Embed Size (px)
Citation preview

THE JOURNAL OF BIOLCKXAL CHEMISTRY 0 1994 by The American Society for Biochemistry and Molecular Biology, Inc.
Vol. 269, No. 15, Issue of April 15, pp. 11147-11154, 1994 Printed in U.S.A.
Measurements of ATP Binding on the Large Cytoplasmic Loop of the Sarcoplasmic Reticulum Ca2+-ATPase Overexpressed in Escherichia coli*
(Received for publication, November 23, 1993, and in revised form, December 28, 1993)
Marie-Jo MoutinSQ, Martine CuillelS, Catherine RapinS, Roger MirasS, Marielle Angefl, Anne-Marie Lomprhn, and Yves Dupont8 From the j5aboratoire de Biophysique Moleculaire et Cellulaire, URA 520 d u Centre National de la Recherche Scientifique, Commissariat a 1’Energie Atomique, Grenoble, France and the llznstitut National de la Sante et de la Recherche Medicale U-275-LOA, Ecole Nationale Superieure de Techniques Auancees, Ecole Polytechnique, Centre de IYuette, Palaiseau, France
The large cytoplasmic loop of the sarcoplasmic reticu- lum Ca2+-ATPase (LCL), situated between Lys3’@ and Phe”, is believed to contain both its phosphorylation and ATP binding domains. A cDNA fragment coding for this amino acid sequence was generated in vitro and cloned in vector pQE8 which allowed the overexpres- sion in Escherichia coli of this Ca2+-ATPase domain fused with a cluster of 6 histidines at its NH, terminus. The fusion protein produced in an insoluble form within bacteria was solubilized in 4 M urea, purified on immo- bilized Ni2+, and then renatured by elimination of urea. More than 4 mg of purified renatured fusion protein was obtained from 500 ml of culture.
ATP binding on the refolded protein was demon- strated by two methods: 1) detection of ATP-induced in- trinsic fluorescence change and 2) binding of the fluo- rescent ATP analogue 2’,3’-0-(2,4,6-trinitrophenyl)- adenosine-5‘-triphosphate (TNP-ATP) and its chase by ATP. It is shown that the LCL protein has one single TNP-ATP binding site having a dissociation constant (Kd) of 1.6-1.9 p ~ . Both methods yielded a Kd for ATP around 200 p~. Binding of other nucleotides was de- tected with a sequence of Kd identical to that found for native Ca2+-ATPase: ATP < ADP < GTP < AMP < ITP.
A Mg2‘ binding site was also found on the LCL protein (K, = 100 p~ at pH 7.2). The fluorescence of bound TNP- ATP was found to be highly dependent on Mg2‘ binding on this site.
~ ~~ ~ ~~~~~~~~~~
The ATPase transporting calcium (Ca’+-ATPase) within skel- etal muscle sarcoplasmic reticulum (SR)’ is a member of a class of transmembrane proteins which form a characteristic phos- phoprotein intermediate during their reaction cycle (for review see Inesi (19851, Green and MacLennan (1989), and Bigelow
SC1-0170-C) The costs of publication of this article were defrayed in *This work was supported in part by a grant from the EEC (no
part by the payment of page charges. This article must therefore be hereby marked “aduertisernent” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
5 To whom correspondence and reprint requests should be addressed: Laboratoire de Biophysique Moleculaire et Cellulaire, DBMS-BMC,
54-87. CEA, F-38054 Grenoble Cedex 9. Tel.: 33-76-88-55-93; Fax: 33-76-88-
The abbreviations used are: SR, sarcoplasmic reticulum; LCL. large cytoplasmic loop; nt +n, nucleotide number ( n ) beginning from AofATG of the first Met in the sequence; IPTG, isopropyl-1-thio-p-D galactopy- ranoside; PAGE, polyacrylamide gel electrophoresis; NTA, nitriloacetic acid; PCR, polymerase chain reaction; MOPS, 3-(N-morpholino)pro- panesulfonic acid; GHis, cluster of six histidines; TNP-ATP, 2’,3’-0- (2,4,6-trinitrophenyI)adenosine-5‘-triphosphate; PBS, phosphate-buff- ered saline.
and Inesi (1992)). SR Ca2+-ATPase has been one of the most extensively studied P-type cation pumps owing to its natural abundance and homogeneity in SR membranes. Now, most of the enzymes of this family have been cloned and sequenced (for review see Serrano (1988), Green and MacLennan (19891, and Serrano and Portillo (1990)). A model for their structure was proposed on the basis of protein sequences, affinity-labeled sites, and, more recently, site-directed mutagenesis and elec- tron microscopy (for recent reviews see Green and Stokes (1992) and Inesi and Kirtley (1992)). Most of the results were obtained on the SR Ca*+-ATPase.
The SR Ca2+-ATPase consists of a single polypeptidic chain of 994 amino acid residues, Their most likely arrangement com- prises a small and a large cytoplasmic loop separating three clusters of transmembrane segments (MacLennan et al., 1985; Brand1 et al., 1986; Green and MacLennan, 1989). By affinity labeling experiments several functional sites have been iden- tified in the large cytoplasmic loop, which contains one-third of the amino acids of the protein (Bigelow and Inesi, 1992). More- over, this large region of the protein includes five of the seven most highly conserved sections of P-type ATPase sequences. The NH,-terminal part of the large cytoplasmic loop comprises the aspartic acid residue (Asp351) that is phosphorylated by ATP. Its COOH terminus has several sites labeled by nucleotide analogues. In between is a region labeled by fluorescein iso- thiocyanate (at Lys515) in competition with ATP. The whole large cytoplasmic loop was proposed to subdivided into three domains: the phosphorylation domain, the nucleotide binding domain, and the central domain which is the region of interac- tion between the NH,- and COOH-terminal parts (Green and MacLennan, 1989; Green and Stokes, 1992).
Prediction of the structure of the ATP binding domain has been made using information derived from affinity labeling and from mutagenesis experiments performed by the use of tran- sient expression of the entire SR Ca2+-ATPase in COS-1 cells (Maruyama et al., 1989; Clarke et al., 1990). However, this expression system used until now to test the structure-function of the entire protein did not allow the direct measurement of ATP and Mg2‘ binding, but rather gave indirect information because of the small quantity of mutated protein obtained (Maruyama et al., 1989; Toyofutu et al., 1992). Up to now, the exact fold of the catalytic site still remains uncertain (Green and Stokes, 1992). The localization of the Mg2‘ binding site(s) involved in phosphorylation and ATP binding also remains un- known.
In this paper, we describe the production of an isolated soluble protein corresponding to the large cytoplasmic loop (LCL) of the SR Ca2+-ATPase. Using intrinsic fluorescence and 2’,3’-0-(2,4,6-trinitrophenyl)adenosine-5’-triphosphate (TNP-
11147

11148 ATP Binding on the Isolated Nucleotide Domain of Ca2'-ATPase
tion for expression of the 6His-LCL FIG. 1. Plasmid pKAEL construc-
fusion protein. Plasmid pKAEL was
(Qiagen), a cDNA fragment obtained by constructed by inserting in plasmid pQE8
PCR coding for the factor %-specific cleavage site and just downstream for the large cytoplasmic loop (LCL: Lys3" to Phe7,0) of the SR Ca2+-ATPase from fast
tor; RBS, ribosome binding site; GHis, se- skeletal muscle. Prl l o p , promoter opera-
quence coding for a cluster of 6 histidines; To, terminator; nt +n, nucleotide number ( n ) beginning from A of ATG of the first Met in the sequence of SERCA1.
4 nt +2220 Hind111
Bgl IIBamH I
Factor Xa + nt +985
LCL
Sph I (nt +1793)
ATP) binding, we show that this fragment of the protein is able to bind ATP with high affinity and is sensitive to Mg2+. These first results open the way of mutagenesis experiments on the LCL protein that should be particularly useful to provide in- formation on the structure of the ATP binding domain of P-type ATPases.
