Embed Size (px)
Citation preview

Chemical Physics 392 (2012) 90–95
Contents lists available at SciVerse ScienceDirect
Chemical Physics
journal homepage: www.elsevier .com/locate /chemphys
Magnetism in disjoint/non-disjoint composite bands
Masashi HatanakaDepartment of Green and Sustainable Chemistry, School of Engineering, Tokyo Denki University, 2-2 Kanda Nishiki-cho, Chiyoda-ku, Tokyo 101-8457, Japan
a r t i c l e i n f o a b s t r a c t
Article history:Received 27 July 2011In final form 18 October 2011Available online 6 November 2011
Keywords:Organic ferromagnetsNon-bonding orbitalsWannier functionsDisjoint/non-disjointComposite bands
0301-0104/$ - see front matter � 2011 Elsevier B.V. Adoi:10.1016/j.chemphys.2011.10.026
E-mail address: [email protected]
Non-bonding degenerate systems with disjoint/non-disjoint composite bands are suggested. In these sys-tems, one of the non-bonding bands is disjoint, and another is non-disjoint type. Whereas the Wannierfunctions in the former bands span no common atoms, the Wannier functions in the latter bands spancommon atoms. Regardless of the disjoint bands, these systems are predicted to be ferromagnetic dueto non-trivial exchange interactions in the non-disjoint bands. The ferromagnetic interactions are sup-ported by DFT calculations.
� 2011 Elsevier B.V. All rights reserved.
1. Introduction
Non-Kekulé polymers are promising candidates for organic fer-romagnets [1–4]. They have non-bonding crystal orbitals (NBCOs),which are degenerate within Hückel level of theory. 1 in Fig. 1 isthe simplest non-Kekulé polymer. This compound has been theo-retically well analyzed as a ferromagnetic polymer. Several work-ers predicted the spin-polarization structure of this polymer[2,5], and the ground state has been predicted to be ferromagneticby UHF (unrestricted Hartree–Fock) method [2]. UHF description ofthe valence electrons corresponds to the classical Heisenberg mod-el. There appears spin alternation between the adjacent carbonsites, and ferrimagnetic ground states are predicted by countingthe number of up and down spins [6]. In the field of physics, sucha situation is known as bipartite lattices with ferrimagnetism. Inmost cases, difference of number of up and down spins is identicalto the number of non-bonding orbitals. Then, from the Hund’s rule,spin quantum number S of the ground state is predicted byS = (NC � 2ND)/2, where NC and ND are the number of carbon atomicsites and number of formal double bonds [7]. The Heisenberg mod-el can be also applied to p–p stacking organic-radical assemblies[8], and many high-spin organic molecules have been synthesizedand characterized by magnetic measurements. However, in 1977,Borden and Davidson found another spin-preference rule basedon localized orbital methods [9]. That is, for a given biradical, theNBMOs (non-bonding molecular orbitals) are classified into twogroups. When they span no common atoms (disjoint type), the sys-tem is singlet. When they span common atoms (non-disjoint type),the system is triplet due to relative reduction of Coulomb repulsion
ll rights reserved.
in the ionic terms of the total wavefunctions. This rule has beenwidely applied to many biradicals [10], and the mathematical as-pect of the NBMO choice was formulated by Aoki and Imamura[11]. Nowadays, the Borden–Davidson rule is better than the con-ventional Heisenberg model in that some exceptions for the spin-polarization rule (seen in so-called doubly disjoint systems) is alsoexplained by the localized method [10]. In general, spin-quantumnumber predicted by the simple Hund’s rule is larger than thatby valence bond theory (Heisenberg model), and some violationsof Hund’s rule have been overcome by the Borden-Davidson rule.The comparative studies on such high-spin preference have beendone by Klein and coworkers [12–16].
