Embed Size (px)
Citation preview

LDL-Activated p38 in Endothelial Cells Is Mediated by RasYi Zhu, Hailing Liao, Nanping Wang, Kuo-Sheng Ma, Lynne K. Verna, John Y.-J. Shyy,
Shu Chien, Michael B. Stemerman
Abstract—Endothelial dysfunction is a major atherogenic proinflammatory event. LDL causes the activation andphenotypic changes of cultured vascular endothelial cells (ECs). We previously reported that LDL activates c-Jun andAP-1 in ECs. In this study, we demonstrated that p38–ATF-2 is activated by LDL in human ECs and that this activationis mediated by Ras. When ECs are incubated with LDL in pathophysiological concentrations, the p38-mediated ATF-2phosphorylation and ATF-2 transactivation are increased in a time- and dose-dependent manner. To elucidate theupstream mechanism in LDL-activated p38 in ECs, we demonstrate that LDL increases Ras translocation from thecytoplasm to the cellular membrane, with concurrent increases in Ras binding activity to GST–Raf-1. Overexpressionof RasN17, a dominant negative mutant of Ras, attenuates the LDL-induced increases in (1) phosphorylation of ATF-2,(2) phosphorylation of c-Jun, (3) AP-1 binding, and (4) AP-1–driven luciferase activity. To study the effect of p38 inthe regulation of an LDL targeting gene, we show that a specific p38 inhibitor attenuates LDL-induced E-selectin at themRNA level. Thus, LDL activates both p38 and JNK signaling pathways through Ras activation, and furthermore, theseevents may play an important role in LDL-induced endothelial activation.(Arterioscler Thromb Vasc Biol. 2001;21:1159-1164.)
Key Words: p38 n ATF-2 n Ras n LDL n ECs
Endothelial dysfunction is one of the earliest proinflam-matory vascular events leading to atherosclerosis.1 Na-
tive LDL has been implicated in initiating endothelial cell(EC) dysfunction.2 When incubated with LDL, ECs in cultureare activated, and several genes involved in atherogenesis,including E-selectin, intercellular adhesion molecule(ICAM)-1, and vascular cell adhesion molecule (VCAM)-1,are upregulated.3–6 EC activation by LDL involves mobiliza-tion of calcium, activation of protein kinases, and an increasein the transactivation of AP-1 transcription factor.3,4,7–9 Theeffects of LDL on intracellular signal transduction leading toEC activation, however, have been studied only to a limitedextent.
In response to stimulation by mitogens, cytokines, UVirradiation, and other environmental stresses, membrane-associated small GTPase molecules, eg, p21-Ras, are acti-vated. Ras cycles between an active GTP-bound and aninactive GDP-bound state, functioning as a molecular switchin response to cell-activating stimuli. Ras can trigger at least3 diverging mitogen-activated protein kinase
(MAPK) cascades. The first cascade is mediated by Raf-1activation and transmits signals through MAPK/extracellularsignal-regulated kinase (ERK) kinase
(MEK)1/2 to activate ERK. The second cascade operatesthrough MEKK1 and JNKK to activate c-Jun NH2-terminalkinases (JNKs). The third cascade leads to p38 activation.Efficient activation of p38 requires phosphorylation of Thr-
180 and Tyr-182. At least 3 Thr/Thy kinases (MKK3,MKK4/SEK1, and MKK6) phosphorylate and activate p38.This leads to the activation of multiple transcription factors,such as ATF-2 and CHOP, that induce the expression ofproinflammatory genes, such as E-selectin.10
ATF-2 can form heterodimers with c-Jun, which positivelyregulate c-Jun promoter activity.11 Recently, we found thatLDL increases AP-1 activity and activates the JNK–c-Junpathway but not the ERK/c-Fos pathway in human ECs.8,9
This report was designed to study whether LDL activates thep38–ATF-2 pathway in ECs and to elucidate the upstreamsignaling involved. Our study demonstrates that LDL en-hances Ras translocation to membrane and Ras activation inhuman umbilical vein ECs (HUVECs) and that these eventsactivate both p38–ATF-2 and JNK–c-Jun signaling path-ways. Furthermore, we show that LDL-induced E-selectin isblocked by a specific p38 inhibitor.
MethodsCell Culture and LDL IsolationHUVECs were isolated and maintained as described previously.8 Allexperiments were performed with cells up to passage 3 in ECmedium and cultured to confluence before LDL treatment. LDL wasisolated from nonfrozen human plasma as described.8,9 The LDLpreparations contained,0.0005 U endotoxin/mg cholesterol asdetermined by the chromogenicLimulustest (BioWhittaker). For allstudies, LDL was used at a final cholesterol culture concentration of240 mg/dL (6.24 mmol/L).
Received March 15, 2001; revision accepted April 27, 2001.From the Division of Biomedical Sciences, University of California, Riverside, and the Department of Bioengineering and Whitaker Institute of
Biomedical Engineering, University of California, San Diego, La Jolla (S.C.), Calif.Correspondence to Yi Zhu, MD, Division of Biomedical Sciences, University of California, Riverside, CA 92521. E-mail [email protected]© 2001 American Heart Association, Inc.