MATERIALS AND METHODS Screening Procedure-Four-hundred thousand independent clones
from a neonatal rabbit skeletal fast-twitch muscle cDNA library con- structed in hzap I1 vector (Stratagene) were simultaneously screened with two SR Ca2+-ATPase cDNA probes. Probe 1 corresponded to a coding portion of the rat heart Ca2'-ATPase cDNA(RH117, n t +265 to n t +1446; Lompre et al. 1989). Hybridized a t low stringency (0.5 x SSC, 0.1% SDS, 65 "C) (1 x SSC = 0.15 M sodium chloride and 0.015 M sodium citrate), it allowed to select the Ca2+-ATPase clones which extended to the 5' end portion of the cDNA. Probe 2 corresponded to the most 3' PuuII fragment of the neonatal fast-twitch Ca2+-ATPase. This later probe was hybridized and washed a t high stringency (0.2 x SSC, 0.1% SDS at 65 "C) and allowed to identify the clones specific for the fast Ca2+-ATPase isoform (SERCA1). Both probes were labeled by the ran- dom priming method with [CX-~~PI~ATP (3000 Ci/mmol, Amersham) us- ing a random primer hexamer (Promega) and the Klenow enzyme (Boehringer Mannheim). The clones which reacted with the two probes after three rounds of screening were purified. Two families of clones were isolated: one extending from nt +780 to +2787 and the other from n t +500 t o + E 4 0 of SERCA1. Clone 3 (see below) was a member of the former family. The pBluescript plasmid DNA from the isolated clones was then excised by co-infection with the helper phage R408 (Strat- agene), and inserts were characterized by restriction endonuclease mapping and by sequencing on both strands using the Sequenase kit (United State Biochemical Corp.).
DNA Amplification by Polymerase Chain Reaction-A portion of the Ca2+-ATPase cDNA previously isolated (2 ng of purified DNAfrom clone 3, n t +780 to n t +2787) was amplified by polymerase chain reaction (PCR) in the presence of a 2 p~ concentration of PCR primer pair (each of which contained an endonuclease restriction site, see below), 200 concentrations of the four deoxynucleotide triphosphates, and 5 units of Taq polymerase (Boehringer Mannheim) in 100 pl of reaction medium. Forty PCR cycles of 3 min at 95 "C, 2 min a t 45 "C, and 2 min a t 74 "C were performed in the Minicycler PTC-150 (M.J. Research Inc). The products were analyzed by agarose gel electrophoresis.
PCR primers (synthesized on an Applied Biosystems Synthesizer) are as follows.
Primer 5 ' : 5"GGC AGA TCT ATC GAA GGT AGA AAG AAC GCC ATC GTG A 0 0
BqlII Factor Xa SERCAl
Primer 3': 5"CCA AGC TTA GAA GTT GTC GTC CGC CA
Hind1 I I stop
SERCAl
Cloning Strategy in the pQE Vector: Construction of plLAEL-The
1.2-kilobase pair PCR product was purified on agarose gel, digested with the restriction endonuclease, and subcloned into the BamHI- and HindIII-cut arms of pQE8 (Qiagen) to generate pKAEL (Fig. 1). Recom- binant plasmid was used to transform XL1 blue strain of Escherichia coli. The E. coli transformants were selected on LB agar containing ampicillin (100 pg/ml). Clones obtained were screened with a 612-nt 35S-dATP-radiolabeled probe (PpuM1-Sphl fragment from clone 3) and directly tested by PCR in their ability to allow the production of the 1.2-kilobase pair DNA fragment. Plasmid DNA of positive clones was then analyzed by restriction endonuclease mapping and finally se- quenced. In order to reduce the number of nucleotides to be sequenced, the PpuM1-Sphl fragment of the chosen clones was firstly exchanged with the same fragment coming from clone 3 which was entirely se- quenced in the first part of this work.
Overexpression of LCL in E. coli: Extraction with Urea-Engineered pKAEL was used to transform M15 E. coli strain (Qiagen) already transformed by pREP4 plasmid (Qiagen). PREP4 is a repression plas- mid containing genes for the Lac1 and for the neomycin phosphotrans- ferase which confers kanamycine resistance. The E. coli transformants were selected on LB agar containing ampicillin (100 pg/ml) and kana- mycin (25 pg/ml). For monitoring induction of gene expression, an over- night culture of 2 ml was prepared, and 500 pl of this culture was used to inoculate 500 ml of LB medium enriched with 10 nm KC1 and 4 mM MgSO, and containing 100 pg/ml ampicillin and 25 pg/ml kanamycin. Expression was induced a t a cell density corresponding to an optical density of 0.8-1 at 600 nm by adding 2 nm IPTG (isopropyl-1-thio-P-o- galactopyranoside). The bacteria were further grown within 2 h, until an optical density of 2 at 600 nm was raised.
Cells were then collected, suspended in 40 ml of the lysis buffer (50 mM NaH,PO,, 300 nm NaC1, 5 mM EDTA, and 1 m phenylmethane- sulfonyl fluoride), and vigorously sonicated at 0 "C. The lysate was adjusted to 10 nm MgCI, and treated with Dk4s.e I (1 pg/ml) for 15 min at 37 "C. It was then centrifuged 10 min a t 13,000 x g and the super- natant discarded (SO). The pellet was resuspended in 20 ml of 2 M urea, 10 mM Tris.HC1,lOO mM NaH,PO, at pH 8 and incubated 20 min at 0 "C. After 10 min centrifugation at 13,000 x g (elimination of the superna- tant Sl) , the pellet was again washed with the same solution and centrifuged (elimination of S2). This last pellet was finally suspended in the 20 ml of the "extraction medium" containing 4 M urea, 10 nm Tris.HC1, 100 mM NaH,PO, at pH 8 and incubated overnight a t 4 "C. After centrifuging twice 10 min at 13,000 x g the supernatant (S3) con- taining the extracted LCL protein was loaded onto the affinity column.
protein, we have used an NTA-Ni2+ (NTA = nitriloacetic acid) adsorbent Purification and Renaturation Procedures-To purify the engineered
purchased from Qiagen and prepared according to the manufacturer's protocol. Briefly, 10 ml of this adsorbent were packed into a chromatog- raphy column and equilibrated with the extraction medium (see above). The 4 M urea extract was then loaded and the column was washed with 50 ml of the extraction medium and 30 ml of the same medium but buffered a t pH 6.3. Fusion proteins were then eluted at pH 4 3 . Col- lected fractions were analyzed on SDS-PAGE (polyacrylamide gel per- formed in the presence of sodium dodecyl sulfate, see below)
Immediately after purification, 6His-LCL fractions were neutralized using Tris.HC1 and were combined. Two protocols were then employed to refold the protein.

ATP Binding on the Isolated Nucleotide Domain of Ca2+-ATPase 11149
In the first protocol, refolding was initiated by a progressive elimi- nation of urea. Therefore, LCL proteins were dialyzed during 20 h against a solution containing decreasing concentrations of urea and 100 m~ KC], 20 m~ MOPS a t pH 7.0 in an 1 to 100 volume ratio. The dialysate was then centrifuged a t 360,000 x g for 1 h to eliminate the unfolded product. Refolded soluble LCL were finally concentrated on Macrosep 30K (Filtron), rapidly frozen in liquid nitrogen in 300 mM saccharose, 100 m~ KCl, 20 m~ MOPS at pH 7.0, and conserved until used for their characterization.
In the second protocol, refolding was performed by a large dilution of the protein in a medium that did not contain urea. First, the purified protein was dialyzed twice in a 1 to 35 volume ratio against 4 M urea, 100 mM KCl, 20 m~ MOPS at pH 7.2 (in order to eliminate phosphate). It was then concentrated 10-fold by ultracentrifugation on YM30 mem- brane (Amicon Corp.). Refolding was initiated by a 1 to 100 dilution of LCL in 20% saccharose, 200 m~ KC], 20 mM MOPS at pH 7.2 and 4 "C. The protein was stored at 4 "C under continuous stirring within 15 h and then centrifuged a t 360,000 x g for 1 h to eliminate the unfolded product. It was finally concentrated on YM 30 membrane and conserved in liquid nitrogen until used.
Protein Characterization-Protein concentration was determined by the Folin-Lowry method (Zak and Cohen, 1961). Bovine serum albumin was used as reference.
Denaturing polyacrylamide gels (SDS-PAGE) were performed ac- cording to the Laemmli procedure (Laemmli, 1970). Stacking and run- ning gels contained 5 and 15% polyacrylamide, respectively. The peptide bands were stained with Coomassie Blue.
For immunoblotting, proteins were electrotransferred from SDS- PAGE to nitrocellulose membrane (Bio-Rad). Membranes were first incubated for 1 h at room temperature with 0.05% Tween 20 and 3% bovine serum albumin in PBS (PBS: 137 m~ NaCl, 2.7 mM KCl, 4.3 mM N%HPO,, 1.4 m~ KH,PO,) and then incubated for 3 h at room tem- perature in the same medium but containing a Y200- or 1/500-fold dilution of guinea pig anti-rabbit skeletal muscle SR Ca2+-ATPase an- tiserum. Preparation and characterization of the antiserum have been described previously (Enoufet al., 1988). Five washes with 0.05% Tween 20 in PBS were then performed. Bound antibodies were detected using '251-labeled G protein (Amersham) and by autoradiography. Membranes were incubated for 2 h at room temperature with the '251-protein (0.04 pCi/cm2 of membrane) and then washed with 0.05% Tween 20 in PBS and with PBS alone. Membranes were dried a t 60 "C before autoradiog- raphy.
