Embed Size (px)
Citation preview

Location of the Glucuronosyltransferase Domain in the Heparan Sulfate Copolymerase EXT1 by
Analysis of Chinese Hamster Ovary Cell Mutants
(glycosyltransferases/glycosaminoglycans/exostosins/multiple hereditary exostosis)
Ge Wei†* , Xiaomei Bai†, Karen J. Bame‡, Thomas I. Koshy§¶, Patricia G. Spear§, and Jeffrey D.
Esko†#
†Department of Cellular and Molecular Medicine, Glycobiology Research and Training
Program, University of California, San Diego, La Jolla, California, 92093-0687
‡Division of Molecular Biology and Biochemistry, School of Biological Sciences, University of
Missouri-Kansas City, Kansas City, MO 64110
§Department of Microbiology-Immunology, Northwestern University Medical School, Chicago,
IL 60611
*On leave from the Department of Biochemistry and Molecular Genetics, University of
Alabama, Birmingham, AL 35294.
¶Present address: Abbott Diagnostics Division, Abbott Laboratories, 100 Abbott Park Road,
Abbott Park, IL 60064-6016.
Copyright 2000 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on June 22, 2000 as Manuscript M002990200 by guest on February 13, 2018
http://ww
w.jbc.org/
Dow
nloaded from

# For additional information, contact: Jeffrey D. Esko, University of California-San Diego,
Department of Cellular & Molecular Medicine, Glycobiology Research and Training Program,
9500 Gilman Drive, CMM-East Room 1054, La Jolla, CA 92093-0687 858/822-1100 FAX:
858/534-5611 e-mail: [email protected]
Wei et al. EXT1 mutants
2
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

SUMMARY
Heparan sulfate formation occurs by the copolymerization of glucuronic acid (GlcA) and N-
acetylglucosamine (GlcNAc) residues. Recent studies have shown that these reactions are
catalyzed by a copolymerase encoded by EXT1 and EXT2, members of the exostosin family of
putative tumor suppressors linked to Hereditary Multiple Exostoses (HME). Previously, we
identified a collection of Chinese hamster ovary cell mutants (pgsD) that failed to make heparan
sulfate (Lidholt et al. (1992) Proc. Natl. Acad. Sci. USA 89: 2267-2271). Here, we show that
pgsD mutants contain mutations that either alter GlcA transferase activity selectively, or that
affect both GlcNAc and GlcA transferase activities. Expression of EXT1 corrects the
deficiencies in the mutants, whereas EXT2 and the related EXT-like cDNAs do not. Analysis of
the EXT1 mutant alleles revealed clustered missense mutations in a domain that included a
D/EXD/E motif thought to bind the nucleotide sugar from studies of other transferases. These
findings provide insight into the location of the GlcA transferase subdomain of the enzyme and
indicate that loss of the GlcA transferase domain may be sufficient to cause HME.
Wei et al. EXT1 mutants
3
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

INTRODUCTION
Heparan sulfate functions in a wide variety of events during cell differentiation and tissue
morphogenesis by binding and activating a number of growth factors, adhesion molecules, and
enzymes (1,2). Its assembly initiates by the formation of an oligosaccharide primer consisting of -
GlcNAcα1-3GlcAβ1-3Galβ1-3Galβ1-4Xylβ-O- attached to specific serine residues in a
proteoglycan core protein
(3). This primer serves as a substrate for the copolymerase that adds alternating β1-4 linked GlcA
and α1-4 linked GlcNAc residues, resulting in chains ~100 or more sugar units in length. Over
the years, several groups have studied these reactions in vitro with the aim of defining the
specificity and overall characteristics of the transferase(s) (early literature reviewed in (4); also see
(5-7)). Several years ago, a set of heparan sulfate deficient mutants of Chinese hamster ovary cells
were isolated and through indirect genetic analysis, it became clear that the two transferases were
in fact encoded by a single genetic locus, designated pgsD (8). Subsequently, the copolymerase
was purified to near homogeneity and the isolated 70 kDa protein was shown to have both
GlcNAc and GlcA transferase activities (9). These genetic and biochemical findings indicated that
the copolymerase appeared to be a bifunctional protein with two active sites.
Hereditary multiple exostosis (HME) is a dominant genetic disorder (10) resulting in the
formation of generally benign cartilage-capped outgrowths from various bones (11-13). Positional
cloning experiments have identified three relevant genetic loci, designated EXT1, EXT2 and
EXT3 located at 8q24.1, 11p11-13, and 19p respectively (14-16). By primary DNA sequence , EXT1
and EXT2 exhibit about 35% homology and are predicted to encode Type II trans-membrane
Wei et al. EXT1 mutants
4
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

proteins. In addition, three other loci designated EXTL1/R3, EXTL2 and EXTL3/R1 have been
identified by hybridization and sequence homology searches (17-20), but no clinical cases of HME have
been mapped to these loci. Mutations at EXT1 and EXT2 coupled with loss of heterozygosity
correlates with chondrosarcoma in some cases, suggesting that the wild-type alleles may
normally behave like tumor suppressors (21,22). The EXT gene family appears to be widely
distributed, with orthologs in C. elegans (rib-1/rib-2) and Drosophila (tout-velu).
The connection between the heparan sulfate copolymerase and EXT was established by
McCormick et al. (23), who cloned a cDNA with homology to EXT1 based on its ability to restore
viral susceptibility to a cell line selected for resistance to herpes simplex virus and found to be
defective for GAG biosynthesis (24,25). This finding was followed by a report that recombinant forms
of EXT1 and EXT2 have GlcNAc and GlcA transferase activities (26). Recent studies suggest that
EXT1 and EXT2 may actually collaborate to form a functional oligomeric complex in the Golgi,
the site where heparan sulfate polymerization is thought to occur (27). Mutations in the EXT1 locus
in Drosophila (tout velu), cultured cells, and mice results in loss of heparan sulfate in vivo,
providing strong evidence that EXT1 is indeed the copolymerase responsible for heparan sulfate
formation (8,23,28,29).
Here, we report the expression cloning of hamster EXT1 by complementation of pgsD
strains. Ectopic expression of EXT1 fully corrects the enzymatic and heparan sulfate
deficiencies in the mutants, whereas introduction of EXT2 and the related EXT-like cDNAs fail
to complement the defect in pgsD cells. We also show that pgsD mutants contain mutations in
EXT1 and that two classes of alleles exist based on differential activity of the GlcNAc and GlcA
transferases in vitro. Sequencing the mutant alleles indicate that the GlcA transferase domain
Wei et al. EXT1 mutants
5
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

most likely resides in the N-terminal portion of the protein.
EXPERIMENTAL PROCEDURES
Cell Culture. CHO cells (CHO-K1) were obtained from the American Type Culture
Collection (CCL-61). One class of pgsD mutants (strains 623, 677, 108, 115, 6, 154, 625p, and
803) were isolated by replica plating and colony autoradiography, using 35SO4 incorporation to
identify clones that produced less proteoglycans (8,30). H661 was discovered by in situ enzymatic
assay of GlcN N-sulfotransferase activity in mutagenized clones replica plated to polyester cloth
(30). This strain failed to produce endogenous substrate for the enzyme (31). HVR101 and HVR104
were isolated from mutagenized CHO-K1 cells by selecting for cells that survived exposure to
herpes simplex virus type 2, which is cytotoxic to wild type cells (32), followed by screening for
colonies that incorporated 35SO4 but were resistant to virus infection. The purity of each strain
was ensured by its isolation from cultures containing only mutant colonies. Cells were
maintained in Ham’s F12 medium (33) (Hyclone Laboratories) supplemented with 7.5% (vol/vol)
fetal bovine serum (Hyclone Laboratories), 100 µg/ml streptomycin sulfate, and 100 units/ml
penicillin G. Cells were switched to sulfate-deficient medium (34) with 7.5% dialyzed fetal bovine
serum and 100 unit/ml penicillin G for radiolabeling studies.
Purification of GAG chains. Cells were labeled for 24 hr with 10 µCi/ml of H235SO4
(1325 Ci/mmol, DuPont NEN) in sulfate-free medium. Radiolabeled GAG chains were isolated
by anion-exchange chromatography on DEAE-Sephacel as described previously (31) and separated
by anion-exchange HPLC on a 7.5 mm inner diameter x 7.5 cm column of DEAE-3SW
Wei et al. EXT1 mutants
6
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

