Embed Size (px)
Citation preview

J. Pathol. 188: 63–68 (1999)
KERATINOCYTE APOPTOSIS AND p53 EXPRESSION INCUTANEOUS LUPUS AND DERMATOMYOSITIS
. 1*, ̃ 1, 1, . 1, 2 . -3
1Servicio de Reumatología, Hospital 12 de Octubre, Madrid, Spain2Servicio de Anatomía Patológica, Hospital 12 de Octubre, Madrid, Spain
3Servicio de Reumatología, Complejo Hospitalario Universitario de Santiago, Santiago de Compostela, Spain
SUMMARY
Keratinocyte apoptosis may be induced by ultraviolet-B radiation and represents a potential source of fragmented autoantigens inautoimmune diseases. This study investigates whether excessive keratinocyte apoptosis occurs in the skin lesions of cutaneous lupus(CLE) and dermatomyositis (DM) and the potential mechanisms responsible for this phenomenon. Skin biopsies have been studied from19 patients with CLE and DM, eight with scleroderma, and five healthy controls. Apoptosis was detected by in situ end-labelling offragmented DNA. The expression of Bcl-2, PCNA, p53, and Ki-67 proteins was studied by immunohistochemistry. In DM and CLEskin, the number of apoptotic keratinocytes was significantly increased (p=0·008) compared with normal skin. In both diseases, a largeaccumulation of apoptotic keratinocytes and apoptotic bodies was present in the disrupted basal zone. Unlike normal skin, a largenumber of keratinocytes, particularly those morphologically apoptotic, expressed p53 protein. A significant increase in the number ofproliferating Ki-67 positive (p=0·0007) and PCNA-positive (p=0·0008) nuclei was also observed. In both CLE and DM, exaggeratedand inappropriate keratinocyte apoptosis occurs. It is associated with increased expression of p53 and PCNA. This suggests that normalsolar radiation alone or in combination with additional local factors induces DNA damage and excessive keratinocyte apoptosis in theseautoimmune diseases of the skin. Apoptosis can mediate the severe epidermal lesions observed in both diseases and the release offragmented autoantigens into the dermis. Copyright ? 1999 John Wiley & Sons, Ltd.
KEY WORDS—apoptosis; p53; UV radiation; keratinocytes; skin; systemic lupus erythematosus; dermatomyositis; autoimmune diseases
*Correspondence to: Jose L. Pablos, Servico de Reumatologia,Hospital 12 de Octubre, 28041 Madrid, Spain. E-mail:[email protected].
Contract/grant sponsor: Ministerio de Sanidad y Consumo (Spain);Contract/grant number: FIS 98/0356.
Contract/grant sponsor: Ministerio de Educación y Cultura (Spain);Contract/grant number: PM 96/0028.
INTRODUCTION
Dermatomyositis (DM) and systemic lupus erythema-tosus (SLE) are systemic autoimmune diseases withprominent skin involvement. DM is almost invariablyassociated with inflammatory muscle disease, andcutaneous lupus erythematosus (CLE) may occur as amanifestation of SLE or localized to the skin.1,2 In theaffected skin of both DM and CLE, there are non-specific dermal perivascular infiltrates of activatedmacrophages and T-helper lymphocytes,3,4 and dermal–epidermal deposits of immunoglobulins and comp-lement.5,6 These are relatively non-specific findings thatcan be observed in other immune-mediated skin dis-eases. The epidermal lesions are somewhat more specificand can be of help in histopathological diagnosis. Areasof destruction of basal keratinocytes leading to degen-eration and liquefaction of the epidermal basal zone arecharacteristic of both diseases, and evolution to poikilo-dermatous skin with severe epidermal atrophy mayoccur in chronic lesions.1,7
The pathogenesis of the skin lesions of DM and CLEis not fully understood. However, the preferential
CCC 0022–3417/99/060063–06$17.50Copyright ? 1999 John Wiley & Sons, Ltd.