Arterioscler Thromb Vasc Biol.is available at http://www.atvbaha.org
1159
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from

Western Blotting AnalysisCellular membrane and cytosolic proteins from whole-cell lysatewere isolated as previously described.12 Western analyses withantibodies against H-Ras (Transduction Laboratories) or Anti-ACTIVE MAPK pAb antibody (Promega) were performed aspreviously described.9,12
Affinity Precipitation/Immunoblot ofActivated RasGST fusion protein, corresponding to the human Ras-binding do-main (RBD, residues 1 to 149) of Raf-1 bound to glutathioneagarose, was from Upstate Biotechnology. The procedure used tomeasure Ras binding on GST–Raf-1 RBD was described by com-pany protocol (Upstate Biotechnology).
ATF-2 and c-Jun Phosphorylation AssayThe bacterial expression vector GST–ATF-2 (residues 1 to 109)13
was provided by R.J. Davis (University of Massachusetts MedicalSchool). A GST–ATF-2 fusion protein was isolated and purified.The assay for c-Jun phosphorylation was performed as described.9
The procedures for the ATF-2 phosphorylation assay were similar tothose for c-Jun, except that the cell extracts were incubated withGST–ATF-2. In the study of the role of p38 on ATF-2 phosphory-lation, p38 protein was immunoprecipitated with an antibody againstp38. Then, the immunoprecipitated p38 was incubated with agarose-bound GST–ATF-2 in a kinase buffer.14
Recombinant AdenovirusesThe recombinant adenovirus Ad-RasN17 encoding for RasN17 wasconstructed as described previously.15 The adenoviruses wereplaque-purified, expanded and titrated in 293 cells, and purified bycesium chloride methods.16 For adenoviral infection, confluentHUVECs were exposed to adenoviral vectors (Ad-RasN17 or Ad-b-gal as control) at a multiplicity of infection of 100 to 500 for 2hours. After the viruses had been washed out, HUVECs werecontinuously incubated for 18 to 24 hours before the treatment.16
Plasmids and TransfectionFor transactivation experiments, we used the Targefect transfectionmethod (Targeting Systems). The in vivo trans-reporting system waspurchased from Stratagene. This system includes a pFA–c-Jun,pFC–ATF-2, pFA2-Elk, or pFA-CHOP (CHOP is a transcriptionfactor specific response to p38 activation) as an activator plasmid,and a reporter plasmid (pFR-Luc). pFC-dbd plasmid and pFC-MEKK plasmid were used as a negative and positive control,respectively. pRSV-b-gal was cotransfected as a transfection control.After 24 hours of LDL or phorbol 12-myristate 13-acetate (PMA)exposure, samples were collected and assayed for luciferase activity.The results were normalized againstb-galactosidase.9
Electrophoretic Mobility Shift AssayAfter infection by the adenoviral construct Ad-RasN17, HUVECmonolayers on 100-mm dishes were exposed to 240 mg/dL of LDLfor 6 hours or to 50 ng/mL PMA for 2 hours.8 Nuclear extracts wereprepared, and an electrophoretic mobility shift assay was performedwith consensus sequences for AP-1 and nuclear factor (NF)-kB asdescribed.8
Northern HybridizationTotal RNA isolation and Northern analysis for hABC1 and vonWillebrand factor (vWF) expression were performed.8 The probes ofE-selectin and vWF cDNA were labeled with [a-32P]dCTP byDECApriming (Ambion) as previously described.6
StatisticsQuantitative data were expressed as mean6SEM. Statistical signif-icance of the data was evaluated by Student’st test. Probabilityvalues ofP,0.05 were considered significant. For nonquantitativedata, the results were expressed as representative of$3 independentexperiments.
ResultsLDL Increases p38 Activity in ECsWe have previously shown that LDL activates JNK/c-Jun, butnot ERK/c-Fos, in ECs and proposed that Ras may play animportant role in LDL-induced EC activation.8,9 As anothermajor MAPK pathway induced by Ras, p38/ATF-2 activationby LDL in ECs was investigated in this study. ConfluentHUVECs were exposed to LDL in concentrations up to 240mg/dL for various times. Cell lysates were collected fromvarious samples for ATF-2 phosphorylation assay. As shownin Figure 1A, LDL increases ATF-2 phosphorylation 30minutes after LDL exposure, reaching a peak at 2 hours. Toconfirm the involvement of p38 in ATF-2 phosphorylation,we immunoprecipitated p38 proteins with anti-p38 antibodyand then used the precipitated p38 to perform the kinaseactivity assay. A similar pattern of ATF-2 phosphorylationwas obtained with the p38-immunoprecipitated samples com-pared with whole-cell lysates. Exposure of ECs to differentconcentrations of LDL caused increased p38 activity in adose-dependent fashion, with incremental increases over theconcentration range of 160 to 240 mg/dL (Figure 1B).Therefore, LDL at a concentration of 240 mg/dL was used inthe rest of the experiments.
LDL Increases ATF-2 and c-Jun Transactivationin ECsTo further investigate whether LDL activates ATF-2 transac-tivation in ECs as a consequence of p38 activation, ECs werecotransfected with activator plasmids pFC–ATF-2, pFA-CHOP, pFA–c-Jun, or pFA2-Elk, and the reporter plasmidpFR-Luc. pFC-MEKK plasmid was used as a positive controlin the groups of pFC–ATF-2, pFA-CHOP, and pFA–c-Juntransfection. PMA was a positive control for pFA2-Elktransfection. pFC-dbd plasmid containing the DNA bindingdomain of the yeast GAL4 but lacking any activation domainserved as negative control. Eighteen hours after transfection,cells were exposed to LDL for 24 hours, and samples were
Figure 1. Effect of LDL on ATF-2 phosphorylation. A, HUVECswere incubated with 240 mg/dL LDL for different periods of timeas indicated. B, HUVECs were incubated with different concen-trations of LDL for 2 hours. Cellular protein was extracted in alysis buffer. The cell extracts were incubated with agarose-bound GST–ATF-2 (A and B, top). The p38 proteins in theextracts were immunoprecipitated by an anti-p38 antibody, andthen the precipitated p38 was incubated with agarose-boundGST–ATF-2 (A and B, bottom). After being pelleted and washed,the GST–ATF-2 beads were resuspended and incubated in akinase buffer containing [32P]ATP. The samples were then sepa-rated by SDS-polyacrylamide gel electrophoresis and furtheranalyzed by autoradiography. Data are representative of 3 inde-pendent experiments.
