Embed Size (px)
Citation preview

Latent LytM at 1.3 A Resolution
Sergey G. Odintsov1,2†, Izabela Sabala1,2†, Malgorzata Marcyjaniak1,2
and Matthias Bochtler1,2*
1International Institute ofMolecular and Cell Biology, ul.Trojdena 4, 02-109 WarsawPoland
2Max-Planck-Institute forMolecular Cell Biology andGenetics, Pfotenhauerstr. 10801309 Dresden, Germany
LytM, an autolysin from Staphylococcus aureus, is a Zn2þ-dependent glycyl-glycine endopeptidase with a characteristic HxH motif that belongs to thelysostaphin-type (MEROPS M23/37) of metallopeptidases. Here, wepresent the 1.3 A crystal structure of LytM, the first structure of a lyso-staphin-type peptidase. In the LytM structure, the Zn2þ is tetrahedrallycoordinated by the side-chains of N117, H210, D214 and H293, the secondhistidine of the HxH motif. Although close to the active-site, H291, thefirst histidine of the HxH motif, is not directly involved in Zn2þ-coordi-nation, and there is no water molecule in the coordination sphere of theZn2þ, suggesting that the crystal structure shows a latent form of theenzyme. Although LytM has not previously been considered as a pro-enzyme, we show that a truncated version of LytM that lacks theN-terminal part with the poorly conserved Zn2þ ligand N117 has muchhigher specific activity than full-length enzyme. This observation is con-sistent with the known removal of profragments in other lysostaphin-type proteins and with a prior observation of an active LytM degradationfragment in S. aureus supernatant. The “asparagine switch” in LytM isanalogous to the “cysteine switch” in pro-matrix metalloproteases.
q 2003 Elsevier Ltd. All rights reserved.
Keywords: LytM; proenzyme; metallopeptidase; peptidoglycan hydrolase;Staphylococcus aureus*Corresponding author
Introduction
Metallopeptidases can often be recognised by thepresence of a short conserved signature sequencecontaining histidine and glutamate residues. Themost common motif is HExxH (“zinzins”), butother motifs such as HxxEH (“inverzincins”),HxxE (“carboxypeptidase family”) and HxH (e.g.lysostaphin-like) have also been described.1
HxH metalloproteases of the lysostaphin-type ofpeptidases occur in bacteriophages, in Gram-positive and in Gram-negative bacteria. Onemember sequence has also been found in Anophelesgambiae, the African malaria mosquito.2 Theproteases from bacteriophages and Gram-positivebacteria have been studied most, and were shownto have a preference for peptides containing polygly-cine residues, especially Gly-Gly-Xaa, where Xaa isany aliphatic hydrophobic residue.3 This specificityis consistent with their physiological role: lysosta-
phin-like peptidases from bacteriophages andGram-positive bacteria cleave polyglycine cross-bridges in the peptidoglycan of Gram-positive bac-terial cells.3 Their role in Gram-negative bacteria isless clear: some peptidases, like b-lytic proteasefrom Achromobacter lyticus, target cell walls of Gram-positive bacteria, possibly providing a competitiveadvantage to the producer organism.4 Others seemto have additional roles, like LasA from Pseudomonasaeruginosa that is believed to participate in host elas-tin degradation.5
Lysostaphin-type peptidases from Gram-positivebacteria appear to share the affinity for glycine-richpeptides. Nevertheless, some of them showremarkable specificity. For example, it is knownthat Staphylococcus simulans cell walls are resistantto lysostaphin, which they produce, and that thisresistance is mediated by an increase in the serineand a decrease in the glycine content of thebacterial cell wall.6 In many cases, the presence oflysostaphin-type proteins is coupled with thepresence of a self-resistance mechanism. A goodexample is the millericin B operon fromStreptococcus milleri: the operon encodes millericinB, a peptidoglycan hydrolase, and several otherproteins that add a leucine to the polyglycine
0022-2836/$ - see front matter q 2003 Elsevier Ltd. All rights reserved.
† S.O. and I.S. contributed equally to this work.Abbreviation used: E64, trans-epoxysuccinyl-L-
leucylamido-(4-guanidino)butane.E-mail address of the corresponding author:
doi:10.1016/j.jmb.2003.11.009 J. Mol. Biol. (2004) 335, 775–785

precursor and thus contribute to self-protection ofthe producer strain.7
Although such arrangements are common, thereare also many cases of lysostaphin-like peptido-glycan hydrolases that can degrade the cell walls ofthe producer organism at least in vitro. Such peptido-glycan hydrolases are known as autolysins.8
Although functional redundancy in autolysins hasmade it difficult to assign specific functions to indi-vidual enzymes, autolysins are believed to beinvolved in vegetative growth, peptidoglycan matu-ration, cell wall expansion, cell wall turnover andprotein secretion.8
LytM is an autolysin that was originallyidentified in an autolysis defective mutant ofStaphylococcus aureus.9 Based on its similarity tolysostaphin, the enzyme would be expected to bespecific for glycine-rich sequences, a conclusionthat is supported by the experimental data.Ramadurai et al.10 report that the enzyme releasesfree amino groups, but not reducing sugars fromstaphylococcal cell walls, supporting its classifi-cation as a protease. Moreover, it degrades thecell walls from S. carnosus, but not those fromMicrococcus luteus, leading the authors to speculatethat the enzyme is a glycyl-glycine endopeptidase.LytM activity can easily be assayed byzymography with purified cell walls as the sub-strate. Intriguingly, LytM preparations from bothnative and recombinant sources have beenreported to give rise to three bands in zymography,one band corresponding to the full-length enzyme,and two bands of lower molecular mass that wereconsidered as degradation products.9
LytM and other lysostaphin-type proteins do notshare significant sequence similarity with otherpeptidase families of known structure. In spite ofthe widespread in vitro use of lysostaphin for lysis ofS. aureus cells,11 and although lysostaphin has proveneffective against meticillin-resistant S. aureus strainsin various animal models,12 no crystal or NMR struc-ture for any lysostaphin-type protein is available. Toour knowledge, even the complete set of Zn-ligandshas not been determined.