(TosoHaas, Montgomeryville, PA). The column was equilibrated in 10 mM KH2PO4 buffer (pH
6.0) containing 0.2% (w/v) Zwittergent 3-12 and 0.2 M NaCl. GAGs were eluted with a linear
gradient of NaCl (0.2-1 M) in the same buffer using a flow rate of 1 ml/min and by increasing
the NaCl concentration by 10 mM/min. The effluent from the column was monitored for
radioactivity with an in-line radioactivity detector (Radiomatic Flo One/beta, Packard
Instruments) with sampling rates every 6 sec. The data was averaged over one minute intervals.
Enzyme assays. GlcNAc transferase (UDP-GlcNAc:N-acetylheparosan α1-4N-acetyl-
D-glucosaminyltransferase and GlcA transferase (UDP-GlcA:N-acetylheparosan β1-4-D-
glucuronosyltransferase) activities were assayed using oligosaccharide acceptors prepared from
the capsular polysaccharide of Escherichia coli K5 (35). Briefly, the polysaccharide was partially N-
deacetylated with hydrazine and subjected to deaminative cleavage with nitrous acid at pH 3.9 (36)
The resulting mixture of oligosaccharides averaged 16 residues in length with GlcA at their non-
reducing termini (average mass = 3200 Da) (8,37). This mixture was used as substrate for GlcNAc
transferase assays. Small portions (0.6-1.0 mg) were digested as needed with β-D-
glucuronidase from bovine liver (Sigma, G-0501, 0.5 mg) in 0.1 M sodium acetate buffer, pH
5.0, at 37ÚC overnight. The chains were separated from the released GlcA by gel exclusion
chromatography on a PD-10 column (Pharmacia), lyophilized, and used as substrate for GlcA
transferase.
Confluent monolayers of CHO cells were scraped with a rubber policeman in
homogenization buffer (0.25 M sucrose, 20 mM Tris-Cl, pH 7.4, 1 mM phenylmethylsulfonyl
fluoride, 1 µg/ml each leupeptin and pepstatin A) and sonicated by two 1-sec pulses at output
setting 3 on a 550 Sonic Dismembrator (Fisher Scientific) equipped with a microtip. Each 25-µl
Wei et al. EXT1 mutants
7
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

reaction for GlcNAc transferase contained 25 mM 3-[N-morpholino]propanesulfonic acid
(MOPS), pH 6.5, 20 mM MnCl2, 0.3% Triton X-100 (w/v), 1 mM UDP-[6-3H]GlcNAc (220
Ci/mole), 25 µg cleaved N-acetylheparosan and 10-20 µg of cell homogenate protein. To assay
GlcA transferase, each 25-µl reaction contained 25 mM 2-[N-morpholino]ethanesulfonic acid
(MES), pH 5.5, 20 mM MnCl2, 0.03% Triton X-100 (w/v), 2 mM UDP-[1-3H]GlcA (250
Ci/mole), 70 µg β-glucuronidase-treated N-acetylheparosan and 10-20 µg of cell homogenate
protein. Product formation was proportional to time for over 4 hr at 37ÚC, but reactions were
routinely terminated after 3 hours by boiling or by adding dilute acid. Products were isolated by
DEAE-Sephacel chromatography (Pharmacia), mixed with chondroitin sulfate carrier (1 mg),
precipitated in 80% ethanol, and quantified by liquid scintillation spectrometry. UDP-[1-
3H]GlcA was synthesized from D-[1-3H]Glucose (10 Ci/mmol; NEN Life Science Products) as
described (38,39).
To determine the apparent Km values for UDP-GlcNAc and UDP-GlcA, the nucleotide
sugars were added in increasing concentrations to a fixed amount of N-acetylheparosan
oligosaccharides (0.8 mg/ml and 2 mg/ml, respectively). The signal obtained at each
concentration of UDP-sugar in the absence of acceptor was <10% of that obtained in its
presence, and these values were subtracted. Increasing concentrations of cleaved N-
acetylheparosan were added to determine the Km for the acceptors while holding the nucleotide
sugar concentration at 1 mM and 2 mM for the GlcNAc and GlcA transferase reactions,
respectively. The data was fitted by a nonlinear least-squares algorithm (SigmaPlot 4.11, Jandel
Scientific) to the Michaelis-Menten rate equation (40). The reactions were assumed to be pseudo
Wei et al. EXT1 mutants
8
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

first-order since the second substrate was kept at a constant value. Optimal concentrations of
detergent and divalent cations were determined for each reaction. The pH was varied by using a
mixture of MOPS and MES buffers (25 mM each).
Cloning the pgsD locus. Approximately 1 x 107 pgsD-H661 cells were transfected with
a mixture of 500 µg of a wild-type CHO-K1 cDNA library prepared in pcDNA1 (Invitrogen)
and 1.8 mg of pMAMneo (Clontech) using Lipofectin (Life Technologies, Inc.) under conditions
recommended by the manufacturer. Stable transfectants were selected by adding 0.4 mg/ml
geneticin sulfate (G-418) (Life Technologies, Inc.) to the medium, and positive clones were
screened by replica plating and 125I-FGF-2 binding (41). Two clones that bound 125I-FGF were
identified in this way (461 and 477-1) and purified by two rounds of subcloning. Genomic
DNA was then isolated from clone 477-1 and used to transfect ~1.2 x 107 pgsD-H661 cells (200
µg genomic DNA and 6.8 µg of pSV2neo [Clontech]). Secondary transfectants were selected by
resistance to G-418 and screened by replica plating and binding to 125I-FGF-2, yielding one
positive clone (5607). The genomic DNA was also partially digested with Sau3A I (Life
Technologies, Inc.) and fractionated by centrifugation through a 10%-40% (w/vol) sucrose
gradient. Fragments of 9-23 kb were pooled and a genomic library was constructed using
Lambda FIX II/XhoI partial Fill-in Vector Kit and Gigapack III Gold Cloning Kit (Stratagene).
~2 x 106 plaques were screened using a 32P-labeled probe prepared from the CMV-promoter
region of pcDNA1 (BamHI-NheI fragment) as a probe. Two positive phages 13-1-2 and 26-5-
1 were identified, and 13-1-2 was further characterized. A 7 kb-Not I fragment in 13-1-2 was
subcloned into pcDNA3 and sequenced by BWG-Biotech, Inc. (High point, North Carolina).
Wei et al. EXT1 mutants
9
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

This clone contained a full-length copy of EXT1 (Genbank accession number AF252858).
Transfection of CHO cells. Plasmid or lambda phage DNA was introduced into mutant
cells using lipofectAMINE (Life Technologies, Inc.) according to the manufacturer’s
instructions. Two days after transfection, cells were labeled with 10 µCi/ml of 35SO4 for 24 hr in
sulfate-deficient medium, and the GAGs were isolated and analyzed by anion-exchange HPLC.
To isolate stable transfectants expressing murine mEXT1 and mEXT2, cells were transfected
with pCDNA3-mEXT1-myc/His and pCDNA3-mEXT2-myc/His (gifts from Frank Tufaro,
University of British Columbia, Canada) as above. After two days, cells were replated in 100-
mm dishes and treated with 0.4 mg/ml G-418 for 5 days. Cells then were overlaid with
Whatman filter paper No. 50 and glass beads in order to obtain discreet colonies (30). The medium
was replenished once, and after 5 days, single colonies were isolated with cloning rings. Positive
clones were screened by flow cytometry analysis using biotinylated FGF-2 (38) and confirmed by
Western blot analysis using anti-myc antibodies (Invitrogen).
Full length forms of mutant EXT1 cDNAs were isolated by RT-PCR as described below.
The PCR products were digested by EcoRI and BamHI and inserted into the homologous sites in
pFLAG-CMV2 (Sigma) to produce FLAG-tagged EXT1. Wild-type and mutant pgsD cells
were transfected with the cDNAs, and the cells were analyzed by flow cytometry using biotin-
FGF-2 as probe (38).
Expression of EXT-like cDNA in pgsD mutants. Human EXTL1 cDNA was PCR
cloned from a human smooth muscle first-strand cDNA library (Clontech) using the following
two sets of primers: hEXTL1-803-5, 5’-CCTGCTCTTCCTGCTTGCTG-3’ and hEXTL1-2297-3, 5’-
CTTCCTGTGCCCATCAATGAC-3’, and hEXTL1-2164-5, 5’-TGAAGCTCATCCAGGCGGT-3’ and hEXTL
Wei et al. EXT1 mutants
10
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