involvement of sun-exposed areas, the worsening oflesions, and the systemic flares of SLE after solar orultraviolet (UV) radiation exposure point to photoexpo-sure as an important element in the pathogenesis of CLEand DM.8,9 The mechanisms of photosensitivity havebeen more extensively studied in CLE, where it has beenproposed that UV radiation may cause exacerbation oflocal and systemic autoimmunity by inducing changes inthe expression and antibody binding of keratinocyteautoantigens.10,11
Exposure of normal keratinocytes in vivo (sunburncells) and in vitro to UV-B radiation induces DNAdamage and apoptosis.12–14 During keratinocyte apop-tosis, nucleosomes and many other autoantigenicnucleoproteins are clustered and specifically fragmentedin the surface blebs of apoptotic cells.14 Thus, apoptoticdebris may be the source of specifically fragmented andpotentially immunogenic autoantigens.14–16 Based onthese observations, Casciola-Rosen et al. hypothesizedthat UV-induced keratinocyte apoptosis may initiateand drive the development of autoimmunity under per-missive conditions.14,17 Nevertheless, keratinocyte apop-tosis occurs physiologically in the skin, associated withthe terminal differentiation of cells becoming incorpor-ated in the stratum corneum.18,19 In keratinocytes andother cell types, UV-induced DNA damage is followedby p53 expression, which may induce either growtharrest, allowing for DNA repair, or apoptosis.20 Prolif-erating cell nuclear antigen (PCNA) expression in non-proliferating nuclei due to nucleotide excision repair isanother marker of this process.21
Received 20 April 1998Revised 22 July 1998
Accepted 8 December 1998

64 J. L. PABLOS ET AL.
In the present work, we investigated whether there isabnormal keratinocyte apoptosis in the lesional skin ofCLE and DM. Unlike in normal skin controls, wedemonstrate that exaggerated and inappropriate kerati-nocyte apoptosis occurs in DM and CLE lesions. It isassociated with p53 expression, increased proliferation,and PCNA expression. This suggests that normal solarradiation, alone or in combination with additionallocal factors, induces DNA damage and excessivekeratinocyte apoptosis in these autoimmune skindiseases.
MATERIALS AND METHODS
Skin biopsies
Skin biopsies were taken from clinically active lesionsof eight DM and 11 CLE (discoid lesions in all cases)patients. All but one were from photoexposed areas. Thepatients were Caucasians with an age range from 21 to76 years (mean 42&17 years). A previous history ofphotosensitivity was present in five CLE and two DMpatients. At the time of biopsy, three CLE patients hadsystemic manifestations of SLE and four DM patientshad myopathy.
Five normal skin biopsies were taken from disease-free edges of surgically excised benign epidermal naevi,or from surgical cosmetic interventions of normal skin.In all cases, biopsies were from photoexposed skin ofindividuals with an age range from 21 to 57 years (mean32&16 years). To exclude non-specific consequences ofdermal inflammation, we also studied nine skin biopsiesfrom active lesions of patients with localized or systemicscleroderma, all containing dermal inflammatory infil-trates. Their ages ranged from 27 to 73 years (mean47&13 years). Formalin-fixed, paraffin-embedded skinsections of all biopsies were used for the followingstudies.
Immunohistochemistry
Antigen retrieval was performed by microwave heat-ing (3#5 min at 750 W in 1 m EDTA, pH 8) priorto p53 and Ki-67 staining. Staining was performedfollowing the indirect avidin–biotin horseradishperoxidase method for PCNA, Bcl-2, and Ki-67, andalkaline phosphatase for p53 (ABC standard, VectorLaboratories, Inc., Burlingham, CA, U.S.A.). Colourwas developed with diaminobenzidine or fast red(Vector Laboratories, Inc.) respectively. The followingantibodies and dilutions were used: anti-PCNA clonePC10 at 1:1000 (Boehringer Mannheim, Mannheim,Germany), anti-Ki-67 clone MM-1 at 1:50 (Novocastra,Newcastle upon Tyne, U.K.), a specific rabbit poly-clonal anti-Bcl-2 (N-19) at 1:500 (Santa CruzBiotechnology, Santa Cruz, CA, U.S.A.), and the anti-p53 monoclonal DO-1 recognizing the wild-type protein,at 1:20 dilution (Santa Cruz Biotechnology). Controlswith normal serum instead of primary antibodywere included. Sections were counterstained withhaematoxylin.
Copyright ? 1999 John Wiley & Sons, Ltd.
In situ detection of apoptotic cells
We performed the terminal transferase mediateddUTP end labelling technique (TUNEL) as described byGavrileli et al., with minor modifications.22 Sectionswere permeabilized with 0·5 per cent Triton X-100 inPBS for 10 min at room temperature. The labellingmixture consisted of 30 m Trizma base (pH 7·2),140 m sodium cacodylate, 1 m cobalt chloride,3 n dATP, and 0·3 n FITC-12-dUTP containing0·3 U/ìl of terminal deoxynucleotidyl transferase (TdT)(Boehringer Mannheim). It was applied to the sectionsfor 30 min at 37)C in a humid chamber. The reactionwas stopped by adding 0·5 EDTA. Sections werewashed three times in PBS and freshly observed byfluorescence microscopy. Control sections omitting TdTwere included. In some cases, sections were furtherdeveloped with an alkaline phosphatase-conjugatedanti-fluorescein antibody and NBT/BCIP as substrate.
