Embed Size (px)
Citation preview

Miscibility and Surface Characterization of aPoly(vinylidene fluoride)-Poly(vinyl methyl ketone)Blend by Inverse Gas Chromatography
ALI AL-GHAMDI,* ZEKI Y. AL-SAIGH
Department of Chemistry and Geology, Columbus State University, 4225 University Avenue, Columbus, Georgia 31907
Received 26 August 1999; revised 24 January 2000; accepted 28 January 2000
ABSTRACT: The miscibility of blends of semicrystalline poly(vinylidene fluoride)(PVF2)and poly(vinyl methyl ketone) (PVMK) along with surface characterization were inves-tigated using the inverse gas chromatography method (IGC), over a range of blendcompositions and temperatures. Three chemically different families, alkanes, acetates,and alcohols, were utilized for this study. The values of the PVF2-PVMK interactionparameters were found to be slightly positive for most of the solutes used, althoughsome degree of miscibility was found at all compositions. Miscibility was greatest at a50:50 w/w composition of the blend. The interaction parameters obtained from IGC arein excellent agreement with those obtained using calorimetry on the same blends. Thecalculated molar heat of sorption of alkanes, acetates, and alcohols into the blend layerreveal the impact of the combination of dispersive and hydrogen bonding forces on theinteraction of solutes with the blend’s backbone. The dispersive component of the surfaceenergy was found to range from 18.70–64.30 mJ/m2 in the temperature range of 82–163°C. A comparison of the blend’s surface energy with that of mercury and other polymersis given. © 2000 John Wiley & Sons, Inc. J Polym Sci B: Polym Phys 38: 1155–1166, 2000Keywords: blend of poly(vinylidene fluoride); poly(vinyl methyl ketone); inverse gaschromatography; miscibility; interaction coefficients; surface energy
INTRODUCTION
Polymer blends are a unique class of materialsthat are easily obtained by blending two ho-mopolymers. Polymer blends are often called “al-loys” and provide unique properties that cannotbe obtained through a single homopolymer. Thekey factor in the characterization of polymerblends is the miscibility. Blends can be completelymiscible, partially miscible, or immiscible. Blendsof poly(vinylidene fluoride) (PVF2) are importantcommercially because PVF2 has been used in
many applications, such as in the electronics in-dustry. PVF2 is a highly polar polymer that pos-sesses ferroelectrical (piezoelectrical and pyro-electrical) properties,1 and it has an interestingmorphology below its melting point.2 It has beenshown that poly(vinyl methyl ketone) (PVMK) isan attractive diluent in blends, particularly whenconducting polymers are used.3 Blending PVMKwith conducting polypyrrole has improved its me-chanical properties without causing loss of con-ductivity.
We have examined the application of inversegas chromatography (IGC) to PVF2 below andabove its melting point. 2 It has been shown thatIGC is more selective than differential scanningcalorimetry (DSC) in obtaining the degree of crys-tallinity of PVF2.2 We were able to report thedegree of crystallinity (29–64%) and specific vol-
* Present address: King Abdulaziz Military Academy, P.O.Box 73040, Riyadh 11538, Saudi Arabia
Correspondence to: Z. Y. Al-Saigh (E-mail: [email protected])Journal of Polymer Science: Part B: Polymer Physics, Vol. 38, 1155–1166 (2000)© 2000 John Wiley & Sons, Inc.
1155

umes of PVF2 at any single temperature in therange of (80–160 °C). Also, it was found that IGCyielded interaction parameters for the miscibilityof blends containing PVF2 comparable to the datareported by DSC.4
Paul et al.5 have reported on the miscibilityof PVF2 with a number of homopolymers usingthe DSC method. The miscibility is based on theidea that specific interactions between chemicalgroups of complementary dissimilarity in twopolymers should produce an exothermic contri-bution to the heat of mixing, and such interac-tions might lead to miscibility for a large num-ber of properly selected pairs. They investigatedthe miscibility of PVF2 and PVMK using DSC,melting point depression and cloud point obser-vations. However, until recently, thermody-namic studies of polymer blends were limited bythe number of available methods. IGC hasemerged to be more selective than DSC in ob-taining accurate information on miscibility, be-cause DSC simply categorizes polymer blendsystems as miscible or immiscible depending onwhether a single Tg is observed or not. Thus,DSC does not provide any thermodynamic in-formation about the strength of the interactionbetween components, or any quantitative rank-ing systems according to the degree of interac-tion.
It is the purpose of this article to examine themiscibility of PVF2-PVMK over a range of tem-peratures near to the PVF2 melting temperature.PVMK was selected as a proton-acceptor polymerto blend with PVF2 in order to obtain a miscibleblend. We will show that IGC can provide detailedthermodynamic information about the strength ofthe interactions between the polymer pair (PVF2-PVMK) at a range of weight fractions and tem-peratures, and between the blend and solutes. Wewill extend the IGC studies to examine the sur-face characterization of the blend and to calculatethe dispersive component of the surface energy ata range of temperatures. IGC has been applied inthe past to several blend systems: PVF2-poly(methyl methacrylate),6,7 PVF2-poly(ethyl methac-rylate),4 PVF2-poly(ethyl acrylate),8 PVF2- poly-styrene.9 To date, IGC has not been applied to thePVF2-PVMK system. However, the same blendwas characterized using DSC and FTIR.10 Also,we reported recently that IGC was successfullyapplied to the characterization of PVF2,2 andPVMK11 as homopolymers.