3074-3, 5’-AGGAGAAGGTAAGGCCACGC-3’. PCR conditions were: 95ºC for 3 min, 35 cycles of 95ºC
for 45 sec, 50ºC for 45 sec and 72ºC for 2.5 min, and 72ºC for 10 min using pfu polymerase
(Stratagene). Both PCR fragments were cloned into pPCR-Script Amp SK(+) plasmid
(Stratagene), cut with BglII and NotI, and cloned into the Not I site of pcDNA3.1(-) to obtain a
full-length cDNA. The correct orientation of the clone was determined by EcoRI digestion.
To clone human EXTL2 cDNA, the PCR primers were 5’-
GGCTGGCCCTACTGCAATC-3’ and 5’-cgcatcctcgagaaGCTACTCAAATGCCAAGC-3’
(the lower-case letters stand for non-coding sequence and the underlined sequence refers to an
XhoI site). The PCR conditions were similar to those described above. The PCR product was
cloned into pPCR-Script Amp SK(+) plasmid (Stratagene). The insert with the correct
orientation was cloned into the NotI and BamHI sites of pcDNA3.1(-).
The original cDNA clone of human EXTL3 in pBluescript II SK(+) (KIAA0519) was a
gift from Dr. Takahiro Ngase (Kazusa DNA Research Institute, Kisarazu, Chiba 292 Japan). The
full-length cDNA was cloned into the XhoI and NotI sites of pcDNA3.1(-).
pcDNA3.1-hEXTL1, pcDNA3.1-hEXTL2, and pcDNA3.1-hEXTL3 were transfected
into pgsD-677, pgsD-H661 and pgsD-623. At least 8 stable transfectants from each mutant for
each cDNA were picked. Expression of the human cDNAs in the stable transfectants was
confirmed by Northern blot analysis. Expression of heparan sulfate was examined by flow
cytometry using biotin-FGF-2 (41).
Northern Analysis. mRNA was isolated from cells using QuickPrep Micro mRNA
Purification Kit (Pharmacia Biotech). The mRNA was denatured at 65 oC in a solution of 50%
formamide (v/v), 6% formaldehyde (v/v), 20 mM MOPS (pH 7.0), electrophoresed in 1.0%
Wei et al. EXT1 mutants
11
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

agarose containing 6% formaldehyde (v/v), and transferred passively for 18 hr to a Nytran plus
nylon membrane (Schleicher & Schuell). The blotted RNA was crosslinked by UV irradiation
and then prehybridized for 2 h at 42oC in a solution containing 50% formamide (v/v), 20 mM
sodium phosphate (pH 6.8), 5 X SSC, 1X Denhardt’s solution, 1% SDS (w/v), 5% (w/v) dextran
sulfate and 100 µg/ml denatured salmon sperm DNA. Double- stranded DNA probes were
labeled with [32P]dCTP by random oligonucleotide primers (Prime-IT II labeling kit,
Stratagene) using full length cDNAs as templates. The probe was purified by Elute-tip
(Schleicher &Schuell) before hybridization. Hybridization was carried out at 42 oC overnight in
prehybridization buffer containing ~1 x 106 cpm /ml of the 32P-labeled probe. The membrane
was washed two times with 2X SSC, 0.1% SDS for 30 min at 42 oC, and then two times with 0.2
X SSC, 0.1% SDS for 30 min at 65 oC. Hybridization was visualized by phosphoimaging
(Storm 860, Molecular Dynamics) after overnight exposure.
PCR analysis of EXT1 mutations. mRNA from mutant and wild-type CHO cells was
purified as described above. First-strand cDNA was produced using random primers in
SuperscriptTM Preamplification System according to the manufacturer’s protocol (Life
Technologies, Inc.). To determine the mutations in pgsD-HVR104, 623, H661, and 677, two
PCR fragments of ~1 kb each, which together covered the entire coding sequence, were isolated
from each cell line. PCR primers were: 5-CCTCTTGACCCAGGC-AGGACACATG-5 and 5-
GCGTGAGCGGATCTGCATTGGGAAG-3, 5-TGCCACTTTCTGTCTGGTTCC-3 and 5-
ATGACGGCAGCTTGGTTCC-3. PCR was carried out with pfu polymerase (Clontech; 25
cycles at 94ÚC for 1 min, 55ÚC for 1min, and 72ÚC for 2 min, followed by a final incubation at
Wei et al. EXT1 mutants
12
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

72ÚC for 7 min). The PCR products were cloned into pCR-Script Amp SK(+) (Stratagene). At
least four clones from each PCR product were sequenced on both strands by the dideoxy chain
termination method using Taq polymerase (dye terminator cycle sequencing, Perkin-Elmer) with
a DNA automatic sequencer (ABI PRISM genetic analyzer). For other pgsD mutants, full-
length EXT1 cDNAs were isolated by RT-PCR using the following primers: 5’-
cgcgaattcaCAGGCCAAAAAACGCGAGGGCATC-3’ and 5’-gtggatcccTCAAAGCCGTTCAATGTCTC-3’
(lower case letters stand for non-cDNA sequences and the underlined sequences are EcoRI and
BamHI sites, respectively). The sequences were determined directly from these PCR products.
RESULTS
Two classes of pgsD mutants exist. Several heparan sulfate deficient CHO cell mutants
were previously isolated and characterized (8). In addition, new mutants have been identified based
on the their inability to produce substrate for GlcN N-sulfotransferase (31), failure to bind to FGF-2
(41), or by their resistance to herpes simplex virus(24). The latter assay is based on the observation that
HSV utilizes heparan sulfate as a co-receptor for attachment and fusion of the viral envelope
with the plasma membrane (32,42-44). Based on cell hybridization studies, all of these mutants belong to a
single complementation group that was originally designated pgsD (8).
PgsD mutants have mutations that affect the copolymerase responsible for the alternating
addition of α1-4 linked GlcNAc and β1-4-linked GlcA units to nascent heparan sulfate chains.
These reactions are catalyzed by a bifunctional protein that contains both GlcNAc transferase
and GlcA transferase activities. Previous studies showed that the mutation in strain 677 affected
both enzyme activities, which provided the first evidence that both catalytic sites resided in a
Wei et al. EXT1 mutants
13
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