Quantitation and statistical analysis
To compare the numbers of immunostained orTUNEL-labelled cells in normal and diseased skin, theabsolute number of labelled nuclei per field at #200magnification was counted. This covers an area ofepidermis that includes 38&1·6 basal cells. At least fiverandomly selected fields of each of three sectionsper biopsy were counted and the mean was used forstatistical analysis. Means and standard deviationswere calculated and compared by the Wilcoxontwo-sample test.
RESULTS
Apoptotic keratinocytes were detected by TUNEL inthe epidermis of all normal and diseased skin sections bystrong fluorescence labelling or alkaline phosphaseimmunostaining that contrasted with a weak back-ground of normal nuclei (Fig. 1). In normal skin,apoptotic cells were mainly granular layer cells, scarceand confined to upper epidermal layers, as previouslydescribed.18 Apoptotic nuclei had a flattened or pycnoticappearance, but nuclear fragmentation or apoptoticbodies were not observed (Figs 1A and 1B). In CLE andDM skin, the numbers of apoptotic cells were signifi-cantly larger than in controls (p=0·0008; Table I).Increased numbers of apoptotic nuclei were mainlyobserved in two areas. First, in their normal location inthe upper epidermal layers, and second, in basal andsuprabasal layers, where they were never observed innormal skin (Figs 1C–1F). In contrast with normal skin,nuclear fragmentation and apoptotic bodies were fre-quently observed. Apoptotic bodies were abundantalong the basal cell layer, particularly in areas withsevere liquefaction (Figs 1C and 1E). To explorewhether the above changes were non-specific conse-quences of dermal inflammation, skin samples of local-ized or systemic scleroderma containing inflammatoryinfiltrates and taken from similarly photoexposed areaswere TUNEL-stained. The number and pattern of
J. Pathol. 188: 63–68 (1999)
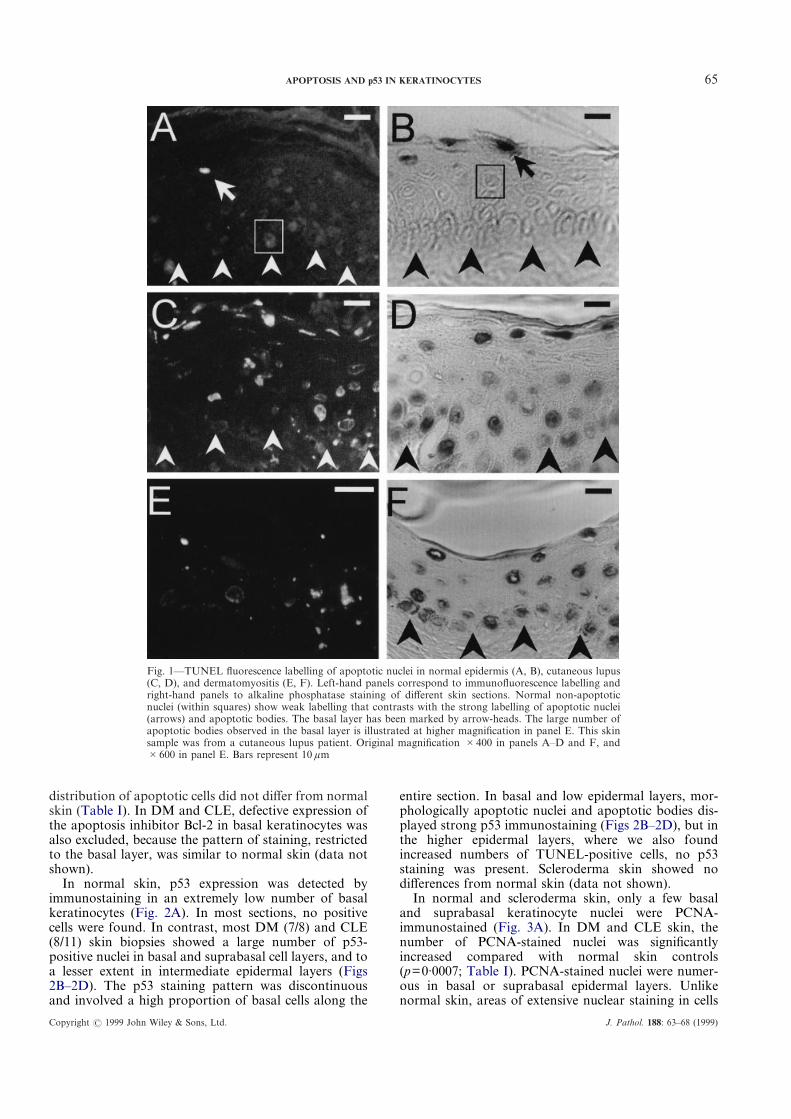
65APOPTOSIS AND p53 IN KERATINOCYTES
Fig. 1—TUNEL fluorescence labelling of apoptotic nuclei in normal epidermis (A, B), cutaneous lupus(C, D), and dermatomyositis (E, F). Left-hand panels correspond to immunofluorescence labelling andright-hand panels to alkaline phosphatase staining of different skin sections. Normal non-apoptoticnuclei (within squares) show weak labelling that contrasts with the strong labelling of apoptotic nuclei(arrows) and apoptotic bodies. The basal layer has been marked by arrow-heads. The large number ofapoptotic bodies observed in the basal layer is illustrated at higher magnification in panel E. This skinsample was from a cutaneous lupus patient. Original magnification #400 in panels A–D and F, and#600 in panel E. Bars represent 10 ìm