Recently, a localized-orbital method using Wannier functionswas found as another guiding principle for determination of themagnetic properties of non-bonding bands [17–20]. The theoreti-cal proof is based on variational principle for the lowest- and high-est-spin states [20]. That is, for a given band, there exists a properWannier transformation which leads to minimal exchange inte-grals, and the Wannier functions determine the magnetic proper-ties of the system [20]. While disjoint Wannier functions span nocommon atoms, non-disjoint Wannier functions span commonatoms. The former leads to antiferromagnetic, and the latter leadsto ferromagnetic ground states. This is expansion of the Borden-Davidson rule toward extended degenerate systems. Indeed, 1 inFig. 1 has non-disjoint Wannier functions [17]. Poly(alpha,3-ben-zyldiyl) (so-called poly-(m-phenylene)) (2 in Fig. 1) is also awell-known ferromagnetic polymer, of which Wannier functionsspan common atoms within the adjacent unit cells [17]. On theother hand, poly(1,3-cyclobutadienediyl) (so-called poly(cyclobut-adiene)) (3 in Fig. 1) is an antiferromagnetic polymer, becauseeach Wannier function localizes at only one unit cell [17]. The

1
2
3
4 5
N
N
N
N N
Ferromagnetic
Ferromagnetic
Antiferromagnetic
Ferromagnetic Antiferromagnetic
=HC
H2C CH2
Fig. 1. Molecular structures of the simplest non-Kekulé polymer (1), poly(m-phenylene) (2), poly(cyclobutadiene) (3), allyl-radical assemblies (4 and 5).
6
O O O7
NH
O
NH
NH
O
NH
NH
O
NH
8
NH
O
NH
NH
O
NH
NH
O
NH
-2e/unit
8'
N
N
N
Fig. 2. Molecular structures of disjoint/non-disjoint composite systems. 6 hasphenylene and trimethylenemethane skeletons, and 7 is the oxallyl analogue. 8 is amore realizable analogue with ureido groups. 80 is the oxidized form of 8.
M. Hatanaka / Chemical Physics 392 (2012) 90–95 91
spin-preference rule using Wannier functions has been applied tonot only p-conjugate systems but also p–p-stacking systems suchas allyl radical assemblies. 4 in Fig. 1 is an allyl-radical assemblywith ferromagnetic interactions [19]. 5 in Fig. 1 is an allyl-radicalassembly with antiferromagnetic interactions [19]. While 4 hasnon-disjoint Wannier functions, 5 has disjoint Wannier functions.In particular, 5 is an interesting exception for the spin-polarizationrule. That is, while Heisenberg model or McConnell rule predictsferrimagnetic spin alternations, the ground state is anitferromag-netic, regardless of the classical spin-alternation structure. Thus,Wannier analysis of polyradicals has been well applied to designfor many organic ferromagnets. Indeed, very high-spin polyradicalshave been synthesized by using non-Kekulé skeletons with non-disjoint orbitals [21–25].
If polyradicals or radical assemblies have plural radical centersin their unit cells, there should be composite bands near the fron-tier levels. After proper orbital mixing, each band may be disjointor non-disjoint. Exchange interactions in disjoint bands are zero,and usually lead to antiferromagnetism due to the through-spaceinteractions between the adjacent cells. On the other hand, non-disjoint bands lead to ferromagnetism due to the significantly po-sitive exchange interactions. As special cases, composite flat-bandsystems in which disjoint/non-disjoint bands coexist are theoreti-cally interesting in that the spin preference is intuitively not so evi-
dent. In this article, non-Kekulé polymers having both disjoint andnon-disjoint bands are suggested. 6 in Fig. 2 is a non-Kekulé poly-mer of which skeleton consists of mixture of 1 and 2. 7 in Fig. 2 isan oxallyl analogue of 6, and 8 is a ureido analogue of 6. We notethat 8 can be properly oxidized to 80, which is isoelectronic with 6and 7. In view of organic synthesis, non-Kekulé systems with het-ero atoms are promising in that the chemical stability is quite high.Oxallyls are important high-spin intermediates of organic synthe-sis [26,27], and ureidos including phenyl-ureido skeleton like 8 isfamiliar with us as urea derivatives [28]. When ureidos are oxi-dized by proper oxidants, high-spin systems are probably obtained,though further stabilizations by substituent effects are needed,similar to phenyl-substituted aminium high-spin polymers [29].Indeed, as fundamental analyses, high-spin stabilities of urea dica-tion [30] and biuret trication [31] have been established by CASSCFmethod. In polymers 6, 7, and 80, there appear two independentNBCO bands, and each band can be transformed into Wannierfunctions. As shown below, one of the NBCO band is disjoint, whichspans no common atoms, and another NBCO band is non-disjoint,which spans common atoms within the adjacent cells. We studythe electronic structures of them, and predict the spin preferencein view of exchange interactions between the Wannier functions.