single protein (8). In order to characterize the other mutants in the collection, GlcNAc and GlcA
transferase assays in CHO cell homogenates were optimized with respect to buffer, pH,
detergent, oligosaccharide acceptors and nucleotide sugar donors. In general the enzyme activity
was dependent on detergent, but did not vary significantly in the range of 0.03 - 1%. Both
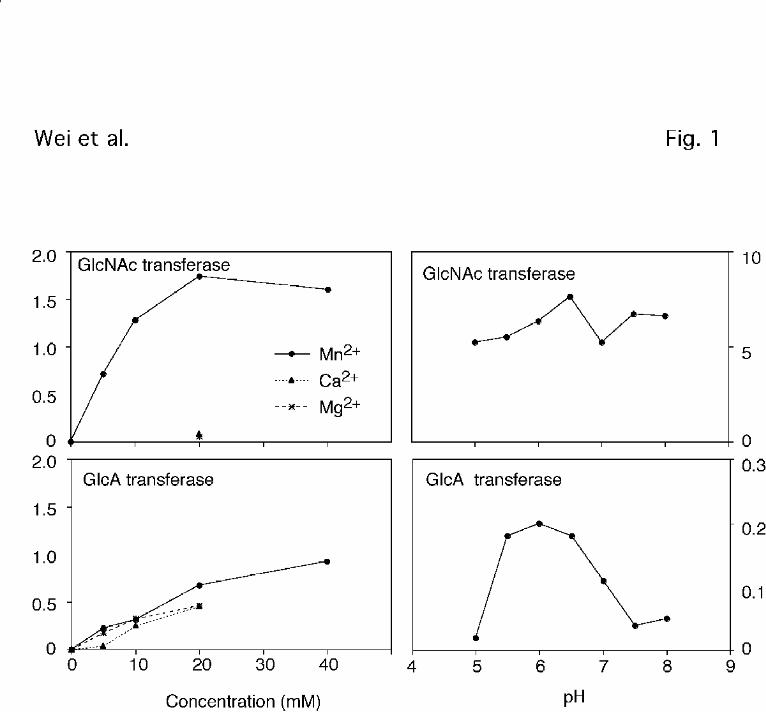
reactions also were dependent on added divalent cations. The GlcNAc transferase showed an
absolute preference for Mn2+ over Ca2+ or Mg2+ (Fig. 1), as previously reported for enzyme
solubilized from murine mastocytoma (6). The GlcA transferase reaction showed less selectivity
for divalent cations. The pH dependence also varied for the two activities. The GlcNAc
transferase reaction proceeded across a broad pH range (pH 5-8), whereas the GlcA transferase
reaction showed an optimum at pH 5.5-6.5 (Fig. 1). Although the shape of the curves differ
from previously published data , the overall conclusions are the same (6).
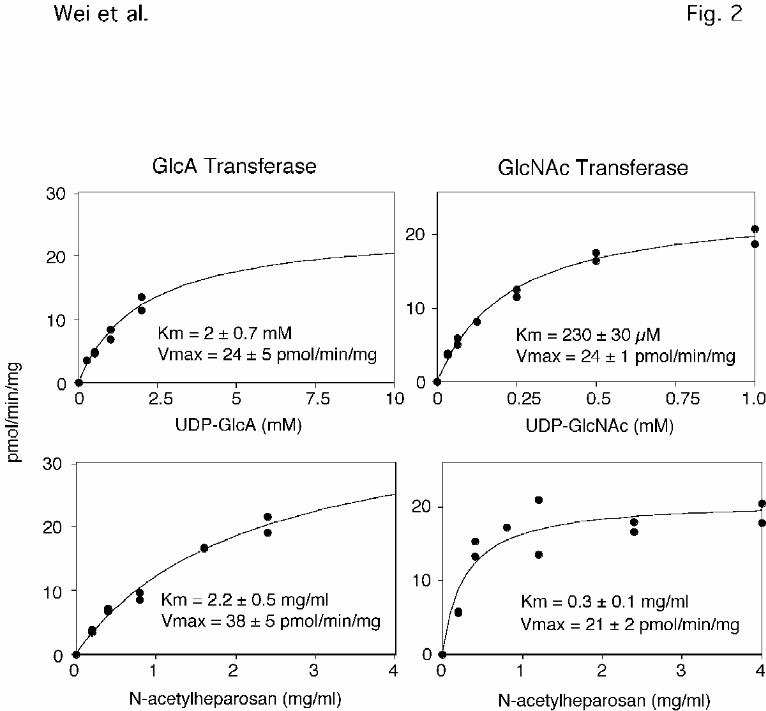
The enzyme activities differed significantly in their catalytic efficiency, as measured by
the estimated value of Vmax/Km (Fig. 2). The apparent Km values for UDP-GlcNAc and N-
acetylheparosan oligosaccharides for the GlcNAc transferase reaction were 230±30 µM and
0.3±0.1 mg/ml (~95 µM), respectively. The extrapolated values of Vmax were 24 ± 1 and 21 ± 2
pmol/min/mg, respectively. For the GlcA transferase, the apparent Km values for UDP-GlcA
and N-acetylheparosan oligosaccharides were 2.0 ± 0.7 mM and 2.2 ± 0.5 mg/ml (~0.7 mM),
respectively, and the values of Vmax were 24 ± 5 and 38 ± 5 pmol/min/mg, respectively. These
rates adequately account for the overall synthesis of heparan sulfate in CHO cells (~5 nmol/mg
cell protein/generation)2. Given these values, the apparent catalytic efficiency (Vmax/Km) for
the GlcNAc transferase was 10-fold greater than for GlcA transferase. However, under
Wei et al. EXT1 mutants
14
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

conditions of saturating amounts of donor and acceptor, the maximal velocities were comparable.
Previous studies also showed that the GlcNAc transferase had a lower Km for the acceptor
compared to the GlcA transferase (6). No attempt was made to examine how activity depended on
size of the oligosaccharide acceptor.
Using the optimized assay conditions, the activities of GlcA and GlcNAc transferases
were measured in 20 different pgsD mutants and the behavior of representative strains is shown
in Table 1. Based on differences in activity, the mutants fell into two subgroups. Strains in
group 1D have normal or elevated GlcNAc transferase activity but reduced GlcA transferase
activity (e.g., strains 108, 115, 154, 623, HVR 101, and HVR104). In contrast, strains in Group
2D have reduced levels of both enzymes (677, H661, 6, 803). One of the isolates also exhibited
partial reduction in both activities (625p). These results suggested that the strains bore different
mutant alleles and that the catalytic site for GlcA transferase may be located in a discrete region
of the protein that was susceptible to mutagenesis. Alternatively, the selective loss of GlcA
transferase activity could reflect an ancillary role for pgsD in activating GlcA transferase. In
either case, it became desirable to clone the pgsD gene in order to study this domain and the
overall organization of the encoded protein.
Initial attempts to clone the pgsD locus based on transient expression of cDNA libraries
and correction of the heparan sulfate deficiency were unsuccessful (45). However, we were able to
isolate a stable transfectant that regained the capacity to produce heparan sulfate. A genomic
clone containing the correcting activity was eventually isolated from a lambda library
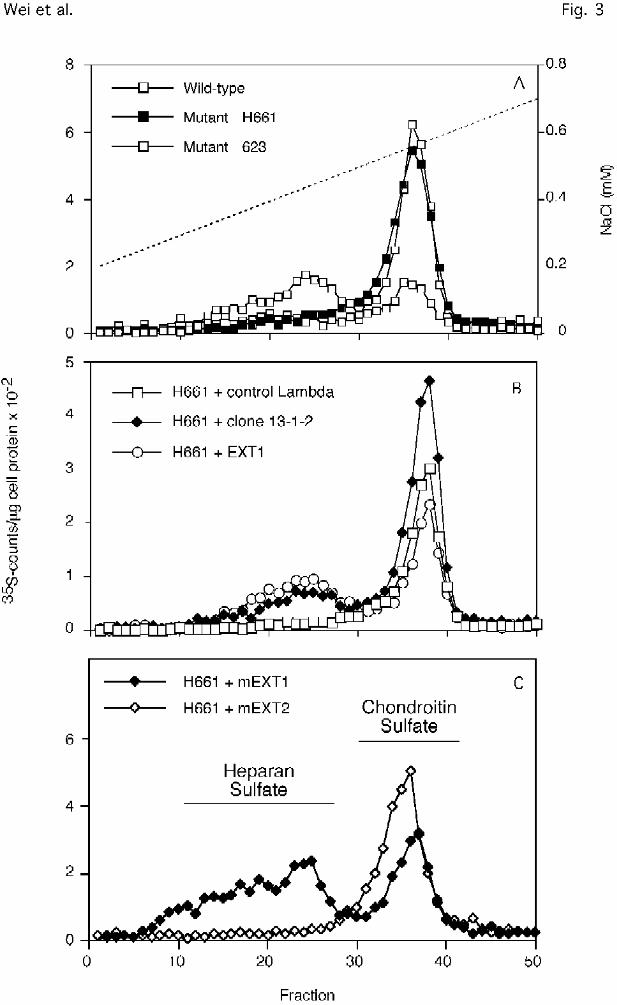
(Experimental Procedures) and reintroduction of the clone into the mutant corrected the heparan
sulfate deficiency (Fig. 3B). Direct sequencing showed that it contained a full-length copy of
Wei et al. EXT1 mutants
15
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

the hamster homolog of EXT1, which had been cloned from the mouse as this work was in
progress (23). Hamster EXT1 is 98% identical to human and mouse EXT1, with most of the
variations occurring in the putative stem region separating the catalytic domains from the
transmembrane segment (Fig. 4). Two possible N-glycosylation sites (marked by circles) as
well as the cytoplasmic and transmembrane domains are conserved in all three species. The high
degree of conservation of EXT1 suggests that it plays an important essential function, a
conclusion supported by recent reports showing that EXT1 null mutants in mice exhibit
embryonic lethality (29) and corresponding mutations in the Drosophila homolog (tout-velu) cause
developmental defects (46,47).
Only EXT1 corrects the pgsD deficiency. EXT1 is a member of family of proteins,
whose members include EXT2, EXTL1/R3, EXTL2 and EXTL3/R1. Lind et al have reported
that both EXT1 and EXT2 contain enzyme activities corresponding to heparan sulfate GlcNAc
transferase and GlcA transferase (26). However, inspection of the published data indicates that
neither enzyme reaction was optimized with respect to donor or acceptor and that the increase in
activity in mutant cells transfected with EXT2 was low. Furthermore, recent work indicates that
EXT1 and EXT2 proteins may form oligomers, which would make analysis of recombinant
proteins produced in animal cells somewhat problematic (27) . To gain further insight into this
problem, murine EXT1 and EXT2 and human EXTL1/R3, EXTL2 and EXTL3/R1 were
introduced into H661 cells, and stable transfectants were isolated. As shown in Fig.3C, only
EXT1 corrected the heparan sulfate deficiency in the mutant. This was confirmed by examining
numerous transfectants of different pgsD strains with the various cDNAs (16/30 drug resistant
clones from 5 different transfections showed correction by EXT1, whereas 0/30 clones
Wei et al. EXT1 mutants
16
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