DATA REDUCTION
Thermodynamics of Polymer Blends
The key term in the calculation of any thermody-namic quantity using IGC is the specific retentionvolume Vg
o measured directly from chromato-graphic parameters. Vg
o is commonly used to de-scribe the elution behavior of solutes, and it isdefined as:
Vgo 5 Dt
Fw
273.15Tr
32
SPi
PoD 2
2 1
SPi
PoD 3
2 1(1)
Here, Dt 5 ts 2 tm is the difference between theretention time of the solute ts and of an unre-tained solute (marker) tm. F is the flow rate of thecarrier gas measured at room temperature Tr, wis the mass of the stationary phase, and Pi and Poare the inlet and outlet pressures, respectively,and are used for the correction for the compress-ibility of the carrier gas. The compressibility fac-tor J can be derived using the last term in eq 1. Ina chromatographic column, a fast equilibrium be-tween the stationary and mobile phases usuallydevelops. As a result, Vg
o measured from the chro-matographic quantities is amenable to thermody-namic interpretation. However, if the columntemperature is not at least 50 °C above the glass-transition temperature of the polymer, Vg
o is notamenable for thermodynamic interpretationowing to surface adsorption and kinetic effects. Vg
o
values obtained by using 0% loading columns canbe subtracted from Vg
o values obtained from col-umns with polymer loadings. The net Vg
o valuesare then amenable for thermodynamic calcula-tions. This procedure will eliminate any contribu-tion to the net Vg
o by the solid support.When a polymer is coated onto the solid sup-
port, the interaction parameters of the solute–polymer can then be calculated, using Vg
o mea-sured according to eq 1, as follows:
x12 5 ln273.15Rn2
VgoV1P1
o 2 1 1V1
M2n22
B11 2 V1
RT P1o
(2)
Equation 2 is used routinely for calculation of x12from IGC experiments. Here x12 is the polymer–solute interaction parameter, where 1 denotes thesolute and 2 denotes the polymer; n2 is the specific
1156 AL-GHAMDI AND AL-SAIGH

volume of the polymer at column temperature T;M1 is the molecular weight of the solute; V1 is themolar volume of the solute; R is the gas constant;and B11 is the second virial coefficient of the sol-ute in the gaseous state.
For polymer blends containing two semicrys-talline polymers such as PVF2-PVMK, the mor-phology of the polymer is more complex as com-pared to an amorphous polymer. Above the melt-ing point, both phases will be at a melt state.However, below the melting point, the structureof the polymer contains two domains, amorphousand crystalline. Therefore, the chromatographicretention involves two mechanisms, one causedby the interaction of the solute with the amor-phous domain, and the other caused by the inter-action of solutes with the crystalline surface.
In a polymer blend, the key term in the misci-bility of a polymer–polymer pair is the free energyof mixing, DGm.
DGm 5 DHm 2 TDSm (3)
where DSm is the combinatorial entropy of mixingand DHm is the molar heat of mixing. Flory12
attributed the combinatorial entropy of mixing tothe mixing of the segments on a lattice of fixedvolume. Because entropy depends on volume, anadditional contribution to the entropy of mixingmay be necessary in eq 3. Sanchez et al.13 havedeveloped a theory to allow for this effect by con-sidering that all mixtures obey the equation ofstate when appropriate reducing parameters,such as pressure and temperature, are used forvolume. Other “equation-of-state” theories of mix-tures yield a combinatorial entropy of mixing sim-ilar to that of Flory. However, the combinatorialentropy becomes negligible as the molecularweight of the polymer becomes high. Therefore, incase of high molecular weight polymers, only thevalue of DHm describes the miscibility of the poly-mer pairs. Flory and Huggins first introduced thevolume fraction term fi in their theory, whichdescribed polymer solutions with reasonable suc-cess.12 The free energy of mixing as described bythe Flory–Huggins theory, is:
DGmix 5 RT$n1ln f1 1 n2ln f2 1 n1f2x12% (4)
Where ni is the number of moles of the ith com-ponent, RT has its usual meaning, and x12 is aparameter that is inversely proportional to abso-lute temperature. Parameter x12 is the same pa-
rameter introduced in eq 2 where it is an enthal-pic contact parameter. The two logarithmic termsrepresent the (combinatorial) entropy of mixing.While the sign of the combinatorial entropy al-ways favors mixing, it is clear that its magnitudeis greatly diminished as molar volumes becomevery large. Thus, at high molecular weights, onlya negative x23 parameter satisfies the conditionfor miscibility of a polymer blend.
Utilizing the specific retention volume, Vgo, the
polymer–polymer interaction coefficient, x23 canbe derived. When a polymer pair is used as astationary (liquid) phase in a chromatographiccolumn, subscripts 2 and 3 will be used to repre-sent polymers 2 and 3, respectively. Subscript 1refers to the test solute. The interaction betweenthe two polymers is expressed in terms of the freeenergy of mixing DGmix, which has the same formas eq 3, only the subscripts change to 2 and 3. Thefirst two (entropic) terms in this equation arenegligible for polymer blends. Thus, for a polymerblend to be miscible (DGmix being negative), x23must be negative. When considering IGC of poly-mer blends, the free energy of mixing must bewritten for a three-component system. It is usu-ally expressed as:
DGmix 5 RT@n1ln f1 1 n2ln f2 1 n3ln f3
1 n1f2x12 1 n1f3x13 1 n2f3x23# (5)
Equation 5 is generally satisfactory for nonpolarmixtures. Unless the interaction coefficients x arelargely independent of the blend in eq 2, n2 shouldbe replaced by (w2n2 1 w3n3), where w2 and w3 arethe weight fractions and n2 and n3 are the corre-sponding specific volumes of the two polymers inthe blend. Thus, one can easily derive eq 6:
ln273.15R~w2n2 1 w3n3!