NH,-terminal amino acids sequence analysis was performed on an automated Applied Biosystems gas-phase sequenator (477A) equipped with an on-line phenylthiohydantoin-derivative analyzer (model 120A).
Optical Measurements-Intrinsic fluorescence spectra were meas- ured using a SLM-8000 spectrofluorimeter.
Measurements at fixed wavelength were performed a t room tempera- ture in a 1 x 1-cm fluorescence cuvette under continuous stirring using the MOS-200 optical system (Bio-Logic S. A,, Claix, France). Reagents were injected with Hamilton syringes in small volumes (2-20 pl). Ex- citation was provided using a xenon-mercury arc lamp (100 or 150 watts) and a monochromator. Emitted light was detected a t 90".
For TNP-ATP fluorescence measurements the monochromator was set at 410 nm, and the emitted light was filtered by a Balzers K4 filter (52&570 nm) and a cut-off filter with 50% transmission a t 495 nm. Inner filter effects due to TNP-ATP absorbance were corrected a t all concentrations up to 50 p~ TNP-ATP. This was performed by lineariza- tion of the TNP-ATP concentration dependence of the observed fluores- cence of free TNP-ATP. From these calibrations the effective absorbance of TNP-ATP was obtained in each of the experimental condition used. It was subsequently used to correct the fluorescence measurements in the presence of proteins.
For intrinsic fluorescence measurements the excitation monochro- mator was set at 297 nm and the emitted light was filtered by a high- pass cut-off filter with 50% transmission at 335 nm and a black filter to eliminate stray light above 400 nm. TNP-ATP was purchased from Molecular Probes, Inc.
ATPase Actiuity-The assay was performed at 35 "C in 100 pl with 100 pg/ml 6His-LCL, 20 m v MOPS, 100 m~ KC1 a t pH 7.0 in the presence of either 0 or 5 m~ free M e . The reaction was started by addition of 5 mM ATP and stopped at various times (between 0 and 24 h) by addition of 300 pl of 1% SDS (w/v). Inorganic phosphate concen- tration was evaluated by spectrophotometric measurement a t 740 nm after 15 min of a color development carried out with 250 pl of ammo- nium molybdate reagent and 250 pl of Elon reagent, according to Fiske and Subbarow (1925), as modified by Dufour et al. (1988). Spontaneous
hydrolysis in the same conditions, but without enzyme, were also meas- ured and used for correction.
RESULTS
Our overall aim was to study the ATP binding domain of the CaZ+-ATPase. Therefore we have developed a method to pro- duce high quantities of an isolated soluble protein representing the LCL of the SR Ca2+-ATPase of fast twitch skeletal muscle, from lysine 329 to phenylalanine 740.
Construction of the E. coli Expression Vector pKAEL-The plasmid pQE8 was used for the construction of pKAEL which allowed the overexpression in E. coli cells of the LCL protein fused with an histidine cluster (6His tag) at its NH, terminus. Plasmid pQE8 contains a regulable promoter/operator (element N250PSN250P29 from T5), a synthetic ribosomal binding site (RBSII), and a 6His tag sequence preceding a cloning site or- ganized as indicated in Fig. 1. I t also encodes p-lactamase, which confers ampicillin resistance. Its promoter/operator is repressed in the presence of the lac repressor (Lac11 and can be induced by the addition of IPTG. By fusing the recombinant protein with a 6His tag, the purification protocol was reduced to a single chromatographic step which was performed on a metal chelate adsorbent (Hochuli et al., 1988; Hochuli, 1990) as explained below.
The plasmid pKAEL was constructed as shown in Fig. 1 by inserting in pQE8 a cDNA fragment containing the sequence of LCL just downstream from the sequence of the factor Xa-spe- cific cleavage site. The LCL sequence was obtained by PCR amplification of the plasmid DNA of clone 3 isolated as de- scribed under "Materials and Methods." Its insert was entirely sequenced before using for PCR amplification. Four silent mu- tations were found in its sequence compared with that of SERCAl (MacLennan et al., 1985; Brand1 et al., 1986).
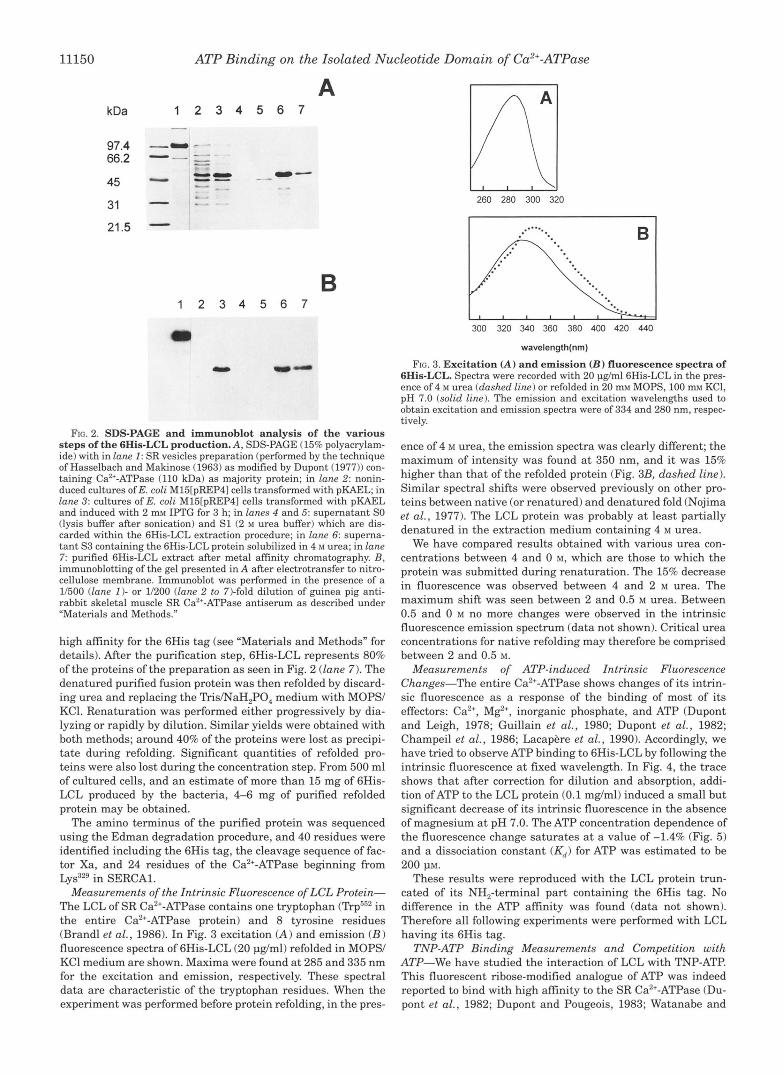
Production of the 6His-LCL Fusion Protein-Fig. 2A shows an SDS-PAGE of the proteins of a noninduced extract of cells and that of cells induced with 2 mM IPTG within 3 h (lune 2 and 3 ) . Immunoblotting of the gel of Fig. 2A using an antiserum specific for SR Ca2+-ATPase is seen in Fig. 2B. The antiserum reacts specifically with SR vesicles Ca2+-ATPase (lane 1 ) and also with the large band at around 50 kDa in induced cells (lane 3 ). This apparent molecular mass corresponds well to that cal- culated for the 6His-LCL protein (46,975 Da). Densitometry scans of the gel show that E. coli cells synthesize LCL at a very high level; the recombinant protein represents about 35% of the total cellular proteins.
The proteins profiles and immunoblotting of the few steps of the extraction and purification protocols developed for the en- gineered protein are also illustrated in Fig. 2. Despite its most probable cytoplasmic location in the entire protein (MacLennan et al., 19851, the LCL fragment of the SR Ca2+-ATPase produced was insoluble in E. coli cells. After disruption of E. coli cells by sonication and then centrifugation, the protein remained in the pellet as can be observed on SDS-PAGE (Fig. 2, lune 5 ) . Other experimental conditions of overexpression were tested includ- ing those described by Blackwell and Horgan (1991) in which induced culture is performed at 25 "C in the presence of sorbi- tol. LCL was always found insoluble. The high level of expres- sion may explained that this foreign protein aggregate within cytoplasm of E. coli cells (Marston, 1986). We have tried various media in order to solubilize the LCL in the pellet after sonica- tion and the less drastic conditions were retained. In the ex- traction protocol currently used, two washes in 2 M urea were performed in order to eliminate some of the contaminating proteins, then the protein of interest was solubilized in 4 M urea (Fig. 2, lane 6).
