Embed Size (px)
Citation preview

Journal of the Neurological Sciences 315 (2012) 160–163
Contents lists available at SciVerse ScienceDirect
Journal of the Neurological Sciences
j ourna l homepage: www.e lsev ie r .com/ locate / jns
Short communication
Mitochondrial recessive ataxia syndrome mimicking dominant spinocerebellar ataxia
Eino J.H. Palin a, Anna H. Hakonen a, Mari Korpela b, Anders Paetau c, Anu Suomalainen a,b,⁎a Research Programs Unit, Molecular Neurology, University of Helsinki, Helsinki, Finlandb Department of Neurology, Helsinki University Central Hospital, Helsinki, Finlandc Department of Pathology, Helsinki University Central Hospital, Helsinki, Finland
⁎ Corresponding author at: Biomedicum-Helsinki, ReseNeurology, r. c523B, P.O. Box 63, University of HelsTel.: +358 947171965; fax: +358 947171964.
E-mail addresses: [email protected] (E.J.H. Palin)(A.H. Hakonen), [email protected] (M. Korpela), [email protected] (A. Suomalainen).
0022-510X/$ – see front matter © 2011 Elsevier B.V. Aldoi:10.1016/j.jns.2011.11.028
a b s t r a c t
a r t i c l e i n f oArticle history:Received 8 September 2011Received in revised form 16 November 2011Accepted 22 November 2011Available online 12 December 2011
Keywords:MIRASPOLGSpinocerebellar ataxiamtDNA
We studied the genetic background of a family with SCA, showing dominant inheritance and anticipation.Muscle histology, POLG1 gene sequence, neuropathology and mitochondrial DNA analyses in a mother anda son showed typical findings for a mitochondrial disorder, and both were shown to be homozygous for a re-cessive POLG1mutation, underlying mitochondrial recessive ataxia syndrome, MIRAS. The healthy father wasa heterozygous carrier for the same mutation. Recessively inherited MIRAS mutations should be tested indominantly inherited SCAs cases of unknown cause, as the high carrier frequency of MIRAS may result intwo independent introductions of the mutant allele in the family and thereby mimic dominant inheritance.
© 2011 Elsevier B.V. All rights reserved.
1. Introduction
Autosomal dominant cerebellar ataxias [ADCA or spinocerebellarataxias (SCAs)]— are the most common subgroup of dominant hered-itary ataxias [1], which are most often caused by polyglutamineexpansions. However, diagnosis is typically based on clinical charac-teristics, family history and brain imaging; genetic diagnosis isfound in a minority of cases [2]. SCAs show wide clinical variability,but unsteady ataxic gait, clumsiness and dysarthria, beginning typi-cally at 20's or 30's, are their common hallmarks. Anticipation inage of disease onset is typical for disorders with polyglutamineexpansion.
Mitochondrial dysfunction has shown to be an important cause ofSCAs, often combined with sensory neuropathy and epilepsy. Frie-dreich's ataxia is caused by recessive frataxin mutations, associatedwith defective mitochondrial iron metabolism [3], whereas mito-chondrial recessive ataxia syndrome (MIRAS; or MSCA-E; mitochon-drial SCA with epilepsy) and infantile-onset SCA are caused byrecessive nuclear gene mutations in mitochondrial DNA (mtDNA)maintenance proteins [4–7]. Here we present a mitochondrial SCA
arch Programs Unit, Molecularinki, 00290 Helsinki, Finland.
, [email protected]@hus.fi (A. Paetau),
l rights reserved.
family, with MIRAS-like symptoms, but autosomal dominant-like in-heritance pattern and age of onset suggestive of anticipation.
2. Materials and methods
2.1. Patients
The study was approved by Helsinki University Central HospitalEthical Review Board, and the subjects gave their written informedconsent.
P1 (mother) developed gait disturbance by the age of 35, progres-sing to increasing clumsiness in lower extremities, dysarthria, diplo-pia, and occasional amnesia at the age of 44. She developed ataxia,slight polyneuropathy, and external ophthalmoplegia. At the ageof 46 she had slightly increased plasma creatine kinase levels(178–261 U/l, reference 0–150 U/l) and symmetrical cerebellar pe-duncular white matter signal intensity increase in brain MRI. Thefirst epileptic seizure, requiring treatment by general anesthesia, oc-curred at the age of 55, after which she was hospitalized permanently.From her 30's, she received psychiatric care due to anxiety and de-pression. A neuropsychological examination revealed decrease in vi-sual reasoning and memory functions. She deceased at the age of 56due to pneumonia and pulmonary embolism.