VgoV1P1
o 2 1 2B11 2 V1
RT P1o
5 f2Fx12 2V1
M2n2G 1 f3Fx13 2
V1
M3n3G
2V1
V2f2f3x23 (6)
Usually the parameter x239 is introduced as:
x923 5 SV1
V2Dx23 (7)
Comparison of eqs 2 and 7 suggests that to obtainx23 for blends, x12 and x13 should be known. Thus,
CHARACTERIZATION OF A PVF2-PVMK BLEND 1157

three columns are usually prepared—two for ho-mopolymers and a third for the blend. The threecolumns should be studied under identical condi-tions of column temperature, carrier gas flowrate, inlet pressure, and solutes used. This keepsall auxiliary parameters (P1
o, T, M2, M3, V1, n2, n3,and B11) identical for the three experiments, anda combination of eq 2 (taken twice for two ho-mopolymers) and eq 7 for the blend will yield:
x923 5
lnVg,blend
o
W2n2 1 W3n32 f2ln
Vg,2o
n22 f3ln
Vg,3o
n3
f2f3(8)
Here, the second subscript of Vgo identifies the
nature of the column. From eq 8, x239 may becalculated even for solutes for which the param-eters P1
o, B11, and V1 are not known or are knownwith insufficient accuracy. Munk et al.14 showedthat x23 values evaluated from experiments de-pended on the chemical nature of the solute, incontradiction to theory. To correct for this effect,Munk introduced a phenomenological relation de-veloped by Munk and Al-Saigh15 and suggestedby Pouchly et al.16
SURFACE ENERGY OF POLYMERS
To quantify the interaction of solute in the gas-eous form with the polymer layer, the surfaceenergy, gs, may be obtained. The surface energydescribes interactions due to dispersive forces or acombination of dispersive forces with H-bondingor with dipole–dipole forces. Fowkes17 first re-ported this method of characterization. Fowkes17
determined the surface energy of several compo-nents. Generally, the contribution of dispersiveforces and all other types of forces can be ex-pressed as the energy of adhesion as follows:
ga 5 gd 1 gsp (9)
where gd is the contribution of dispersive forcesand gsp is the contribution of specific interactionforces such as H-bonding, dipole–dipole, acid-base, and so forth.
The IGC method was successfully applied inrecent years to determine the surface propertiesof divided solids.18–28 From gas chromatographicmeasurements, Vg
o is determined by using eq 1. Vgo
relates to the equilibrium constant K between the
adsorbed solute and the polymer surface as fol-lows:
Vgo 5 KA (10)
where K is the surface partition coefficient and Ais the total surface area of the polymer powder inthe chromatographic column. Thermodynami-cally, the molar free energy of adsorption, DG1
a, ofsolute on the polymer layer can be related to Vg
o bythe following relationship:
DG1a 5 2RT ln Vg
o 1 C (11)
where C is a constant depending on A. eq 9 relatesthe energy of adhesion to the free energy of ad-sorption as follows:
RT ln Vgo 1 C 5 2NaÎgs
dgid (12)
Where gsd and gi
d are the dispersive components ofthe solid surface and the interactive solutesphase, respectively. N is Avogadro’s number anda is the area of the adsorbed molecules (solutes).
In IGC experiments, a series of interactive sol-utes, such as alkanes, can be injected into thechromatographic column in order to determinethe dispersive surface energy, gs
d. A plot of G1a or
(RT ln Vgo) versus the number of carbons in the
alkane chain can be meaningful, because such aplot is linear and the slope of the straight line willaccount for the incremental contribution of DG1
a.The molar enthalpy of adsorption can also be
calculated from DG1a as follows:
DH1a 5 2T2
d
dT SDG1a
T D (13)
Combining eqs 12 and 13 yields the dispersivesurface energy as follows:
gsd 5
14gCH2
HDGaCH2
NaCH2J 2
(14)
Where gCH2 is the surface energy of a hydrocar-bon consisting only of n-alkanes, aCH2 is the areaof one OCH2O group. Equation 14 usually teststhe IGC method for obtaining the dispersive sur-face energy of polymers.
1158 AL-GHAMDI AND AL-SAIGH

EXPERIMENTAL
Materials
In order to establish the strength of the PVF2-PVMK interaction and to obtain information re-garding the strength and type of interactionforces between the blend and the gaseous mobilephase, we have selected three chemically differ-ent families of solutes in this work. Vanishinglysmall amounts of a series of alkanes, acetates,and alcohols were injected into the chromato-graphic column. These solutes will probe the dis-persive, dipole, and hydrogen bonding interac-tions with the blend backbone. A total of sixteensolutes, chromatographic grade, were purchasedfrom Aldrich Chemical Co. Their purity waschecked by gas chromatography prior to use.PVF2 was obtained from Aldrich Chemical Co. ina powder form. Its molecular weight was deter-mined earlier as 250,000 by gel permeation chro-matography.2 PVMK, also supplied by AldrichChemical Co., was in a powder form and had anaverage molecular weight of 500,000 as measuredby Aldrich using GPC.
The melting temperature of PVMK was deter-mined by Aldrich as 160 °C. DSC measurementswere performed on PVMK using modulated DSC(TA Instrument), which revealed a Tg value of 35°C. Chromatographic support, Chromosorb W(AW-DMCS treated, 60/80 mesh) was obtainedfrom Analabs. Chromatographic columns weremade in the laboratory from 5-ft-long, copper tub-ing, 1/4 inch in o.d. All copper columns werewashed with methanol and annealed for severalhours before use. Five chromatographic columnswere prepared. Table I shows the description ofthese columns. Five weight fractions of PVF2 andPVMK were used, ranging from 0 to 100% PVF2.The resulting load of the blend on the column wasabout 7% in all columns. Blending was achievedby dissolving PVF2 and PVMK in hot acetonitrile
and then depositing the solution onto the solidsupport using the method reported earlier.4
INSTRUMENTATIONS AND PROCEDURE
Chromatographic measurements were made us-ing a modified Hewlett Packard 5730A gas chro-matograph equipped with a thermal conductivitydetector. The chromatograph was modified to al-low continuous monitoring of the carrier gas flowrate, the inlet and outlet pressure, and the col-umn temperature. These modifications along withthe complete chromatographic procedure were re-ported in our earlier publication.4 Continuousmonitoring is important because it reduces exper-imental error significantly in the four measurableparameters mentioned in eq 1. This procedureyielded better-controlled measurable quantities.The monitored parameters are usually measuredover a period of seven hours and then their valuesare averaged. Because the blend used in thisstudy contains semicrystalline polymers, every ef-fort was made to avoid recrystallization of bothPVF2 and PVMK by keeping the chromatographoperational at all times. During the course of theexperiments, the oven temperature was uni-formly increased until a complete set of data wasobtained. Control of the mass of the blend in thestationary phase has been modified and a newmethod for coating the polymer was developedand recently reported.4 A flow rate of 8 mL/minwas used throughout this work in order to elimi-nate the effect of flow rate (kinetic) on Vg
o values.From our previous experiments, the flow rate ofthe carrier gas (nitrogen in this case) between0–10 mL/min had no significant effect on the re-tention volumes. Flow rates above 10 mL/minmay cause a considerable error in the retentionvolumes, particularly if helium is used as a car-rier gas.29
Table I. Stationary Phase and Columns Description
ColumnNo.