1160 Arterioscler Thromb Vasc Biol. July 2001
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from
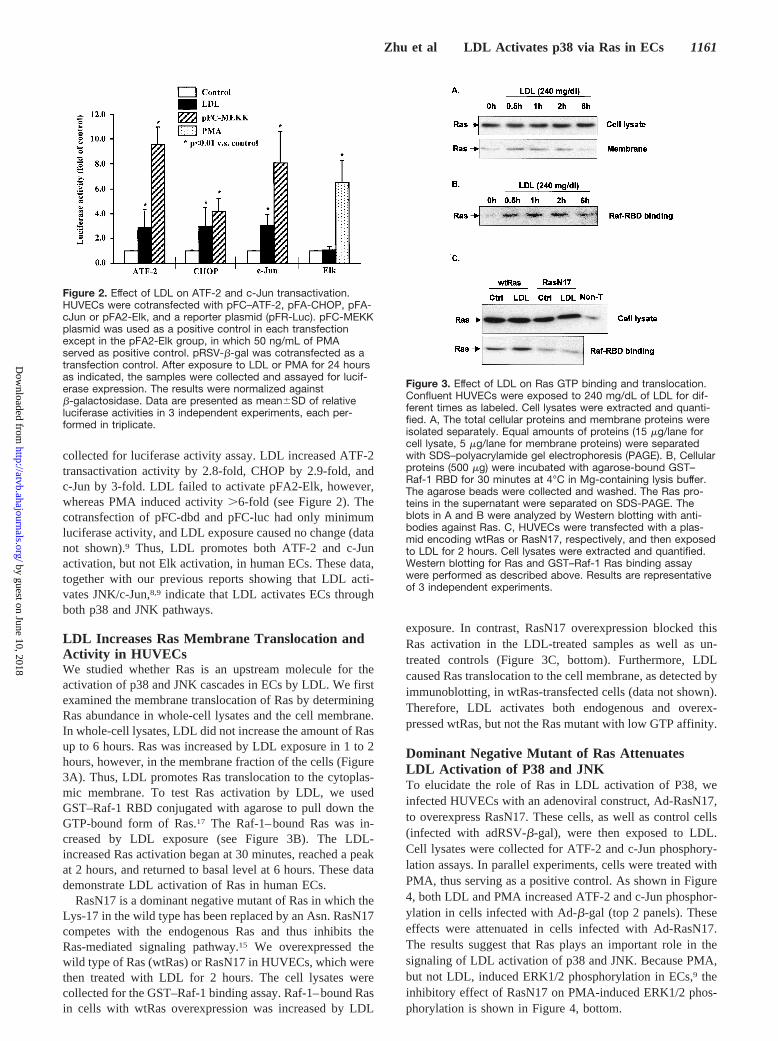
collected for luciferase activity assay. LDL increased ATF-2transactivation activity by 2.8-fold, CHOP by 2.9-fold, andc-Jun by 3-fold. LDL failed to activate pFA2-Elk, however,whereas PMA induced activity.6-fold (see Figure 2). Thecotransfection of pFC-dbd and pFC-luc had only minimumluciferase activity, and LDL exposure caused no change (datanot shown).9 Thus, LDL promotes both ATF-2 and c-Junactivation, but not Elk activation, in human ECs. These data,together with our previous reports showing that LDL acti-vates JNK/c-Jun,8,9 indicate that LDL activates ECs throughboth p38 and JNK pathways.
LDL Increases Ras Membrane Translocation andActivity in HUVECsWe studied whether Ras is an upstream molecule for theactivation of p38 and JNK cascades in ECs by LDL. We firstexamined the membrane translocation of Ras by determiningRas abundance in whole-cell lysates and the cell membrane.In whole-cell lysates, LDL did not increase the amount of Rasup to 6 hours. Ras was increased by LDL exposure in 1 to 2hours, however, in the membrane fraction of the cells (Figure3A). Thus, LDL promotes Ras translocation to the cytoplas-mic membrane. To test Ras activation by LDL, we usedGST–Raf-1 RBD conjugated with agarose to pull down theGTP-bound form of Ras.17 The Raf-1–bound Ras was in-creased by LDL exposure (see Figure 3B). The LDL-increased Ras activation began at 30 minutes, reached a peakat 2 hours, and returned to basal level at 6 hours. These datademonstrate LDL activation of Ras in human ECs.
RasN17 is a dominant negative mutant of Ras in which theLys-17 in the wild type has been replaced by an Asn. RasN17competes with the endogenous Ras and thus inhibits theRas-mediated signaling pathway.15 We overexpressed thewild type of Ras (wtRas) or RasN17 in HUVECs, which werethen treated with LDL for 2 hours. The cell lysates werecollected for the GST–Raf-1 binding assay. Raf-1–bound Rasin cells with wtRas overexpression was increased by LDL
exposure. In contrast, RasN17 overexpression blocked thisRas activation in the LDL-treated samples as well as un-treated controls (Figure 3C, bottom). Furthermore, LDLcaused Ras translocation to the cell membrane, as detected byimmunoblotting, in wtRas-transfected cells (data not shown).Therefore, LDL activates both endogenous and overex-pressed wtRas, but not the Ras mutant with low GTP affinity.