The fusion protein was then purified in the presence of urea on an Ni2+-NTAresine (nickel-nitriloacetic acid-agarose) having
11150
kDa
97.4 66.2
45
31
21.5
ATP Binding on the Isolated Nucleotide Domain of Ca2’-ATPase
A 1 2 3 4 5 6 7
B 1 2 3 4 5 6 7
FIG. 2. SDS-PAGE and immunoblot analysis of the various steps of the 6His-LCL production. A, SDS-PAGE (15% polyacrylam- ide) with in lane 1: SR vesicles preparation (performed by the technique of Hasselbach and Makinose (1963) as modified by Dupont (1977)) con- taining Ca”-ATPase (110 kDa) as majority protein; in lane 2: nonin- duced cultures of E. coli M15tpREP41 cells transformed with pKAEL; in lane 3: cultures of E. coli M15[pREP41 cells transformed with pKAEL and induced with 2 mM IPTG for 3 h; in lanes 4 and 5 supernatant SO (lysis buffer after sonication) and S1 (2 M urea buffer) which are dis- carded within the 6His-LCL extraction procedure; in lane 6: superna- tant S3 containing the 6His-LCL protein solubilized in 4 M urea; in lane 7 purified 6His-LCL extract after metal affinity chromatography. B, immunoblotting of the gel presented in A after electrotransfer to nitro- cellulose membrane. Immunoblot was performed in the presence of a 1/500 (lane I ) - or 1/200 (lane 2 to 7)-fold dilution of guinea pig anti- rabbit skeletal muscle SR Ca2+-ATPase antiserum as described under “Materials and Methods.”
high affinity for the 6His tag (see “Materials and Methods” for details). After the purification step, 6His-LCL represents 80% of the proteins of the preparation as seen in Fig. 2 (lane 7). The denatured purified fusion protein was then refolded by discard- ing urea and replacing the Tris/NaH,PO, medium with MOPS/ KCI. Renaturation was performed either progressively by dia- lyzing or rapidly by dilution. Similar yields were obtained with both methods; around 40% of the proteins were lost as precipi- tate during refolding. Significant quantities of refolded pro- teins were also lost during the concentration step. From 500 ml of cultured cells, and an estimate of more than 15 mg of 6His- LCL produced by the bacteria, 4-6 mg of purified refolded protein may be obtained.
The amino terminus of the purified protein was sequenced using the Edman degradation procedure, and 40 residues were identified including the 6His tag, the cleavage sequence of fac- tor Xa, and 24 residues of the Ca2+-ATPase beginning from Lys3” in SERCA1.
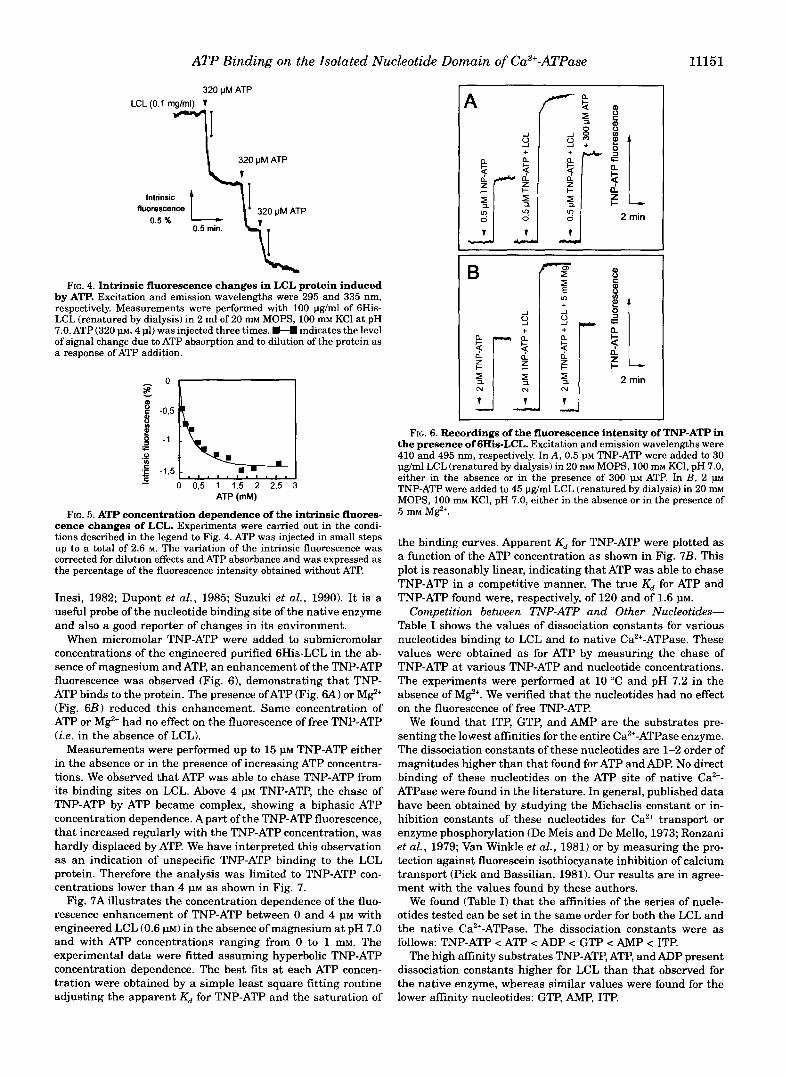
Measurements of the Intrinsic Fluorescence of LCL Protein- The LCL of SR Ca2+-ATPase contains one tryptophan ( T q P 2 in the entire Ca2+-ATPase protein) and 8 tyrosine residues (Brandl et al., 1986). In Fig. 3 excitation (A) and emission (B) fluorescence spectra of 6His-LCL (20 pg/ml) refolded in MOPS/ KC1 medium are shown. Maxima were found at 285 and 335 nm for the excitation and emission, respectively. These spectral data are characteristic of the tryptophan residues. When the experiment was performed before protein refolding, in the pres-
260 280 300 320
300 320 340 360 380 400 420 440
wavelength(nm)
FIG. 3. Excitation (A) and emission (I?) fluorescence spectra of 6His-LCL. Spectra were recorded with 20 pg/mlGHis-LCL in the pres- ence of 4 M urea (dashed line) or refolded in 20 mM MOPS, 100 m~ KCI, pH 7.0 (solid line). The emission and excitation wavelengths used to obtain excitation and emission spectra were of 334 and 280 nm, respec- tively.
ence of 4 M urea, the emission spectra was clearly different; the maximum of intensity was found at 350 nm, and it was 15% higher than that of the refolded protein (Fig. 3B, dashed line). Similar spectral shifts were observed previously on other pro- teins between native (or renatured) and denatured fold (Nojima et al., 1977). The LCL protein was probably at least partially denatured in the extraction medium containing 4 M urea.
We have compared results obtained with various urea con- centrations between 4 and 0 M, which are those to which the protein was submitted during renaturation. The 15% decrease in fluorescence was observed between 4 and 2 M urea. The maximum shift was seen between 2 and 0.5 M urea. Between 0.5 and 0 M no more changes were observed in the intrinsic fluorescence emission spectrum (data not shown). Critical urea concentrations for native refolding may therefore be comprised between 2 and 0.5 M.
Measurements of ATP-induced Zntrinsic Fluorescence Changes-The entire Ca2’-ATPase shows changes of its intrin- sic fluorescence as a response of the binding of most of its effectors: Ca2+, M e , inorganic phosphate, and ATP (Dupont and Leigh, 1978; Guillain et al., 1980; Dupont et al., 1982; Champeil et al., 1986; Lacapere et al., 1990). Accordingly, we have tried to observe ATP binding to 6His-LCL by following the intrinsic fluorescence at fixed wavelength. In Fig. 4, the trace shows that after correction for dilution and absorption, addi- tion of ATP to the LCL protein (0.1 mg/ml) induced a small but significant decrease of its intrinsic fluorescence in the absence of magnesium at pH 7.0. The ATP concentration dependence of the fluorescence change saturates at a value of -1.4% (Fig. 5) and a dissociation constant (K,) for ATP was estimated to be
These results were reproduced with the LCL protein trun- cated of its NH,-terminal part containing the 6His tag. No difference in the ATP affinity was found (data not shown). Therefore all following experiments were performed with LCL having its 6His tag.