Here, we present the 1.3 A crystal structure of full-length LytM, the first structure of a lysostaphin-typemetallopeptidase. We report the complete set of Zn2þ
ligands in LytM, and because this set deviates fromprior speculations in the literature, confirm their roleby site-directed mutagenesis. Finally, we demon-strate that a truncated form of LytM that roughly cor-responds to the mature form of other lysostaphin-related proteases4,5,13 has much higher specificactivity than full-length protein, strongly suggestingthat the lytM gene encodes a preproenzyme.
Results
LytM expression, purificationand crystallisation
Consistent with the extracellular location of the
protein, the gene for LytM encodes an export sig-nal at the N terminus. According to SignalP,14 thisleader sequence comprises the first 23 amino acidresidues. For heterologous expression in Escherichiacoli, a cleavage site two residues downstream of thephysiological cleavage site is predicted. Here,LytM was expressed with a histidine tag upstreamof the natural LytM export sequence. Remarkably,the histidine-tag modified leader sequence wascleaved in E. coli, resulting in a protein with theN-terminal sequence AETTN… as shown by ESImass spectrometry and protein fingerprinting, con-sistent with the SignalP prediction. The proteinwas purified and crystallised as described inMaterials and Methods. The structure was thensolved by multiple wavelength anomalous diffrac-tion (MAD) on a single selenomethionine-contain-ing crystal exploiting the anomalous signal fromboth the zinc and the selenium atoms.
LytM fold
The crystal structure shows that LytM is a two-domain protein. The N-terminal domain (shownat the bottom of Figure 1) is not conserved inlysostaphin-type proteins. Its ordered part inLytM comprises residues 45 to roughly 98 and canbe described as a mixed b-sheet that is formed bytwo b-hairpins connected by an a-helix. TheN-domain makes only very limited contacts withthe C-domain, and none of its residues is close tothe metal centre.
The C-domain (shown at the top of Figure 1)comprises residues 99–316 and can be dividedinto two ordered regions that are located upstreamand downstream of the disordered segment fromresidue 147 to residue 182. The region upstream ofthe disordered segment is poorly conserved andmostly coiled with the exception of one helix. Itanchors only one of the four Zn2þ ligands, N117(dotted lines in Figure 1). In stark contrast, theregion downstream of the disordered segment iswell-conserved and rich in secondary structureelements (continuous lines in Figure 1). Its fold isarranged around a central, six-stranded anti-parallel b-sheet. In Richardson nomenclature,15 thetopology of this sheet can be described as þ1,þ4x, 21, 21, 21. With the exception of N117, allligands to the Zn2þ are anchored on this sheet orin surrounding loops. A second, much smallerantiparallel b-sheet runs essentially parallel withthe main b-sheet, but none of its residuescontributes to the metal centre (continuous lines inFigure 1).
The zinc-binding site
Although no exogenous Zn2þ was added torecombinant LytM, the Zn2þ site in the crystals isfully occupied. The identification of the metal ionas Zn2þ was confirmed by X-ray fluorescence thatshowed the expected absorption maximum at theK-edge of Zn2þ at 1.283 A. Contrary to the common
776 Latent LytM

assumption that both histidine residues of the HxHsignature sequence are ligands for the Zn2þ,2 allatoms in the imidazole ring of the first histidine ofthe HxH motif are at least 4 A away from the Zn2þ
and thus clearly not directly involved in Zn2þ
chelation.As shown in Figure 2, the Zn2þ in our crystals is
tetrahedrally coordinated by N117, H210, D214and H293, the second histidine residue of theHxH motif. Both histidine residues that act asligands for the Zn2þ donate a hydrogen bond fromthe imidazole nitrogen that does not contact themetal. This common, so-called elec-His-Zn motiforients the imidazole ring in space and is believedto make it more basic and thus a better ligand tothe metal.16 In H210, the imidazole N1 coordinatesthe Zn2þ, and the Nd donates a hydrogen bond tothe carbonyl oxygen of P200. In H293, it is the Nd
that coordinates the Zn2þ, and the N1 that donatesa hydrogen bond to the oxygen of the terminal car-boxamide of Q295 (see Figure 2). As elec-His-Znmotifs are thought to be favourable, Q295 shouldbe conserved. This is indeed the case, but with aremarkable twist: in a substantial number of lyso-staphin-type proteins, Q295 is replaced with gluta-mic acid that we presume to be in the sameconformation and to act as a hydrogen bondacceptor. As residue Q295 is located just two resi-dues downstream of the HxH motif, the signaturesequence is better described as HxHxQ/E.
LytM latency
The most unusual feature of the LytM crystalstructure is the lack of an ordered water moleculein the coordination sphere of the Zn2þ. At 1.3 A
Figure 1. Stereo Ca-trace of the LytM structure. The N and C-domains are located at the bottom and top, respectively.Residues of the N-domain and of the C-domain upstream of the disordered segment are represented by dotted lines(45–146). The Ca-trace for residues downstream of the disordered segment (183–314) is presented as drawn lines andcorresponds roughly to the active protease. Side-chains for some important active-site residues are also presented.
Figure 2. Stereo view of the Zn2þ-binding site in LytM. The main-chain trace is drawn with bold lines, side-chains arepresented with thin lines. The signature sequence HxHxE/Q runs from left to right in the foreground, and containsH291, H293 and Q295. Interactions between the Zn2þ and its ligands are shown with broken lines, and hydrogenbonds that are donated from histidine residues 210, 291 and 293 are presented as dotted lines.
Latent LytM 777

resolution, the absence of electron density for anordered water molecule that could ligand the Zn2þ
is significant, all the more so since the tetrahedralgeometry of Zn2þ coordination by four amino acidligands leaves no space for a water molecule orsubstrate carbonyl oxygen to contact the Zn2þ
directly. Thus, the metal centre cannot be catalyticin the form found in the crystal structure.
