés esetben az elválasztott és keresett komponensek színesek (pl. klorofill, festékanyagok, stb.) ám ez a ritkább eset ezért különböző eljárásokat kell alkalmazni a láthatóvá tételhez: UV-fényben vizsgáljuk a lemezt, vagy a keresett vegyületcsoportokra alkalmazható reagensekkel (előhívószer) lepermetezzük a lemezt.
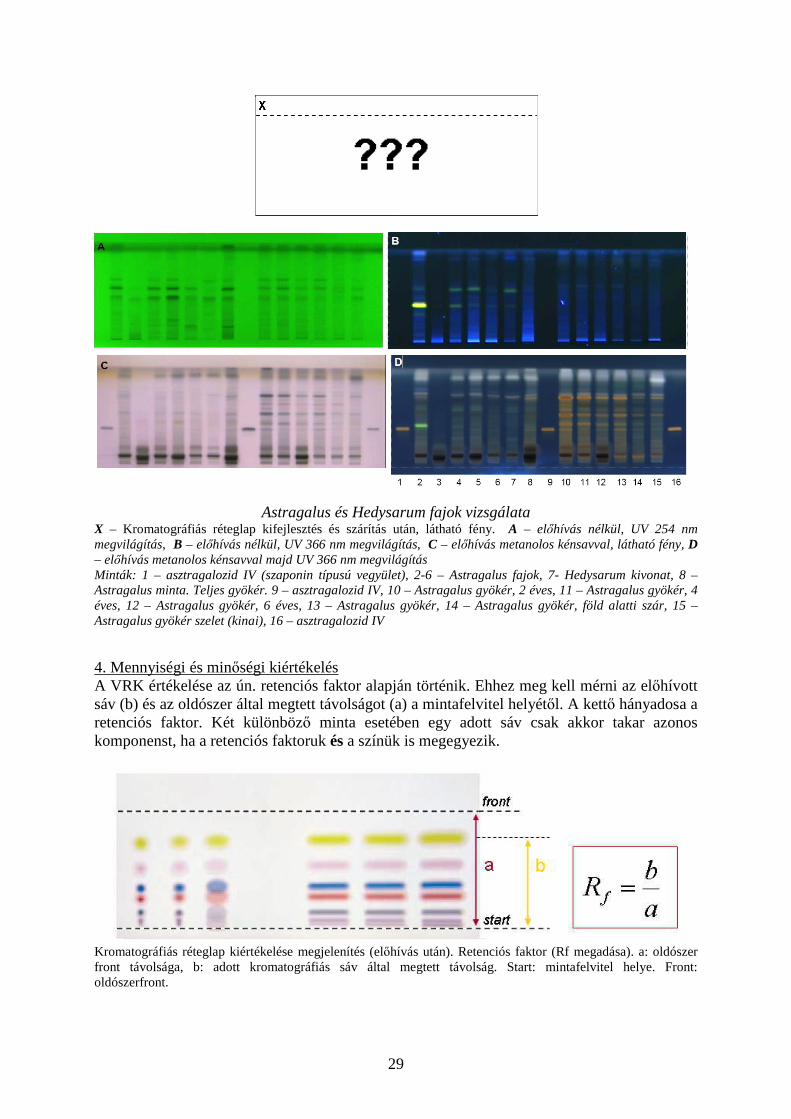
29
Astragalus és Hedysarum fajok vizsgálata X – Kromatográfiás réteglap kifejlesztés és szárítás után, látható fény. A – előhívás nélkül, UV 254 nm megvilágítás, B – előhívás nélkül, UV 366 nm megvilágítás, C – előhívás metanolos kénsavval, látható fény, D – előhívás metanolos kénsavval majd UV 366 nm megvilágítás Minták: 1 – asztragalozid IV (szaponin típusú vegyület), 2-6 – Astragalus fajok, 7- Hedysarum kivonat, 8 – Astragalus minta. Teljes gyökér. 9 – asztragalozid IV, 10 – Astragalus gyökér, 2 éves, 11 – Astragalus gyökér, 4 éves, 12 – Astragalus gyökér, 6 éves, 13 – Astragalus gyökér, 14 – Astragalus gyökér, föld alatti szár, 15 – Astragalus gyökér szelet (kinai), 16 – asztragalozid IV 4. Mennyiségi és minőségi kiértékelés A VRK értékelése az ún. retenciós faktor alapján történik. Ehhez meg kell mérni az előhívott sáv (b) és az oldószer által megtett távolságot (a) a mintafelvitel helyétől. A kettő hányadosa a retenciós faktor. Két különböző minta esetében egy adott sáv csak akkor takar azonos komponenst, ha a retenciós faktoruk és a színük is megegyezik.