2. Theoretical aspects
Wannier analyses of 6–8 can be done within Hückel approxima-tion, similar to 1–5 [20]. First, the non-bonding Bloch functions uk
are obtained as functions of wavenumber k:
uk ¼1ffiffiffiffiNp
XN
l
Xn
r
expðiklÞCrðkÞvl;r ; ð1Þ
where N and n are number of unit cells and atomic sites inside thecell, respectively. vl,r is the rth atomic site in lth cell, and Cr(k) isthe cell-periodic moiety of Bloch coefficients obtained by the usual

92 M. Hatanaka / Chemical Physics 392 (2012) 90–95
secular equation. Wannier function localized at the mth cell is ex-pressed by:
wv ¼XN
l
Xn
r
arðl� mÞvl;r ; ð2Þ
where
arðl� mÞ ¼ 12p
Z p
�pexpfiðl� mÞkgCrðkÞdk; ð3Þ
s ¼ l� m: ð4Þ
In general, Cr(k) cannot be determined due to the phase factor ex-p{i � d(k)}. However, we can choose d(k) so that Cr(k) � exp{i � d(k)}becomes symmetric with respect to k. That is, we newly use C0rðkÞ:
C 0rðkÞ ¼CrðkÞ þ CrðkÞ�
2¼ CrðkÞ þ Crð�kÞ
2ð5Þ
instead of Cr(k). This is real part of Cr(k). Wannier functions deducedfrom each C0rðkÞ are symmetric with respect to s, as is easily con-firmed by Eq. (3). That is, ar(s) = ar(�s). These are the best Wanneirfunctions, which minimizes the exchange integrals between thenon-bonding electrons. The proof of this theorem is given by Fourierexpansion of the phase factor [20]. We note that the best Wannierfunctions should be renormalized by proper scalars, because Wan-nier functions derived from Eq. (5) are not always normalized tounity. Within the simple Hückel approximation, we can adopt thefollowing renormalization factor:
C 0 ¼ 1ffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiffiPs
PrjarðsÞj2
r : ð6Þ
In this article, band structure of 6 was first deduced by simpleHückel crystal orbital method, and Wannier transform was donefor the non-bonding bands. Next, band structures of 6, 7, and 8were calculated by extended Hückel crystal orbital method, inwhich the ionization potentials of H (1s), C (2s), C (2p), N (2s), N(2p), O (2s), O (2p) were set to be 13.60, 21.43, 11.42, 26.00,13.40, 35.30, 17.76 eV, respectively. The Slater l values (effectivenuclear charge divided by the principal quantum number) for H,C, N and O were set to be 1.30, 1.625, 1.95, and 2.275, respectively.For simplicity, CAC, CAN, C@O, CAH, and NAH bond lengths wereset to be 1.40, 1.40, 1.20, 1.09, and 1.01 Å, respectively, and all thebond angles were fixed to be 120�. Planarity of all the molecularskeletons was conserved for simplicity. The overlap integrals were
k
Fig. 3. Band structure of 6 at Hückel level of theory.
taken into account within three times as long as the period a. Theprogram for the band calculation was written by the author andperformed on a personal computer. DFT analyses were performedfor oligomers of 6, 7, and 80 under restricted VWN/6-31G⁄ levelof theory [32] by using GAMESS [33].