As the evidence in the literature for a metal-dependent mechanism of lysostaphin-type pro-teins activity is firm10,17 and because we could notfind any alternative proteolytic dyad or triad, wefocused on the possibility of structural rearrange-ments of the active-site for peptidase activation.Several lines of evidence suggested that the aspara-gine residue N117 was most likely to be involvedin the changes. Firstly, systematic databasesearches show that histidine is the most commonligand for Zn2þ, followed by glutamic acid, asparticacid and cysteine.18 In contrast, asparagine is anunusual ligand for a catalytic Zn2þ site. Secondly,the two Zn2þ chelating histidine residues in LytMare strongly conserved, the aspartic acid ismoderately conserved, and the asparagine is notconserved at all (alignment not shown). Thirdly,the asparagine is the only Zn2þ ligand that islocated upstream of a large, 40 residue segment inthe crystal structure that is disordered, suggestingthat N117 itself could be easily moved withoutdisrupting structural integrity of the protein.Finally and most importantly, a profragmentroughly comprising the residues upstream andinside the disordered sequence in our crystal struc-ture is known to be cleaved off in other lyso-staphin-type peptidases (alignments not shown).4,19
Further supporting this idea, we noted thatdegradation fragments in our LytM preparationsappeared to be active (data not shown). Catalyti-cally active degradation products in LytMpreparations from both native and recombinantsources have been noted before.9 Based on theirmigration behaviour in zymography, Ramaduraiet al. assigned molecular masses of 19 kDa and22 kDa.9 Consistent with their report, we also finddegradation products in our recombinant prep-arations from E. coli. However, mass spectrometryyields molecular masses of 14.8 kDa for the domi-nant fragment, substantially less than assigned byRamadurai et al.9 and implying a cleavage sitedownstream of the asparagine Zn2þ ligand.
Experimental LytM activation
We therefore hypothesized that a C-terminalfragment of LytM that lacks the asparagine ligandto the Zn2þ should be more active than the wild-type. For the definition of the C-terminal fragment,we took guidance from the crystal structure andexpressed the fragment of LytM downstream ofthe disordered region in our crystals with anN-terminal histidine tag. The resulting proteincomprises residues 185–316 of the wild-typesequence that together have a molecular mass of14.4 kDa. A slightly longer, 14.7 kDa fragmentcould be generated by trypsin-mediated cleavageof full-length LytM. Cleavage occurs betweenK179 and A180, consistent with the preference oftrypsin for basic residues in P1 position of the sub-strate. As a control, the complete C-domain (LytM99-316) that contains all four amino acid ligands tothe Zn2þ was also tested (for an overview of theconstructs, see Figure 3).
The results of a thin layer chromatography(TLC)-based pentaglycine degradation assay withequimolar amounts of the different proteins arepresented in Figure 4A. Under assay conditions,full-length LytM has no detectable activity (seelane LytM full-length). The same is true of the trun-cated protein that lacks the entire N-domain, butstill contains the full C-domain including theasparagine ligand N117 (see lane LytM 99-316). Inmarked contrast, the protein that begins just down-stream of the disordered region in LytM and lacksN117 shows clear activity and seems to split penta-glycine into di- and triglycine (see lane LytM 185-316). The same result is obtained when LytM isprocessed with trypsin (see lane LytM þ trypsin).A control reaction confirms that trypsin itself isinactive against the pentaglycine substrate, asexpected from its specificity (not shown).
Gratifyingly, the active truncation mutant LytM185-316 shows the inhibitor sensitivity that wouldbe expected for a metallopeptidase (see Figure 4B).10 mM EDTA, 1 mM 1,10-phenanthroline, glycinehydroxamate the divalent metal ions Zn2þ andHg2þ inhibit the activity. In contrast, the epoxide-based cysteine protease inhibitor E64 has noeffect, and PMSF inhibits only partially. It hasbeen stated in the literature that (NH4)2SO4 andglucosamine inhibit the activity of LytM.10 Con-sistent with this report, 10 mM of either (NH4)2SO4
Figure 3. Overview of the domainorganisation of LytM and of thetruncation constructs that were gen-erated for activity tests. Domainboundaries are indicated with longvertical bars. Grey boxes indicateordered regions in the crystal struc-ture. On the right, the molecularmasses of the different constructsare listed.
778 Latent LytM

or N-acetylglucosamine, the variant ofglucosamine that is part of the muramic acidpeptidoglycan backbone, cause substantial LytMinhibition, although a clear residual activityremains in our hands.
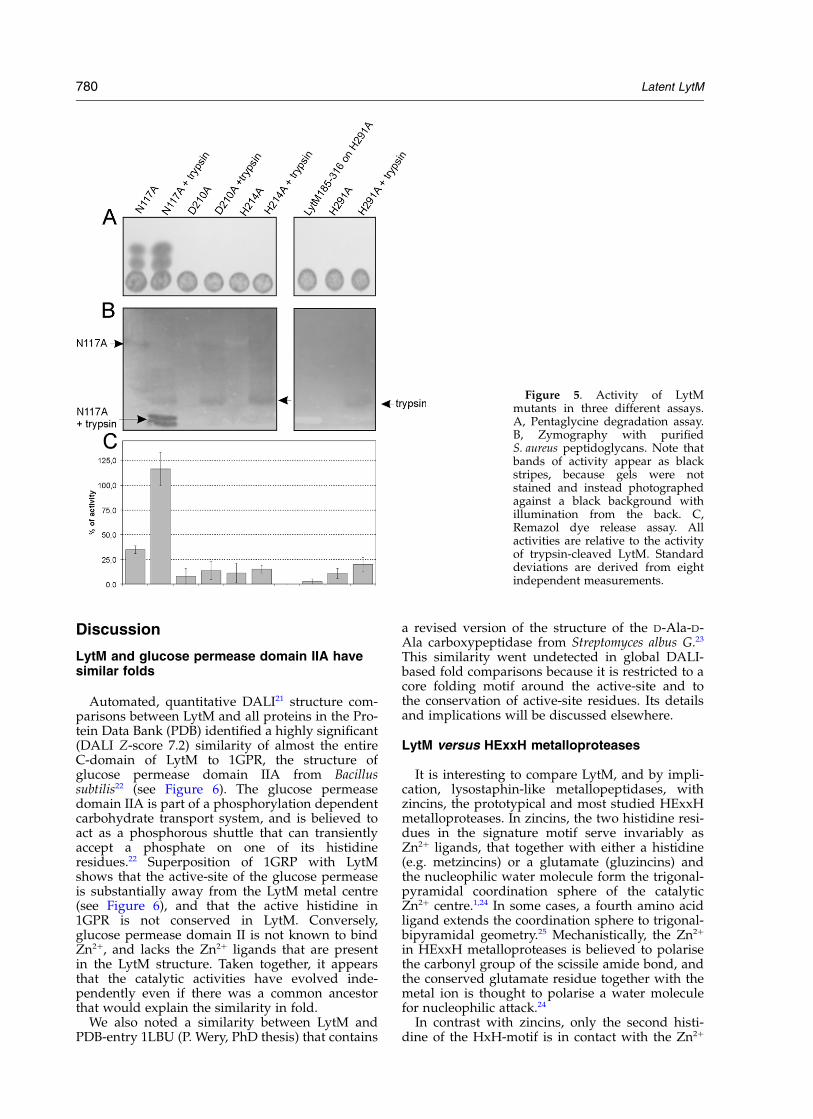
Mutagenesis of the Zn21 chelating asparagine