distribution of apoptotic cells did not differ from normalskin (Table I). In DM and CLE, defective expression ofthe apoptosis inhibitor Bcl-2 in basal keratinocytes wasalso excluded, because the pattern of staining, restrictedto the basal layer, was similar to normal skin (data notshown).
In normal skin, p53 expression was detected byimmunostaining in an extremely low number of basalkeratinocytes (Fig. 2A). In most sections, no positivecells were found. In contrast, most DM (7/8) and CLE(8/11) skin biopsies showed a large number of p53-positive nuclei in basal and suprabasal cell layers, and toa lesser extent in intermediate epidermal layers (Figs2B–2D). The p53 staining pattern was discontinuousand involved a high proportion of basal cells along the
Copyright ? 1999 John Wiley & Sons, Ltd.
entire section. In basal and low epidermal layers, mor-phologically apoptotic nuclei and apoptotic bodies dis-played strong p53 immunostaining (Figs 2B–2D), but inthe higher epidermal layers, where we also foundincreased numbers of TUNEL-positive cells, no p53staining was present. Scleroderma skin showed nodifferences from normal skin (data not shown).
In normal and scleroderma skin, only a few basaland suprabasal keratinocyte nuclei were PCNA-immunostained (Fig. 3A). In DM and CLE skin, thenumber of PCNA-stained nuclei was significantlyincreased compared with normal skin controls(p=0·0007; Table I). PCNA-stained nuclei were numer-ous in basal or suprabasal epidermal layers. Unlikenormal skin, areas of extensive nuclear staining in cells
J. Pathol. 188: 63–68 (1999)

66 J. L. PABLOS ET AL.
of the higher epidermal layers were also observed (Fig.3C). In scleroderma, no significant differences fromnormal skin were found (p=0·41).
Since PCNA expression may increase due to either cellproliferation or alternative mechanisms such as DNAexcision repair, we examined the expression of thespecific proliferation marker Ki-67.23 In DM and CLE,we also found a significant increase in the number ofKi-67-positive nuclei compared with normal skin(p=0·0008; Table I). Ki-67-positive nuclei were presentonly in basal and suprabasal layers. In scleroderma skin,the number of Ki-67-positive nuclei was not significantlydifferent from normal skin (p=0·67; Table I).
Table I—Numbers of TUNEL-, PCNA-, and Ki-67-stainednuclei in skin sections of DM, CLE, and normal or sclero-derma controls
TUNEL PCNA Ki-67
CLE (n=11) 25·7&14·1 15·7&6·3 7·3&3·6DM (n=8) 33·3&14·5 13·5&5·4 7·2&3·4DM+CLE (n=19) 28·9&14·4* 14·8&5·9† 7·3&3·5‡Normal controls (n=5) 2·6&1·5 3·0&2·1 0·8&0·8Scleroderma (n=9) 3·7&2·0 2·3&1·6 1·3&1·0
Data represent means&standard deviations.*p=0·0008 vs. controls.†p=0·0007 vs. controls.‡p=0·0008 vs. controls.Differences in TUNEL, PCNA, and Ki-67 scores were also signifi-
cant in separate analyses of DM or CLE groups versus normalcontrols.