3. Results and discussions
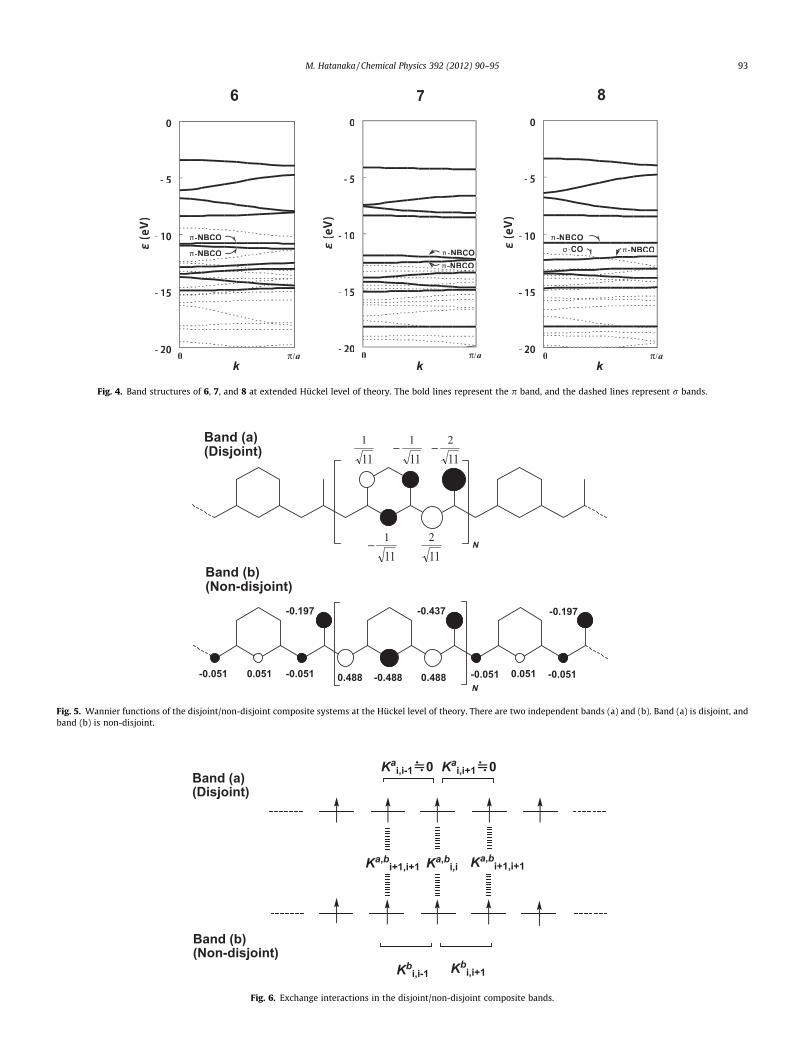
Fig. 3 shows band structure of 6 at the Hückel level of theory.The energy e is in unit of absolute value of b (resonance integral)with respect to the non-bonding level a. We note that the NBCOswith eigenvalue a (Coulomb integral) are doubly overlapped, asdepicted by the central bold line. These two bands correspond tothe plural band components, say, (a) and (b). Fig. 4 shows bandstructure of 6, 7 and 8 at extended Hückel level of theory. The boldlines represent p bands, and dashed lines represent r bands. Thequalitative shapes of dispersions resemble that of the Hückel levelof theory. We see that the NBCOs are split into two bands with nar-row gaps, which results from non-neighbor interactions and het-ero-atom perturbation. As a rough estimation, when the orbitalgaps between the two orbitals are enough smaller than the Cou-lomb integrals J between them, high-spin ground states are ex-pected [34]. Energy crossing of the two non-bonding bands isforbidden due to the p symmetries. Therefore, for realization of fer-romagnetic ground states, the mean band gap |De| should be smal-ler than the Coulomb integral J between the two ith Wannierfunctions. Taking into account of Wannier coefficients in the band(a) and (b), this criterion can be described as:
Dejj < Ja;bi;i ffi
Xunitcell
r
aar ð0Þ
2abr ð0Þ
2ðrr rrj Þ; ð7Þ
where (rr|rr) is the one-centered electron repulsion on the rth site.The superscripts and subscripts in J represent the correspondingbands and location of the unit cells, respectively. As shown later,this inequality is well satisfied for 6, 7, and 80, because the energysplittings between their NBMOs are at most 0.5–1 eV (10–20 kcal/mol). All the Wannier functions should be singly occupied, and fer-romagnetic ground states are highly expected. For actual analysis ofnearly degenerate cases, we had better examine the magnitude ofexchange integrals, rather than Coulomb integrals, becauseclosed-singlet states can be properly transformed into open-shellstates by configuration interactions. Then, degree of high-spin pref-erence is approximately evaluated only by exchange integrals be-tween the singly occupied orbitals. The high-spin preference canbe also proved directly by DFT calculations on the oligomer of eachunit.
Fig. 5 shows Wannier function of 6. We note that there existtwo independent bands (a) and (b) at non-bonding level due tothe two radical centers. The amplitudes of each band are not deter-mined uniquely within one-electron model due to the degeneracy.However, within each band, there exists a set of Wannier functionswhich minimize the exchange integrals, and the resultant Wannierfunctions are identical to the maximally localized Wannier func-tions. By using these Wannier functions, we can estimate the Cou-lomb integral J between the two ith Wannier functions. Supposingthat (rr|rr) is ca. 11 eV for carbon and charged nitrogen,J ¼ 0:178ðrrjrrÞ ; 45 kcal=mol >j De j. Thus, inequality (7) is wellsatisfied for 6, 7, and 80. Strictly speaking, we should transformthe two composite bands simultaneously so as to minimize the to-tal exchange integrals. However, it is evident from the amplitudepattern that any Wannier transformation cannot make them bothdisjoint simultaneously, and thus, the best Wannier functions al-ways consist of disjoint and non-disjoint pair. Actually, if the Blochcoefficients C0rðkÞ are independent of k, the corresponding Wannierfunctions should be disjoint [20]. In the present system, it can be
k k k
Fig. 4. Band structures of 6, 7, and 8 at extended Hückel level of theory. The bold lines represent the p band, and the dashed lines represent r bands.