P2 (Father) is a 63-year-old male, with no history of neurologicalsymptoms, but who has type-2 diabetes and hypertension.
P3 (Son) is a 41 year old male, who had gait disturbance sincechildhood. Early onset suggested anticipation. In his 20's he devel-oped photophobia and general clumsiness and benign paroxysmalpositional vertigo. From the age of 37 he has had unspecific sensory
161E.J.H. Palin et al. / Journal of the Neurological Sciences 315 (2012) 160–163
polyneuropathy, confirmed by electromyography. He had severalsimple partial seizures at the age of 39, and has mild anxiety and de-pression. Occasionally he has had increased levels of plasma creatinekinase (336–571 U/l, reference 50–400 U/l) and plasma gamma-glutamyl transpeptidase (131 U/l, reference 10–80 U/l). Brain MRIrevealed symmetrical increased white-matter signal intensities incerebellar peduncles.
P3 has a healthy brother.No SCA mutation screening was done on either patient, as this was
not in routine clinical use at the time of diagnosis. No consanguinitywas reported by the family members.
2.2. DNA analysis
DNA extraction from muscle samples, POLG1 mutation analysis,long-range PCR analysis of mtDNA, real-time quantitative PCR ofmtDNA and mtDNA point mutation load analysis were done as previ-ously [8, 9], with cytochrome B and APP genes used as controls inqPCR. Mutation frequency was determined scoring every mutation
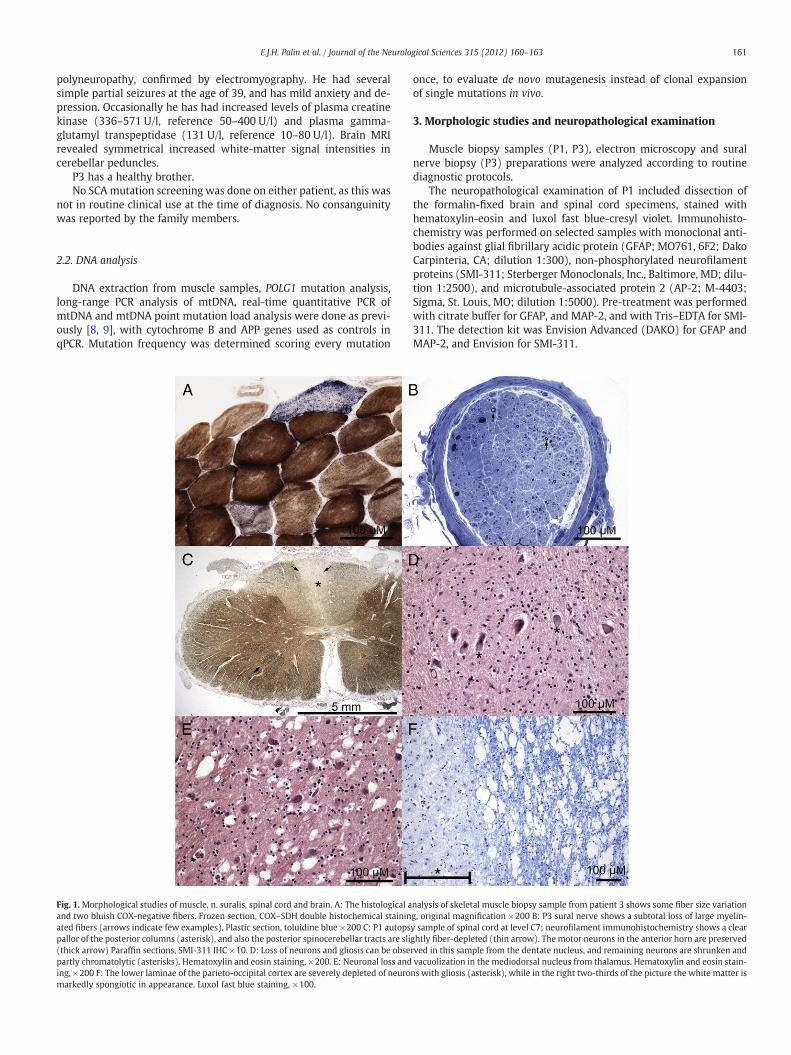
Fig. 1.Morphological studies of muscle, n. suralis, spinal cord and brain. A: The histological aand two bluish COX-negative fibers. Frozen section, COX–SDH double histochemical staininated fibers (arrows indicate few examples). Plastic section, toluidine blue ×200 C: P1 autopspallor of the posterior columns (asterisk), and also the posterior spinocerebellar tracts are sli(thick arrow) Paraffin sections, SMI-311 IHC ×10. D: Loss of neurons and gliosis can be obsepartly chromatolytic (asterisks). Hematoxylin and eosin staining, ×200. E: Neuronal loss anding, ×200 F: The lower laminae of the parieto-occipital cortex are severely depleted of neuromarkedly spongiotic in appearance. Luxol fast blue staining, ×100.