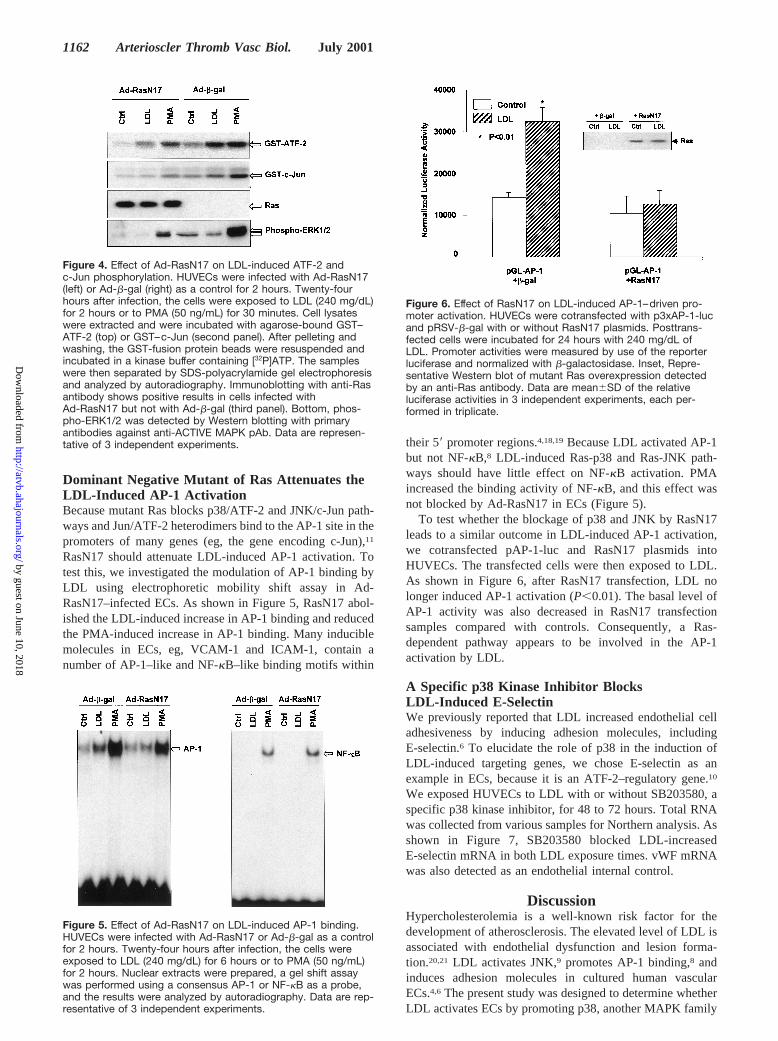
Dominant Negative Mutant of Ras AttenuatesLDL Activation of P38 and JNKTo elucidate the role of Ras in LDL activation of P38, weinfected HUVECs with an adenoviral construct, Ad-RasN17,to overexpress RasN17. These cells, as well as control cells(infected with adRSV-b-gal), were then exposed to LDL.Cell lysates were collected for ATF-2 and c-Jun phosphory-lation assays. In parallel experiments, cells were treated withPMA, thus serving as a positive control. As shown in Figure4, both LDL and PMA increased ATF-2 and c-Jun phosphor-ylation in cells infected with Ad-b-gal (top 2 panels). Theseeffects were attenuated in cells infected with Ad-RasN17.The results suggest that Ras plays an important role in thesignaling of LDL activation of p38 and JNK. Because PMA,but not LDL, induced ERK1/2 phosphorylation in ECs,9 theinhibitory effect of RasN17 on PMA-induced ERK1/2 phos-phorylation is shown in Figure 4, bottom.
Figure 2. Effect of LDL on ATF-2 and c-Jun transactivation.HUVECs were cotransfected with pFC–ATF-2, pFA-CHOP, pFA-cJun or pFA2-Elk, and a reporter plasmid (pFR-Luc). pFC-MEKKplasmid was used as a positive control in each transfectionexcept in the pFA2-Elk group, in which 50 ng/mL of PMAserved as positive control. pRSV-b-gal was cotransfected as atransfection control. After exposure to LDL or PMA for 24 hoursas indicated, the samples were collected and assayed for lucif-erase expression. The results were normalized againstb-galactosidase. Data are presented as mean6SD of relativeluciferase activities in 3 independent experiments, each per-formed in triplicate.
Figure 3. Effect of LDL on Ras GTP binding and translocation.Confluent HUVECs were exposed to 240 mg/dL of LDL for dif-ferent times as labeled. Cell lysates were extracted and quanti-fied. A, The total cellular proteins and membrane proteins wereisolated separately. Equal amounts of proteins (15 mg/lane forcell lysate, 5 mg/lane for membrane proteins) were separatedwith SDS–polyacrylamide gel electrophoresis (PAGE). B, Cellularproteins (500 mg) were incubated with agarose-bound GST–Raf-1 RBD for 30 minutes at 4°C in Mg-containing lysis buffer.The agarose beads were collected and washed. The Ras pro-teins in the supernatant were separated on SDS-PAGE. Theblots in A and B were analyzed by Western blotting with anti-bodies against Ras. C, HUVECs were transfected with a plas-mid encoding wtRas or RasN17, respectively, and then exposedto LDL for 2 hours. Cell lysates were extracted and quantified.Western blotting for Ras and GST–Raf-1 Ras binding assaywere performed as described above. Results are representativeof 3 independent experiments.