Band (a) (Disjoint)
Band (b)(Non-disjoint)
11
1
11
1−
11
1−
11
2−
11
2
0.488 -0.488 0.488
-0.437
-0.051 0.051 -0.051
-0.197 -0.197
-0.051 0.051 -0.051
N
N
Fig. 5. Wannier functions of the disjoint/non-disjoint composite systems at the Hückel level of theory. There are two independent bands (a) and (b). Band (a) is disjoint, andband (b) is non-disjoint.
Band (b)(Non-disjoint)
Band (a)(Disjoint)
Kbi,i+1
Ka,bi,i
Kbi,i-1
Kai,i-1 0
Ka,bi+1,i+1Ka,b
i+1,i+1
Kai,i+1 0
Fig. 6. Exchange interactions in the disjoint/non-disjoint composite bands.
M. Hatanaka / Chemical Physics 392 (2012) 90–95 93
0
50
100
150
0 2 4 6N
EL-E
H(k
cal/m
ol)
6
7
8'
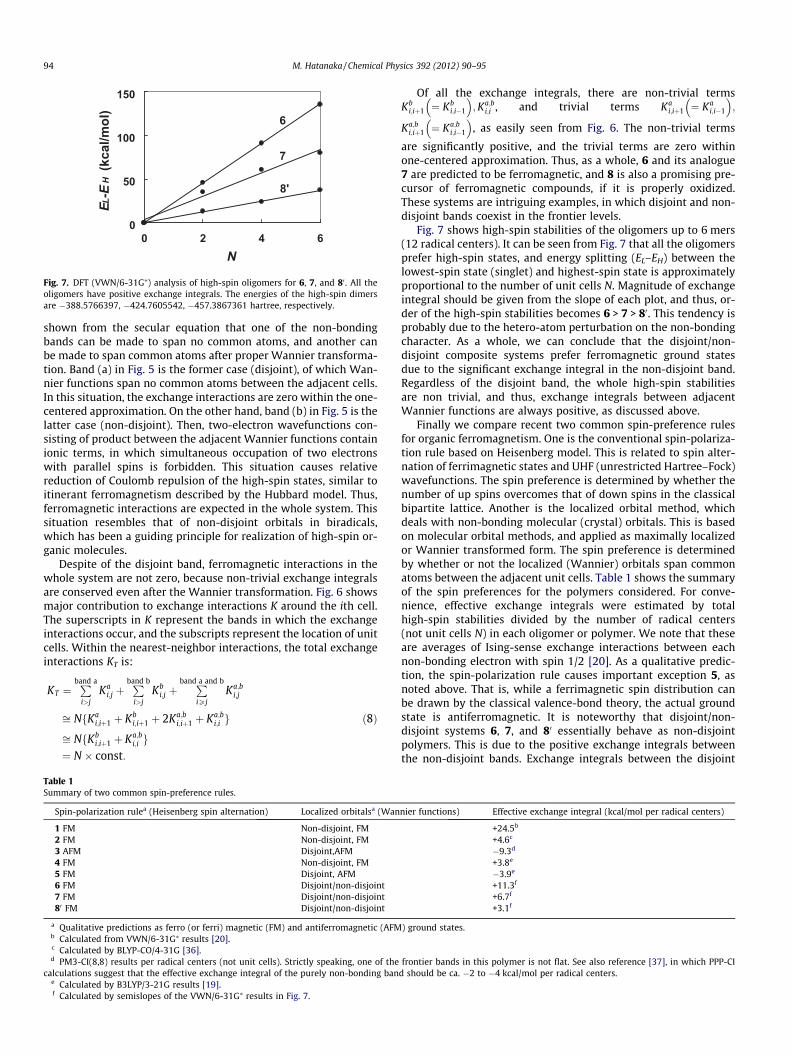
Fig. 7. DFT (VWN/6-31G⁄) analysis of high-spin oligomers for 6, 7, and 80 . All theoligomers have positive exchange integrals. The energies of the high-spin dimersare �388.5766397, �424.7605542, �457.3867361 hartree, respectively.