once, to evaluate de novo mutagenesis instead of clonal expansionof single mutations in vivo.
3. Morphologic studies and neuropathological examination
Muscle biopsy samples (P1, P3), electron microscopy and suralnerve biopsy (P3) preparations were analyzed according to routinediagnostic protocols.
The neuropathological examination of P1 included dissection ofthe formalin-fixed brain and spinal cord specimens, stained withhematoxylin-eosin and luxol fast blue-cresyl violet. Immunohisto-chemistry was performed on selected samples with monoclonal anti-bodies against glial fibrillary acidic protein (GFAP; MO761, 6F2; DakoCarpinteria, CA; dilution 1:300), non-phosphorylated neurofilamentproteins (SMI-311; Sterberger Monoclonals, Inc., Baltimore, MD; dilu-tion 1:2500), and microtubule-associated protein 2 (AP-2; M-4403;Sigma, St. Louis, MO; dilution 1:5000). Pre-treatment was performedwith citrate buffer for GFAP, and MAP-2, and with Tris–EDTA for SMI-311. The detection kit was Envision Advanced (DAKO) for GFAP andMAP-2, and Envision for SMI-311.
nalysis of skeletal muscle biopsy sample from patient 3 shows some fiber size variationg, original magnification ×200 B: P3 sural nerve shows a subtotal loss of large myelin-y sample of spinal cord at level C7; neurofilament immunohistochemistry shows a clearghtly fiber-depleted (thin arrow). The motor neurons in the anterior horn are preservedrved in this sample from the dentate nucleus, and remaining neurons are shrunken andvacuolization in the mediodorsal nucleus from thalamus. Hematoxylin and eosin stain-ns with gliosis (asterisk), while in the right two-thirds of the picture the white matter is

Fig. 2. Mitochondrial DNA integrity. A: MtDNA copy number in skeletal muscle sam-ples, relative to nuclear gene APP, percentage. C1, 72 year old healthy female; C2, a52 year old male with autosomal dominant progressive external ophthalmoplegy,caused by mutation in POLG1; P1 and P3 MIRAS patients. Quantitative PCR analysis,mtDNA value compared to single-copy nuclear gene APP. Values relative to C1. B:MtDNA point mutation load per 10,000 bp, skeletal muscle. Samples as in A. MtDNAsubstitution mutations are shown in light gray, and insertion/deletion mutations indark gray. On average, we analyzed ~20,000 nt for D-loop region and 25,000 nt for cy-tochrome B region per patient.
162 E.J.H. Palin et al. / Journal of the Neurological Sciences 315 (2012) 160–163
4. Results
4.1. Sequencing of POLG1
We sequenced all exons in POLG1 from P1, P2 and P3. Both P1(mother) and P3(son) were homozygous for the mutations c.2243G→C in cis with c.3428A→G causing W748S and E1143Gamino acid changes in the polypeptide (from now on the mutationis called W748S+E1143G). P2 (father), who had no neurologicalsymptoms was heterozygous for W748S+E1143G.