DISCUSSION
Autoantibodies against nucleosomes and differentnuclear proteins are the hallmark of SLE and othernon-organ-specific autoimmune diseases. Why there is abreakdown of tolerance to this restricted group ofautoantigens is unknown. Recently, a susceptibility tospecific cleavage by apoptosis-dependent nucleases andproteases of these autoantigens has been described.14–17
This has focused our attention on apoptotic cells aspotential immunogens. Apoptosis, however, occursphysiologically in many normal tissues. If apoptosis isan immunizing event in autoimmune disease, it shouldbe excessive or aberrant to provide the necessary con-centration of autoantigens to induce a T-cell-drivenhumoral response.24 It should also happen in a favour-able immune context and in a genetically appropriatehost. The ability of UV radiation to induce keratinocyteapoptosis and the effects of solar exposure on the courseof autoimmune diseases such as SLE or DM point to theskin as one of the relevant places to study thisabnormality.12–14 In both diseases, severe epidermal cellloss characterizes chronic lesions.1,3 We have foundextensive keratinocyte apoptosis in the skin lesions ofCLE and DM. Furthermore, many apoptotic bodieswere concentrated in areas of severe basal disruption,readily available to antigen-presenting cells in an inflam-
Copyright ? 1999 John Wiley & Sons, Ltd.
matory environment. Interestingly, a recent study hasshown that apoptotic keratinocyte blebs are removedthrough direct binding to C1q complement factor, in-dependently of antibodies.25 This provides a rationalefor the strong link between C1q deficiency and thedevelopment of SLE, in the context of keratinocyteapoptosis as a source of autoantigens. Since many othergenetic defects of complement factors (C1r, C1s, C2, andC4) are also associated with SLE,26 the possible partici-pation of these complement factors in the removal ofapoptotic fragments warrants further studies.
A low rate of proliferation of basal cells makes up forthe apoptotic loss of keratinocytes reaching terminaldifferentiation during normal epidermal homeostasis. Inmost cell types, proteins of the Bcl-2 family regulate thesusceptibility of cells to apoptosis. In epidermis, pro-and anti-apoptotic Bcl-2 family members have a typicalgradient of expression that favours apoptosis as the cellsreach the higher epidermal layers.27,28 Inflammatoryskin lesions such as psoriasis or lichen planus arecharacterized by increased rates of proliferation andapoptosis of keratinocytes, which are probably relatedto decreased Bcl-2 expression.29 Defective Bcl-2 expres-sion in basal keratinocytes of DM or CLE was notobserved in our study. Nor did keratinocyte apoptosisseem to be a non-specific consequence of dermal inflam-matory cell infiltration, because it was not present in theskin lesions of scleroderma. It is of note that the nuclearautoantigens recognized by scleroderma autoantibodiesare not cleaved by apoptosis dependent-proteases.30