transfected with EXT2 and 0/8 clones each for the EXT-like sequences regained heparan sulfate
synthesis). Northern blots confirmed that many of the stably transfected lines expressed the
integrated cDNAs (data not shown). Enzymatic assay of extracts prepared from EXT1-
transfectants showed that both enzyme activities were recovered (38 ± 6 pmol/min/mg for GlcA
transferase and 19 ± 0.2 pmol/min/mg for GlcNAc transferase). Transiently expressing both
mEXT1 and mEXT2 simultaneously in H661 did not restore heparan sulfate synthesis to a
greater extent than EXT1 alone (data not shown). Thus, only EXT1 appears to have the ability to
correct the pgsD deficiency.
PgsD strains contain point mutations in EXT1. The correction of pgsD mutants by EXT1
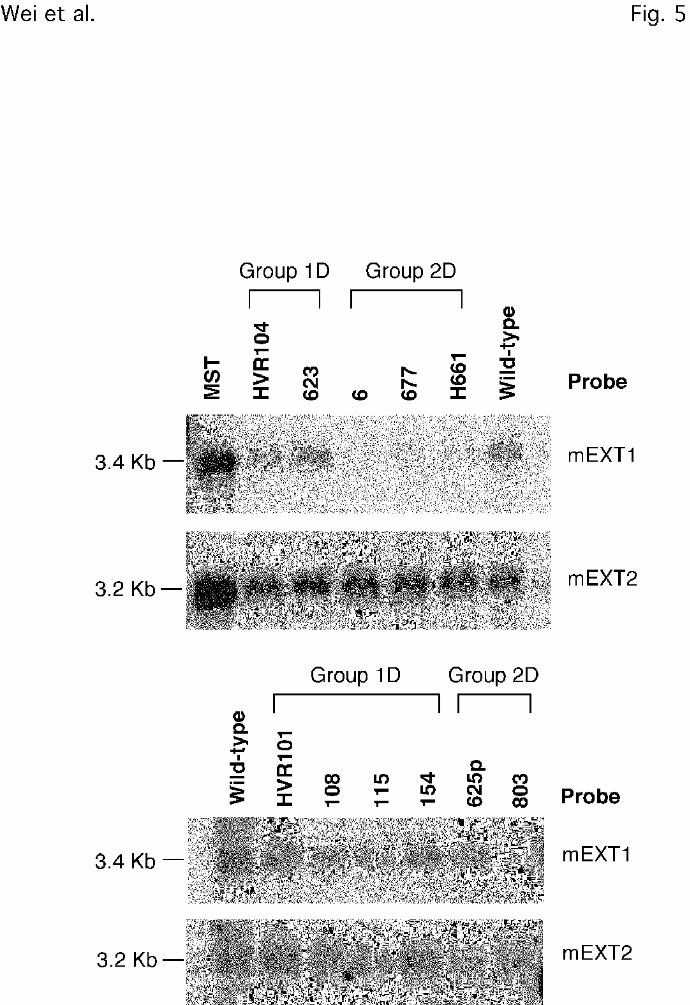
cDNA suggested that they contained mutations in the EXT1 locus. Northern blot analysis of
poly(A+) mRNA from the various mutants showed that most of the strains in Group 2D had
greatly diminished levels of EXT1 message, consistent with their missing both GlcNAc and
GlcA transferase activities (Fig. 5). A partial mutant designated 625p expresses ~3-fold less
heparan sulfate than wild-type cells, a partial reduction of GlcNAc and GlcA transferase
activities, and mRNA for EXT1. In contrast, Group 1D mutants, which have reduced GlcA
transferase activity and normal GlcNAc transferase activity, expressed EXT1 mRNA at levels
comparable to wild-type CHO cells (Fig. 5). These findings suggested that Group 1D mutants
probably contained point mutations that affected only the GlcA transferase domain.
To test this possibility, full length PCR products of EXT1 were prepared and sequenced
from mutant and wild-type cells. As shown in Table 2, strains in Group 1D contain missense
mutations that resulted in substitutions at different residues, indicating the independent origin of
the various mutants. Many of the mutations were non-conservative, but two of them changed
Wei et al. EXT1 mutants
17
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Arg residues to Lys, suggesting that these two positions are very important for activity. All of
the group 1D mutations cluster towards the N-terminal side of the central region of the protein,
surrounding a DXD motif at residue 313-315 (or EYE at residues 318-320), which is conserved
in the active site of many glycosyltransferases (48-50). Three group 2D mutants yielded products by
RT-PCR analysis, and sequencing revealed Gln398Stop in H661. Mutations have not been
found in the other two strains, suggesting that they may have mutations in promoter or regulatory
sequences.
To confirm that the mutations in EXT1 inactivated the catalytic activity, cDNAs
containing each mutation were generated and introduced into mutant cells. The mutant cDNAs
did not correct the heparan sulfate deficiency nor did they restore enzyme activity in vitro. These
constructs also did not suppress heparan sulfate formation when introduced into wildtype cells,
indicating that they did not exhibit dominant-negative effects (data not shown).
DISCUSSION
Recently, the GlcNAc and GlcA transferases involved in heparan sulfate biosynthesis
were shown to be encoded by EXT1, and possibly EXT2 (23,26). McCormick et al. discovered EXT1
while seeking genes that could restore susceptibility to herpes simplex virus-1 infection in
mutant sog9 cells, which fail to produce GAGs (24). These investigators also showed that EXT1
constructs containing the missense mutations present in the mutants did not restore susceptibility
to HSV-1 infection, demonstrating that the defects most likely caused a loss of functional
enzyme. Subsequently, Lind et al. reported the cloning of bovine heparan sulfate copolymerase
based on sequencing the purified enzyme and found that it was homologous to EXT2 (26). EXT1
Wei et al. EXT1 mutants
18
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

and EXT2 apparently can form a heterooligomeric complexes based on co-immunoprecipitation
and colocalization studies in transfected cells (27). Other glycosyltransferases have been reported to
consist of multiple subunits (51,52), but in these cases the function of the subunits have been established
by genetic as well as biochemical criteria. Thus, mutants are needed in EXT2 to rigorously
establish its role in heparan sulfate biosynthesis. In this regard, it is interesting that none of the
CHO or L-cell mutants altered in heparan sulfate formation have defects in EXT2, suggesting
that it may play non-essential role in the assembly process, perhaps by acting as a chaperone for
EXT1. Evidence suggesting that the localization of EXT1 in the Golgi may depend on
expression of EXT2 is consistent with this idea (27).
Six members of the EXT gene family have now been discovered. EXT1-3 have been
assigned to human chromosome 8q24.1, 11p11-13 and 19p, respectively, (14,21,53-57), whereas EXTL1/R3,
EXTL2, and EXTL3/R1 were discovered by homology screens (17-20). Recently, Kitagawa et al.
suggested that EXTL2 has GlcNAc transferase activity responsible for the addition of the first
α-GlcNAc residue in heparan sulfate biosynthesis, -GlcNAcα1-3GlcAβ1-3,Galβ1-3Galβ1-
4Xylβ-O-
(58), but more detailed kinetic studies of the protein are needed to confirm this idea (37,59). Northern blots
of mRNAs from CHO cells do not detect EXTL2 transcripts, suggesting that perhaps one of the
other EXT loci may fulfill this role (data not shown). The function of the other EXT proteins is
unknown, but their homology to EXT1 and EXT2 suggest that they might have related activities.
However, none of them appear to function as a copolymerase, since introduction into the CHO
cell mutants does not restore heparan sulfate biosynthesis.
Our studies of the Group 1D pgsD mutants show that most of strains have point
Wei et al. EXT1 mutants
19
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