94 M. Hatanaka / Chemical Physics 392 (2012) 90–95
shown from the secular equation that one of the non-bondingbands can be made to span no common atoms, and another canbe made to span common atoms after proper Wannier transforma-tion. Band (a) in Fig. 5 is the former case (disjoint), of which Wan-nier functions span no common atoms between the adjacent cells.In this situation, the exchange interactions are zero within the one-centered approximation. On the other hand, band (b) in Fig. 5 is thelatter case (non-disjoint). Then, two-electron wavefunctions con-sisting of product between the adjacent Wannier functions containionic terms, in which simultaneous occupation of two electronswith parallel spins is forbidden. This situation causes relativereduction of Coulomb repulsion of the high-spin states, similar toitinerant ferromagnetism described by the Hubbard model. Thus,ferromagnetic interactions are expected in the whole system. Thissituation resembles that of non-disjoint orbitals in biradicals,which has been a guiding principle for realization of high-spin or-ganic molecules.
Despite of the disjoint band, ferromagnetic interactions in thewhole system are not zero, because non-trivial exchange integralsare conserved even after the Wannier transformation. Fig. 6 showsmajor contribution to exchange interactions K around the ith cell.The superscripts in K represent the bands in which the exchangeinteractions occur, and the subscripts represent the location of unitcells. Within the nearest-neighbor interactions, the total exchangeinteractions KT is:
KT ¼Pband a
i>jKa
i;j þPband b
i>jKb
i;j þPband a and b
iPjKa;b
i;j
ffi NfKai;iþ1 þ Kb
i;iþ1 þ 2Ka;bi;iþ1 þ Ka;b
i;i g
ffi NfKbi;iþ1 þ Ka;b
i;i g¼ N � const:
ð8Þ
Table 1Summary of two common spin-preference rules.
Spin-polarization rulea (Heisenberg spin alternation) Localized orbitalsa (Wan
1 FM Non-disjoint, FM2 FM Non-disjoint, FM3 AFM Disjoint,AFM4 FM Non-disjoint, FM5 FM Disjoint, AFM6 FM Disjoint/non-disjoint7 FM Disjoint/non-disjoint80 FM Disjoint/non-disjoint
a Qualitative predictions as ferro (or ferri) magnetic (FM) and antiferromagnetic (AFMb Calculated from VWN/6-31G⁄ results [20].c Calculated by BLYP-CO/4-31G [36].d PM3-CI(8,8) results per radical centers (not unit cells). Strictly speaking, one of the
calculations suggest that the effective exchange integral of the purely non-bonding bane Calculated by B3LYP/3-21G results [19].f Calculated by semislopes of the VWN/6-31G⁄ results in Fig. 7.
Of all the exchange integrals, there are non-trivial termsKb
i;iþ1 ¼ Kbi;i�1
� �;Ka;b
i;i , and trivial terms Kai;iþ1 ¼ Ka
i;i�1
� �;
Ka;bi;iþ1 ¼ Ka;b
i;i�1
� �, as easily seen from Fig. 6. The non-trivial terms
are significantly positive, and the trivial terms are zero withinone-centered approximation. Thus, as a whole, 6 and its analogue7 are predicted to be ferromagnetic, and 8 is also a promising pre-cursor of ferromagnetic compounds, if it is properly oxidized.These systems are intriguing examples, in which disjoint and non-disjoint bands coexist in the frontier levels.
Fig. 7 shows high-spin stabilities of the oligomers up to 6 mers(12 radical centers). It can be seen from Fig. 7 that all the oligomersprefer high-spin states, and energy splitting (EL–EH) between thelowest-spin state (singlet) and highest-spin state is approximatelyproportional to the number of unit cells N. Magnitude of exchangeintegral should be given from the slope of each plot, and thus, or-der of the high-spin stabilities becomes 6 > 7 > 80. This tendency isprobably due to the hetero-atom perturbation on the non-bondingcharacter. As a whole, we can conclude that the disjoint/non-disjoint composite systems prefer ferromagnetic ground statesdue to the significant exchange integral in the non-disjoint band.Regardless of the disjoint band, the whole high-spin stabilitiesare non trivial, and thus, exchange integrals between adjacentWannier functions are always positive, as discussed above.