TNP-ATP Binding Measurements and Competition with ATP-We have studied the interaction of LCL with TNP-ATP. This fluorescent ribose-modified analogue of ATP was indeed reported to bind with high affinity to the SR Ca2+-ATPase (Du- pont et al., 1982; Dupont and Pougeois, 1983; Watanabe and
200 pM.
ATP Binding on the Isolated Nucleotide Domain of Ca2'-ATPase
320 MM ATP LCL (0.1 mo/ml) t
\ 320 MMATP
Intrinsic fluorescence
0.5 %
FIG. 4. Intrinsic fluorescence changes in LCL protein induced by ATP. Excitation and emission wavelengths were 295 and 335 nm, respectively. Measurements were performed with 100 pg/ml of 6His- LCL (renatured by dialysis) in 2 ml of 20 m~ MOPS, 100 m~ KC1 at pH 7.0. ATP (320 p, 4 pl) was injected three times. H indicates the level of signal change due to ATP absorption and to dilution of the protein as a response of ATP addition.
FIG. 5. ATP concentration dependence of the intrinsic fluores- cence changes of LCL. Experiments were carried out in the condi- tions described in the legend to Fig. 4. ATP was injected in small steps up to a total of 2.6 M. The variation of the intrinsic fluorescence was corrected for dilution effects and ATP absorbance and was expressed as the percentage of the fluorescence intensity obtained without ATP.
Inesi, 1982; Dupont et al., 1985; Suzuki et al., 1990). I t is a useful probe of the nucleotide binding site of the native enzyme and also a good reporter of changes in its environment.
When micromolar TNP-ATP were added to submicromolar concentrations of the engineered purified 6His-LCL in the ab- sence of magnesium and ATP, an enhancement of the TNP-ATP fluorescence was observed (Fig. 61, demonstrating that TNP- ATP binds to the protein. The presence ofATP (Fig. 6A or M e (Fig. 6B) reduced this enhancement. Same concentration of ATP or Mg2' had no effect on the fluorescence of free TNP-ATP (i.e. in the absence of LCL).
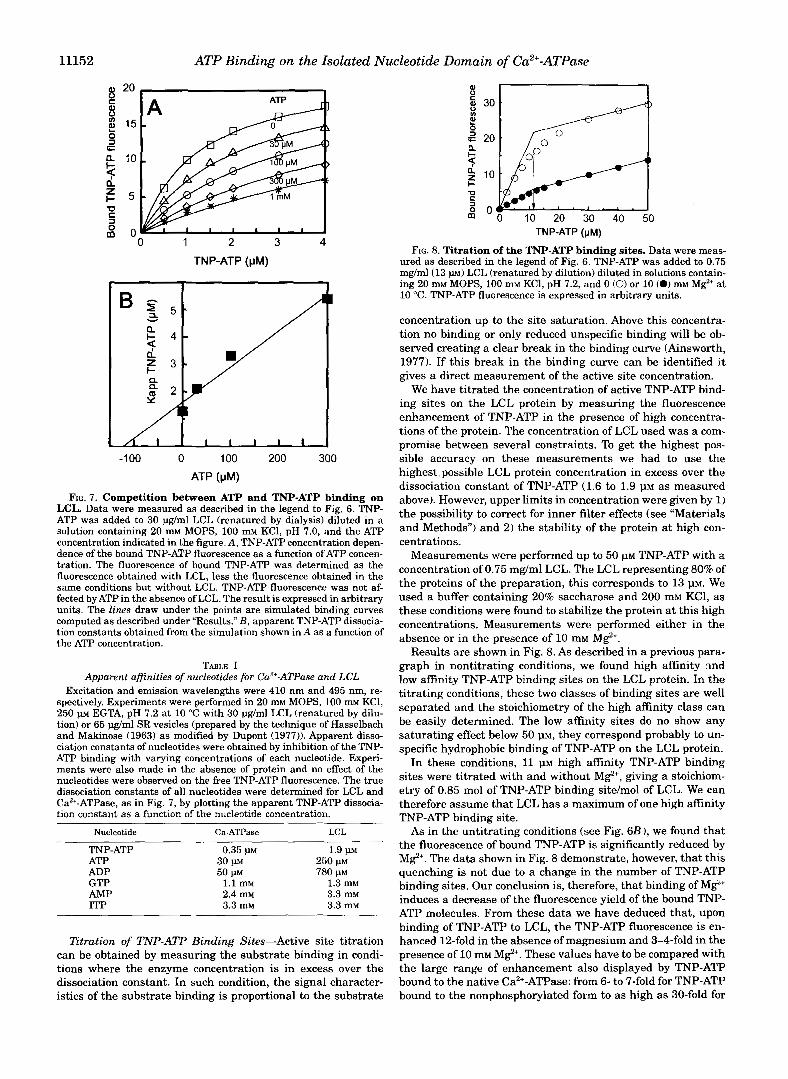
Measurements were performed up to 15 1.1~ TNP-ATP either in the absence or in the presence of increasing ATP concentra- tions. We observed that ATP was able to chase TNP-ATP from its binding sites on LCL. Above 4 PM TNP-ATP, the chase of TNP-ATP by ATP became complex, showing a biphasic ATP concentration dependence. A part of the TNP-ATP fluorescence, that increased regularly with the TNP-ATP concentration, was hardly displaced by ATP. We have interpreted this observation as an indication of unspecific TNP-ATP binding to the LCL protein. Therefore the analysis was limited to TNP-ATP con- centrations lower than 4 p~ as shown in Fig. 7.
Fig. 7A illustrates the concentration dependence of the fluo- rescence enhancement of TNP-ATP between 0 and 4 p~ with engineered LCL (0.6 p ~ ) in the absence of magnesium at pH 7.0 and with ATP concentrations ranging from 0 to 1 mM. The experimental data were fitted assuming hyperbolic TNP-ATP concentration dependence. The best fits at each ATP concen- tration were obtained by a simple least square fitting routine adjusting the apparent Kd for TNP-ATP and the saturation of
A
h- n.
? 0
B
1
I! +
4 5, N N
11151
FIG. 6. Recordings of the fluorescence intensity of TNP-ATP in the presence of 6His-LCL. Excitation and emission wavelengths were 410 and 495 nm, respectively. In A, 0.5 p~ TNP-ATP were added to 30 pg/ml LCL (renatured by dialysis) in 20 m~ MOPS, 100 mM KCl, pH 7.0, either in the absence or in the presence of 300 p~ ATP. In B , 2 p TNP-ATP were added to 45 pg/ml LCL (renatured by dialysis) in 20 m~ MOPS, 100 m~ KCl, pH 7.0, either in the absence or in the presence of 5 mM M e .
the binding curves. Apparent Kd for TNP-ATP were plotted as a function of the ATP concentration as shown in Fig. 7B. This plot is reasonably linear, indicating that ATP was able to chase TNP-ATP in a competitive manner. The true Kd for ATP and TNP-ATP found were, respectively, of 120 and of 1.6 p ~ .
Competition between TNP-ATP and Other Nucleotides- Table I shows the values of dissociation constants for various nucleotides binding to LCL and to native Ca2+-ATPase. These values were obtained as for ATP by measuring the chase of TNP-ATP at various TNP-ATP and nucleotide concentrations. The experiments were performed at 10 "C and pH 7.2 in the absence of M e . We verified that the nucleotides had no effect on the fluorescence of free TNP-ATP.
We found that ITP, GTP, and AMP are the substrates pre- senting the lowest affinities for the entire Ca2+-ATPase enzyme. The dissociation constants of these nucleotides are 1-2 order of magnitudes higher than that found for ATP and ADP. No direct binding of these nucleotides on the ATP site of native Ca2+- ATPase were found in the literature. In general, published data have been obtained by studying the Michaelis constant or in- hibition constants of these nucleotides for Ca2+ transport or enzyme phosphorylation (De Meis and De Mello, 1973; Ronzani et al., 1979; Van Winkle et al., 1981) or by measuring the pro- tection against fluorescein isothiocyanate inhibition of calcium transport (Pick and Bassilian, 1981). Our results are in agree- ment with the values found by these authors.
We found (Table I) that the affinities of the series of nucle- otides tested can be set in the same order for both the LCL and the native Ca2+-ATPase. The dissociation constants were as
The high affinity substrates TNP-ATP, ATP, and ADP present dissociation constants higher for LCL than that observed for the native enzyme, whereas similar values were found for the lower affinity nucleotides: GTP, AMP, ITP.
follows: TNP-ATP < ATP < ADP < GTP < AMP < ITP.