Zhu et al LDL Activates p38 via Ras in ECs 1161
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from
Dominant Negative Mutant of Ras Attenuates theLDL-Induced AP-1 ActivationBecause mutant Ras blocks p38/ATF-2 and JNK/c-Jun path-ways and Jun/ATF-2 heterodimers bind to the AP-1 site in thepromoters of many genes (eg, the gene encoding c-Jun),11
RasN17 should attenuate LDL-induced AP-1 activation. Totest this, we investigated the modulation of AP-1 binding byLDL using electrophoretic mobility shift assay in Ad-RasN17–infected ECs. As shown in Figure 5, RasN17 abol-ished the LDL-induced increase in AP-1 binding and reducedthe PMA-induced increase in AP-1 binding. Many induciblemolecules in ECs, eg, VCAM-1 and ICAM-1, contain anumber of AP-1–like and NF-kB–like binding motifs within
their 59promoter regions.4,18,19Because LDL activated AP-1but not NF-kB,8 LDL-induced Ras-p38 and Ras-JNK path-ways should have little effect on NF-kB activation. PMAincreased the binding activity of NF-kB, and this effect wasnot blocked by Ad-RasN17 in ECs (Figure 5).
To test whether the blockage of p38 and JNK by RasN17leads to a similar outcome in LDL-induced AP-1 activation,we cotransfected pAP-1-luc and RasN17 plasmids intoHUVECs. The transfected cells were then exposed to LDL.As shown in Figure 6, after RasN17 transfection, LDL nolonger induced AP-1 activation (P,0.01). The basal level ofAP-1 activity was also decreased in RasN17 transfectionsamples compared with controls. Consequently, a Ras-dependent pathway appears to be involved in the AP-1activation by LDL.
A Specific p38 Kinase Inhibitor BlocksLDL-Induced E-SelectinWe previously reported that LDL increased endothelial celladhesiveness by inducing adhesion molecules, includingE-selectin.6 To elucidate the role of p38 in the induction ofLDL-induced targeting genes, we chose E-selectin as anexample in ECs, because it is an ATF-2–regulatory gene.10
We exposed HUVECs to LDL with or without SB203580, aspecific p38 kinase inhibitor, for 48 to 72 hours. Total RNAwas collected from various samples for Northern analysis. Asshown in Figure 7, SB203580 blocked LDL-increasedE-selectin mRNA in both LDL exposure times. vWF mRNAwas also detected as an endothelial internal control.
DiscussionHypercholesterolemia is a well-known risk factor for thedevelopment of atherosclerosis. The elevated level of LDL isassociated with endothelial dysfunction and lesion forma-tion.20,21 LDL activates JNK,9 promotes AP-1 binding,8 andinduces adhesion molecules in cultured human vascularECs.4,6 The present study was designed to determine whetherLDL activates ECs by promoting p38, another MAPK family
Figure 4. Effect of Ad-RasN17 on LDL-induced ATF-2 andc-Jun phosphorylation. HUVECs were infected with Ad-RasN17(left) or Ad-b-gal (right) as a control for 2 hours. Twenty-fourhours after infection, the cells were exposed to LDL (240 mg/dL)for 2 hours or to PMA (50 ng/mL) for 30 minutes. Cell lysateswere extracted and were incubated with agarose-bound GST–ATF-2 (top) or GST–c-Jun (second panel). After pelleting andwashing, the GST-fusion protein beads were resuspended andincubated in a kinase buffer containing [32P]ATP. The sampleswere then separated by SDS-polyacrylamide gel electrophoresisand analyzed by autoradiography. Immunoblotting with anti-Rasantibody shows positive results in cells infected withAd-RasN17 but not with Ad-b-gal (third panel). Bottom, phos-pho-ERK1/2 was detected by Western blotting with primaryantibodies against anti-ACTIVE MAPK pAb. Data are represen-tative of 3 independent experiments.
Figure 5. Effect of Ad-RasN17 on LDL-induced AP-1 binding.HUVECs were infected with Ad-RasN17 or Ad-b-gal as a controlfor 2 hours. Twenty-four hours after infection, the cells wereexposed to LDL (240 mg/dL) for 6 hours or to PMA (50 ng/mL)for 2 hours. Nuclear extracts were prepared, a gel shift assaywas performed using a consensus AP-1 or NF-kB as a probe,and the results were analyzed by autoradiography. Data are rep-resentative of 3 independent experiments.
Figure 6. Effect of RasN17 on LDL-induced AP-1–driven pro-moter activation. HUVECs were cotransfected with p3xAP-1-lucand pRSV-b-gal with or without RasN17 plasmids. Posttrans-fected cells were incubated for 24 hours with 240 mg/dL ofLDL. Promoter activities were measured by use of the reporterluciferase and normalized with b-galactosidase. Inset, Repre-sentative Western blot of mutant Ras overexpression detectedby an anti-Ras antibody. Data are mean6SD of the relativeluciferase activities in 3 independent experiments, each per-formed in triplicate.