Kromatográfiás réteglap kiértékelése megjelenítés (előhívás után). Retenciós faktor (Rf megadása). a: oldószer front távolsága, b: adott kromatográfiás sáv által megtett távolság. Start: mintafelvitel helye. Front: oldószerfront.

30
Faanyagok azonosítása vékonyréteg kromatográfiával Feladat Különböző fajok faanyagainak azonosítása a faanyag (geszt) extraktumának vékonyréteg kromatográfiás elválasztása, előhívása és a kapott kromatogram minőségi kiértékelése alapján. Extrakció 0.10 g darált és szitált faanyagot vagy furnért (0.2-0.63 mm-es szemcse frakcióból) egy Eppendorf-csőbe bemérünk, majd 1 ml 80%-os metanol oldattal ultrahangos fürdőn folyamatosan extraháljuk 30 percig. Az extrakumot Whatman GF/A üvegszálas szűrőpapíron megszűrjük és az így kapott szűrletet alkalmazzuk a vékonyréteg kromatográfiás analízishez. Mintafelvitel Állófázisként 20 cm x 10 cm-es méretű, finomszemcsés (HPTLC minőségű) szilikagél réteget használunk. Az extraktumból 30 µlitert kapillárissal a rétegre felviszünk. A réteglap aljától 6 mm-re sávszerűen vigyük fel a mintákat kb 15-20 mm hosszúságban. A réteglap bal szélétől 15 mm-re kezdjük a mintafelvitelt, az egyes sávok közt legalább 10 mm helyet hagyjunk. Ügyeljünk, hogy az üvegkapilláris ne sértse fel a réteget. A mintafelvitel végén jelöljük be a kívánt kifejlesztési távolságot a réteglapon (6 cm) valamint az egyes felvitt minták számát/nevét ! Kifejlesztés A kifejlesztés telítetlen gőzterű, felszálló kádban történik. A kád egyik vályújába töltsünk 3 ml mozgófázist (87:3:10 etilacetát:víz:hangyasav elegy) fedjük le a kádat,majd hagyjuk kb. 10 percig, hogy a gőztér telítődjön. Ezután a másik vályúba is töltsünk 1.5 ml mozgófázist és ebbe helyezzük bele a réteglapot. A kifejlesztési távolság a lap aljától számítva 6 cm legyen! A kifejlesztés végeztével vegyük ki a réteglapot a kádból és hideg vagy langyos levegőáram segítségével addig szárítsuk, amíg oldószermentes nem lesz !
Felszálló kifejlesztő kád Előhívás A réteglapot egyenletesen fújjuk le a megadott előhívószerrel, ezután vizsgáljuk látható valamint ultraibolya fénynél (254 nm és 366 nm). A kapott kromatogramokat dokumentáljuk fényképpel is !