PVF2
wt/gPVMK
wt/gPVF2
wt %Support
wt/gLoad
%Column Length,
cm
1 0.5585 0 1.0000 8.0010 6.98 1522 0.2802 0.2806 0.4996 7.9000 7.10 1523 0.3747 0.1236 0.7519 7.9578 6.26 1524 0.1253 0.3750 0.2504 7.8990 6.33 1525 0 0.5727 0 7.9597 7.19 152
CHARACTERIZATION OF A PVF2-PVMK BLEND 1159
The chromatographic modifications were ex-tended to include a completely automated datahandling system. An analog/digital data acquisi-tion board (IEEE-488) in the form of a Keithlydigital multimeter was interfaced with a personalcomputer containing a second IEEE-488 board.This allowed for precise measurements of the re-tention times of the solutes injected into the chro-matographic column. The chromatographic signalwas analyzed as a function of time, and the datawas stored for further thermodynamic calcula-tions, which were performed by the PC. The re-tention volumes of solutes on a zero loading col-umn (support only) were stored in a separate fileand interpolated over a wide range of tempera-tures. These retention volumes were then sub-tracted from those measured on loaded columns.This procedure was used to correct for the effect ofthe “inert” solid support on the retention volumes.This automated system was fast and ideal forroutine IGC measurements.
RESULTS AND DISCUSSION
Retention Diagrams
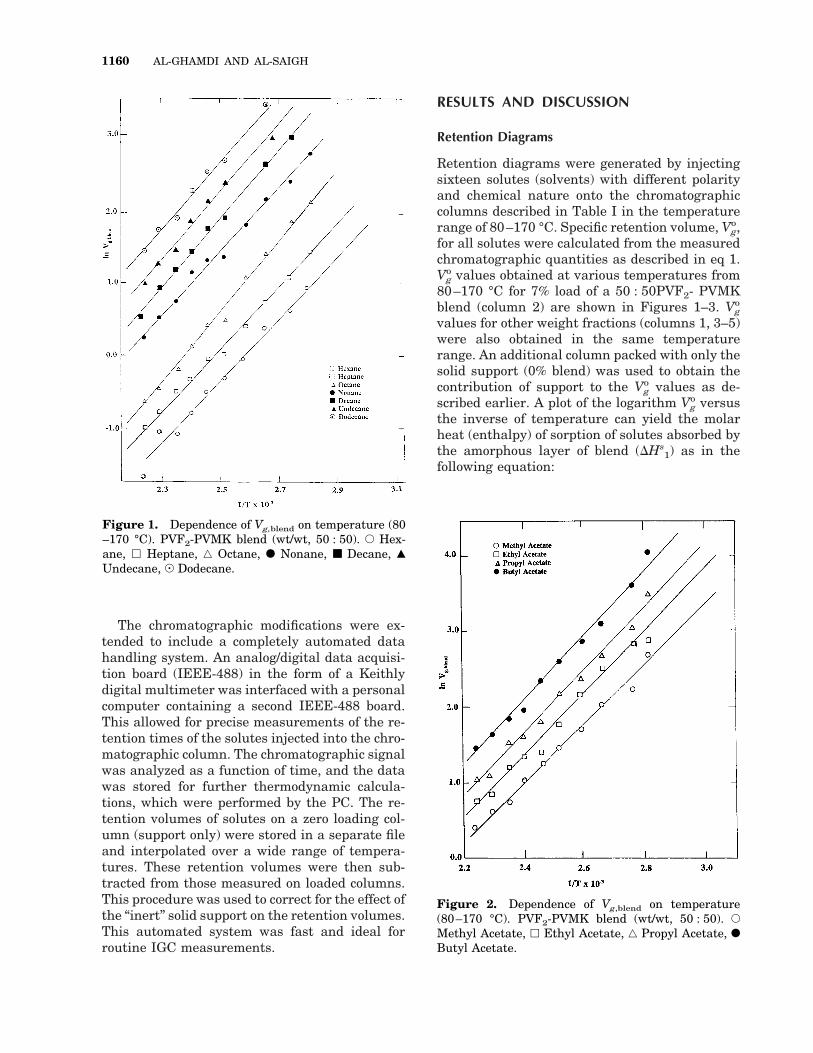
Retention diagrams were generated by injectingsixteen solutes (solvents) with different polarityand chemical nature onto the chromatographiccolumns described in Table I in the temperaturerange of 80–170 °C. Specific retention volume, Vg
o,for all solutes were calculated from the measuredchromatographic quantities as described in eq 1.Vg
o values obtained at various temperatures from80–170 °C for 7% load of a 50 : 50PVF2- PVMKblend (column 2) are shown in Figures 1–3. Vg
o
values for other weight fractions (columns 1, 3–5)were also obtained in the same temperaturerange. An additional column packed with only thesolid support (0% blend) was used to obtain thecontribution of support to the Vg
o values as de-scribed earlier. A plot of the logarithm Vg
o versusthe inverse of temperature can yield the molarheat (enthalpy) of sorption of solutes absorbed bythe amorphous layer of blend (DHs
1) as in thefollowing equation:
Figure 1. Dependence of Vg,blend on temperature (80–170 °C). PVF2-PVMK blend (wt/wt, 50 : 50). E Hex-ane, h Heptane, ‚ Octane, F Nonane, ■ Decane, Œ
Undecane, J Dodecane.