4.2. Morphologic studies and neuropathological findings
The muscle samples from P1 and P3 exhibited similar morpholo-gy: minor variation in fiber size and a few COX-negative fibers(Fig. 1A). In electron microscopy performed on P1's muscle, mainlysubsarcolemmal mitochondria were increased in number with somesize-variation, but no ‘parking lot’ inclusions (data not shown). Thesural nerve from P3 demonstrated a subtotal loss of large myelinatedfibers; severe axonal neuropathy (Fig. 1B). The posterior columns ofspinal cord of P1 (especially the gracile) and the posterior spinocere-bellar tracts were atrophic (Fig. 1C). Clarke's dorsal nuclei showedsome neuronal loss, while anterior horns or intermediolateral col-umns were spared. Cerebellum showed slight, patchy dropout ofPurkinje cells, but the dentate nucleus was severely atrophic(Fig. 1D). Neuronal loss was seen in the inferior olives, substantianigra and the mediodorsal thalamic nucleus, the latter showing alsospongiotic vacuolization, suggestive for transsynaptic degeneration(Fig. 1E). Especially the parieto-occipital region of the subcorticalwhite matter was spongiotic, with cortical neuronal loss and gliosis(Fig. 1F).
4.3. MtDNA analyses
Muscle mtDNA amount of P1 and P3 was reduced, 33% (±15.9 SD)and 69% (±16.1 SD), respectively, when compared to healthy controlmuscle (Fig. 2A), but no indication of multiple mtDNA deletions wasseen in long-range PCR, designed to enrich mtDNA deleted forms(data not shown). MtDNA variant load in D-loop region was higherin P1, but not in P3 (Fig. 2B). P1 and P3 shared some D-loop variants,which were not seen in healthy controls or in mtDNA variant data-bases, suggesting maternal inheritance. Cytochrome-b gene showedsimilar amount of variants in the patients and the control subject.
5. Discussion
MIRAS is a common cause of inherited ataxia in Europe, with car-rier frequencies of the common mutations (leading to amino acidchanges W748S+E1143G or A467T) ranging from 0.6% of the popu-lation in Belgium to 0.8% in Finland or 2% in Norway, but with limitedamount or no patients in Italy and UK [4, 6, 7, 10, 11] Here we presenta family in which the mother and son had SCA, consistent with dom-inant inheritance pattern. The son had an earlier age of disease-onsetthan his mother, interpreted as dominant SCA with anticipation. Afew histological changes suggestive for a mitochondrial disorderand epileptic attacks, unusual for SCA, led to testing of MIRAS muta-tion. Both the mother and the son were shown to carry a homozygousmutation of recessive MIRAS. The surprising dominant-like inheri-tance of a recessive disorder could have been explained by uniparen-tal disomy, heterozygote manifestation of the mother, or by secondintroduction of a POLG1 mutation into the family, which is a welldocumented incident in neurological diseases [12]. Indeed, the fatherwas shown to be heterozygous for the same change. This finding em-phasizes the importance of testing MIRAS mutations even in familiesshowing dominant-like SCA, as the high population frequency of
POLG1 mutations may result in several introductions of mutations inapparently non-consanguineous families [13].
Anticipation is a typical feature of polyglutamine repeat-expansiondisorders, including those causing dominant SCAs [1]. Such ananticipation-like situation has been reported previously in a familywith a mitochondrial myopathy, Parkinsonism and early menopause,caused by a dominant POLG1 mutation [8, 14]. Anticipation-like phe-nomenon could be explained by a diagnostic bias: a patient, whoseparent suffered from a progressive neurological disorder, may haveattracted medical attention for mild symptoms, and got an early diag-nosis. However, in the case of POLG1mutations, affecting mtDNA syn-thesis, mtDNA point mutations could accumulate in the ovum of theaffected mother, which could be transmitted to the offspring and af-fect the manifestation of the phenotype. We did not, however, detectincreased point mutation load in the son of this pedigree, suggestingcontribution of other protective or predisposing factors underlyingvariability in age of manifestation. We conclude that the anticipationseen in the P3 is probably due to diagnostic bias.
The neuropathologic pattern of changes in the deceased patientindicated highly specific sensory nervous system manifestation: Se-verely affected peripheral sensory nerves and ascending tracts, withmore recent changes observed centrally, perhaps representing trans-synaptic degeneration. This raises a possibility that the initial symp-toms would be linked to degeneration of the longest sensory axons,which would then affect the sensory tract in a retrograde manner.