To our knowledge, an asparagine-mediatedlatency mechanism is novel in metalloproteases.To test our prediction that the asparagine has aninhibitory role, we replaced the asparagine Zn2þ
ligand with alanine. Consistent with our expec-tations, we find that the full-length mutant hasactivity in three different functional assays. Theactivity is robust in the TLC pentaglycine degra-dation assay (see Figure 5A), and clearly abovebackground in zymography and the Remazol dyeassays (see Figure 5B and C). We assume that thehigher substrate concentration in the TLC assayhelps to displace the mutant inhibitory region ofthe enzyme. For the zymography and Remazoldye assays, the substrate concentration is muchlower, and the active-site is probably still partiallyoccluded by the N-terminal part of the enzyme,even though the specific interaction with the Zn2þ
has been disrupted. Trypsin cleavage enhances theactivity of the N117A mutant to the level observedwith cleaved wild-type enzyme (see Figure 5C,note that all activities are relative to cleaved wild-type enzyme). This is expected, because theC-terminal cleavage products from wild-typeenzyme and mutant enzyme are identical.
Mutagenesis of Zn21 chelating residues inactive LytM
The three Zn2þ ligands that we expect to bepresent in active enzyme, H210, D214 and H293,are conserved in lysostaphin-type sequences, butall three occur outside the YxHx(11)Vx(12/20)Gx(5-6)H10,20 signature motif for lysostaphin-type peptidases, and two of them, H210 and D214,
have not been considered as Zn2þ ligands beforeto the best of our knowledge. Instead, it was specu-lated that the two histidine residues of the HxHsignature motif would contact the Zn2þ directly,2
both because of an analogy with carboanhydrase4
and because the first histidine of the HxH motifwas shown to be essential for activity.19
To test the relevance of the LytM structure for thesituation in solution, we mutated the three Zn2þ
ligands individually to alanine in the context offull-length LytM. All three constructs could beexpressed, and two of them, H210A and D214Ayielded soluble proteins. Consistent with ourexpectations, the mutant proteins were inactiveboth before and after the trypsin cleavage step inthe pentaglycine (see Figure 5A, left panel), zymo-graphy (see Figure 5B, left panel) and Remazoldye (see Figure 5C) assays. The third mutant,H293A was expressed in good yield, but wasentirely insoluble. An attempt to express theH293A mutation in the context of truncatedLytM 185-316 again gave insoluble protein, clearlyshowing the importance of H293 for foldingand supporting a role for this residue in Zn2þ
chelation.
Mutagenesis of the first histidine of theHxH motif
The first histidine of the HxH motif, H291, isclearly not a ligand for the Zn2þ in the LytM crystalstructure. Nevertheless, its strong conservationamong lysostaphin-type peptidases clearlysuggests a functional role for this residue. To testthis hypothesis, we replaced this histidine with ala-nine. Consistent with our expectation, the resultantLytM mutant is devoid of activity in the penta-glycine assay (see Figure 5A, right panel) and inzymography (see Figure 5B, right panel), bothbefore and after truncation with trypsin. Consistentwith these results, at best residual activity isobserved in the Remazol dye assay (see Figure 5C).
Figure 4. Thin-layer chromatography pentaglycine digestion assay. A, Comparison of the activities of full-lengthLytM, the C-domain of LytM 99-316, the region downstream of the disordered segment of LytM 185-316 and of tryp-sin-cleaved LytM. B, Inhibitor sensitivity of truncated LytM 185-316. Unlike all other inhibitors, glycine hydroxamatestains with ninhydrin. Therefore, a control lane “glycine hydroxamate” (without the þ sign) with glycine hydroxamateonly has been included in the panel. The lane þ glycine hydroxamate (with the þ sign) shows the result of mixing theinhibitor with enzyme and substrate, just as for all other inhibitors.
Latent LytM 779
Discussion
LytM and glucose permease domain IIA havesimilar folds
Automated, quantitative DALI21 structure com-parisons between LytM and all proteins in the Pro-tein Data Bank (PDB) identified a highly significant(DALI Z-score 7.2) similarity of almost the entireC-domain of LytM to 1GPR, the structure ofglucose permease domain IIA from Bacillussubtilis22 (see Figure 6). The glucose permeasedomain IIA is part of a phosphorylation dependentcarbohydrate transport system, and is believed toact as a phosphorous shuttle that can transientlyaccept a phosphate on one of its histidineresidues.22 Superposition of 1GRP with LytMshows that the active-site of the glucose permeaseis substantially away from the LytM metal centre(see Figure 6), and that the active histidine in1GPR is not conserved in LytM. Conversely,glucose permease domain II is not known to bindZn2þ, and lacks the Zn2þ ligands that are presentin the LytM structure. Taken together, it appearsthat the catalytic activities have evolved inde-pendently even if there was a common ancestorthat would explain the similarity in fold.