1162 Arterioscler Thromb Vasc Biol. July 2001
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from

member, and whether Ras mediates LDL activation of thep38 pathway. Our findings demonstrate that (1) LDL acti-vates the p38-mediated signaling pathway, (2) LDL increasesRas membrane translocation and activation, (3) a dominantnegative mutant of Ras attenuates LDL-induced ATF-2 andc-Jun phosphorylation and AP-1 activation, and (4) a specificp38 kinase inhibitor attenuates LDL-induced induction ofE-selectin at the mRNA level.
Originally identified as a proto-oncogene, Ras has beenbroadly studied for its function and regulation. There is littleinformation, however, on the physiological and pathophysi-ological roles of Ras in cardiovascular cells, especially inendothelium exposed to lipoprotein. As a small GTPasemolecule, Ras moves to the plasma membrane after thesynthesis in free cytoplasmic ribosomes and the farnesylationof its posttranslational C-terminal. Ras activation can triggerat least 3 diverging MAPK cascades, ie, p38, JNK, and ERK.Distinct intracellular MAPK signaling cascades are differen-tially activated to orchestrate transcriptional activation inresponse to various extracellular stimuli. LDL activates themembrane-associated Ras in human ECs, which in turnpreferentially activates p38 and JNK, the 2 MAPKs particu-larly involved in stress responses. Both native LDL andoxidized LDL have been found to stimulate p44/42 and p38MAPKs in smooth muscle cells and microphages.22–25 Incontrast, neither native LDL nor Cu21-oxidized LDL stimu-lates ERK activity in bovine aortic ECs.25 It is not clear whyLDL causes this differential activation of MAPKs in ECs.Fluid shearing of vascular ECs increases the JNK activity by.10-fold but activates ERK to a much lesser degree.26
Osmotic pressure and UV irradiation also selectively activatethe JNK pathway but not the ERK pathway.27,28 Ras canactivate both Raf-1 and MEKKs. Raf-1 activates ERK but notJNK/p38, whereas MEKKs activate JNK/p38 but not ERK.29
Thus, LDL-stimulated Ras may activate MEKKs to a greaterextent than Raf-1. Alternatively, other LDL-induced signal-ing pathways may inhibit Raf-1 signaling. cAMP inhibitionof ERK activation by preventing Ras-dependent activation ofRaf-1 has been reported.30 Indeed, LDL increased cAMP-responsive element–binding protein binding and protein ki-nase A activation in ECs.8 Recently, the phosphatidylinositol3-kinase (PI3K)–Akt pathway was shown to inhibit theRaf-MEK-ERK pathway through Akt phosphorylation ofRaf-1.31,32 The involvement of the PI3K-Akt pathway in theLDL-mediated EC activation remains to be studied. Regard-ing upstream signaling, Cdc42 and Rac-1, in addition to Ras,can also selectively activate JNK and p38.33,34 Thus, LDLmay also activate JNK/p38 through Rho family GTPase-mediated signal transduction.
The guanine nucleotide exchange factors are important inRas activation in response to growth factors, shear stress, andcytokines. In many instances, docking proteins such asGrb2/Sos are necessary for Ras activation. Sos is a cytoplas-mic Ras-GEF that is constitutively associated with the adap-tor protein Grb2. When Grb2 interacts with a tyrosine-phos-phorylated membrane receptor, it positions Sos at the plasmamembrane where it can promote activation of Ras. Alterna-tively, a tyrosine-phosphorylated Shc may serve as a bait forGrb2 docking and Sos activation. We detected no membranetranslocation of these docking proteins in HUVECs at up to 6hours of LDL exposure (Zhu et al, unpublished observation).Thus, LDL may activate Ras in ECs through a processindependent of growth factor activation. Conversely, theelevated cellular cholesterol level resulting from LDL expo-sure could promote the membrane translocation of caveolin-1and Ras, which is a caveolin-binding signaling molecule, intocaveolae in human ECs.12 Thus, caveolin may play a role inLDL-mediated Ras activation.
E-selectin is an important adhesion molecule that is tran-siently and specifically expressed in ECs on stimulation withcytokines and LDL.6,35 E-selectin can serve as a marker forendothelial activation. ATF2 was reported to play an impor-tant role in the activation of the E-selectin promoter.10 LDLincreases E-selectin at both the mRNA and protein levels6,8
and can increase the E-selectin promoter activity (Zhu et al,unpublished observation). Here, we report that a p38 kinaseinhibitor attenuates LDL-induced E-selectin at the mRNAlevel. Thus, it appears likely that LDL activates the E-selectinpromoter in ECs through the ATF-2–dependent mechanism.The blocking effects of RasN17 on ATF-2 and c-Jun phos-phorylation and AP-1 transactivation provide further evi-dence for the involvement of Ras in this transcriptionalactivation.
Collectively, the results of the present and previous studiesshow that LDL activates ECs predominantly through Ras-JNK/p38 signaling pathways. This supports our workinghypothesis that LDL perturbs the cell membrane and activatesmembrane-associated proteins (eg, Ras), which, in turn,activate the JNK/p38 pathways. Subsequently, c-Jun andATF-2 are activated and form a c-Jun/ATF-2 dimer to inducec-Jun expression via the TRE site at the Jun promoter.Accumulation of c-Jun by de novo synthesis consistentlyelevates the level of AP-1, which further chronically inducesother specific cellular genes regulated by AP-1 (eg, ICAM-1and E-selectin), leading to EC activation.