Figure 2. Dependence of Vg,blend on temperature(80–170 °C). PVF2-PVMK blend (wt/wt, 50 : 50). E
Methyl Acetate, h Ethyl Acetate, ‚ Propyl Acetate, FButyl Acetate.
1160 AL-GHAMDI AND AL-SAIGH

DH1s 5 2R
d~ln Vgo!
dS 1TD
(15)
Data are lacking in Figures 1–3 for volatile sol-utes such as pentane at high temperatures andfor high boiling point solutes, such as dodecane, atlower temperatures, because of the condensationof these solutes on the column. Figures 1–3 showthat linear relationships exist between ln Vg
o and1/T for the alkanes, acetates, and alcohols. Themolar heats of sorption, determined from theslope of these lines, are given in Table II, whichshows a comparison of the molar heat of sorptionof the pure PVF2, PVMK, and their blend. Thesorption process involves the transfer of the sol-ute molecules from the vapor phase into theamorphous part of the blend. This process isstrongly depended on the blend-solute interac-tion, x12, and therefore the heat of sorption asso-ciated with this process depends on the interac-tion, too. It is evident from Table II that theblend-solute interactions fall in between thePVF2-solute and PVMK-solute interactions formost of the alkanes. The PVF2-PVMK blend sur-face contains a mixture of dispersive and dipole–
dipole sites, and dipole–dipole interactions areexpected. Plots of molar heat of sorption versusthe number of carbon atoms in the alkanes, ace-tates, and alcohol series are shown in Figure 4.The lines shown here, slopes of 25.50, 215.32,and 222.74 kcal/mol, for alkanes, acetates, andalcohols, respectively. These values indicate thecontribution of additional CH2 groups in the threefamilies’ backbone. The contribution in the alco-hol family is the most exothermic followed by theacetates. This exothermicity shows the effect ofthe combination of dispersive with the dipole andhydrogen bonding attraction forces. Upon com-paring these data with the CH2 contribution inPVF2-alkanes (20.44 kcal/mol), and PVF2-ace-tates, (20.73 kcal/mol), we note that blending ofPVF2-PVMK significantly increases the contribu-tion of CH2 groups to the molar sorption of sol-utes.
Thermodynamic Parameters
The PVF2-PVMK interaction parameters werecalculated according to eq 8 for all weight frac-tions and in the temperature range 100–160 °C.Table III shows the alkane-blend interaction pa-rameters, which are plotted versus the weightfraction of PVF2 in Figure 5. It appears from thesedata that miscibility of the blend is maximized ata 50 : 50 mixture. Although x23 for alkane-blendsystems are slightly positive, Tables IV and Vshow x23 values are closer to zero or negative at
Table II. Molar Heat of Sorption, DH1a, (Kcal/mol)
for PVF2, PVMK, and Their Blend, 80–170 °C
Solute PVF2 PVMK Blend
Pentane – – –Hexane – – 28.45Heptane – 28.15 28.70Octane 22.63 28.57 28.84Nonane 23.69 210.03 29.81Decane 23.25 210.38 29.37Undecane 24.33 211.17 29.77Dodecane 24.11 212.47 29.31Methyl Acetate 26.81 25.99 28.22Ethyl Acetate 28.31 24.53 28.57Propyl Acetate 28.57 24.46 29.32Butyl Acetate 28.58 – 29.41Methanol – 29.23 29.27Ethanol – 29.46 29.04Propanol – 24.00 29.04Butanol – – 29.93
Figure 3. Dependence of Vg,blend on temperature(80–170 °C). PVF2-PVMK blend (wt/wt, 50:50). E
Methanol, h Ethanol, ‚ Propanol, F Butanol.
CHARACTERIZATION OF A PVF2-PVMK BLEND 1161

higher temperatures for the acetates and alco-hols. Methanol showed the most exothermic valueof x23 than any other solute. Our data agree withresults obtained by Paul et al.5 that PVF2 is mis-cible with PVMK at all weight fractions. How-ever, our data do not show that the two polymersare highly miscible due to the slightly positivevalues of x23. Paul et al. found that PVF2 inter-acted strongly with PVAc, PMMA, and PMA, andless strongly with PVMK. Although, Paul et al.obtained a negative interaction energy density forthe same blend using DSC, IGC did not reproducethe DSC results due to the effect of the chemicalnature of solutes on x23. The miscibility arisesfrom the interaction of the carbonyl group inPVMK with PVF2 segments. Table IV shows thepolymer–polymer interaction parameters as afunction of temperature using acetates. This table
Table III. Interaction Parameters, x12, x13, x23, as a Function of Composition andTemperature for Alkane-PVF2, Alkane PVMK, and Alkanes-Blend Systems
Alkanes x12 x13 x23 50 : 50 Blend x23 75 : 25 Blend Temperature
C6 2.06 – 0.23 0.76 100°CC7 2.42 3.86 0.17 0.53C8 2.92 3.81 0.20 0.49C9 3.39 3.73 0.28 0.50C10 3.65 3.83 0.31 0.51C11 4.22 3.81 0.31 0.70C12 – – – –
C6 1.79 – 0.30 0.955 120°CC7 2.15 4.00 0.30 0.76C8 2.58 3.60 0.24 0.61C9 3.04 3.79 0.37 0.58C10 3.33 3.74 0.39 0.51C11 3.60 3.80 0.40 0.51C12 – 3.86 0.29 0.45
C6 1.71 – 0.54 0.79 140°CC7 2.04 3.91 0.42 0.66C8 2.53 3.45 0.34 0.57C9 3.08 3.60 0.39 0.53C10 3.45 3.80 0.28 0.50C11 3.66 3.58 0.24 0.49C12 4.07 3.75 0.28 0.43
C6 1.77 – – 0.74 160°CC7 2.19 3.05 0.33 0.67C8 2.63 3.12 0.38 0.65C9 3.04 3.20 0.33 0.61C10 3.53 3.54 0.37 0.58C11 3.34 3.68 0.40 0.56C12 3.06 3.99 0.36 0.24
Figure 4. Family plot, the molar heat of sorption inKcal/mol versus number of carbons in the solute’s back-bone.