mutations clustered in the central part of EXT1 in close proximity to each other. McCormick et
al have shown that two of the human mutant alleles of EXT1 in this region (G339D and R340C)
lack the ability to correct the EXT1 defect in sog9 cells, and when immunoprecipitated with
EXT2 the complex lacked GlcA transferase activity (27). Taken together, the data suggests that this
central region defines the active site of the GlcA transferase domain. The location of the
GlcNAc transferase domain is unknown. Although it is tempting to speculate that subdomains of
EXT1 may fold independently and give rise to functional transferase subsites on the
copolymerase, more mutants are needed to define these domains. We have not yet found any
pgsD alleles that selectively inhibit GlcNAc transferase activity without affecting GlcA
transferase, suggesting that mutations in this region may destabilize the overall structure of the
protein. We also cannot exclude the possibility that the GlcA transferase activity may depend on
association of EXT1 with another component, for example another EXT. The apparent
activation of transferase activity seen when EXT1 and EXT2 are co-expressed in mouse L-cells
is consistent with this idea (27).
Mutations in EXT1 and EXT2 are associated with hereditary multiple exostosis (HME) (10,12,13,60,61).
However, the relationship between loss of heparan sulfate synthesis seen in the mutants and
HME in humans is unclear. HME is characterized by cartilage-capped bony outgrowths at the
growth plates of bones. The trait is inherited in an autosomal dominant fashion, suggesting that
loss of a single copy is sufficient to observe exostosis. Chondrosarcoma in HME patients often
correlates with loss of heterozygosity at the locus, suggesting that the wild-type allele may
normally act as a tumor suppressor. Recently, a Drosophila homologue of EXT1 (tout-velu) was
found to be required for the diffusion of Hedgehog, and tout velu mutants lack heparan sulfate (28,46).
Wei et al. EXT1 mutants
20
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Since Hedgehog homologs (Indian Hedgehog) as well as heparin-binding growth factors (e.g.,
FGFs) are involved in vertebrate cartilage and bone development (62), it is intriguing to speculate
that a decrease of heparan sulfate in HME interferes with normal signaling events mediated by
these factors. Many mutations in human EXT1 from HME patients occur in the region defined
by the mutant CHO alleles(13), suggesting that loss of just the GlcA transferase domain is sufficient
to cause disease.
Further studies are needed to establish that exostosis is actually caused by diminished
heparan sulfate assembly. Mice containing a null allele at the EXT1 locus show severe
developmental anomalies very early in development, around E6.5 (29). ES cells derived from
homozygous null embryos completely lack heparan sulfate, indicating that EXT1 is the primary
route for generating heparan sulfate in both vertebrates and in flies (28,29)Interestingly, ES cells
derived from heterozygous null animals contain reduced amounts of heparan sulfate and about
30-50% of the normal levels of GlcNAc and GlcA transferase activities (29). However,
heterozygous null animals have no obvious growth defects, and cartilage-capped outgrowths
reminiscent of HME are not present. Although this observation may reflect differences in bone
growth between species, it could also point to other functions for the EXT gene products.
Perhaps, the phenotype may be related to gain-of-function changes induced by the missense or
splicing mutations known to occur at the EXT1 locus in HME patients. The availability of null
mice creates the possibility of producing transgenic animals containing the mutant alleles
described here and in HME patients.
Wei et al. EXT1 mutants
21
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

References
1. Lander, A. D. and Selleck, S. B. (2000) J.Cell Biol. 148, 227-232
2. Lander, A., Natako, H., Selleck, S. B., Turnbull, J. E., and Coath, C. (1999) Cell surface
proteoglycans in signaling and development, Ed., Human Frontiers of Science, Strassbourg
3. Esko, J. D. and Zhang, L. (1996) Curr.Opin.Struct.Biol. 6, 663-670
4. Rodén, L. (1980) Structure and metabolism of connective tissue proteoglycans. In Lennarz,
W. J., editor. The Biochemistry of Glycoproteins and Proteoglycans, Plenum Press, New
York
5. Kusche, M., Hannesson, H. H., and Lindahl, U. (1991) Biochem.J. 275, 151-158
6. Lidholt, K. and Lindahl, U. (1992) Biochem.J. 287, 21-29
7. Lidholt, K., Fjelstad, M., Jann, K., and Lindahl, U. (1994) Carbohydr.Res. 255, 87-101
8. Lidholt, K., Weinke, J. L., Kiser, C. S., Lugemwa, F. N., Bame, K. J., Cheifetz, S.,
Massagué, J., Lindahl, U., and Esko, J. D. (1992) Proc.Natl.Acad.Sci.USA 89, 2267-2271
9. Lind, T., Lindahl, U., and Lidholt, K. (1993) J.Biol.Chem. 268, 20705-20708
10. Legeai-Mallet, L., Margaritte-Jeannin, P., Lemdani, M., Le Merrer, M., Plauchu, H.,
Maroteaux, P., Munnich, A., and Clerget-Darpoux, F. (1997) Hum.Genet. 99, 298-302
11. Wells, D. E., Hill, A., Lin, X., Ahn, J., Brown, N., and Wagner, M. J. (1997) Hum.Genet.
Wei et al. EXT1 mutants
22
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

99, 612-615
12. Hecht, J. T., Hogue, D., Wang, Y., Blanton, S. H., Wagner, M., Strong, L. C., Raskind, W.,
Hansen, M. F., and Wells, D. (1997) Am.J.Hum.Genet. 60, 80-86
13. Wuyts, W., Van Hul, W., De Boulle, K., Hendrickx, J., Bakker, E., Vanhoenacker, F.,
Mollica, F., Ludecke, H. J., Sayli, B. S., Pazzaglia, U. E., Mortier, G., Hamel, B., Conrad,
E. U., Matsushita, M., Raskind, W. H., and Willems, P. J. (1998) Am.J.Hum.Genet. 62,
346-354
14. Le Merrer, M., Legeai-Mallet, L., Jeannin, P. M., Horsthemke, B., Schinzel, A., Plauchu,
H., Toutain, A., Achard, F., Munnich, A., and Maroteaux, P. (1994) Hum.Mol.Genet. 3,
717-722
15. Stickens, D., Clines, G., Burbee, D., Ramos, P., Thomas, S., Hogue, D., Hecht, J. T.,
Lovett, M., and Evans, G. A. (1996) Nat.Genet. 14, 25-32
16. Wuyts, W., Van Hul, W., Wauters, J., Nemtsova, M., Reyniers, E., Van Hul, E. V., De
Boulle, K., de Vries, B. B., Hendrickx, J., Herrygers, I., Bossuyt, P., Balemans, W.,
Fransen, E., Vits, L., Coucke, P., Nowak, N. J., Shows, T. B., Mallet, L., van den
Ouweland, A. M., McGaughran, J., Halley, D. J., and Willems, P. J. (1996)
Hum.Mol.Genet. 5, 1547-1557
17. Van Hul, W., Wuyts, W., Hendrickx, J., Speleman, F., Wauters, J., De Boulle, K., Van
Roy, N., Bossuyt, P., and Willems, P. J. (1998) Genomics. 47, 230-237
Wei et al. EXT1 mutants
23
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

18. Wise, C. A., Clines, G. A., Massa, H., Trask, B. J., and Lovett, M. (1997) Genome.Res. 7,
10-16
19. Wuyts, W., Van Hul, W., Hendrickx, J., Speleman, F., Wauters, J., De Boulle, K., Van
Roy, N., Van Agtmael, T., Bossuyt, P., and Willems, P. J. (1997) Eur.J.Hum.Genet. 5,
382-389
20. Saito, T., Seki, N., Yamauchi, M., Tsuji, S., Hayashi, A., Kozuma, S., and Hori, T. (1998)
Biochem.Biophys.Res.Commun. 243, 61-66
21. Raskind, W. H., Conrad, E. U., Chansky, H., and Matsushita, M. (1995) Am.J.Hum.Genet.
56, 1132-1139
22. Hecht, J. T., Hogue, D., Strong, L. C., Hansen, M. F., Blanton, S. H., and Wagner, M.
(1995) Am.J.Hum.Genet. 56, 1125-1131
23. McCormick, C., Leduc, Y., Martindale, D., Mattison, K., Esford, L. E., Dyer, A. P., and
Tufaro, F. (1998) Nat.Genet. 19, 158-161
24. Banfield, B. W., Leduc, Y., Esford, L., Schubert, K., and Tufaro, F. (1995) J.Virology 69,
3290-3298
25. Banfield, B. W., Leduc, Y., Esford, L., Visalli, R. J., Brandt, C. R., and Tufaro, F. (1995)
Virology 208, 531-539
26. Lind, T., Tufaro, F., McCormick, C., Lindahl, U., and Lidholt, K. (1998) J.Biol.Chem. 273,
26265-26268
Wei et al. EXT1 mutants
24
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