p53 protein is another important regulator of epider-mal cell dynamics. Behaving as a tumour suppressorgene,20,31 its expression is critical in restraining cellproliferation after genetic damage. Expression of p53protein does not occur in normal skin. Expression of p53in skin may be due to two different mechanisms. First,DNA damage induced by UV radiation may transientlyincrease the expression of normal p53 protein.32 Second,p53 mutations induce permanent expression of non-functional p53 protein.33 Small numbers of epidermalcells expressing mutated p53 and clustered in smallpatches (clones) can be observed in normal individualsand increase with age, particularly in photoexposedareas.33 Epidermal p53 expression is known to happenin normal skin, after experimental or solar UVradiation-induced DNA damage.12 In this case, p53expression involves not only basal and suprabasalkeratinocytes, but also cells in the upper layers, in amosaic pattern.12,34 It is accompanied by increasedexpression of PCNA, a protein involved in unscheduledDNA synthesis during DNA repair.21 In our study, thepattern of p53 staining points to DNA damage as one ofthe potential mechanisms inducing wild-type p53 expres-sion in CLE and DM. The increase in PCNA-positivekeratinocytes above the suprabasal layer also supportsthis view, although mechanisms different from DNAdamage may also induce PCNA expression innon-proliferating cells in inflammatory diseases.35
The normal cellular effect of p53 can be either growtharrest, through the expression of the cyclin-dependentkinase inhibitor p21, or apoptosis.20 In the presentcase, increased apoptosis seems to be the p53-induced
J. Pathol. 188: 63–68 (1999)
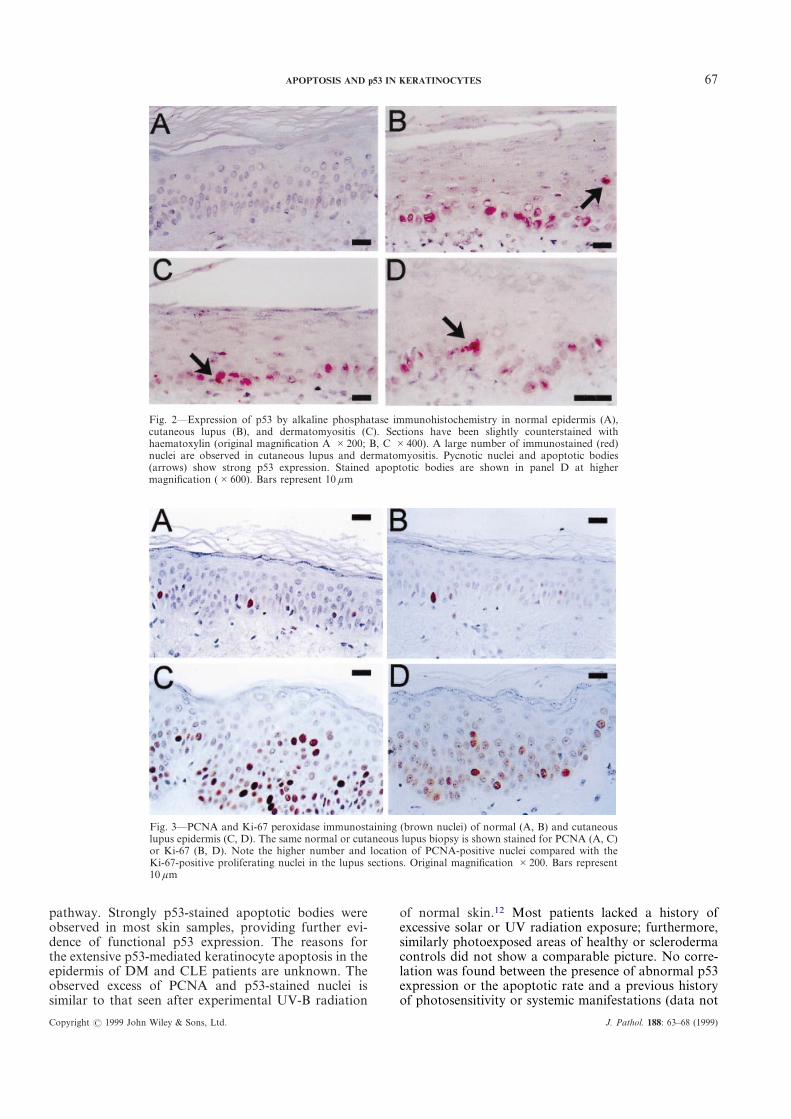
67APOPTOSIS AND p53 IN KERATINOCYTES
Fig. 2—Expression of p53 by alkaline phosphatase immunohistochemistry in normal epidermis (A),cutaneous lupus (B), and dermatomyositis (C). Sections have been slightly counterstained withhaematoxylin (original magnification A #200; B, C #400). A large number of immunostained (red)nuclei are observed in cutaneous lupus and dermatomyositis. Pycnotic nuclei and apoptotic bodies(arrows) show strong p53 expression. Stained apoptotic bodies are shown in panel D at highermagnification (#600). Bars represent 10 ìm
Fig. 3—PCNA and Ki-67 peroxidase immunostaining (brown nuclei) of normal (A, B) and cutaneouslupus epidermis (C, D). The same normal or cutaneous lupus biopsy is shown stained for PCNA (A, C)or Ki-67 (B, D). Note the higher number and location of PCNA-positive nuclei compared with theKi-67-positive proliferating nuclei in the lupus sections. Original magnification #200. Bars represent10 ìm
pathway. Strongly p53-stained apoptotic bodies wereobserved in most skin samples, providing further evi-dence of functional p53 expression. The reasons forthe extensive p53-mediated keratinocyte apoptosis in theepidermis of DM and CLE patients are unknown. Theobserved excess of PCNA and p53-stained nuclei issimilar to that seen after experimental UV-B radiation
Copyright ? 1999 John Wiley & Sons, Ltd.
of normal skin.12 Most patients lacked a history ofexcessive solar or UV radiation exposure; furthermore,similarly photoexposed areas of healthy or sclerodermacontrols did not show a comparable picture. No corre-lation was found between the presence of abnormal p53expression or the apoptotic rate and a previous historyof photosensitivity or systemic manifestations (data not
J. Pathol. 188: 63–68 (1999)

68 J. L. PABLOS ET AL.
shown). Increased susceptibility of keratinocyte nuclei tonormal levels of UV-B radiation might explain ourfindings. Several studies suggest that inflammatorycytokines such as interferon-gamma or tumour necrosisfactor may enhance p53 expression and apoptosis inkeratinocytes.36,37 They could potentially amplify thep53 response to UV-B radiation in keratinocytesexposed to dermal inflammation in autoimmune skindiseases.