1162 AL-GHAMDI AND AL-SAIGH

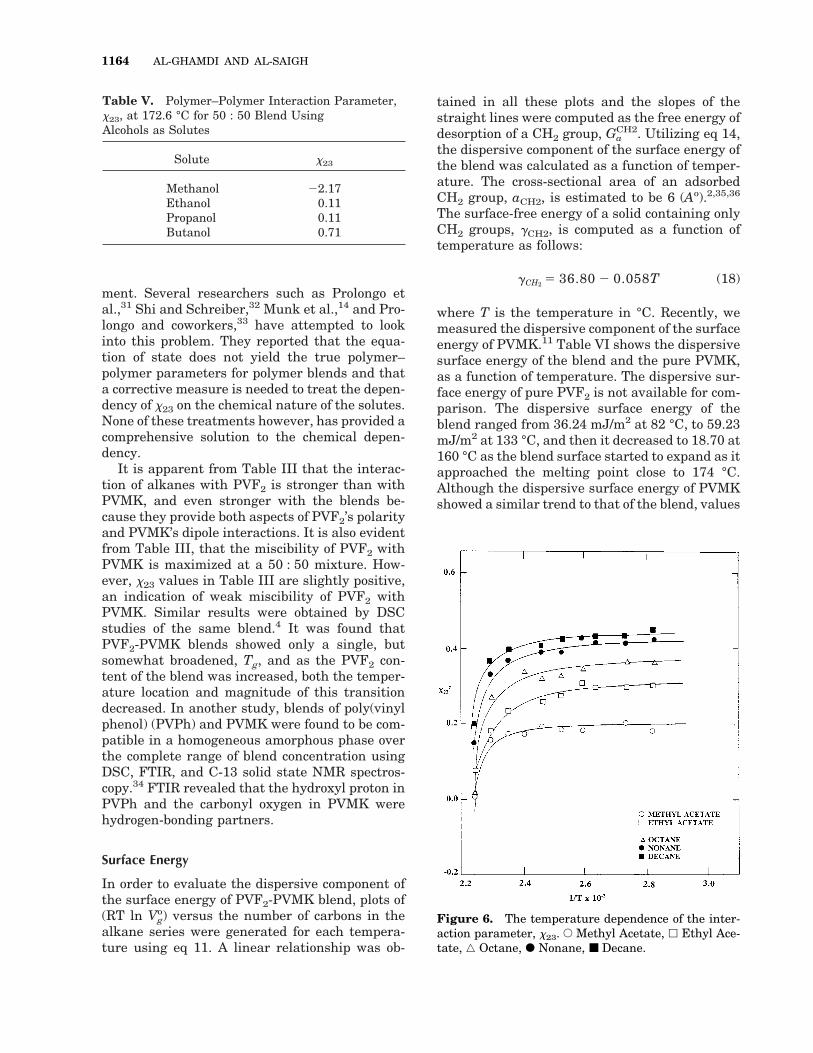
also confirms that the two polymers are nothighly miscible. However, the extent of miscibilityis increased at higher temperatures, particularlyat 163.2 °C at which negative values of x23 wereobtained. Table V shows the interaction parame-ters at 172.60 °C with various alcohols as solute.Although we were not able to conduct experi-ments using alcohols at lower temperature, TableV shows a good correlation of the interaction pa-rameters relative to alkanes and acetates.
Wolf et al.30 suggested that the temperaturedependence of x at different concentrations dem-
onstrates that the heat of dilution generally in-creases with increasing weight fraction, whereasthe noncombinatorial entropy of dilution de-creases. This may lead to a linear interdepen-dence of the enthalpy and entropy part of x. Thesecan be obtained from the following equations:
xH 5DH1
RTf22 5
1T 1
dx
d1T2 (16)
and
xs 5 x 2 xH 5 2DS1
R
Rf22 (17)
where xH and xS are the enthalpy and entropycontributions to the interaction parameters. DH1is the molar heat of dilution, DS1
R, is the entropy ofdilution and f2 is the volume fraction of PVF2.Figure 6 shows the temperature dependence of xof selected solutes. All these plots have interceptsclose to zero as the contribution of entropy ofdilution to the interaction parameters. However,the slopes of the lines are small and in the orderof 0.0063, 0.0017, 0.0069, 0.0063, 0.014 cal/mol foroctane, nonane, decane, and methyl and ethylacetate, respectively.
It is clear from Tables III, IV, and V that x23values differ from one solute to another. Thisdependence of x23 on the chemical nature of sol-utes has been observed and discussed in recentyears. It is our conclusion that this dependence isnot a result of the IGC technique, but rather dueto the inadequacy of polymer solution thermody-namic theories. From the late 1980s to thepresent, several groups have recognized that thedependence of the polymer–solute and polymer–polymer interaction parameters are real. Effortswere made to introduce new treatments to thepolymer solution theories and extreme care needsto be exercised for data interpretation and treat-
Figure 5. The effect of the weight fraction of PVF2 onthe interaction parameter, x23 at various tempera-tures. E Heptane, h Octane, ‚ Nonane, F Decane, ŒDecane, ■ Dodecane.
Table IV. Polymer–Polymer Interaction Parameter, x23, as a Function of Temperature in °C for 50 : 50 BlendUsing Acetates as Solutes
Solutes 82.4 92.5 113.1 123.2 132.8 142.8 152.0 163.2
Methyl Acetate 0.41 0.29 0.39 0.09 0.44 0.79 0.29 20.69Ethyl Acetate 0.55 0.52 0.41 20.02 0.37 0.72 0.52 20.43Propyl Acetate 1.11 1.01 0.63 0.165 0.51 0.79 0.62 20.17Butyl Acetate 1.22 1.19 0.65 0.43 0.60 0.81 0.74 0.29
CHARACTERIZATION OF A PVF2-PVMK BLEND 1163
ment. Several researchers such as Prolongo etal.,31 Shi and Schreiber,32 Munk et al.,14 and Pro-longo and coworkers,33 have attempted to lookinto this problem. They reported that the equa-tion of state does not yield the true polymer–polymer parameters for polymer blends and thata corrective measure is needed to treat the depen-dency of x23 on the chemical nature of the solutes.None of these treatments however, has provided acomprehensive solution to the chemical depen-dency.