11152 ATP Binding on the Isolated Nucleotide Domain of Ca2'-ATPase
8 20 c A ATP
p 10
2 E 5 U c 3
TNP-ATP (PM)
I
-1 00 0 100 200 300
ATP (PM)
FIG. 7. Competition between ATP and TNP-ATP binding on LCL. Data were measured as described in the legend to Fig. 6. TNP- ATP was added to 30 pg/ml LCL (renatured by dialysis) diluted in a solution containing 20 mM MOPS, 100 mM KC1, pH 7.0, and the ATP concentration indicated in the figure. A, TNP-ATP concentration depen- dence of the bound TNP-ATP fluorescence as a function of ATP concen- tration. The fluorescence of bound TNP-ATP was determined as the fluorescence obtained with LCL, less the fluorescence obtained in the same conditions but without LCL. TNP-ATP fluorescence was not af- fected by ATP in the absence of LCL. The result is expressed in arbitrary
computed as described under "Results." B , apparent TNP-ATP dissocia- units. The lines draw under the points are simulated binding curves
tion constants obtained from the simulation shown in A as a function of the ATP concentration.
TABLE I Apparent affinities of nucleotides for Ca2+-ATPase and LCL
Excitation and emission wavelengths were 410 nm and 495 nm, re- spectively. Experiments were performed in 20 m~ MOPS, 100 m~ KCl, 250 p EGTA, pH 7.2 at 10 "C with 30 pg/ml LCL (renatured by dilu- tion) or 65 pg/ml SR vesicles (prepared by the technique of Hasselbach and Makinose (1963) as modified by Dupont (1977)). Apparent disso- ciation constants of nucleotides were obtained by inhibition of the TNP- ATP binding with varying concentrations of each nucleotide. Experi- ments were also made in the absence of protein and no effect of the nucleotides were observed on the free TNP-ATP fluorescence. The true dissociation constants of all nucleotides were determined for LCL and CaZ'-ATPase, as in Fig. 7, by plotting the apparent TNP-ATP dissocia- tion constant as a function of the nucleotide Concentration.
Nucleotide Ca-ATPase LCL
TNP-ATP 0.35 PM ATP ADP GTP 1.1 mM AMP 2.4 m~
1.3 mM
ITP 3.3 mM
3.3 mM 3.3 mM
1.9 1" 30 w 250 p~ 50 w 780 p~
Ztration of TNP-ATP Binding Sites-Active site titration can be obtained by measuring the substrate binding in condi- tions where the enzyme concentration is in excess over the dissociation constant. In such condition, the signal character- istics of the substrate binding is proportional to the substrate
r n I I
0 10 20 30 40 50 TNP-ATP (pM)
FIG. 8. Titration of the TNP-ATP binding sites. Data were meas- ured as described in the legend of Fig. 6. TNP-ATP was added to 0.75 mg/ml(13 p ~ ) LCL (renatured by dilution) diluted in solutions contain- ing 20 m~ MOPS, 100 m~ KCl, pH 7.2, and 0 (0) or 10 (0) m~ Mg2' a t 10 "C. TNP-ATP fluorescence is expressed in arbitrary units.
concentration up to the site saturation. Above this concentra- tion no binding or only reduced unspecific binding will be ob- served creating a clear break in the binding curve (Ainsworth, 1977). If this break in the binding curve can be identified it gives a direct measurement of the active site concentration.
We have titrated the concentration of active TNP-ATP bind- ing sites on the LCL protein by measuring the fluorescence enhancement of TNP-ATP in the presence of high concentra- tions of the protein. The concentration of LCL used was a com- promise between several constraints. To get the highest pos- sible accuracy on these measurements we had to use the highest.possible LCL protein concentration in excess over the dissociation constant of TNP-ATP (1.6 to 1.9 PM as measured above). However, upper limits in concentration were given by 1) the possibility to correct for inner filter effects (see "Materials and Methods") and 2) the stability of the protein a t high con- centrations.
Measurements were performed up to 50 p~ TNP-ATP with a concentration of 0.75 mg/ml LCL. The LCL representing 80% of the proteins of the preparation, this corresponds to 13 VM. We used a buffer containing 20% saccharose and 200 mM KCl, as these conditions were found to stabilize the protein at this high concentrations. Measurements were performed either in the absence or in the presence of 10 mM M e .
Results are shown in Fig. 8. As described in a previous para- graph in nontitrating conditions, we found high affinity nnd low affinity TNP-ATP binding sites on the LCL protein. In the titrating conditions, these two classes of binding sites are well separated and the stoichiometry of the high affinity class can be easily determined. The low affinity sites do no show any saturating effect below 50 p ~ , they correspond probably to un- specific hydrophobic binding of TNP-ATP on the LCL protein.
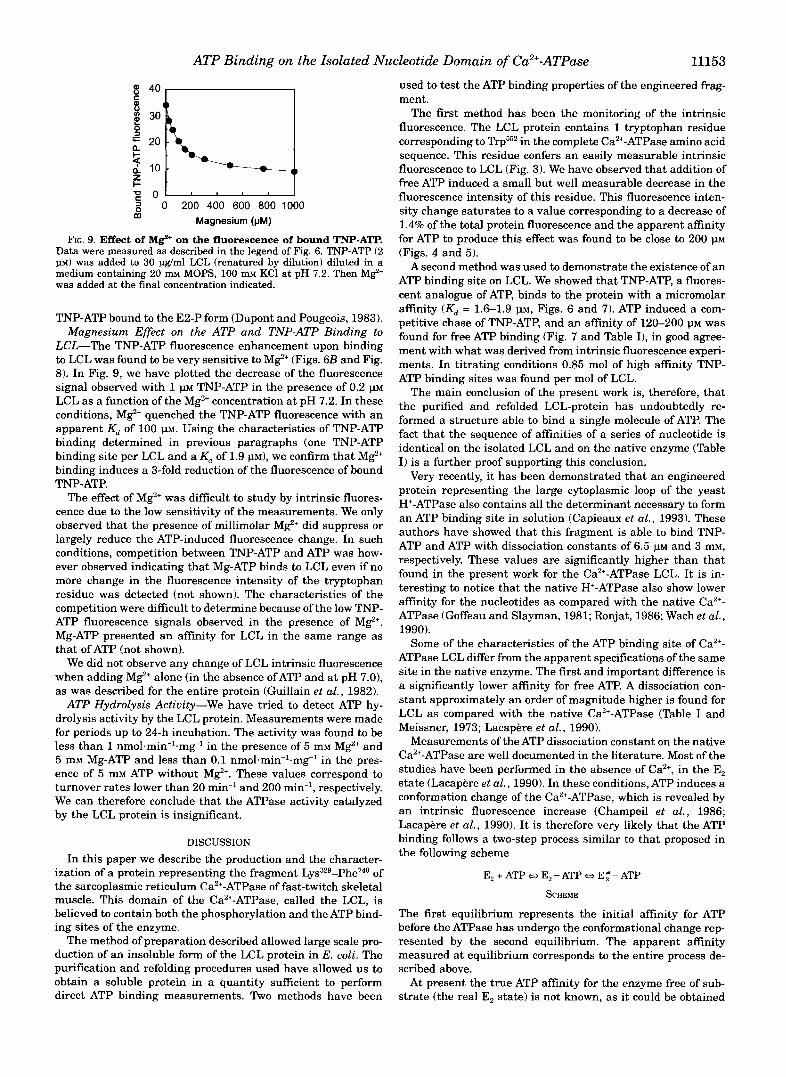
In these conditions, 11 PM high affinity TNP-ATP binding sites were titrated with and without M e , giving a stoichiom- etry of 0.85 mol of TNP-ATP binding site/mol of LCL. We can therefore assume that LCL has a maximum of one high affinity TNP-ATP binding site.
As in the untitrating conditions (see Fig. 6 B ) , we found that the fluorescence of bound TNP-ATP is significantly reduced by M e . The data shown in Fig. 8 demonstrate, however, that this quenching is not due to a change in the number of TNP-ATP binding sites. Our conclusion is, therefore, that binding of Mg2' induces a decrease of the fluorescence yield of the bound TNP- ATP molecules. From these data we have deduced that, upon binding of TNP-ATP to LCL, the TNP-ATP fluorescence is en- hanced 12-fold in the absence of magnesium and 3-4-fold in the presence of 10 mM M e . These values have to be compared with the large range of enhancement also displayed by TNP-ATP bound to the native Ca2+-ATPase: from 6- to 7-fold for TNP-ATP bound to the nonphosphorylated form to as high as 30-fold for
ATP Binding on the Isolated Nucleotide Domain of Ca2+-ATPase 11153
0 200 400 600 800 1000 Magnesium (pM) m
FIG. 9. Effect of M e on the fluorescence of bound TNP-ATP. Data were measured as described in the legend of Fig. 6. TNP-ATP (2 11~) was added to 30 pg/ml LCL (renatured by dilution) diluted in a medium containing 20 m MOPS, 100 m KC1 at pH 7.2. Then Mg2' was added at the final concentration indicated.