We also noted a similarity between LytM andPDB-entry 1LBU (P. Wery, PhD thesis) that contains
a revised version of the structure of the D-Ala-D-Ala carboxypeptidase from Streptomyces albus G.23
This similarity went undetected in global DALI-based fold comparisons because it is restricted to acore folding motif around the active-site and tothe conservation of active-site residues. Its detailsand implications will be discussed elsewhere.
LytM versus HExxH metalloproteases
It is interesting to compare LytM, and by impli-cation, lysostaphin-like metallopeptidases, withzincins, the prototypical and most studied HExxHmetalloproteases. In zincins, the two histidine resi-dues in the signature motif serve invariably asZn2þ ligands, that together with either a histidine(e.g. metzincins) or a glutamate (gluzincins) andthe nucleophilic water molecule form the trigonal-pyramidal coordination sphere of the catalyticZn2þ centre.1,24 In some cases, a fourth amino acidligand extends the coordination sphere to trigonal-bipyramidal geometry.25 Mechanistically, the Zn2þ
in HExxH metalloproteases is believed to polarisethe carbonyl group of the scissile amide bond, andthe conserved glutamate residue together with themetal ion is thought to polarise a water moleculefor nucleophilic attack.24
In contrast with zincins, only the second histi-dine of the HxH-motif is in contact with the Zn2þ
Figure 5. Activity of LytMmutants in three different assays.A, Pentaglycine degradation assay.B, Zymography with purifiedS. aureus peptidoglycans. Note thatbands of activity appear as blackstripes, because gels were notstained and instead photographedagainst a black background withillumination from the back. C,Remazol dye release assay. Allactivities are relative to the activityof trypsin-cleaved LytM. Standarddeviations are derived from eightindependent measurements.
780 Latent LytM

in LytM. The strong, although not universal con-servation of the first histidine of this motif clearlysuggests a catalytic role for this residue. In supportof this hypothesis, we have shown that replace-ment of the first histidine of the HxH motif withalanine abolishes LytM activity in three differentassays. The LytM result is consistent with a pre-vious mutagenesis study on LasA, another lyso-staphin-type peptidase, which concluded thatH120 of LasA, the equivalent of H291 of LytM andthe first histidine of the HxH motif, is essential foractivity.19 Several interpretations of this result arepossible: as LytM contains no glutamate residue inthe active-site, it is tempting to speculate that thefirst conserved histidine of the HxH motif is thefunctional equivalent of the glutamate in HExxHmetalloproteases and thus has a role in the acti-vation of the incoming water molecule. However,we caution that another conserved histidine inlysostaphin-type peptidases, H260 in LytM, isequally close to the metal centre and could alsoplay this role. In the absence of a crystal structureof active LytM in complex with a substrate ana-logue, other less likely roles for one or both ofthese histidine residues remain possible, forexample in the stabilisation of a reactionintermediate.
An asparagine switch?
Like LytM, matrix metalloproteases (MMPs) aresecreted in a latent form. MMP activation occursby a variety of disparate means, including proteasetreatment, conformational perturbants such assodium dodecyl sulphate, heavy metals such as
Hg2þ and sulphhydryl-alkylating agents such asN-ethylmaleimide.26 All these activation mechan-isms were traced back to the displacement of acysteine residue of the profragment from the cata-lytic Zn2þ centre, an activation mechanism thatvan Wart and Birkedal-Hansen termed the“cysteine switch”.26 The cysteine switch mechan-ism has been confirmed by structural studies. Inthe proforms of both latent gelatinase A (pro-MMP-2)27 and latent stromelysin-I (pro-MMP-3),28
the sulphur of a cysteine of the profragment isdirectly liganded to the catalytic Zn2þ. In themature forms, the place of the cysteine sulphurs istaken by a solvent molecule or by a Zn2þ chelatinggroup of an inhibitor.28,29 Because of the analogywith the cysteine switch in MMPs, we suggest theterm “asparagine switch” for the latency mechan-ism of LytM. The poor conservation of the aspara-gine in lysostaphin-like enzymes makes it likelythat residues other than asparagine can act as theswitch in other family members, so that thebroader latency mechanism of lysostaphin-typepeptidases will eventually turn out to be a “Zn2þ
ligand switch”.
Is full-length LytM active?
The prior literature on LytM consistentlyassumes that the full-length enzyme is the activespecies.9,10 Experimentally, Ramadurai et al. haveshown that their LytM preparations give rise tothree bands in zymography, one corresponding tothe full length enzyme and two bands at 19 kDaand 22 kDa that they identify as degradationproducts.9 To our knowledge, these authors never
Figure 6. Superposition of the Ca traces of LytM (red lines) and glucose permease domain IIA (blue lines). The corre-spondence between the two proteins extends essentially over the entire C-terminal domain of LytM. In both structures,every tenth residue is marked with a dot. The Zn2þ in LytM is shown as a red sphere with black centre, and the histi-dine in glucose permease domain IIA that can be phosphorylated is drawn in ball-and-stick representation.
Latent LytM 781

quantified the specific activity of the different pep-tidase forms. In our hands, the full-length proteinhas residual activity at best, and only the truncatedenzyme yields a robust zymography signal (datanot shown). The published zymography resultswith equally intense bands for all three forms ofprotease could thus be due to a large amount ofLytM and a small amount of degraded enzyme.Alternatively, it is possible that activation occurredduring the renaturation step after denaturing gelelectrophoresis, although this does not happen toan appreciable extent in our hands. We also notedthat the specific activity of full-length LytM prep-arations grows over time, especially in samplesthat were contaminated with other proteins fromE. coli. Gratifyingly, this increase in activity corre-lates with increasing amounts of degraded LytMin these preparations.
Is LytM processed in vivo?
The much higher specific activity of truncatedLytM suggests that the enzyme could be processedin vivo, an assumption that would be consistentwith the known processing to activate other lyso-staphin-type proteins.4,5,13 Nevertheless, the con-clusion is not inevitable. LytM is thought to be anautolysin with a role in actively growing anddividing cells.10 As the enzyme acts on the pro-ducer organism, a tight control of its activityshould therefore be desirable, and transientactivation could happen through mechanismsother than proteolysis. It would be conceivablethat full-length LytM is an enzyme with a “lid”that is displaced by certain substrates only,although the near-complete lack of activity of theenzyme in our assays does not support this con-clusion. The disordered LytM region in the crystalstructure and the processing of LytM by the non-physiological activator trypsin and by proteasecontaminations from the heterologous host E. coliall suggest that many proteases from S. aureuswould probably also activate LytM in vitro. Thus,to fully confirm the profragment hypothesis, theexact cleavage that occurs in vivo will have to bereproduced in vitro. Alternatively, the profragmenthypothesis could be proven by a knockout thatabolishes LytM processing in vivo. Work in thisdirection is under way.