It is apparent from Table III that the interac-tion of alkanes with PVF2 is stronger than withPVMK, and even stronger with the blends be-cause they provide both aspects of PVF2’s polarityand PVMK’s dipole interactions. It is also evidentfrom Table III, that the miscibility of PVF2 withPVMK is maximized at a 50 : 50 mixture. How-ever, x23 values in Table III are slightly positive,an indication of weak miscibility of PVF2 withPVMK. Similar results were obtained by DSCstudies of the same blend.4 It was found thatPVF2-PVMK blends showed only a single, butsomewhat broadened, Tg, and as the PVF2 con-tent of the blend was increased, both the temper-ature location and magnitude of this transitiondecreased. In another study, blends of poly(vinylphenol) (PVPh) and PVMK were found to be com-patible in a homogeneous amorphous phase overthe complete range of blend concentration usingDSC, FTIR, and C-13 solid state NMR spectros-copy.34 FTIR revealed that the hydroxyl proton inPVPh and the carbonyl oxygen in PVMK werehydrogen-bonding partners.
Surface Energy
In order to evaluate the dispersive component ofthe surface energy of PVF2-PVMK blend, plots of(RT ln Vg
o) versus the number of carbons in thealkane series were generated for each tempera-ture using eq 11. A linear relationship was ob-
tained in all these plots and the slopes of thestraight lines were computed as the free energy ofdesorption of a CH2 group, Ga
CH2. Utilizing eq 14,the dispersive component of the surface energy ofthe blend was calculated as a function of temper-ature. The cross-sectional area of an adsorbedCH2 group, aCH2, is estimated to be 6 (Ao).2,35,36
The surface-free energy of a solid containing onlyCH2 groups, gCH2, is computed as a function oftemperature as follows:
gCH2 5 36.80 2 0.058T (18)
where T is the temperature in °C. Recently, wemeasured the dispersive component of the surfaceenergy of PVMK.11 Table VI shows the dispersivesurface energy of the blend and the pure PVMK,as a function of temperature. The dispersive sur-face energy of pure PVF2 is not available for com-parison. The dispersive surface energy of theblend ranged from 36.24 mJ/m2 at 82 °C, to 59.23mJ/m2 at 133 °C, and then it decreased to 18.70 at160 °C as the blend surface started to expand as itapproached the melting point close to 174 °C.Although the dispersive surface energy of PVMKshowed a similar trend to that of the blend, values
Table V. Polymer–Polymer Interaction Parameter,x23, at 172.6 °C for 50 : 50 Blend UsingAlcohols as Solutes
Solute x23
Methanol 22.17Ethanol 0.11Propanol 0.11Butanol 0.71
Figure 6. The temperature dependence of the inter-action parameter, x23. E Methyl Acetate, h Ethyl Ace-tate, ‚ Octane, F Nonane, ■ Decane.
1164 AL-GHAMDI AND AL-SAIGH

are generally lower than those for the blend. It isevident that the addition of PVF2 to PVMK has atmost temperatures increased the dispersive sur-face energy. This is in agreement with earlierresults reported by Papirer et al.37 who observeda decrease of the surface energy of treated CaCO3with increasing temperature. At these tempera-tures, the surface of the blend expands. Thus, thesurface energy decreases and allows the vapor topenetrate the surface. Similar observations werereported on a silica carrying alkyl grafts.38
It is of interest to compare values for surfaceenergy of the blend with values of other polymers
obtained by methods such as NMR and FTIR.Table VII shows comparative data on the surfaceenergies of various polymers relative to that ofmercury. It is clear that the blend’s surface en-ergy value obtained at 77–80 °C, is between val-ues for nonpolar and highly polar polymers. Thesurface energy values listed in Table VII corre-spond well with the types of interaction sitesavailable in the polymer backbone. Comparingthese data with the surface energy of conductingpolymers,39,40 the surface energy of the blend isclose to that of Cl-doped Ppy.39 The dispersivecomponent to the surface energy of several poly-pyrroles is in the range of 30–60 mJ/m2, whereasconducting polypyrroles have a much higher sur-face energy, 106 mJ/m2.39,40
REFERENCES AND NOTES
1. Kawai, H. Jpn J Appl Phys 1969, 8, 975.2. Chain, C. T.; Al-Saigh, Z. Y. Macromolecules 1989,
22, 2974.3. Wang, H. L.; Fernandez, J. E. Macromolecules
1992, 25, 6179.4. Al-Saigh, Z. Y.; Chen, P. Macromolecules 1991, 24,
3788.5. (a) Berstein, R. E.; Wahrmund, D. C.; Barlow,
J. W.; Paul, D. R. Polymer Eng Sci 1978, 18, 1220;(b) Paul, D. R.; Barlow, J. W.; Berstein, R. E.;Wahrmund, D. C. Polymer Eng Sci 1978,18, 1225.
6. DiPaola-Bananyi, G.; Fletcher, S. J.; Degre, P. Mac-romolecules, 1982, 15, 885.
7. DiPaola-Baranyi, G. Inverse Gas Chromatography,Characterization of Polymers and Other Materials;Lloyed, D. R.; Ward, T. C.; Schreiber, H. P., Eds.;American Chemical Society Symposium Series 391:Washington, DC, 1989; p 108.