Finally we compare recent two common spin-preference rulesfor organic ferromagnetism. One is the conventional spin-polariza-tion rule based on Heisenberg model. This is related to spin alter-nation of ferrimagnetic states and UHF (unrestricted Hartree–Fock)wavefunctions. The spin preference is determined by whether thenumber of up spins overcomes that of down spins in the classicalbipartite lattice. Another is the localized orbital method, whichdeals with non-bonding molecular (crystal) orbitals. This is basedon molecular orbital methods, and applied as maximally localizedor Wannier transformed form. The spin preference is determinedby whether or not the localized (Wannier) orbitals span commonatoms between the adjacent unit cells. Table 1 shows the summaryof the spin preferences for the polymers considered. For conve-nience, effective exchange integrals were estimated by totalhigh-spin stabilities divided by the number of radical centers(not unit cells N) in each oligomer or polymer. We note that theseare averages of Ising-sense exchange interactions between eachnon-bonding electron with spin 1/2 [20]. As a qualitative predic-tion, the spin-polarization rule causes important exception 5, asnoted above. That is, while a ferrimagnetic spin distribution canbe drawn by the classical valence-bond theory, the actual groundstate is antiferromagnetic. It is noteworthy that disjoint/non-disjoint systems 6, 7, and 80 essentially behave as non-disjointpolymers. This is due to the positive exchange integrals betweenthe non-disjoint bands. Exchange integrals between the disjoint
nier functions) Effective exchange integral (kcal/mol per radical centers)
+24.5b
+4.6c
�9.3d
+3.8e
�3.9e
+11.3f
+6.7f
+3.1f
) ground states.
frontier bands in this polymer is not flat. See also reference [37], in which PPP-CId should be ca. �2 to �4 kcal/mol per radical centers.

M. Hatanaka / Chemical Physics 392 (2012) 90–95 95
and non-disjoint bands are also positive. Thus, as a whole, twopredictions of the spin preference coincide only when at leastone non-bonding band has non-disjoint character. This suggestsus that Wannier analysis on organic ferromagnetism is better thanthe conventional spin-polarization rule.
Single-molecule magnets require negative zero-field splittingparameter D and giant spin-quantum number S. For example,Christou et al. found single-molecule magnets using transitionmetals with negative D [35]. Similarly, for a given polymer chain,the zero-field splitting parameter D is desired to be negative. Inaddition, each chain should be packed so that intermolecular ex-change integral is positive. This requires intermolecular linkagesbetween the starred and unstarred atoms. This has been alreadypointed out by the author in the reference [20]. These linkagesshould cause ferromagnetic interactions between each polymerchain due to the three dimensional non-disjoint Wannier func-tions. To our knowledge, this is the most promising packing to real-ize ferromagnetic interactions of three-dimensional non-bondingsystems. In organic systems, D usually results from dipole–dipoleinteractions, and difference in intra- and intermolecular interac-tions probably causes significant anisotropy. Thus, these polymersare useful for molecular designing of organic ferromagnetism withcomposite bands.
4. Conclusion
Disjoint/non-disjoint composite bands were suggested asintriguing electronic structures. Ferromagnetic polymers with dis-joint/non-disjoint composite bands were newly proposed based onhydrocarbons, oxallyls, and ureidos. Whereas the Wannier func-tions in the disjoint band span no common atoms, the Wannierfunctions in the non-disjoint band span common atoms. Afterany Wannier transformation, there remain non-trivial exchangeinteractions within the non-disjoint bands, and between disjoint/non-disjoint bands. Thus, regardless of the disjoint bands, thesesystems are predicted to be ferromagnetic, which were supportedby DFT calculations.
References
[1] N. Mataga, Theor. Chim. Acta 10 (1968) 372.[2] N. Tyutyulkov, P. Schuster, O. Polansky, Theor. Chim. Acta 63 (1983) 291.[3] N.N. Tyutyulkov, S.C. Karabunarliev, Int. J. Quantum. Chem. 29 (1986) 1325.[4] F. Dietz, N. Tyutyulkov, Chem. Phys. 264 (2001) 37.[5] A.A. Ovchinnikov, V.O. Cheranovskii, Theor. Exp. Chem. 17 (1981) 6.[6] A.A. Ovchinnikov, Theor. Chim. Acta 47 (1978) 297.[7] H.C. Longuet-Higgins, J. Chem. Phys. 18 (1950) 265.[8] H.M. McConnell, J. Chem. Phys. 39 (1963) 1910.[9] W.T. Borden, E.R. Davidson, J. Am. Chem. Soc. 99 (1977) 4587.