31
Lefújó egység vékonyréteg kromatográfiás előhíváshoz
Kiértékelés A rétegre felvitt standard minták segítségével azonosítsuk be a vizsgált mintánkat (az azonos színű és retenciós faktorú kromatográfiás sávok alapján). Az azonosítás helyességét erősítsük meg különböző megvilágítási módozatok mellett is. Nagy hatékonyságú folyadék kromatográfia A folyadék kromatográfia elve A nagy hatékonyságú folyadék kromatográfia (HPLC) olyan elválasztási technika, amelyben a porózus állófázist egy oszlopba töltjük és a mozgófázist egy pumpa segítségével áramoltatjuk a mozgófázison keresztül állandó optimális áramlási sebességgel. Az elválasztandó mintát egy injektor segítségével impulzusszerűen az oszlop elejére, az áramló mozgófázisba adagoljuk, ahol azok teljesen feloldódnak a mozgófázisban. A minta komponensei az álló- és mozgófázisok közti eltérő megoszlási tulajdonságuk (ie. kémiai szerkezetük) miatt eltérő sebességgel fognak végighaladni a kromatográfiás oszlopon és eltérő visszatartási (retenciós) idő után mosódnak le (eluálódnak) az oszlopról. Az elválasztott komponenseket az oszlop után kapcsolt detektorral érzékeljük, mely az oszlopról távozó mozgófázis valamely tulajdonságának (fényabszorpció, törésmutató, fluoreszcencia, stb.) mérése alapján érzékeli az eluálódó vegyület minőségét és mennyiségét. Az állófázis polaritása alapján megkülönböztetünk normál (poláros) és fordított (apoláros) fázisú tölteteket. Alkalmazhatók még ioncserés és méretkizárásos elven működő állófázisok is a minta összetételétől és az analitikai feladattól függően. A mozgófázist célszerűen az állófázishoz illetve az elválasztandó komponenseknek megfelelően kell megválasztani.

32
A HPLC mérőrendszer felépítése. A: mozgófázis tartályok; B: keverő szelep és gradienspumpa; C: Túlnyomás szabályozó; D: pulzáláscsökkentő; E: keverőkamra; F: mintabemérő szelep; G: kromatográfiás oszlop (állófázissal töltve); H. vezérlőegység; I: detektor (pl. UV fotométer); J: számítógép interfész (A/D konverter); K: számítógép; L: nyomtató A HPLC analízis lépései 1. Minta injektálása A vizsgálandó homogén folyadékelegy adott térfogatnyi mennyiségét (1-20 µliter) injektáló szelep segítségével impulzusszerűen az áramló mozgófázisba juttatjuk. 1. Elválasztás Az elválasztás adott, konstans térfogatáram mellett történik. A mozgófázis kényszeráramlását az állófázison keresztül nagy nyomású pumpa segítségével lehet megvalósítani. Az áramlás során létrejövő magas nyomásesés (100-1000 bar) következtében a hőmérséklet megemelkedhet (áramló mozgófázis súrlódása az állófázison). A hőmérséklet befolyásolja a megoszlási egyensúlyokat, ti. az elválasztás hatékonyságát, ezért a kromatográfiás rendszert termosztálni szükséges. 2. Detektálás, kiértékelés Az oszlopról lekerülő mozgófázis oldva tartalmazza az egyes elválasztott vegyületeket. A mozgófázis valamely tulajdonságát folyamatosan mérve a benne oldott komponens minőségével és mennyiségével arányos jelet kapunk. A detektorok működési elv alapján lehetnek destruktív (anyagok elbomlanak - pl.: ionizáció elvén működő tömeg szelektív detektorok, származékképző detektorok) és nem destruktív (azonosított anyagok nem bomlanak el – pl: fényabszorciós, fluoreszcens, RI index detektorok) jellegűek. Az egyes vegyületek a kromatogramon csúcsok formájában képeződnek le. A csúcs retenciós ideje az adott komponens minőségére, a csúcs magassága illetve a csúcs alatti terület (intergál) a mennyiségére jellemző.