8. Galin, M.; Maslinko, L. Eur Polym J 1987, 23, 923.
Table VII. Comparative Data on gsd in mJ/m2 of
Several Polymers, Blend, and Mercury
Polymer gsd mJ/m2
Temperature°C Reference
PVF2-PVMKBlend (50: 50) 36.24 82 This Work
PEO 11.04 77 41PVMK 26.47 77 11Hg 200.00 20 42PVC 41.50 20 43PMMA 40.00 20 43
Polypropylene 28.90 44Polyurethane 20.30 44Polyethylene 33.10 44Cl-doped Ppy* 42.00 39NO3-doped
Ppy 61.20 40Fe (CN)6
4-dopedPpy 106.00 40
PTEF 19.00 20 43
* Ppy is polypyrrole.
Table VI. Dispersive Surface Energies of PVMK, 50 : 50 PVF2-PVMK Blend and gCH2
Temperature, °CPVMK Surface Energy,
gsd, mJ/m2
PVF2-PVMK SurfaceEnergy, gs
d, mJ/m2CH2 Surface Energy
gCH2, mJ/m2
82 28.46 36.24 16.1892 29.21 29.72 15.59
103 31.59 58.30 14.99113 31.50 64.30 14.40123 29.87 52.77 13.82133 28.52 59.23 13.26143 29.08 29.01 12.68152 23.92 18.90 12.14163 21.2 18.70 11.49
CHARACTERIZATION OF A PVF2-PVMK BLEND 1165

9. Morales, E.; Herrero, C. R.; Acosta, J. L. Polym Bull1992, 29, 401.
10. Qin, C.; Belfiore, L.A. Polym Prepr 1990, 3(1), 369.11. Al-Saigh, Z. Y. Polym Int 1996, 40, 25.12. Flory, P. J. Principles of Polymer Chemistry; Cor-
nell University: Ithaca, NY, 1953.13. Sanchez, I. C.; Lacombe, R. H. Macromolecules
1978, 11, 1145.14. Munk, P.; Hattam, P.; Abdual-Azim, A.; Du, Q.
Makromol Chem Macromol Symp 1990, 38, 205.15. Al-Saigh, Z. Y.; Munk, P. Macromolecules 1984, 17,
803.16. Pouchly, J.; Zivny, A.; Solc, K. J Polym Sci Part C,
1968, 23, 245.17. Fowkes, F. M.; Ind Eng Chem Prod Res Dev 1967,
56, 40.18. Balard, H.; Papirer, E. Prog Org Coat 1993, 22, 1.19. Papirer, E.; Schultz, J.; Turchi, C. Eur Polym J
1984, 20, 1155.20. Papirer, E.; Schultz, J.; Jagiello, J.; Baeza, R.;
Clauss, F. Chemically Modified Surfaces; Mottola,H. A.; Steinmetz, J. R., Eds.; Elsevier: Amsterdam,1992; p 334.
21. Kuczinski, J.; Papirer, E. Eur Polym J 1991, 27,653.
22. Papirer, E; Roland, P.; Nardin, M.; Balard, H. JColloid Interface Sci 1986, 113, 62.
23. Papirer, E.; Balard, H.; Vidal, A. Eur Polym J 1988,24, 783.
24. Ligner, G.; Vidal, A.; Balard, H.; Papirer, E. J Col-loid Interface Sci 1989, 133, 200.
25. Filbert, C. Diplome d’ Etudes Approfondies, Uni-versite de Haute Alsace, Mulhouse, France, 1989.
26. Papirer, E.; Ligner, G.; Vidal, A.; Balard, H.;Mauss, F. In Chemically Modified Oxide Surfaces;
Leyden, E.; Collins, W. T., Eds.; Gordon andBreach: New York, 1990; p 361.
27. Papirer, E.; Eckhardt, A.; Muller, F.; Yvon, J. JMater Sci 1990, 25, 5109.
28. Chehimi, M. M.; Abel, M.-L.; Pigois-Landureau, E.;Delamar, M. Synth Met 1993, 60, 183.
29. Card, T. W.; Al-Saigh, Z. Y.; Munk, P. J Chro-matogr 1984, 301, 261.
30. Petri, H. M.; Wolf, B. A. Macromolecules 1994, 27,2714.
31. Prolongo, M. G.; Masegosa, R. M.; Horta, A. Mac-romolecules 1989, 22, 4346.
32. Shi, Z. H.; Schreiber, H. P. Macromolecules 1989,24, 3522.
33. Lezcano, E. G.; Coll, C.S.; Prolongo, M. G. Macro-molecules 1992, 25, 6849.
34. Qin, C.; Pires, A. T. N.; Belfiore, L. A. Macromole-cules 1991, 24, 666.
35. Dorris, M. G.; Gray, D. G. J Colloid Interface Sci1980, 77, 353.
36. Schmitt, P.; Koerper, E.; Schultz, J.; Papirer, E.Chromatographia 1988, 25, 786.
37. Schmitt, P.; Koerper, E.; Schultz, J.; Papirer, E.Chromatographia 1988, 25, 786.
38. Saint Flour, C.; Papirer, E. J Colloid Interface Sci1983, 91, 69.
39. Chehimi, M. M.; Abel, M.-L.; Pigois-Landureau, E.;Delamar, M. Synth Met 1993, 60, 183.
40. Chehimi, M. M.; Pigois-Landureau, E.; Delamar,M. M. J Chim Phys 1992, 89, 1173.
41. Al-Saigh, Z. Y. Polymer 1999, 40, 3479.42. Fowkes, F. M. Ind Eng Chem 1964, 561, 40.43. Wu, S. Polymer Interface and Adhesion; Marcel
Dekker: New York, 1982.44. Bonnerup, C.; Gatenholm, P. J Adhes Sci Technol
1993, 7, 247.
1166 AL-GHAMDI AND AL-SAIGH















![Pore Structure Characterization of Poly(vinylidene ...carbonlett.org/Upload/files/CARBONLETT/[236-242]-07.pdf · Pore Structure Characterization of Poly(vinylidene chloride)-](https://img.pdfslide.net/doc/110x75/5c361c8209d3f2fc4d8b79cf/pore-structure-characterization-of-polyvinylidene-236-242-07pdf-pore.jpg)