TNP-ATP bound to the E2-P form (Dupont and Pougeois, 1983). Magnesium Effect on the ATP and TNP-ATP Binding to
LCL-The TNP-ATP fluorescence enhancement upon binding to LCL was found to be very sensitive to MgZ' (Figs. 6B and Fig. 8). In Fig. 9, we have plotted the decrease of the fluorescence signal observed with 1 m TNP-ATP in the presence of 0.2 PM LCL as a function of the MgZ' concentration at pH 7.2. In these conditions, MgZ' quenched the TNP-ATP fluorescence with an apparent Kd of 100 1". Using the characteristics of TNP-ATP binding determined in previous paragraphs (one TNP-ATP binding site per LCL and a Kd of 1.9 PM), we confirm that Mg2' binding induces a 3-fold reduction of the fluorescence of bound
The effect of Mg2' was difficult to study by intrinsic fluores- cence due to the low sensitivity of the measurements. We only observed that the presence of millimolar MgZ' did suppress or largely reduce the ATP-induced fluorescence change. In such conditions, competition between TNP-ATP and ATP was how- ever observed indicating that Mg-ATP binds to LCL even if no more change in the fluorescence intensity of the tryptophan residue was detected (not shown). The characteristics of the competition were difficult to determine because of the low TNP- ATP fluorescence signals observed in the presence of Mg2'. Mg-ATP presented an affinity for LCL in the same range as that of ATP (not shown).
We did not observe any change of LCL intrinsic fluorescence when adding Mg2' alone (in the absence ofATP and at pH 7.0), as was described for the entire protein (Guillain et al . , 1982).
ATP Hydrolysis Activity-We have tried to detect ATP hy- drolysis activity by the LCL protein. Measurements were made for periods up to 24-h incubation. The activity was found to be less than 1 nmol.min".mg" in the presence of 5 mM MgZ' and 5 mM Mg-ATP and less than 0.1 nmol.min".mg-l in the pres- ence of 5 mM ATP without M$+. These values correspond to turnover rates lower than 20 min" and 200 min", respectively. We can therefore conclude that the ATPase activity catalyzed by the LCL protein is insignificant.
TNP-ATP.
DISCUSSION
In this paper we describe the production and the character- ization of a protein representing the fragment Lys329-Phe740 of the sarcoplasmic reticulum Ca2+-ATPase of fast-twitch skeletal muscle. This domain of the Ca2+-ATPase, called the LCL, is believed to contain both the phosphorylation and the ATP bind- ing sites of the enzyme.
The method of preparation described allowed large scale pro- duction of an insoluble form of the LCL protein in E. coli. The purification and refolding procedures used have allowed us to obtain a soluble protein in a quantity sufficient to perform direct ATP binding measurements. Two methods have been
used to test the ATP binding properties of the engineered frag- ment.
The first method has been the monitoring of the intrinsic fluorescence. The LCL protein contains 1 tryptophan residue corresponding to in the complete Ca2+-ATPase amino acid sequence. This residue confers an easily measurable intrinsic fluorescence to LCL (Fig. 3). We have observed that addition of free ATP induced a small but well measurable decrease in the fluorescence intensity of this residue. This fluorescence inten- sity change saturates to a value corresponding to a decrease of 1.4% of the total protein fluorescence and the apparent affinity for ATP to produce this effect was found to be close to 200 PM (Figs. 4 and 5).
A second method was used to demonstrate the existence of an ATP binding site on LCL. We showed that TNP-ATP, a fluores- cent analogue of ATP, binds to the protein with a micromolar affinity (Kd = 1.6-1.9 p ~ , Figs. 6 and 7). ATP induced a com- petitive chase of TNP-ATP, and an affinity of 120-200 l,m was found for free ATP binding (Fig. 7 and Table I), in good agree- ment with what was derived from intrinsic fluorescence experi- ments. In titrating conditions 0.85 mol of high affinity TNP- ATP binding sites was found per mol of LCL.
The main conclusion of the present work is, therefore, that the purified and refolded LCL-protein has undoubtedly re- formed a structure able to bind a single molecule of ATP. The fact that the sequence of affinities of a series of nucleotide is identical on the isolated LCL and on the native enzyme (Table I) is a further proof supporting this conclusion.
Very recently, it has been demonstrated that an engineered protein representing the large cytoplasmic loop of the yeast H+-ATPase also contains all the determinant necessary to form an ATP binding site in solution (Capieaux et al., 1993). These authors have showed that this fragment is able to bind TNP- ATP and ATP with dissociation constants of 6.5 VM and 3 mM, respectively. These values are significantly higher than that found in the present work for the Ca2+-ATPase LCL. It is in- teresting to notice that the native H+-ATPase also show lower affinity for the nucleotides as compared with the native CaZ+- ATPase (Goffeau and Slayman, 1981; Ronjat, 1986; Wach et al . , 1990).
Some of the characteristics of the ATP binding site of Ca2+- ATPase LCL differ from the apparent specifications of the same site in the native enzyme. The first and important difference is a significantly lower affinity for free ATP. A dissociation con- stant approximately an order of magnitude higher is found for LCL as compared with the native Ca2+-ATPase (Table I and Meissner, 1973; Lacapere et al . , 1990).
Measurements of the ATP dissociation constant on the native Ca2+-ATPase are well documented in the literature. Most of the studies have been performed in the absence of Ca", in the E, state (Lacapere et al . , 1990). In these conditions, ATP induces a conformation change of the Ca2+-ATPase, which is revealed by an intrinsic fluorescence increase (Champeil et al., 1986; Lacapere et al., 1990). I t is therefore very likely that the ATP binding follows a two-step process similar to that proposed in the following scheme
E2+ATPoE,-ATP-Ef-ATP
SCHEME
The first equilibrium represents the initial affinity for ATP before the ATPase has undergo the conformational change rep- resented by the second equilibrium. The apparent affinity measured a t equilibrium corresponds to the entire process de- scribed above.
At present the true ATP affinity for the enzyme free of sub- strate (the real E, state) is not known, as it could be obtained

11154 ATP Binding on the Isolated Nucleotide Domain of Ca2+-ATPase
only before the ATP-induced conformational change has devel- oped. Some indication of it can be found in a recent study by DeJesus et al. (1993). These authors have shown that ATP is able to bind to the Ca2+-ATPase enzyme frozen in the E, state by thapsigargin. Binding of ATP and thapsigargin are uncom- petitive and the dissociation constant found for ATP lies in the range of 200 VM, a value very similar to that found in the present work for the LCL protein.
It is conceivable that, because it is not linked to the mem- brane moiety of the Ca2+-ATPase, the LCL protein is unable to undergo the conformation change in response of ATP binding that would makes its affinity for ATP to increase.
The possibility of existence of a low affinity form of the nucle- otide binding site has been proposed or discussed in many instances in the literature. It is based on the well documented complex modulation of the ATPase activity by ATP (De Meis and De Mello, 1973; Dupont, 1977) and on the observation of low affinity ATP binding sites as measured using fluorescein iso- thiocyanate (Pick, 1981) or TNPnucleotides (Dupont et al., 1985; Bishop et al., 1987). These authors have given convincing evi- dence for the existence of a low affinity or "regulatory" form of the ATP binding, most probably while the enzyme is in its phos- phorylated form (Bishop et al., 1987; Champeil et al., 1988).
Another interesting observation in the present work is the absence of significant effect of Mg2' on the dissociation constant of ATP for LCL protein, whereas this ion is known to increase in most conditions the affinity of ATP for the native Ca2+- ATPase.
The effect of Mg2' on the specifications of the ATP binding site of the native Ca2+-ATPase is complex and not entirely un- derstood. At submillimolar concentrations Mg2' induces an in- crease of the affinity for ATP, which has been proposed to be a consequence of the formation of Mg-ATP; at millimolar concen- trations, Mg2' ions induce a lowering of the ATP affinity, which has been proposed to be a consequence of the formation of Mg,-ATP (Lacapere et al., 1990).
We have shown that Mg2' binds to the LCL with an affinity of 100 p~ (at pH 7.2). The main effect of this binding is a reduction of the fluorescence yield of the bound TNP-ATP. In addition, the LCL protein intrinsic fluorescence change in- duced by ATP binding becomes vanishingly small in the pres- ence of Mg2'.