[10] W.T. Borden, Mol. Cryst. Liq. Cryst. 232 (1993) 195.[11] Y. Aoki, A. Imamura, Int. J. Quantum Chem. 74 (1999) 491.[12] S.A. Alexander, D.J. Klein, J. Am. Chem. Soc. 110 (1988) 3401.[13] D.J. Klein, Pure Appl. Chem. 55 (1983) 299.[14] D.J. Klein, S.A. Alexander, in: R.B. King, D.H. Rouvray (Eds.), Graph Theory and
Topology in Chemistry, vol. 51, Elsevier, Amsterdam, The Netherlands, 1987, p.404.
[15] D.J. Klein, S.A. Alexander, Mol. Cryst. Liq. Cryst. 232 (1993) 219.[16] D.J. Klein, C.J. Nelin, S. Alexander, F.E. Matsen, J. Chem. Phys. 77 (1982) 3101.[17] M. Hatanaka, R. Shiba, Bull. Chem. Soc. Jpn. 80 (2007) 2342.[18] M. Hatanaka, R. Shiba, Bull. Chem. Soc. Jpn. 81 (2008) 460.[19] M. Hatanaka, R. Shiba, Bull. Chem. Soc. Jpn. 81 (2008) 966.[20] M. Hatanaka, Theor. Chem. Acc. 129 (2011) 151.[21] H. Iwamura, Adv. Phys. Org. Chem. 26 (1990) 179.[22] H. Iwamura, Proc. Jpn. Acad. Ser. B 81 (2005) 233.[23] A. Rajca, Chem. Rev. 94 (1994) 871.[24] S. Rajca, A. Rajca, J. Solid State. Chem. 159 (2001) 460.[25] A. Rajca, J. Wongsriratanakul, S. Rajca, Science 294 (2001) 1503.[26] M.B. Coolidge, K. Yamashita, K. Morokuma, W.T. Borden, J. Am. Chem. Soc. 112
(1990) 1751.[27] A.S. Ichimura, P.M. Lahti, A.R. Matlin, J. Am. Chem. Soc. 112 (1990) 2868.[28] F. Lortie, S. Boileau, L. Bouteiller, C. Chassenieux, B. Demé, G. Ducouret, M.
Jalabert, F. Lauprêtre, P. Terech, Langmuir 18 (2002) 7218.[29] H. Murata, D. Miyajima, R. Takada, H. Nishide, Polym. J. 37 (2005) 818.[30] M. Hatanaka, Comput. Theor. Chem. 968 (2011) 22.[31] M. Hatanaka, R. Shiba, J. Comp. Chem. Jpn. 5 (2006) 171.[32] S.H. Vosko, L. Wilk, M. Nusair, Can. J. Phys. 58 (1980) 1200.[33] GAMESS Version 12, M.W. Schmidt, K.K. Baldridge, J.A. Boatz, S.T. Elbert, M.S.
Gordon, J.H. Jensen, S. Koseki, N. Matsunaga, K.A. Nguyen, S. Su, T.L. Windus, M.Dupuis, J.A. Montgomery Jr., J. Comput. Chem. 14 (1993) 1347.
[34] T.A. Albright, J.K. Burdett, M.H. Whangbo, Orbital Interactions in Chemistry,Wiley & Sons, New York, 1985. Chapter 8.
[35] M. Murugesu, S. Takahashi, A. Wilson, K.A. Abboud, W. Wernsdorfer, S. Hill, G.Christou, Inorg. Chem. 47 (2008) 9459.
[36] M. Mitani, Y. Takano, Y. Yoshioka, K. Yamaguchi, J. Chem. Phys. 111 (1999)1309.
[37] J. Pranata, D.A. Dougherty, J. Am. Chem. Soc. 109 (1987) 1621.



















![[ACM-ICPC] Disjoint Set](https://img.pdfslide.net/doc/110x75/554ba5c8b4c905ae618b4ec4/acm-icpc-disjoint-set.jpg)