27. McCormick, C., Duncan, G., Goutsos, K. T., and Tufaro, F. (2000)
Proc.Natl.Acad.Sci.USA 97, 668-673
28. Toyoda, H., Kinoshita-Toyoda, A., and Selleck, S. B. (2000) J.Biol.Chem. 275, 2269-
2275
29. Lin, X., Wei, G., Shi, Z., Dryer, L., Esko, J. D., Wells, D. E., and Matzuk, M. M. (2000)
Development in press,
30. Esko, J. D. (1989) Meth.in Cell Biol. 32, 387-422
31. Bame, K. J. and Esko, J. D. (1989) J.Biol.Chem. 264, 8059-8065
32. Shieh, M.-T., WuDunn, D., Montgomery, R. I., Esko, J. D., and Spear, P. G. (1992) J.Cell
Biol. 116, 1273-1281
33. Ham, R. G. (1965) Proc.Natl.Acad.Sci.USA 53, 288-293
34. Esko, J. D., Elgavish, A., Prasthofer, T., Taylor, W. H., and Weinke, J. L. (1986)
J.Biol.Chem. 261, 15725-15733
35. Vann, W. F., Schmidt, M. A., Jann, B., and Jann, K. (1981) Eur.J.Biochem. 116, 359-364
36. Shively, J. E. and Conrad, H. E. (1976) Biochemistry 15, 3932-3942
37. Fritz, T. A., Gabb, M. M., Wei, G., and Esko, J. D. (1994) J.Biol.Chem. 269, 28809-28814
38. Wei, G., Bai, X. M., Sarkar, A. K., and Esko, J. D. (1999) J.Biol.Chem. 274, 7857-7864
Wei et al. EXT1 mutants
25
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

39. Bai, X. M., Wei, G., Sinha, A., and Esko, J. D. (1999) J.Biol.Chem. 274, 13017-13024
40. Michaelis, L. and Menten, M. L. (1913) Biochem.Z. 49, 333
41. Bai, X. M. and Esko, J. D. (1996) J.Biol.Chem. 271, 17711-17717
42. Shieh, M. T. and Spear, P. G. (1994) J.Virol. 68, 1224-1228
43. WuDunn, D. and Spear, P. G. (1989) J.Virol. 63, 52-58
44. Shukla, D., Liu, J., Blaiklock, P., Shworak, N. W., Bai, X. M., Esko, J. D., Cohen, G. H.,
Eisenberg, R. J., Rosenberg, R. D., and Spear, P. G. (1999) Cell 99, 13-22
45. Fukuda, M., Bierhuizen, M. F. A., and Nakayama, J. (1996) Glycobiology 6, 683-689
46. Bellaiche, Y., The, I., and Perrimon, N. (1998) Nature 394, 85-88
47. The, I., Bellaiche, Y., and Perrimon, N. (1999) Mol.Cell 4, 633-639
48. Busch, C., Hofmann, F., Selzer, J., Munro, S., Jeckel, D., and Aktories, K. (1998) J Biol
Chem 273. 273, 19566-72
49. Wiggins, C. A. R. and Munro, S. (1998) Proc.Natl.Acad.Sci.USA 95, 7945-7950
50. Gastinel, L. N., Cambillau, C., and Bourne, Y. (1999) EMBO J. 18, 3546-3557
51. Silberstein, S. and Gilmore, R. (1996) FASEB J. 10, 849-858
52. Watanabe, R., Inoue, N., Westfall, B., Taron, C. H., Orlean, P., Takeda, J., and Kinoshita,
T. (1998) EMBO J. 17, 877-885
Wei et al. EXT1 mutants
26
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

53. Máñez, S., Recio, M. D., Gil, I., Gómez, C., Giner, R. M., Waterman, P. G., and Ríos, J. L.
(1999) J.Nat.Prod. 62, 601-604
54. Ahn, J., Ludecke, H. J., Lindow, S., Horton, W. A., Lee, B., Wagner, M. J., Horsthemke,
B., and Wells, D. E. (1995) Nat.Genet. 11, 137-143
55. Blanton, S. H., Hogue, D., Wagner, M., Wells, D., Young, I. D., and Hecht, J. T. (1996)
Am.J.Med.Genet. 62, 150-159
56. Wuyts, W., Ramlakhan, S., Van Hul, W., Hecht, J. T., van den Ouweland, A. M., Raskind,
W. H., Hofstede, F. C., Reyniers, E., Wells, D. E., and de Vries, B. (1995)
Am.J.Hum.Genet. 57, 382-387
57. Wu, Y. Q., Heutink, P., de Vries, B. B., Sandkuijl, L. A., van den Ouweland, A. M.,
Niermeijer, M. F., Galjaard, H., Reyniers, E., Willems, P. J., and Halley, D. J. (1994)
Hum.Mol.Genet. 3, 167-171
58. Kitagawa, H., Shimakawa, H., and Sugahara, K. (1999) J.Biol.Chem. 274, 13933-13937
59. Fritz, T. A., Agrawal, P. K., Esko, J. D., and Krishna, N. R. (1997) Glycobiology 7, 587-
595
60. Legeai-Mallet, L., Munnich, A., Maroteaux, P., and Le Merrer, M. (1997) Clin.Genet. 52,
12-16
61. Solomon, L. (1964) Am.J.Hum.Genet. 16, 351-363
Wei et al. EXT1 mutants
27
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

62. Ferguson, C. M., Miclau, T., Hu, D., Alpern, E., and Helms, J. A. (1998)
Ann.N.Y.Acad.Sci. 857, 33-42
63. Montgomery, R. I., Lidholt, K., Flay, N. W., Liang, J., Vertel, B., Lindahl, U., and Esko, J.
D. (1992) Proc.Natl.Acad.Sci.USA 89, 11327-11331
Wei et al. EXT1 mutants
28
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

1 Abbreviations – GAG, glycosaminoglycan; CHO, Chinese hamster ovary; PBS, phosphate-
buffered saline;; TCA, trichloroacetic acid; HEPES, 4-(2-hydroxyethyl)-1-piperazine-
ethanesulfonic acid; HPLC, high pressure liquid chromatography; GlcA transferase, UDP-
glucuronic acid:GlcNAc β1-4 glucuronosyltransferase; GlcNAc transferase, UDP-N-
acetylglucosamine:GlcA α1-4glucosaminyltransferase
2 CHO cells make ~1 µg of heparan sulfate/mg cell protein. The calculation assumes a doubling
time of ~12 hours, a turnover rate of 300%/generation, and a molecular mass of 50,000 Da for
the heparan sulfate chains.
Acknowledgments
This work was supported by grants GM33063 (J.D.E.) and AI36293 (P.S.) from the National
Institutes of Health. We thank the Kazusa DNA Research Institute for providing EXTL3 cDNA
and Dr. Carol Wise (University of Texas, Southwestern Medical Center) for providing EXTL1
subclones
Wei et al. EXT1 mutants
29
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

Figure legends
Figure 1. Divalent cation dependence and pH profiles for GlcNAc and GlcA transferase
activities. Cell extracts prepared from wild-type CHO cells were assayed for GlcNAc and GlcA
transferase activities using N-acetylheparosan as substrate and either UDP-GlcNAc or UDP-
GlcA as donor (Experimental Procedures). The concentration of divalent cations and pH was
varied in order to find the preferred concentration.
Figure 2. Substrate saturation curves for GlcNAc and GlcA transferase activities. Cell extracts
prepared from wild-type CHO cells were assayed for GlcNAc and GlcA transferase activities
using N-acetylheparosan as substrate and either UDP-GlcNAc or UDP-GlcA as donor
(Experimental Procedures). The individual data sets were analyzed for best fit to the Michaelis-
Menten equation. Saturation of the GlcA transferase was difficult to measure due to the high
background in the assay when the donor concentration was raised above 2 mM. The average
length of the oligosaccharide acceptor was 16 sugar units (average mass of 3200 Da).
Figure 3. Anion-exchange HPLC of GAGs from CHO cells. The various cell lines were labeled
with 35SO4 (10 µCi/ml) for 24 hr, and the radiolabeled GAGs in the cells and the media were
isolated and analyzed by anion-exchange HPLC (Experimental Procedures). The amount of
radioactivity in each fraction was normalized to the equivalent amount of cell protein that had
been analyzed. A, wild type, mutant pgsD-623 (Group 1D) and mutant pgsD-H661 (Group
2D). B, Mutant H661 transiently transfected with lambda clone 13-1-2, CHO EXT1 cDNA, or
Wei et al. EXT1 mutants
30
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