Figure 7. Effect of p38 inhibitor on LDL-induced E-selectinmRNA in ECs. Confluent HUVECs were exposed to 240 mg/dLof LDL with or without p38 inhibitor SB203580 (10 mmol/L) for48 to 72 hours as labeled. RNA was isolated, and 15-mg sam-ples of total RNA were resolved by gel electrophoresis and thenhybridized with [a-32P]-labeled E-selectin or vWF cDNA aslabeled. Results shown are representative of 3 independentexperiments. Resulting hybridization bands were quantified bydensitometry and normalized against vWF. Relative density isexpressed as a percentage of basic control (lane 1).
Zhu et al LDL Activates p38 via Ras in ECs 1163
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from

AcknowledgmentsThis study was supported in part by NIH grant HL-43023 (toM.B.S.), HL-60789 (to J.Y.-J.S.), and American Heart Association,Western States Affiliate grant 98-252 (to Y.Z.). J.Y.-J.S. is anEstablished Investigator of the American Heart Association.
References1. Albelda SM, Smith CW, Ward PA. Adhesion molecules and inflam-
matory injury.FASEB J. 1994;8:504–512.2. Parthasarathy S, Steinberg D, Witztum JL. The role of oxidized low-
density lipoproteins in the pathogenesis of atherosclerosis.Annu RevMed. 1992;43:219–225.
3. Allen S, Khan S, Al-Mohanna F, Batten P, Yacoub M. Native low densitylipoprotein-induced calcium transients trigger VCAM-1 and E-selectinexpression in cultured human vascular endothelial cells.J Clin Invest.1998;101:1064–1075.
4. Lin JH, Zhu Y, Liao HL, Kobari Y, Groszek L, Stemerman MB.Induction of vascular cell adhesion molecule-1 by low-densitylipoprotein.Atherosclerosis. 1996;127:185–194.
5. Haller H, Schaper D, Ziegler W, Philipp S, Kuhlmann M, Distler A, LuftFC. Low-density lipoprotein induces vascular adhesion moleculeexpression on human endothelial cells.Hypertension. 1995;25:511–516.
6. Smalley DM, Lin JH, Curtis ML, Kobari Y, Stemerman MB, PritchardKAJ. Native LDL increases endothelial cell adhesiveness by inducingintercellular adhesion molecule-1.Arterioscler Thromb Vasc Biol. 1996;16:585–590.
7. Ko Y, Totzke G, Seewald S, Schmitz U, Schiermeyer B, Meyer zuBrickwedde MK, Vetter H, Sachinidis A. Native low-density lipoprotein(LDL) induces the expression of the early growth response gene-1 inhuman umbilical arterial endothelial cells.Eur J Cell Biol. 1995;68:306–312.
8. Zhu Y, Lin JH, Liao HL, Friedli OJ, Verna L, Marten NW, Straus DS,Stemerman MB. LDL induces transcription factor activator protein-1 inhuman endothelial cells.Arterioscler Thromb Vasc Biol. 1998;18:473–480.
9. Zhu Y, Liao HL, Wang N, Friedli OJ, Verna L, Stemerman MB. Low-density lipoprotein activates Jun N-terminal kinase (JNK) in humanendothelial cells.Biochim Biophys Acta. 1999;1436:557–564.
10. De LL, Johnson DR, Whitley MZ, Collins T, Pober JS. cAMP and tumornecrosis factor competitively regulate transcriptional activation throughand nuclear factor binding to the cAMP-responsive element/activatingtranscription factor element of the endothelial leukocyte adhesionmolecule-1 (E-selectin) promoter.J Biol Chem. 1994;269:19193–19196.
11. van Dam H, Duyndam M, Rottier R, Bosch A, de Vries-Smits L, HerrlichP, Zantema A, Angel P, van der Eb AJ. Heterodimer formation of cJunand ATF-2 is responsible for induction of c-jun by the 243 amino acidadenovirus E1A protein.EMBO J. 1993;12:479–487.
12. Zhu Y, Liao HL, Wang N, Yuan Y, Ma K-S, Verna L, Stemerman MB.Low-density lipoprotein promotes caveolin-1 and Ras translocation tocaveolae: role of cholesterol in endothelial signaling.Arterioscler ThrombVasc Biol. 2000;20:2465–2470.
13. Gupta S, Campbell D, Derijard B, Davis RJ. Transcription factor ATF2regulation by the JNK signal transduction pathway.Science. 1995;267:389–393.
14. Whitmarsh AJ, Yang SH, Su MS, Sharrocks AD, Davis RJ. Role of p38and JNK mitogen-activated protein kinases in the activation of ternarycomplex factors.Mol Cell Biol. 1997;17:2360–2371.
15. Jin G, Wu C-H, Li YS, Hu J, Shyy JY, Chien S. Effects of active andnegative mutants of Ras on rat arterial neointima formation.J Surg Res.2000;94:124–132.
16. Wang N, Verna L, Hardy S, Forsayeth JR, Zhu Y, Stemerman MB.Adenovirus-mediated overexpression of c-Jun and c-Fos induces inter-cellular adhesion molecule-1 and monocyte chemoattractant protein-1 in
human endothelial cells.Arterioscler Thromb Vasc Biol. 1999;19:2078–2084.
17. de RJ, Bos JL. Minimal Ras-binding domain of Raf1 can be used as anactivation-specific probe for Ras.Oncogene. 1997;14:623–625.
18. Degitz K, Li LJ, Caughman SW. Cloning and characterization of the59-transcriptional regulatory region of the human intercellular adhesionmolecule 1 gene.J Biol Chem. 1991;266:14024–14030.