Materials and Methods
Cloning, protein expression and purification
LytM sequences from various strains of S. aureus havebeen deposited in sequence databases, and two alterna-tive start codons have been assigned.9,30 We favour thesecond ATG as the start codon because of the presenceof a Shine-Dalgarno sequence ten nucleotides upstreamand assume for counting purposes that the translationproduct has the sequence MKKL… and refer to themethionine upstream of the two lysine residues as resi-due 1. Standard PCR techniques were used to amplifythe lytM gene from genomic DNA of S. aureus strainNCTC8325. The gene was cloned into a derivative ofpET15b, named pET15bmod, that lacks the originalEco RI site and contains a newly introduced Eco RI sitein place of the original thrombin cleavage site. For pro-tein expression, BL21 (DE3) cells carrying the plasmidwere grown to an A600 of 0.7, induced with 1 mM IPTGand shifted to 28 8C for up to six hours. For expressionof the selenomethionine variant of the protein, the plas-mid was transformed into BL834 (DE3), the methionineauxotrophic parent strain of BL21 (DE3). A small scaleovernight culture was grown in NMM medium contain-ing 0.3 mM L-methionine, and used to inoculate the fullscale culture in NMM containing 0.3 mM D,L-seleno-methionine. Cells were grown to an A600 of 1.5, andkept at 28 8C for six hours after induction with 1 mMIPTG.31
Cells were harvested and resuspended in buffer A(20 mM Tris (pH 7.5), 50 mM NaCl). After sonificationand clarificiation of the lysate, the supernatant wasapplied to a DEAE Sepharose FF column (AmershamPharmacia, 30 ml column volume) equilibrated in bufferA. The protein was recovered 90% pure in the flowthrough. After concentration (Amicon 10 kDa cut-offregenerated cellulose filters), the protein was subjectedto a gel filtration step on Sephacryl S-300 HR (AmershamPharmacia) in 5 mM Tris (pH 7.5). LytM migrated as amonomer. The selenomethionine variant behaved indis-tinguishably from the wild-type. Therefore, massspectrometry was used to confirm the incorporation ofselenomethionine prior to synchrotron data collection.
Crystallisation
Crystals were grown in sitting drops at 21 8C byequilibrating a 1:1 mixture of protein (around 40 mg/mlin 5 mM Tris, pH 7.5) and reservoir buffer againstreservoir buffer containing 170 mM ammoniumsulphate, 25.5% (w/v) PEG 8K and 15% (v/v) glycerol.Crystals appeared after three or four days, belongedto space group P12(1)1 with cell constants
Table 1. Data collection and refinement statistics
Data collection statistics Refinement statistics
Space group P12(1)1 R-factor (%) 16.4a (A) 45.33 R-free (%) 18.1b (A) 53.23 rmsd bond distance (A) 0.01c (A) 51.60 rmsd angles (deg.) 1.3b 104.02 B (isotropic) from Wilson 12.7Independent reflections 54 733Resolution (A) 1.3 Ramachandran core (%) 92.3Completeness (%) 94 Ramachandran additionally all (%) 7.7Rsym (%) (last shell in brackets) 6.2 (17.4) Ramachandran generously all (%) 0.0I=s (last shell in brackets) 6.4 (3.5) Ramachandran disallowed (%) 0.0
782 Latent LytM

45.33 A £ 53.23 A £ 51.60 A, 908 £ 104.028 £ 908 and con-tained one molecule of LytM per asymmetric unit. Theycould be flash-frozen directly from mother liquor anddiffracted in-house to about 1.7 A and to 1.3 A on BL2,BESSY, Berlin. For the deposited dataset, the high-resolution pass was collected on BESSY and the low-resolution pass in-house on a separate specimen. Thecrystallographic quality factors for the combined datasetare excellent and are summarised in Table 1.
Structure determination
The structure was determined by two-wavelengthMAD on a single selenomethionine crystal, collectingdata at the absorption maximum of Se (l ¼ 0:979857 �A;f 0exp ¼ 27:8; f 0exp ¼ 4:3) and Zn (l ¼ 1:282713 �A; f 0exp ¼27:2; f 0exp ¼ 4:0) to 1.9 A resolution. Anomalous differ-ence Patterson maps showed very clear contrast forboth absorption maxima, with one Zn-site standing outin the l ¼ 1:282713 �A anomalous map. Cross-phasingthe anomalous data at the selenium absorption edgewith phases derived from the Zn-site identified seleniumpositions in the anomalous difference Fourier map withvery clear signal, but sites on both hands were stillpresent. Selenium–selenium cross-vectors in the l ¼0:979857 �A anomalous difference Patterson map wereused to split the set of candidate selenium positions intotwo sets with consistent hand, giving four possibleheavy atom arrangements for the phasing procedure:two choices for the hand of the Zn site, and then twochoices each for the hand of the selenium sites. Onechoice led to a very clear map that could be interpretedautomatically by the ARP/WARP procedure, while allthree other choices gave uninterpretable maps. The finalmodel comprises 234 residues, namely residues 45–146and residues 183–314 (the initiator methionine is takenas residue 1). This implies that 19 residues that arechemically present at the N terminus of our moleculeare disordered. The same applies for the 36 residues147–182 and for the two most C-terminal residues. Inspite of the rather large number of disordered residues,Rcryst and Rfree at 1.3 A resolution are satisfactory (seeTable 1). The model has reasonable stereochemistry andthe sites of methionine sulphur atoms are consistentwith the selenium positions used for phasing, oncemore confirming the sequence assignment.