163E.J.H. Palin et al. / Journal of the Neurological Sciences 315 (2012) 160–163
In conclusion, recessive MIRAS may mimic dominant SCAs, due tohigh population frequency of the mutations. Correct diagnosis in thecase of MIRAS is especially important, as these patients may developepilepsy at any stage of the disease, and their valproate treatmentleads to life-threatening acute liver damage, requiring liver transplan-tation [15].
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
Wewish to thank Markus Innilä and Anu Harju for technical assis-tance, and the following funding sources that enabled the study:Sigrid Juselius Foundation, Academy of Finland, University ofHelsinki, Helsinki University Central Hospital (for AS) and The FinnishParkinson Foundation (for EP).
References
[1] Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions andbeyond. Lancet Neurol 2010;9:885–94.
[2] Fogel B, Perlman S. An approach to the patient with late-onset cerebellar ataxia.Nat Clin Pract Neurol 2006;2:629–35 quiz 1 p following 35–35.
[3] Pandolfo M, Pastore A. The pathogenesis of Friedreich ataxia and the structure andfunction of frataxin. J Neurol 2009;256(Suppl 1):9–17.
[4] Hakonen AH, Heiskanen S, Juvonen V, Lappalainen I, Luoma PT, Rantamaki M,et al. Mitochondrial DNA polymerase W748S mutation: a common cause of auto-somal recessive ataxia with ancient European origin. Am J Hum Genet 2005;77:430–41.
[5] Nikali K, Suomalainen A, Saharinen J, Kuokkanen M, Spelbrink JN, Lönnqvist T,et al. Infantile onset spinocerebellar ataxia is caused by recessive mutations in mi-tochondrial proteins Twinkle and Twinky. HumMol Genet 2005;14(20):2981–90.
[6] Van Goethem G, Luoma P, Rantamäki M, Al Memar A, Kaakkola S, Hackman P,et al. POLG mutations in neurodegenerative disorders with ataxia but no muscleinvolvement. Neurology 2004;63:1251–7.
[7] Winterthun S, Ferrari G, He L, Taylor RW, Zeviani M, Turnbull DM, et al. Autosomalrecessive mitochondrial ataxic syndrome due to mitochondrial polymerasegamma mutations. Neurology 2005;64:1204–8.
[8] Luoma P, Melberg A, Rinne JO, Kaukonen JA, Nupponen NN, Chalmers RM, et al.Parkinsonism, premature menopause, and mitochondrial DNA polymerasegamma mutations: clinical and molecular genetics study. Lancet 2004;364:875–82.
[9] Hakonen AH, Goffart S, Marjavaara S, Paetau A, Cooper H, Mattila K, et al. Infantile-onset spinocerebellar ataxia and mitochondrial recessive ataxia syndrome are as-sociated with neuronal complex I defect and mtDNA depletion. Hum Mol Genet2008;17:3822–35.
[10] Cagnoli C, Brussino A, Di Gregorio E, Caroppo P, Stola S, Dragone E, et al. Mutationsin the POLG1 gene are not a relevant cause of cerebellar ataxia in Italy. J Neurol2008;255:1079–80.
[11] Craig K, Ferrari G, Tiangyou W, Hudson G, Gellera C, Zeviani M, et al. The A467Tand W748S POLG substitutions are a rare cause of adult-onset ataxia in Europe.Brain : a journal of neurology 2007;130:E69 author reply E70.
[12] Lücking CB, Bonifati V, Periquet M, Vanacore N, Brice A, Meco G. Pseudo-dominantinheritance and exon 2 triplication in a family with parkin gene mutations. Neu-rology 2001;57:924–7.
[13] Kurt B, Jaeken J, Van Hove J, Lagae L, Löfgren A, Everman DB, et al. A novel POLGgene mutation in 4 children with Alpers-like hepatocerebral syndromes. Archivesof Neurology 2010;67:239–44.
[14] Melberg A, Arnell H, Dahl N, Stålberg E, Raininko R, Oldfors A, et al. Anticipation ofautosomal dominant progressive external ophthalmoplegia with hypogonadism.Muscle Nerve 1996;19:1561–9.
[15] Ferrari G, Lamantea E, Donati A, Filosto M, Briem E, Carrara F, et al. Infantile hepa-tocerebral syndromes associated with mutations in the mitochondrial DNApolymerase-gammaA. Brain 2005;128:723–31.





