control lambda; C, Clones of H661 stably transfected with mouse cDNAs of EXT1 or EXT2.
The elution positions for heparan sulfate and chondroitin sulfate was established by selective
enzymatic digestion of the chains and are indicated by the bars in panel C.
Figure 4. Primary sequence of hamster, human and mouse EXT1. The shaded segment at the N-
terminus is the putative transmembrane domain. Two potential attachment sites for Asn-linked
glycans are circled. Individual amino acids altered in the mutants are shaded and described in
greater detail in Table 2. The underlined tripeptides indicates the positions of DXD and a EXE
motif possibly involved in nucleotide sugar binding.
Figure 5. Northern blot analysis of EXT1 and EXT2. Approximately 5 µg of poly(A+) RNA
from each strain was separated by electrophoresis, blotted to nylon membranes, and probed with
radiolabeled EXT1 or EXT2 cDNA probes (Experimental Procedures). MST refers to a
mastocytoma derived cell line that produces heparin (63).
Wei et al. EXT1 mutants
31
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from
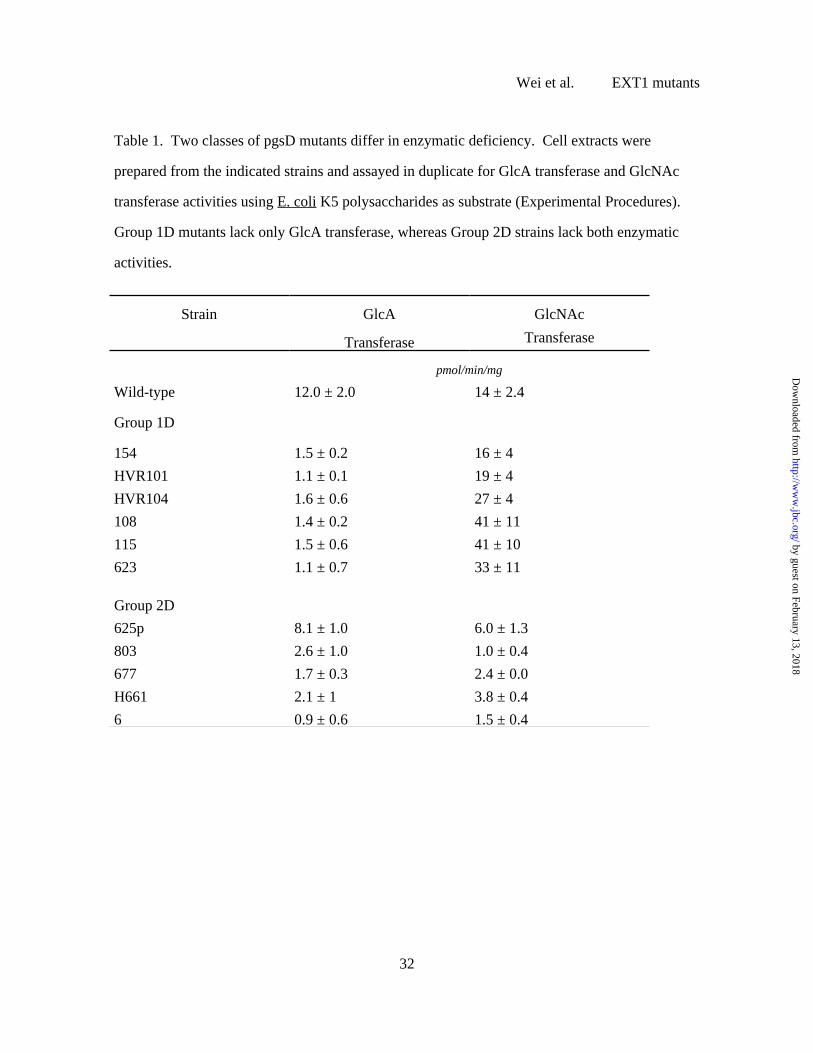
Table 1. Two classes of pgsD mutants differ in enzymatic deficiency. Cell extracts were
prepared from the indicated strains and assayed in duplicate for GlcA transferase and GlcNAc
transferase activities using E. coli K5 polysaccharides as substrate (Experimental Procedures).
Group 1D mutants lack only GlcA transferase, whereas Group 2D strains lack both enzymatic
activities.
Strain GlcA
Transferase
GlcNAc
Transferase
pmol/min/mg
Wild-type 12.0 ± 2.0 14 ± 2.4
Group 1D
154 1.5 ± 0.2 16 ± 4
HVR101 1.1 ± 0.1 19 ± 4
HVR104 1.6 ± 0.6 27 ± 4
108 1.4 ± 0.2 41 ± 11
115 1.5 ± 0.6 41 ± 10
623 1.1 ± 0.7 33 ± 11
Group 2D
625p 8.1 ± 1.0 6.0 ± 1.3
803 2.6 ± 1.0 1.0 ± 0.4
677 1.7 ± 0.3 2.4 ± 0.0
H661 2.1 ± 1 3.8 ± 0.4
6 0.9 ± 0.6 1.5 ± 0.4
Wei et al. EXT1 mutants
32
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from
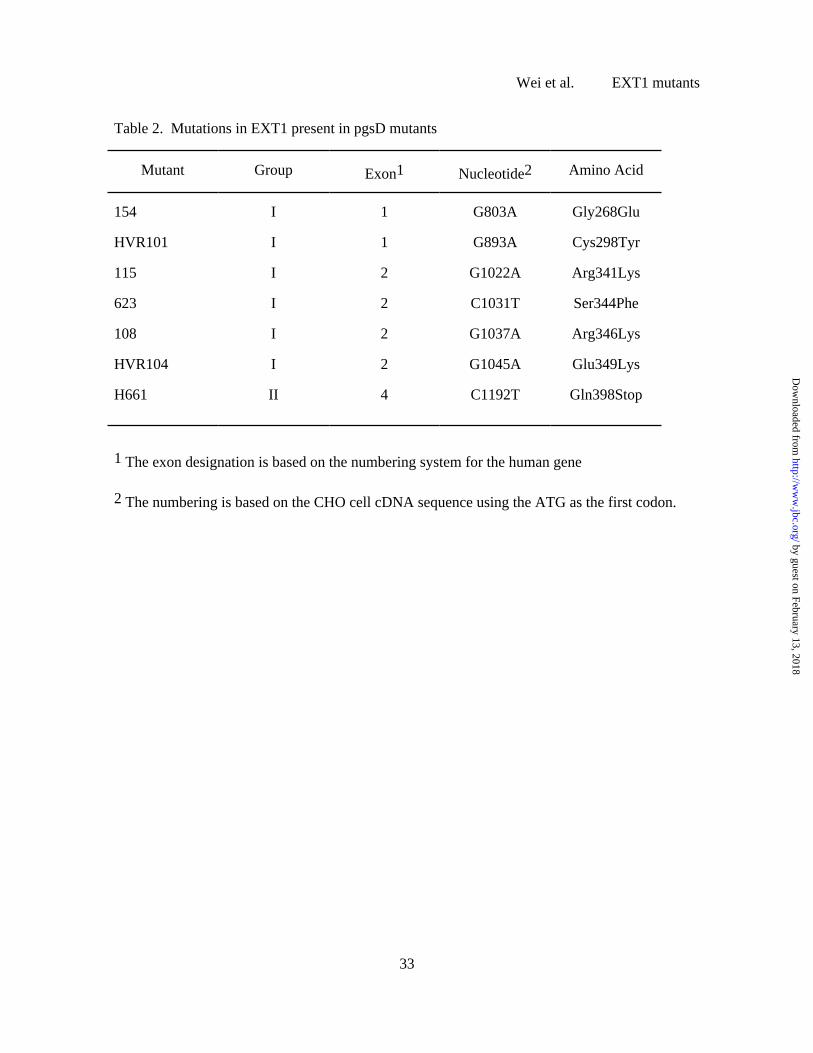
Table 2. Mutations in EXT1 present in pgsD mutants
Mutant Group Exon1 Nucleotide2 Amino Acid
154 I 1 G803A Gly268Glu
HVR101 I 1 G893A Cys298Tyr
115 I 2 G1022A Arg341Lys
623 I 2 C1031T Ser344Phe
108 I 2 G1037A Arg346Lys
HVR104 I 2 G1045A Glu349Lys
H661 II 4 C1192T Gln398Stop
1 The exon designation is based on the numbering system for the human gene
2 The numbering is based on the CHO cell cDNA sequence using the ATG as the first codon.
Wei et al. EXT1 mutants
33
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from

EskoGe Wei, Xiaomei Bai, Karen J. Bame, Thomas I. Koshy, Patricia G. Spear and Jeffrey D.
(EXT1) by analysis of Chinese hamster ovary cell mutantsLocation of the glucuronosyltransferase domain in the heparan sulfate copolymerase
published online June 22, 2000J. Biol. Chem.
10.1074/jbc.M002990200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on February 13, 2018http://w
ww
.jbc.org/D
ownloaded from