A HPLC mér őrendszer felépítése

33
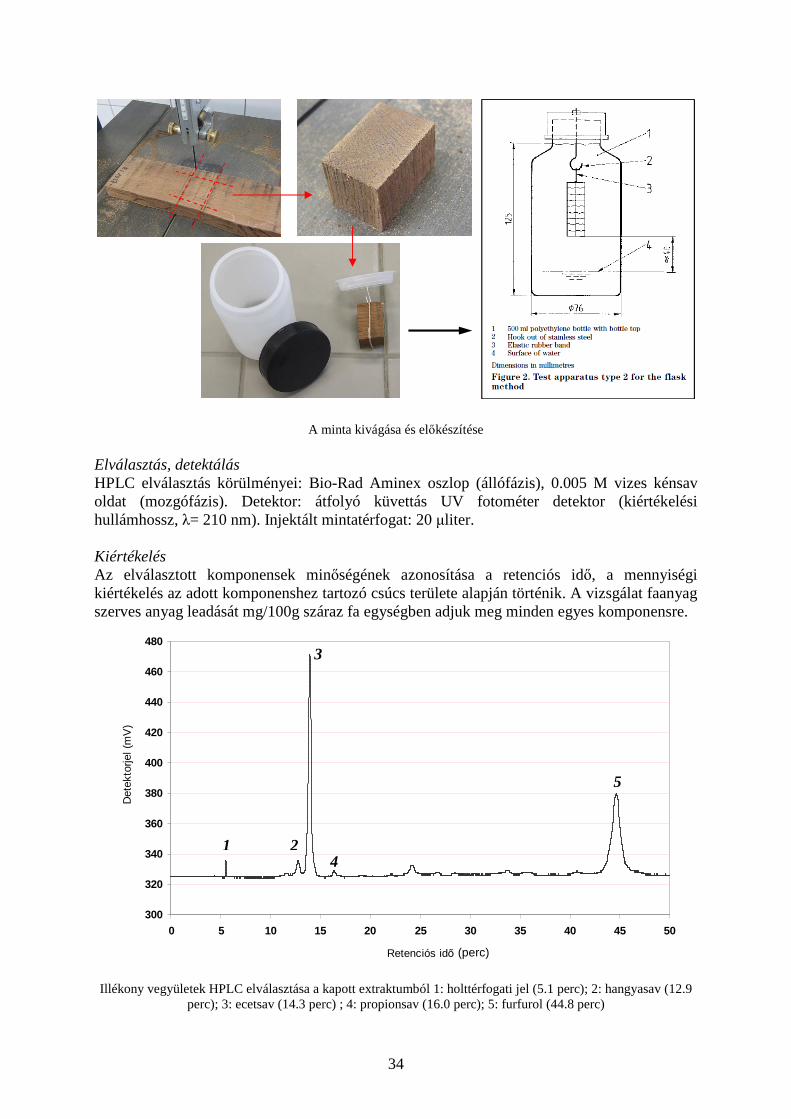
HPLC kromatogram és az elválasztás körülményei Termikusan kezelt faanyagok illékony anyag leadásának mérése Feladat A kezelt faanyagok, falemezek illetve forgácslapok esetében egészségügyi, minőségi és technológiai szempontból egyaránt fontos a különböző a termékből felszabaduló illékony vegyületek minőségének és mennyiségének vizsgálata. A termikusan kezelt faanyagok illékony sav- és aldehid leadásának vizsgálata az EN 717-3, 1996 szabvány alapján. Azonosítsuk a leadott szerves komponenseket és adjuk meg a leadási érték átlagát és a mérés tapasztalati szórását minden egyes azonosított komponensre! Minta előkészítés, extrakció A szabványnak megfelelően 2.5 cm x 2.5 cm x 4 cm méretű fatestet kivágunk a vizsgált lap közepéből. A fatestet 500 ml-es zárható polietilén edénybe helyezzük melybe előzőleg 50 ml desztillált vizet öntöttünk. Az edényt lezárjuk és 24 órán keresztül 40 oC-on tartjuk. Ez alatt a fából emittálódó vízben jól oldódó poláros karakterű illékony szerves anyagok (többnyire savak és aldehidek) a vízben elnyelődnek (abszorbeálódnak). Az inkubációs idő leteltével a folyadékot leöntjük és szűrjük, a kapott folyadékból a sav és aldehid tartalom közvetlenül folyadékkromatográfiásan meghatározható. Az alkalmazott előkészítési és extrakciós módszer előnye, hogy a külső körülmények betartásával jól reprodukálható illetve a faanyagból csak a vizsgált vegyületek kivonása történik meg, egyéb járulékos komponenseké és mátrixanyagoké nem. Ez a tény az elválasztást és kromatográfiás kiértékelést rendkívül megkönnyíti.