In recent work, Forge et al. (1993) have given more details on the Mg2' binding possibilities to the Ca2+-ATPase. These au- thors proposed that, depending on the Ca2+-ATPase conforma- tion, Mg2+ can bind to the protein catalytic site with dissocia- tion constants varying from 250 PM to 20 mM.
We propose that Mg2' may induce a change in the conforma- tion of the ATP binding site of the LCL Ca2+-ATPase. It is plausible that this change masks the increase of affinity due to the change of substrate (ATP to Mg-ATP) observed on the native enzyme. As a results of Mg2' binding or release, the LCL protein may have distinct ATP binding site polarities explaining the fluorescence change of bound TNP-ATP. Such important con- formation changes of the nucleotide binding site accompanied by change in its polarity were already described on the native Ca2+- ATPase (Watanabe and Inesi, 1982; Dupont and Pougeois, 1983; Davidson and Berman, 1987; Ferreira and Verjovski-Almeida, 1989). Interestingly the same TNP-ATP fluorescence quenching by magnesium has been found for the isolated cytoplasmic loop of the yeast H+-ATPase (Capieaw et al., 1993).
By allowing, for the first time, the direct measurement of ATP binding to an engineered fragment of the SR Ca2+-ATPase, we provide the basis for a very attractive approach to the study
of the ATP binding domain of the P-type ATPases. Such studies will involve amino acid substitutions by site-directed mutagen- esis in the LCL cDNA, overexpression of the mutants, and study of their properties. They would certainly improve our understanding of molecular details on phosphorylation, ATP, and Mg2' sites.
Acknowledgments-We thank Etienne Capieaux for helpful discus- sions; we thank Mathilde Vingon and Jerome Garin for the sequencing of amino terminus of the 6His-LCL protein; we thank Jean-Paul Issar-
MacLennan for providing SERCAl cDNA probe. tel for the nucleotidic primers synthesis; A,". Lompre thanks Dr.
REFERENCES
Ainsworth, S. (1977) in Steady-state Enzyme Kinetics, pp. 38-40, Unwin Brothers
Bigelow, D. J., and Inesi, G. (1992) Biochim. Biophys. Acta 1113,323-338 Bishop, J. E., Al-Shawi, M. K., and Inesi, G. (1987) J. Biol. Chem. 262,4658-4663 Blackwell, J. R., and Horgan, R. (1991) FEBS Lett. 295, 10-12 Brandl, C. J., Green, N. M., Korczak, B., and MacLennan, D. H. (1986) Cell 44,
Capieaux, E., Rapin, C., Dupont, Y., and Goffeau, A. (19931 J. Biol. Chem. 268,
Champeil, P., Le Maire, M., Moller, J. V., Riollet, S., Guillain, F., and Green, N. M.
Champeil, P., Riollet, S., Orlowski, S., Guillain, F., Seehregts, C. J., and McIntosh,
Clarke, D. M., Loo, T. W., and MacLennan, D. H. (1990) J. Biol. Chem. 265,
Davidson, G . A., and Berman, M. C. (1987) J. Biol. Chem. 262, 7041-7046 DeJesus, F., Girardet, J.-L., and Dupont, Y. (1993) FEBS Lett. 332,229-232 De Meis, L., and Fialho De Mello, M. C. (1973) J. Biol. Chem. 248, 3691-3701 Dufour, J. P., Amory, A,, and Goffeau, A. (1988) Methods Enzymol. 258,513-533 Dupont, Y. (1977) Eur. J. Biochem. 72, 185-190 Dupont, Y., and Leigh, J . B. (1978) Nature 273,396-398 Dupont, Y., and Pougeois, R. (1983) FEBS Lett. 156,93-98 Dupont, Y., Chapron, Y., and Pougeois, R. (19821 Biochem. Biophys. Res. Commun.
Dupont, Y., Pougeois, R., Ronjat, M., Vejovsky-Almeida, S. (1985) J. Biol. Chem.
Enouf, J . , Lompre, A.. M., Bredoux, R., Bourdeau, N., De la Bastie, D., and Levy-
Ferreira, S. T., and Vejovski-Almeida, S. (1989) J . Biol. Chem. 264, 15392-15397 Fiske, C. H., and Suhharow, Y. (1925) J. Biol. Chem. 66, 375-400
Goffeau, A,, and Slayman, C . W. (1981) Biochim. Biophys. Acta 639, 197-223 Forge, V., Mintz, E., and Guillain, F. (1993) J. Biol. Chem. 268, 10953-10960
Green, N. M., and MacLennan, D. H. (1989) Biochem. Soc. nans. 17,8194322 Green, N. M., and Stokes, D. L. (1992) Acta Physiol. Scand. 146, 5 9 4 8 Guillain, F., Gingold, M. P., Biischlen, S., and Champeil, P. (1980) J. Biol. Chem.
Guillain, F., Gingold, M. P., and Champeil, P. (1982)J. Biol. Chem. 257,7366-7371 Hasselbach, W., and Makinose, M. (1963) Biochem. J. 339,94111 Hochuli, E. (1990) Gen. Eng. 12,87-98 Hochuli, E., Banwarth, W., Loheli, H., Gentz, R., and Stiiher, D. (1988) Bioi
Inesi, G. (1985) Annu. Reu. Physiol. 47, 573-601 Inesi, G., and Kirtley, M. R. (1992) J. Bioenerg. Biomembr. 24,271-283 Lacapere, J. J., Bennett, N., Dupont, Y., and Guillain, F. (1990) J. Biol. Chem. 265,
Laemmli, U. K. (1970) Nature 227, 680-685 Lompre, A. M., De la Bastie, D., Boheler, K. R., and Schwartz, K. (1989) FEBS Lett.
MacLennan, D. H., Brandl, C. J., Korczak, B., and Green, N. M. (1985) Nature 22,
Marston, F. A. 0. (1986) Biochern. J. 240, 1-12 Maruyama, K., Clarke, D. M., Fujii, J., Inesi, G., Loo, T. W., and MacLennan, D. H.
(1989) J. Bid. Chem. 264,1303%13042 Meissner, G. (1973) Biochim. Biophys. Acta 298, 906-926 Nojima, H., Ikai, A,, Oshima, T., and Noda, H. (1977) J. Mol. B i d . 116, 429442 Pick, U. (1981) Eur. J. Biochem. 121,187-195 Pick, U., and Bassilian, S. (1981) FEBS Lett. 123, 127-130 Ronjat, M. (1986) Ph.D. thesis, University of Grenohle, Grenoble, France
Serrano, R. (1988) Biochim. Biophys. Acta 947, 1-28 Ronzani, N., Migala, A,, and Hasselhach, W. (1979) Eur. J . Biochem. 101,593-606
Serrano, R., and Portillo, F. (1990) Biochim. Biophys. Acta 1018, 195-199 Suzuki, H., Kubota, T., Kubo, K., and Kanazawa, T. (1990) Biochemistry 29,7040-
Tovofuku. T.. Kurzvdlowski. K., Lvtton, J.. and MacLennan, D. H. (1992) J. B i d .
Ltd., The Gresham Press, Old Woking, Surrey, Great Britain
597-607
21895-21900
(1986) FEBS Lett. 206,8698
D. B. (1988) J. Biol. Chem. 263, 12288-12294
22223-22227
106, 1272-1279
260,7241-7249
Toledano, S. (1988) J. Biol. Chem. 263, 13922-13929
255,2072-2076
Technology, 1321-1325
348-353
249 ,3541
696-700
7045 . .
Van Winkle, W. B., Tate, C. A,, Bick, R. J., and Entman, M. L. (1981) J. Biol. Chem.
Wach, A,, Ahlers, J., and Graher, P. (19901 Eur. J. Biochem. 189,675482 Watanahe. T.. and Inesi. G . (1982) J. Biol. Chem. 257, 11510-11516
"Chem.'267, 14490-14496
256,2268-2274
Zak, B., and Cohen, J. (1961) J. Clin. Chim. Acta 6, 665-670














![The COMATOSE ATP-Binding Cassette Transporter Is · The COMATOSE ATP-Binding Cassette Transporter Is Required for Full Fertility in Arabidopsis1[W][OA] Steven Footitt2, Daniela Dietrich,](https://img.pdfslide.net/doc/110x75/5e3fcb8fadfcd6003a2272db/the-comatose-atp-binding-cassette-transporter-is-the-comatose-atp-binding-cassette.jpg)