Mutagenesis
LytM mutants N117A, H210A, D214A and H293Awere generated by PCR-based site-directed mutagenesisaccording to the Stratagene protocol with Pfu TurboDNA polymerase (Stratagene). Truncated versions ofLytM were generated by cloning corresponding PCRfragments into the expression vector pET15mod. Theresulting constructs LytM 99-316 and LytM 185-316started at A99 and H185, respectively. As the H293Amutation in the context of full-length LytM yieldedinsoluble protein, this mutation was also introduced inthe context of the LytM 185-316 construct, but unfortu-nately again yielded only insoluble protein. Full-lengthsoluble point mutants were purified as the wild-typeprotein. Truncated versions were expressed with anN-terminal histidine tag, applied in buffer A to Ni–NTA agarose (Quiagen), washed with 50 mM imidazolein buffer A and eluted with 300 mM imidazole, again inbuffer A and subsequently subjected to a gel filtrationstep on Sephacryl S-300 HR (Amersham Pharmacia).
Trypsin digest
Proteins (20 mg) were digested with 0.1 unit of trypsin(Sigma) for ten minutes at 37 8C. The reaction wasstopped by adding benzamidine to the final concen-tration of 5 mM.
Peptidoglycan isolation
Peptidoglycans were isolated from S. aureusATCC25923. Autoclaved cells were washed twice inbuffer A and then incubated in 4% SDS, first at roomtemperature for 90 minutes and then at 100 8C for 20minutes. Insoluble material was extensively washedwith buffer A and finally resuspended in 10% trichloro-acetic acid (TCA) and incubated at 4 8C for 48 hourswith gentle shaking. Harvested insoluble material wasthen treated with 2 mg/ml of Pronase (Sigma) in bufferA for one hour at 60 8C. After extensive washing in thesame buffer, peptidoglycans were resuspended in bufferA and used for further experiments or stored at 220 8C.
Zymography
The enzyme was assayed for activity on a 12% (w/v)polyacrylamide-sodium dodecyl sulphate gel containing0.2% (w/v) peptidoglycans isolated from S. aureus asdescribed above. About 3 mg of the analysed proteinwas loaded on each lane. The gels were incubated fortwo to four hours at 37 8C in 20 mM Tris–HCl (pH 7.5),containing 2.5% Triton X-100 to permit protein renatura-tion. Lytic zones appeared as clear bands within theopaque gel. For photos, the gel was held on a glassplate against a sheet of black paper, with illuminationfrom the back.
TLC
Pentaglycine (5 mM, Sigma) was incubated for fivehours at 37 8C with enzyme samples (1 mg/ml) in a totalvolume of 20 ml in 10 mM Tris–HCl buffer (pH 7.5).Subsequently, 5 ml samples of the reaction were appliedon a TLC plate covered with Silica Gel 60 (Merck) anddeveloped with a mixture of n-butanol:acetic acid:water(4:1:1, by vol.). To visualise the spots, the plate wassprayed with 0.2% ninhydrin in ethanol and heated to100 8C.
Remazol dye release assay
The Remazol dye release assay was done as describedby Zhou et al.32 Briefly, Remazol Brilliant Blue (RBB,Sigma) derivatives were prepared by suspendinginsoluble peptidoglycans in 250 mM NaOH containing20 mM RBB and incubating them first at 37 8C for sixhours and then at 4 8C for 12 hours. Dyed productswere washed extensively until a colourless supernatantwas obtained. The dye release assays were done for 12hours at 37 8C in buffer A, at a substrate concentrationof 3 mg/ml and at an enzyme concentration of 0.05 mg/ml. Reactions were stopped by adding half the volumeof 96% ethanol, insoluble material was removed by cen-trifugation and the absorbance was measured at 595 nm.
Atomic coordinates
Structure factors and coordinates have been deposited
Latent LytM 783

in the PDB and will be available on publication underaccession code 1QWY.
Acknowledgements
Melanie Stefan is gratefully acknowledged forhelp with the assays during a summer internshipin our laboratory. We are also grateful to the staffof beamline BL2/BESSY, Berlin for generousallocation of beam time and assistance duringdata collection. This work was done with financialsupport from KBN, decision 1789/E-529/SPB/5.PR UE/DZ 600/2002-2005 and from the Com-mission of the European Communities, specificRTD program “Quality of Life and Managementof Living Resources”, QLRT-2001-01250, “Novelnon-antibiotic treatment of staphylococcaldiseases”.
References
1. Hooper, N. M. (1994). Families of zinc metallo-proteases. FEBS Letters, 354, 1–6.
2. Rawlings, N. D., O’Brien, E. & Barrett, A. J. (2002).MEROPS: the protease database. Nucl. Acids Res. 30,343–346.
3. Barrett, A. J., Rawlings, N. D. & Woessner, J. F. (1998).Handbook of Proteolytic Enzymes, Academic Press,London.
4. Li, S. L., Norioka, S. & Sakiyama, F. (1990). Molecularcloning and nucleotide sequence of the beta-lyticprotease gene from Achromobacter lyticus. J. Bacteriol.172, 6506–6511.
5. Peters, J. E. & Galloway, D. R. (1990). Purificationand characterization of an active fragment of theLasA protein from Pseudomonas aeruginosa: enhance-ment of elastase activity. J. Bacteriol. 172, 2236–2240.
6. DeHart, H. P., Heath, H. E., Heath, L. S., LeBlanc,P. A. & Sloan, G. L. (1995). The lysostaphin endo-peptidase resistance gene (epr) specifies modificationof peptidoglycan cross bridges in Staphylococcussimulans and Staphylococcus aureus. Appl. Environ.Microbiol. 61, 1475–1479.
7. Beukes, M. & Hastings, J. W. (2001). Self-protectionagainst cell wall hydrolysis in Streptococcus milleriNMSCC 061 and analysis of the millericin B operon.Appl. Environ. Microbiol. 67, 3888–3896.
8. Smith, T. J., Blackman, S. A. & Foster, S. J. (2000).Autolysins of Bacillus subtilis: multiple enzymeswith multiple functions. Microbiology, 146, 249–262.