Retenciós id ő (perc)
Detektor jel (mV)
34
A minta kivágása és előkészítése Elválasztás, detektálás HPLC elválasztás körülményei: Bio-Rad Aminex oszlop (állófázis), 0.005 M vizes kénsav oldat (mozgófázis). Detektor: átfolyó küvettás UV fotométer detektor (kiértékelési hullámhossz, λ= 210 nm). Injektált mintatérfogat: 20 µliter. Kiértékelés Az elválasztott komponensek minőségének azonosítása a retenciós idő, a mennyiségi kiértékelés az adott komponenshez tartozó csúcs területe alapján történik. A vizsgálat faanyag szerves anyag leadását mg/100g száraz fa egységben adjuk meg minden egyes komponensre.
300
320
340
360
380
400
420
440
460
480
0 5 10 15 20 25 30 35 40 45 50
Retenciós idő
Det
ekto
rjel (
mV
)
Illékony vegyületek HPLC elválasztása a kapott extraktumból 1: holttérfogati jel (5.1 perc); 2: hangyasav (12.9 perc); 3: ecetsav (14.3 perc) ; 4: propionsav (16.0 perc); 5: furfurol (44.8 perc)
1 2
3
4
5
(perc)

35
1. A retenciós idők alapján azonosítsuk az oldatban található komponenseket! 2. Mintánként 3 párhuzamos mérést végezve határozza meg az egyes komponensek csúcsterületeit! Meghatározandó minden egyes komponens esetében a mért három párhuzamos mérésből az adott komponens leadási értékének átlaga és a mérés tapasztalati szórása. 2.1 Egy adott komponensre a mért csúcsterület (mV*sec) és az extraktumban előforduló koncentráció (mg/liter) közti összefüggések (kalibrációs kiértékelő egyenes egyenlete) a következők:
- Ecetsav: c = 0.9931 * csúcsterület + 1.0231 (R2 = 0.9917) - Hangyasav: c = 0.6045 * csúcsterület + 0.7656 (R2 = 0.9997) - Propionsav: c = 0.9341 * csúcsterület + 2.3601 (R2 = 0.9920) - Furfurol: c = 0.13289 * csúcsterület + 1.1024 (R2 = 0.9991) (R2 – a kalibrációs egyenes határozottsági foka) Egy adott komponensre (pl. ecetsav) a leadást a következő képlettel számoljuk:
(0.9931 1.0231) 50100
1001000 ( )
100
ecetsav
TL
NG
⋅ + ⋅= ⋅ −⋅ ⋅
Ahol:
Lecetsav : a minta ecetsav leadása a vizsgált körülmények között (mg/100 g száraz fa) T : az ecetsav csúcsterülete (mVsec) G : bemért faminta tömege (g) N : faminta nedvesség tartalma (%)
2.2 A tapasztalati szórás kiszámolása (pl. ecetsav esetében) a következő képlettel történik:
2 2 2_ 1 _ 2 _ 3( ) ( ) ( )
1ecetsav átlag ecetsav ecetsav átlag ecetsav ecetsav átlag ecetsavL L L L L L
sn
− + − + −=
−
Ahol: Lecetsav_átlag : a minta ecetsav leadásának átlaga (mg/100 g száraz fa) Lecetsav1,2,3 : a minta ecetsav leadása, az egyes párhuzamos mérések (mg/100 g száraz fa) s : tapasztalati szórás (mg/100g száraz fa) n : párhuzamos mérések száma (n=3)

36
Felhasznált irodalmak
• Németh K. - A Faanyag degradációja • Németh K. – Faanyagkémia • Camag application notes HPTLC identification of Astragalus and Hedysarium (www.camag.ch)
• Camag Bibliographic Service: Parameters of Planar Chromatography
(www.camag.ch)




