ACKNOWLEDGEMENTS
This work was supported by grants FIS 98/0356 fromthe Ministerio de Sanidad y Consumo and PM 96/0028from the Ministerio de Educación y Cultura (Spain).
REFERENCES1. Provost TT, Watson R. Pathogenesis of cutaneous manifestations of lupus
erythematosus. In: Wallace DJ, Hahn D, eds. Dubois’ Lupus Erythemato-sus. 4th edn. Philadelphia: Lea & Febiger, 1993: 279–301.
2. Bradley WG, Tandan R. Inflammatory diseases of muscle. In: Kelley WN,Harris ED, Jr Ruddy S, Sledge CB, eds. Textbook of Rheumatology. 3rdedn. Philadelphia: W B Saunders, 1989: 1263–1287.
3. Andrews BS, Schenk A, Barr R, Friou G, Mirick G, Ross P. Immuno-pathology of cutaneous human lupus erythematosus defined by murinemonoclonal antibodies. J Am Acad Dermatol 1986; 15: 474–481.
4. Hausmann G, Herrero C, Cid MC, Casademont J, Lecha M, Mascaro JM.Immunopathologic study of skin lesions in dermatomyositis. J Am AcadDermatol 1991; 25: 225–230.
5. Burnham TK, Neblett TR, Fine G. Application of fluorescent antibodytechnique to the investigation of lupus erythematosus and various derma-toses. J Invest Dermatol 1963; 41: 451–456.
6. Nesbitt LT Jr. Cutaneous immunofluorescence in dermatomyositis. Int JDermatol 1980; 19: 270–278.
7. Janis JF, Winkelmann RK. Histopathology of the skin in dermatomyositis:a histopathologic study of 55 cases. Arch Dermatol 1968; 97: 640–650.
8. Cheong WK, Hughes RV, Norris PG, Hawk JL. Cutaneous photosensi-tivity in dermatomyositis. Br J Dermatol 1984; 131: 205–208.
9. Cohen MR, Isenberg A. Ultraviolet irradiation in systemic lupus erythema-tosus: friend or foe? Br J Rheumatol 1996; 35: 1002–1007.
10. Furukawa F, Kashihara-Sawami M, Lyons MB, Norris DA. Binding ofantibodies to the extractable nuclear antigens of SS-A/Ro and SS-B/La isinduced on the surface of human keratinocytes by ultraviolet light (UVL):implications for the pathogenesis of photosensitive cutaneous lupus. J InvestDermatol 1990; 94: 77–85.
11. Golan TD, Elkon KB, Gharavi AE, Krueger JG. Enhanced membranebinding of autoantibodies to cultured keratinocytes of systemic lupuserythematosus patients after ultraviolet B/ultraviolet A radiation. J ClinInvest 1992; 90: 1067–1076.
12. Coates PJ, Save V, Ansari B, Hall PA. Demonstration of DNA damage/repair in individual cells using in situ end labelling: association of p53 withsites of DNA damage. J Pathol 1995; 176: 19–26.
13. Schwarz A, Bhardwaj R, Aragane Y, et al. Ultraviolet-B-induced apoptosisof keratinocytes: evidence for partial involvement of tumor necrosis factor-alpha in the formation of sunburn cells. J Invest Dermatol 1995; 104:922–927.
14. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemiclupus erythematosus are clustered in two populations of surface structureson apoptotic keratinocytes. J Exp Med 1994; 179: 1317–1330.
Copyright ? 1999 John Wiley & Sons, Ltd.
15. Casciola-Rosen LA, Anhalt GJ, Rosen A. DNA-dependent protein kinaseis one of a subset of autoantigens specifically cleaved early during apoptosis.J Exp Med 1995; 182: 1625–1634.
16. Casiano CA, Martin SJ, Green DR, Tan EM. Selective cleavage of nuclearautoantigens during CD95 (Fas/APO-1)-mediated T cell apoptosis. J ExpMed 1996; 184: 765–770.
17. Casciola-Rosen L, Rosen A. Ultraviolet light-induced keratinocyteapoptosis: a potential mechanism for the induction of skin lesions andautoantibody production in LE. Lupus 1997; 6: 175–180.