9. Ramadurai, L. & Jayaswal, R. K. (1997). Molecularcloning, sequencing, and expression of lytM, aunique autolytic gene of Staphylococcus aureus.J. Bacteriol. 179, 3625–3631.
10. Ramadurai, L., Lockwood, K. J., Nadakavukaren,M. J. & Jayaswal, R. K. (1999). Characterization of achromosomally encoded glycylglycine endo-peptidase of Staphylococcus aureus. Microbiology, 145,801–808.
11. Zygmunt, W. A., Browder, H. P. & Tavormina, P. A.(1967). Lytic action of lysostaphin on susceptible andresistant strains of Staphylococcus aureus. Can.J. Microbiol. 13, 845–853.
12. Climo, M. W., Patron, R. L., Goldstein, B. P. & Archer,
G. L. (1998). Lysostaphin treatment of experimentalmethicillin-resistant Staphylococcus aureus aorticvalve endocarditis. Antimicrob. Agents Chemother. 42,1355–1360.
13. Thumm, G. & Gotz, F. (1997). Studies on prolyso-staphin processing and characterization of the lyso-staphin immunity factor (Lif) of Staphylococcussimulans biovar staphylolyticus. Mol. Microbiol. 23,1251–1265.
14. Nielsen, H., Engelbrecht, J., Brunak, S. & von Heijne,G. (1997). Identification of prokaryotic andeukaryotic signal peptides and prediction of theircleavage sites. Protein Eng. 10, 1–6.
15. Richardson, J. S. (1977). beta-Sheet topology and therelatedness of proteins. Nature, 268, 495–500.
16. Alberts, I. L., Nadassy, K. & Wodak, S. J. (1998).Analysis of zinc binding sites in protein crystalstructures. Protein Sci. 7, 1700–1716.
17. Kessler, E., Safrin, M., Abrams, W. R., Rosenbloom, J.& Ohman, D. E. (1997). Inhibitors and specificity ofPseudomonas aeruginosa LasA. J. Biol. Chem. 272,9884–9889.
18. Vallee, B. L. & Auld, D. S. (1990). Zinc coordination,function and structure of zinc enzymes and otherproteins. Biochemistry, 29, 5647–5659.
19. Gustin, J. K., Kessler, E. & Ohman, D. E. (1996). Asubstitution at His-120 in the LasA protease ofPseudomonas aeruginosa blocks enzymatic activitywithout affecting propeptide processing or extra-cellular secretion. J. Bacteriol. 178, 6608–6617.
20. Sugai, M., Fujiwara, T., Akiyama, T., Ohara, M.,Komatsuzawa, H., Inoue, S. & Suginaka, H. (1997).Purification and molecular characterization of glycyl-glycine endopeptidase produced by Staphylococcuscapitis EPK1. J. Bacteriol. 179, 1193–1202.
21. Holm, L. & Sander, C. (1995). Dali: a network tool forprotein structure comparison. Trends Biochem. Sci. 20,478–480.
22. Liao, D. I., Kapadia, G., Reddy, P., Saier, M. H., Jr,Reizer, J. & Herzberg, O. (1991). Structure of the IIAdomain of the glucose permease of Bacillus subtilis at2.2 A resolution. Biochemistry, 30, 9583–9594.
23. Dideberg, O., Charlier, P., Dive, G., Joris, B., Frere,J. M. & Ghuysen, J. M. (1982). Structure of aZn2 þ -containing D-alanyl-D-alanine-cleaving car-boxypeptidase at 2.5 A resolution. Nature, 299,469–470.
24. Bode, W., Gomis-Ruth, F. X. & Stockler, W. (1993).Astacins, serralysins, snake venom and matrixmetalloproteinases exhibit identical zinc-bindingenvironments (HEXXHXXGXXH and Met-turn)and topologies and should be grouped into acommon family, the “metzincins”. FEBS Letters, 331,134–140.
25. Stocker, W. & Bode, W. (1995). Structural features of asuperfamily of zinc-endopeptidases: the metzincins.Curr. Opin. Struct. Biol. 5, 383–390.
26. Van Wart, H. E. & Birkedal-Hansen, H. (1990). Thecysteine switch: a principle of regulation of metallo-proteinase activity with potential applicability to theentire matrix metalloproteinase gene family. Proc.Natl Acad. Sci. USA, 87, 5578–5582.
27. Morgunova, E., Tuuttila, A., Bergmann, U., Isupov,M., Lindqvist, Y., Schneider, G. & Tryggvason, K.(1999). Structure of human pro-matrix metallo-proteinase-2: activation mechanism revealed. Science,284, 1667–1670.
28. Becker, J. W., Marcy, A. I., Rokosz, L. L., Axel, M. G.,Burbaum, J. J., Fitzgerald, P. M. et al. (1995).
784 Latent LytM

Stromelysin-1: three-dimensional structure of theinhibited catalytic domain and of the C-truncatedproenzyme. Protein Sci. 4, 1966–1976.
29. Feng, Y., Likos, J. J., Zhu, L., Woodward, H., Munie,G., McDonald, J. J. et al. (2002). Solution structureand backbone dynamics of the catalytic domain ofmatrix metalloproteinase-2 complexed with ahydroxamic acid inhibitor. Biochim. Biophys. Acta,1598, 10–23.
30. Baba, T., Takeuchi, F., Kuroda, M., Yuzawa, H., Aoki,K., Oguchi, A. et al. (2002). Genome and virulence
determinants of high virulence community-acquiredMRSA. Lancet, 359, 1819–1827.
31. Budisa, N., Steipe, B., Demange, P., Eckerskorn, C.,Kellermann, J. & Huber, R. (1995). High-level bio-synthetic substitution of methionine in proteins byits analogs 2-aminohexanoic acid, selenomethionine,telluromethionine and ethionine in Escherichia coli.Eur. J. Biochem. 230, 788–796.
32. Zhou, R., Chen, S. & Recsei, P. (1988). A dye releaseassay for determination of lysostaphin activity. Anal.Biochem. 171, 141–144.
Edited by R. Huber
(Received 11 September 2003; received in revised form 5 November 2003; accepted 7 November 2003)
Latent LytM 785




