19. Shyy JY, Lin MC, Han J, Lu Y, Petrime M, Chien S. The cis-actingphorbol ester “12-O-tetradecanoylphorbol 13-acetate”-responsiveelement is involved in shear stress-induced monocyte chemotactic protein1 gene expression.Proc Natl Acad Sci U S A. 1995;92:8069–8073.
20. Cybulsky MI, Gimbrone MAJ. Endothelial expression of a mononuclearleukocyte adhesion molecule during atherogenesis.Science. 1991;251:788–791.
21. Li H, Cybulsky MI, Gimbrone MA, Libby P. An atherogenic diet rapidlyinduces VCAM-1, a cytokine-regulatable mononuclear leukocyteadhesion molecule, in rabbit aortic endothelium.Arterioscler Thromb.1993;13:197–204.
22. Sachinidis A, Kettenhofen R, Seewald S, Gouni-Berthold I, Schmitz U,Seul C, Ko Y, Vetter H. Evidence that lipoproteins are carriers ofbioactive factors.Arterioscler Thromb Vasc Biol. 1999;19:2412–2421.
23. Metzler B, Li C, Hu Y, Sturm G, Ghaffari-Tabrizi N, Xu Q. LDLstimulates mitogen-activated protein kinase phosphatase-1 expression,independent of LDL receptors, in vascular smooth muscle cells.Arte-rioscler Thromb Vasc Biol. 1999;19:1862–1871.
24. Auge N, Escargueil-Blanc I, Lajoie-Mazenc I, Suc I, Andrieu-Abadie N,Pieraggi MT, Chatelut M, Thiers JC, Jaffrezou JP, Laurent G, Levade T,Negre-Salvayre A, Salvayre R. Potential role for ceramide in mitogen-activated protein kinase activation and proliferation of vascular smoothmuscle cells induced by oxidized low density lipoprotein.J Biol Chem.1998;273:12893–12900.
25. Kusuhara M, Chait A, Cader A, Berk BC. Oxidized LDL stimulatesmitogen-activated protein kinases in smooth muscle cells and macro-phages.Arterioscler Thromb Vasc Biol. 1997;17:141–148.
26. Li YS, Shyy JY, Li S, Lee J, Su B, Karin M, Chien S. The Ras-JNKpathway is involved in shear-induced gene expression.Mol Cell Biol.1996;16:5947–5954.
27. Galcheva-Gargova Z, Derijard B, Wu IH, Davis RJ. An osmosensingsignal transduction pathway in mammalian cells.Science. 1994;265:806–808.
28. Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of anoncoprotein- and UV-responsive protein kinase that binds and potentiatesthe c-Jun activation domain.Genes Dev. 1993;7:2135–2148.
29. Minden A, Lin A, McMahon M, Lange-Carter C, Derijard B, Davis RJ,Johnson GL, Karin M. Differential activation of ERK and JNK mitogen-activated protein kinases by Raf-1 and MEKK.Science. 1994;266:1719–1723.
30. Cook SJ, McCormick F. Inhibition by cAMP of Ras-dependent activationof Raf. Science. 1993;262:1069–1072.
31. Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K,Moelling K, Yancopoulos GD, Glass DJ. Differentiation stage-specificinhibition of the Raf-MEK-ERK pathway by Akt.Science. 1999;286:1738–1741.
32. Zimmermann S, Moelling K. Phosphorylation and regulation of Raf byAkt (protein kinase B).Science. 1999;286:1741–1744.
33. Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of theJNK signaling cascade and c-Jun transcriptional activity by the smallGTPases Rac and Cdc42Hs.Cell. 1995;81:1147–1157.
34. Bagrodia S, Derijard B, Davis RJ, Cerione RA. Cdc42 and PAK-mediatedsignaling leads to Jun kinase and p38 mitogen-activated protein kinaseactivation.J Biol Chem. 1995;270:27995–27998.
35. Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA.Interleukin-1 activation of vascular endothelium: effects on procoagulantactivity and leukocyte adhesion.Am J Pathol. 1985;121:394–403.
1164 Arterioscler Thromb Vasc Biol. July 2001
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from

Chien and Michael B. StemermanYi Zhu, Hailing Liao, Nanping Wang, Kuo-Sheng Ma, Lynne K. Verna, John Y.-J. Shyy, Shu
LDL-Activated p38 in Endothelial Cells Is Mediated by Ras
Print ISSN: 1079-5642. Online ISSN: 1524-4636 Copyright © 2001 American Heart Association, Inc. All rights reserved.
Greenville Avenue, Dallas, TX 75231is published by the American Heart Association, 7272Arteriosclerosis, Thrombosis, and Vascular Biology
doi: 10.1161/hq0701.0924732001;21:1159-1164Arterioscler Thromb Vasc Biol.
http://atvb.ahajournals.org/content/21/7/1159World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://atvb.ahajournals.org//subscriptions/
at: is onlineArteriosclerosis, Thrombosis, and Vascular Biology Information about subscribing to Subscriptions:
http://www.lww.com/reprints
Information about reprints can be found online at: Reprints:
document. Question and AnswerPermissions and Rightspage under Services. Further information about this process is available in the
which permission is being requested is located, click Request Permissions in the middle column of the WebCopyright Clearance Center, not the Editorial Office. Once the online version of the published article for
can be obtained via RightsLink, a service of theArteriosclerosis, Thrombosis, and Vascular Biologyin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 10, 2018http://atvb.ahajournals.org/
Dow
nloaded from





