18. Polakowska RR, Piacentini M, Bartlett R, Goldsmith LA, Haake AR.Apoptosis in human skin development: morphogenesis, periderm, and stemcells. Dev Dynam 1994; 199: 176–188.
19. Maruoka Y, Harada H, Mitsuyasu T, et al. Keratinocytes become termi-nally differentiated in a process involving programmed cell death. BiochemBiophys Res Commun 1997; 238: 886–890.
20. Smith ML, Fornace AJ Jr. p53-mediated protective responses to UVradiation. Proc Natl Acad Sci U S A 1997; 94: 12 255–12 257.
21. Aboussekhra A, Wood RD. Detection of nucleotide excision repair inci-sions in human fibroblasts by immunostaining for PCNA. Exp Cell Res1995; 221: 326–332.
22. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed celldeath in situ specific labelling of nuclear DNA fragmentation. J Cell Biol1992; 119: 493–501.
23. Smith MD, Healy E, Thompson V, Morley A, Rees JL. Use of in situdetection of histone mRNA in the assessment of epidermal proliferation:comparison with the Ki-67 antigen and BrdU incorporation. Br J Dermatol1995; 132: 359–366.
24. Radic MZ, Weigert M. Genetic and structural evidence for antigen selectionof anti-DNA antibodies. Annu Rev Immunol 1994; 12: 487–520.
25. Korb LC, Ahearn JM. C1q binds directly and specifically to surface blebs ofapoptotic human keratinocytes. Complement deficiency and systemic lupuserythematosus revisited. J Immunol 1997; 158: 4525–4528.
26. Schur PH. Complement and systemic lupus erythematosus. In: Wallace DJ,Hahn D, eds. Dubois’ Lupus Erythematosus. 4th edn, Philadelphia: Lea &Febiger, 1993: 120–127.
27. Hockenbery DM, Zutter M, Hickey W, Nahm M, Korsmeyer SJ. BCL2protein is topographically restricted in tissues characterized by apoptoticcell death. Proc Natl Acad Sci U S A 1991; 88: 6961–6965.
28. Krajewski S, Krajewska M, Shabaik A, Miyashita T, Wang HG, Reed JC.Immunohistochemical determination of in vivo distribution of Bax, adominant inhibitor of Bcl-2. Am J Pathol 1994; 145: 1323–1336.
29. Bianchi L, Farrace MG, Nini G, Piacentini M. Abnormal Bcl-2 and ‘Tissue’transglutaminase expression in psoriatic skin. J Invest Dermatol 1994; 103:829–833.
30. Casciola-Rosen LA, Anhalt GJ, Rosen A. Scleroderma autoantigens areuniquely fragmented by metal-catalyszed oxidation reactions: implicationsfor pathogenesis. J Exp Med 1997; 185: 71–79.
31. Lane DP. Cancer: p53 guardian of the genome. Nature 1992; 358: 15–16.32. Hall PA, McKee PH, Menage HD, Dover R, Lane DP. High levels of p53
protein in UV-irradiated normal human skin. Oncogene 1993; 8: 203–207.33. Jonason AS, Kunala S, Price GJ, et al. Frequent clones of p53-mutated
keratinocytes in normal human skin. Proc Natl Acad Sci U S A 1996; 93:14 025–14 029.
34. Ren ZP, Ponten F, Nister M, Ponten J. Two distinct p53 immunohisto-chemical patterns in human squamous-cell skin cancer, precursors andnormal epidermis. Int J Cancer (Pred Oncol) 1996; 69: 174–179.
35. Harrison RF, Reynolds GM, Rowlands DC. Immunohistochemical evi-dence for the expression of proliferating cell nuclear antigen (PCNA) bynon-proliferating hepatocytes adjacent to metastatic tumours and in inflam-matory conditions. J Pathol 1993; 171: 115–122.
36. Brysk MM, Selvanayagam P, Arany I, Brysk H, Tyring SK, Rajaraman S.Induction of apoptotic nuclei by interferon-gamma and by predesquamin incultured keratinocytes. J Interferon Cytokine Res 1995; 15: 1029–1035.
37. Schwarz A, Bhardwaj R, Aragane Y, et al. Ultraviolet-B-induced apoptosisof keratinocytes: evidence for partial involvement of tumor necrosis factor-alpha in the formation of sunburn cells. J Invest Dermatol 1995; 104:922–927.
J. Pathol. 188: 63–68 (1999)





























